

Abstract
Objective
To determine whether familial aggregation of status epilepticus (SE) occurs in a large cohort of familial common epilepsies.
Methods
We used the Epilepsy Phenome/Genome Project dataset, which consisted of 2,197 participants in 1,043 family units with ≥2 members having a common generalized or nonacquired focal epilepsy (NAFE). We identified participants with a history of traditionally defined SE (TSE) (seizures ≥30 minutes) and operationally defined SE (OSE) (seizures ≥10 minutes) by chart review. We assessed familial aggregation of TSE and OSE using χ2 analysis and generalized estimating equations (GEE).
Results
One hundred fifty-five (7%) participants in 1,043 families had ≥1 episodes of TSE. Two hundred fifty (11%) had ≥1 episodes of OSE. In a χ2 analysis, the number of family units with ≥2 members having TSE (odds ratio [OR] 4.79, 95% confidence interval [CI] 2.56–8.97) or OSE (OR 4.23, 95% CI 2.67–6.70) was greater than expected by chance. In GEE models adjusted for sex, broad epilepsy class (GE or NAFE), age at onset, and duration of epilepsy, TSE in a proband predicted TSE in a first-degree relative (OR 2.79, 95% CI 1.24–6.22), and OSE in a proband predicted OSE in a first-degree relative (OR 2.91, 95% CI 1.65–5.15). The results remained significant in models addressing epilepsy severity by incorporating the number of antiseizure medications used or epilepsy surgery.
Conclusions
TSE and OSE showed robust familial aggregation in a cohort of familial epilepsy independently of epilepsy severity or class, suggesting that genetic factors contribute to SE independently of the genetic cause of these epilepsies.
ClinicalTrials.gov identifier:
The traditional definition of status epilepticus (TSE) is a single seizure or series of seizures without intervening recovery of consciousness lasting ≥30 minutes.1,2 A widely accepted, operational definition of status epilepticus (OSE) reduces the duration to 5 to 10 minutes, which recognizes the need for rapid treatment.1,2 It is a common neurological emergency that may be increasing in frequency,3 has significant morbidity and mortality,3–5 and is costly.6
Limited understanding of the underlying pathophysiology hampers efforts to improve outcomes and to reduce costs associated with status epilepticus (SE).7 Few studies have focused on the mechanisms by which seizures end compared to other aspects of epilepsy.8 Most studies examining seizure termination have examined the cellular and network changes that occur during a seizure.8–10 An alternative, complementary, but less commonly used approach is to identify genetic variants that predispose to SE.11 A recent study showed that GluA1 knockout mice are less susceptible to SE than wild-type mice, although knockout and wild-type mice are equally susceptible to seizures.12 These results suggest that the genetic determinants for seizure initiation and termination may be distinct. A twin study and an analysis of genetic mutations associated with SE provide preliminary evidence for this hypothesis in humans, although these studies did not conclusively demonstrate that the genetic predispositions to epilepsy and SE are separate.11,13,14
To obtain additional support for distinct genetic mechanisms for seizure initiation and termination, we hypothesized that genetic mechanisms contribute to SE in patients with common epilepsies independently of the genetic causes of epilepsy. We tested this hypothesis by examining within-family aggregation of SE among participants in the Epilepsy Phenome/Genome Project (EPGP) using an approach similar to those in prior studies investigating familial epilepsy characteristics in this dataset.15–17
Methods
EPGP description
EPGP was a multi-institutional, cross-sectional effort that created a database with detailed phenotypic data paired with banked DNA samples from persons with common and rare epilepsies.18 A collaboration involving 27 international academic epilepsy centers, EPGP used multiple recruitment methods to enroll 5,445 participants from November 2007 until its close in May 2014.19
Standard protocol approvals, registrations, and patient consents
The Institutional Review Board at each center approved the protocol. All participants or their legal guardians provided written informed consent. This trial was registered at ClinicalTrials.gov as NCT00552045.
Participants
EPGP included different epilepsy types, and the data presented here come from family units consisting of first-degree relatives with a common generalized (GE) or nonacquired focal (NAFE) broad epilepsy class. Some families contributed >2 members such as 3 siblings or a sibling pair and a parent. Eligibility requirements for all participants included being ≥4 weeks old at enrollment, having had ≥2 unprovoked seizures or 1 unprovoked seizure with an epileptiform EEG, being <45 years old at the time of the first unprovoked seizure, and having high-quality clinical and laboratory data as assessed by the enrolling clinician.18 Exclusion criteria included febrile seizures only; an antecedent cause of epilepsy before the first unprovoked seizure; microcephaly; a recognized genetic syndrome, chromosomal abnormality, or pathogenic variant in a known epilepsy gene; moderate to severe developmental delay before epilepsy onset; autism; gestational age <32 weeks; or being an identical twin of a participant. EPGP did not require genetic testing before enrollment, but all who did and had a positive result were approved by the Adjudications Core.
EPGP collected phenotypic data such as seizure history, seizure types, febrile seizures, medical history, family history, and basic demographic information and assigned each participant an epilepsy type.18 The data used for this study were obtained from a medical record abstraction completed by the site principal investigator (PI) during a formal chart review. Other data collected included digital EEG files and digital brain MRI files or reports when digital EEG and MRI files were not available. The EPGP EEG and Imaging Cores reviewed the EEGs, MRIs, and relevant medical records. The site PI assigned a final diagnosis based on all the collected data. Two members of the EPGP Data Review Core independently reviewed a subset of cases to ensure data quality and consistency. The participants included here had complete data that the EPGP Data Review Core had validated.
The analyses reported here included participants classified as having GE, NAFE, NAFE + GE, or unclassifiable epilepsy.18 Those with GE had clinical seizures with a generalized onset, a normal brain MRI if done, and an EEG showing generalized epileptiform activity with a normal posterior dominant rhythm. Those with NAFE had seizures with a semiology consistent with a focal onset and a normal MRI or one showing mesial temporal sclerosis or focal cortical dysplasia. If the MRI was normal, those with NAFE could have a normal EEG or one showing a focal interictal abnormality or a focal seizure. However, participants having NAFE with a normal MRI and normal EEG required approval by the Adjudications Core. A brain MRI was not required of participants with self-limited epilepsy with centrotemporal spikes and an EEG consistent with this syndrome. Participants whose broad epilepsy class could not be determined were categorized as unclassifiable. First-degree relatives did not necessarily have the same epilepsy type.
Outcome variables
The phenotypic information collected through EPGP included information on TSE (seizure ≥30 minutes) and OSE (seizure ≥10 minutes). We included both TSE and OSE because of the trend toward decreasing the minimum duration required for SE.1,2 During medical record abstraction, the site PI determined whether each participant had TSE or a OSE, the date and duration of each episode of TSE and OSE if known, and whether the episodes were observed by medical personnel. The site PI could not determine whether 12% (274 of 2,197) of the participants had TSE and whether 13% (291 of 2,197) of the participants had OSE because the medical records lacked sufficient data. We considered these participants not to have had seizures of this duration in the analysis. The number of antiseizure medications used and a history of epilepsy surgery were not abstracted for participants enrolled after October 2011 to decrease the phenotyping workload (table). This change was one of several implemented to allow the study to meet enrollment goals and projected timelines for phenotyping.18
Table.
Participant characteristics

Covariates/predictors
We calculated the age at onset by subtracting the participant's year of birth from the year of the first afebrile seizure. Epilepsy duration at enrollment was determined by subtracting the year of the first afebrile seizure from the year the participant was enrolled in EPGP. Both the age at onset and epilepsy duration variables were measured in whole number years. We recoded the epilepsy class variable (GE, NAFE, or NAFE + GE) to create a dichotomous (yes/no) NAFE variable. Participants with NAFE or NAFE + GE were classified as having NAFE. Similar results were obtained if participants with NAFE alone were classified as having NAFE, and those with NAFE + GE were not included in the NAFE group (data not shown). A continuous variable containing the number of antiseizure medications taken was recoded into a dichotomous variable indicating whether a participant had taken >2 antiseizure medications vs ≤2 on the basis of the formal definition of drug-resistant epilepsy.20 A documented history of epilepsy surgery also was coded into a dichotomous variable.
Statistical analysis
All analyses were conducted with SAS software for Windows version 9.4 (SAS Institute Inc, Cary, NC) or R 3.3.2 (R Foundation for Statistical Computing, Vienna, Austria).21,22 Medians and interquartile ranges were reported for continuous variables. The Wald χ2 statistic (Proc GENMOD, SAS) and the Wilcoxon rank-sum test for clustered data (clusrank, R) were used to assess differences between participants with and without TSE or OSE. These methods have the advantage of accounting for the nonindependence of family members. Throughout this study, the OSE group included all those with TSE.
Familial aggregation was assessed with 2 methods. We used a χ2 analysis to determine whether the number of family units in which the proband and a first-degree relative had TSE or OSE was greater than expected according to the prevalence of TSE and OSE in the EPGP study population. The first participant recruited in a family was designated the proband; if multiple participants enrolled simultaneously, the member with the most complete data was designated the proband.16 For some families, >1 first-degree relative was enrolled. The first-degree relative closest in age to the proband was selected for this analysis.
In the second approach, we assessed familial aggregation of TSE and OSE using a dataset containing all first-degree relatives of probands. The outcome was defined as the presence of TSE or OSE (separate models) in a first-degree relative with the primary exposure defined as the presence of TSE or OSE in the proband. We used generalized estimating equations (GEE) with a binomial distribution, logit link function, and an exchangeable correlation structure to account for the nonindependence of observations due to clustering of participants within families. Sex, age at onset, epilepsy duration, broad epilepsy class, number of antiseizure medications used, and history of epilepsy surgery were included as covariates in adjusted models. The number of antiseizure medications used and epilepsy surgery were included as indicators of epilepsy severity.
Data availability
The dataset containing the deidentified participant data analyzed for this study is available from the corresponding author on request.
Results
Participant characteristics
Of the 5,445 participants enrolled in EPGP, 2,197 had NAFE, GE, both, or an unclassified epilepsy (table and figure 1). These 2,197 participants belonged to 1,043 family units, of which 946 enrolled 2 relatives (91% of units), 85 enrolled 3 relatives (8%), 11 enrolled 4 relatives (1%), and 1 enrolled 6 relatives. Thus, 47% (1,043 of 2,197) of the participants were designated as probands. In relationship to the proband, 59% (682 of 1,154) of the participants were siblings, 38% (433 of 1,154) were parents, and 2% (23 of 1,154) were children.
Figure 1. EPGP flowchart.

EPGP = Epilepsy Phenome/Genome Project; GE = generalized epilepsy; IS = infantile spasms; LGS = Lennox-Gastaut syndrome; NAFE = nonacquired focal epilepsy; OSE = operationally defined status epilepticus; PMG = polymicrogyria; PVNH = periventricular nodular heterotopia; TSE = traditionally defined status epilepticus.
TSE and OSE were relatively rare in the EPGP cohort. Of the 2,197, 155 (7%) participants had ≥1 episodes of TSE, of whom 88 had ≥1 episodes observed by medical personnel (table). Two hundred fifty (11%), including all with TSE, had ≥1 episodes of OSE, of whom 117 had ≥1 observed episodes of OSE (table).
Forty-nine percent of the participants with TSE and 51% of the participants with OSE were male compared to 42% of those without TSE or OSE (table). Participants with TSE or OSE also had an earlier age at epilepsy onset, a greater proportion classified as NAFE, a higher incidence of treatment with >2 antiseizure medications, a more frequent history of epilepsy surgery, and a greater likelihood of having had a febrile seizure than those who did not have TSE or OSE (table and figures 2 and 3). Those with TSE had a longer duration of epilepsy at enrollment than those who did not (figures 2 and 3), but the same was not true for participants with OSE. The first episode of OSE occurred before or in the year of diagnosis in 69% (136 of 197). Of the participants with TSE, 51% had NAFE only compared to 29% of those without TSE, and 33% had GE only compared to 50% of those without SE (table). The participants with OSE showed a similar epilepsy class distribution.
Figure 2. Probability distribution plots of select variables for participants with and without TSE.

(A) Age at epilepsy onset (years) (nwith TSE = 133, nwithout TSE = 1,586, z = 7.218, p < 0.001), (B) epilepsy duration (years) (nwith TSE = 133, nwithout TSE = 1,581, z = −2.560, p = 0.011), and (C) number of antiseizure medications used (nwith TSE = 75, nwithout TSE = 925, z = −6.880, p < 0.001). The clustered Wilcoxon rank-sum test was used for all statistical tests. TSE = traditionally defined status epilepticus.
Figure 3. Probability distribution plots of select variables for participants with and without OSE.

(A) Age at epilepsy onset (years) (nwith OSE = 218, nwithout OSE = 1,500, z = 9.531, p < 0.001), (B) epilepsy duration (years) (nwith OSE = 218, nwithout OSE = 1,496, p = 0.897), and (C) number of antiseizure medications used (nwith OSE = 124, nwithout OSE = 876, z = −5.132, p < 0.001). The clustered Wilcoxon rank-sum test was used for all statistical tests. OSE = operationally defined status epilepticus.
Chi-square analysis
Under the hypothesis of familial aggregation of SE, we predicted the number of families with ≥2 members with TSE or OSE to be greater than expected by chance. Sixteen of 1,043 family units had a proband and ≥1 first-degree relative with TSE, which was greater than the 5.11 expected from the prevalence of TSE in the EPGP data set (odds ratio [OR] 4.78, 95% confidence interval [CI] 2.56–8.97). In these 16 family units having 2 first-degree relatives with TSE, the 2 relatives did not always have the same epilepsy type; only 10 of 16 were concordant for broad epilepsy class, and only 7 of 10 were concordant for epilepsy syndrome. In 35 of 1,043 family units, a proband and ≥1 first-degree relatives had OSE, which exceeded the 12.6 predicted on the basis of the prevalence of OSE in the study (OR 4.23, 95% CI 2.67–6.70). In these families, 15 of 35 were concordant for broad epilepsy class, and 10 of 15 were concordant for epilepsy syndrome. For comparison, relatives were concordant for broad epilepsy class in 62% (590 of 946) of the family units enrolling only 2 relatives.
GEE analysis
The GEE analysis included the 1,138 participants classified as siblings, parents, or children of probands. If genetic mechanisms contribute to SE, a first-degree relative of a proband with TSE or OSE would be more likely to have at least 1 episode of TSE or OSE than a first-degree relative of a proband who did not have TSE or OSE. However, other factors in some family units such as a clustering of male individuals, an earlier age at onset, a longer duration of epilepsy, a clustering of NAFE, or more severe epilepsy may influence the within-family aggregation of TSE and OSE.
To control for the possible influence of familial characteristics associated with SE, we used multivariable GEE models including as sex, age at onset, epilepsy duration, and having NAFE as covariates (n = 783). In the adjusted TSE model, TSE in the proband predicted TSE in a first-degree relative (OR 2.79, 95% CI 1.24–6.27) (figure 4A). Similarly, OSE in the proband predicted OSE in a first-degree relative (OR 2.91, 95% CI 1.65–5.15) (figure 4B). Earlier age at epilepsy onset and NAFE were also significant predictors of TSE and OSE in these models, but sex and epilepsy duration were not significant. In other adjusted models, TSE in the proband predicted OSE in a first-degree relative (OR 3.19, 95% CI 1.64–6.19). However, OSE in the proband did not predict TSE in a first-degree relative (OR 1.90, 95% CI 0.91–3.96), possibly because the TSE group is only a subset of the OSE group.
Figure 4. ORs for risk factors for TSE and OSE.
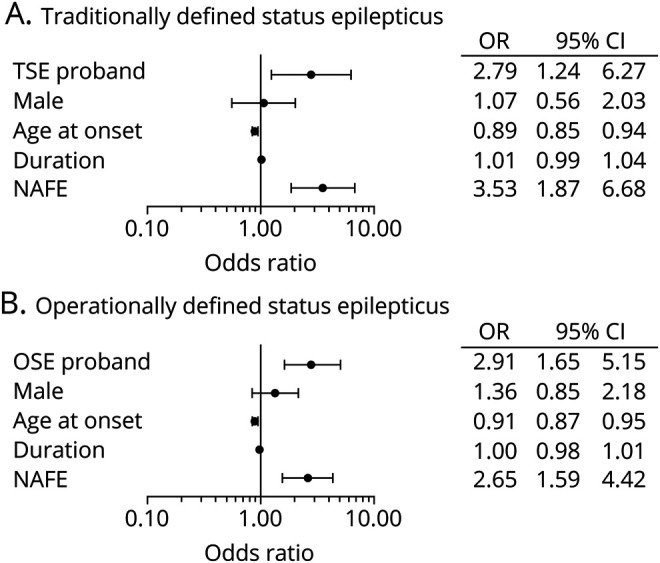
Odds ratios (ORs) for potential risk factors for (A) traditionally defined status epilepticus (TSE) and (B) operationally defined status epilepticus (OSE) were calculated with a generalized estimating equations model. TSE proband and OSE proband indicate the adjusted OR for a first-degree relative having TSE or OSE if the proband has TSE or OSE. The other independent variables apply to the first-degree relative with age at epilepsy diagnosis (age at onset) and epilepsy duration (duration) being continuous variables and the others being binary. Sample size = 783 in panels A and B. CI = confidence interval; NAFE = nonacquired focal epilepsy.
We also elected to examine the effects of other commonly used clinical indicators of epilepsy severity in our TSE and OSE GEE models. First, we added the number of antiseizure medications used (>2 vs ≤2) to the adjusted TSE and OSE models described above.20 With the addition of the antiseizure medication variable to the model, the OR for TSE in first-degree family members of probands with vs without TSE was 4.37 (95% CI 1.30–14.7) (n = 385) (figure 5A), and the OR for OSE in first-degree family members of probands with vs without OSE was 3.26 (95% CI 1.41–7.50) (n = 385) (figure 6A). Models including a positive history of epilepsy surgery as a measure of severity provided similar results. The OR for TSE in a first-degree relative of probands with vs without TSE was 4.43 (95% CI 1.48–13.3) (n = 396) (figure 5B), and the OR for OSE in a first-degree relative of probands with vs without OSE was 3.26 (95% CI 1.40–7.60) (n = 396) (figure 6B). The wider CIs in the GEE analyses including number of antiseizure medications and a history of epilepsy surgery reflect a smaller sample size because of missing data (see Methods and table). The only other significant covariate for TSE in these models was the number of antiseizure medications. Age at epilepsy onset and the number of antiseizure medications were significant covariates for OSE.
Figure 5. ORs for risk factors for TSE with adjustment for epilepsy severity.
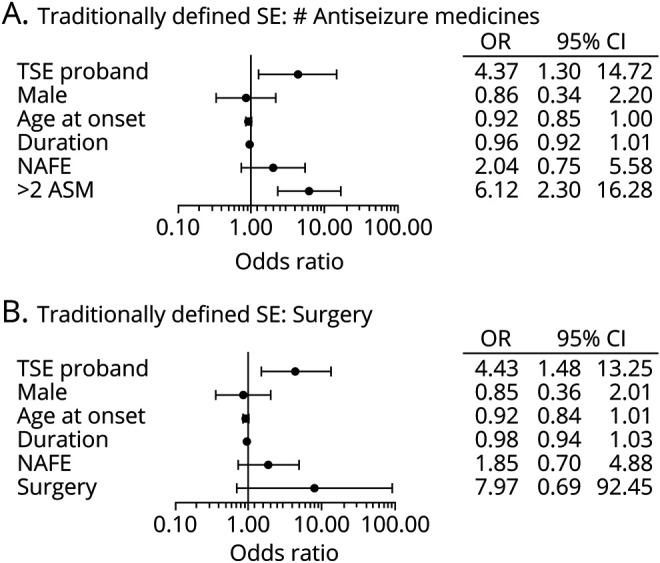
Odds ratios (ORs) for potential risk factors for traditionally defined status epilepticus (TSE) were calculated with a generalized estimating equations model including (A) number of the antiseizure medications (ASMs) used or (B) epilepsy surgery. TSE proband indicates the adjusted OR for a first-degree relative having TSE if the proband has TSE. The other independent variables apply to the first-degree relative with age at epilepsy diagnosis (age at onset) and epilepsy duration (duration) being continuous variables and the others being binary. Sample size = 385 in panel A and 396 in panel B. CI = confidence interval; NAFE = nonacquired focal epilepsy; SE = status epilepticus; surgery = epilepsy surgery.
Figure 6. ORs for risk factors for OSE with adjustment for epilepsy severity.
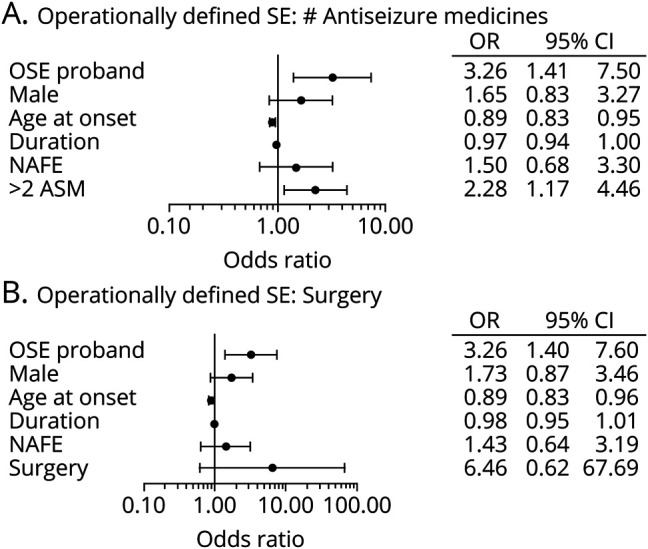
ORs for potential risk factors for operationally defined status epilepticus (OSE) were calculated with a generalized estimating equations model including (A) number of the antiseizure medications (ASMs) used or (B) epilepsy surgery. OSE proband indicates the adjusted OR for a first-degree relative having OSE if the proband has OSE. The other independent variables apply to the first-degree relative with age at epilepsy diagnosis (age at onset) and epilepsy duration (duration) being continuous variables and the others being binary. Sample size = 385 in panel A and 396 in panel B. CI = confidence interval; NAFE = nonacquired focal epilepsy; SE = status epilepticus; surgery = epilepsy surgery.
Discussion
Our primary finding is that SE, whether defined traditionally or operationally, in families with common epilepsies shows familial aggregation independently of broad epilepsy class and epilepsy severity in a well-phenotyped, large cohort of familial epilepsy. The EPGP database is unique in its size and the detail of participant phenotyping. The participants with TSE and OSE have characteristics similar to those observed in prior studies,23,24 and their characteristics are consistent with those reported previously for sex and broad epilepsy class.25,26 The nature of this cohort broadens the applicability of the findings because EPGP did not selectively enroll families with TSE or OSE, which were not part of the inclusion or exclusion criteria. A prior study in 332 twin pairs with seizures found a higher concordance rate for SE in monozygotic than in dizygotic twins.13,14 Our finding of familial aggregation of SE in 1,043 families in a separate familial cohort strengthens the possibility of a genetic contribution to SE in patients with epilepsy. Furthermore, this familial aggregation is independent of seizure susceptibility and severity, suggesting that it is independent of the genetic cause of epilepsy in these families.
The proportion of male individuals among those with TSE or OSE was higher than the proportion of male individuals in the entire EPGP cohort. After adjustment for the female predominance in the EPGP cohort, the TSE and OSE groups had a 15% excess of male individuals, which is similar to that in population-based studies of SE from all causes in developed countries.26 The female predominance in the entire EPGP cohort may reflect the relatively high proportion of GEs, which are more common in female individuals.17,27,28 This female predominance is unusual because studies reporting a sex difference in epilepsy prevalence typically find it to be slightly higher among male individuals.29,30
Although NAFE is more common than GE,31 EPGP included more participants with GE than NAFE. The high proportion of GE in EPGP is expected because the familial risk of epilepsy is greater in GE than NAFE.17,32 The overall prevalence of SE and the prevalence of SE among those with NAFE being twice as high as those with GE in this study are similar to those reported previously.25,33–35
The predominance of male individuals and those with NAFE among participants with TSE and OSE does not account for the familial aggregation of TSE and OSE in the EPGP database based on the GEE analysis, but the familial aggregation of other clinical characteristics could underlie the finding. For example, monozygotic twins show greater concordance than dizygotic twins for febrile SE.36 This observation raises the possibility that the familial aggregation of TSE and OSE reflects the familial aggregation of febrile SE. However, the exclusion of those with febrile seizures ≥15 minutes did not alter the results in the χ2 or unadjusted GEE analysis (data not shown). We did not include prolonged febrile seizures as a covariate in the adjusted models for the following reasons: (1) all participants have epilepsy, making the distinction between a febrile seizure and an epileptic seizure in the setting of a fever difficult; (2) common factors may predispose those with febrile seizures and those with epilepsy to have SE; (3) the number of febrile seizures of uncertain length (table) and the difficulty in determining whether participants with prolonged febrile seizures were classified as having OSE on the basis of a prolonged febrile seizure alone were concerns; and (4) some with a febrile seizure ≥15 minutes may not have had TSE as defined here.
Another possible explanation for our results is that TSE and OSE are simply markers for epilepsy severity because SE is a predictor of drug-resistant epilepsy,37 although they may not predict outcome in drug-resistant epilepsy.38 Despite epilepsy surgery being more common among those with TSE and OSE, it did not account for the familial aggregation of TSE and OSE. Similarly, the number of antiseizure medications used did not eliminate familial aggregation. This result supports the argument of others that SE is not just a marker for severe epilepsy.11,39
Despite the extensive efforts to characterize the participants in EPGP well,18 this study used medical records, which limited detailed characterization of some participants and may have resulted in misclassification of participants. The 17% of participants whose broad epilepsy type could not be classified is one such shortcoming, as recognized in other familial studies of epilepsy.32 We do not know the semiology of the SE, although most episodes of SE likely were generalized convulsive seizures because they are easy to recognize. The limited numbers of participants classified as having a specific epilepsy syndrome preclude a syndrome-based analysis of SE. We also do not know whether they were on a maintenance antiseizure medication at the time of the SE, although most had their first episode of SE early during the course of their disease, as others have shown previously.33,34
Other factors that may have influenced our results are important to recognize. A diagnosis of SE in this cross-sectional study depended on documentation in the medical record by clinicians who may have missed SE because emergency department clinicians are known to underestimate seizure duration and not recognize SE.40 Recognizing nonconvulsive seizures after a convulsive seizure and distinguishing the ictal from postictal state in those with focal seizures with impaired awareness are diagnostically challenging for clinicians and families. Some participants may not yet have had SE but will in the future. The number of such participants may be relatively small because the probability of a first episode of SE in children appears to plateau at 7.5% at 7 to 8 years after diagnosis,25 which is near the median duration of epilepsy in this study. In addition, controlling for duration of epilepsy at enrollment did not eliminate the familial aggregation of SE despite the longer duration of epilepsy in these participants. We also do not know how poor adherence to the treatment regimen or antiseizure medication levels affected the occurrence of SE in the EPGP population. Any underreporting of SE in this sample, including the omission of 5- to 10-minute-long seizures, which would meet the new definition of SE,1,2 may have reduced the robustness of our findings. Alternatively, there may have been reporting bias because family members who had observed SE may be more likely to recognize SE, including nonconvulsive events. Finally, some psychogenic nonepileptic seizures may have inadvertently been included in this cohort. Although having a family member with epilepsy is a known risk factor for psychogenic nonepileptic seizures, the stringent phenotyping for study inclusion should have minimized this confounding factor.41
Recognition of the familial aggregation for SE is valuable clinically and in guiding future research in epilepsy and SE. This finding is helpful in counseling patients and their families about their risk for SE. This familial aggregation was independent of broad epilepsy class, just as the age at epilepsy onset is independent of broad epilepsy class.42 This observation may reflect a shared environmental or epigenetic factor,11,43 and future studies should investigate the interaction between environmental factors and genetics in determining the risk for SE. The familial aggregation found here also suggests that SE may help define a subgroup of patients with shared epilepsy susceptibility genes that do not necessarily predispose to a specific seizure or epilepsy type, as already proposed by an earlier study using the EPGP dataset.17 This result underscores the importance of detailed phenotyping to the identification of these genes.15,16 Our findings further suggest that distinct factors may be responsible for seizure termination and predisposition. The identification of factors responsible for seizure termination, which have received little attention,8–10 may provide insight into clinical presentations that occur in patients without active epilepsy such as new-onset refractory SE and febrile infection-related epilepsy syndrome, a subcategory of new-onset refractory SE .44 More important, their identification may lead to a better understanding of the mechanisms underlying seizure termination and to novel therapies for preventing and treating this common neurologic emergency.
Acknowledgment
The authors recognize the invaluable contributions of the individuals with epilepsy and their families who participated in EPGP. They thank the EPGP clinical coordinators, site PIs, neurologists, and support staff for their assistance with recruitment, data management, and phenotyping. They appreciate the efforts of the EPGP Community Referral Network composed of healthcare professionals not paid by EPGP who referred eligible families to EPGP.
Glossary
- CI
confidence interval
- EPGP
Epilepsy Phenome/Genome Project
- GE
generalized epilepsy
- GEE
generalized estimating equations
- NAFE
nonacquired focal epilepsy
- OE
operationally defined SE
- OR
odds ratio
- PI
principal investigator
- SE
status epilepticus
- TSE
traditionally defined SE
Appendix 1. Authors
Appendix 2. Coinvestigators

Footnotes
Editorial, page 667
Study funding
Study funded by the NIH (U01NS053998).
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Chen JW, Wasterlain CG. Status epilepticus: pathophysiology and management in adults. Lancet Neurol 2006;5:246–256. [DOI] [PubMed] [Google Scholar]
- 2.Trinka E, Cock H, Hesdorffer D, et al. A definition and classification of status epilepticus: report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015;56:1515–1523. [DOI] [PubMed] [Google Scholar]
- 3.Dham BS, Hunter K, Rincon F. The epidemiology of status epilepticus in the United States. Neurocrit Care 2014;20:476–483. [DOI] [PubMed] [Google Scholar]
- 4.Sculier C, Gainza-Lein M, Sanchez Fernandez I, Loddenkemper T. Long-term outcomes of status epilepticus: a critical assessment. Epilepsia 2018;59(suppl):155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neligan A, Noyce AJ, Gosavi TD, Shorvon SD, Kohler S, Walker MC. Change in mortality of generalized convulsive status epilepticus in high-income countries over time: a systematic review and meta-analysis. JAMA Neurol 2019;76:897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanchez Fernandez I, Loddenkemper T. Estimating the cost of admissions related to convulsive status epilepticus in the United States of America. Seizure 2018;61:186–198. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez Fernandez I, Abend NS, Agadi S, et al. Gaps and opportunities in refractory status epilepticus research in children: a multi-center approach by the Pediatric Status Epilepticus Research Group (pSERG). Seizure 2014;23:87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loscher W, Kohling R. Functional, metabolic, and synaptic changes after seizures as potential targets for antiepileptic therapy. Epilepsy Behav 2010;19:105–113. [DOI] [PubMed] [Google Scholar]
- 9.Kohling R. Prolonged seizures: what are the mechanisms that predispose or cease to be protective? A review of animal data. Epileptic Disord 2014;16(suppl):23–36. [DOI] [PubMed] [Google Scholar]
- 10.Zubler F, Steimer A, Gast H, Schindler KA. Seizure termination. Int Rev Neurobiol 2014;114:187–207. [DOI] [PubMed] [Google Scholar]
- 11.Bhatnagar M, Shorvon S. Genetic mutations associated with status epilepticus. Epilepsy Behav 2015;49:104–110. [DOI] [PubMed] [Google Scholar]
- 12.Adotevi N, Lewczuk E, Sun H, et al. α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor plasticity sustains severe, fatal status epilepticus. Ann Neurol 2020;87:84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corey LA, Pellock JM, Boggs JG, Miller LL, DeLorenzo RJ. Evidence for a genetic predisposition for status epilepticus. Neurology 1998;50:558–560. [DOI] [PubMed] [Google Scholar]
- 14.Corey LA, Pellock JM, DeLorenzo RJ. Status epilepticus in a population-based Virginia twin sample. Epilepsia 2004;45:159–165. [DOI] [PubMed] [Google Scholar]
- 15.Winawer MR, Shih J, Beck ES, Hunter JE, Epstein MP; EPGP Investigators. Genetic effects on sleep/wake variation of seizures. Epilepsia 2016;57:557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tobochnik S, Fahlstrom R, Shain C, Winawer MR; EPGP Investigators. Familial aggregation of focal seizure semiology in the Epilepsy Phenome/Genome Project. Neurology 2017;89:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Epi4K Consortium. Phenotypic analysis of 303 multiplex families with common epilepsies. Brain 2017;140:2144–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.EPGP Collaborative; Abou-Khalil BA, Alldredge B, Bautista J, et al. The Epilepsy Phenome/Genome Project. Clin Trials 2013;10:568–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGovern K, Karn CF, Fox K; EPGP Investigators. Surpassing the target: how a recruitment campaign transformed the participant accrual trajectory in the Epilepsy Phenome/Genome Project. Clin Transl Sci 2015;8:518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010;51:1069–1077. [DOI] [PubMed] [Google Scholar]
- 21.R Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 22.Y Jiang. clusrank: Wilcoxon Rank Sum Test for Clustered Data: R package version 0.1-1. Vienna: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 23.DeLorenzo RJ, Garnett LK, Towne AR, et al. Comparison of status epilepticus with prolonged seizure episodes lasting from 10 to 29 minutes. Epilepsia 1999;40:164–169. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez Fernandez I, Vendrame M, Kapur K, et al. Comparison of pediatric patients with status epilepticus lasting 5-29 min versus ≥30 min. Epilepsy Behav 2014;37:1–6. [DOI] [PubMed] [Google Scholar]
- 25.Berg AT, Shinnar S, Testa FM, et al. Status epilepticus after the initial diagnosis of epilepsy in children. Neurology 2004;63:1027–1034. [DOI] [PubMed] [Google Scholar]
- 26.Neligan A, Shorvon SD. Frequency and prognosis of convulsive status epilepticus of different causes: a systematic review. Arch Neurol 2010;67:931–940. [DOI] [PubMed] [Google Scholar]
- 27.Christensen J, Kjeldsen MJ, Andersen H, Friis ML, Sidenius P. Gender differences in epilepsy. Epilepsia 2005;46:956–960. [DOI] [PubMed] [Google Scholar]
- 28.McHugh JC, Delanty N. Epidemiology and classification of epilepsy: gender comparisons. Int Rev Neurobiol 2008;83:11–26. [DOI] [PubMed] [Google Scholar]
- 29.Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the “common” neurologic disorders? Neurology 2007;68:326–337. [DOI] [PubMed] [Google Scholar]
- 30.Banerjee PN, Filippi D, Allen Hauser W. The descriptive epidemiology of epilepsy: a review. Epilepsy Res 2009;85:31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olafsson E, Ludvigsson P, Gudmundsson G, Hesdorffer D, Kjartansson O, Hauser WA. Incidence of unprovoked seizures and epilepsy in Iceland and assessment of the epilepsy syndrome classification: a prospective study. Lancet Neurol 2005;4:627–634. [DOI] [PubMed] [Google Scholar]
- 32.Peljto AL, Barker-Cummings C, Vasoli VM, et al. Familial risk of epilepsy: a population-based study. Brain 2014;137:795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sillanpaa M, Shinnar S. Status epilepticus in a population-based cohort with childhood-onset epilepsy in Finland. Ann Neurol 2002;52:303–310. [DOI] [PubMed] [Google Scholar]
- 34.Stroink H, Geerts AT, van Donselaar CA, et al. Status epilepticus in children with epilepsy: Dutch study of epilepsy in childhood. Epilepsia 2007;48:1708–1715. [DOI] [PubMed] [Google Scholar]
- 35.Malek AM, Wilson DA, Martz GU, et al. Mortality following status epilepticus in persons with and without epilepsy. Seizure 2016;42:7–13. [DOI] [PubMed] [Google Scholar]
- 36.Eckhaus J, Lawrence KM, Helbig I, et al. Genetics of febrile seizure subtypes and syndromes: a twin study. Epilepsy Res 2013;105:103–109. [DOI] [PubMed] [Google Scholar]
- 37.Berg AT, Levy SR, Novotny EJ, Shinnar S. Predictors of intractable epilepsy in childhood: a case-control study. Epilepsia 1996;37:24–30. [DOI] [PubMed] [Google Scholar]
- 38.Callaghan B, Schlesinger M, Rodemer W, et al. Remission and relapse in a drug-resistant epilepsy population followed prospectively. Epilepsia 2011;52:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shorvon S, Trinka E. Regulatory aspects of status epilepticus. Epilepsia 2018;59(suppl):128–134. [DOI] [PubMed] [Google Scholar]
- 40.Hesdorffer DC, Shinnar S, Lewis DV, et al. Design and phenomenology of the FEBSTAT study. Epilepsia 2012;53:1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vincentiis S, Valente KD, Thome-Souza S, Kuczinsky E, Fiore LA, Negrao N. Risk factors for psychogenic nonepileptic seizures in children and adolescents with epilepsy. Epilepsy Behav 2006;8:294–298. [DOI] [PubMed] [Google Scholar]
- 42.Ellis CA, Churilov L, Epstein MP, et al. Epilepsy in families: age at onset is a familial trait, independent of syndrome. Ann Neurol 2019;86:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henshall DC. Epigenetic changes in status epilepticus. Epilepsia 2018;59(suppl):82–86. [DOI] [PubMed] [Google Scholar]
- 44.Hirsch LJ, Gaspard N, van Baalen A, et al. Proposed consensus definitions for new-onset refractory status epilepticus (NORSE), febrile infection-related epilepsy syndrome (FIRES), and related conditions. Epilepsia 2018;59:739–744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset containing the deidentified participant data analyzed for this study is available from the corresponding author on request.