

Abstract
A major obstacle to obtaining relevant results in cancer vaccination has been the lack of identification of immunogenic antigens. Dendritic cell (DC)-based cancer vaccines used preventively may afford protection against tumor inoculation, but the effect of antigen choice on anti-tumor protection is not clear. When using irradiated syngeneic tumor cells to load DCs, tumor self-antigens are provided, including tumor-associated antigens (TAAs) and neoantigens generated by tumor mutations. On the other hand, allogeneic tumor cells could only supply shared TAAs. To assess the advantages of each source in protective vaccination, we analyzed in C57BL/6 mice the effect of loading DCs with irradiated syngeneic B16-F1 or allogeneic Cloudman melanoma cells; both cell lines were characterized by whole exome sequencing and RNAseq. Tumor cell components from the two irradiated cell lines were efficiently internalized by DCs, and transported to MHC-class II positive tubulovesicular compartments (MIICs). DCs loaded with allogeneic irradiated Cloudman cells (DC-ApoNecALLO) induced a partially effective anti-melanoma protection, although Cloudman and B16-F1 cells share the expression of melanocyte differentiation antigens (MDAs), cancer-testis antigens (CTAs) and other TAAs. DCs loaded with syngeneic B16-F1 cells (DC-ApoNecSYN) established a more potent and long-lasting protection and induced a humoral anti-B16F1 response, thus suggesting that neoepitopes are needed for inducing long-lasting protection.
Keywords: Anti-melanoma vaccination, Dendritic cells, Tumor-associated antigens, Neoepitopes, Allogeneic cells, Syngeneic cells
1. Introduction
For the past decades, clinical trials on cancer vaccines have given modest results (reviewed in [1]). One of the major obstacles has been the inability to clearly identify relevant immunogenic antigens.
In melanoma, immunogenic TAAs have been described, including melanocyte differentiation antigens (MDAs) such as Melan-A, Pmel/gp100 and Tyr [2-4], and Cancer-Testis-Antigens (CTAs) [5-7]. On the other hand, human melanoma is the cancer with the highest somatic mutation prevalence [8]. Non-synonymous mutations in the coding regions of the genome can give rise to neoepitopes, given that mutated proteins are expressed and their peptides presented by the patient’s MHC-I and MHC-II molecules to specific CD8+ and CD4+ T lymphocytes [9]. In the context of cancer immunotherapy, neoepitope-specific T lymphocytes have not been clonally deleted and should be able to mount an effective immune response [9,10]. Neoantigen reactive T cells have been identified in several cancers, including melanoma [11,12] and personalized cancer vaccines targeting neoantigens are already being tested in clinical trials [13]. DCs play a key role in vaccination, as they are capable of capturing, processing and presenting antigens to naïve T cells. When allogeneic tumor cells are used as source of antigen in DC-based vaccination, MDAs like Pmel and Melan-A are efficiently cross-presented to specific CD8 T cells [14] and measurable immune responses against such MDAs were observed in patients [15].
We have previously developed an experimental preventive anti-melanoma vaccine consisting of DCs loaded with syngeneic irradiated murine melanoma cells (DC-ApoNec vaccine), which provides long-term anti-B16-F1 melanoma protection [16,17]. Using this system, we have now analyzed in C57BL/6 mice the effect of a syngeneic (H-2b haplotype) versus an allogeneic (H-2d haplotype) melanoma antigen source in DC-based preventive vaccination. DCs were loaded with irradiated syngeneic B16-F1 cells (DC-ApoNec SYN) or allogeneic Cloudman melanoma cells (DC-ApoNec ALLO). If neoepitopes were the only antigens to trigger immune protection, then only DC-ApoNec SYN would be a successful vaccine. If, on the contrary, TAAs shared by allogeneic tumors are also useful antigens, vaccination with DC-ApoNec ALLO would have some efficacy. We performed high throughput exome and RNA sequencing to establish the antigenic repertoire of both cell lines. We demonstrate that while vaccination with DC-ApoNec ALLO induces partially effective protection, DC-ApoNec SYN establishes a more potent and long-lasting protection.
2. Material and methods
2.1. Mice and tumor cell lines
Eight-12-week-old male mice maintained in pathogen-free conditions were used. C57BL/6J mice were purchased from the Fundación Instituto Leloir (Buenos Aires, Argentina) and MHC-II-GFP mice were kindly provided by the Hidde Ploegh Laborator [18]. Studies were performed in accordance to protocols approved by the Animal Care and Use Committee of the Fundación Instituto Leloir and of the University of Pennsylvania and CHOP.
B16-F1 cell line was purchased from the ATCC and cultured in DMEM (Sigma) 4,5 g/l glucose containing 10% FBS (Natocor, Argentina). Cloudman S91 cell line was purchased from the ATCC and cultured in DMEM 4,5 g/l glucose containing 15% FBS. H5V cell line (Istituto di Ricerche Farmacologiche Mario Negri, Milan) was cultured in DMEM 1 g/l glucose containing 10% FBS. 4T1 cell line was generously provided by Dr. Osvaldo Podhajcer (Fundación Instituto Leloir) and cultured in RPMI (Sigma) containing 10% FBS. Cells were maintained in culture for no >10 passages in 100 U/ml penicillin and 100 μg/ml streptomycin (Rochet) supplemented media and were periodically tested negative for mycoplasma.
2.2. Quantitative real-time PCR
mRNA levels were quantified as previously described [19]. Briefly, PCR runs were carried out using SYBR Universal Master Mix (Applied Biosystems, Carlsbad, CA, USA), and relative expression levels were determined by the ΔΔCt method [20] using actb gene expression to normalize all samples (Ct: Threshold Cycle). The primers used are listed in Table 1.
Table 1.
Primer sequences for qRT-PCR.
| Gene | Forward | Reverse |
|---|---|---|
| Actb | TGCCCTGAGGCTCTTTTCCAGC | ACGCAGCTCAGTAACAGTCCGC |
| Tyr | CCTAACTTACTCAGCCCAGC | AGAGCGGTATGAAAGGAACC |
| Dct | CCTGGCCAAGAAGAGTATCC | CACGTCACACTCGTTCTTCC |
| Pmel | CGGATGGTCAGGTTATCTGG | ATGGTGAAGGTTGAACTGGC |
| Mlana | CCTCAAGGGAAAGATGCTCA | GCCTTAAAGCGGAAGTGTGA |
2.3. Induction of apoptosis and necrosis
B16-F1 and Cloudman cells were γ-irradiated (70 Gy, Siemens lineal accelerator, Instituto Alexander Fleming, Buenos Aires) and stored in liquid nitrogen. Cells were thawed and cultured using the corresponding culture media for each cell line. Non-adherent and adherent cells were collected. Apoptosis and necrosis were determined with FITC-Annexin V (AnV) – propidium iodide (PI) Apoptosis Detection Kit I (BD Biosciences) following the manufacturer’s recommendations, measured by flow cytometry (FACSCalibur Flow Cytometer, Becton Dickinson) and analyzed using FlowJo 7.6 software.
2.4. Isolation and culture of bone marrow-derived DCs
Bone marrow DC progenitors were obtained from the femurs and tibias of 8–12-week-old C57BL/6 mice and differentiated to DCs in RPMI containing 0.02 μg/ml recombinant murine GM-CSF (Peprotech, Mexico), as previously described [17].
2.5. Incorporation of ApoNec cells by DCs and immunofluorescence analysis of maturation markers
ApoNec cells were stained with PKH26 (Sigma) following the manufacturer’s recommendations and cultured with DCs for 24 h at 37 °C followed by 1 h at 4 °C incubation with anti-CD11c-PE-cy7 antibody (Ab) (Clone HL3, BD biosciences). Immunofluorescence was assessed by flow cytometry and analyzed using FlowJo 7.6 software.
Non-stained ApoNec cells were cultured with DCs for 24 h at 37 °C, incubated for 15 min at 4 °C with mouse Fc-block, and incubated for another 45 min at 4 °C with PE-anti-mouse CD11c (CloneHL3), FITC-anti-mouse I-Ab (Clone AF6-120.1) or FITC-anti-mouse CD86 (Clone GL.1) (all Abs from BD Biosciences). Cells were then washed with PBS and fixed with 2% PFA. Immunofluorescence was quantified by flow cytometry and data analyzed using FlowJo 7.6 software.
2.6. Live cell imaging
MHC-II –GFP DCs were adhered ON to poly-L-lysine–coated glass-bottom 35-mm culture dishes (MatTek, Ashland, MA), co-cultured for 3 or 7 h on a 1:1 ratio with PKH26 pre-stained ApoNec cells (Sigma) and visualized as described in [21].
2.7. Transmission electron microscopy
DCs were co-cultured with ApoNec cells in a 1:1 ratio for 24 h at 37 °C, detached with DMEM, washed with 0.1 M pH 7.4 phosphate buffer, fixed for 4 h at 4 °C in 2.5% glutaraldehyde, washed again, and post-fixed first in osmium tetroxide and then in uranyl acetate prior to dehydration. Cells were visualized using a Zeiss EM 10C transmission electron microscope (LANAIS, Buenos Aires, Argentina).
2.8. Confocal microscopy
DCs were adhered ON to glass-bottomed plates, and co-cultured for 6 h with Celltrace violet-stained ApoNec cells on a 1:1 ratio, fixed with 2% PFA and permeabilized with Permwash (BD biosciences) and sequentially stained with rat-anti-Lamp1 Ab (clone 1D4B) and anti-rat-Alexa488 Ab in 0.1% saponin in PBS for 1 h. Stained cells were visualized using a BioVision spinning disk system on a Leica DMI8 microscope, and images were captured on a Hamamatsu Orca Flash 4.0 V2 CMOS camera and Visiview software (Dept. of Pathology & Laboratory Medicine, Children’s Hospital of Philadelphia). ImageJ (National Institutes of Health) was used for the posterior analysis of the images.
2.9. DC-ApoNec preventive vaccination
DC-ApoNec vaccine was prepared and administered as described in [17]. Briefly, DCs and ApoNec cells were co-cultured in a 1:1 ratio for 24 h and resuspended in PBS. Four doses of 2 × 105 DC-ApoNec were administrated s.c. every 14 days in the inguinal region of the same flank. Two weeks after, 1.5 × 104 viable B16-F1 cells were administered s.c. on the contralateral flank to the vaccination site. Mice were monitored every two days for tumor growth by palpation, and tumor size was measured with a Vernier caliper. Animals were euthanized when tumors displayed ulceration or a size ≥500 mm3.
2.10. Humoral response
At days 28-34th after tumor challenge, blood samples were obtained by sub-mandibular bleeding and serum stored at −80 °C until used.
To determine the presence of serum Abs by FACS, 2 × 105 B16-F1 cells were incubated with 10% normal goat serum, centrifuged 5 min at 300 g at 4 °C, incubated for 60 min at 4 °C with serum dilutions, washed with PBS, incubated 30 min with a 1/100 dilution of RPE-goat anti-mouse Immunoglobulins (R0480, Dako), washed with PBS and fixed with 2% PFA. Mean Fluorescence Intensities (MFI) were analyzed in a FACScalibur cytometer.
For ELISA, B16-F1 cells and C57Bl/6 splenocyte monolayers on 96-well cell culture plates were fixed with 4% PFA for 15 min at room temperature, blocked with 100 μl 1% BSA in PBS for 1 h, incubated overnight at 4 °C with 75 μl of 1/200 serum dilutions in 0.1% BSA in PBS, washed 3 times with 150 μl 0.1% BSA in PBS, incubated with 100 μl 1/10000 dilution in 0.1% BSA in PBS of HRP-AffiniPure F (ab′)2 Fragment Goat Anti-Mouse IgG (H + L) (Jackson ImmunoResearch) and washed 3 times with 150 μl 0.1% BSA in PBS. TMB (Sigma) was used to detect HRP activity, and the reaction was stopped with 2 M H2SO4. Absorbance at 450 nm was assessed.
2.11. Western blot analysis
50 μg protein of B16-F1 or skeletal muscle lysates (used as control, data not shown) were run on 8% SDS–PAGE gels, transferred to nitrocellulose membranes, blocked for 1 h at room temperature, incubated overnight at 4 °C with a 1/100 serum dilution from protected mice (4 mice pooled per group), washed, and stained with peroxidase-conjugated rabbit anti mouse IgG + A + M specific secondary antibody (Zymed) for 1 h at room temperature. The bound antibody was detected using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Scientific) according to the manufacturer’s protocol. Imaging was performed on an LAS 500 scanner (GE).
2.12. IFN-γ enzyme-linked immunospot assay (ELISPOT)
Splenocytes were seeded (1 × 106) in 24-well plates in 1 ml of complete medium consisting of RPMI containing 10% FBS, 2 mM glutamine, 100 U/mL penicillin, 100 μg/ml streptomycin, 10 mM HEPES, 50 μM 2-mercaptoethanol and 100 U/mL of human IL-2 (Laboratorio Pablo Cassará SRL, Argentina). B16-F1 lysate was added (lysate from 70,000 B16-F1 cells/1 × 106 splenocytes) and they were cultured at 37 °C for 12 days. Every 3 days, fresh complete medium with IL-2 was added. Murine IFN-γ Elispot Set (BD Biosciences) was used and manufacturer’s instructions were followed. Harvested splenocytes from 12-day culture were added to the IFN-γ-coated plates (2 × 105 cells/well) and cultured with complete medium with B16-F1 lysate for 18 h at 37 °C. As positive control 20 ng/ml PMA + 1 μg/ml Ionomycin was used (data not shown). Spots were visualized by adding 50 μl/well of AEC Substrate (BD Biosciences) for 2 min. Substrate reaction was stopped by washing the plate with deionized water. Plates were scanned using an AID iSPOT ELR088IFL analyzer.
2.13. High throughput sequencing
Snap-frozen pellets were prepared from C57Bl/6 splenocytes, B16-F1, Cloudman and 4T1 cells. DNA and RNA extraction, sequencing and standard bioinformatic analysis were all performed by BGI Tech Solutions (Hong Kong). Whole exome sequencing: Agilent SureSelect Mouse Exon (50 Mb), 50× coverage, and then sequencing (PE150). RNAseq: Illumina HiSeq400 100PE sequencing platform, >20 million reads/sample. The Rnaseq information was graphed using Plotly. The MHC-I binding predictions were made using the IEDB analysis resource Consensus tool [22] which combines predictions from ANN aka NetMHC (4.0) [23-25], SMM [26] and Comblib [27]. The predicted output is given in units of IC50 nM. FPKM values of the analyzed genes are included on Supplementary Table II. The raw data is available at the Geo Database online repository, series record GSE109268 provides access to the data.
2.14. Statistical methods
GraphPad Prism version 5.00 Software (San Diego, USA) and Infostat version 2016 (Córdoba, Argentina) were used to graph data and perform statistical analysis.
3. Results
3.1. B16-F1 and Cloudman melanoma cells are efficient sources of MDAs.
We investigated the expression of MDAs by the syngeneic B16-F1 (H-2b haplotype) and the allogeneic Cloudman (H-2d haplotype) melanoma cell lines. Messenger RNA levels were quantified by qRT-PCR (Fig. 1A). Both melanoma cell lines expressed Tyr, Dct (Trp-2), Mlana (Melan-A) and Pmel, and no significant differences in expression were observed between the two cell lines. Furthermore, both underwent apoptosis after 70 Gy irradiation, with >50% of irradiated cells staining positive for Annexin V after 48 h of culture (Fig. 1B) and displaying lack of proliferative capacity colony forming assay in soft agar (data not shown). Thus, 48-hour post-irradiation cells (ApoNec) were later used to load DCs.
Fig. 1.
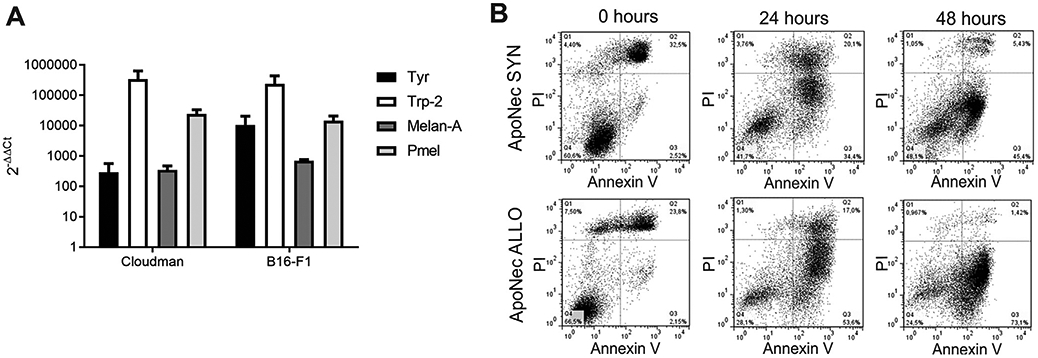
MDA expression and apoptosis induction after γ-irradiation. (A) mRNA levels for MDAs were assessed by qRT-PCR in B16-F1 and Cloudman cells. The 2−ΔΔCt values were calculated using B16-F1 and Cloudman Cts compared to H5V endothelial cells Cts. Actb was used as reference gene. Mean ± SD from two independent experiments is shown. (Studenťs t test, p > 0.05). (B) B16-F1 and Cloudman cells were γ-irradiated (70 Gy) and cultured for 0, 24 or 48 h. They were then stained with AnV-FITC and IP, and percentages of apoptotic/necrotic cells were assessed by flow cytometry.
3.2. B16-F1 and Cloudman irradiated cells are incorporated by CD11c+ cells and are transported within MIIC
B16-F1 and Cloudman ApoNec cells were labelled with PKH26 and co-cultured in vitro with immature DCs at 37 °C or 4 °C (unspecific binding). After 24 h, 60.3 ± 21.1% and 71.0 ± 19.4% of CD11c+ cells internalized cell components from B16-F1 ApoNec or Cloudman ApoNec cells respectively (Fig. 2A). Cloudman ApoNec loaded DCs (DC-ApoNec ALLO) significantly upregulated surface MHC-II compared to B16-F1 ApoNec loaded DCs (DC-ApoNec SYN), while surface CD86 expression was not affected (Fig. 2B).
Fig. 2.

Internalization of ApoNec cells by DCs. (A) ApoNec SYN or ApoNec ALLO cells were stained with PKH26 and cultured with DCs for 24 h at 37 °C or 4 °C (unspecific binding). DCs were stained with anti-CD11c Ab, and PKH26 incorporation in CD11c+ cells was assessed by flow cytometry. Three independent experiments were performed, dotplots from a representative experiment are shown. The percentage of incorporation of ApoNec cells by DCs was assessed as the percentage of CD11c+ PKH26+ cells/CD11c+ cells. Mean ± SD from three independent experiments is shown. (Studenťs t test, α = 0.05, n = 3, p > 0.05). (B) MHC-II and CD86 MFI on DC-ApoNec was assessed by staining with anti-CD11c Ab and either anti-I-Ab or anti-CD86 antibody and analyzed by flow cytometry. Lipopolysaccharide (LPS)-treated DCs were used as positive control. Student’s t test was used to compare DC-ApoNec SYN and DC-ApoNec ALLO (α = 0.05, n = 3, *p < 0.05). (C) Transmission electron microscopy of DC-ApoNec SYN and DC-ApoNec ALLO (i) DC showing membrane ruffles and multiple endocytic/phagocytic compartments, some loaded with pigment (7000×). (ii) Endocytic/phagocytic compartments containing pigment granules (white arrows) shown at higher magnification (50,000×). (iii) DC showing membrane ruffles and endocytic/phagocytic compartments (12,000×). (iv) Macropinosomes (black arrows) shown at higher magnification (50,000×). (D) DCs were cultured for 6 hs with Celltrace violet-stained ApoNec SYN or ApoNec ALLO (red). Then, lysosomes in DCs were stained using anti-Lamp-1 Ab (green) and visualized using confocal microscopy.
We also analyzed by electron microscopy morphologic features of DC-ApoNec SYN and DC-ApoNecALLO (Fig. 2C). In DC-ApoNecSYN and DC-ApoNec ALLO we observed numerous endocytic/phagocytic compartments, some containing melanin (Fig. 2C). ApoNec cell-derived material was also probably incorporated by macropinocytosis, as membrane ruffling could be observed in DCs and flocculent material could be observed in intracellular compartments (Fig. 2C iv). After 6 h of co-culture, ApoNec material localized to vesicles that were labeled for lysosomal associated membrane protein 1 (LAMP1) vesicles in DCs (Fig. 2D), suggesting that the majority of ApoNec-containing endosomes/phagosomes had matured and fused with lysosomes.
Peptide loading on MHC-II molecules occurs within MHC-II-containing compartments (MIIC)[28]. MHC-II GFP-expressing DCs were used to identify MIIC and their interaction with incorporated ApoNec material (Fig. 3). We observed ApoNec material within MIIC at 3 and 7 h of co-culture, and are also present within extensive dynamic tubules that emerged from these compartments (Movies S1-S4); such tubules have been shown to facilitate either cell surface expression of peptide-MHC-II complexes from endolysosomes [29,30] or mixing of phagosome contents for enhanced MHC class II presentation from phagosomes [31].
Fig. 3.

MIIC in DC-ApoNec cells. ApoNec SYN or ApoNec ALLO were stained with PKH26 and cultured with MHC-II GFP-expressing DCs. At 3 or 7 h of co-culture DCs were visualized by spinning-disk confocal microscopy.
3.3. Partially effective melanoma protection is induced by vaccination with DCs loaded with Cloudman ApoNec cells
It has been previously demonstrated that vaccination with DC-ApoNec SYN is effective and long-lasting [16]. To test whether DC-ApoNec ALLO could also elicit effective long-lasting melanoma protection, four s.c. doses of 2 × 105 DC-ApoNec ALLO or DC-ApoNec SYN were administered to C57Bl/6 mice, one every 14 days. Unloaded DCs were administered as control of unspecific immune activation, and PBS (vehicle) was administered as negative control. Fourteen days after completion of the vaccination scheme, 1.5 × 104 viable B16-F1 cells were s.c. injected, and the animals were monitored for tumor growth.
DC-ApoNecSYN vaccination induced complete protection, with 100% of vaccinated animals remaining tumor-free 10 weeks after tumor challenge (Fig. 4A). By comparison, DC-ApoNec ALLO vaccination induced partially effective anti-melanoma protection, as 60% of the animals remained tumor-free, with a significant difference in protection between DC-ApoNec SYN and DC-ApoNec ALLO vaccinations (p < 0.05). Tumor-free animals were rechallenged two months after the first tumor challenge with a s.c. injection of 1.5 × 104 viable B16-F1 cells and observed for an additional 60 days. DC-ApoNec SYN vaccinated mice remained tumor-free, while there was a significant decrease in protection in mice vaccinated with DC-ApoNec ALLO (Fig. 4B).
Fig. 4.

Tumor protection elicited by DC-ApoNec SYN and DC-ApoNec ALLO vaccination. (A) Kaplan Meier analysis showing the percentage of tumor-free animals following tumor challenge: four s.c. doses of DC-ApoNec SYN or DC-ApoNec ALLO vaccines were administered (one dose every 14 days). Fourteen days after completion of the vaccination scheme, 1.5 × 104 viable B16-F1 cells were administered s.c., and the animals were monitored for tumor growth for the following 60 days. One representative experiment of two independent experiments is shown. Statistical analysis: Logrank Test was performed for each group against the control group (PBS) (n = 10, α = 0.05, *p < 0.05, ***p< 0.001). (B) Kaplan Meier analysis showing the percentage of tumor-free animals following tumor rechallenge: 2 months after B16-F1 administration, tumor-free animals were rechallenged with another s.c. injection of 1.5 × 104 viable B16-F1 cells and the animals were monitored for tumor growth for the following 10 weeks. Statistical analysis: Logrank Test (6 ≤ n ≤ 9, α = 0.05, p < 0.05) (C) Anti-B16-F1 Abs induced by vaccination. B16-F1 viable cells were incubated with sera obtained one month after tumor challenge (1:200, 1:400, 1:800, 1:1600 and 1:3200 dilutions) and then incubated with RPE-anti-mouse immunoglobulins Ab. Geo Mean RPE-Fluorescence Intensities were assessed by flow cytometry. Mice protected by DC-ApoNecALLO vaccination (P) or non-protected (NP) were analyzed as separate groups. Sera obtained from unvaccinated/unchallenged mice were used as control. Mean ± SD is shown (3 ≤ n ≤ 5, α = 0.05). Geo mean values between treatments (1:200 serum dilution) were compared using the nonparametric Kruskal Wallis Test, and as Post-Test, Dunn’s Multiple Comparison Test was performed to compare each treatment against control sera. (D) Serum anti-B16-F1 IgG induced by vaccination by ELISA. B16-F1 cells were plated on 96-well cell culture plates, fixed with 4% PFA and incubated with 1/200 serum dilutions obtained from vaccinated and protected animals. HRP-AffiniPure F(ab’)2 Fragment Goat Anti-Mouse IgG (H + L) (Jackson ImmunoResearch) 1/10,000 dilution was used as secondary Ab. Absorbance at 450 nm was assessed. Statistical analysis was performed using Kruskal Wallis non-parametric test and Dunńs Multiple Comparison Test was used to compare each treatment against PBS (α = 0.05, n = 4) (E) Western blot analysis of B16F1 cell lysate with serum from PBS-treated mice, DC-ApoNecSYN or DC-ApoNecALLO vaccinated and protected mice, with detection using HRP- rabbit anti mouse IgG + A + M specific secondary antibody. Shown is one representative of two independent experiments. (F) IFN-y ELISPOT. Splenocytes (Sp) were obtained from vaccinated animals 10 weeks following tumor challenge, or at the time of sacrifice in the non-protected animals, pooled (n = 4), and cultured for 12 days with medium containing B16-F1 lysate. ELISPOT was performed by incubating the Sp with B16-F1 lysate for 24 hs. As positive control we incubated the Sp with 20 ng/ml PMA + 1 μg/ml Ionomycin (not shown). Three independent experiments were performed. Elispot plates and counts (in white) shown correspond to a representative experiment.
3.4. Humoral and cellular response to B16-F1 cells is induced by DC-ApoNec SYN vaccination
In order to analyze if an anti-B16-F1 humoral response targeting surface antigens was induced by DC-ApoNec vaccination, sera were obtained one month after tumor challenge to assess anti-B16-F1 Abs. For this, viable B16-F1 cells were incubated with increasing serum dilutions, followed by incubation with a PE-conjugated Ab specific for mouse immunoglobulins. Immunofluorescence was analyzed by flow cytometry (Fig. 4C). Compared to control sera from PBS and DCs-treated mice, DC-ApoNec SYN induced a significant anti-B16-F1 humoral response (p < 0.05). In contrast, there was no significant humoral anti-B16-F1 response with DC-ApoNec ALLO vaccination (p > 0.05). Additionally, we analyzed by ELISA the induction of anti-B16-F1 IgG antibodies and observed again that only DC-ApoNec SYN induced a significant response (p < 0.01) (Fig. 4D). We did not observe serum reactivity from vaccinated mice towards normal C57Bl/6 cells (data not shown). We performed Western blots to analyze the pattern of anti-B16-F1 antigens recognized by sera from protected mice, and observed that the majority of the proteins recognized were not shared between DC-ApoNec SYN and DC-ApoNec ALLO sera (Fig. 4E). There were two bands of approximately 150 kDa detected by both DC-ApoNec SYN and DC-ApoNec ALLO sera.
Additionally, we isolated splenocytes from vaccinated animals 10 weeks following tumor challenge or at the time of sacrifice in the non-protected animals, cultured them for 12 days with medium containing B16-F1 lysate, and then performed an IFN-y ELISPOT assay adding B16-F1 lysate for 18 h. We observed higher IFN-y production by splenocytes from animals vaccinated with DC-ApoNec SYN than from those vaccinated with DC-ApoNec ALLO or controls (Fig. 4F).
3.5. High throughput genomic and transcriptomic analysis of B16-F1 and Cloudman cell lines
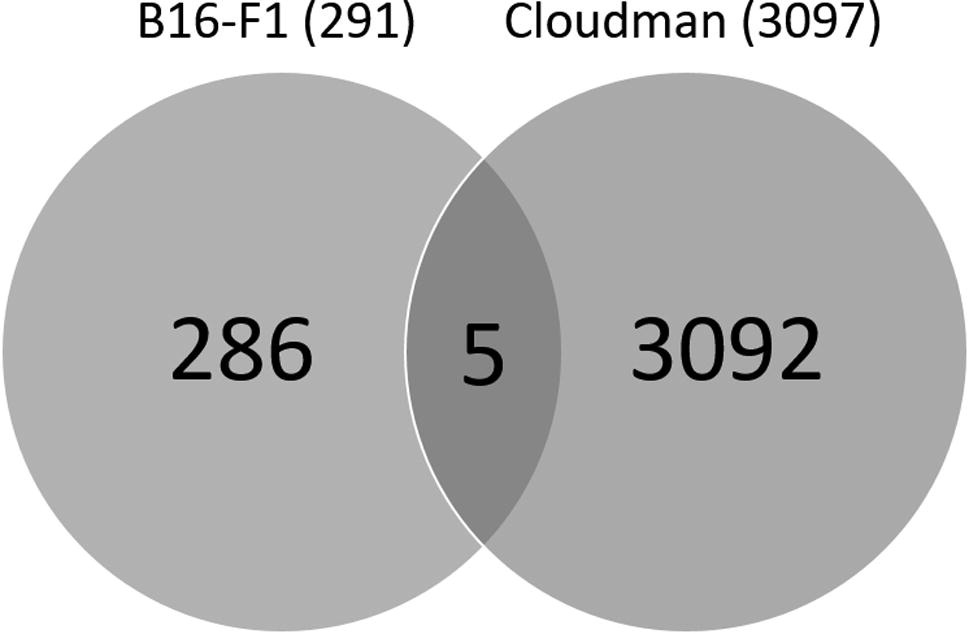
To determine the antigenic repertoire of B16-F1 and Cloudman cells, and of 4T1 cells as a non-melanocytic control, we analyzed their exome sequence and their transcriptome. To identify tumor-specific mutations B16-F1 cells were compared to normal C57Bl/6 splenocytes. 1041 new nonsynonymous somatic point mutations were identified, with 291 of them in expressed genes. Of such mutated proteins, only 5 were shared by B16-F1 and Cloudman cells (Supplementary Figure 1 and Supplementary Table I) and have poor predicted H-2 binding affinities, so they have a low probability to be efficiently presented to CD8 T cells in C57Bl/6 mice. Thus, it is unlikely that anti-melanoma protection provided by DC-ApoNec ALLO was due to such shared mutated proteins. The rest of the 286 mutated B16-F1 proteins could provide neoepitope candidates not shared by Cloudman cells. We have also detected 12 frameshift variant insertions and deletions in B16-F1 expressed genes that could provide neoepitope candidates, and are not shared by Cloudman cells.
We investigated by RNAseq analysis if there were other TAAs being expressed by B16-F1 and Cloudman cells that could be involved in anti-melanoma protection (Fig. 5). As expected, B16-F1 and Cloudman shared very high expression of MDAs. Interestingly, they both shared the expression of other TAAs as Spag9 and Maged2 and several CTAs, such as Odf2, Kif2c, Ddx20 and Lrwd1 (Supplementary Table II).
Fig. 5.

Antigenic profile of the cell lines as determined by RNAseq. Relative mRNA expression (log10 (FPKM + 1)) by B16-F1 and Cloudman melanoma cells. As non-melanocytic allogeneic cell line mammary carcinoma 4T1 cells were used. MDAs (dark grey), CTAs (medium grey) and other TAAs (light grey).
4. Discussion and conclusions
Preventive vaccination with DC-ApoNec SYN has proven to induce a significantly higher long-term protection than vaccination with DC-ApoNecALLO. We determined by transcriptome analysis that Cloudman cells are a good source of melanoma TAAs, including CTAs and high levels of MDAs. On the contrary, they do not share B16-F1 neoepitopes for MHC class I presentation, as the few identified shared mutations do not have strong predicted MHC-I binding affinities. We therefore conclude that shared MDAs, CTAs or other TAAs, are only capable of eliciting partial anti-melanoma protection. If we consider that with DC-ApoNec ALLO vaccination 60% of the animals remain tumor-free after the first tumor challenge, and of these 56% remain tumor-free after the second challenge, only 34% overall protection is achieved compared to 100% protection induced by DC-ApoNecSYN. These data would support the conclusion that shared TAAs are not capable of inducing long-term protection and that self-antigens/neoepitopes present in syngeneic cells are necessary to induce a potent long-lasting anti-melanoma protection.
B16-F1 and Cloudman melanoma cell lines undergo apoptosis after 70 Gy irradiation and are efficiently internalized by bone-marrow derived DCs. ApoNec material localizes to mature endolysomes, phagosomes and derived tubules that also harbor MHC-II molecules in DCs, suggesting that ApoNec-derived material can be efficiently processed for T-cell presentation. The only difference observed was a significant upregulation of surface MHC-II in DCs loaded with allogeneic cells, but this was not accompanied by upregulation of other costimulatory molecules as CD86, and ultimately did not induce a potent anti-tumoral protection.
It is noteworthy that DC-ApoNec SYN vaccination induces a very high level of anti-B16-F1 IgG Abs. Although vaccination in cancer aims to generate a long-lasting response dependent on antigen-specific CD8 T cells that generate cytotoxic T lymphocytes capable of killing tumor cells [32], it has also become evident from studies with anti-TAA monclonal Abs that these collaborate with T cells to amplify anti-tumor responses. Ab effector-mediated tumor cell killing potentially leading to antigen spreading and an inflammatory reprogramming of the tumor microenvironment have been described [33]. We observe that vaccination with DC-ApoNec SYN significantly induces IgG Abs directed towards B16-F1 proteins. Nonetheless, by Western blot we also detected some reactivity of DC-ApoNec ALLO sera towards B16-F1 antigens, and most of the bands recognized are not shared between DC-ApoNec SYN and DC-ApoNec ALLO vaccinated mice. It would be important to determine the lytic effect of these Abs against B16-F1 tumor cells, which could be particularly interesting when targeting cell-surface antigens.
RNAseq analysis was performed to determine the antigenic repertoire shared between B16-F1 and Cloudman melanoma cells lines. Apart from MDAs, we searched for other TAAs that could explain the partial protection induced by DC-ApoNec ALLO vaccination. The expression of Cancer-testis antigens (CTAs) are restricted in healthy adults to male germ cells and placenta, but are ectopically expressed in many human tumors [34]. Thus, CTAs could be considered tumor-specific antigens capable of inducing cancer-restricted immune responses [35], and could be more immunogenic than MDAs, which are constitutively present in melanocytes. In support of this notion, B16-F1 and Cloudman cells share the expression of testis-restricted CTAs like Odf2, Kif2c, Ddx20, Lrwd1, Boris, Acrbp and Tsga10, which provide epitopes with strong predicted binding affinities. Most of these CTAs have cytoplasmic or nuclear expression, so they could be only involved in CD8 T cell responses. Many known human CTAs have not been analyzed in this work as testis-restricted CTAs differ between humans and mice, as shown when analyzing RNA profiling data sets generated by the mouse ENCODE project.
This work suggests that TAAs and CTAs shared by B16-F1 and Cloudman tumor cells constitute partially effective target antigens for DC-based anti-melanoma preventive vaccination. The higher level of protection achieved by loading DCs with B16-F1 melanoma cells suggests that neoepitopes are needed for inducing long-lasting protection. It has recently become clear that mutated neoantigens are an important target of antitumor T cell responses, but unfortunately not all tumors have a high/relevant number of mutations, and they are typically patient-specific. Thus, further study of shared tumor antigens needs to be developed as an alternative immunotherapeutic approach.
Supplementary Material
{kind=link}
Acknowledgements
Collaboration with Dr. Michael Marks and Dr. Adriana Mantegazza at the Childreńs Hospital of Philadelphia was possible thanks to the travel fellowship awarded to Dr. Soledad Mac Keon by ‘The Company of Biologists’, Disease Models and Mechanisms (http://dmm.biologists.org), Disease Models and Mechanisms Travelling Fellowship – DMM – 161119. This work benefited from the Immune Epitope Database and Analysis Resource (http://www.iedb.org).
Funding
This work was supported by grants from the following institutions: Agencia para el Desarrollo Científico y Tecnológico (PRESTAMO BID PICT 2014-1932), Fundación SALES and Fundación María Calderón de la Barca, Argentina.
Abbreviations:
- Ab
antibody
- ALLO
allogeneic
- AnV
Annexin V
- ApoNec
48-hour cultured 70Gy irradiated cells (Apoptotic and Necrotic cells)
- CTAs
cancer-testis antigens
- DC
dendritic cell
- DC-ApoNec
dendritic cells loaded with ApoNec cells during 24-hour co-culture
- MDAs
melanocyte differentiation antigens
- MFI
mean fluorescence intensity
- MIIC
MHC-class II positive tubulovesicular compartments
- PI
propidium iodide
- s.c.
subcutaneous
- SYN
syngeneic
- TAAs
tumor-associated antigens
Footnotes
Declaration of Competing Interest
None.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.vaccine.2019.07.018.
References
- [1].Melero I et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol 2014;11(9):509–24. [DOI] [PubMed] [Google Scholar]
- [2].Wolfel T et al. Two tyrosinase nonapeptides recognized on HLA-A2 melanomas by autologous cytolytic T lymphocytes. Eur J Immunol 1994;24(3):759–64. [DOI] [PubMed] [Google Scholar]
- [3].Coulie PG et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med 1994;180(1):35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kawakami Y et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA 1994;91(14):6458–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].van der Bruggen P et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991;254(5038):1643–7. [DOI] [PubMed] [Google Scholar]
- [6].Gaugler B et al. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J Exp Med 1994;179(3):921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Boel P et al. BAGE: a new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity 1995;2(2):167–75. [DOI] [PubMed] [Google Scholar]
- [8].Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Castle JC et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012;72(5):1081–91. [DOI] [PubMed] [Google Scholar]
- [10].Kreiter S et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015;520(7549):692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Robbins PF et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013;19(6):747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].van Rooij N et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 2013;31(32): e439–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bobisse S et al. Neoantigen-based cancer immunotherapy. Ann Transl Med 2016;4(14):262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].von Euw EM et al. Monocyte-derived dendritic cells loaded with a mixture of apoptotic/necrotic melanoma cells efficiently cross-present gp100 and MART-1 antigens to specific CD8(+) T lymphocytes. J Transl Med 2007;5:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].von Euw EM et al. A phase I clinical study of vaccination of melanoma patients with dendritic cells loaded with allogeneic apoptotic/necrotic melanoma cells. Analysis of toxicity and immune response to the vaccine and of IL-10 −1082 promoter genotype as predictor of disease progression. J Transl Med 2008;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Goldszmid RS et al. Dendritic cells charged with apoptotic tumor cells induce long-lived protective CD4+ and CD8+ T cell immunity against B16 melanoma. J Immunol 2003;171(11):5940–7. [DOI] [PubMed] [Google Scholar]
- [17].Mac Keon S et al. Vaccination with dendritic cells charged with apoptotic/necrotic B16 melanoma induces the formation of subcutaneous lymphoid tissue. Vaccine 2010;28(51):8162–8. [DOI] [PubMed] [Google Scholar]
- [18].Bogyo M, Ploegh HL. Antigen presentation. A protease draws first blood. Nature 1998;396(6712):625–7. [DOI] [PubMed] [Google Scholar]
- [19].Arriaga JM et al. Metallothionein expression in colorectal cancer: relevance of different isoforms for tumor progression and patient survival. Hum Pathol 2012;43(2):197–208. [DOI] [PubMed] [Google Scholar]
- [20].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001;25(4):402–8. [DOI] [PubMed] [Google Scholar]
- [21].Mantegazza AR et al. Increased autophagic sequestration in adaptor protein-3 deficient dendritic cells limits inflammasome activity and impairs antibacterial immunity. PLoS Pathog 2017;13(12):e1006785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim Y et al. Immune epitope database analysis resource. Nucl Acids Res 2012;40(Web Server issue):W525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nielsen M et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 2003;12(5):1007–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lundegaard C et al. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res 2008;36(Web Server issue):W509–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 2016;32 (4):511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinf 2005;6:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sidney J et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res 2008;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Neefjes JJ et al. The biosynthetic pathway of MHC class II but not class I molecules intersects the endocytic route. Cell 1990;61(1):171–83. [DOI] [PubMed] [Google Scholar]
- [29].Chow A et al. Dendritic cell maturation triggers retrograde MHC class II transport from lysosomes to the plasma membrane. Nature 2002;418(6901):988–94. [DOI] [PubMed] [Google Scholar]
- [30].Boes M et al. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature 2002;418(6901):983–8. [DOI] [PubMed] [Google Scholar]
- [31].Mantegazza AR et al. TLR-dependent phagosome tubulation in dendritic cells promotes phagosome cross-talk to optimize MHC-II antigen presentation. Proc Natl Acad Sci USA 2014;111(43):15508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013;39(1):38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wittrup KD. Antitumor antibodies can drive therapeutic T cell responses. Trends Cancer 2017;3(9):615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fratta E et al. The biology of cancer testis antigens: putative function, regulation and therapeutic potential. Mol Oncol 2011;5(2):164–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget 2015;6(18):15772–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.