

Abstract
Objective
To determine whether years of education and the ε4 risk allele at APOE influence β-amyloid (Aβ) pathology similarly in asymptomatic individuals with a family history of sporadic Alzheimer disease (AD) and presymptomatic autosomal dominant AD mutation carriers.
Methods
We analyzed cross-sectional data from 106 asymptomatic individuals with a parental history of sporadic AD (PREVENT-AD cohort; age 67.28 ± 4.72 years) and 117 presymptomatic autosomal dominant AD mutation carriers (DIAN cohort; age 35.04 ± 9.43 years). All participants underwent structural MRI and Aβ-PET imaging. In each cohort we investigated the influence of years of education, APOE ε4 status, and their interaction on Aβ-PET.
Results
Asymptomatic individuals with a parental history of sporadic AD showed increased Aβ burden associated with APOE ε4 carriage and lower level of education, but no interaction between these. Presymptomatic mutation carriers of autosomal dominant AD showed no relation between APOE ε4 and Aβ burden, but increasing level of education was associated with reduced Aβ burden. The association between educational attainment and Aβ burden was similar in the 2 cohorts.
Conclusions
While the APOE ε4 allele confers increased tendency toward Aβ accumulation in sporadic AD only, protective environmental factors, like increased education, may promote brain resistance against Aβ pathology in both sporadic and autosomal dominant AD.
Risk of Alzheimer disease (AD) in its sporadic form (sAD) has been suggested to be influenced by both environmental (e.g., education) and genetic (e.g., APOE ε4) factors.1 Autosomal dominant AD (ADAD) is characterized by an early onset, typically between the 30s to 50s, due to a mutated gene (presenilin1 [PSEN1], presenilin2 [PSEN2], amyloid precursor protein [APP]) involved in β-amyloid (Aβ) production.2 Because the causal mutations are fully penetrant, ADAD is considered a privileged opportunity to evaluate preclinical AD and the cascade of biomarker changes leading to AD dementia.3–5 The effects of ADAD mutations are often assumed to be nonmodifiable; therefore, few studies have evaluated the influence of potential modifiers on ADAD trajectories.6–8 While Aβ deposition has been associated with cognitive decline in both sAD9 and ADAD,10 it remains unclear whether Aβ accumulation is comparably influenced by environmental and genetic factors in the preclinical phase of the 2 variants.
We sought to evaluate whether factors known to affect sAD risk also affect presymptomatic ADAD mutation carriers. We examined whether level of education and APOE ε4 status are associated with Aβ burden similarly in asymptomatic individuals at risk of sAD and in presymptomatic ADAD mutation carriers.
Methods
Participants
We studied 106 asymptomatic elderly individuals with a parental history of sAD from the PREVENT-AD cohort and 125 presymptomatic ADAD mutation carriers from the DIAN network.
Presymptomatic Evaluation of Experimental or Novel Treatments for AD (PREVENT-AD) is a single-center longitudinal cohort of cognitively normal older adults at elevated risk of AD, as described elsewhere.11 Data analyzed here came from PREVENT-AD data release 5 (November 30, 2017). Briefly, between September 2011 and November 2017, PREVENT-AD enrolled 399 cognitively normal older individuals having at least one parent or multiple siblings with sAD. Inclusion criteria included (1) age 60 and above, or 55 and above for individuals less than 15 years younger than their index relative’s age at symptom onset, (2) normal cognition, and (3) no history of major neurologic or psychiatric disease. Normal cognition was defined as a Clinical Dementia Rating (CDR)12 score of 0 and Montreal Cognitive Assessment13 score ≥24. In the few cases of ambiguous results, participants were evaluated further with a more extensive neuropsychological test battery, which was carefully reviewed by neuropsychologists and physicians to ensure normal cognition. At each follow-up, cognitive examinations were also reviewed and individuals whose performance was questionable were further evaluated with an extensive neuropsychological battery. Only individuals who did not show cognitive decline according to the norms and the clinician review were kept in the study. Participants underwent clinical and cognitive examinations, blood tests, and MRI annually. Between February 2017 and April 2018, a subset of the cohort underwent an Aβ-PET scan using 18F-NAV4694. Participants with a parental history of sAD who were less than 15 years from the age of their parent at symptom onset (irrespective of their actual age) and for whom structural MRI and Aβ-PET were available were included in the analyses, resulting in a sample of 106 individuals.
The multisite longitudinal Dominantly Inherited Alzheimer Network (DIAN) study enrolls individuals age 18 and older who have a biological parent bearing a mutation responsible for ADAD. All participants undergo clinical and cognitive assessment, genetic testing, and imaging (MRI and PET, including a 11C Pittsburgh compound B (PiB) PET to quantify Aβ pathology). We studied baseline data from 125 cognitively normal (i.e., presymptomatic, defined as a CDR global score of 0)6 mutation carriers archived in the DIAN data freeze 10 (January 2009 to May 2016) having both structural MRI and Aβ-PET of suitable quality for investigation.
We estimated expected years to symptom onset (EYO)3 in the 2 cohorts, as previously described. During the medical interview, PREVENT-AD participants were asked to report the age of the parent at which the family observed significant cognitive/memory changes. Each participant's age at assessment was subtracted from his or her parent's age at symptom onset.14,15 In DIAN, EYO was calculated using (1) the mean age at dementia onset of the specific family mutation (calculated by integrating the age at onset reported in the literature across individuals with the same specific mutation)5,16 and (2) the parental age at onset as a reference (determined using semi-structured interview in which family members were asked about the age at first progressive cognitive decline).3 When family mutation age at onset was not available (18% of cases in the present study), the parental age at onset was used (see reference 5 for similar procedure).
Table.
Demographics

Standard protocol approvals, registrations, and patient consents
PREVENT-AD and DIAN studies were approved by regional ethics committees (institutional review board at McGill University for PREVENT-AD, Washington University Human Research Protection Office and local institutional review boards of the participating sites for DIAN) and all participants gave written informed consent to the study prior to participation.
Genetics
APOE genotype in PREVENT-AD was determined using the PyroMark Q96 pyrosequencer (Qiagen, Toronto, Canada) and the following primers: rs429358_amplification_forward 5′-ACGGCTGTCCAAGGAGCT G-3′, rs429358_amplification_reverse_biotinylated 5′-CACCTCGCCGCGGTACTG-3′, rs429358_sequencing 5′-CGGACATGGAGGACG-3′, rs7412_amplification_forward 5′-CTCCGCGATGCCGATGAC-3′, rs7412_amplification_reverse_biotinylated 5′-CCCCGGCCTGGTACACTG-3′, and rs7412_sequencing 5′-CGATGACCTGCAGAAG-3′.14 DIAN genotyping was performed by the DIAN Genetics Core at Washington University using PCR-based amplification of the appropriate exon followed by Sanger sequencing.3,18 DIAN determined APOE genotype using an ABI predesigned real-time TaqMan assay.
Image acquisition
T1-weighted images were acquired for PREVENT-AD using a Siemens (Munich, Germany) 3T scanner at the Brain Imaging Center of the Douglas Mental Health University Institute (Montréal, Canada) following Alzheimer's Disease Neuroimaging Initiative (ADNI; adni-info.org)19 protocol: sagittal, repetition time 2300 ms, echo time 2.98 ms, flip angle 9°, 176 slices, voxel size = 1 × 1 × 1 mm3, field of view 256 mm2. Aβ-PET scans were performed at the Montreal Neurological Institute (MNI; Montréal, Canada) on a Siemens high-resolution research tomograph at a mean interval of 257 ± 138 days from the MRI session. A 30-minute PET acquisition scan started 40 minutes after intravenous injection of ∼6 mCi of 18F-NAV4694. Transmission scans were acquired for attenuation correction.
DIAN acquired T1-weighted MRI scans on 3T scanners (multiple sites) by applying ADNI parameters and procedures.3,4,20 Aβ-PET scans were acquired at different centers after intravenous injection of 8 to 18 mCi of 11C-PiB. Some participants underwent a 70-minute full dynamic acquisition following the time of injection, while the remainder underwent a 30-minute scan after a 40-minute rest period. Transmission scans were obtained for attenuation correction.
Image processing
PREVENT-AD and DIAN data were processed similarly. T1-weighted MRI data were processed using FreeSurfer software version 5.3 (surfer.nmr.mgh.harvard.edu/). Images were segmented and parceled in native space using the Desikan-Killiany atlas.21 T1-weighted MRIs were further processed using the Segment routine of Statistical Parametric Mapping version 12 (SPM12; Wellcome Department of Imaging Neuroscience, London, UK), implemented in MATLAB (MathWorks, Natick, MA). Briefly, images were segmented, normalized to the MNI template, and modulated for nonlinear warping. MRI data were only used for PET preprocessing.
For PET processing, we applied a common pipeline (github.com/villeneuvelab/vlpp) to the 40- to 70-minute postinjection frames of all individuals, exclusively, to obtain a uniform scanning window. Aβ-PET images were realigned onto their respective MRI, masked to remove the scalp and CSF in an attempt to avoid contamination by nongray or nonwhite matter voxels, and smoothed using a full width at half maximum Gaussian kernel of 8mm. Resulting images were scaled using whole cerebellum uptake values (whole cerebellum was preferred to cerebellum gray matter to account better for white matter off-target binding variability between tracers). Global neocortical Aβ burden was quantified by extracting, in native space, the mean standardized uptake value ratio (SUVR) of the frontal, temporal, parietal, and posterior cingulate cortex of the Desikan-Killiany atlas.17 Furthermore, bilateral precuneus SUVR was extracted, because this region appears to be among the most sensitive to changes in both ADAD4 and sAD.17,22 Finally, bilateral striatum SUVR was also extracted because it shows early accumulation in ADAD23,24 and might reflect pathology severity in sAD.25 To allow for voxelwise analyses, scaled PET images were normalized to the MNI template by applying the normalization parameters of their respective T1-weighted images (see above). Finally, PET scans were reprocessed following the Centiloid guidelines to allow between-cohort comparisons. Full processing pipelines are fully described elsewhere.26,27 Briefly (and after validation of the implementation of the Centiloid pipeline in our laboratory using the Global Alzheimer's Association Interactive Network [GAAIN] data), 50- to 70-minute postinjection frames were averaged and registered to their respective MRIs. T1-weighted image normalization parameters (see above) were used to move the PET images into the MNI space. Images were then scaled using the whole cerebellum reference volume of interest (VOI) downloaded from the GAAIN website (gaain.org/centiloid-project). The cortical SUVR was finally extracted in the cortex VOI from the same source. To obtain the Centiloid values, SUVRs were modified using the equation provided in the previous publication: Centiloid value = 100 × (PiB SUVR − 1.009)/1.067 for PiB-PET images26 and Centiloid value = 100 × (NAV4694 SUVR − 1.028)/1.174 for NAV4694-PET images.27
All images underwent strict visual quality control through the processing procedure. Images showing questionable preprocessing, including normalization, were removed from the analyses. All PREVENT-AD participants passed quality control, while 8 DIAN participants were removed due to quality control failure, reducing the DIAN sample to 117 individuals.
Statistical analysis
Analyses were performed using the Statistical Package for Social Sciences (SPSS) version 22, and results were considered significant at p < 0.05. General linear models (GLMs) were run separately in PREVENT-AD and DIAN cohorts. We assessed the association of years of education and APOE ε4 status with the mean neocortical SUVRs in both cohorts, adjusting for age, sex, and MMSE, as well as site and mutation type (PSEN1/PSEN2/APP) for DIAN. We then tested for interaction between years of education and APOE ε4 status. All analyses were repeated using the precuneus and striatum SUVRs.17,22–25 In the case of significant findings in both cohorts, we further explored the relative influence of these factors on Aβ load in preclinical ADAD vs sAD, conducting similar GLMs on standardized Centiloid values26,27 instead of SUVRs, incorporating cohort (PREVENT-AD vs DIAN) as a covariate of interest, and assessing for the interaction between these factors (education and/or APOE ε4 status) and cohort.
Significant results were further explored voxel-wise using SPM12 to assess the regional distribution of these effects. Results were considered significant at p < 0.005 uncorrected with a minimum cluster size of 200.
Complementary GLMs, including the same covariates except MMSE, were run on the mean neocortical SUVRs to assess modulation of the association between EYO and Aβ burden by years of education and APOE ε4 status (i.e., EYO × education and EYO × APOE ε4 status interactions).
The confounding effect of reverse causation on the findings involving education (i.e., ongoing pathology preventing achievement of higher levels of education) was also explored. To do so we investigated the possibility, in DIAN, of a simple cooccurrence of early school abandonment and early Aβ due to an early expected age at onset by assessing the association between parental and specific family mutation age at onset, education level, and Aβ burden. We also assessed the role of age and genetic predisposition on educational achievements by testing the association between (1) age and APOE ε4 status with years of education in both cohorts and (2) the presence of ADAD mutations and years of education in DIAN, taking advantage of an independent set of mutation noncarriers from DIAN.
Data availability
Qualified researchers may obtain de-identified imaging and clinical PREVENT-AD data used for this study from the corresponding author and the study team upon reasonable request. De-identified imaging and clinical data from the DIAN study used in the preparation of this manuscript are available upon request to qualified researchers. Information can be found at dian.wustl.edu/our-research/observational-study/dian-observational-study-investigator-resources/data-request-terms-and-instructions/.
Results
Study participants
Participants' demographics are detailed in the table. DIAN presymptomatic mutation carriers were younger than PREVENT-AD asymptomatic individuals, and were further away from their expected onset (i.e., lower EYO). Proportion of women was higher in PREVENT-AD when compared to DIAN. The cohorts did not differ in terms of years of education, proportion of APOE ε4 carriers, or Mini-Mental State Examination (MMSE) performance. The proportion of Aβ-positive individuals (determined by Gaussian mixture models in each cohort)17 was higher in DIAN than in PREVENT-AD.
Years of education and APOE ε4 status in each cohort
In PREVENT-AD, higher educational attainment was associated with lower mean neocortical SUVR (F1,99 = 4.04, p = 0.05), while APOE ε4 carriers had a higher mean neocortical SUVR than noncarriers (F1,99 = 6.63, p = 0.01). There was no education × APOE ε4 status interaction on the mean neocortical SUVR (F1,98 = 1.44, p = 0.23; figure 1, A–C). Of note, controlling for the delay between MRI and PET did not affect these effects (education: F1,98 = 4.32, p = 0.04; APOE ε4 status: F1,98 = 5.57, p = 0.02; education × APOE ε4 status interaction: F1,97 = 1.86, p = 0.18). Effects of education and APOE ε4 status were also replicated when using nonparametric tests (education: Spearman ρ = −0.22, p = 0.03; APOE ε4 status: Mann-Whitney U = 882, p = 0.003; residuals were computed to control for age, sex, and MMSE scores). Results were similar when focusing on the precuneus SUVR (education: F1,99 = 4.35, p = 0.04; APOE ε4 status: F1,99 = 5.20, p = 0.03; education × APOE ε4 status interaction: F1,98 < 1, p = 0.32), but only the association with APOE ε4 status remained for the striatum (education: F1,99 = 1.21, p = 0.28; APOE ε4 status: F1,99 = 5.33, p = 0.02; education × APOE ε4 status interaction: F1,98 = 2.22, p = 0.14). Voxel-wise analyses suggested that the association with education was most apparent in the lateral temporal and parietal regions, the left lateral prefrontal and right orbitofrontal cortices, and the precuneus (figure 2A). APOE status effect was localized mainly in the medial and lateral prefrontal regions, extending to the precuneus (figure 2B).
Figure 1. Association between years of education, APOE ε4 status, and their interaction on β-amyloid (Aβ) pathology.

Years of education (A, D), APOE ε4 status (B, E), and their interactive effects (C, F) on Aβ pathology in asymptomatic individuals with a parental history of sporadic Alzheimer disease (AD) (Presymptomatic Evaluation of Experimental or Novel Treatments for AD [PREVENT-AD], A–C) and presymptomatic autosomal dominant AD mutation carriers (Dominantly Inherited Alzheimer Network [DIAN], D–F). Raw data (i.e., unadjusted standardized uptake value ratio [SUVR]) are plotted. Solid lines represent estimated regression lines; dotted lines represent 95% confidence intervals. Statistical values were obtained using general linear models controlling for age, sex, and Mini-Mental State Examination, as well as mutation type and site in DIAN.
Figure 2. Voxelwise associations among years of education, APOE ε4 status, and β-amyloid (Aβ) pathology.

Visualization of the voxelwise association between Aβ pathology and years of education (A, C) and APOE ε4 status (B, D) in asymptomatic individuals with a parental history of sporadic Alzheimer disease (AD) (Presymptomatic Evaluation of Experimental or Novel Treatments for AD [PREVENT-AD]; A, B) and presymptomatic autosomal dominant AD mutation carriers (Dominantly Inherited Alzheimer Network [DIAN]; C, D). Statistical maps thresholded at p < 0.005 uncorrected with a minimum cluster size of 200.
Analyses in DIAN presymptomatic ADAD mutation carriers revealed that the mean neocortical SUVR was associated with years of education (F1,94 = 5.04, p = 0.03) such that a higher level of education was associated with lower Aβ burden in presymptomatic ADAD mutation carriers. There was no association of Aβ burden with APOE ε4 status (F1,94 < 1, p = 0.39) or education × APOE ε4 interaction (F1,93 < 1, p = 0.33; figure 1, D–F). Nonparametric tests provided similar effects of education and APOE ε4 status (education: Spearman ρ = −0.18, p = 0.05; APOE ε4 status: Mann-Whitney U = 1,301, p = 0.40; residuals were computed to control for age, sex, MMSE scores, site, and mutation). Results were similar when focusing on the precuneus SUVR (education: F1,94 = 4.56, p = 0.04; APOE ε4 status: F1,94 = 1.42, p = 0.25; education × APOE ε4 interaction: F1,93 < 1, p = 0.73), while no effect survived when looking at the striatum SUVR (education: F1,94 < 1, p = 0.34; APOE ε4 status: F1,94 = 1.49, p = 0.23; education × APOE ε4 interaction: F1,93 < 1, p = 96). Voxel-wise analyses revealed that the effect of education was largely distributed, higher education being associated with lower binding in the bilateral prefrontal rostral, lateral frontal, parietal, and right temporal regions as well as in the posterior cingulate and precuneus; while no voxel-wise association was found with APOE ε4 (figure 2, C–D). To ensure that the apparent difference of APOE ε4 association with Aβ burden in PREVENT-AD and DIAN was not attributable to limited power in the latter, we repeated analyses after removing covariates specific to DIAN (i.e., site and mutation type). Results remained similar.
Comparison between cohorts
In an attempt to compare the magnitude of the effect of years of education found in PREVENT-AD and DIAN, we standardized Aβ-PET values following the Centiloid recommendations. The previous analyses were repeated using Centiloid values instead of SUVRs and results remained similar. There was no years of education × cohort interaction (F1,212 < 1, p = 0.70; figure 3), suggesting that the influence of education on Aβ pathology is relatively comparable between these 2 cohorts of cognitively unimpaired individuals.
Figure 3. Comparison of the association between education and β-amyloid (Aβ) pathology in Presymptomatic Evaluation of Experimental and Novel Treatments for AD (PREVENT-AD) vs Dominantly Inherited Alzheimer Network (DIAN).
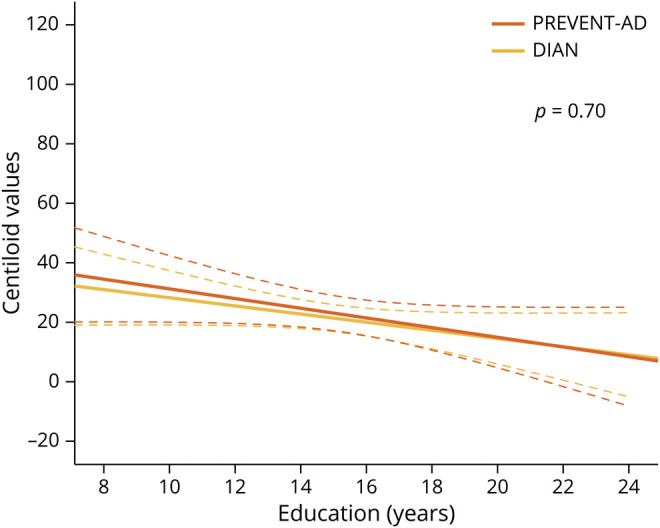
Direct comparison of the effect of years of education on Aβ burden between individuals with a parental history of sporadic Alzheimer disease (AD) (PREVENT-AD) and presymptomatic autosomal dominant AD mutation carriers (DIAN) using standardized Centiloid values. Raw data (i.e., unadjusted standardized uptake value ratio) are plotted. Solid lines represent estimated regression lines; dotted lines represent 95% confidence intervals. Statistical values were obtained using general linear models controlling for age, sex, Mini-Mental State Examination, and APOE ε4 status.
Complementary analyses
Complementary analyses were performed to further understand the effect of education and APOE ε4, and more specifically whether they were able to modulate the relationship between the mean neocortical SUVR and EYO in the 2 cohorts, separately. To do so, we assessed in a first step the relationship between EYO and the mean neocortical SUVR for each cohort. In a second step, we evaluated the EYO × education and EYO × APOE ε4 status interactions. Due to power issues, the triple-way interaction (i.e., EYO × education × APOE ε4 status) was not tested. All analyses were done using GLMs, controlling for age and sex and, for DIAN only, site and mutation type.
In PREVENT-AD, the sporadic parental EYO was related to the mean neocortical SUVR (F1,102 = 4.06, p = 0.05; figure 4A), but neither education nor APOE ε4 status modulated this relationship (F1,100 = 1.24, p = 0.27 and F1,100 = 2.14, p = 0.15 for the education × EYO and APOE ε4 × EYO interactions, respectively; figure 4, B–C). Results remained relatively similar when additionally controlling for the delay between MRI and PET (sporadic parental EYO [pEYO]: F1,101 = 3.29, p = 0.07; pEYO × education interaction: F1,99 < 1, p = 0.38; pEYO × APOE status interaction: F1,99 = 2.16, p = 0.15).
Figure 4. Estimated years to symptom onset (EYO) and its interactive effects with education and APOE ε4 status on β-amyloid (Aβ) pathology.

EYO (A, D) and its interaction with years of education (B, E) and APOE ε4 status (C, F) on Aβ pathology in asymptomatic individuals with a parental history of sporadic Alzheimer disease (AD) (Presymptomatic Evaluation of Experimental or Novel Treatments for AD [PREVENT-AD], A–C) and presymptomatic autosomal dominant AD mutation carriers (Dominantly Inherited Alzheimer Network [DIAN], D–F). Raw data (i.e., unadjusted standardized uptake value ratio) are plotted. Solid lines represent estimated regression lines; dotted lines represent 95% confidence intervals. Statistical values were obtained using general linear models controlling for age and sex, as well as mutation type and site in DIAN. Years of education was entered as a continuous variable in all analyses, but for visualization purposes individuals are categorized in low and high education according to the median number of years of education of the group (B, E).
In DIAN, the global neocortical SUVR increased as a function of the mean family mutation EYO (F1,97 = 15.41, p < .001; figure 4D). As was the case in the PREVENT-AD, neither education nor APOE ε4 status interacted with EYO to moderate this relationship (F1,95 < 1, p = 0.56 and F1,95 < 1, p = 0.87 for the education × EYO and APOE ε4 × EYO interactions, respectively; figure 4, E–F). Using the parental age at onset (instead of the mean age at onset of the family-specific mutation) to calculate EYO, the relationship between the mean neocortical SUVR and parental EYO was marginal (F1,97 = 2.93, p = 0.09). Neither the education × EYO interaction nor the APOE ε4 × EYO interaction had an effect on Aβ burden (F1,94 < 1, p = 0.54 and F1,95 < 1, p = 0.76, respectively).
Finally, to explore the potential confounding effect of reverse causation on the association between years of education and Aβ pathology, we tested the association between parental and mutation-specific age at onset, education level, and Aβ burden. Parental and family mutation age at onset were associated with years of education but not with mean neocortical SUVR (data available from Dryad, e-Supplement: doi.org/10.5061/dryad.ttdz08kth). We also assessed the associations of age and genetic mutations carriage (APOE ε4 or ADAD mutation) with educational achievement. Age, APOE ε4, and ADAD mutations carriage were not associated with lower educational achievements in our samples (data available from Dryad, e-Supplement, tables e-1 and e-2: doi.org/10.5061/dryad.ttdz08kth).
Discussion
Increasing efforts have been made toward understanding the preclinical phase of AD and identifying effective prevention strategies.28 Given its predictability and the low prevalence of age-related confounders ADAD is considered a privileged condition to study the preclinical phase of the disease.3 However, it remains unclear whether the preclinical phase of ADAD can be influenced by environmental and genetic factors, as seems to be the case in sAD. Our results suggest that higher education may delay Aβ accumulation similarly in both variants of the disease, while APOE ε4 allele seems to influence Aβ burden in people at risk of sAD only.
Education has been widely used as a proxy of cognitive reserve and related, in cognitively normal individuals, to a lower incidence of AD dementia29 and lower expression of AD biomarkers,30–32 including lower levels of Aβ burden.33,34 Higher levels of education might thus promote brain resistance (i.e., capacity to avoid brain pathology).35 Our study showed that the effect of education notably affects the precuneus, which is among the most sensitive brain regions, and potentially one of the earliest regions to show pathologic Aβ accumulation in sAD and ADAD.4,5,17,22 By contrast, the striatum, which shows Aβ pathology early in ADAD23,24 and might help staging Aβ severity in sAD,25,36 did not show such an effect. While this remains to be further understood in ADAD, the absence of influence of education in the striatum in sAD might be due to the fact that education protects against early accumulation, and does not influence later stages of amyloidosis.
Using the Centiloid scale, we were able to compare the effect of education between the 2 cohorts. Although the conversion in Centiloid values needs to be taken carefully as this standardization might be imperfect, our findings suggest that education has a similar effect on Aβ deposition in asymptomatic individuals at risk of sAD and presymptomatic ADAD mutation carriers. Therefore, enriched environment might protect against or delay Aβ accumulation, even in mutation carriers who are known to have a very aggressive form of the disease. This is in line with recent studies showing an association between physical activity and Aβ-PET in DIAN ADAD mutation carriers,6,37 this effect being further characterized by an EYO × exercise interaction when restricting analyses to Aβ-positive presymptomatic mutation carriers.6 Another study, from the Antioquia cohort (Colombia), found that education was protective against the onset of cognitive impairment in PSEN1 E280A mutation carriers, a higher level of education being able to delay their onset by 3 years.38 Thus healthier lifestyle seems able to influence Aβ pathology and delay AD onset, even in the presence of fully penetrant mutations responsible for ADAD.
To further investigate this question, we next assessed if education modulates the association between EYO and Aβ accumulation by testing the interaction between education and EYO on Aβ burden. While EYO was associated with higher Aβ burden in the 2 cohorts,3,4,14,15 education did not modulate this relationship. The absence of interaction between EYO and education suggests that higher education could delay Aβ pathology, without affecting its rate of accumulation once the process has started. In this scenario, the strength of the association between Aβ burden and EYO would be similar in individuals with high and low education, but the ones with higher education would be able to resist pathology36 (start accumulating Aβ burden later), which might in turn allow delay of the actual age at onset, as previously highlighted in the Columbian cohort.38 Alternatively, this absence of interaction could suggest that the presence of genetic mutation or Aβ pathology prevents higher educational achievements. Thus lower education would simply cooccur with, or be due to, the presence of Aβ pathology. Whereas we cannot completely rule out the possibility of reversed causation, we found insufficient evidence for a simple cooccurrence of early parental/specific family mutation age of onset–Aβ burden and early school dropout, and no direct effect of mutations (APOE ε4 or ADAD) or age on educational achievements, suggesting that age and genetic mutations cannot fully account for our findings. Finally, this a priori negative finding could also be related to the nature of EYO, which is only an approximation of time to symptom onset. The differential relationship between EYO and Aβ burden in DIAN according to whether this index is calculated based on the family mutation mean age at onset or the parental age at onset supports this idea. EYO is also limited by the imprecision of the measurement itself. As age at parental onset was directly reported by the participants in each cohort, this index is likely to be contaminated by bias recall. Follow-up studies, using the actual age of conversion, will help determine whether the association found between education and Aβ burden were related to a delayed age at conversion in both cohorts.
In sAD, a large body of research has evidenced that not only environmental but also genetic factors influence the disease progression. Thus the role of APOE ε4 genotype on AD risk and its association with earlier and higher Aβ accumulation in cognitively normal older individuals has already been widely shown.39 The influence of APOE ε4 in ADAD is less clear. Some studies suggested that APOE ε4 influences ADAD progression7,40–42 while others did not.6,8,16,38,43–45 Our results support this latter group of findings. The fact that this population already carries a gene that massively disrupts Aβ production might reduce the influence of any other genes, like APOE ε4, involved in amyloidosis. APOE ε4 status, like education, did not modulate the relationship between EYO and Aβ pathology. While the limited role of APOE ε4 in ADAD is consistent with this finding, such an interaction in individuals at risk of sAD has been published previously by our group, when using CSF Aβ1-42 rather than Aβ-PET (overlap of 35 participants between the 2 studies).14 Longitudinal studies are needed to further understand these results.
Neither in PREVENT-AD nor in DIAN did the effect of APOE ε4 status interact with education on Aβ burden. While this is in line with previous findings in ADAD,6,38 some studies in cognitively normal elderly individuals, assessing either the effect of other lifestyle factors (i.e., cognitive or physical activity) on Aβ burden or the effect of education on other AD biomarkers, highlighted a stronger effect of education in APOE ε4 carriers when compared to noncarriers.46–49 It has been hypothesized that APOE ε4 noncarriers have a lower sensitivity to environmental factors; alternatively, a lack of variability might prevent finding strong effects in this group. In the present study, all PREVENT-AD participants were already at risk of sAD, which might have provided sufficient sensitivity or variability to detect an effect of environmental factors in APOE ε4 noncarriers. Whether the absence of an education × APOE ε4 status interaction on Aβ burden is generalizable to other cohorts or is due to our cohort's preexisting risk will need further evaluation.
Some limitations should be acknowledged. First, important differences persist preventing the direct comparison of the 2 cohorts (i.e., uncertainty that PREVENT-AD participants will develop sAD later on, use of different Aβ-PET tracers, multisite vs single-center cohorts, different age range of the 2 cohorts). Even if it might be imperfect, the Centiloid standardization used to compare directly the effect of education on Aβ burden between the 2 cohorts circumvents some of these limitations. The sample size remains relatively small and it did not allow us to assess triple-way interactions (education × APOE ε4 × EYO) or further divide the sample according, for instance, to their Aβ status. Evaluating the influence of education in Aβ-positive vs Aβ-negative individuals would notably allow us to better characterize the effect of education. We used years of education as a measure of modifiable factors. Whereas our objective was more exploratory than mechanistic, it is important to acknowledge that the use of education alone prevents clear interpretation of the specific role of education. First, education differs according to various social factors, including generations, country, ethnicity/race, and socioeconomic status, making it difficult to consider years of education to be rigorously identical across individuals. The relative homogeneity of the 2 cohorts (e.g., mainly Western world, White individuals) suggests that the influence of these factors is limited in the present study, but also limits the generalization of the results to other racial/ethnic groups. In addition, higher education is likely to be associated with other positive lifestyle factors (e.g., better socioeconomic status, lifetime cognitive activity, diet) or nonmodifiable protective factors (e.g., more efficient neural processing may be correlated with pursuing advanced education), all being potential factors underlying resistance to Aβ pathology. Future studies, with appropriate designs, should focus on teasing apart whether the protective effects are actually attributable to years of education or whether they are due to early-life socioeconomic status, biological predisposition, or later-life exposures associated with higher educational ascertainment. Although some of these education correlates might be modifiable, others might not, which would limit the effect of education as a target for interventions. Finally, the cross-sectional nature of the present study prevents us from drawing a strong conclusion on the causal relationship between years of education and Aβ pathology in preclinical AD. Future studies, including longitudinal ones, should be designed to assess these questions and understand further the influence of genetic and environmental factors on the preclinical phase of ADAD and sAD.
Overall, our results suggest that a higher level of education might delay Aβ burden in both variants, suggesting the existence of reserve mechanisms not only in individuals at risk of sAD, but also in individuals carrying aggressive ADAD variants. By contrast, while APOE ε4 is strongly related to Aβ burden in asymptomatic individuals at risk of sAD, it does not influence Aβ load in preclinical ADAD. These results add to the growing literature suggesting that preventive strategies targeting environmental/modifiable risk factors could prevent, or at least delay, AD.50
Acknowledgment
The authors thank the members of the Villeneuve Lab, J. Tremblay-Mercier, A. Labonté, D. Dea, C. Madjar, and the PREVENT-AD center for participant recruitment, data acquisition, and data management (a complete listing of PREVENT-AD Research Group members can be found in the PREVENT-AD database: preventad.loris.ca/acknowledgements/acknowledgements.php?date=[2018-04-30]); the members of the Brain Imaging Center of the Douglas Mental University Health Institute for MRI acquisitions; the members of the Cyclotron and PET units of the Montreal Neurological Institute for PET tracer production and acquisitions; K. Paumier, R. Hornbeck, P. Wang, and S. Flores for their help in DIAN data preparation; all the centers involved in DIAN data acquisition and DIAN research and support staff; and the participants and their families. This article was reviewed by DIAN study investigators for scientific content and consistency of data interpretation with previous DIAN study publications.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADAD
autosomal dominant Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- CDR
Clinical Dementia Rating
- DIAN
Dominantly Inherited Alzheimer Network
- EYO
expected years to symptom onset
- GAAIN
Global Alzheimer's Association Interactive Network
- GLM
general linear model
- MMSE
Mini-Mental State Examination
- MNI
Montreal Neurological Institute
- pEYO
parental expected years to symptom onset
- PiB
Pittsburgh compound B
- PREVENT-AD
Presymptomatic Evaluation of Experimental or Novel Treatments for AD
- sAD
sporadic Alzheimer disease
- SUVR
standardized uptake value ratio
- VOI
volume of interest
Appendix. Authors
Study funding
This work was supported by 2 Canada Research Chairs (S.V., J.C.S.B.), a Canadian Institutes of Health Research project grant PJT-148963 (S.V.), a Canada Fund for Innovation (S.V.), an Alzheimer's Association Research Grant NIRG-397028 (S.V.), the Lemaire foundation (J.P., S.V.), the J.L. Levesque Foundation (J.P.), a joint Alzheimer Society of Canada and Brain Canada Research grant NIG-17-08 (S.V.), a StoP-AD fellowship (J.G.), a Quebec Bio-Imaging Network scholarship (J.G.), and a joint FRQ-S and Alzheimer Society of Canada scholarship (A.P.B.). PREVENT-AD was funded by a $13.5 million, 7-year public–private partnership using funds provided by McGill University, the Fonds de Recherche du Québec–Santé (FRQ-S), an unrestricted research grant from Pfizer Canada, the Levesque Foundation, the Douglas Hospital Research Centre and Foundation, the Government of Canada, the Canada Fund for Innovation, and Genome Quebec Innovation Center (J.C.S.B., J.P.). Data collection and sharing for this project was also supported by The Dominantly Inherited Alzheimer's Network (DIAN, U19AG032438) funded by the National Institute on Aging (NIA), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI).
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/Nhttps://n.neurology.org/lookup/doi/10.1212/WNL.0000000000010314 for full disclosures.
References
- 1.Chételat G. Alzheimer disease: Aβ-independent processes: rethinking preclinical AD. Nat Rev Neurol 2013;9:123–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 2010;23:213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bateman RJ, Xiong C, Benzinger TLS, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benzinger TLS, Blazey T, Jack CR, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci USA 2013;110:E4502–E4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon BA, Blazey TM, Su Y, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol 2018;17:241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer's disease. Alzheimers Dement 2017;13:1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wijsman EM, Daw EW, Yu X, et al. APOE and other loci affect age-at-onset in Alzheimer's disease families with PS2 mutation. Am J Med Genet B Neuropsychiatr Genet 2005;132B:14–20. [DOI] [PubMed] [Google Scholar]
- 8.Van Broeckhoven C, Backhovens H, Cruts M, et al. APOE genotype does not modulate age of onset in families with chromosome 14 encoded Alzheimer's disease. Neurosci Lett 1994;169:179–180. [DOI] [PubMed] [Google Scholar]
- 9.Clark LR, Racine AM, Koscik RL, et al. Beta-amyloid and cognitive decline in late middle age: findings from the Wisconsin Registry for Alzheimer's Prevention Study. Alzheimers Dement 2016;12:805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang F, Gordon BA, Ryman DC, et al. Cerebral amyloidosis associated with cognitive decline in autosomal dominant Alzheimer disease. Neurology 2015;85:790–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breitner JCS, Poirier J, Etienne PE, Group JMLFTP-AR. Rationale and structure for a new center for studies on prevention of Alzheimer's disease (StoP-AD). 2016. Available at: search.datacite.org/works/10.14283/JPAD.2016.121. Accessed July 4, 2017. [DOI] [PubMed]
- 12.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412. [DOI] [PubMed] [Google Scholar]
- 13.Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699. [DOI] [PubMed] [Google Scholar]
- 14.Villeneuve S, Vogel JW, Gonneaud J, et al. Proximity to parental symptom onset and amyloid-β burden in sporadic Alzheimer disease. JAMA Neurol 2018;75:608–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogel JW, Vachon-Presseau E, Pichet Binette A, et al. Brain properties predict proximity to symptom onset in sporadic Alzheimer's disease. Brain 2018;141:1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryman DC, Acosta-Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 2014;83:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, et al. Existing Pittsburgh compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain 2015;138:2020–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fagan AM, Xiong C, Jasielec MS, et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer's disease. Sci Transl Med 2014;6:226ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jack CR, Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008;27:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris JC, Aisen PS, Bateman RJ, et al. Developing an international network for Alzheimer research: the Dominantly Inherited Alzheimer Network. Clin Investig 2012;2:975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 2006;31:968–980. [DOI] [PubMed] [Google Scholar]
- 22.Palmqvist S, Schöll M, Strandberg O, et al. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat Commun 2017;8:1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villemagne VL, Ataka S, Mizuno T, et al. High striatal amyloid beta-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol 2009;66:1537–1544. [DOI] [PubMed] [Google Scholar]
- 24.Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci 2007;27:6174–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanseeuw BJ, Betensky RA, Mormino EC, et al. PET staging of amyloidosis using striatum. Alzheimers Dement 2018;14:1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klunk WE, Koeppe RA, Price JC, et al. The Centiloid project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement 2015;11:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rowe CC, Jones G, Doré V, et al. Standardized expression of 18F-NAV4694 and 11C-PiB β-amyloid PET results with the Centiloid scale. J Nucl Med 2016;57:1233–1237. [DOI] [PubMed] [Google Scholar]
- 28.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association Workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng X, D'Arcy C. Education and dementia in the context of the cognitive reserve hypothesis: a systematic review with meta-analyses and qualitative analyses. PLoS One 2012;7:e38268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arenaza-Urquijo EM, Landeau B, La Joie R, et al. Relationships between years of education and gray matter volume, metabolism and functional connectivity in healthy elders. Neuroimage 2013;83:450–457. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Julkunen V, Paajanen T, et al. Education increases reserve against Alzheimer's disease: evidence from structural MRI analysis. Neuroradiology 2012;54:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim JP, Seo SW, Shin HY, et al. Effects of education on aging-related cortical thinning among cognitively normal individuals. Neurology 2015;85:806–812. [DOI] [PubMed] [Google Scholar]
- 33.Arenaza-Urquijo EM, Bejanin A, Gonneaud J, et al. Association between educational attainment and amyloid deposition across the spectrum from normal cognition to dementia: neuroimaging evidence for protection and compensation. Neurobiol Aging 2017;59:72–79. [DOI] [PubMed] [Google Scholar]
- 34.Landau SM, Marks SM, Mormino EC, et al. Association of lifetime cognitive engagement and low β-amyloid deposition. Arch Neurol 2012;69:623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arenaza-Urquijo EM, Vemuri P. Resistance vs resilience to Alzheimer disease: clarifying terminology for preclinical studies. Neurology 2018;90:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho SH, Shin JH, Jang H, et al. Amyloid involvement in subcortical regions predicts cognitive decline. Eur J Nucl Med Mol Imaging 2018;45:2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Müller S, Preische O, Sohrabi HR, et al. Relationship between physical activity, cognition, and Alzheimer pathology in autosomal dominant Alzheimer's disease. Alzheimers Dement 2018;14:1427–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aguirre-Acevedo DC, Lopera F, Henao E, et al. Cognitive decline in a Colombian kindred with autosomal dominant Alzheimer disease: a retrospective cohort study. JAMA Neurol 2016;73:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fouquet M, Besson FL, Gonneaud J, La Joie R, Chételat G. Imaging brain effects of APOE4 in cognitively normal individuals across the lifespan. Neuropsychol Rev 2014;24:290–299. [DOI] [PubMed] [Google Scholar]
- 40.Pastor P, Roe CM, Villegas A, et al. Apolipoprotein E epsilon4 modifies Alzheimer's disease onset in an E280A PS1 kindred. Ann Neurol 2003;54:163–169. [DOI] [PubMed] [Google Scholar]
- 41.Oxtoby NP, Young AL, Cash DM, et al. Data-driven models of dominantly-inherited Alzheimer's disease progression. Brain 2018;141:1529–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim YY, Hassenstab J, Cruchaga C, et al. BDNF Val66Met moderates memory impairment, hippocampal function and tau in preclinical autosomal dominant Alzheimer's disease. Brain 2016;139:2766–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carmona-Iragui M, Balasa M, Benejam B, et al. Cerebral amyloid angiopathy in down syndrome and sporadic and autosomal-dominant Alzheimer's disease. Alzheimers Dement 2017;13:1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee S, Zimmerman ME, Narkhede A, et al. White matter hyperintensities and the mediating role of cerebral amyloid angiopathy in dominantly-inherited Alzheimer's disease. PLoS One 2018;13:e0195838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ringman JM, Diaz-Olavarrieta C, Rodriguez Y, et al. Neuropsychological function in nondemented carriers of presenilin-1 mutations. Neurology 2005;65:552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wirth M, Villeneuve S, La Joie R, Marks SM, Jagust WJ. Gene-environment interactions: lifetime cognitive activity, APOE genotype, and β-amyloid burden. J Neurosci 2014;34:8612–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arenaza-Urquijo EM, Gonneaud J, Fouquet M, et al. Interaction between years of education and APOE ε4 status on frontal and temporal metabolism. Neurology 2015;85:1392–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Head D, Bugg JM, Goate AM, et al. Exercise engagement as a moderator of the effects of APOE genotype on Amyloid deposition. Arch Neurol 2012;69:636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-β plasma and brain levels: results from the Australian Imaging, Biomarkers and Lifestyle Study of Ageing. Mol Psychiatry 2013;18:875–881. [DOI] [PubMed] [Google Scholar]
- 50.Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet 2017;390:2673–2734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Qualified researchers may obtain de-identified imaging and clinical PREVENT-AD data used for this study from the corresponding author and the study team upon reasonable request. De-identified imaging and clinical data from the DIAN study used in the preparation of this manuscript are available upon request to qualified researchers. Information can be found at dian.wustl.edu/our-research/observational-study/dian-observational-study-investigator-resources/data-request-terms-and-instructions/.