

Abstract
Objective
To gather natural history data to better understand the changing course of type 2 Gaucher disease (GD2) in order to guide future interventional protocols.
Methods
A structured interview was conducted with parents of living or deceased patients with GD2. Retrospective information obtained included disease presentation, progression, medical and surgical history, medications, family history, management, complications, and cause of death, as well as the impact of disease on families.
Results
Data from 23 patients were analyzed (20 deceased and 3 living), showing a mean age at death of 19.2 months, ranging from 3 to 55 months. Fourteen patients were treated with enzyme replacement therapy, 2 were treated with substrate reduction therapy, and 3 underwent bone marrow transplantation. Five patients received ambroxol and one was on N-acetylcysteine, both considered experimental treatments. Fifteen patients had gastrostomy tubes placed; 10 underwent tracheostomies. Neurologic disease manifestations included choking episodes, myoclonic jerks, autonomic dysfunction, apnea, seizures, and diminished blinking, all of which worsened as disease progressed.
Conclusions
Current available therapies appear to prolong life but do not alter neurologic manifestations. Despite aggressive therapeutic interventions, GD2 remains a progressive disorder with a devastating prognosis that may benefit from new treatment approaches.
Gaucher disease (GD) is an autosomal recessive lysosomal storage disorder caused by deficient glucocerebrosidase due to pathogenic biallelic variants in GBA1. It is classically divided into 3 types based on the presence and severity of neurologic progression: non-neuronopathic type 1 (GD1), acute neuronopathic type 2 (GD2), and chronic neuronopathic type 3 (GD3),1,2 although considerable clinical heterogeneity is seen within each type. The symptoms of GD1 range from asymptomatic to greatly affected with hepatosplenomegaly, cytopenias, and bone involvement.3,4 With GD3, manifestations include visceral involvement, myoclonic epilepsy, ataxia, learning deficits or dementia, and impaired horizontal saccadic ocular movements.3,4 Enzyme replacement therapy (ERT) effectively treats the visceral manifestations but it does not cross the blood–brain barrier. There is no validated effective therapy for the neurologic manifestations of GD2 and GD3.5
GD2, the most severe and progressive form, leads to death in infancy or early childhood. Initially thought to be stereotypic, in presentation, the phenotype now includes hydrops fetalis, congenital ichthyosis, hepatosplenomegaly, thrombocytopenia, anemia, and failure to thrive with progressive deterioration in swallow function.6–8 In the last decade, advances in diagnostic methods combined with more aggressive treatment and access to experimental therapies have prolonged the survival of such infants. This study aimed to delineate the changing clinical course of GD2, in order to provide a better understanding of the current natural history. This data are essential when designing future interventional protocols and will help to identify treatment goals as well as milestones to measure efficacy more accurately.
Methods
Standard protocol approvals, registrations, and patient consents
Twenty-three patients with GD2 were enrolled by informed parental consent in this natural history protocol (NCT00001215) approved by the National Human Genome Research Institute Institutional Review Board at the NIH.
Patients
A cohort of 23 living or deceased patients with clinically confirmed GD2 were identified from patient databases, patient support groups, and referring physicians.
Methods
A structured telephone interview consisting of standardized and in-depth medical questions was conducted with caregivers of affected individuals. Medical records were reviewed based on availability.
A 5-page questionnaire was developed and used during the telephone/in-person interview addressing a wide range of topics, beginning with general demographics and a detailed family history. Details of disease presentation were elicited including the age at presentation, initial symptoms, diagnostic odyssey, and method used to confirm diagnosis. The progression of disease was reviewed including extent of organ involvement, developmental milestones, eye findings, and neurologic manifestations. Parents were asked about imaging studies, biopsies, interventions, surgeries, and disease/procedural complications. We specifically inquired about feeding methods, formulas, food consistency, weight gain, and use of feeding tubes. Respiratory issues were elicited, including history of tracheostomy or need for ventilatory support, including the indications. Hospitalizations and medications were reviewed.
Additional questions focused on the consequences of the child's disease on family members including marital stress, custody issues, separations, insurance issues, mental health involvement, and whether counseling was offered to the family. These questions were not part of a validated questionnaire or scale. The age, cause, and circumstances surrounding death, if applicable, were discussed, as well as details of hospice care, including reasons for initiating the process. If an autopsy was performed, parents were asked to share the reports. Demographic and clinical data were summarized with graphic statistics and frequencies and percentages of discrete variables. Genotyping was performed by bidirectional Sanger sequencing of all 11 exons and flanking intronic regions of GBA1 as described previously.9
Data availability
Anonymized data will be shared by request from any qualified investigator.
Results
Demographics
Of the 23 patients, 15 (65.3%) were female and 8 (34.7%) were male (table 1). Five patients (21.7%) had a family history of GD2, including 4 sets of siblings. The majority of the cohort was Caucasian, but African American, Native American, Hispanic, and South Asian ancestries were also reported.
Table 1.
Demographics, family history, and prenatal and postnatal history

Prenatal and postnatal history
Twenty-one patients (91.3%) were born at term (table 1). Two (8.7%) were preterm twins weighing 1.45 kg at birth, whose mother had premature rupture of membranes and an emergency caesarean section at 28 weeks gestation. The birth weights of the other 21 infants ranged from 2.4 to 4.5 kg, with 14 weighing over 3 kg. Immediate postnatal manifestations included jaundice (34.7%), thrombocytopenia (26%), difficulty establishing feeds (26%), liver and/or spleen enlargement (17.4%), stridor (13%), congenital ichthyosis (13%), choking episodes (13%), gastroesophageal reflux (GER) (4.3%), and transient tachypnea of the newborn (4.3%). Ten (44%) patients were transferred to a referral hospital or newborn intensive care unit during the neonatal period.
Diagnosis of GD2
Sixteen patients (69.5%) presented with disease-related findings at birth including thrombocytopenia (30.4%), feeding issues (30.4%), failure to thrive (FTT) (30.4%), liver and/or spleen enlargement (21.7%), choking episodes (21.7%), jaundice (21.7%), apnea (17.4%), stridor (17.4%), collodion baby phenotype (13%), fussiness (8.7%), GER (8.7%), strabismus (8.7%), poor suck/swallow (4.3%), and vasovagal episodes, where the infant was reported to have “turned red and stopped breathing” (4.3%). The other 7 patients presented at 1–6 months of age, with symptoms including strabismus (21.7%), FTT (21.7%), fussiness (13%), choking episodes (8.7%), GER (8.7%), splenomegaly (4.3%), feeding issues (4.3%), developmental delay (4.3%), and hypertonia (4.3%) (table 2). The age at diagnosis ranged from 1 week to 15 months (mean 5.7 months). Of the 23 patients, 20 are deceased (87%), with 3 currently alive (13%) as of January 2020. The mean age at death was 19.2 months, with a range from 3 to 55 months. Regardless of age at first manifestation, there was often a considerable diagnostic delay, ranging from 1 week to 11 months (mean of 5 months). While ultimately the diagnosis was confirmed by molecular analyses on each patient, bone marrow biopsies were performed on 7 (30.4%). One patient (21) was identified during a pilot newborn screening project.
Table 2.
Diagnosis of GD2

Patient genotypes
The individual genotypes reported in table 2 are listed using a conventional numbering system without the 39 amino acid GBA1 leader sequence. Wide genotypic heterogeneity was observed. While L444P was the most common individual mutation, no patient was homozygous for L444P. Six patients (26%) had an L444P allele with a recombinant allele that included L444P. D409H was encountered in 5 patients (21.7%), G202R and IVS2+1G>A in 2 each (8.7%), and the twins shared genotype G416C/R296Q. Of note, in 2 cases, a mutant allele was not identified in the mother, presumably as a result of somatic mosaicism or a new mutation as previously reported.10,11
Systemic manifestations
Three infants (13%) were described as “collodion” babies because of congenital ichthyosis, and 1 (4.3%) had peeling of skin at birth (table 3). Twelve babies (52.1%) were reported to have thrombocytopenia, and anemia was mentioned in 6 (26.1%). Organomegaly, usually both hepatomegaly and splenomegaly, was reported in 19 (83%), and 4 (17.4%) had a history of jaundice. While cardiac manifestations were not common, murmurs were reported in 4 (17.4%), and 1 underwent surgery for a vascular ring.
Table 3.
Different manifestations and interventions

Diagnostic evaluations included MRI or CT study of the brain, performed in 15 patients (65%). Abdomen imaging was performed in 21 patients (91%): ultrasound in 18, CT in 4, and MRI in 3. Barium swallow studies were performed in 10 patients (43.4%). One patient had an MRI of the chest, 1 underwent an echocardiogram, and 1 had a spine MRI and a DEXA scan.
Respiratory involvement was universal. Difficulties in handling secretions, apnea, stridor, choking episodes, cyanotic episodes, or laryngospasms were described in every child (100%). While these manifestations were secondary to brainstem involvement, they eventually led to complications due to aspiration. Ten patients (43.5%) had tracheostomies due to respiratory compromise.
FTT was observed in all affected infants (100%). This was directly related to problems with swallowing, choking, and/or food aversion. Tube feeding was required at some point for 20 of the infants (87%). Gastrostomy tubes (G-tubes) were surgically placed in 15 (65.2%). However, patient 5 was reportedly eating pureed food and crackers until his death. GER was described in 17 patients (74%).
General fussiness was described in almost all patients. Fevers and excessive sweating were described in 14 infants (61%). Reactions to anesthesia were reported in 5 (21.7%); 4 patients had difficulty being weaned off anesthesia and 1 was described to have a high tolerance to anesthesia. Two of the older infants were noted to have scoliosis and/or kyphosis.
Developmental milestones
All patients were reported to have developmental delay (100%). Developmental milestones achieved varied widely. A social smile was described in all (100%). However, age at achieving this milestone ranged from 1 to 6 months. Verbal development also differed greatly. Twelve infants (52.2%) did not babble or talk. Nine children were described as babbling (39.1%). Five children (21.7%) were able to say words like “mama,” “dada,” or “Elmo,” although onset of language was delayed. None progressed to say more than a few words.
Twenty of the infants (87%) were described as being able to hold their head up. Patient 3 had developmental regression and lost this skill. Twelve infants (52.1%) were able to roll over at least once. Nine infants (39.1%) were able to sit supported at some point, 2 crawled (8.7%), and 3 (13%) could stand or use a walker. Two patients were able to take steps with assistance, one being patient 22, described in a series of children who fell into an intermediate phenotype between GD2 and GD3.12
Neurologic and neuro-ophthalmologic manifestations
One of the major neurologic symptoms described was abnormal tone, which was seen in all the patients (100%). Hypotonia was reported in 8 patients (34.8%), while 15 (65.2%) had hypertonia, which was variable in severity. The range included generalized hypertonia, neck/truncal hypertonia, truncal and lower extremity hypertonia, or opisthotonos. Patient 4 had such pronounced hypertonia that she was initially diagnosed with stiff-person syndrome. Poor suck and swallow reflex were reported in 16 patients (69.6%). Fifteen infants (65.2%) developed myoclonus, and other forms of seizures were mentioned in 10 (43.5%). Seventeen (74%) had an EEG performed at some point during their course, 9 of which were abnormal. Strabismus was a common symptom, reported in 18 patients (78.3%). This was often the initial finding prior to diagnosis. Four infants (17.4%) were noted to have oculomotor apraxia. Decreased blinking was a subjective finding noted by parents of 18 children (78.3%).
Therapy and management
The decision to treat these affected infants reflected parental and physician preferences (table 4). ERT was utilized in 14 infants (60.9%). Ten of the patients (43.5%) had long-term central venous access established in the form of a port-a-cath or Broviac line as noted in table 4. The age at ERT initiation ranged from several weeks to 13 months (figure 1). Six infants (26.1%) started infusions after 7 months of age. The duration of ERT treatment for these patients varied, ranging from 1 to 54 months. Among the 14 patients treated with ERT, parents reported clinical improvement in 10 (71.4%). Seven parents (50%) reported that their children appeared to be more comfortable, 7 (50%) reported improvement of organomegaly, 2 patients (14.3%) gained weight, and 3 patients (21.4%) had improved complete blood counts. One family (7.1%) reported a decrease in vasovagal episodes. Three parents (21.4%) did not appreciate any clinical response resulting from ERT. Among the 9 patients who did not receive ERT, the age at death ranged from 3 to 17 months (mean 9.8 months). The age at death ranged between 8 and 55 months (mean 26.9) for the 11 treated infants who died. As of January 2020, there are 3 living children who are all receiving ERT with ambroxol who are aged 74, 17, and 27 months.
Table 4.
Treatment and management

Figure 1. Graphic rendition of clinical course over time.

Summary of the clinical course from birth of 23 children with type 2 Gaucher disease demonstrating the disease duration and the treatments and interventions attempted. The shaded regions indicate the use of enzyme replacement therapy (ERT) (light blue), substrate reduction therapy (SRT) + ERT (salmon), ambroxol (dark blue), and ERT + ambroxol (blue–green).
Two patients received miglustat, a substrate reduction therapy (SRT), in addition to ERT. Patient 7 received the drug for 1 month until age 16 weeks. Patient 8 was started on SRT at 1 year of age, 6 months after initiating ERT, continuing until his death 24 months later. Ambroxol was administered in addition to ERT in 3 cases and 2 infants received ambroxol as monotherapy. Bone marrow transplantation (BMT) was attempted in 3 children (patients 7, 10, and 22). Patient 7 underwent BMT twice, at 12 and 16 weeks. Each of the transplanted children also received ERT at some point. The transplanted patients 7, 10, and 22 died at 15, 55, and 51 months, respectively. Patients 7 and 10 died during sleep; patient 22 died from a cardiac arrest posttransplant.
Other classes of medications were administered for symptom management (table 4). Fifteen infants (65.2%) received histamine-2 blockers or proton pump inhibitors to treat GER. Erythromycin, lactulose, metoclopramide, or cisapride were given for gastrointestinal motility issues. Most infants were treated with antiepileptic medications, including cannabidiol oil. Two patients were given carbidopa/levodopa with or without bromocriptine for abnormal movements. Glycopyrrolate, atropine drops, and onabotulinumtoxinA injections were administered to control secretions, while baclofen was given for muscle spasms. One infant was treated with N-acetylcysteine with the thought that its antioxidant and anti-inflammatory activities might improve neurologic symptoms. Gabapentin, morphine, hydromorphone, methadone, and fentanyl were used for pain management. One infant was treated with propranolol for autonomic dysfunction and another with phenobarbital for laryngeal spasms.
The 2 most common surgical interventions were G-tube placement and tracheostomy, which both had a correlation with longevity (figure 1). With the exception of patient 5, no patient without a feeding tube survived past 9 months. Patient 11 received total parenteral nutrition (TPN) for 150 days after his bone marrow transplant. Of the 10 infants who survived past 16 months, 7 (70%) required tracheostomies. Ventilated patients often had difficulty being extubated, and several required a tracheostomy after hospitalizations for respiratory-related illnesses.
Hospitalizations and complications
All patients were hospitalized at some point during their disease course. Eight infants (34.8%) were admitted for extended evaluations immediately or within 1 week of birth. The premature twins went directly to the neonatal intensive care unit and never went home. Most patients were hospitalized prior to, or at time of diagnosis, usually for an evaluation due to FTT or choking episodes. Eighteen patients (78.2%) had at least 3 prolonged hospitalizations, ranging from a week to months, frequently requiring admissions to the intensive care unit for mechanical ventilation. Frequent indications for recurrent hospitalizations included respiratory-related illnesses, tracheitis, sepsis, seizures, abdominal complications, and surgical procedures.
End of life
The age at death among the 20 deceased patients ranged from 3 to 55 months. Hospice care was provided for 12 patients (60%), although duration of hospice care varied from days to over 4 months. Several patients received around-the-clock nursing care. Twelve babies (60%) died at home, while the other 8 patients (40%) died in the hospital.
The cause of death was listed as respiratory failure in 15 cases (75%), cardiac arrest in 2 (10%), progressive brainstem dysfunction in 1 (5%), and unexplained deaths that occurred during sleep in 2 (10%). Of the 15 cases of respiratory failure, 7 were due to worsening seizures, 2 were caused by complications of pneumonia, 2 were caused by hypoxic events, 2 were caused by frequent apneas, 1 was due to worsening tracheitis, and 1 was due to acute respiratory distress syndrome secondary to a fat embolism caused by a fracture.
Autopsies were performed on 3 of the patients (9, 18, and 22). The neuropathology autopsy report for patient 9 revealed changes consistent with the clinical diagnosis of GD2. There was neuronal loss, predominantly in calcarine cortex (with Gaucher cells) and hippocampal C4 sector. There was moderate neuronal loss seen in thalamus, CA3 hippocampal sector, several structures of the brainstem, and dentate nucleus. Perivascular Gaucher cells were seen in the subcortical white matter. Mild cerebral atrophy with generalized gliosis, mild atrophy of interhemispheric commissures, and mild atrophy of the cortical spinal tracts were described.
Patient 18 died at age 5.5 months. She was placed on hospice care soon after her diagnosis at age 4 months and she did not receive ERT, gastrostomy, or tracheostomy. The cause of death was listed as CNS involvement by GD2. She was found to have extensive involvement with accumulation of Gaucher cells in liver, spleen, lymph nodes, and bone marrow. Her spleen weighed 16 times the mean for her age. Bilateral pulmonary consolidation was noted. The neuropathology report described perivenular and cortical Gaucher cells, gliosis, and neuronal loss.
The autopsy on patient 22 reported the cause of death as acute bilateral pulmonary edema and acute lung injury of unknown cause with subacute right-sided heart failure. The liver showed marked congestion with multifocal involvement by residual GD despite the BMT. Multifocal evidence of disease was found in the spleen, thymus, lymph nodes, and bone marrow. The CNS showed no evidence of Gaucher cells.
Of the 7 infants who survived for more than 24 months, 6 were born after 2008. The seventh (22) was a child considered to have the intermediate phenotype who died after BMT.
Psychosocial
To better understand how parents and families of patients were affected, data related to family separations, insurance issues, mental health issues, and genetic counseling were collected. Two families reported separations, in one case after having 2 affected children and 1 affected fetus. In the second set, the mother of the patient did not feel adequately supported. Nine families reported insurance issues, including difficulties receiving coverage for initiating ERT, home nursing, hospice care, and emergency medical technician costs. Twelve families reported mental health issues: 3 families reported needing antidepressive/anxiolytic medication, 9 families reported one or both parents being diagnosed with depression, and 6 families reported requiring counseling. Fourteen families reported difficulty communicating with medical staff, especially pertaining to the diagnosis, management, and prognosis of GD2. They believed most physicians were unfamiliar with this condition and did not have current or accurate information. For example, one mother reported she had a hysterectomy in her 20s because she was informed there was a 90% chance of having another child with GD2.
Discussion
GD2, the rarest form of the disease, has been recognized since 1927.13 From the start, it was described as a uniformly devastating and progressive disease, with death during the first year of life. By 1972, a series of 67 patients were summarized, describing infants who appeared normal at birth, but subsequently developed hepatosplenomegaly, regression of developmental milestones, and arrest of growth. The age at death was reported as being between 1 month and 2 years, with a mean of 9 months.14 In the past few decades, there has been a greater appreciation of the degree of phenotypic heterogeneity in GD2.15 This was prompted in part by the generation of a neonatal lethal murine model of GD16,17 and the recognition of analogous infants who died either prenatally or shortly after birth.18 This neonatal form of GD was typically associated with hydrops fetalis18,19 or a collodion baby phenotype.20 On the other end of the spectrum, 9 infants were described with an intermediate phenotype characterized by a later onset of manifestations and age at diagnosis (range 8–20 months, mean 12.6).12
In this work, we sought to establish the current natural history of patients with GD2 in the first world setting, including the United States, Canada, and New Zealand. Although the disorder was well described in the 20th century,14,15 our clinical perception is that several factors recently have affected the clinical course of these patients. By performing this retrospective study of 22 infants born in the 21st century, we aimed to provide an updated description of GD2, which will be particularly valuable as new therapies become available.
The study demonstrated that different interventions have contributed to disease duration in patients with GD2 (figure 1). In 1992, the first effective therapy for GD, ERT, became available,21 but because it did not cross the blood–brain barrier, it was not effective for CNS manifestations in neuronopathic GD,22,23 even when administered intrathecally24 or from birth.25 The use of ERT in GD2 was initially discouraged, but in recent years it has been offered to many families, as described here. Two brain-permeable drugs, miglustat and ambroxol, have been prescribed for some children, despite no confirmed indication or efficacy. In a phase II randomized open-label clinical trial of miglustat, it was not found to influence neurologic involvement in patients with GD3.26,27 There are several case reports suggesting that ambroxol, a commonly used expectorant, may affect the neurologic manifestations of GD3,28,29 but no clinical trials have been conducted and there are no data available for use in GD2. BMT are now more widely performed in infants, and 3 children in this series were transplanted.
In addition to new therapeutics, improved supportive care has become more widely available in the past decades. Home NG/NJ feeding, TPN and nutritional supplements are frequently used for gravely ill infants. Home ventilation is now also possible.
The mean age at death among these patients was 19 months, substantially longer than the 9 months described prior to the era of ERT. However, it is misleading to emphasize the mean age, as the surveyed population includes 2 groups, one comprising children who received aggressive interventions, and a second who were treated conservatively. Also, due to improved awareness of GD2 in the newborn period, infants now diagnosed and managed with multiple interventions may have died without a diagnosis in earlier years.
Examining the infants who died before age 16 months separately from those who lived longer than 16 months was particularly informative. Remarkably, the percentage of infants with symptoms at birth or within the first 2 months, and the number diagnosed by age 6 months, was similar in both groups. The major difference was the number of interventions performed. Seven patients who lived longer than 16 months received tracheostomies (64%) as compared to 3 infants in the first group (25%). Ten of the 11 infants in the older groups had ports placed for prolonged infusions of ERT as opposed to only 1 in the younger group. Furthermore, G-tubes were placed in 10 of the 11 (90%) from group 2 vs 4 of 12 (25%) in the first group. BMT may have also prolonged life, as the 3 transplanted patients died at 15, 55, and 51 months. Thus, overall, longevity was likely more correlated with management than initial disease burden.
The 2 major decisions influencing the length of disease duration were whether to begin ERT and whether to perform a tracheostomy. Eight infants never received ERT and 3 were treated with ERT for less than 2 months. Of these 11 children, the mean age at death was 11 months, which was not different from that reported in the pre-ERT era (figure 2). Among those who received ERT for longer than 2 months, mean survival is at least 30 months. In addition, tracheostomy delayed death. Survival among the children who received tracheostomies ranged from 10 to 64+ months with a mean of over 27 months. The longest-lived children tended to have both ERT and tracheostomies. However, 3 of the children who did not undergo tracheostomy lived for over 2 years. Two received bone marrow transplants and the other received weekly ERT with ambroxol, but had several ICU admissions requiring intubation.
Figure 2. Kaplan-Meier plot showing the impact of enzyme replacement therapy (ERT) on survival.
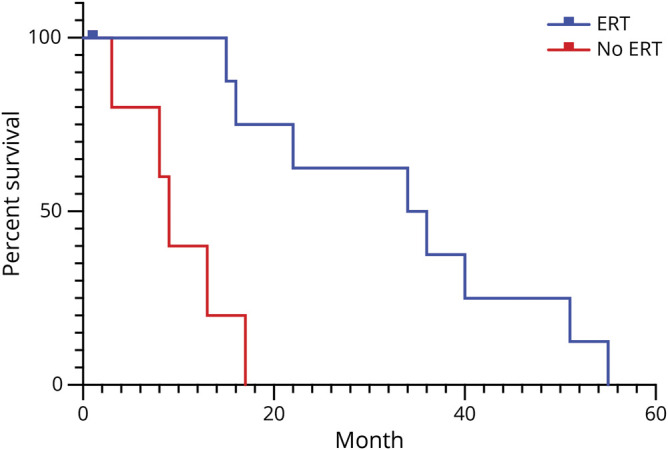
Kaplan-Meier plots showing survival of infants with Gaucher disease who did (blue) or did not (red) receive more than 2 months of ERT.
In this study, genotype was not particularly helpful in establishing prognosis. There was a considerable genotypic heterogeneity, rendering it difficult to establish the effect of specific mutations. Seven infants had mutation L444P together with a recombinant allele, yet their phenotypes and course were varied.
Several new therapeutic strategies are being pursued for neuronopathic GD.5,30 Promising animal data regarding the efficacy of gene therapy in murine models of GD2 haven been reported.31 An adeno-associated virus-directed gene therapy clinical trial for neuronopathic GD was announced recently.32 Several different types of glucocerebrosidase chaperones in addition to ambroxol are being pursued, with the hope that they will be efficacious for neuronopathic GD.33–36 Newer brain-permeable forms of substrate reduction therapy are in trial in adults with GD3 and may undergo trials in patients with GD2.37 N-acetylcysteine, used as an adjuvant therapy in neurodegenerative disorders due to its antioxidant and anti-inflammatory activity, was tried in one patient.38 It remains to be determined whether any of these strategies will successfully reverse or arrest neurodegeneration in this disorder.
One goal of this study was to find specific clinical findings that might be ameliorated by future therapies targeting the CNS, and several were identified. A universal problem was an abnormal swallow, which can be detected and monitored following 5 parameters during a fluoroscopic swallow study.8 Weight gain and intake should be followed closely, as well as symptoms of GER, as improved growth measures might reflect efficacy. Developmental milestones were always markedly delayed, and thus careful periodic developmental assessments could reflect improvement. Airway compromise manifesting as stridor, apnea, choking, and laryngospasm were extremely common and should be followed specifically. Abnormalities on EEGs and brainstem somatosensory evoked potentials were not always seen, but they are not expected to improve without CNS penetrant therapy, and thus may be another measure to monitor. Control of seizures could be a parameter for monitoring. However, in our patient cohort, seizure activity was considered to be the most challenging to control. Given the complexity of presentation, data pertaining to type of seizure and age at onset were not captured. Eye findings were almost universal, and thus longitudinal neuro-ophthalmologic evaluations are warranted. The observation that many of the patients had a decreased blink rate is interesting and may provide an easily ascertained efficacy measure. Improvement in brainstem function might also be reflected by fewer cases of pneumonia, less suctioning, and alleviating the need for tracheostomies. Monitoring the ability to handle secretions and the need for suctioning may be a useful measure. Extreme fussiness, cyanotic episodes, and temperature dysregulation with high fevers were reported frequently. These symptoms should be followed during any future therapeutic trial and may complicate assessment of side effects and off-target effects of future treatments.
It is not clear whether future therapies will replace ERT or be administered concurrently with ERT. In the absence of ERT, it is important to monitor blood counts and liver and spleen size at regular intervals. Other biomarkers like glucosyl sphingosine levels may also prove useful in assessing disease burden.39
Treatment can alter the natural history of a lethal disease, but it can also uncover aspects of the pathophysiology that were never previously appreciated. In the case of GD3, chronic treatment has uncovered the development of Gaucheroma,40 cystic bone lesions,41 and learning disabilities.42 During any future clinical trials, it will be essential to carefully phenotype other organs to determine whether there might be other currently unappreciated manifestations of GD2.
Acknowledgment
The authors thank the participants and the referring physicians.
Glossary
- BMT
bone marrow transplantation
- ERT
enzyme replacement therapy
- FTT
failure to thrive
- GD
Gaucher disease
- GD1
Gaucher disease type 1
- GD2
Gaucher disease type 2
- GD3
Gaucher disease type 3
- GER
gastroesophageal reflux
- SRT
substrate reduction therapy
- TPN
total parenteral nutrition
Appendix. Authors
Study funding
This research was supported by the Intramural Research Programs of the National Human Genome Research Institute and the NIH.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Lal TR, Sidransky E. The spectrum of neurological manifestations associated with Gaucher disease. Diseases 2017;5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiss K, Gonzalez AN, Lopez G, Pedoeim L, Groden C, Sidransky E. The clinical management of type 2 Gaucher disease. Mol Genet Metab 2015;114:110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab 2004;83:6–15. [DOI] [PubMed] [Google Scholar]
- 4.Sidransky E. Gaucher disease: insights from a rare Mendelian disorder. Discov Med 2012;14:273–281. [PMC free article] [PubMed] [Google Scholar]
- 5.Gary SE, Ryan E, Steward AM, Sidransky E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev Endocrinol Metab 2018;13:107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mignot C, Doummar D, Maire I, et al. Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev 2006;28:39–48. [DOI] [PubMed] [Google Scholar]
- 7.Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 2017;18:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seehra G, Solomon B, Ryan E, et al. Five-parameter evaluation of dysphagia: a novel prognostic scale for assessing neurological decline in Gaucher disease type 2. Mol Genet Metab 2019;127:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stone DL, Tayebi N, Orvisky E, et al. Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease. Hum Mutat 2000;15:181–188. [DOI] [PubMed] [Google Scholar]
- 10.Hagege E, Grey RJ, Lopez G, et al. Type 2 Gaucher disease in an infant despite a normal maternal glucocerebrosidase gene. Am J Med Genet A 2017;173:3211–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saranjam H, Chopra SS, Levy H, et al. A germline or de novo mutation in two families with Gaucher disease: implications for recessive disorders. Eur J Hum Genet 2012;21:115–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goker-Alpan O, Schiffmann R, Park JK, et al. Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. J Pediatr 2003;143:273–276. [DOI] [PubMed] [Google Scholar]
- 13.Oberling C, Woringer P. La maladie de Gaucher chez la nourrisson. Rev Fr Pédiatr 1927;3:475–532. [Google Scholar]
- 14.Frederickson DS, Sloan HR. Glucosylceramide lipidoses: Gaucher’s disease. In: Stanbury JB, Wyngaarden JB, Frederickson DS, eds. The Metabolic Basis of Inherited Disease. New York: McGraw-Hill; 1972:730–759. [Google Scholar]
- 15.Tayebi N, Stone DL, Sidransky E. Type 2 Gaucher disease: an expanding phenotype. Mol Genet Metab 1999;68:209–219. [DOI] [PubMed] [Google Scholar]
- 16.Tybulewicz VLJ, Tremblay ML, Lamarca, et al. Animal model of Gaucher disease from targeted disruption of the mouse glucocerebrosidase gene. Nature 1992;357:407–410. [DOI] [PubMed] [Google Scholar]
- 17.Sidransky E, Sherer DM, Ginns EI. Gaucher disease in the neonate: a distinct Gaucher phenotype is analogous to a mouse model created by targeted disruption of the glucocerebrosidase gene. Pediatr Res 1992;32:494–498. [DOI] [PubMed] [Google Scholar]
- 18.Stone DL, Diggelen OPV, Klerk JBD, et al. Is the perinatal lethal form of Gaucher disease more common than classic type 2 Gaucher disease? Eur J Hum Genet 1999;7:505–509. [DOI] [PubMed] [Google Scholar]
- 19.Stone DL, Sidransky E. Hydrops fetalis: lysosomal storage disorders in extremis. Adv Pediatr 1999;46:409–440. [PubMed] [Google Scholar]
- 20.Stone DL. Type 2 Gaucher disease: the collodion baby phenotype revisited. Arch Dis Child Fetal Neonatal Ed 2000;82:F163–F166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency: macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med 1991;324:1464–1470. [DOI] [PubMed] [Google Scholar]
- 22.Erikson A, Johansson K, Månsson J, et al. Enzyme replacement therapy of infantile Gaucher disease. Neuropediatrics 1993;24:237–238. [DOI] [PubMed] [Google Scholar]
- 23.Bove K, Daugherty C, Grabowski G. Pathological findings in Gaucher disease type 2 patients following enzyme therapy. Hum Pathol 1995;26:1040–1045. [DOI] [PubMed] [Google Scholar]
- 24.Bembi BCG, Zanatta M, Bottega M, et al. Cerebrospinal-fluid infusion of Alglucerase in the treatment of acute neuronopathic Gaucher disease. Pediatr Res 1995;38:425. [Google Scholar]
- 25.Prows CA, Sanchez N, Daugherty C, Grabowski GA. Gaucher disease: enzyme therapy in the acute neuronopathic variant. Am J Med Genet 1997;71:16–21. [PubMed] [Google Scholar]
- 26.Schiffmann R, Fitzgibbon EJ, Harris C, et al. Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol 2008;64:514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altarescu G, Hill S, Wiggs E, et al. The efficacy of enzyme replacement therapy in patients with chronic neuronopathic Gaucher's disease. J Pediatr 2001;138:539–547. [DOI] [PubMed] [Google Scholar]
- 28.Kim YM, Yum MS, Heo SH, et al. Pharmacologic properties of high-dose ambroxol in four patients with Gaucher disease and myoclonic epilepsy. J Med Genet 2020;57:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narita A, Shirai K, Itamura S, et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: a pilot study. Ann Clin Transl Neurol 2016;3:200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mistry PK, Lopez G, Schiffmann R, Barton NW, Weinreb NJ, Sidransky E. Gaucher disease: progress and ongoing challenges. Mol Genet Metab 2017;120:8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Massaro G, Mattar CNZ, Wong AMS, et al. Fetal gene therapy for neurodegenerative disease of infants. Nat Med 2018;24:1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prevail Therapeutics Announces IND Active for PR001 for Treatment of Neuronopathic Gaucher Disease. Available at: biospace.com/article/releases/prevail-therapeutics-announces-ind-active-for-pr001-for-treatment-of-neuronopathic-gaucher-disease/. Accessed January 8, 2020.
- 33.Ivanova MM, Changsila E, Turgut A, Goker-Alpan O. Individualized screening for chaperone activity in Gaucher disease using multiple patient derived primary cell lines. Am J Transl Res 2018;10:3750–3761. [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar DT, Iyer S, Christy JP, et al. A comparative computational approach toward pharmacological chaperones (NN-DNJ and ambroxol) on N370S and L444P mutations causing Gaucher’s disease. Adv Protein Chem Struct Biol 2019;114:315–339. [DOI] [PubMed] [Google Scholar]
- 35.Fog CK, Zago P, Malini E, et al. The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine 2018;38:142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aflaki E, Borger DK, Moaven N, et al. A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J Neurosci 2016;36:7441–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.GZ/SAR402671 in Combination With Cerezyme in Adult Patients With Gaucher Disease Type 3 (LEAP). Available at: clinicaltrials.gov/ct2/show/NCT02843035?cond=Gaucher+disease+type+3&draw=2&rank=1. Accessed January 8, 2020.
- 38.Holmay MJ, Terpstra M, Coles LD, et al. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin Neuropharmacol 2013;36:103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hurvitz N, Dinur T, Becker-Cohen M, et al. Glucosylsphingosine (lyso-Gb1) as a biomarker for monitoring treated and untreated children with Gaucher disease. Int J Mol Sci 2019;20:3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivanova M, Limgala RP, Changsila E, Kamath R, Ioanou C, Goker-Alpan O. Gaucheromas: when macrophages promote tumor formation and dissemination. Blood Cell Mol Dis 2018;68:100–105. [DOI] [PubMed] [Google Scholar]
- 41.Oppenheim IM, Canon AM, Barcenas W, et al. Bilateral symmetrical cortical osteolytic lesions in two patients with Gaucher disease. Skeletal Radiol 2011;40:1611–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steward AM, Wiggs E, Lindstrom T, et al. Variation in cognitive function over time in Gaucher disease type 3. Neurology 2019;93:e2272–e2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator.