

Abstract
Objective
To determine whether treatment with escitalopram compared with placebo would lower CSF β-amyloid 42 (Aβ42) levels.
Rationale
Serotonin signaling suppresses Aβ42 in animal models of Alzheimer disease (AD) and young healthy humans. In a prospective study in older adults, we examined dose and treatment duration effects of escitalopram.
Methods
Using lumbar punctures to sample CSF levels before and after a course of escitalopram treatment, cognitively normal older adults (n = 114) were assigned to placebo, 20 mg escitalopram × 2 weeks, 20 mg escitalopram × 8 weeks, or 30 mg escitalopram × 8 weeks; CSF sampled pretreatment and posttreatment and within-subject percent change in Aβ42 was used as the primary outcome in subsequent analyses.
Results
An overall 9.4% greater reduction in CSF Aβ42 was found in escitalopram-treated compared with placebo-treated groups (p < 0.001, 95% confidence interval [CI] 4.9%–14.2%, d = 0.81). Positive baseline Aβ status (CSF Aβ42 levels <250 pg/mL) was associated with smaller Aβ42 reduction (p = 0.006, 95% CI −16.7% to 0.5%, d = −0.52) compared with negative baseline amyloid status (CSF Aβ42 levels >250 pg/mL).
Conclusions
Short-term longitudinal doses of escitalopram decreased CSF Aβ42 in cognitively normal older adults, the target group for AD prevention.
Clinicaltrials.gov identifier
Classification of evidence
This study provides Class II evidence that for cognitively normal older adults, escitalopram decreases CSF Aβ42.
The ongoing challenge of Alzheimer disease (AD) has motivated major efforts to disentangle the pathophysiology and discover treatments for this disorder. Several studies have demonstrated a link among serotonin, AD, and β-amyloid (Aβ), manifested by a reduction of serotonin receptor levels1,2 in AD and decreased Aβ production in vitro with activation of the serotonin receptor. Treatment with serotonin or serotonin receptor agonists activates intracellular signaling cascades.3–5 In vivo modulation of serotonin levels shows a consistent effect, with single-dose treatment using selective serotonin reuptake inhibitors (SSRIs) in young APP/PS1 mice reducing Aβ levels in the brain interstitial fluid (ISF) by 25% and chronic SSRI treatment reducing Aβ plaque load by 50%.6 The effects of serotonin on lowering Aβ were demonstrated in humans in retrospective6 and prospective studies.7 Retrospective studies showed that people with a history of SSRI treatment for depression had a significant reduction in amyloid plaque binding in PET imaging studies.6 Acute human prospective CSF studies7 following a single dose of an SSRI demonstrated a significant reduction of newly formed Aβ in CSF in young cognitively normal people. In the current study, we tested the hypothesis that treatment with the SSRI escitalopram would prospectively lower Aβ levels in cognitively normal older adults.
Methods
Standard protocol approvals, registrations, and patient consents
This study was approved by institutional review boards at the University of Pennsylvania (Penn) and Washington University (WU) and all participants gave written consent.
Primary research question
We conducted a prospective study to examine dose and treatment duration effects of escitalopram, using lumbar punctures (LPs) to sample CSF levels before and after a course of escitalopram treatment. Our primary hypothesis was that compared with placebo, the overall treatment effect of escitalopram is to lower CSF Aβ42 levels. We included 2 different doses of escitalopram and 2 different treatment durations. In post hoc comparisons, we further divided participants by CSF baseline status into amyloid-positive and amyloid-negative based on baseline CSF Aβ42 measurements8 and examined how escitalopram-lowering effects on Aβ42 differed across amyloid status.
Classification of evidence
This study provides Class II evidence that for cognitively normal older adults, escitalopram decreases CSF Aβ42.
CSF studies
A double-blind, placebo-controlled study was performed to determine the CNS effect of 2 doses and 2 different durations of treatment with escitalopram. Participants were recruited from the community by public service announcements and advertisements and were paid for their time. Healthy 50- to 84-year-old male and female volunteers were invited to participate, screened, and enrolled. Exclusion criteria included any serious or unstable medical illness including neurologic disease and use of antidepressants within the past 2 years. All participants were screened by a physician at Penn (Y.I.S.) or WU (B.J.S.) to exclude neurologic and psychiatric illness, including major depression (all Hamilton Depression Rating Scale9 scores ≤7), and screened by the Clinical Dementia Rating scale (CDR) to exclude those who did not score within the cognitively normal range (CDR = 0).10 Following screening, participants underwent LP and then were assigned to 1 of 4 treatment arms: 20 mg/d escitalopram × 2 weeks; 20 mg/d escitalopram × 8 weeks; 30 mg/d escitalopram × 8 weeks; or placebo (×2 weeks or 8 weeks), depending on the treatment arm being compared. Treatment group was assigned by a clinical coordinator not involved in the study. All CSF samples were collected in the morning after an overnight fast. Only water was permitted until the LP was completed. The LPs were performed with a 22–24 gauge Sprotte spinal needle. Approximately 20 mL of CSF was collected into two 14-mL polypropylene tubes, combined into one 30-mL polypropylene tube, gently mixed, and then 0.5 mL aliquots were pipetted into labeled polypropylene tubes on ice and frozen at −80°C. Aβ42 levels were measured using Luminex (Austin, TX) immunoassay (LMX) and mass spectrometry (MS). A second LP was performed within a day of finishing the treatment course.
CSF immunoassay
We used both LMX and MS to determine Aβ levels; LMX was the primary method to determine outcome. The multiplex xMAP Luminex platform was used with Fujirebio (INNO-BIA AlzBio 3; Ghent, Belgium) immunoassay kit–based reagents. The kit reagents include a mixture of 3 xMAP color-coded carboxylated microspheres, each containing a bead set coupled with well-characterized capture monoclonal antibodies (see reference 8 for details). All samples were analyzed in duplicate in each run. A result was defined as the arithmetic mean of duplicate results. All assays were performed at the Penn Biomarker Research Laboratory. Assays are specific for Aβ1–42 (4D7A3; bead region 56) and were used with a vial with analyte-specific biotinylated detector monoclonal antibodies (3D6 for Aβ1–42). Monoclonal antibodies are used in the assay with production processes assurance of lot-to-lot consistency. In the Alzheimer's Disease Neuroimaging Initiative cross-site test–retest performance across a range of concentration values, the % coefficient of variation [CV] was 5.7% for Aβ1–42.11 The CSF quality control pools were included in each analytical run to help determine acceptability of each run. Full details of the implementation of the INNO-BIA AlzBio3 immunoassay on the Luminex analytical platform are described elsewhere.12
MS determination of CSF Aβ42 and Aβ40 in CSF using 2D ultraperformance liquid chromatography (UPLC) tandem MS
Human synthetic peptides Aβ1–42 and Aβ1–40 together with their internal standards (isotopically labeled, nitrogen-15) Aβ1–42 and Aβ1–40, all with trifluoroacetate as counter ion, were purchased from r-Peptide (Watkinsville, GA). Accuracy of the weight of this material is based on aminoacid analysis of the standards used for UPLC analyses of each lot of material. Spiking solutions for each calibrator and quality control (QC) sample contain both Aβ peptides in one mixture. Following pretreatment with high-concentration guanidine hydrochloride, to release Aβ peptides from complexes with other proteins and Aβ oligomers, samples were loaded on Oasis MCX micro-elution mixed-bed ion exchange.
2D-UPLC-MS-MS system characteristics
Analysis of Aβ peptides was carried out on Xevo TQ-S triple quadrupole mass spectrometer (Waters, Milford, MA) with an electrospray probe in an ion source, interfaced with an ACQUITY Ultra Performance LC system (Waters) including sample manager, 2 pumps, and a column oven with 2 columns.
Precision and accuracy performance of MS
For 3 QC samples shown to perform equivalently to human CSF (Korecka 2014), the mean accuracy for Aβ1–42 and Aβ1–40 ranged from 91.7% to 100% and mean precision (% CV) ranged from 5.2% to 10.1% over a range of 5 to 9 days and 3 different lots of the standard Aβ1–42 and one for Aβ1–40. Further details are shown in reference 13. All MS analyses were performed at the Penn Biomarker Research Laboratory.
Plasma and CSF determination of escitalopram levels
All CSF and plasma levels of escitalopram and 2 metabolites (desmethylcitalopram [desmet] and didesmethylcitalopram [dides]) were measured using a previously published liquid chromatographic method.14 A fluorescence detector set at 235 nm excit and 300 nm emiss produced clean chromatograms with no interference from other drugs or endogenous material. The method was validated from 300 ng/mL to the lower limit of quantitation (2.5 ng/mL) resulting in an intraday variation of no more than 10% for all 3 compounds at 7 concentrations of the calibration curve. Interday variation did not exceed 6.8% for the 3 quality controls over 16 consecutive days. Change in CSF Aβ1–42 was correlated with CSF escitalopram levels using a Pearson correlation.
Study design
In designing the study, to estimate power to detect a 15% decrease in Aβ concentrations estimated from our mouse data (equivalent to 10 mg escitalopram in the current study), a t test was used to estimate the power to compare the mean differences of Aβ concentrations between the placebo group and escitalopram group. To achieve 81% power with a 2-tailed t test (unpaired) and 5% significance level, a mean difference of 0.38 (or 15% reduction from the placebo group to the treatment group) can be detected with SD of 0.39 for a sample of 36 participants, where 18 participants are in the placebo group and 18 in the escitalopram group. To increase power, the 10 mg escitalopram group was dropped and the doses increased to 20 mg. Initially there had been 3 groups planned and recruited (placebo, 20 mg escitalopram × 2 weeks, and 20 mg escitalopram × 8 weeks), with a sample size of 30 per group, allowing the detection of a moderate effect using a one-way analysis of variance with 3 groups. However, in order to investigate a dose relationship as well as a time duration effect, we added a fourth group, 30 mg escitalopram × 8 weeks, beginning in the third year of the 5-year grant and prior to any data analysis. We recruited this fourth group at a 2:1 ratio, in order to over-recruit for this group, with 2 participants recruited into the 30 mg group for every one participant split among the other 3 groups. Thus, while the study initially randomized participants, the overall design was a cohort exposure study.
Statistical analyses of CSF data
Demographics and baseline amyloid status were summarized by treatment group. χ2 tests were used to assess differences in sex and race distributions across treatment groups. Differences in age between treatment and placebo groups, as well as across treatment doses, were assessed using 95% confidence intervals (CIs) for the mean in each group. For each outcome measure (Aβ42 from Luminex), percent change was computed as 100 × (follow-up − baseline)/baseline. Overall average percent changes in the placebo and combined doses treatment group were reported with standard errors. Analysis of covariance (ANCOVA) was used to study the overall effect of escitalopram on average percent change for each outcome measure, combining all doses into a single treatment group and adjusting for age and sex as covariates. The main effect of baseline amyloid status and the interaction between status and treatment group were included in the analysis of Aβ42 to study whether baseline amyloid status was a moderator of the effect of escitalopram. In addition to percent changes, we also studied the raw differences in Aβ42 by treatment and amyloid status, subtracting the pretreatment values from the post-treatment values. The motivation for this was to ensure the effects were not driven by including the baseline value in the denominator of the outcome. Baseline amyloid status was defined as amyloid-positive (+) (CSF Aβ42 <250 pg/mL) or -negative (−) (CSF Aβ42 ≥250 pg/mL).8 Next, ANCOVA was used to study the effect of escitalopram dose on average percent change for each outcome measure, adjusting for age and sex as covariates. An F test was performed to test the global null of equivalent means among the placebo and 3 escitalopram dose groups. Following rejection of the global null, all pairwise comparisons among the 4 groups were performed using Tukey honest significant difference to control the family-wise type I error at 5%. The same analyses were conducted for the MS results. The model for Aβ40 included main effects of treatment group, age, and sex. All analyses included the available data. There was no interpolation for missing data. To help quantify effect sizes of interest for significant group mean differences, unadjusted Cohen d was calculated.
Data availability
Data have been deposited in aggregate into Clintrials.gov (NCT02161458), including study protocol and statistical analysis plan. In addition, upon request, individual de-identified CSF data will be shared. This will include sex, age binned in 5-year increments, baseline and post-treatment CSF Aβ42 levels, and treatment modality. Data will be accessible to principal investigators at academic institutions or industry affiliates with a research protocol for analyzing the data. Requests must be made in writing to the corresponding author and will be reviewed by the Penn–WU publications committee. Data will be available for 3 years.
Results
Escitalopram effect on Aβ42 levels in CSF
There were 241 people screened for the study. Following screening, 117 participants met criteria, consented, and were enrolled into one of the 4 treatment arms. Three participants were excluded from analyses due to (1) missing baseline Aβ42 level (male, 20 mg 8 weeks), (2) missing follow-up Aβ42 level (male, placebo), or (3) missing age (male, placebo). See the table for demographic characteristics and baseline amyloid status by treatment group. As shown in the table, sex (number of women) (χ2 = 3.31, df = 3, p = 0.35) and race (White, Black, or Other) (χ2 = 10.75, df = 9, p = 0.29) did not differ among the groups. Mean age did not differ between the placebo and escitalopram group when all doses were combined, but the CI for mean age of the 20 mg/8 weeks group was nonoverlapping with the 20 mg/2 weeks group, which motivated us to adjust for age in all analyses. As discussed below, in post hoc analyses, baseline brain amyloid (+) status (low CSF Aβ42 levels) differed among the groups. Among 8 participants, there were 13 side effects during the study: among participants on escitalopram there were 2 with increased sleepiness, 1 with both increased sleepiness and chest pain, 1 with decreased libido, 1 with headache, dizziness, and syncopal episode during LP, 1 with laryngitis, and 1 with nausea. Among participants on placebo, 1 had change in sleep pattern, sexual side effects, and dry mouth.
Table.
Treatment

Luminex determination of CSF Aβ42 levels
There was a significant effect of escitalopram when combining all doses (F1,109 = 16.64, p < 0.001), with the escitalopram group showing a greater percent reduction in CSF Aβ42 concentrations than the placebo group. Overall, there was a 9.4% point difference (95% CI 4.9%–14.2%, d = 0.81) in reduction between the treated and placebo groups. The combined doses escitalopram group experienced a 6.0% (SEM 1.2%) decrease on average and the placebo group experienced a 3.5% (SEM 2.2%) increase in Aβ42 concentrations (figure 1A). The global null test for equivalence of means across placebo and all doses was rejected (F3,107 = 6.26, p < 0.001), and post hoc pairwise tests indicated that the placebo group had a significantly different mean percent change than the 20 mg/8 weeks and 30 mg/8 weeks groups; the 95% Tukey CIs for the difference (placebo − escitalopram) were 3.4–19.5, adjusted p = 0.002, d = 0.94, and 2.0–17.1, adjusted p = 0.007, d = 0.69, respectively, reflecting the observed mean decrease in Aβ42 percent change in the 2 escitalopram groups and mean increase in the placebo group. Results are displayed in figure 1B. The treatment group by baseline amyloid status interaction was not significant (F1,107 = 0.216, p = 0.643), indicating that amyloid status was not a treatment effect modifier. However, on average, the baseline amyloid-positive group had significantly less percent reduction in Aβ42, i.e., poorer outcome, than the amyloid-negative group (F1,107 = 7.97, p = 0.006, 95% CI −16.7% to 0.5%, d = −0.52).
Figure 1. Effect of escitalopram on CSF β-amyloid (Aβ)42.
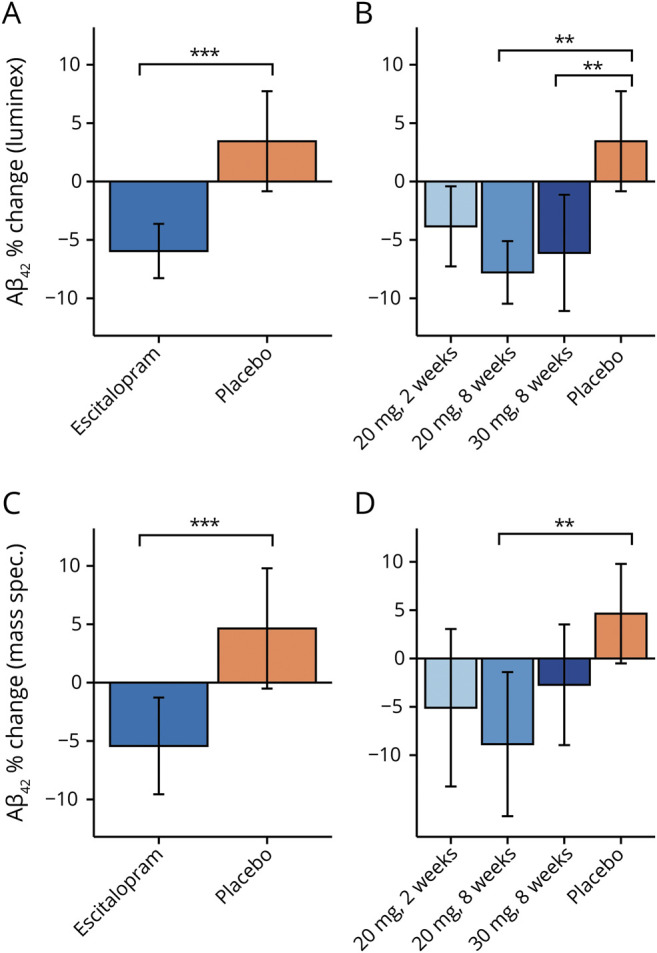
(A) Using the Luminex assay, the combined escitalopram group demonstrated a greater percent reduction in Aβ42 than the placebo group, with an overall difference between groups of 9.4% (F1,109 = 16.64, p < 0.001, 95% confidence interval [CI] 4.9%–14.2%, d = 0.81). On average, there was a 6.0% (SEM 1.2%) reduction of Aβ42 in the escitalopram-treated group (blue bar) vs a 3.5% (SEM 2.2%) increase in the placebo-treated group (orange bar). (B) The 20 mg 2 weeks escitalopram group (light blue bar) had a 3.8% (SEM 1.8%) reduction in Aβ42 on average (not significant compared to placebo). For the 20 mg × 8 weeks group (medium blue bar), there was a 7.8% (SEM 1.4%) average reduction. For the 30 mg × 8 weeks escitalopram group (dark blue bar), the average reduction was 6.1% (SEM 2.5%), compared with an average increase of 3.5% (2.2%) in the placebo-treated group (orange bar; Tukey adjusted p = 0.002, 95% CI 3.4%–19.5%, d = 0.94 and p = 0.007, 95% CI 2.0%–17.1%, d = 0.69, comparing placebo to the 20 mg × 8 weeks and 30 mg × 8 weeks groups, respectively). (C) Using mass spectrometry, similar results were found: overall difference in average percent change between groups of 11.1% (F1,109 = 11.593, p < 0.001, 95% CI [18.5%–4.9%, d = 0.60). See Results for details. (D) For the 20 mg × 8 weeks group (medium blue bar), there was a 10.5% (SEM 3.6%) average reduction that was significantly different than the placebo group (Tukey adjusted p = 0.001, 95% CI 5.5%–29.0%, d = 0.89). See Results for other details. All p values resulted from 2-sided statistical tests and statistical significance was set at *p < 0.05, **p < 0.01, and ***p < 0.001.
Effects of escitalopram on CSF Aβ42 levels determined by MS
Overall, there was an 11.1 percentage point difference in treatment effect between the escitalopram and placebo-treated groups, similar to the results using the Luminex assay. The placebo group experienced a 5.2% (SEM 2.6%) increase in Aβ42 and the combined doses escitalopram group experienced a 5.9% (SEM 2.1%) decrease on average. ANCOVA indicated that the escitalopram group had significantly greater percent reduction in Aβ42 than the placebo group (F1,109 = 11.593, p < 0.001, 95% CI 18.5%–4.9%, d = 0.60) (figure 1C). The global null test for equivalence of means across placebo and all doses was rejected (F3,107 = 5.41, p = 0.002), and post hoc pairwise tests indicated that the 20 mg/8 weeks escitalopram group had a significantly greater mean percent decrease than the placebo group (average difference with placebo group = 10.5%, SEM 3.6%). The 95% Tukey CI for the difference (placebo − escitalopram) was 5.5%–29.0% (adjusted p = 0.001, d = 0.89), reflecting the observed mean decrease in the treatment group and increase in Aβ42 percent change in the placebo group (adjusted p = 0.001). Results are displayed in figure 1D. The interaction term between treatment and amyloid status was not significant (F1,107 = 0.095, p = 0.758), but in the model without the interaction term, the effect of amyloid status at baseline was significant (F1,108 = 4.92, p = 0.029, 95% CI 1.0–17.2, d = 0.35), with the amyloid-positive group experiencing less percent reduction in Aβ42.
Effect of baseline amyloid status on escitalopram treatment effect
On average (figure 2), amyloid-negative escitalopram-treated (combined doses) patients had a 7.3% (SEM 1.2%) decrease; amyloid-positive escitalopram-treated patients had a 2.1% (SEM 2.9%) decrease; amyloid-negative placebo-treated patients had a 1.2% (SEM 2.2%) increase; amyloid-positive placebo-treated patients had a 9.5% (SEM 5.5%) increase. Participants who were amyloid positive at baseline (pooled) had significantly less percent reduction in Aβ42 (F1,107 = 7.968, p = 0.006, 95% CI −16.7% to −0.5%, d = −0.52), controlling for age, sex, and treatment group (not shown).
Figure 2. Baseline amyloid status changes the effect of escitalopram on reduction in CSF β-amyloid (Aβ)42.
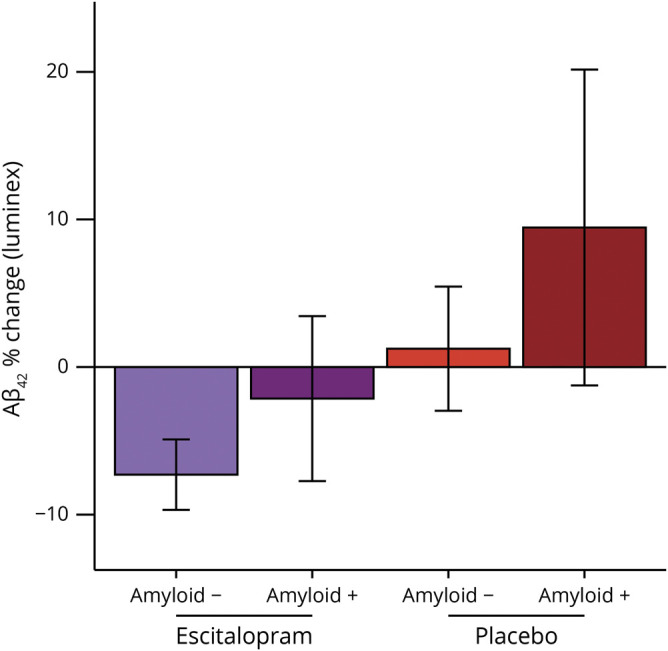
Participants who were amyloid positive at baseline (pooled) had significantly less percent reduction in Aβ42 (F1,107 = 7.968, p = 0.006, 95% confidence interval −16.7% to −0.5%, d = −0.52), controlling for age, sex, and treatment group (not pictured). On average (pictured), amyloid-negative escitalopram-treated (combined doses) patients had a 7.3% (SEM 1.2%) decrease; amyloid-positive escitalopram-treated patients had a 2.1% (SEM 2.9%) decrease; amyloid-negative placebo-treated patients had a 1.2% (SEM 2.2%) increase; amyloid-positive placebo-treated patients had a 9.5% (SEM 5.5%) increase.
Correlation between escitalopram levels and decrease in Aβ42
The correlation between plasma drug (escitalopram) levels and decrease in Aβ42 was not significant (n = 54, r = −0.05, p = 0.72). There was not a significant correlation between CSF drug levels and decrease in Aβ42 concentrations (n = 28, r = −0.25, p = 0.19). However, plasma and CSF escitalopram levels were significantly correlated (n = 28, r = 0.91, p < 0.001), supporting the presence of escitalopram in the CSF.
Escitalopram effect on Aβ40 levels in CSF
There were 20 participants missing baseline Aβ40 and 1 participant missing the follow-up Aβ40 measure. Using the complete cases with both measurements, the placebo group experienced a 3.7% (1.6%) increase in Aβ40 and the combined doses escitalopram group experienced a 0.7% (1.5%) decrease on average. The main effect of treatment group was not significant (F1,89 = 3.74, p = 0.057, 95% CI −10.0% to 0.1%, d = 0.39), controlling for age and sex. The global null test for equivalence of means across placebo and all doses was not rejected (F3,87 = 2.18, p = 0.096), so post hoc pairwise tests were not performed. The effect of Aβ on percent Aβ40 reduction was not significant (F1,87 = 0.01, p = 0.788, 95% CI −1.6% to 16.9%, d = 0.04).
Effect of escitalopram on Aβ42/Aβ40
The Aβ42/Aβ40 ratio has been suggested as superior to Aβ42 alone for identifying patients with AD and a recent review15 of the evidence concluded that the ratio provides the most sensitive and stable measure for predicting conversion to AD. We computed the effect of escitalopram on the Aβ 42/40 ratio. The escitalopram group (combining all doses) had a significantly greater decrease in Aβ 42/40 ratio compared to the placebo group (F1,89 = 7.39, p = 0.008, 95% CI 0.005–0.032, d = 0.61). The overall change in the escitalopram group was −0.015 (SEM 0.0005), while the placebo group had a change of 0.003 (SEM 0.0008).
Discussion
In a prospective study of CSF Aβ42 measurements, we show that escitalopram administered at 2 different doses (20 mg and 30 mg) and for either 2 weeks or 8 weeks was associated with a decrease in the level of CSF Aβ42 as measured by both the Luminex platform and by MS with a large effect size of 0.81 for the combined escitalopram doses in the Luminex determination. Importantly, this effect was seen in cognitively normal older adults (age >50 years), the target age group for an AD prevention strategy. Complementary rodent studies16 showed that the administration of escitalopram 5 mg/kg/d (roughly comparable to a 24 mg dose in a 60 kg human)17 blocked the growth of existing amyloid plaques and significantly reduced the appearance of new plaques, compared to vehicle-treated animals. Further, these companion rodent studies16 found an acute dose–response effect of escitalopram on brain ISF Aβ. These studies also showed that for the chiral drug citalopram, there was no effect of (R)-citalopram, the inactive enantiomer; only an effect of the (S)-citalopram enantiomer (also known as escitalopram) was noted. Chronic administration of escitalopram in rodent studies16 decreased the accumulation of amyloid plaque in hippocampus and cortex, similarly to effects we previously reported using citalopram.6,7 The mechanism for this reduction is through serotonin (5-hydroxytryptamine [5-HT]) receptor subtypes 5-HT4, 5-HT6, and 5-HT7, to activate a signaling pathway that culminates in an upregulation of α-secretase activity to suppress Aβ42 generation.18
Reducing Aβ42 concentrations and maintaining low concentrations with any agent, including SSRIs, may mitigate the deleterious effects of cortical Aβ42 deposition by limiting the rate of toxic Aβ species formation. As part of pathogenesis, early in the disease process soluble Aβ monomers aggregate into soluble Aβ oligomers and insoluble Aβ plaques, both with adverse effects on synaptic transmission and neuronal toxicity.19 The transformation of the normal soluble Aβ species into these toxic conformations is concentration dependent, with high Aβ concentrations more likely to aggregate than lower Aβ concentrations.20 In animal models, even small decreases (12%–25%) in Aβ42 concentration, as measured in the CSF, have been associated with a substantial lowering of plaque formation.21,22 It might seem paradoxical to lower CSF concentrations given that CSF Aβ42 is lower in AD and that lower CSF Aβ42 is correlated with higher brain plaque binding23 in PET studies. However, recent mouse and human studies24 using 13C in SILK (stable isotope labeling kinetics) technology visualized CSF Aβ incorporation into individual brain plaques, showing that it was concentration dependent. Further, individuals with autosomal dominant AD or Down syndrome who have a 20%–50%25 increase in Aβ42 levels develop AD pathology and symptoms up to 20–30 years before patients with typical sporadic AD.
Our previous human study found a 23% decrease in CSF Aβ concentration following acute single dose administration of 60 mg of the SSRI citalopram. In the current study, conducted in older adults rather than in a young population, the maximal decrease was only 10%. While both SSRIs (citalopram and escitalopram) have similar effects, the magnitude of those changes differed in the current study, perhaps due to study differences. First, the usual dose of escitalopram, 20 mg/d, used in the current study given the older age of participants, has a dose equivalence 30% less than the dose of citalopram used in the previous study. Second, among those who received treatment with the higher 30 mg/d dose (which resulted in less reduction in CSF Aβ concentration than the 20 mg/d dose), there were double the number of participants who were amyloid-positive at baseline in comparison to the placebo or 20 mg groups. The smaller decrease in Aβ may be due in part to the important effect we have shown in the current study of amyloid-positive status on decreasing the escitalopram effect on lowering Aβ. Thus in amyloid-positive participants who have a higher load of plaque Aβ, given that CSF Aβ and plaque Aβ are in equilibrium, this may reflect the buffering capacity of amyloid plaques on CSF Aβ,23 supported by a recent study revealing active changes in incorporation of Aβ into plaques.24 In future studies, this could be resolved by using 13C labeling techniques to label newly formed protein, as we had done previously7 to directly measure change in Aβ production. In the current study, however, we elected to sample CSF by LP determination, due to the greater burden in older patients of an indwelling catheter vs LP. An additional limitation of the study was the adaptive and nonrandom assignment to the treatment arms when we added a fourth arm, the 30 mg dose, which could have resulted in unmeasured confounding in the results, for example, the higher number of amyloid-positive participants in the 30 mg arm. Future studies will be needed to validate these SSRI effects. Finally, the expected relationship between plasma or CSF escitalopram levels with change in Aβ42 was not observed. One possible explanation is that the effect of SSRIs on Aβ is long lived and separated from minute-to-minute serotonin signaling, which could explain why there is not a 1:1 relationship between drug levels and Aβ levels. Another possibility is that the effect is not direct but rather mediated through another factor. One such factor that has been proposed is sleep modulating the Aβ42 levels,26 although participants in this trial did not report a change in sleep.
Development of safe and effective therapeutic approaches that can reduce Aβ levels even modestly may prevent pathologic amyloid accumulation and the subsequent cascade of neuronal damage, which could prevent or slow progression to symptomatic AD. A limitation of the current study is the lack of CSF tau measurements. In designing future studies using the new National Institute on Aging–Alzheimer’s Association terminology for AD, defined as the presence of amyloid and tau in the brain, a primary prevention study would involve recruiting amyloid-negative participants and treating them over the course of years or decades. It is important to note that reducing the rate of plaque accumulation is unlikely to be a viable treatment strategy for individuals who have already developed dementia, as there is little evidence that progression of their disease is dependent on the progression of their amyloid deposition. Thus, some studies27 but not all28 find that SSRIs do not slow the course of AD or improve the cognitive function of patients with AD. To the extent that many people in the amyloid-negative group may never develop brain Aβ deposition, it will be critical to determine how biomarker changes can be used to indicate a need for initiating treatment. It remains to be seen whether the relatively modest reduction in CSF Aβ42 shown in the current study could translate to clinical benefits. If a greater reduction could be demonstrated for the higher 30 mg dose, or for a longer duration of exposure, or for a different SSRI more specific for the relevant 5-HT subtypes, the feasibility might be higher. In considering higher doses in an older adult population, caution is warranted given potential cardiac effects including increased QTc interval. In previous studies,7,18 we demonstrated that the SSRI mechanism of action operated through ERK via binding to the 5-HT4, 5-HT6, and 5-HT7 receptors. This offers the opportunity for developing drugs with higher affinity for these receptors as agents for reducing CSF Aβ42 and plaque accumulation and could lead to new therapeutic preventive strategies as the next step in advancing a prevention strategy. We also note that while a strategy aimed at decreasing Aβ42 is important, as a single target this may not be sufficient to address AD pathology and combination therapies with multiple targets may be required.
Acknowledgment
The authors thank the study participants.
Glossary
- 5-HT
5-hydroxytryptamine
- Aβ
β-amyloid
- AD
Alzheimer disease
- ANCOVA
analysis of covariance
- CDR
Clinical Dementia Rating
- CI
confidence interval
- CV
coefficient of variation
- ISF
interstitial fluid
- LMX
Luminex immunoassay
- LP
lumbar puncture
- MS
mass spectrometry
- Penn
University of Pennsylvania
- QC
quality control
- SSRI
selective serotonin reuptake inhibitor
- UPLC
ultraperformance liquid chromatography
- WU
Washington University
Appendix. Authors
Footnotes
Study funding
This work was supported by R01AG041502 (Y.I.S.) and by the Knight Alzheimer's Disease Research Center at Washington University: P50AG005681 (J.C.M.), P01AG003991 (J.C.M.), and P01AG026276 (J.C.M.).
Disclosure
R.F. Suckow is listed as a coinventor on several issued patents and applications with AstraZeneca, though no compensation has been received or anticipated. The issued patents and applications have no relevance to the work presented in this article. R.F. Suckow is also a shareholder with Johnson & Johnson. J.C. Morris receives support from Eli Lilly/Avid Radiopharmaceuticals and is funded by NIH grants P50AG005681, P01AG003991, P01AG026276, and UF1AG032438. Neither J.C. Morris nor his family owns stock or has equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company. A.M. Fagan receives research support from the National Institute on Aging at the NIH, the DIAN-TU Pharma Consortium, Fujirebio, and Roche Diagnostics. She is a member of the Scientific Advisory Boards for AbbVie, Genentech, and Roche Diagnostics, and provides consultation for Araclon/Grifols, DiamiR, and DiademRes. There are no conflicts with the present study. Y.I. Sheline, B.J. Snider, J.C. Beer, D. Seok, J.-M. Lee, T. Waligorska, M. Korecka, I. Aselcioglu, L.M. Shaw, and J.R. Cirrito report no disclosures relevant to this manuscript. Go to Neurology.org/Nhttps://n.neurology.org/lookup/doi/10.1212/WNL.0000000000010725 for full disclosures.
References
- 1.Arai H, Kosaka K, Iizuka R. Changes of biogenic amines and their metabolites in postmortem brains from patients with Alzheimer-type dementia. J Neurochem 1984;43:388–393. [DOI] [PubMed] [Google Scholar]
- 2.Reynolds GP, Mason SL, Meldrum A, et al. 5-Hydroxytryptamine (5-HT)4 receptors in post mortem human brain tissue: distribution, pharmacology and effects of neurodegenerative diseases. Br J Pharmacol 1995;114:993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arjona AA, Pooler AM, Lee RK, Wurtman RJ. Effect of a 5-HT(2C) serotonin agonist, dexnorfenfluramine, on amyloid precursor protein metabolism in guinea pigs. Brain Res 2002;951:135–140. [DOI] [PubMed] [Google Scholar]
- 4.Nitsch RM, Deng M, Growdon JH, Wurtman RJ. Serotonin 5-HT2a and 5-HT2c receptors stimulate amyloid precursor protein ectodomain secretion. J Biol Chem 1996;271:4188–4194. [DOI] [PubMed] [Google Scholar]
- 5.Robert SJ, Zugaza JL, Fischmeister R, Gardier AM, Lezoualc'h F. The human serotonin 5-HT4 receptor regulates secretion of non-amyloidogenic precursor protein. J Biol Chem 2001;276:44881–44888. [DOI] [PubMed] [Google Scholar]
- 6.Cirrito JR, Disabato BM, Restivo JL, et al. Serotonin signaling is associated with lower amyloid-beta levels and plaques in transgenic mice and humans. Proc Natl Acad Sci USA 2011;108:14968–14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheline YI, West T, Yarasheski K, et al. An antidepressant decreases CSF Abeta production in healthy individuals and in transgenic AD mice. Sci Transl Med 2014;6:236re234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaw LM, Waligorska T, Fields L, et al. Derivation of cutoffs for the Elecsys(R) amyloid beta (1-42) assay in Alzheimer's disease. Alzheimers Dement 2018;10:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960;23:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 11.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol 2011;121:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olsson A, Vanderstichele H, Andreasen N, et al. Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem 2005;51:336–345. [DOI] [PubMed] [Google Scholar]
- 13.Korecka M, Waligorska T, Figurski M, et al. Qualification of a surrogate matrix-based absolute quantification method for amyloid-beta(4)(2) in human cerebrospinal fluid using 2D UPLC-tandem mass spectrometry. J Alzheimers Dis 2014;41:441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oyehaug E, Ostensen ET, Salvesen B. Determination of the antidepressant agent citalopram and metabolites in plasma by liquid chromatography with fluorescence detection. J Chromatogr 1982;227:129–135. [DOI] [PubMed] [Google Scholar]
- 15.Hansson O, Lehmann S, Otto M, Zetterberg H, Lewczuk P. Advantages and disadvantages of the use of the CSF amyloid beta (Abeta) 42/40 ratio in the diagnosis of Alzheimer's disease. Alzheimers Res Ther 2019;11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cirrito JR, Wallace CE, Yan P, et al. Effect of escitalopram on Aβ levels and plaque load in an Alzheimer mouse model. Neurology 2020;95:e2666–e2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008;22:659–661. [DOI] [PubMed] [Google Scholar]
- 18.Fisher JR, Wallace CE, Tripoli DL, Sheline YI, Cirrito JR. Redundant Gs-coupled serotonin receptors regulate amyloid-beta metabolism in vivo. Mol Neurodegener 2016;11:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith LM, Strittmatter SM. Binding sites for amyloid-beta oligomers and synaptic toxicity. Cold Spring Harb Perspect Med 2017;7:a024075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lomakin A, Teplow DB, Kirschner DA, Benedek GB. Kinetic theory of fibrillogenesis of amyloid beta-protein. Proc Natl Acad Sci USA 1997;94:7942–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McConlogue L, Buttini M, Anderson JP, et al. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem 2007;282:26326–26334. [DOI] [PubMed] [Google Scholar]
- 22.Yan P, Bero AW, Cirrito JR, et al. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci 2009;29:10706–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol 2006;59:512–519. [DOI] [PubMed] [Google Scholar]
- 24.Wildburger NC, Gyngard F, Guillermier C, et al. Amyloid-beta plaques in clinical Alzheimer's disease brain incorporate stable isotope tracer in vivo and exhibit nanoscale heterogeneity. Front Neurol 2018;9:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carmona-Iragui M, Videla L, Lleo A, Fortea J. Down syndrome, Alzheimer disease, and cerebral amyloid angiopathy: the complex triangle of brain amyloidosis. Dev Neurobiol 2019;79:716–737. [DOI] [PubMed] [Google Scholar]
- 26.Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 2016;354:1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones HE, Joshi A, Shenkin S, Mead GE. The effect of treatment with selective serotonin reuptake inhibitors in comparison to placebo in the progression of dementia: a systematic review and meta-analysis. Age Ageing 2016;45:448–456. [DOI] [PubMed] [Google Scholar]
- 28.Brendel M, Sauerbeck J, Greven S, et al. Serotonin selective reuptake inhibitor treatment improves cognition and grey matter atrophy but not amyloid burden during two-year follow-up in mild cognitive impairment and Alzheimer's disease patients with depressive symptoms. J Alzheimers Dis 2018;65:793–806. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data have been deposited in aggregate into Clintrials.gov (NCT02161458), including study protocol and statistical analysis plan. In addition, upon request, individual de-identified CSF data will be shared. This will include sex, age binned in 5-year increments, baseline and post-treatment CSF Aβ42 levels, and treatment modality. Data will be accessible to principal investigators at academic institutions or industry affiliates with a research protocol for analyzing the data. Requests must be made in writing to the corresponding author and will be reviewed by the Penn–WU publications committee. Data will be available for 3 years.