

Abstract
Background
Several neurotransmitter receptors activate signaling pathways that alter processing of the amyloid precursor protein (APP) into β-amyloid (Aβ). Serotonin signaling through a subset of serotonin receptors suppresses Aβ generation. We proposed that escitalopram, the most specific selective serotonin reuptake inhibitor (SSRI) that inhibits the serotonin transporter SERT, would suppress Aβ levels in mice.
Objectives
We hypothesized that acute treatment with escitalopram would reduce Aβ generation, which would be reflected chronically with a significant reduction in Aβ plaque load.
Methods
We performed in vivo microdialysis and in vivo 2-photon imaging to assess changes in brain interstitial fluid (ISF) Aβ and Aβ plaque size over time, respectively, in the APP/presenilin 1 mouse model of Alzheimer disease treated with vehicle or escitalopram. We also chronically treated mice with escitalopram to determine the effect on plaques histologically.
Results
Escitalopram acutely reduced ISF Aβ by 25% by increasing α-secretase cleavage of APP. Chronic administration of escitalopram significantly reduced plaque load by 28% and 34% at 2.5 and 5 mg/d, respectively. Escitalopram at 5 mg/kg did not remove existing plaques, but completely arrested individual plaque growth over time.
Conclusions
Escitalopram significantly reduced Aβ in mice, similar to previous findings in humans treated with acute dosing of an SSRI.
Alzheimer disease (AD) is the most common cause of dementia, with an incidence that doubles every 5 years after 65 years of age and typically leads to death within 7–8 years of diagnosis.1 The prevalence across all world regions is projected to increase dramatically in the next decades to 130 million patients by midcentury, unless preventive measures are developed.2 AD is characterized by the accumulation of β-amyloid (Aβ) plaques and oligomers as well as tau neurofibrillary tangles. Aggregation of the Aβ peptide is concentration-dependent, with high levels being much more likely to form higher-ordered, toxic species. Amyloid plaques and soluble Aβ oligomers exist within the brain extracellular space. Whereas the initial aggregation seed may form intracellularly or extracellularly,3,4 it appears that soluble Aβ within the brain extracellular space, or interstitial fluid (ISF), is one source that contributes to these toxic species.5,6 Consequently, mechanisms that regulate Aβ levels could be therapeutic targets to maintain low levels of the peptide to reduce or eliminate toxic Aβ species from the brain.
Aβ levels are regulated by multiple mechanisms. Synaptic transmission leads to Aβ generation at or near the presynaptic terminal, which is then released into the ISF.7–9 Activation of certain neurotransmitter receptors, such as M1 muscarinic acetylcholine receptors,10 NMDA receptors,11,12 and AMPA receptors,13,14 can alter amyloid precursor protein (APP) processing, which affects Aβ generation. Direct infusion of serotonin into the brain, or treatment with a selective serotonin reuptake inhibitor (SSRI) antidepressant, also decreases Aβ acutely in brain by reducing Aβ production with no obvious effect on Aβ clearance.15 Our group and others have found in mice and humans that NMDA and Gαs-coupled serotonin receptors 5-HT4, 5-HT6, and 5-HT7 activate the ERK signaling cascade,16 which increases α-secretase enzymatic activity to reduce Aβ levels.11,12,15–19
Long-term reductions in Aβ levels in many cases reduces the aggregation of peptide into plaques in mice and humans. Chronic SSRI treatment over the course of 4 months in APP/presenilin 1 (PS1) mice reduced Aβ plaque load in the hippocampus and cortex by 50%.15 Similar reductions in Aβ were demonstrated by SSRI treatment in the 3xTg AD mouse model.20 In a retrospective study, humans with a history of SSRI use to treat depression had less Pittsburgh compound B binding in the brain15 and acute studies in humans demonstrated a significant reduction in CSF Aβ in young, cognitively normal participants following a single dose of an SSRI. Similarly, in this issue of Neurology®, Sheline et al.21 demonstrate that subchronic dosing of escitalopram for 2 weeks or 8 weeks also suppresses CSF Aβ levels significantly.
SSRI antidepressants are commonly used Food and Drug Administration–approved drugs with a reasonable safety record with long-term use. Given that serotonin signaling is one synaptic mechanism that regulates Aβ, we proposed that the SSRI escitalopram, the most selective drug in this class for blocking serotonin reuptake as opposed to norepinephrine,22 would suppress brain Aβ levels and reduce plaque load in mice. In the current study, we determined the acute effects of escitalopram on APP processing and Aβ metabolism in APP transgenic mice using in vivo microdialysis. In addition, we prospectively determined the chronic effects of escitalopram on plaque burden and individual plaque growth over time.
Methods
Mouse studies
We bred APP/PS1+/− hemizygous mice23 to wild-type C3H/B6 mice (Jackson Labs, Bar Harbor, Maine). These mice harbored the PS1ΔE9 deletion and the human APP Swedish mutation, inserted into a single locus. Male and female littermate mice were equally distributed between all experimental groups. For multiphoton studies, 6-month-old mice were randomly entered into 3 study arms: (1) escitalopram 5 mg/kg, (2) escitalopram 2.5 mg/kg, or (3) vehicle (2% DMSO in normal saline), injected intraperitoneally, QD for 28 days. Multiphoton imaging was performed on the day before the first injection, and after the last injection (see below).
Standard protocol approvals
All experimental procedures involving animals were performed in accordance with guidelines established by the Animal Studies Committee at Washington University.
In vivo microdialysis
In vivo microdialysis to assess brain ISF Aβ in the hippocampus of awake, freely moving APP/PS1+/− mice was performed similarly to previously described methods.15,24 This technique samples soluble molecules within the extracellular fluid that are smaller than 38 kilodaltons, the molecular weight cutoff of the probe membrane. Aβ capable of entering the probe has been termed exchangeable Aβ or eAβ.
Under isoflurane volatile anesthetic, guide cannula (BR-style, Bioanalytical Systems, Indianapolis, IN) were cemented above the left hippocampus (3.1 mm behind bregma, 2.5 mm lateral to midline, and 1.2 mm below dura at a 12° angle). Two-millimeter microdialysis probes were inserted through the guides so the membrane was contained entirely within the hippocampus (BR-2, 30-kilodalton MWCO membrane, Bioanalytical Systems). Microdialysis perfusion buffer was artificial CSF (perfusion buffer in mM: 1.3 CaCl2, 1.2 MgSO4, 3 KCl, 0.4 KH2PO4, 25 NaHCO3, and 122 NaCl, pH 7.35) containing 15% bovine serum albumin (Sigma, St. Louis, MO) that was filtered through a 0.22 μM membrane. Flow rate was a constant 1.0 μL/minute. Samples were collected every 60–90 minutes with a refrigerated fraction collector into polypropylene tubes and assessed for Aβx-40 by ELISA at the completion of each experiment. Basal concentrations of ISF Aβ were defined as the mean concentration of Aβ over the 9 hours preceding drug treatment. Once basal ISF Aβ concentrations were established, APP/PS1 mice were administered either vehicle (phosphate-buffered saline [PBS]) or citalopram (R, S mixture), escitalopram 2.5 mg/kg, escitalopram 5 mg/kg, or R-citalopram. After drug treatment, ISF Aβ concentrations were sampled every 60 minutes for an additional 24 hours, then all samples were assayed for Aβ concentration by sandwich ELISA. All ISF Aβ concentrations were normalized to the basal Aβ concentration in each mouse.
Secretase assays
Enzymatic activity of α-secretase and β-secretase was measured using FRET-based cleavage assays (R&D Systems, Minneapolis, MN). Hippocampal tissue was isolated from 3-month-old APP/PS1 hemizygous mice treated with escitalopram (5 mg/kg) or vehicle (PBS) and killed 8 hours later. Tissue was lysed in Cell Extraction Buffer (Novagen, Madison, WI) by mechanical homogenization. Cell extracts were incubated with secretase-specific peptides conjugated to the reporter molecules EDANS and DABCYL for 15–30 minutes. EDANS fluorescence was read on a Cytation 5-microtiter plate reader (BioTek, Winooski, VT).
Aβ sandwich ELISA
ISF Aβ concentrations were assessed using sandwich ELISAs as described.15 Briefly, a mouse anti-Aβ40 antibody (mHJ2) or mouse anti-Aβ42 antibody (mHJ7.4) was used to capture and a biotinylated central domain antibody (mHJ5.1) was used to detect, followed by streptavidin-poly-HRP-40 (Fitzgerald Industries, Concord, MA). All ELISA assays were developed using Super Slow ELISA TMB (Sigma) and absorbance read on a Bio-Tek Epoch plate reader at 650 nm. The standard curve for each assay utilized synthetic human Aβ1-40 peptide (American Peptide, Sunnyvale, CA).
Chronic escitalopram administration
Beginning at 4 months of age, APP-PS1 hemizygous female mice were administered normal drinking water (vehicle) or 5 mg/kg/d escitalopram or 2.5 mg/kg/d escitalopram in drinking water for a total of 4 months. Littermate mice were divided equally between the treatment groups. Mice were housed 3–5 animals per cage. Volume of water drunk per cage and animal body weight were tracked throughout the study and did not differ between cages or treatment groups. At 8 months of age, mice were killed and CSF drawn from the cisterna magna25 followed by transcardial perfusion of chilled PBS with 0.3% heparin. One hemisphere of the brain was postfixed overnight in 4% paraformaldehyde followed by processing for histologic analysis of Aβ plaque burden. The other hemisphere had the hippocampus and cortex micro-dissected, then snap frozen on dry ice for future biochemical analysis of brain Aβ levels.
Tissue extraction of Aβ
To evaluate various pools of brain Aβ, we performed a sequential extraction of tissue with PBS, 1% triton X-100 in PBS, then 5M guanidine to grossly assess the extracellular-enriched fraction, the membrane-bound and intracellular fraction, and the insoluble fraction, respectively. All lysis buffers were chilled to 4°C and contained protease inhibitors without EDTA. Tissue was lysed at a 1:10 wet weight/volume ratio. PBS and Triton X-100 extractions were performed by mechanical dounce homogenization while the guanidine extraction was performed with sonication to maximally solubilize remaining Aβ within the tissue. Tissue was spun in a microcentrifuge at 21,000 g for 15 minutes at 4°C following each extraction. Aβ1-40 and Aβ1-42 were measured by sandwich ELISA and normalized to total protein in each sample, as determined by a micro-BCA assay (Peirce, Rockford, IL).
Quantitative analyses of Aβ deposition
Brain hemispheres were placed in 30% sucrose before freezing and cutting on a freezing sliding microtome. Serial coronal sections of the brain at 50-μm intervals were collected from the rostral anterior commissure to caudal hippocampus as landmarks. Sections were stained with biotinylated human-specific anti-Aβ antibody, mHJ3.4 (a gift from Dr. David Holtzman, Washington University, St. Louis, MO). Stained brain sections were scanned with a NanoZoomer slide scanner (Hamamatsu Photonics, Japan). For quantitative analyses of mHJ3.4-biotin staining, scanned images were exported with NDP viewer software (Hamamatsu Photonics) and converted to 8-bit grayscale using ACDSee Pro 3 software (ACD Systems, Victoria, Canada). Converted images were thresholded to highlight plaques and then analyzed by the Analyze Particles function in ImageJ software (NIH).26 Identified objects after thresholding were individually inspected to confirm the object as a plaque or not. Three brain sections per mouse, each separated by 300 μm, were used for quantification. These sections correspond roughly to sections at Bregma −1.7, −2.0, and −2.3 mm in the mouse brain atlas. The average of 3 sections was used to represent a plaque load for each mouse. For analysis of Aβ plaque in the cortex, the cortex immediately dorsal to the hippocampus was assessed. All analyses were performed in a blinded manner by 2 independent researchers. The data that were statistically analyzed and plotted represents the average plaque load per section between the 2 researchers.
Cranial window surgery
Thinned-skull cranial windows were prepared on the day of the first multiphoton imaging session as described previously.6 Briefly, mice were anesthetized under volatile isoflurane and the skin and periosteum were removed to expose the skull. A high-speed drill and microsurgical blade (Surgistar) were used to thin the skull until transparent and flexible. Two thinned-skull windows (each 0.8–1.0 mm in diameter) over each hemisphere were prepared in each animal.
In vivo multiphoton microscopy
Six-month-old APP/PS1 mice were treated with escitalopram (n = 4 per group) or vehicle (n = 5) for 28 days. To quantify growth of individual amyloid plaques, longitudinally intravital multiphoton imaging was used, as described previously.27 Briefly, mice were injected intraperitoneally with the fluorescent amyloid-binding compound methoxy-X04 (5 mg/mL) 24 hours prior to each imaging session.28 Animals were mounted on a custom-built stereotaxic apparatus and a small ring of molten bone wax was applied to the skull surrounding the perimeter of the window to create a water immersion chamber. The cranial window was centered under the objective lens on a 2-photon microscope (LSM 510 META NLO system [Carl Zeiss Inc., Oberkochen, Germany] with a Cameleon Ti: Sapphire laser [Coherent Inc., Santa Clara, CA]). Two-photon fluorescence was generated with 750 nm excitation and fluorescence emission was detected at 435–485 nm. A 10× water-immersion objective (numerical aperture 0.33, Zeiss) was used to create a site map during initial imaging and a 40× water-immersion objective (numerical aperture 0.75, Zeiss) was used for high-resolution quantification of individual amyloid plaques. Incremental z-stack image series (step distance = 10 and 5 μm under 10× and 40× objectives, respectively) were acquired from the skull surface to approximately 200 μm into cortex.
To determine the effect of escitalopram on amyloid plaque formation and growth, the same sites for each animal were imaged on day 0 and day 28. Collapsed z-stack images for each individual plaque were captured. The plaque radius and intensity was determined using SigmaScan Pro Image Analysis Software (Systat Software) with a preset threshold (threshold = mean + 4 × SD). Plaques were excluded from analysis if they were located at the edge of the window, exhibited fluorescence intensity less than the mean intensity of an adjacent background region, or if the image acquisition was affected by motion artifacts from heartbeat or respiration.
Statistical analyses for mouse studies
Statistically significant differences were determined using Prism statistical software (Graph Prism 8, San Diego, CA). All data were presented as mean ± SEM. For data analysis, 1-way analysis of variance with the Dunnett multiple comparison post test was applied. All p values resulted from 2-sided statistical tests and statistical significance was set at *p < 0.05, **p < 0.01, and ***p < 0.001.
Data availability
Raw and analyzed data will be shared upon written request to the corresponding author from any qualified investigator.
Results
Comparison of citalopram, escitalopram, and R-citalopram in brain ISF Aβ levels in mice
Citalopram is a mixture of 2 enantiomers: S-citalopram, the active molecule, and R-citalopram, the inactive molecule. Escitalopram consists only of the active enantiomer, whereas R-citalopram should be an inactive component of citalopram. In theory, citalopram should have comparable efficacies as half the dose of escitalopram. We sought to determine the effect of each of these compounds on ISF Aβ42 levels at comparable “active doses.”
Mice treated with citalopram (R,S mixture) and escitalopram had a gradual reduction in ISF Aβ40 and Aβ42 whereas R-citalopram and vehicle produced no change (figure 1, A and B). At the end of the 24 hours sampling, 10 mg/kg citalopram significantly reduced ISF Aβ40 by 25.6% ± 6.1% (mean ± SEM; n = 4, p = 0.006, 95% CI 7.9%, 41.6%), while 5 mg/kg escitalopram, a comparable dose of the active enantiomer, reduced ISF Aβ40 by 20.2% ± 4.1% (n = 6, p = 0.014, 95% CI 4.1%, 34.7%). Citalopram and escitalopram were not significantly different from each other, however (p = 0.668, 95% CI −21.6%, 11.0%). R-citalopram, the inactive enantiomer of citalopram, had no effect on change in ISF Aβ levels (n = 5, p = 0.923, 95% CI −24.9%, 16.8%) compared to vehicle (figure 1B). Aβ42 was altered similarly to Aβ40 (figure 1B). Citalopram reduced Aβ42 by 21.8% ± 2.8% (n = 5, p < 0.0001, 95% CI 12.1%, 29.7%) while escitalopram reduced Aβ42 by 26.8% ± 2.5% (n = 5, p < 0.0001, 95% CI 17.1%, 34.8%). Each drug had a similar effect on both Aβ40 and Aβ42 levels.
Figure 1. Escitalopram acutely reduces brain interstitial fluid (ISF) β-amyloid (Aβ) levels over a 24-hour period.

(A) Mice treated with citalopram (R,S mixture) and escitalopram had a gradual reduction in ISF Aβ whereas R-citalopram and vehicle produced no change. At the end of 24 hours sampling, citalopram significantly reduced ISF Aβ40 by 25.3% ± 6.1% (mean ± SEM; n = 4, p < 0.01) and escitalopram reduced Aβ by 20.2% ± 4.1% (n = 6, p < 0.01). R-Citalopram had no effect on ISF Aβ levels (n = 5) compared to vehicle. (B) Citalopram reduced Aβ42 by 21.8% ± 2.8% (n = 5, p < 0.0001) while escitalopram reduced ISF Aβ42 by 26.8 ± 2.5% (n = 5, p < 0.0001). R-citalopram had no effect on Aβ42 levels. Data are presented as mean ± SEM. All p values resulted from 2-sided statistical tests and statistical significance was set at *p < 0.05, **p < 0.01, and ***p < 0.001.
Escitalopram increases α-secretase activity
Three-month-old APP/PS1 mice were administered 5 mg/kg escitalopram or vehicle, killed 8 hours later, and brains processed for biochemistry. The hippocampus was lysed and then measured by enzymatic activity of α-secretase and β-secretase using a FRET-based cleavage assay. Following escitalopram treatment, α-secretase activity significantly increased by 51.1% ± 3.3% (n = 8, p < 0.001, 95% CI 19.8%, 42.1%), whereas β-secretase activity only changed by 10.8% ± 3.5% (n = 8, p = 0.212, 95% CI −4.0%, 16.6%) compared to vehicle-treated mice (figure 2).
Figure 2. Escitalopram increased α-secretase enzymatic activity.
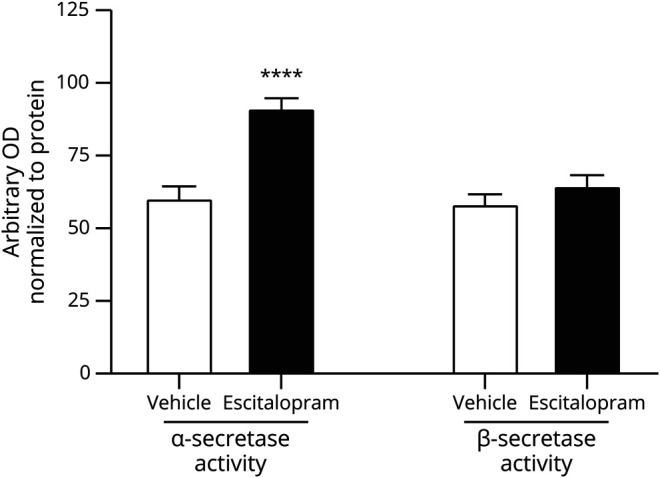
Amyloid precursor protein (APP)/presenilin 1 (PS1) mice were administered 5 mg/kg escitalopram, then killed 8 hours later. Enzymatic activity of α-secretase and β-secretase were measured in the hippocampus. α-Secretase activity significantly increased by 51.1% ± 3.3% (n = 8, p < 0.001) while β-secretase activity changed insignificantly by 10.8% ± 3.5% (n = 8, p = 0.212) compared to vehicle-treated mice. Data presented as mean ± SEM.
Chronic escitalopram effect on brain plaque load in mice
APP/PS1 mice, starting at 4 months of age prior to plaque pathology, were administered plain water or escitalopram at 2.5 mg/kg and 5 mg/kg in drinking water for 4 months. At 8 months of age, mice were killed to assess Aβ aggregation. As assessed histologically, escitalopram at both doses significantly reduced plaque burden within the brains of these mice compared to littermate controls that drank only water (figure 3A with representative histology images). Hippocampal plaque load was significantly reduced by 28.7% ± 0.05% (p = 0.029, 95% CI 0.022%, 0.43%) and 34.4% ± 0.05% (p = 0.009, 95% CI 0.067%, 0.48%) for escitalopram 2.5 and 5 mg/d, respectively (figure 3A, n = 8 per group). The observed reduction in the escitalopram 2.5 and 5 mg/kg groups did not differ significantly from each other (p = 0.849, 95% CI −0.16%, 0.25%).
Figure 3. Chronic administration of escitalopram reduced β-amyloid (Aβ) plaque load.

(A) Hippocampal plaque load for both Aβ40 and Aβ42 were significantly reduced for escitalopram 2.5 mg/d and 5 mg/d, respectively (n = 8 per group). Representative images at 5× magnification. (B) Levels of insoluble Aβ40 and Aβ42 were significantly reduced in the 2.5 mg/kg escitalopram group and in the 5 mg/kg escitalopram group, respectively (n = 8 per group). All p values resulted from 2-sided statistical tests and statistical significance was set at *p < 0.05, **p < 0.01, and ***p < 0.001.
The contralateral brain regions were processed for biochemical analysis of Aβ protein levels. PBS-soluble and Triton X-100-soluble Aβ levels did not change significantly in escitalopram-treated mice (data not shown); however, levels of insoluble Aβ40 and Aβ42 extracted with 5M guanidine were significantly reduced by 42.7% ± 5.6% (p = 0.005, 95% CI 40.4%, 241.5%) and 56.3% ± 3.4% (p = 0.041, 95% CI 11.4%, 588.1%) in the 2.5 mg/kg escitalopram group, respectively, and by 43.8% ± 6.9% (p < 0.001, 95% CI 85.3%, 286.4%) and 69.0% ± 3.0% (p < 0.001, 95% CI 322.7%, 899.4%) in the 5 mg/kg escitalopram group, respectively (figure 3B, n = 8 per group). Though plaque load histologically and insoluble Aβ biochemically measure overlapping pools of Aβ, we propose that the discrepancy in significance between the analyses is that by its nature quantification by histology is less precise than biochemically using an ELISA. There was no change in CSF Aβ40 and Aβ42 levels at 5 mg/kg escitalopram (Aβ40 p = 0.357, 95% CI −15.6%, 65.5%; Aβ42 p = 0.215, 95% CI −28.0%, 30.4%).
Escitalopram arrests individual plaque growth assessed by in vivo 2-photon microscopy
During a 28-day interval, plaques in vehicle-treated mice grew 116.8 μm2, while plaques in mice treated with 2.5 mg/kg escitalopram grew an average 65.5 μm2. In both vehicle and 2.5 mg groups, the average size of plaques was significantly larger at day 28 compared to day 0 (p < 0.001, 0 days vehicle 341 μm2 vs 28 days vehicle 458 μm2 [382, 534]; and p < 0.05, 2.5 mg/kg escitalopram 0 days 387 μm2 vs 2.5 mg/kg escitalopram 28 days 452 μm2 [348, 556], paired t test), respectively. However, in mice treated with 5 mg/kg escitalopram, mean plaque size at day 28 was not significantly different from that at day 0 (p = 0.83, 0 days 404 μm2 vs 28 days 399 μm2 [343, 456], paired t test), suggesting that plaques did not grow. The rate of plaque growth in the vehicle group was significantly higher than that in the 5 mg/kg escitalopram group (p = 0.002, vehicle 190% [140, 241] vs 5 mg/kg escitalopram 119% [101, 137]), but not that of the 2.5 mg/kg escitalopram group (p = 0.391, vehicle 190% [140, 241] vs 2.5 mg/kg escitalopram 140% [110,170]; figure 4, A and B). Furthermore, treatment with 5 mg/kg escitalopram reduced the number of newly appearing plaques compared with vehicle (p = 0.03, vehicle 9.6 plaques [8.2, 10.9] vs 5 mg/kg escitalopram 3 plaques [1.34, 4.68]), but had no effect on the number of disappearing plaques (p = 0.47, vehicle 1.1 [0.224, 2.04] vs 5 mg/kg escitalopram 2.2 [1.29, 3.09]), whereas 2.5 mg/kg escitalopram treatment had no effect on either the number of appearing plaques compared with vehicle (p = 0.07, vehicle 9.6 [8.2, 10.9] vs 2.5 mg/kg escitalopram 7.6 [4.26, 11]) or the number of disappearing plaques (p = 0.47, vehicle 1.1 [0.224, 2.04] vs 2.5 mg/kg escitalopram 1.0 [0.144, 1.89]; figure 4C). There was not sufficient cerebral amyloid angiopathy in the image captures to analyze vascular Aβ changes in response to escitalopram.
Figure 4. Escitalopram over 4 weeks completely arrested individual plaque growth.

Six-month-old amyloid precursor protein (APP)/presenilin 1 (PS1) mice were treated with escitalopram at doses of 2.5 or 5 mg/kg per day or vehicle IP for 28 days (vehicle: n = 11 mice, 104 plaques; escitalopram 2.5 mg/kg: n = 5 mice, 40 plaques; escitalopram 5 mg/kg: n = 4 mice, 66 plaques) and imaged using 2-photon microscopy. (A) Representative multiphoton micrographs (20× magnification) of individual amyloid plaques in the cortex of APP/PS1 mice (before [0 days] and 28 days after treatment. P = plaque. (B) 5 mg/kg escitalopram and 10 mg/kg escitalopram attenuated the growth of preexisting plaques. (C) Escitalopram 5 mg/kg and escitalopram 10 mg/kg reduced the appearance of new plaques. All p values resulted from 1-way analysis of variance with Dunnett multiple comparison post statistical tests and statistical significance was set at *p < 0.05, **p < 0.01, and ***p < 0.001.
Discussion
Rodent studies showed that the chronic administration of escitalopram 5 mg/kg/d blocked the growth of existing amyloid plaques and significantly reduced the appearance of new plaques, compared to vehicle-treated animals. This dose is roughly comparable to a 24 mg dose in a 60 kg human,29 similar to the 10 mg and 20 mg doses often prescribed to individuals. Importantly, the rates that a bolus dose of drug is absorbed when given intraperitoneally in mice vs when given orally in humans will differ, so direct comparison of dosages can be difficult. Further, escitalopram had a rapid and sustained acute effect on ISF Aβ40 and Aβ42 levels. Importantly, studies showed that there was no effect of R-citalopram, the inactive enantiomer; only an effect of S-citalopram (also known as escitalopram). This complements a study in cognitively normal, elderly individuals whereby 8 weeks of escitalopram reduced CSF Aβ levels by almost 10%.30
Escitalopram had similar effects on both ISF Aβ40 and Aβ42 in terms of both magnitude of change as well as kinetics. Interestingly, in humans, escitalopram reduced CSF Aβ42 but not Aβ40.21 Treatment with the SSRI increased enzymatic activity of α-secretase without a change in β-secretase activity. This is similar to previous findings demonstrating that serotonin receptors activate the ERK signaling pathway to increase nonamyloidogenic processing of APP.15 Interestingly, there was a dose-dependent change in Aβ in only select measures; overall plaque load, as assessed histologically, did not differ between the 2.5 and 5.0 mg/kg treatment groups, whereas guanidine-extractable Aβ42 levels were different, with a trend for a difference in Aβ40. While plaque load and an insoluble extraction of tissue measure similar pools of Aβ, guanidine-extractable Aβ in tissue consists of more than just plaque (e.g., Aβ inside small vesicles that are not easily lysed in detergent), which appears to be more mobile and reduced in escitalopram-treated mice.
Chronic reductions in Aβ levels, either by suppressing production or enhancing clearance, can have profound influences on the amount of Aβ that accumulates as plaques, as well as associated synaptic and neuritic pathologies. Formation of these toxic conformations from the normal soluble Aβ species is concentration-dependent, with high Aβ concentrations much more likely to aggregate than lower Aβ concentrations.29 Even small decreases in Aβ concentration have been associated with a substantial lowering of plaque burden. A 12%–25% decrease in soluble Aβ concentration in animal models produced a substantial plaque reduction.6,31 In humans, Aβ levels that are 20%–50% higher, such as in familial AD or Down syndrome, produce pathology and symptoms decades before patients with sporadic AD. In our prior studies with the SSRI citalopram, soluble Aβ concentration was reduced by 25% with a concurrent 78% reduction in new plaque formation in mice,19 whereas in our current rodent studies, we found a reduction in new plaque formation of 34%. So while reducing Aβ will likely limit plaques within the brain, a complete reduction may not be required.
Independent of medications, depression appears to increase the risk of developing AD.32–34 Several retrospective studies have found that SSRIs reduce the risk of AD symptoms in individuals with depression,35–37 while individuals with depression taking SSRIs are at lower risk of AD compared to untreated individuals with depression, but still are at greater risk compared to controls without depression. Though acute studies in humans have demonstrated that SSRIs can reduce Aβ levels,19 prospective trials are still necessary to determine whether SSRIs, or other serotonin modulators, are directly responsible for the reduction in amyloid plaque load in human brain.
Numerous clinical trials using γ-secretase inhibitors, β-secretase inhibitors, and anti-Aβ vaccinations have been conducted to suppress Aβ levels. The initial trials treated individuals who already exhibited mild to moderate behavioral symptoms of AD and failed to produce a measureable improvement in cognition. Later studies treated individuals who were cognitively normal, but at risk of developing AD within a few years. These trials also failed to show cognitive benefit. Given the failure of numerous Aβ targeted therapeutics, it remains possible that targeting Aβ may not be beneficial to AD. Alternatively, however, these studies may have failed because they were started too late in the course of the disease; Aβ and tau had already caused substantial cell death, which could not be reversed. As such, if targeting Aβ as a therapeutic intervention, treatment may need to be started much earlier, meaning that individuals may need to be on a drug for a decade or more before changes could be detected. A drug will need to be safe and tolerated for that period of time. SSRIs are not innocuous, but millions of individuals take them for decades with generally manageable side effects. Of the 15 serotonin receptor subtypes, only 5-HT4, 5-HT6, and 5-HT7 receptors are responsible for suppressing Aβ levels following SSRI administration16; thus it may be feasible to develop more targeted agents to lower Aβ levels.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- APP
amyloid precursor protein
- ISF
interstitial fluid
- PBS
phosphate-buffered saline
- PS1
presenilin 1
- SSRI
selective serotonin reuptake inhibitor
Appendix. Authors
Footnotes
See page 858
Study funding
This work was supported by NIH/NINDS R01 NS094692 (J.R.C.), NIH/NIA P50 AG00568 (J.R.C.), NIH/NINDS P01 NS074969 (J.R.C.), NIH/NIA R01 AG064902 (J.R.C.), R01 NS094692 (J.-M.L.), R21 AG05533301 (J.-M.L.), and R01 AG041502 (Y.I.S.) and by the Knight Alzheimer's Disease Research Center at Washington University. The Article Processing Charge was funded by NIH/NINDS R01 NS094692 (J.R.C.).
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/Nhttps://n.neurology.org/lookup/doi/10.1212/WNL.0000000000010733 for full disclosures.
References
- 1.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003;60:1119–1122. [DOI] [PubMed] [Google Scholar]
- 2.Shah H, Albanese E, Duggan C, et al. Research priorities to reduce the global burden of dementia by 2025. Lancet Neurol 2016;15:1285–1294. [DOI] [PubMed] [Google Scholar]
- 3.Gouras GK, Tsai J, Naslund J, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 2000;156:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer-Luehmann M, Stalder M, Herzig MC, et al. Extracellular amyloid formation and associated pathology in neural grafts. Nat Neurosci 2003;6:370–377. [DOI] [PubMed] [Google Scholar]
- 5.Bero AW, Yan P, Roh JH, et al. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci 2011;14:750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan P, Bero AW, Cirrito JR, et al. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci 2009;29:10706–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cirrito JR, Deane R, Fagan AM, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 2005;115:3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cirrito JR, Stewart F, Mennerick S, Holtzman D. Synaptic transmission dynamically modulates interstitial fluid amyloid-β levels. In: Selkoe D, Triller A, Christen Y, eds. Synaptic Plasticity and the Mechanism of Alzheimer's Disease. Berlin: Springer; 2008. [Google Scholar]
- 9.Kamenetz F, Tomita T, Hsieh H, et al. APP processing and synaptic function. Neuron 2003;37:925–937. [DOI] [PubMed] [Google Scholar]
- 10.Nitsch RM, Deng M, Tennis M, Schoenfeld D, Growdon JH. The selective muscarinic M1 agonist AF102B decreases levels of total Abeta in cerebrospinal fluid of patients with Alzheimer's disease. Ann Neurol 2000;48:913–918. [PubMed] [Google Scholar]
- 11.Verges DK, Restivo JL, Goebel WD, Holtzman DM, Cirrito JR. Opposing synaptic regulation of amyloid-beta metabolism by NMDA receptors in vivo. J Neurosci 2011;31:11328–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoey SE, Williams RJ, Perkinton MS. Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J Neurosci 2009;29:4442–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hettinger JC, Lee H, Bu G, Holtzman DM, Cirrito JR. AMPA-ergic regulation of amyloid-beta levels in an Alzheimer's disease mouse model. Mol Neurodegener 2018;13:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoey SE, Buonocore F, Cox CJ, Hammond VJ, Perkinton MS, Williams RJ. AMPA receptor activation promotes non-amyloidogenic amyloid precursor protein processing and suppresses neuronal amyloid-beta production. PLoS One 2013;8:e78155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cirrito JR, Disabato BM, Restivo JL, et al. Serotonin signaling is associated with lower amyloid-beta levels and plaques in transgenic mice and humans. Proc Natl Acad Sci USA 2011;108:14968–14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisher JR, Wallace CE, Tripoli DL, Sheline YI, Cirrito JR. Redundant Gs-coupled serotonin receptors regulate amyloid-beta metabolism in vivo. Mol Neurodegener 2016;11:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nitsch RM, Deng M, Growdon JH, Wurtman RJ. Serotonin 5-HT2a and 5-HT2c receptors stimulate amyloid precursor protein ectodomain secretion. J Biol Chem 1996;271:4188–4194. [DOI] [PubMed] [Google Scholar]
- 18.Lesne S, Ali C, Gabriel C, et al. NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J Neurosci 2005;25:9367–9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheline YI, West T, Yarasheski K, et al. An antidepressant decreases CSF Abeta production in healthy individuals and in transgenic AD mice. Sci Transl Med 2014;6:236re234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson RL, Guo Z, Halagappa VM, et al. Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp Neurol 2007;205:166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheline YI, Snider BJ, Beer JC, et al. Effect of escitalopram dose and treatment duration on CSF Aβ levels in healthy older adults: a controlled clinical trial. Neurology 2020;95:e2658–e2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez C, Reines EH, Montgomery SA. A comparative review of escitalopram, paroxetine, and sertraline: are they all alike? Int Clin Psychopharmacol 2014;29:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savonenko A, Xu GM, Melnikova T, et al. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer's disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis 2005;18:602–617. [DOI] [PubMed] [Google Scholar]
- 24.Cirrito JR, May PC, O'Dell MA, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci 2003;23:8844–8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeMattos RB, Bales KR, Parsadanian M, et al. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer's disease. J Neurochem 2002;81:229–236. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Onstead L, Randle S, et al. Abeta40 inhibits amyloid deposition in vivo. J Neurosci 2007;27:627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price JL, Morris JC. Tangles and plaques in nondemented aging and "preclinical" Alzheimer's disease. Ann Neurol 1999;45:358–368. [DOI] [PubMed] [Google Scholar]
- 28.Klunk WE, Bacskai BJ, Mathis CA, et al. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol 2002;61:797–805. [DOI] [PubMed] [Google Scholar]
- 29.Lomakin A, Teplow DB, Kirschner DA, Benedek GB. Kinetic theory of fibrillogenesis of amyloid beta-protein. Proc Natl Acad Sci USA 1997;94:7942–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheline YI, Yu M. Linking antidepressant performance with pain network connectivity. Lancet Psychiatry 2019;6:635–636. [DOI] [PubMed] [Google Scholar]
- 31.McConlogue L, Buttini M, Anderson JP, et al. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem 2007;282:26326–26334. [DOI] [PubMed] [Google Scholar]
- 32.Geerlings MI, den Heijer T, Koudstaal PJ, Hofman A, Breteler MM. History of depression, depressive symptoms, and medial temporal lobe atrophy and the risk of Alzheimer disease. Neurology 2008;70:1258–1264. [DOI] [PubMed] [Google Scholar]
- 33.Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D. Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry 2006;63:530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green RC, Cupples LA, Kurz A, et al. Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch Neurol 2003;60:753–759. [DOI] [PubMed] [Google Scholar]
- 35.Geda YE. Blowing hot and cold over depression and cognitive impairment. Neurology 2010;75:12–14. [DOI] [PubMed] [Google Scholar]
- 36.Norum JH, Hart K, Levy FO. Ras-dependent ERK activation by the human G(s)-coupled serotonin receptors 5-HT4(b) and 5-HT7(a). J Biol Chem 2003;278:3098–3104. [DOI] [PubMed] [Google Scholar]
- 37.St George-Hyslop PH, Morris JC. Will anti-amyloid therapies work for Alzheimer's disease? Lancet 2008;372:180–182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw and analyzed data will be shared upon written request to the corresponding author from any qualified investigator.