

Abstract
Objective
To test the hypothesis that we will be able to detect change in motor outcome measures over time in a cohort with mutations in FKRP.
Methods
Individuals with documented FKRP mutations were evaluated annually with a battery of established motor outcome measures including limited quantitative myometry and timed function measures. Results were analyzed using random coefficient regression to determine annual change in each measure. Due to the nonlinear progression through the lifespan of the study participants, pediatric (<19 years) and adult (≥19 years) cohorts were analyzed separately. Effect of genotype was evaluated in each cohort.
Results
Sixty-nine participants (30 pediatric, 44 adult) with at least 2 evaluations were included. There was a small but statistically significant decline in timed motor function measures in both pediatric and adult cohorts. Genotype significantly affected rate of decline in the pediatric but not the adult cohort. Some pediatric patients who are homozygous for the c.826C>A mutation showed improving motor performance in adolescence. Performance on the 10-meter walk/run was highly correlated with other timed function tests.
Conclusions
There is a slow annual decline in motor function in adults with FKRP mutations that can be detected with standard motor outcome measures, while the results in the pediatric population were more variable and affected by genotype. Overall, these analyses provide a framework for development of future clinical trials. The dystroglycanopathies natural history study (Clinical Trial Readiness for the Dystroglycanopathies) may be found on clinicaltrials.gov (NCT00313677).
The dystroglycanopathies comprise a group of muscular dystrophies resulting from hypoglycosylation of α-dystroglycan (α-DG).1,2 They encompass a range of clinical phenotypes, from Walker-Warburg syndrome to adult-onset limb-girdle muscular dystrophy.3 Dystroglycanopathies are also genetically heterogeneous. Mutations in at least 18 different genes can be causative.4,5
One of the most common dystroglycanopathy genes is FKRP, encoding fukutin-related protein.4,6 This protein has been shown to function as a ribitol 5-phosphate transferase, which is necessary for the glycosylation of α-DG.7,8 Several founder mutations have been identified in FKRP, and the most common, c.826A>C (p.Leu276Ile), has been found in high frequency worldwide, particularly in those of European descent.9–11 The homozygous c.826A>C mutation is associated with a more benign phenotype, while the compound heterozygous mutation is typically associated with a more severe phenotype.12–14
There is no specific treatment for this group of disorders and management is symptomatic. Treatment trials are in progress and under development.
We have prospectively followed patients with dystroglycanopathy annually. Here we describe the course of motor function as measured by standard motor outcome measures in a cohort of participants with mutations in FKRP, including children and adults with a wide range of mutations. This natural history data span up to 13 years and will help in the design of future clinical trials.
Methods
Standard protocol approvals, registrations, and patient consents
Institutional review board approval was obtained for all recruitment and data collection for this study. Written informed consent was obtained from all patients or their legal guardians. Additional consent was obtained for review of medical records. The dystroglycanopathy natural history study may be found at clinicaltrials.gov (NCT00313677).
Inclusion criteria
All individuals with a known or suspected dystroglycanopathy were invited to participate in a natural history study with annual follow-up. Inclusion for this analysis is limited to those with documented FKRP mutations, >6 years old, and with at least 2 study evaluations between April 2006 and July 1, 2019. Patients with variants of unknown significance were included in analysis if there was supportive laboratory evidence that the patient had a dystroglycanopathy based on review of clinically obtained muscle biopsy and confirmation of hypoglycosylation by immunohistochemistry,14 elevated creatine kinase, phenotype consistent with a progressive muscular dystrophy, or parental testing to confirm the variants were in trans.
Annual evaluations
Medical history and general examination were completed at enrollment and updated annually for all participants. Medical history was supplemented where possible by review of medical records.
Motor outcome measures
Motor outcome measures were grouped into myometry, timed functional scores, functional scores, and spirometry, summarized below. All measures were collected each year, with the exceptions of the 6-minute walk test (6MWT), which was added in 2009; performance of upper limb (PUL), which was added in 2015; grip strength, which was collected 2006 through 2009 and reinstated in 2016; and Pediatric Evaluation of Disability Inventory (PEDI), which was not collected after 2015.
Physical therapists with clinical trial experience completed the motor testing following standard protocols. These evaluators have shown high reliability testing on these measures in other studies.15–18 Motor function testing protocols were generally consistent with the National Institute of Neurologic Disorders and Stroke (NINDS) Common Data Elements, although this study began before those were available.
Muscle strength
We measured upper extremity and lower extremity strength using the JTech Commander PowerTrack II (2006–2016) and Hoggan Scientific microFET hand‐held myometer (2016–2019) using previously described procedures.19 We assessed strength in bilateral knee flexors, knee extensors, elbow flexors, elbow extensors, and shoulder abductors. Strength was measured in pounds. We performed a minimum of 3 and maximum of 5 assessments for each muscle group.
Grip strength
We measured grip strength using the protocol of the American Society of Hand Therapy.20 Participants were seated with their feet supported, elbow bent to 90°, and wrist at neutral. A Hoggan Scientific microFET grip dynamometer was used to measure force in pounds.
10-Meter timed walk/run (10MW/R)
Time to walk or run 10 meters was assessed by instructing participants to walk or run as fast as possible from a start line to a target 1–2 meters past the 10-meter line. Timing was started when the examiner said “go” and stopped when the patient’s second foot crossed the 10-meter line. Assistive devices were not allowed.21
Time to climb 4 steps (4SC)
Time to ascend 4 standard steps was performed as described by NINDS Common Data Elements, modified so participants were allowed to use railings as needed. Timing was started when the examiner said “go” and stopped when patient assumed an upright position with arms at sides on the top platform of stairs.21,22
Time supine to standing
Patients assumed a starting position of supine on the floor with arms at side and legs straight. Timing was started when the examiner said “go” and stopped when the patient assumed an upright position with arms at sides. Total time to transition, as well as method utilized (i.e., use of hands on body, need for furniture support), were recorded.
6-Minute walk test
The 6MWT was administered using a modified version of the American Thoracic Society method.15
Performance of upper limb
The PUL is an instrument designed to assess upper limb function in ambulant and nonambulant patients with Duchenne muscular dystrophy.23 PUL version 1.2 was administered according to the manual of procedures.
Pediatric Evaluation of Disability Inventory
We used the PEDI to assess functional mobility skills.24
Modified Hammersmith Functional Motor Scale–Extend Gross Motor Function Module (MHFMS-Extend)
We assessed motor skills with the MHFMS-Extend in participants over 2 years of age who were ambulatory.25
Pulmonary function tests
We measured forced vital capacity (FVC) in liters and resulting percent predicted based on age, sex, weight, and height. We measured FVC in sitting and supine using a Renaissance II or MicroLoop spirometer. The best of 3 valid tests for FVC and percent predicted in sitting and supine were reported.
Statistical analysis
In preliminary analysis of the data, there was nonlinear progression through the life span in our cohort. We therefore divided the participants into 2 cohorts, pediatric (>6 to <19 years old) and adult (≥19 years), for separate analysis. The age separation is consistent with common clinical trial design. To include data from those unable to perform a test due to disease, the timed function tests (10MW/R, 4SC, time to rise from supine) were converted to velocity (meters/s, stairs/s, and rise/s, respectively) for analysis. To account for the variation in the number of years of follow-up and intervals between assessments, annual change was determined for each participant, and the estimate of overall mean was determined using random coefficient regression analysis. The same analysis was performed to compare homozygous c.826C>A and other FKRP genotypes. With patients who were no longer able to perform a test due to disease progression, the first 2 instances of being unable to perform were included in the analysis to avoid the influence of inability in the estimate of slope. Correlations between 10MW/R and other outcome measures were determined using correlation analysis for longitudinal paired data.26,27 Nonparametric survival analysis for interval-censored data was used to estimate the cumulative probability over time (age) for the endpoint outcome of inability to perform the specified time task. All analysis was performed using SAS/STAT software, version 14.3 (SAS Institute Inc., Cary, NC).
Data availability
De-identified data are available by request to the corresponding author.
Results
Demographics
Of 86 participants in the natural history study with mutations in FKRP, 69 met the inclusion criteria for this analysis (excluding 16 who had only 1 evaluation and 1 who was <6 years old). Thirty-seven of the final analysis cohort (54%) are homozygous for the common founder mutation, c.826C>A.9 Age at last assessment ranged from 7.9 to 75.5 years, and participants were followed for 0.9–13 years. The pediatric (<19 years old) cohort includes 30 participants, and there are 44 participants in the adult cohort. Five participants were included in both cohorts as they aged out of the pediatric and into the adult cohort. Demographics and genotype summary of these 2 analysis cohorts are summarized in table 1.
Table 1.
Demographics of pediatric and adult cohorts

Tests of motor function
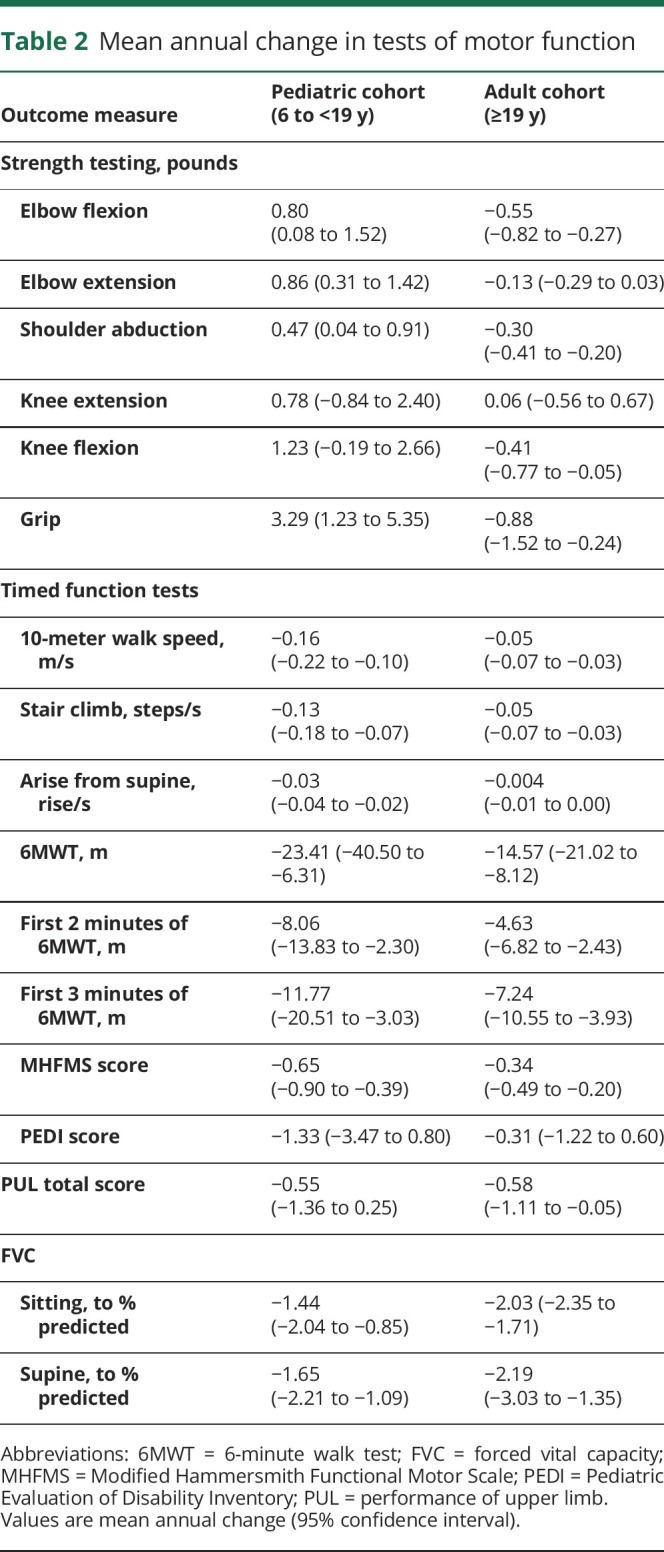
Table 2 summarizes the average annual changes in outcome measures observed in the pediatric and adult cohorts. In the adult cohort, a statistically significant mean annual decline was noted in knee flexor, elbow flexor, and shoulder abductor strength. In the pediatric cohort, muscle strength improved significantly in elbow flexor, elbow extensor, and shoulder abductor strength. Statistically significant decline was seen in the timed function tests (10MW/R, 4SC, arise from supine) in both pediatric and adult cohorts. Total distance at 2 minutes and 3 minutes within the 6MWT were also analyzed, and both showed significant decline across both cohorts. Significant annual decline was also seen in FVC (sitting and supine) and MHFMS. No significant annual change was seen with PEDI in either age cohort. It was noted that PEDI was not sensitive to change in this patient population, and the test was not administered after 2015.
Table 2.
Mean annual change in tests of motor function
We noted the number of participants who were unable to perform, or became unable to perform, timed function tests (10MW/R, 4SC, arise from supine, 6MWT) due to disease progression, summarized in table 3. By age 10, all patients were able to complete 10MW/R, 4SC, and 6MWT, and 9.9% were unable to arise from supine; by age 25, 77.5% of patients were unable to arise from supine, and 23%–25% were unable to complete the other tests. Of patients aged 55–70 years, between 40% and 60% were unable to complete 10MW/R, 4SC, or 6MWT, and 91.9% were unable to arise from supine.
Table 3.
Cumulative percent probability (95% confidence interval) of inability to perform timed function tests

The subpopulation homozygous for the c.826C>A mutation is not represented equally in the pediatric and adult cohorts; 20.0% and 72.7%, respectively (table 1). We therefore performed a subanalysis (by cohort) for each outcome measure to determine whether there was a difference in progression based on genotype, summarized in table 4. In the pediatric group, mean annual change differed significantly by genotype in nearly all measures, except for PUL and both FVC measures. No significant difference in mean annual decline was seen in the adult cohort. The figure illustrates the changes in timed functional outcome measures by age cohort and FKRP genotype.
Table 4.
One-year change in each of the functional motor outcome measures related to age and phenotype

Figure. Illustrative graphs showing change in function over time by FKRP genotype for timed function tests, demonstrating the importance of genotype and age on rate of progression.

4SC = time to climb 4 steps; 6MWT = 6-minute walk test; 10MW = 10-meter walk.
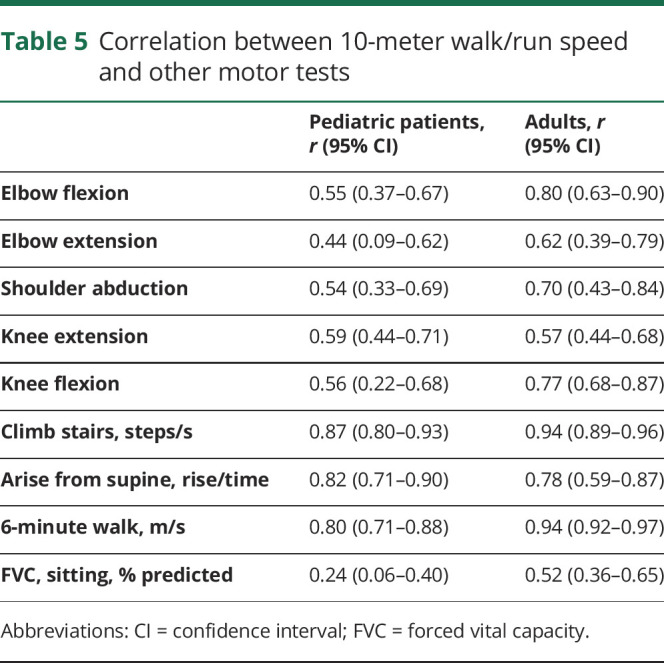
We examined correlations between functional outcomes and the 10MW/R, as well as strength and the 10MW/R, in an effort to identify potentially duplicative measures. Limiting the number of tests participants are asked to perform reduces the burden of study participants. We found that timed function tests were highly correlated with 10MW/R, as shown in table 5. Results of myometry testing were moderately to strongly correlated with 10MW/R, and FVC (sitting) was weakly correlated.
Table 5.
Correlation between 10-meter walk/run speed and other motor tests
Discussion
We present the calculated annual change in motor outcome measures for a diverse cohort of children and adults with muscular dystrophy due to FKRP mutations who were followed longitudinally for up to 13 years. These data demonstrate that there is a significant annual decline in most functional outcome measures. The information presented here will be important in planning future clinical trials.
Whereas this longitudinal study was able to show statistically significant decline in motor function over 1 year, the changes were small. For example, in the 10-meter walk test, a speed of 2.0 meters/s would correspond to a time of 5.0 seconds. Using the pediatric model, speed is predicted to decrease by 0.16 meters/s annually; the following year, the predicted 10-meter walk speed of 1.84 meters/s would correspond to a walk time of 5.43 seconds, an increase of only 0.43 seconds.
We show a strong effect of genotype in patients with FKRP mutations, as has been noted previously. Patients homozygous for the common founder mutation c.826C>A report later age at onset.12–14 There is a skewed genotype representation in the pediatric vs adult cohorts, with the adult cohort including a preponderance of patients homozygous for the common founder mutation c.826C>A. More mildly affected participants often present in adulthood, so are less likely to be followed in the pediatric cohort. In addition, more severely affected patients who presented in childhood might be less likely to participate as adults due to difficulty traveling. In the pediatric cohort, there is a significant difference in rate of disease progression, with those homozygous for c.826C>A showing slower decline. The failure to show a difference between genotype cohorts in adults likely has several contributing factors. First, as noted above, the more severely affected patients (extrapolating from pediatric data, these are more likely to be heterozygous) might not be included, creating a bias toward the more mildly affected patients. In addition, not all novel mutations have the same effect. This is clear when considering other founder mutations. For example, a Mexican founder mutation (c.1387A>G) is associated with a more severe phenotype. Similarly, it is likely that some people with heterozygous mutations have a second mild variant, which would again skew the adult heterozygous population to a milder phenotype.10 Until every variant is characterized, this further complicates clinical trial design.
A European cohort of patients with FKRP mutations has also been followed longitudinally for 6 years.28 This European series (n = 23) was limited to adults (over 18 years) who were ambulatory and homozygous for the c.826A>C mutation, differing from our series, which includes children and those who have varied FKRP genotypes (46.4% of our cohort are not homozygous for the c.826A>C variant). They found no significant decline in motor outcome measures in adults homozygous for the c.826C>A mutation over 1 year. This cohort was recently reexamined and the median change in functional measures over a 6-year follow-up among adult c.826C>A homozygotes was reported.29 We compared these published results with the corresponding subset of our cohort, adults with homozygous c.826C>A mutation, by calculating the estimated 6-year mean difference and corresponding 95% confidence interval (CI) (comparison shown in table 6). The median reported change in 10MW/R, 6MWT, 4SC speed, knee extension strength, and FVC all fell within our projected CI, while the median change in knee flexion strength fell just outside of the 95% CI. This small difference could be due to variation in patient positioning, technique, or type of equipment.
Table 6.
Disease progression over 6 years in the present cohort compared with published results: adults homozygous for c.826C>A mutation

The results of the pediatric group were compared to a 12-month longitudinal study of patients with Duchenne muscular dystrophy.30 The authors reported the mean 1-year change and SD for 10MW/R and 6MWT, stratified into groups according to age and steroid use. Ninety-five percent CIs for the data were calculated. Comparing our pediatric data to the study's >7 years age cohort who had used no or intermittent steroids, we found similarities in these motor outcome tests. For both 10MW/R and 6MWT, the CIs for annual change in the Duchenne study overlapped with the CI for our pediatric cohort, and well as the CIs when stratified by genotype. The Duchenne data were most similar to the mean annual change among compound heterozygotes. This is consistent with the clinical observation that the Duchenne muscular dystrophy phenotype overlaps with more severe forms of limb-girdle muscular dystrophy. A possible limitation to this comparison is that our use of random coefficient regression assumed a linear relationship between age and outcome measures.
These data illustrate at least 2 variables to be taken into consideration in designing clinical trials involving patients with FKRP mutations: age and genotype. Pediatric and adult cohorts showed different rates of decline. Whereas the adult cohort tended toward a stable mean annual change, the pediatric cohort showed a more dramatic decline in timed functional tests. Conversely, there was group improvement in some tests in the pediatric group. This improvement likely reflects the normal growth and development in adolescents in a slowly progressive disease. These factors, together with the variability seen within and between patients, could result in the need for a large patient population in a standard placebo-controlled, 1-year clinical trial. A more homogenous subset could be selected for a clinical trial, although this would complicate enrollment in a rare disease.
Biomarkers sensitive to disease progression will be important for clinical trials in a slowly progressive disorder. Quantitative MRI has shown significant increases in fat replacement in 9 muscles over a 1-year and 6-year period in adult patients homozygous for c.826C>A mutation.29 Imaging was not included in the present study.
Along with the need for biomarkers, minimizing participant burden through less demanding functional outcome measures is desirable. We recognize that values obtained from the first 2 or 3 minutes of a 6MWT cannot be directly compared to a 2- or 3-minute walk test, as patients perform differently depending on how long they expect to walk.31 However, exploratory analysis of 2- and 3-minute walk distances within the 6MWT showed a potential area for limiting participant exertion in performing motor function testing. In both children and adults, both 2-minute and 6-minute walk distances showed a statistically significant annual decrease, and in the pediatric cohort, a significant difference was seen between homozygous individuals and those with other mutations. In addition, we saw excellent correlation among the distances walked in 2, 3, and 6 minutes of the 6MWT. These measures appear promising but will require direct testing.
The correlations between 10MW/R and strength and timed tests were analyzed in an effort to reduce patient burden in study participation. High correlations between 10MW/R and 4SC, arise from supine, and 6MWT were found in both pediatric and adult cohorts, indicating that clinical trials might not require all timed function measures. However, these correlations were not stratified by disease genotype, and as mentioned previously, disease progression varies greatly by FKRP genotype.
Seated FVC (% predicted) showed a significant decline with increasing age in both age cohorts and both genotypes. Although the 10MW/R was highly correlated with timed outcome measures, it was weakly correlated with seated FVC. Analysis of FVC among adult participants who had lost ability to perform the 10MW/R showed a mean annual decline of 2.48%, which is a faster decline in FVC than was seen in the whole adult cohort. The sample size of this group was relatively small, but indicates that FVC, rather than decline in timed outcome measures, could be a measure of disease progression in patients who are no longer ambulatory.
This work has several limitations, many of which are inherent in the investigation of a rare disease. The cohort is relatively small. The work was carried out at a single site; therefore, the cohort is limited to those healthy enough to travel, a source of selection bias. Relative homogeneity of participants in the adult cohort due to travel limitations might not accurately reflect the disease progression among affected adults. In addition, self-selecting participants in natural history studies tend to be highly motivated, and generally receive more intensive medical care. These sources of bias lead to a data pool that might not be representative of the average experience of those with FKRP mutations, and could skew results toward a slower decline.32 As the data were collected over several years for this longitudinal study, there could be drift in approach to motor function testing, but any such changes would affect the whole cohort. This analysis did not control for steroid or supplement use. It is not known if corticosteroids are beneficial, neutral, or detrimental in this generally slowly progressive disorder. The number of patients who used steroids while participating in the study was small (n = 9), and repeating the analysis excluding those on steroids (data not shown) did not significantly alter mean annual change in motor outcome measures. The influence of cardiac function on motor outcome measures was also not directly examined and cardiac testing was not part of the protocol. In general, significant cardiac dysfunction is a late manifestation in those with FKRP mutations and none of the participants in this analysis had physical findings to indicate cardiac failure.33 In addition, the strong correlation between 10MW/R speed and strength measures suggests cardiomyopathy-induced fatigue did not affect performance.
The data presented here, together with results emerging from other groups, provide a strong basis for design of clinical trials in patients with FKRP mutations. We demonstrate that standard motor outcome measures can detect change over time, and both age and genotype should be considered as variables for stratification in clinical trials.
Acknowledgment
The authors thank the University of Iowa Clinical Research Unit for the use of their facilities; the patients and their families; LGMD2i Foundation, Cure LGMD2i Foundation (formerly The Samantha J. Brazzo Foundation), and CureCMD for their annual conference, in association with which some of these data were collected; Linda Lowes, PT, PhD; Megan Iammarino, PT; Wellstone Fellows Jamie Eskuri, Steven McGaughey, Katie Lutz, Braden Jensen, Brianna Brun, Courtney Carlson, and Angela Lee; and Evgenia Folts, Corey McDaniel, Rory Metcalf, Chandra Miller, Amanda Philiph, Madelyn Rasor, and Julia Richarson for their assistance.
Glossary
- 4SC
time to climb 4 steps
- 6MWT
6-minute walk test
- 10MW/R
10-meter timed walk/run
- α-DG
α-dystroglycan
- CI
confidence interval
- FVC
forced vital capacity
- MHFMS-Extend
Modified Hammersmith Functional Motor Scale–Extend Gross Motor Function Module
- NINDS
National Institute of Neurologic Disorders and Stroke
- PEDI
Pediatric Evaluation of Disability Inventory
- PUL
performance of upper limb
Appendix. Authors
Study funding
Study funding was supplied by a Paul D. Wellstone Muscular Dystrophy Cooperative Research Center (MDCRC) grant from the NIH (NIH U54 NS053672).
Disclosure
A.M. Gedlinske reports no disclosures relevant to the manuscript. C.M. Stephan received lodging and travel reimbursements from Pfizer and NIH. S.R.H. Mockler is site clinical evaluator for trials sponsored by Sarepta Therapeutics, Pfizer, PTC, Roche, Acceleron, Italfarmaco, and Catabasis but reports no personal financial interest in any of the above companies. K.M. Laubscher reports no disclosures relevant to the manuscript. K.S. Laubenthal received salary support through the Wellstone grant. C.D. Crockett reports no disclosures relevant to the manuscript. M.B. Zimmerman receives funding from NIH grants 2 P01 HL0496925, 1 R01 HL 119882, 1 R34 AT008349-01, 5 UM1 AR063381-02, 2 U54 NS053672, 1 R01 MG104363-01A1, IU01 DK108334, 1 U54 TR0013564, and 1 R01 CA193249. K.D. Mathews’ research is supported by NIH (NINDS), CDC, and FARA; she is site PI for industry-sponsored trials by Sarepta Therapeutics, Horizon Therapeutics, Pfizer, FibroGen, and PTC; she has received meals, and/or reimbursements, or honoraria to the University of Iowa within the last year from the following: Sarepta Therapeutics, Marathon, BMS, and Genzyme/Sanofi; she reports no personal financial interest in any of the above companies. Go to Neurology.org/N for full disclosures.
References
- 1.Muntoni F, Brockington M, Blake DJ, Torelli S, Brown SC. Defective glycosylation in muscular dystrophy. Lancet 2002;360:1419–1421. [DOI] [PubMed] [Google Scholar]
- 2.Henry MD, Campbell KP. Dystroglycan inside and out. Curr Opin Cell Biol 1999;11:602–607. [DOI] [PubMed] [Google Scholar]
- 3.Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 2002;71:1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohn RD. Dystroglycan: important player in skeletal muscle and beyond. Neuromuscul Disord 2005;15:207–217. [DOI] [PubMed] [Google Scholar]
- 5.Bouchet-Seraphin C, Vuillaumier-Barrot S, Seta N. Dystroglycanopathies: about numerous genes involved in glycosylation of one single glycoprotein. J Neuromuscul Dis 2015;2:27–38. [PubMed] [Google Scholar]
- 6.Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 2001;10:2851–2859. [DOI] [PubMed] [Google Scholar]
- 7.Esapa CT, Benson MA, Schroder JE, et al. Functional requirements for fukutin-related protein in the Golgi apparatus. Hum Mol Genet 2002;11:3319–3331. [DOI] [PubMed] [Google Scholar]
- 8.Kanagawa M, Kobayashi K, Tajiri M, et al. Identification of a post-translational modification with ribitol-phosphate and its defect in muscular dystrophy. Cell Rep 2016;14:2209–2223. [DOI] [PubMed] [Google Scholar]
- 9.Walter MC, Petersen JA, Stucka R, et al. FKRP (826C>A) frequently causes limb-girdle muscular dystrophy in German patients. J Med Genet 2004;41:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee AJ, Jones KA, Butterfield RJ, et al. Clinical, genetic, and pathologic characterization of FKRP Mexican founder mutation c.1387A>G. Neurol Genet 2019;5:e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frosk P, Greenberg CR, Tennese AA, et al. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum Mutat 2005;25:38–44. [DOI] [PubMed] [Google Scholar]
- 12.Mercuri E, Brockington M, Straub V, et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol 2003;53:537–542. [DOI] [PubMed] [Google Scholar]
- 13.Poppe M, Cree L, Bourke J, et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 2003;60:1246–1251. [DOI] [PubMed] [Google Scholar]
- 14.Willer T, Lee H, Lommel M, et al. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat Genet 2012;44:575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDonald CM, Henricson EK, Han JJ, et al. The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve 2010;41:500–510. [DOI] [PubMed] [Google Scholar]
- 16.Berard C, Payan C, Hodgkinson I, Fermanian J; Group MFMCS. A motor function measure for neuromuscular diseases: construction and validation study. Neuromuscul Disord 2005;15:463–470. [DOI] [PubMed] [Google Scholar]
- 17.Iannaccone ST, Hynan LS; American Spinal Muscular Atrophy Randomized Trials Group. Reliability of 4 outcome measures in pediatric spinal muscular atrophy. Arch Neurol 2003;60:1130–1136. [DOI] [PubMed] [Google Scholar]
- 18.Pane M, Mazzone ES, Fanelli L, et al. Reliability of the performance of upper limb assessment in Duchenne muscular dystrophy. Neuromuscul Disord 2014;24:201–206. [DOI] [PubMed] [Google Scholar]
- 19.Beenakker EA, van der Hoeven JH, Fock JM, Maurits NM. Reference values of maximum isometric muscle force obtained in 270 children aged 4-16 years by hand-held dynamometry. Neuromuscul Disord 2001;11:441–446. [DOI] [PubMed] [Google Scholar]
- 20.Fess E, Moran C. Clinical Assessment Recommendations. 1st ed. Chicago: American Society of Hand Therapists; 1981. [Google Scholar]
- 21.Grinnon ST, Miller K, Marler JR, et al. National Institute of Neurological Disorders and Stroke common data element project: approach and methods. Clin Trials 2012;9:322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brooke MH, Griggs RC, Mendell JR, Fenichel GM, Shumate JB, Pellegrino RJ. Clinical trial in Duchenne dystrophy: I: the design of the protocol. Muscle Nerve 1981;4:186–197. [DOI] [PubMed] [Google Scholar]
- 23.Mayhew A, Mazzone ES, Eagle M, et al. Development of the performance of the upper limb module for Duchenne muscular dystrophy. Dev Med Child Neurol 2013;55:1038–1045. [DOI] [PubMed] [Google Scholar]
- 24.Haley SM, Coster WJ, Ludlow LH, Haltiwanger JT, Andrellos PJ. Pediatric Evaluation of Disability Inventory: Development, Standardization, and Administration Manual. Boston: New England Medical Center Publications; 1992. [Google Scholar]
- 25.Krosschell K. MHFMS-Extend Test for Children With Spinal Muscular Atrophy: Gross Motor Module. Elk Grove Village, IL: Cure SMA; 2004. [Google Scholar]
- 26.Hamlett A, Ryan L, Serrano-Trespalacios P, Wolfinger R. Mixed models for assessing correlation in the presence of replication. J Air Waste Manag Assoc 2003;53:442–450. [DOI] [PubMed] [Google Scholar]
- 27.Hamlett A, Ryan L, Wolfinger R. On the use of PROC MIXED to estimate correlation in the presence of repeated measures. Proceedings of the SAS Global Forum 2004 Conference; Montreal, Canada; April 9–12, 2004.
- 28.Willis TA, Hollingsworth KG, Coombs A, et al. Quantitative muscle MRI as an assessment tool for monitoring disease progression in LGMD2I: a multicentre longitudinal study. PLoS One 2013;8:e70993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy AP, Morrow J, Dahlqvist JR, et al. Natural history of limb girdle muscular dystrophy R9 over 6 years: searching for trial endpoints. Ann Clin Transl Neurol 2019;6:1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazzone E, Vasco G, Sormani MP, et al. Functional changes in Duchenne muscular dystrophy: a 12-month longitudinal cohort study. Neurology 2011;77:250–256. [DOI] [PubMed] [Google Scholar]
- 31.McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other clinical endpoints in Duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve 2013;48:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hernan MA, Hernandez-Diaz S, Robins JM. A structural approach to selection bias. Epidemiology 2004;15:615–625. [DOI] [PubMed] [Google Scholar]
- 33.Poppe M, Bourke J, Eagle M, et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann Neurol 2004;56:738–741. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
De-identified data are available by request to the corresponding author.