

Abstract
Smith–Magenis syndrome (SMS) is a genetic disorder caused by haploinsufficiency of the retinoic acid induced 1 (RAI1) gene. In addition to intellectual disabilities, behavioral abnormalities and sleep disturbances, a majority of children with SMS also have significant early-onset obesity. To study the role of RAI1 in obesity, we investigated the growth and obesity phenotype in a mouse model haploinsufficient for Rai1. Data show that Rai1+/− mice are hyperphagic, have an impaired satiety response and have altered abdominal and subcutaneous fat distribution, with Rai1+/− female mice having a higher proportion of abdominal fat when compared with wild-type female mice. Expression analyses revealed that Bdnf (brain-derived neurotrophic factor), a gene previously associated with hyperphagia and obesity, is downregulated in the Rai1+/− mouse hypothalamus, and reporter studies show that RAI1 directly regulates the expression of BDNF. Even though the Rai1+/− mice are significantly obese, serum analyses do not reveal any evidence of metabolic syndrome. Supporting these findings, a caregiver survey revealed that even though a high incidence of abdominal obesity is observed in females with SMS, they did not exhibit a higher incidence of indicators of metabolic syndrome above the general population. We conclude that Rai1 haploinsufficiency represents a single-gene model of obesity with hyperphagia, abnormal fat distribution and altered hypothalamic gene expression associated with satiety, food intake, behavior and obesity. Linking RAI1 and BDNF provides a more thorough understanding of the role of Rai1 in growth and obesity and insight into the complex pathogenicity of obesity, behavior and sex-specific differences in adiposity.
INTRODUCTION
Smith–Magenis syndrome (SMS) is a complex genetic disorder with multisystem involvement caused by haploinsufficiency of the retinoic acid induced 1 (RAI1) gene (1–3). Primary features of SMS include sleep disturbances (attributed to an inverted circadian rhythm), self-injurious behaviors, craniofacial abnormalities, mild-to-severe intellectual disability, brachydactyly, hoarse voice and developmental delays (4). SMS also affects growth and includes a high incidence of childhood obesity. Infants with SMS exhibit hypotonia and feeding difficulties, but the majority of persons with SMS become obese by the time they reach adulthood, with an increased prevalence of hypercholesterolemia (5). A study of 54 children with SMS found that by age 12, females were in the 90th percentile for weight, and by age 14, males were in the 90th percentile. An earlier study found that a majority of SMS patients had lipid values greater than the 95th percentile for age and gender for at least one of the following: total cholesterol, triglycerides and/or low-density lipoproteins (5). Further, in teens and adults with SMS, truncal obesity is common (3).
In a model of SMS, heterozygous Rai1-knockout mice were created on a mixed genetic background and backcrossed to C57Bl/6-Tyrc-Brd (6). These mice carry an insertion of an Escherichiacoli lacZ coding sequence and a neoR expression cassette that deletes 3910 bp of the coding region of Rai1 exon 2 (6). This insertion truncates the Rai1 protein, removing nuclear localization signals and the plant homeo domain (PHD), producing an effectively null allele that does not translocate to the nucleus, which is critical for Rai1 to regulate transcription. Animals that are homozygous for the Rai1 mutation are embryonic lethal by approximately 16 days post-coitum (6). Rai1+/− mice are slightly smaller than their wild-type (WT) littermates at 5 weeks, comparable with control mice at 10 weeks and overweight by 20 weeks (6). They also have behavioral abnormalities, variable penetrance of craniofacial defects and reduced fertility (7–9). Obesity phenotypes and further functional studies characterizing the metabolic profile in these animals, however, have not been studied.
Mouse models have been useful in identifying genetic factors that contribute to hyperphagic behavior and obesity in human disorders, including Bardet–Biedl syndrome (10), prohormone convertase 1 (Pc1) deficiency (11), leptin deficiency (12) and brain-derived neurotrophic factor (Bdnf) haploinsufficiency (13). To understand the role of Rai1/RAI1 in obesity, we analyzed obesity and metabolic profiles in mice and humans haploinsufficient for Rai1/RAI1. We performed the following functional evaluations in Rai1+/− and WT mice: (i) analysis for hyperphagia by observing daily feeding behavior and the satiation response; (ii) analysis of circulating levels of hormones related to feeding behavior, satiety and adiposity [ghrelin, leptin, insulin, peptide YY (PYY), amylin and adiponectin]; and (iii) evaluation of the possible contribution of RAI1 to abdominal and subcutaneous fat deposition. In humans, to characterize obesity in individuals with SMS, an anonymous online survey was created for caregivers that accessed BMI, obesity comorbidities and body shape.
The alteration in the expression of transcription factors may have widespread effects on other genes. The RAI1 protein contains a bipartite nuclear localization signal and a PHD/zinc finger domain (14). It also has a strong homology to the transcription factor TCF20 (15). Given these characteristics, we believe that RAI1 is a dosage-sensitive transcription factor affecting multiple molecular pathways, thus contributing to the complexity of the phenotypes associated with altered gene dosage (16–19). Identification and evaluation of these specific molecular pathways and their associated phenotypes, such as obesity, is key toward understanding the interaction of genes and proteins in these complex disorders.
RESULTS
To characterize feeding in these mouse models, we conducted studies to determine overall food consumption, the proportion of food consumed during the active phase and serum-level analysis of satiety hormones: insulin, PYY, ghrelin and leptin. Temporal assessment of food consumption can be used to determine hyperphagia, with the amount of food consumed during the light or dark phases used to determine whether these animals are modeling ‘night eating’ or eating during the inactive phase. Fasting studies and serum-level studies were used to determine the efficiency of the satiation response after a period of induced hunger, whereas gene expression studies focused toward the identification of genes in key pathways involved in metabolism and behavior.
Isolated feeding studies indicate hyperphagia in adult Rai1+/− mice
Isolated feeding studies in combination with weight assessments were conducted to evaluate growth and the quantity of food consumed per day by Rai1+/− and WT animals aged 5–30 weeks. At 5 weeks of age, male Rai1+/− mice were significantly smaller than WT mice (P = 0.0025, Fig. 1 and Supplementary Material, Fig. S1). By 10 and 15 weeks, no significant weight differences between Rai1+/− and WT mice were observed. However, by 20 weeks of age, male Rai1+/− mice were significantly larger than WT mice (P = 0.0359) and both male and female mice continued to grow substantially larger at 25 and 30 weeks (P = 0.0433 and P < 0.0001, Fig. 1A–C). Older adult Rai1+/− mice (up to 68 weeks) grew as large as 81.5 g before being sacrificed, whereas normal littermates never grew larger than 48.2 g (Supplementary Material, Fig. S3). The feeding behavior of Rai1+/− mice was observed and documented using isolated feeding studies. These studies were age-matched at 15, 20 and 25 weeks for Rai1+/− and WT male mice (Fig. 1C and D). A two-way ANOVA analysis of the average amount of food per day (g) over the weight of the mouse (g) was used to compare each group. Pooled data from multiple isolated feeding experiments show that Rai1+/− male mice were hyperphagic at 20 and 25 weeks (P = 0.0132 and P = 0.031), consuming ∼50% more food than WT littermates and supporting the data that show Rai1+/− mice were significantly larger than WT mice at these ages (Fig. 1C). At 15 weeks, Rai1+/− mice were not larger than WT mice, but they consumed significantly more food per gram of body weight (P = 0.0314) (Fig. 1D), supporting the increased rate of growth during this period (Fig. 1A and B). The rate of weight gain in the Rai1+/− mice was ∼2-fold that of WT over the course of study (Supplementary Material, Fig. S1).
Figure 1.

Growth and feeding behavior in Rai1+/− mice. (A) Body weights for male WT and Rai1+/− mice from 5–30+ weeks. (B) Body weights for female WT and Rai1+/− mice from 5–30+ weeks. (C) Image of Rai1+/− male (right) next to WT male (left) at 20 weeks. (D) Average daily food intake of male Rai1+/− and WT mice. (E) Average daily intake of food (g) over weight of mouse (g). Food intake data are derived from male mice. Data are plotted as mean ± SEM; n≥ 3. *P < 0.05; **P < 0.01; ***P < 0.0001.
Owing to the observed circadian rhythm abnormalities in mice with deletion of Rai1 (18) and the fact that humans with SMS also have sleep disturbance, we performed isolated feeding studies to determine when mice consumed food and specifically whether they were eating during the rest cycle (light phase of the 12 h dark/light cycle). These isolated feeding studies were conducted by taking food measurements every 12 h, shortly after the beginning of each light and dark phase. Male Rai1+/− and WT littermates were studied at 15 and 20 weeks of age. A two-way ANOVA analysis of the proportion of total food consumed during the dark cycle was used to compare each group. At the age of 15 weeks, Rai1+/− mice consumed a significantly higher proportion of their food during the light phase (rest phase) than did WT mice (P = 0.0191, Supplementary Material, Fig. S2). This pattern was not observed at 20 weeks (data not shown).
Fasting feeding studies show reduced satiation in Rai1+/− mice
Hunger was induced in the mice using a 24 h fast to evaluate satiety. Male mice of each genotype (20 weeks old) were isolated in cages with no access to food. After 24 h, serum was collected, blood glucose levels were assessed and food was returned. When food was returned, measurements of food consumption were taken every 2 h for the first 12 h and then every 12 h up to 60 h after the fast (Fig. 2). Food intake after the fast was compared with intake prior to the fast using ANOVA. Data show that WT mice consumed more than their normal intake during the initial 12 h after the fast, but after the initial 12 h post-fast, they experienced a significant drop in consumption when compared with their pre-fasting intake, indicating a satiety response. WT food intake returned to normal by 60 h after the fast (Fig. 2A). However, Rai1+/− mice were observed to have a muted satiation response (Fig. 2B). Food intake never dropped significantly below pre-fasting levels, with mice consuming consistent amounts of food both before and after the fast. These data also suggest that the hyperphagic behavior might be independent of the satiety response.
Figure 2.
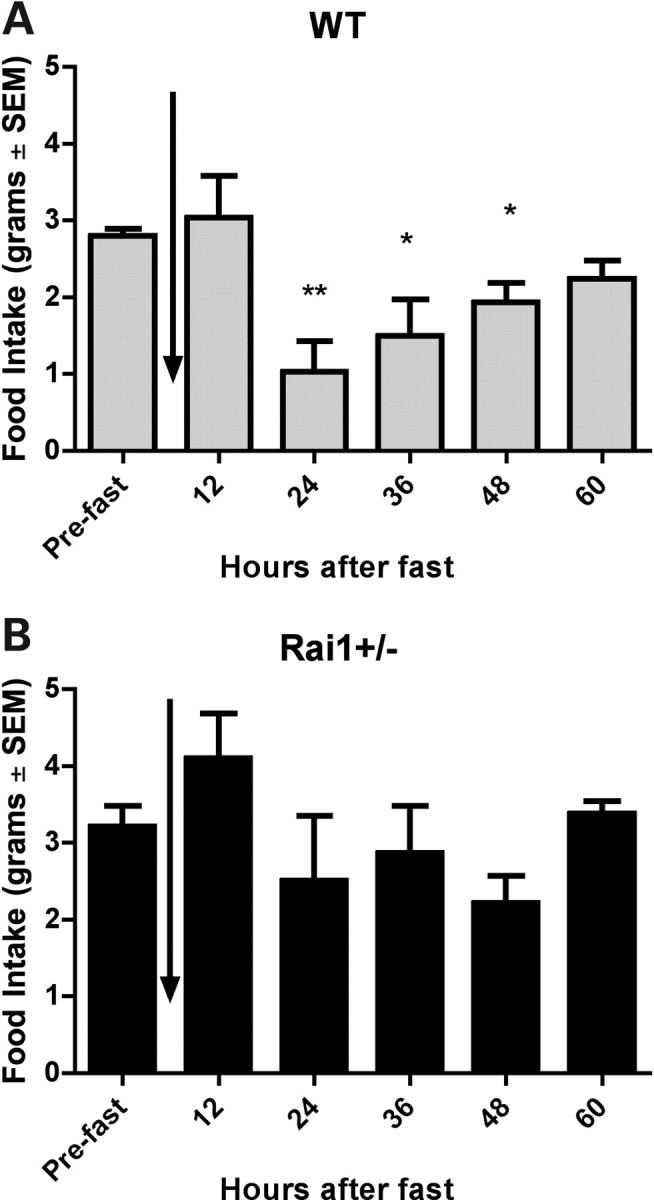
Feeding behavior of Rai1+/− mice after 24 h fast. (A) Food intake for WT mice is shown for pre-fasting and at 12 h intervals post-fasting (B) Food intake for Rai1+/− mice is shown for pre-fasting and at 12 h intervals after 24 h fast. All mice were 15 weeks of age at the time of study. Vertical arrow indicates point of 24 h fast for each graph. Data are plotted as mean ± SEM. *P < 0.05, **P < 0.01.
Increased leptin due to Rai1 haploinsufficiency
To determine the possible effects of the satiety signals leptin and PYY and other related hormones on the phenotype of these mice, fasting serum samples from mice of each genotype were evaluated at the University of Cincinnati Mouse Phenotyping Core. Results show that no differences between genotypes were observed for the fat-secreted hormone adiponectin (Fig. 3A), and similarly, insulin levels were unaltered in the Rai1+/− mice (Fig. 3B). Leptin levels were high in the Rai1+/− mice (P = 0.0368, Fig. 3C) and correlated with the adiposity of the animal. PYY, although not significant, showed a trend toward higher levels in the Rai1+/− mice (Fig. 3D). There was no significant difference between Rai1+/− mice and WT in serum levels of amylin; however, ghrelin did show significance in animals older than 30 weeks of age (P = 0.0059, Fig. 3E and F). No differences in fasting blood glucose levels or IGF1 levels were shown between Rai1+/− and WT mice (Fig. 3G and H), and tests showed no evidence of ketone bodies in urine (data not shown). Although adrenocorticotropic hormone (ACTH) levels (Fig. 3I) were not significantly different between genotypes, corticosterone levels (P = 0.0153, Fig. 3J) were significantly higher in Rai1+/− animals. Since this is a stress-related hormone, the significance of this finding in neurologically impaired mice is not clear. Total cholesterol and triglycerides were also not different in these animals (Fig. 3K and L).
Figure 3.

Serum levels of satiety-related hormones. Fasting serum levels of Rai1+/− mice for adiponectin, insulin, leptin, PYY, amylin, glucose, ghrelin, ACTH, corticosterone, IGF1, total cholesterol and triglycerides. Data show a significant increase in leptin and a trend toward PYY elevation in Rai1+/− mice. Ghrelin levels were significant only in mice >30 weeks of age. Although corticosterone levels were significantly elevated in Rai1+/− mice, ACTH was not different. No significant differences were observed due to age, sex or weight of animals unless indicated. Data represent values from male and female mice and are plotted as mean ± SEM. Leptin is significantly different *P < 0.05, **P < 0.01.
Increased abdominal fat distribution patterns in Rai1+/− mice
Fat distribution was analyzed by the dissection of Rai1+/− and WT mice. Owing to weight differences between WT and Rai1+/− female mice, a significant difference in the amount of fat extracted from Rai1+/ females when compared with WT females was observed (P = 0.0009, Fig. 4A). There was no difference in fat distribution between male WT and Rai1+/− animals (Fig. 4B). WT female mice were shown to have a higher proportion of subcutaneous fat than abdominal fat, whereas female Rai1+/− have a higher proportion of abdominal fat (P < 0.001, Fig. 4C). Histological analysis of adipose tissue sections revealed no significant differences in adipocyte cell size or shape between WT and Rai1+/− animals (Fig. 5), suggesting that more adipocytes are present in the fat tissue in the Rai1+/− mice. As described earlier, white adipose tissue was significantly elevated (by weight) in the Rai1+/− mice, with substantial adipose tissue found in the thoracic cavity of older female mice, in addition to substantial fat stores around all abdominal organs.
Figure 4.
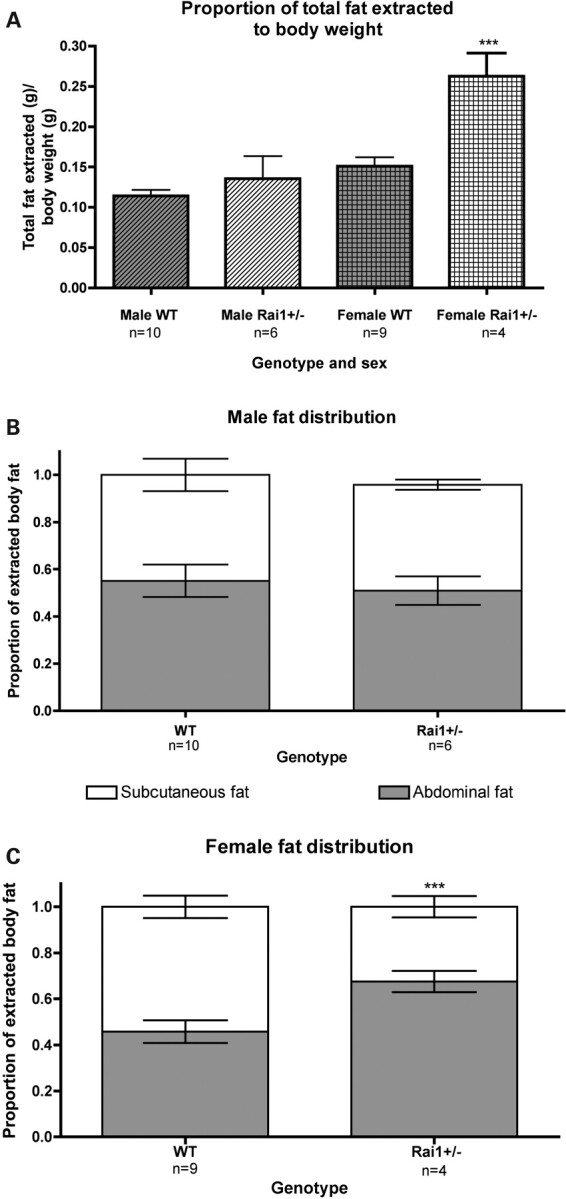
Fat distribution of Rai1+/− mice. (A) Comparison of total fat extracted from sex-matched Rai1+/− and WT mice. Total fat was defined as the weight of the sum of all extracted fat over body weight. (B) Fat distribution in males. Rai1+/− male mice have a lower proportion of abdominal fat than WT mice. (C) Female Rai1+/− animals have an inverted fat deposition pattern compared with males. Female WT mice have a lower proportion of abdominal fat than males, whereas Rai1+/− female mice have a significantly higher proportion of abdominal fat than WT female mice. Data are plotted as mean ± SEM. ***P< 0.001.
Figure 5.

White adipose tissue sections of Rai1+/− mice. Left panel, adipose tissue section from Rai1+/− mouse; right panel, adipose tissue section from WT mouse. Abdominal white adipose tissue was extracted from 40-week-old female mice preserved, sectioned, stained with H & E, and viewed under a light microscope. Analysis of several images using a ×20 objective revealed no significant differences between WT and Rai1+/− adipose tissue with regard to cell size. However, Rai1+/− adipocytes appeared full and were more consistent in size than those observed in WT. No unusual cellular infiltrations were observed.
Body shape and adiposity in persons with RAI1 haploinsufficiency
Although body shape, adiposity and obesity are frequently mentioned by clinicians, no studies have undertaken the analysis of SMS cases regarding BMI, fat deposition or factors associated with metabolic syndrome. Thus, an online survey of SMS caregivers was used to corroborate clinician's notes regarding abdominal obesity and related medical conditions in SMS patients (Table 1; Supplementary Material). Data were collected on 38 individuals with SMS (36 with a RAI1 deletion and 2 with a RAI1 mutation). The mean age of the SMS participant at the time of data collection was 13.5 years (range: 3–51 years), and 58% were female. Of the six adults with SMS (>19 years), two had been diagnosed with type II diabetes, two had high cholesterol and three had high triglycerides. Of the 32 children with SMS (≤19 years), three had high cholesterol and one had high triglycerides. A χ2 analysis revealed a significant difference in male and female reported body fat storage (P = 0.0406). Seventeen individuals (45%) were described as having mostly truncal obesity, with excess weight stored ‘around the waist and stomach areas—very little stored around the legs and buttocks’. Of these 17 individuals, 13 (76%) were female. Ten of the 16 males (62.5%) in the study had uniform fat distribution.
Table 1.
Summary of SMS growth survey
| Parametera |
SMS | |
|---|---|---|
| n | 38 | |
| Mean age (years) | 13.5 ± 9.0 | |
| Range (years) | 3–51 | |
| Sex | 42% male/58% female | |
| Mean BMI | 23.6 ± 7.9 | |
| Mean CDC weight percentile | 74.4 ± 30.0 | |
| >85th percentile for weightb | 55.2% (21/38) | |
| >95th percentile for weightb | 39.4% (15/38) | |
| >85th percentile for weightb; >9 years | 65.2% (15/23) | |
| >95th percentile for weightb; >9 years | 47.8% (11/23) | |
| >85th percentile for weightb; ≤19 years | 53.1% (17/32) | |
| >95th percentile for weightb; ≤19 years | 37.5% (12/32) | |
| Diabetes, type 2c | 5.3% (2/38) | |
| Hypercholesterolemiac | 13.2% (5/38) | |
| High triglyceridesc | 10.5% (4/38) | |
| Fat distributiond | Subcutaneous only | 0 |
| Truncal only | 23.7% (9/38; 7 female, 2 male) | |
| Uniform distribution | 26.3% (10/38; 5 female, 5 male) | |
| No excess weight | 7.8% (3/38; 1 female, 2 male) | |
| Excess uniform distribution | 21.1% (8/38; 3 female, 5 male) | |
| Excess truncal only | 21.1% (8/38; 6 female, 2 male) | |
aAll survey questions are available in Supplementary Material.
bPercentiles were determined using BMI and CDC growth assessment charts.
cDiagnosed by a physician, per parent report.
dFat distribution was reported by the parent/caregiver by selecting a body type with a cartoon and written description (see Supplementary Material).
Population estimates from the Centers for Disease Control show that the prevalence of obesity at ages 6–11 is 17% and at ages 12–19 is 17.3%. Based on our sample, the prevalence of obesity in 32 SMS patients through 19 years of age is 37.5%. Edelman et al. (20) previously reported a higher incidence of obesity in RAI1 mutation patients, likely due to hyperphagia, based on anecdotal reports from parents and physicians. A disproportionate number of SMS females also have a pattern of abdominal fat deposition (Table 1), even though abdominal obesity has a much higher prevalence in males in the general population (21). Abdominal obesity is associated with an increased risk for obesity comorbidities, including hypertension, high triglycerides and diabetes and metabolic syndrome. The incidence of diabetes, hypercholesterolemia and hypertension was not higher for individuals with SMS when compared with the general population (Table 1).
Hypothalamic gene expression is altered by Rai1 haploinsufficiency
Given the significant role that the hypothalamus plays in regulating food intake and satiety, we used expression microarrays to investigate the global expression of genes in the hypothalami of Rai1+/− mice. To identify genes differentially expressed from WT, we used the Illumina custom test for differentiation, which is more useful at estimating variance within a small sample size than the Wilcoxon signed-rank test or Student's t-test. Our analysis detected 23 000 probes for 16 000 genes. In Rai1+/− mice, 92 genes were significantly upregulated (P < 0.05); using a higher stringency (P < 0.01), 5 genes were significantly upregulated. In Rai1+/− mice, 397 genes were significantly downregulated (P < 0.05), whereas using a higher stringency (P < 0.01), 68 genes were significantly downregulated. The top genes downregulated and upregulated by reduced Rai1 expression by differential P-value and fold change are shown in Table 2 (a list of all dysregulated genes are provided in Supplementary Material).
Table 2.
Top dysregulated genes in Rai1+/− mouse hypothalamus
| Gene symbol | Differential P-valuea | Fold change | Gene name | Functions | Associated cellular functions or phenotypesb |
|---|---|---|---|---|---|
| Hoxa5 | <0.0001 | −223.2415 | Homeobox A5 | Transcription factor | Tumorigenesis, anterior–posterior axis |
| Hoxb6 | <0.0001 | −109.6210 | Homeobox B6 | Transcription factor, Antp homeobox family | Embryonic development; anterior–posterior development; leukemia; colon cancer |
| Gh | <0.0001 | −82.7727 | Growth hormone | Somatotropin/PRL hormone family | Growth control, obesity, dwarfism, acromegaly, Kowarski syndrome |
| Pcp2 | <0.0001 | −75.9074 | Purkinje cell protein 2 | GTPase activator, guanine nucleotide exchange factor | Purkinje cell degeneration; cell-type-specific modulator for G protein-mediated cell signaling |
| Prl | <0.0001 | −36.0794 | Prolactin, precursor | Cytokine hormone | Glucocorticoid receptor signaling, hyperinsulinemia; promotes lactation; supports pregnancy |
| Hoxb5 | <0.0001 | −17.3255 | Homeobox B5 | Transcription factor | Lung and gut development; acute myeloid leukemia |
| Slc22a6 | <0.0001 | −15.0388 | Solute carrier family 22, member 6 | Organic anion transporter | Renal sodium-independent organic anion transport and excretion |
| Prg4 | <0.0001 | −12.3189 | Proteoglycan 4 | Chondrocyte-specific proteoglycan | Elastic absorption and energy dissipation of synovial fluid on surface of articular cartilage; prevents protein deposition onto cartilage; mutations result in camptodactyly–arthropathy–coxa vara–pericarditis syndrome |
| Slc6a12 | <0.0001 | −7.1673 | Solute carrier family 6, member 12 | Neurotransmitter transporter | GABA receptor signaling; epilepsy, generalized anxiety disorder |
| Pomc | <0.0001 | −5.2112 | Propiomelanocortin | Melanocortin hormone precursor; 10 peptide hormones produced | Energy homeostasis; immune modulation; adrenocorticotrophin is essential for normal steroidogenesis and maintenance of normal adrenal weight; several distinct melanotropins, lipotropins and endorphins are contained within the adrenocorticotrophin and beta-lipotropin peptides; mutations associated with early-onset obesity, adrenal insufficiency and red hair pigmentation; ACTH stimulates the adrenal glands to release cortisol; MSH increases the pigmentation of skin by increasing melanin production in melanocytes; beta-endorphin and met-enkephalin are endogenous opiates involved in pain homeostasis |
| En1 | <0.0001 | −4.8849 | Engrailed homeobox 1 | Transcription factor | Development; segmentation in Drosophila; pattern formation |
| Myoc | <0.0001 | −4.3210 | Myocilin | Trabecular meshwork glucocorticoid-inducible response protein | Cytoskeletal function, regulation of intraocular pressure in trabecular network; mutation causes hereditary juvenile-onset open-angle glaucoma |
| Dpp4 | 0.0022 | −4.2198 | Dipeptidyl-peptidase 4; DPP-IV; CD26 | Membrane glycoprotein; adenosine deaminase complexing protein-2; T-cell activation antigen CD26 | Intrinsic membrane glycoprotein and a serine exopeptidase that cleaves X-proline dipeptides from the N-terminus of polypeptides; ubiquitous, membrane-bound enzyme that has roles in T-cell activation, nutrition, diabetes, hyperinsulinemia, obesity; metabolism, immune and endocrine systems, bone marrow mobilization, cancer growth and cell adhesion |
| Irx2 | <0.0001 | −3.6987 | Iroquois homeobox 2 | Transcription factor | Pattern formation of vertebrate embryos |
| Slc38a5 | 0.0017 | −3.2475 | Solute carrier family 38, member 5 | Amino acid transporter | Na-coupled transport of neutral amino acids, mental retardation |
| Rbm3 | 0.0038 | 2.04 | RNA-binding motif protein 3, RNP1 | RNA processing, cell cycle | Glycine-rich RNA-binding protein family; encodes a protein with one RNA recognition motif (RRM) domain; expression is induced by cold shock and low oxygen tension |
| Wnk | 0.0060 | 2.21 | Wnk lysine deficient protein kinase 1 | Cytoplasmic serine–threonine kinase expressed in distal nephron | Controls sodium and chloride ion transport by inhibiting the activity of WNK4; may play a role in actin cytoskeletal reorganization |
| Ripk4 | 0.0039 | 2.77 | Receptor-interacting serine–threonine kinase 4 | Serine/threonine protein kinase | Interacts with protein kinase C-delta; can activate NFkappaB; required for keratinocyte differentiation |
| Sgk | 0.0015 | 3.04 | Serum/glucocorticoid-regulated kinase | Cellular stress response | Kinase activates certain potassium, sodium and chloride channels, suggesting an involvement in the regulation of processes such as cell survival, neuronal excitability and renal sodium excretion; high levels of expression may contribute to conditions such as hypertension and diabetic nephropathy |
| Arhgef10 | 0.0025 | 3.94 | Rho guanine nucleotide exchange factor (GEF) 10 | Rho GTPase | May form complex with G proteins and stimulate Rho-dependent signals; role in developmental myelination of peripheral nerves |
aGenes were identified using differential expression P-values and ranked fold change.
bFunctional information from Entrez Gene, RefSeq and Ingenuity. A complete list of dysregulated genes is provided in Supplementary Material.
Ingenuity Pathway Analysis (IPA) software was used to determine which biological pathways involved genes differentially expressed in Rai1+/− mice. A P-value for each pathway was determined using Fisher's exact tests to determine the likelihood of those genes assigned by chance. Considering the obese phenotype observed in Rai1+/− mice and SMS individuals, we examined genes related to obesity. Using IPA for disease and phenotype, we found that 20 genes associated with obesity were differentially expressed in the Rai1+/− mouse hypothalamus (Fig. 6A). Biological pathways altered in Rai1+/− mice include psychological disorders and behavior and neurological development (Fig. 6B and C).
Figure 6.
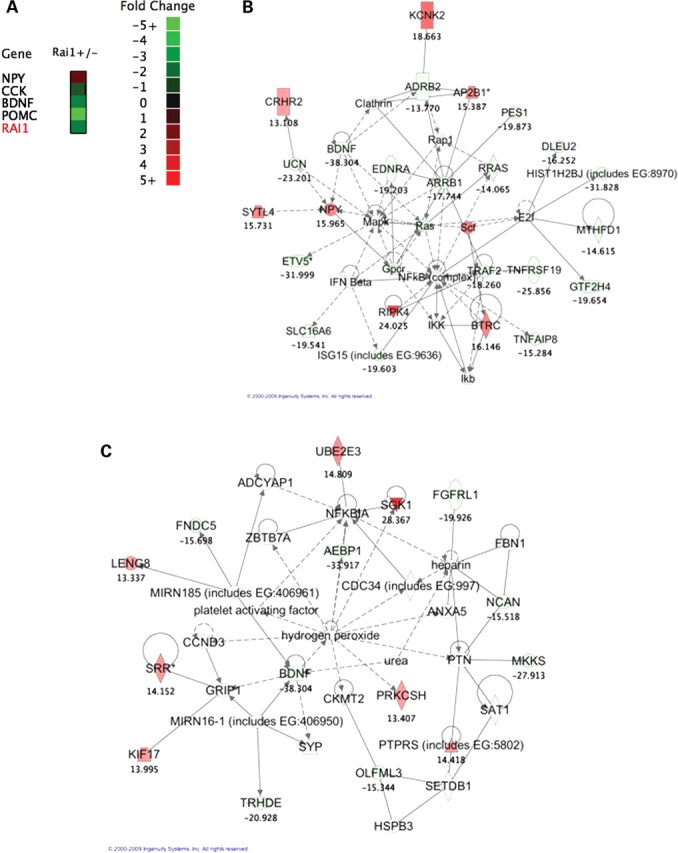
Altered gene expression in the hypothalamus. (A) Heat map of obesity-related gene expression. All genes have a significant differential P-value from WT mice. Obesity function was determined using Ingenuity disease phenotypes and Entrez Gene entries. (B) Psychological disorders and behavioral pathway in Rai1+/− mice. Ingenuity created pathway showing a psychological and behavioral pathway and genes which were altered in Rai1+/− mice. Genes that were differentially expressed from WT in the whole-genome microarray have a fold-change value listed below the gene name. Note the inclusion of confirmed genes BDNF and NPY. IPA score = 39. (C) Nervous system development and cell death pathway in Rai1+/− mice. Ingenuity created pathway showing a nervous system development and cell death pathway and genes which were altered in Rai1+/− mice. Genes that were differentially expressed from WT in the whole-genome microarray have a fold-change value listed below the gene name. Note the inclusion of BDNF. IPA score = 20.
Microrarray data revealed Rai1+/− mice to have an ∼0.6-fold expression of Rai1 compared with WT mice (Supplementary Material), which is muted compared with previous studies which report Rai1+/− mice express less than ∼50% Rai1 than WT mice (6,17). Q-RT–PCR of hypothalamic tissue was used to confirm Rai1 expression in the heterozygous mice and showed ∼40% expression compared with WT (P < 0.0001, Fig. 7A), which is consistent with previous studies.
Figure 7.

Quantitative PCR confirmation of microarray results. WT values are set to 1, and the relative expression in Rai1+/− mouse hypothalamus is shown in arbitrary units. (A) Rai1 expression levels are confirmed and match known genotype expression patterns. (B) No significant alteration of Cck expression in Rai1+/− mice. (C) Npy is not significantly upregulated in Rai1+/−. (D) Bdnf expression is significantly reduced in Rai1+/− mice. (E) Pomc is significantly reduced in Rai1+/−, consistent with expression array findings. (F) Mc4r expression is elevated in Rai1+/−. Graphs represent mean ± SEM (**P< 0.01, ***P< 0.001). All expression data were confirmed from three to six mice of each genotype for every specific gene.
Bdnf expression is reduced in Rai1+/− mice
We selected five genes associated with obesity-associated phenotypes and differentially expressed in the Rai1+/− hypothalamus microarray for gene expression analysis using qPCR: cholecystokinin (Cck), Bdnf and neuropeptide Y (Npy). Expression levels were confirmed using RNA from four Rai1+/− mice and six WT controls (Fig. 7). There were no gender-specific differences in expression for the genes studied. Cck is a brain/gut peptide involved in satiety that regulates food intake and weight gain (22). It can decrease the expression levels of Npy, which are usually co-localized with Cck in the neurons of the arcuate nucleus (23). Cck can also regulate ghrelin expression and has a similar effect as the satiety hormone, leptin (23,24). Microarray data show that Cck was downregulated ∼1.5-fold in Rai1+/− mice compared with WT mice. qPCR showed an insignificant decrease in Rai1+/− mice (Fig. 7B).
NPY is a neurotransmitter located in the neurons of the arcuate nucleus of the hypothalamus and plays an important role in the stimulation of hunger. Ghrelin activates Npy-containing neurons, causing an increase in [Ca2+]i via the phospholipase C and adenylate cyclase-PKA pathways, whereas leptin acts as an agonist via the phosphatidylinositol 3-kinase-PDE3 pathway (25,26). Overexpression of Npy causes an increase in feeding and weight gain (27,28). Loss of Npy expression has been shown to attenuate hyperphagic response to fasting and leptin deficiency (29,30). Microarray data show that Npy was upregulated 1.6-fold in Rai1+/− mice. qPCR showed an insignificant increase in Rai1+/− mice (Fig. 7C).
Bdnf is a neurotrophic growth factor involved in the growth of striatal neurons (31). Reduced expression of BDNF has been identified in depression, schizophrenia and obsessive-compulsive disorder (32–37). Tissue-specific deletion of Bdnf in the ventromedial and dorsomedial hypothalamus was found to induce hyperphagia and obesity (13). Haploinsufficiency for Bdnf in mice was found to cause increased food intake, early-onset obesity, hyperactivity and cognitive impairment (38). Microarray data show that Bdnf was downregulated 2.5-fold in Rai1+/− mice. qPCR confirmed the downregulation observed in Rai1+/− mice (P = 0.0037, Fig. 7D).
Proopiomelanocortin (Pomc) is found to be expressed in many tissues, including the skin, lymphoid system, anterior pituitary and the arcuate nucleus of the hypothalamus (39). Pomc is a precursor of melanocortins that are involved in energy homeostasis such as melanocyte-stimulating hormones (MSHs) and ACTH (40). Mutations in Pomc have been associated with obesity in both human and mice (40,41). Microarray data demonstrated that Pomc was downregulated ∼5.0-fold in Rai1+/− mice. qPCR confirmed the downregulation observed in the Rai1+/− mice (P < 0.0001, Fig. 7E).
The melanocortin 4 receptor (Mc4r) is a G-coupled receptor that interacts with α-MSH and has been shown to be involved with feeding behavior and energy expenditure (42). Mice with deletions of Mc4r exhibit an obesity phenotype, as well as hyperpahgia and hyperleptinemia (42). Expression data shown here reveal that Mc4r expression is upregulated ∼2.25-fold in the Rai1+/− mice (P = 0.0071, Fig. 7F).
RAI1 regulates transcription of BDNF via intronic enhancer element
Utilizing a chromatin immunoprecipitation (ChIP)-chip approach to identify chromatin fragments bound by RAI1, an enhancer element in BDNF intron 1 was identified and cloned (see Materials and Methods). With a luciferase reporter-based system, we examined whether the expression of RAI1 regulates or alters the expression of a luciferase reporter via the cloned BDNF regulatory region. As shown in Figure 8A, overexpression of RAI1 (isoform a) results in ∼2-fold increase in relative luciferase activity, indicating a positive role for RAI1 in the transcription of BDNF via this regulatory region and providing evidence of a role for RAI1 in the regulation of BDNF. Of note, a negative control was used in this assay, a proposed RAI1 isoform c that does not contain the RAI1 bipartite nuclear localization signal or the PHD domain, which are predicted to be necessary for RAI1 transport to the nucleus and induction of transcription, respectively. Figure 8B shows that the RAI1 isoform c protein does not contain the domains required for nuclear localization and thus, should not affect transcription. Further supporting these data, as shown in Figure 8C, RAI1a-GFP localizes to the nucleus, whereas RAI1c-GFP does not. Although these data support RAI1a as a transcriptional regulator directly binding DNA, RAI1c likely plays a role elsewhere in the cell.
Figure 8.

BDNF expression is directly regulated by RAI1 isoform a. (A) HEK293 cells were transfected with RAI1a-GFP or RAI1c-GFP along with the BDNF enhancer region (BDNFluc) and β-gal controls as described in Materials and Methods. Endogenous RAI1-associated gene expression was set to 1.0, and enhanced luciferase activity above background is reported as fold change. Data show that RAI1a enhances expression via the cloned BDNF enhancer region, whereas RAI1c does not. Data are plotted as mean ± SEM; n ≥3. ***P < 0.0001. (B) Predicted gene structures for RAI1 isoforms a and c are shown. RAI1 isoform a is 1906 amino acids (accession NP109590, EAW55692.1), whereas RAI1 isoform c is 966 amino acids (accession AAH21209.1, BC021209). The start site and first 946 amino acids are identical in isoforms a and c, with a unique alternatively spliced region present in isoform c. Exons are indicated by boxes, introns represented by lines and coding regions indicated by gray filled boxes. The following protein domains are indicated: polyQ region (red), nuclear localization signals (blue) and PHD domain (pink). RAI1 isoforms are supported by reported cDNAs. Gene structures are modified from AceView (www.ncbi.nlm.nih.gov) and Genome Browser (www.genome.ucsc.edu). (C) DAPI staining of nuclei transfected with RAI1a-GFP (viewed under DAPI filter, left) and RAI1a-GFP transfected into HEK293 cells (viewed with FITC filter) showing localization of RAI1a to the nucleus. (D) RAI1c-GFP transfected into HEK293 cells and stained with DAPI shows localization of RAI1c to the cytoplasm.
DISCUSSION
We provide unequivocal evidence to show that Rai1 haploinsufficiency affects feeding, satiety and fat deposition patterns in mice and humans. These data show that Rai1+/− mice are hyperphagic and have a delayed satiation response after induced hunger. Obese Rai1+/− mice do not have ketonuria, increased insulin or an elevated fasting glucose, so it is unlikely that they have insulin resistance associated with the observed obesity, nor do they have features consistent with metabolic syndrome. The elevated leptin and PYY levels in these mice indicate that the gut satiety signals are high, which is expected with the high levels of fat in these animals. If leptin and PYY signaling were functioning as we would expect, Rai1+/− mice with an increased serum leptin would have a sensitive satiation response and possibly hypophagia and resistance to obesity (43,44). However, high serum leptin has been identified as an early indication of adult-onset obesity in children (45). Therefore, the obesity observed in these mice is likely due to hyperphagia caused by a muted satiation response possibly due to increased ghrelin levels or leptin resistance (46).
Reduced expression of RAI1 affects adiposity. When we evaluated the fat distributions in Rai1+/− female mice, we found higher proportions of abdominal fat than observed in WT females, whereas Rai1+/− males had less abdominal fat than WT males. This is similar to the significant proportion of females with SMS with reported abdominal adiposity in our survey and consistent with clinical notations from geneticists. The documented abdominal adiposity in female SMS patients theoretically increases the risk for these individuals to develop polycystic ovary syndrome, an endocrine-metabolic disorder that results in increased risk for infertility, obesity, metabolic syndrome, cardiovascular disease; however, these conditions have not been reported in this population. Further evaluation of serum hormones in Rai1+/− mice could be used to determine whether Rai1 affects sex hormone levels and lead to a more comprehensive understanding of the pathogenicity of abdominal fat in females in the general population and to determine whether Rai1/RAI1 haploinsufficiency is possibly protective against these complex medical conditions. Adiponectin levels negatively correlate with high abdominal fat deposition, insulin resistance, diabetes mellitus, coronary heart disease and metabolic syndrome (47–50). Serum adiponectin in Rai1+/− mice was not elevated, and the serum results do not indicate diabetes. These data suggest that RAI1 haploinsufficiency contributes to early-onset obesity and increased abdominal fat deposition in females but does not increase the incidence of abdominal fat-associated insulin resistance and diabetes. Serum data also show that total cholesterol and triglycerides were not different from WT in the Rai1+/− mice, further supporting the absence of metabolic syndrome in this animal model of obesity. There is no conclusive evidence of the pathogenicity associated with increased abdominal fat in these mice. Other obesity comorbidities that should be assessed in humans and mice include cardiovascular risk and blood pressure. There is strong evidence that these comorbidities of abdominal obesity are also affected by alterations in the circadian cycle (51,52). Evaluating cardiovascular health may reveal the downstream effects of the observed adiposity, as well as the relationship between the circadian cycles and obesity comorbidities. A better understanding of the metabolic phenotypes and feeding behaviors in these mice will lead to better understanding of obesity in SMS and the genetic effects on energy homeostasis, which can provide knowledge to improve the health of the general population.
In order to evaluate the downstream gene expression effects of Rai1 haploinsufficiency in mice, we investigated global gene expression levels in the mouse hypothalamus. Our data provide evidence for the role of the hypothalamus in the obesity and satiation phenotypes seen in this mouse model of SMS. A large number of genes were differentially expressed in Rai1+/− mouse hypothalami. Top upregulated and downregulated genes include transcription factors, hormones and neurotransmitters. Many of the genes differentially expressed in this study are associated with obesity. Some genes were found to have phenotypes that relate to those observed in SMS. These may warrant further confirmation and investigation. Growth hormone (GH) and prolactin (PRL) are associated with growth, but hormone levels within SMS cohorts (due to 17p11.2 deletion) were shown to be within normal limits and to follow a normal circadian rhythm (53). Further, analysis of SMS cases due to RAI1 mutation showed only 1 out of 20 cases with documented GH deficiency (54,55). POMC has been associated with severe early-onset obesity (56) and may be an important downstream target of RAI1, and its expression was significantly reduced in the Rai1+/− mice, although Mc4r expression was elevated. Reduced expression of Pomc has been shown to increase food intake and body weight in mice (57). Increased expression of Mc4r is an indication of the activation of the anorexigenic melanocortin system, a regulator of food intake and energy homeostatsis and is consistent with findings in other obese rodent models with letptin resistance showing reduced Pomc expression in association with elevated Mc4r expression (58).
Other phenotypes commonly observed in SMS and associated with genes identified in this study (Supplementary Material) include WNT9B, which is involved in midfacial development; knockout mutations of Wnt9b can model cleft lip in mice (59). Lecithin cholesterol acyltransferase, LCAT, regulates cholesterol transport, and deficiency in mice can cause hepatic overproduction of triglycerides (60). Further, transcription factor 7-like 2, TCF7L2, is involved with insulin signaling in the pancreas (61), and TCF7L2 variants are associated with type II diabetes (62). Gastrulation brain homeobox 2, GBX2, is a transcription factor necessary for hindbrain development (63). Thus, the gene expression microarray data reported here support the role of multiple pathways in the development of the SMS phenotype due to RAI1 haploinsufficiency (Supplementary Material).
Although this study was focused toward genes related to satiety and obesity, these data can be used to identify genes related to observed hypoinsulinemia, cognitive dysfunction and altered molecular, metabolic and circadian mechanisms. Of particular interest are the many genes involved in behavior and psychological disorders. Considering the severe impact of the behavioral phenotypes on the quality of life of SMS patients, these data may be useful in identifying how cognitive function is altered in a complex disorder. Analysis of array data revealed altered expression of key genes involved in obesity and satiety, including Bdnf, which was previously shown to be associated with obesity and hyperphagia in a mouse model. Although Bdnf was not listed among the top dysregulated genes, microarray expression analysis and qPCR revealed ∼2.5-fold reduced expression of the gene Bdnf in Rai1+/− mice in conjunction with altered expression of Rai1 in the hypothalamus. ChIP-chip and reporter studies support direct binding of RAI1 to an intronic Bdnf enhancer as the primary means of regulation. Reduced expression of BDNF has been associated with obesity in mice and humans (38,64,65), and the data we report here document RAI1 as a positive regulator of BDNF expression. Thus, reduced Rai1 expression in mice results in reduced hypothalamic Bdnf expression resulting in an associated suppression of the satiety response and hyperphagia (Fig. 9). BDNF is a neurotropic factor that can affect serotonergic neurotransmission and has been implicated in a number of disorders, including autism, obsessive-compulsive disorder, schizophrenia, Alzheimer disease and eating disorders, among others (66–72). Further supporting the findings in this study linking RAI1 and BDNF are reports showing that haploinsufficiency of BDNF in humans is associated with hyperphagia, obesity and developmental problems (38), whereas mutation of TRKB, the BNDF receptor, is associated with developmental delay and obesity (73).
Figure 9.
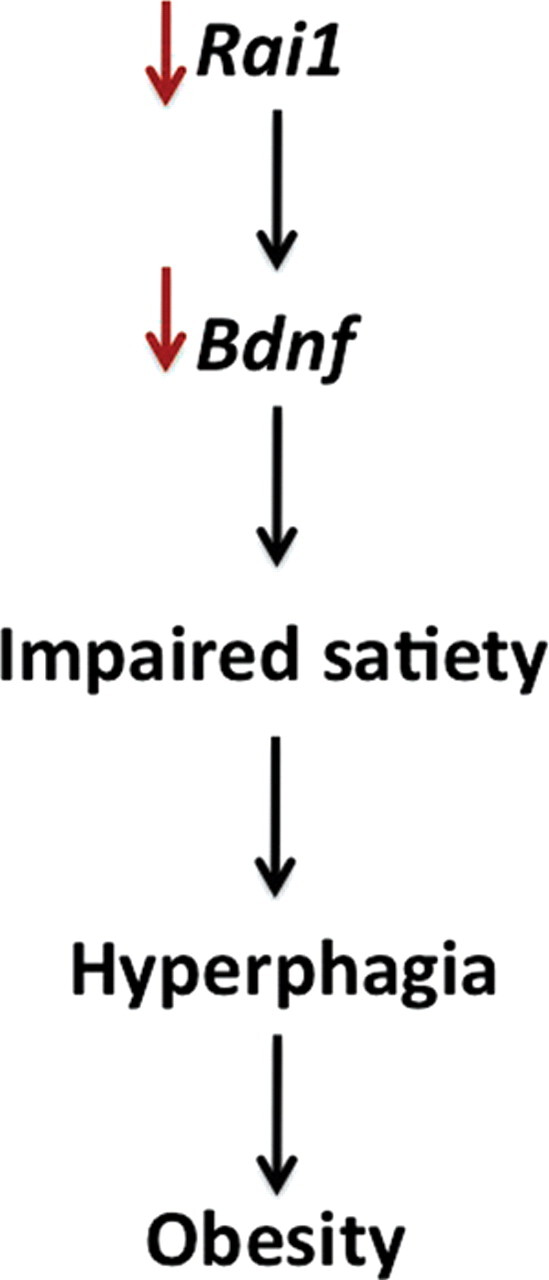
Rai1 haploinsufficiency impairs Bdnf expression, altering the satiety pathway. Data support a direct link between Rai1 and Bdnf that impairs satiety, resulting in hyperphagia, and leading to obesity when Rai1 is haploinsufficient, as shown in the Rai1+/− mouse model and in humans with SMS.
Considering the overlapping behavioral phenotypes of BDNF (38,74) and RAI1 haploinsufficiencies, including hyperphagia, cognitive impairment with global delays, mild–moderate intellectual disabilities with normal memory skills and repetitive behaviors, as well as alterations in the melatonin pathway in SMS, a comprehensive analysis of serotonin uptake and regulation should be studied in these mice. In addition to a more comprehensive understanding of SMS, the data presented in these studies are precursory to a greater characterization of the role of RAI1 in growth, obesity, adiposity and behavior and provide evidence for a significant role in growth, satiety, adiposity and behavior. Our studies indicate that downstream pathways altered by haploinsufficiency of Rai1, including Bdnf, contribute to the phenotypes of SMS and point to the critical importance of optimal gene dosage for normal phenotypic expression and the need to explore BDNF and its related pathways more critically in disorders involving obesity and developmental delay.
MATERIALS AND METHODS
Mice, breeding and maintenance
Mice heterozygous for insertional mutation of Rai1 [previously reported as B6.129S7-Rai1tm1Jrl/J (6) and referred to here as Rai1+/−] were obtained from the Jackson Laboratories, stock 005981, as a fully backcrossed strain (with C57Bl/6J-Tyrc-2J/J). Rai1+/− mice were bred in-house with C57Bl/6J mice. Pups were weaned at 3 weeks. Mice had access to water and a standard breeding chow of Lab Diet 5P06 Prolab RMH 2000 (∼9–10.4% fat) ad libitum. A constant temperature (21°C), humidity (40%) and automatic 12h light/dark cycle were maintained at all times. All tests were performed during the light portion of the cycle except for light-phase feeding studies, which were done using a red light to avoid altering the dark period. All animals were housed in compliance with the guidelines of the IACUC at Virginia Commonwealth University.
Genotyping
DNA was extracted from tails as described previously (16). Rai1+/− genotypes were confirmed using target allele and WT primers as reported in Bi et al. (6).
Isolated feeding studies
Male mice of each genotype (Rai1+/− and WT) were placed into isolated cages and given a pre-weighed amount of food at 5, 10, 15, 20 and 25 weeks. Food was weighed every 24 h for 5 days. Data were compared with age-matched WT littermates.
Light-phase and fasting feeding studies
Male mice of each genotype (Rai1+/− and WT) were placed in isolated cages with a pre-determined amount of food and an unlimited amount of water. Food was weighed every 12 h for 3 days. Night measurements were taken using a red lamp. To fast animals, all food was removed for 24 h, and animals were placed in a new cage to prevent hoarding. After 24 h, a pre-determined amount of food was returned at the beginning of the light phase. Food measurements were taken every 2 h for 6 h after the fast. Measurements continued every 12 h for 3 days. These evaluations were performed at 15 and 20 weeks of age.
Blood glucose
Blood was drawn from a needle prick on the end of the tail and measured using capillary test strips and an Easy Pro glucometer. All glucose samples were taken after 24 h fast.
Serum analysis
Blood was collected from animals via cardiac puncture after CO2-induced unconsciousness. Serum was separated by allowing the blood to clot for 20 min followed by centrifugation at 2000g for 10 min. All serum samples were collected under fasting conditions from the facial vein under general anesthesia with isofluorane at the end of the previously described fasting period. Required additives for each assay were added immediately following blood collection. A 1X protease inhibitor cocktail (Roche) was added for amylin measurements, and the serine protease inhibitor AEBSF [4-(2-amino-ethyl)benzenesulfonyl fluoride hydrochloride; Sigma] was added (0.4 mg/ml) for ghrelin measurements. Serum samples were aliquoted and stored at −20°C. ELISA tests were performed at the University of Cincinnati Mouse Phenotyping Core per standard protocols.
Ketone analysis
Animals were isolated in small container, with no bedding until they urinated, and an Alva Thinz Metabolism Strip No. 25 was dipped into the urine.
Fat distribution patterns in Rai1+/− mice
Fat distribution in Rai1+/− mice was determined via dissection. Male and female mice were selected at 30+ weeks, weighed and euthanized via a CO2 chamber. Intra-abdominal fat pads (gonadal, retroperitoneal and mesenteric) and subcutaneous fat pads (dorsal, inguinal and groin) were collected via dissection and weighed (74). Measurements of both abdominal and subcutaneous fat pads were combined to determine the amount of total fat collected. Data obtained by dissection were consistent with NMR analysis of fat distribution conducted at the University of Cincinnati Mouse Phenotyping Core (data not shown).
Histological analysis
For paraffin histology, abdominal adipose tissues isolated from the same anatomical region from age-matched Rai1+/− and WT females were fixed in 4% paraformaldehyde in phosphate-buffered saline. The tissues were processed using a Tissue-Tek VIP 5 tissue processor (Sakura Finetek USA, Torrance, CA, USA) and then embedded in paraffin blocks. Paraffin blocks were cut into 5 µm sections and then processed using standard deparaffinization and rehydration techniques. Sections were mounted on silanized glass slides and stained with hematoxylin and eosin using a Tissue-Tek Prisma Automated slide stainer (Sakura Finetek USA).
RAI1 participant survey
An anonymous online survey was designed to assess body shape in patients with RAI1 deletions and mutations. The survey was distributed to primary caregivers via the SMS support website PRISMS and the SMS Yahoo! Listserv and was an exempt study approved by the VCU Institutional Review Board. The survey assessed body type by asking the caregiver to select a description which best described the fat deposition pattern for their child. The survey included cartoon depictions of the type of fat deposition, as well as a written description such as ‘My son/daughter tends to store extra weight on arms, legs, hips, and buttocks. Very little weight is stored around the waist.’ The survey addressed the individual's background and included questions regarding gender, age, height, weight, formal diagnosis by a geneticist and the most recent physician visits. The patient's clinical history regarding diabetes, high cholesterol and high triglycerides was also assessed. Percentiles were determined using BMI and CDC growth assessment charts for children aged 2 to 20 years. Results are provided in Table 1. The complete survey is available in Supplementary Material.
Hypothalamus isolation
Male and female 30-week-old WT mice were euthanized by CO2 exposure. Rai1+/− mice were 52+ weeks at the time of death and tissue collection for microarray analysis. Hypothalami were extracted from younger mice for qPCR confirmation of array results. Hypothalami were immediately extracted from mouse brains and stored at −80°C.
RNA isolation
RNA was isolated using the TRIzol reagent/chloroform isolation method. The quality of the isolated RNA was determined using a 260/280 absorbency ratio and assessed using a Thermo Scientific Nanodrop 1000. For microarray studies, equal quantities of RNA were pooled for a WT sample (six male mice), whereas two 52+ week male mice were utilized for the Rai1+/− sample, and data were combined for assessment.
Microarray
Microarray hybridization was performed in the Massey Cancer Center Nucleic Acids Research Facility, DNA Microarray Core, using an Illumina MouseWG-6 v2.0 expression beadchip. The array was read using an Illumina BeadScan confocal scanner and analyzed by Illumina's BeadStudio software. All samples were run in duplicate.
Microarray data analysis
Using the Illumina BeadStudio Software, differential analysis was performed between the Rai1+/− and WT genotypes. Data were normalized using the quantile method. Differential P-values were determined using the Illumina custom analysis. The software was also used to calculate a false discovery rate and to adjust P-values accordingly. Detection P-values <0.05 were used to determine which probes were detected. Heat maps to visualize gene distribution were created using JMP statistical software. Pathway analysis was done using Ingenuity Pathway Analysis for gene and protein interaction. Significant interactions were determined using the Ingenuity Pathway Knowledge Base and a Fisher's exact test for significance for each pathway and biological function.
Real-time quantitative PCR
RNA from hypothalami from male and female mice of each genotype was isolated using the TRIzol reagent/chloroform method and quality assessed by nanodrop. Some individual hypothalamus samples that were used in the pooled samples in the microarray were analyzed for confirmation of array results individually using qPCR. cDNA synthesis was performed using 4 µg of RNA, Invitrogen Superscript 2 reverse transcriptase and random primers according to the manufacturer's instructions. ABI TaqMan probes for mouse mRNA expression were used to detect gene expression for Bdnf, Cck, Npy, Pomc, Mc4r, Rai1 and Gapdh. All samples were run in triplicate in a 10 µl reaction on an ABI Prism 7500 Fast System with Gapdh as an endogenous control.
Creation of plasmids
RAI1aFlag
The human RAI1a coding sequence (accession: NM_030665, beginning at ATG and ending at stop codon) was cloned into pEntr/D-TOPO, using standard manufacturer protocols (Invitrogen, Carlsbad, CA, USA). A 5′ TOPO polylinker was added to cDNA PCR forward primer (CACC) to ensure proper directional cloning. Plasmid DNA was isolated using Fermentus GeneJET™ plasmid mini kit (Burlington, Ontario, Canada) according to standard manufacturer instructions. Inserts were confirmed using RAI1 cDNA primers and sequenced using standard Sanger techniques. Next, RAI1apEntr/D-TOPO was recombined with pDest26Flag, using the standard Gateway protocol provided by Invitrogen to create pDest26RAI1aFlag.
BDNFLuc
The BDNF enhancer element identified in the ChIP-chip assay (described in what follows) was PCR-amplified using Elongase (Invitrogen) following standard manufacturer's protocols (forward primer: 5′-TGCCCGGTATGTACTCCTTC-3′; reverse primer: 5′-CAATTATGCCAGAGGCCAAT-3′) and cloned into StrataClone™ PCR cloning vector, using the standard protocol provided by the manufacturer (Agilent Technologies, Santa Clara, CA, USA) creating BDNFStrata plasmid. The insert was confirmed by standard Sanger sequencing. BDNFStrata and the target vector pGL3pro (Promega Corp., Madison, WI, USA) were digested with KpnI and SacI enzymes (New England Biolabs, Ipswich, MA, USA). The resulting products were gel-purified (Qiagen, Germantown, MD, USA) and insert and target vector were ligated to create BDNFLuc.
RAI1cGFP
RAI1c was created by PCR amplifying the coding sequence (NM_152256.1) as provided by Origene (Rockville, MD, USA). This sequence was cloned into pEntr/D-TOPO in the same manner as stated earlier and recombined with pDest47 (Invitrogen), using standard manufacturer's protocols, creating pRAI1cGFP.
ChIP with microarray
HEK293t cells were transfected with RAI1Flag plasmid as described in what follows by scaling the reaction up for a T75 culture dish. ChIP-chip was processed with mouse IgG Dynabeads (Invitrogen) and monoclonal anti-Flag IgG antibody (Sigma-Aldrich, St Lewis, MO, USA) according to manufacturer's protocols, using Nimblegen ‘ChIP sample preparation protocol v2’ (Roche Nimblegen, Madison, WI, USA). Arrays and initial data were processed by Nimblegen, using the Nimblegen HG18 RefSeq promoter array according to standard manufacturer's protocols. RAI1Flag-binding sites were generated using genomic data points, Peak_Start and Peak_End, from Nimblegen pre-processed data files. A putative BDNF enhancer element was identified in intron 1 of the BDNF gene (Hg18, Chr11:27 680 656–27 681 712 Mb).
Transfections
Human embryonic kidney (HEK293T) cells were maintained in six-well dishes containing Dulbecco's modified Eagle's medium with 10% (v/v) FBS, 2 mml-glutamine and 100 µg/ml of penicillin/streptomycin (Invitrogen) at 37°C in a 5% CO2 incubator. Cells were counted using trypan blue exclusion to ensure >90% viability. Transfections with pUC19, psvβ-Gal, BDNFLuc and RAI1Flag were performed using Lipofectamine™ 2000 (Invitrogen) according to manufacturer's instructions. Briefly, ∼5 × 105 cells were plated in 2.0 ml of growth medium without antibiotics 24 h prior to transfection. A total of 4 µg of total plasmid DNA, using pUC19 plasmid as ‘filler’ DNA, was diluted in 250 µl OptiMEM® reduced serum medium (Invitrogen). Similarly, 10 µl of Lipofectamine™ 2000 was diluted in 250 µl of OptiMEM® reduced serum medium, mixed well and incubated for 5 min. After incubation, diluted plasmid DNA was mixed with diluted Lipofectamine™ 2000 to a total volume of 500 µl and incubated for 20 min, then plasmid:Lipofectamine complexes were added to each well and mixed by rocking. Cells were incubated at 37°C in a 5% CO2 incubator for 24 h.
Luciferase reporter assay
After plasmid DNA transfection and 24 h incubation, HEK293 cells were washed with 2 ml DPBS (Invitrogen), and Tropix Glacto-Light™ (Applied Biosystems, Bedford, MA, USA) standard protocol was used. Briefly, 250 µl of lysis solution was added to each well of the six-well plate and scrapped until all cells were detached. Lysates were collected and centrifuged at 10 000g for 2 min to pellet cell debris. Next, 50 µl of the resulting supernatant was transferred to four wells of a 96-well white luminometer plate. Two wells were treated with 70 µl diluted Galacton® substrate (1:100, Galacton:Reaction buffer dilutent) (Applied Biosystems) and incubated for 30 min. To the two wells that contained the diluted Galacton® substrate, 100 µl of Accelerator(-II) (Applied Biosystems) was added. To the two wells that did not contain the diluted Galacton® substrate, 100 µl of Steady-Glo® Luciferase substrate (Promega Corp.) was added. Each well was read using the Wallac 1420 VICTOR2™ Luminometer (PerkinElmer, Waltham, MA, USA) on a maximum linear scale. Relative luciferase activity from each individual transfection was calculated by dividing the average number of light units read from the wells containing the Steady-Glo® Luciferase substrate by the average number of light units from the wells containing the Galacton® substrate and Accelerator(-II). The equation (ΔLuc/Δβ-Gal = relative luciferase activity) was used. Wells containing pUC19, psvβ-Gal and BDNFLuc were used as baseline luciferase activity. Each experiment was performed independently at least three times in duplicate. P-values were generated by averaging relative luciferase activity from each independent study and performing a two-tailed Student's t-test. Standard deviations were generated using GraphPad.
Statistical analyses
All statistical methods were conducted using JMP software. Mean food consumption at 5, 10, 15, 20 and 25 weeks were compared using pooled data from 12 different isolated feeding studies and dependent on genotype and age. These data were compared among the two genotypes (WT and Rai1+/−) using ANOVA. Food intake after the 24 h fast (taken at 2–48 h) was evaluated using repeated measures ANOVA, with effects for individual mouse, genotype, time point and group time interaction. These data were also evaluated separately for each time point using ANOVA. The average proportion of food consumed at night was evaluated using repeated measures ANOVA.
SUPPLEMENTARY MATERIAL
FUNDING
This project was supported, in part, by funds from Virginia Commonwealth University.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr M. Dance for excellent veterinary care and support throughout this project; Dr E. Wickham and Dr J. Wiley for helpful discussions; The University of Cincinnati Mouse Metabolic Phenotyping Core, Vanderbilt University Mouse Metabolic Phenotyping Core, Ms Meghan MacNeal and Ms Manavi Johri for technical assistance; and Dr J. Almenara, Dr H.D. Massey and C. Minard-Wilson for assistance with histology.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Slager R.E. Newton T.L. Vlangos C.N. Finucane B. Elsea S.H. Mutations in RAI1 associated with Smith–Magenis syndrome. Nat. Genet. 2003;33:466. doi: 10.1038/ng1126. [DOI] [PubMed] [Google Scholar]
- 2.Vlangos C.N. Yim D.K. Elsea S.H. Refinement of the Smith–Magenis syndrome critical region to approximately 950 kb and assessment of 17p11.2 deletions. Are all deletions created equally? Mol. Genet. Metab. 2003;79:134–141. doi: 10.1016/S1096-7192(03)00048-9. [DOI] [PubMed] [Google Scholar]
- 3.Elsea S.H. Girirajan S. Smith–Magenis syndrome. Eur. J. Hum. Genet. 2008;16:412. doi: 10.1038/sj.ejhg.5202009. [DOI] [PubMed] [Google Scholar]
- 4.Greenberg F. Lewis R.A. Potocki L. Glaze D. Parke J. Killian J. Murphy M.A. Williamson D. Brown F. Dutton R. et al. Multi-disciplinary clinical study of Smith–Magenis syndrome (deletion 17p11.2) Am. J. Med. Genet. 1996;62:247–254. doi: 10.1002/(SICI)1096-8628(19960329)62:3<247::AID-AJMG9>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 5.Smith A.C. Gropman A.L. Bailey-Wilson J.E. Goker-Alpan O. Elsea S.H. Blancato J. Lupski J.R. Potocki L. Hypercholesterolemia in children with Smith–Magenis syndrome: del (17) (p11.2p11.2) Genet. Med. 2002;4:118–125. doi: 10.1097/00125817-200205000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Bi W. Ohyama T. Nakamura H. Yan J. Visvanathan J. Justice M.J. Lupski J.R. Inactivation of Rai1 in mice recapitulates phenotypes observed in chromosome engineered mouse models for Smith–Magenis syndrome. Hum. Mol. Genet. 2005;14:983–995. doi: 10.1093/hmg/ddi085. [DOI] [PubMed] [Google Scholar]
- 7.Bi W. Yan J. Shi X. Yuva-Paylor L.A. Antalffy B.A. Goldman A. Yoo J.W. Noebels J.L. Armstrong D.L. Paylor R. et al. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Hum. Mol. Genet. 2007;16:1802–1813. doi: 10.1093/hmg/ddm128. [DOI] [PubMed] [Google Scholar]
- 8.Yan J. Bi W. Lupski J.R. Penetrance of craniofacial anomalies in mouse models of Smith–Magenis syndrome is modified by genomic sequence surrounding Rai1: not all null alleles are alike. Am. J. Hum. Genet. 2007;80:518–525. doi: 10.1086/512043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walz K. Spencer C. Kaasik K. Lee C.C. Lupski J.R. Paylor R. Behavioral characterization of mouse models for Smith–Magenis syndrome and dup(17)(p11.2p11.2) Hum. Mol. Genet. 2004;13:367–378. doi: 10.1093/hmg/ddh044. [DOI] [PubMed] [Google Scholar]
- 10.Rahmouni K. Fath M.A. Seo S. Thedens D.R. Berry C.J. Weiss R. Nishimura D.Y. Sheffield V.C. Leptin resistance contributes to obesity and hypertension in mouse models of Bardet–Biedl syndrome. J. Clin. Invest. 2008;118:1458–1467. doi: 10.1172/JCI32357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lloyd D.J. Bohan S. Gekakis N. Obesity, hyperphagia and increased metabolic efficiency in Pc1 mutant mice. Hum. Mol. Genet. 2006;15:1884–1893. doi: 10.1093/hmg/ddl111. [DOI] [PubMed] [Google Scholar]
- 12.Harris R.B. Zhou J. Redmann S.M., Jr Smagin G.N. Smith S.R. Rodgers E. Zachwieja J.J. A leptin dose–response study in obese (ob/ob) and lean (+/?) mice. Endocrinology. 1998;139:8–19. doi: 10.1210/en.139.1.8. [DOI] [PubMed] [Google Scholar]
- 13.Unger T.J. Calderon G.A. Bradley L.C. Sena-Esteves M. Rios M. Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J. Neurosci. 2007;27:14265–14274. doi: 10.1523/JNEUROSCI.3308-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toulouse A. Rochefort D. Roussel J. Joober R. Rouleau G.A. Molecular cloning and characterization of human RAI1, a gene associated with schizophrenia. Genomics. 2003;82:162. doi: 10.1016/S0888-7543(03)00101-0. [DOI] [PubMed] [Google Scholar]
- 15.Seranski P. Hoff C. Radelof U. Hennig S. Reinhardt R. Schwartz C.E. Heiss N.S. Poustka A. RAI1 is a novel polyglutamine encoding gene that is deleted in Smith–Magenis syndrome patients. Gene. 2001;270:69. doi: 10.1016/S0378-1119(01)00415-2. [DOI] [PubMed] [Google Scholar]
- 16.Girirajan S. Patel N. Slager R.E. Tokarz M.E. Bucan M. Wiley J.L. Elsea S.H. How much is too much? Phenotypic consequences of Rai1 overexpression in mice. Eur. J. Hum. Genet. 2008;16:941. doi: 10.1038/ejhg.2008.21. [DOI] [PubMed] [Google Scholar]
- 17.Girirajan S. Truong H.T. Blanchard C.L. Elsea S.H. A functional network module for Smith–Magenis syndrome. Clin. Genet. 2009;75:364–374. doi: 10.1111/j.1399-0004.2008.01135.x. [DOI] [PubMed] [Google Scholar]
- 18.van der Zwaag B. Franke L. Poot M. Hochstenbach R. Spierenburg H.A. Vorstman J.A. van Daalen E. de Jonge M.V. Verbeek N.E. Brilstra E.H. et al. Gene-network analysis identifies susceptibility genes related to glycobiology in autism. PLoS ONE. 2009;4:e5324. doi: 10.1371/journal.pone.0005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sjottem E. Rekdal C. Svineng G. Johnsen S.S. Klenow H. Uglehus R.D. Johansen T. The ePHD protein SPBP interacts with TopBP1 and together they co-operate to stimulate Ets1-mediated transcription. Nucleic Acids Res. 2007;35:6648–6662. doi: 10.1093/nar/gkm739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edelman E.A. Girirajan S. Finucane B. Patel P.I. Lupski J.R. Smith A.C. Elsea S.H. Gender, genotype, and phenotype differences in Smith–Magenis syndrome: a meta-analysis of 105 cases. Clin. Genet. 2007;71:540–550. doi: 10.1111/j.1399-0004.2007.00815.x. [DOI] [PubMed] [Google Scholar]
- 21.Lemieux S. Prud'homme D. Bouchard C. Tremblay A. Despres J.P. Sex differences in the relation of visceral adipose tissue accumulation to total body fatness. Am. J. Clin. Nutr. 1993;58:463–467. doi: 10.1093/ajcn/58.4.463. [DOI] [PubMed] [Google Scholar]
- 22.Geary N. Endocrine controls of eating: CCK, leptin, and ghrelin. Physiol. Behav. 2004;81:719. doi: 10.1016/j.physbeh.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 23.Brennan I.M. Otto B. Feltrin K.L. Meyer J.H. Horowitz M. Feinle-Bisset C. Intravenous CCK-8, but not GLP-1, suppresses ghrelin and stimulates PYY release in healthy men. Peptides. 2007;28:607. doi: 10.1016/j.peptides.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 24.Moran T.H. Aja S. Ladenheim E.E. Leptin modulation of peripheral controls of meal size. Physiol. Behav. 2006;89:511. doi: 10.1016/j.physbeh.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 25.Kohno D. Nakata M. Maekawa F. Fujiwara K. Maejima Y. Kuramochi M. Shimazaki T. Okano H. Onaka T. Yada T. Leptin suppresses ghrelin-induced activation of neuropeptide Y neurons in the arcuate nucleus via phosphatidylinositol 3-kinase- and phosphodiesterase 3-mediated pathway. Endocrinology. 2007;148:2251–2263. doi: 10.1210/en.2006-1240. [DOI] [PubMed] [Google Scholar]
- 26.Goto M. Arima H. Watanabe M. Hayashi M. Banno R. Sato I. Nagasaki H. Oiso Y. Ghrelin increases neuropeptide Y and agouti-related peptide gene expression in the arcuate nucleus in rat hypothalamic organotypic cultures. Endocrinology. 2006;147:5102–5109. doi: 10.1210/en.2006-0104. [DOI] [PubMed] [Google Scholar]
- 27.Kaga T. Inui A. Okita M. Asakawa A. Ueno N. Kasuga M. Fujimiya M. Nishimura N. Dobashi R. Morimoto Y. et al. Modest overexpression of neuropeptide Y in the brain leads to obesity after high-sucrose feeding. Diabetes. 2001;50:1206–1210. doi: 10.2337/diabetes.50.5.1206. [DOI] [PubMed] [Google Scholar]
- 28.Yang L. Scott K.A. Hyun J. Tamashiro K.L. Tray N. Moran T.H. Bi S. Role of dorsomedial hypothalamic neuropeptide Y in modulating food intake and energy balance. J. Neurosci. 2009;29:179–190. doi: 10.1523/JNEUROSCI.4379-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel H.R. Qi Y. Hawkins E.J. Hileman S.M. Elmquist J.K. Imai Y. Ahima R.S. Neuropeptide Y deficiency attenuates responses to fasting and high-fat diet in obesity-prone mice. Diabetes. 2006;55:3091–3098. doi: 10.2337/db05-0624. [DOI] [PubMed] [Google Scholar]
- 30.Erickson J.C. Hollopeter G. Palmiter R.D. Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science. 1996;274:1704–1707. doi: 10.1126/science.274.5293.1704. [DOI] [PubMed] [Google Scholar]
- 31.Strand A.D. Baquet Z.C. Aragaki A.K. Holmans P. Yang L. Cleren C. Beal M.F. Jones L. Kooperberg C. Olson J.M. et al. Expression profiling of Huntington's disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J. Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duncan L.E. Hutchison K.E. Carey G. Craighead W.E. Variation in brain-derived neurotrophic factor (BDNF) gene is associated with symptoms of depression. J. Affect. Disord. 2009;115:215. doi: 10.1016/j.jad.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Angelucci F. Brene S. Mathe A.A. BDNF in schizophrenia, depression and corresponding animal models. Mol. Psychiatry. 2005;10:345. doi: 10.1038/sj.mp.4001637. [DOI] [PubMed] [Google Scholar]
- 34.Wendland J.R. Kruse M.R. Cromer K.C. Murphy D.L. A large case–control study of common functional SLC6A4 and BDNF variants in obsessive-compulsive disorder. Neuropsychopharmacology. 2007;32:2543. doi: 10.1038/sj.npp.1301394. [DOI] [PubMed] [Google Scholar]
- 35.Kanazawa T. Glatt S.J. Kia-Keating B. Yoneda H. Tsuang M.T. Meta-analysis reveals no association of the Val66Met polymorphism of brain-derived neurotrophic factor with either schizophrenia or bipolar disorder. Psychiatr. Genet. 2007;17:165–170. doi: 10.1097/YPG.0b013e32801da2e2. [DOI] [PubMed] [Google Scholar]
- 36.Neves-Pereira M. Cheung J.K. Pasdar A. Zhang F. Breen G. Yates P. Sinclair M. Crombie C. Walker N. St Clair D.M. BDNF gene is a risk factor for schizophrenia in a Scottish population. Mol. Psychiatry. 2005;10:208. doi: 10.1038/sj.mp.4001575. [DOI] [PubMed] [Google Scholar]
- 37.Katerberg H. Lochner C. Cath D.C. de Jonge P. Bochdanovits Z. Moolman-Smook J.C. Hemmings S.M. Carey P.D. Stein D.J. Sondervan D. et al. The role of the brain-derived neurotrophic factor (BDNF) val66met variant in the phenotypic expression of obsessive-compulsive disorder (OCD) Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009;150B:1050–1062. doi: 10.1002/ajmg.b.30930. [DOI] [PubMed] [Google Scholar]
- 38.Gray J. Yeo G.S. Cox J.J. Morton J. Adlam A.L. Keogh J.M. Yanovski J.A. El Gharbawy A. Han J.C. Tung Y.C. et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55:3366–3371. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bertagna X. Proopiomelanocortin-derived peptides. Endocrinol. Metab. Clin. North Am. 1994;23:467–485. [PubMed] [Google Scholar]
- 40.Coll A.P. Farooqi I.S. Challis B.G. Yeo G.S. O'Rahilly S. Proopiomelanocortin and energy balance: insights from human and murine genetics. J. Clin. Endocrinol. Metab. 2004;89:2557–2562. doi: 10.1210/jc.2004-0428. [DOI] [PubMed] [Google Scholar]
- 41.Krude H. Biebermann H. Luck W. Horn R. Brabant G. Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 1998;19:155–157. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 42.Huszar D. Lynch C.A. Fairchild-Huntress V. Dunmore J.H. Fang Q. Berkemeier L.R. Gu W. Kesterson R.A. Boston B.A. Cone R.D. et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/S0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 43.Batterham R.L. Cowley M.A. Small C.J. Herzog H. Cohen M.A. Dakin C.L. Wren A.M. Brynes A.E. Low M.J. Ghatei M.A. et al. Gut hormone PYY3–36 physiologically inhibits food intake. Nature. 2002;418:650. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- 44.Bjornholm M. Munzberg H. Leshan R.L. Villanueva E.C. Bates S.H. Louis G.W. Jones J.C. Ishida-Takahashi R. Bjorbaek C. Myers M.G., Jr Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J. Clin. Invest. 2007;117:1354–1360. doi: 10.1172/JCI30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fleisch A.F. Agarwal N. Roberts M.D. Han J.C. Theim K.R. Vexler A. Troendle J. Yanovski S.Z. Yanovski J.A. Influence of serum leptin on weight and body fat growth in children at high risk for adult obesity. J. Clin. Endocrinol. Metab. 2007;92:948–954. doi: 10.1210/jc.2006-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munzberg H. Myers M.G. Molecular and anatomical determinants of central leptin resistance. Nat. Neurosci. 2005;8:566. doi: 10.1038/nn1454. [DOI] [PubMed] [Google Scholar]
- 47.Kondo H. Shimomura I. Matsukawa Y. Kumada M. Takahashi M. Matsuda M. Ouchi N. Kihara S. Kawamoto T. Sumitsuji S. et al. Association of adiponectin mutation with type 2 diabetes: a candidate gene for the insulin resistance syndrome. Diabetes. 2002;51:2325–2328. doi: 10.2337/diabetes.51.7.2325. [DOI] [PubMed] [Google Scholar]
- 48.Kumada M. Kihara S. Sumitsuji S. Kawamoto T. Matsumoto S. Ouchi N. Arita Y. Okamoto Y. Shimomura I. Hiraoka H. et al. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler. Thromb. Vasc. Biol. 2003;23:85–89. doi: 10.1161/01.ATV.0000048856.22331.50. [DOI] [PubMed] [Google Scholar]
- 49.Hotta K. Funahashi T. Arita Y. Takahashi M. Matsuda M. Okamoto Y. Iwahashi H. Kuriyama H. Ouchi N. Maeda K. et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000;20:1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 50.Arita Y. Kihara S. Ouchi N. Takahashi M. Maeda K. Miyagawa J. Hotta K. Shimomura I. Nakamura T. Miyaoka K. et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 51.Su W. Guo Z. Randall D.C. Cassis L. Brown D.R. Gong M.C. Hypertension and disrupted blood pressure circadian rhythm in type 2 diabetic db/db mice. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H1634–H1641. doi: 10.1152/ajpheart.00257.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kitamura T. Onishi K. Dohi K. Okinaka T. Ito M. Isaka N. Nakano T. Circadian rhythm of blood pressure is transformed from a dipper to a non-dipper pattern in shift workers with hypertension. J. Hum. Hypertens. 2002;16:193–197. doi: 10.1038/sj.jhh.1001328. [DOI] [PubMed] [Google Scholar]
- 53.De Leersnyder H. Claustrat B. Munnich A. Verloes A. Circadian rhythm disorder in a rare disease: Smith–Magenis syndrome. Mol. Cell. Endocrinol. 2006;252:88. doi: 10.1016/j.mce.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 54.Girirajan S. Vlangos C.N. Szomju B.B. Edelman E. Trevors C.D. Dupuis L. Nezarati M. Bunyan D.J. Elsea S.H. Genotype–phenotype correlation in Smith–Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet. Med. 2006;8:417–427. doi: 10.1097/01.gim.0000228215.32110.89. [DOI] [PubMed] [Google Scholar]
- 55.Girirajan S. Elsas L.J., II Devriendt K. Elsea S.H. RAI1 variations in Smith–Magenis syndrome patients without 17p11.2 deletions. J. Med. Genet. 2005;42:820–828. doi: 10.1136/jmg.2005.031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farooqi I.S. Drop S. Clements A. Keogh J.M. Biernacka J. Lowenbein S. Challis B.G. O'Rahilly S. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes. 2006;55:2549–2553. doi: 10.2337/db06-0214. [DOI] [PubMed] [Google Scholar]
- 57.Challis B.G. Coll A.P. Yeo G.S. Pinnock S.B. Dickson S.L. Thresher R.R. Dixon J. Zahn D. Rochford J.J. White A. et al. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3–36) Proc. Natl Acad. Sci. USA. 2004;101:4695–4700. doi: 10.1073/pnas.0306931101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Enriori P.J. Evans A.E. Sinnayah P. Jobst E.E. Tonelli-Lemos L. Billes S.K. Glavas M.M. Grayson B.E. Perello M. Nillni E.A. et al. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007;5:181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 59.Lan Y. Ryan R.C. Zhang Z. Bullard S.A. Bush J.O. Maltby K.M. Lidral A.C. Jiang R. Expression of Wnt9b and activation of canonical Wnt signaling during midfacial morphogenesis in mice. Dev. Dyn. 2006;235:1448–1454. doi: 10.1002/dvdy.20723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ng D.S. Xie C. Maguire G.F. Zhu X. Ugwu F. Lam E. Connelly P.W. Hypertriglyceridemia in lecithin-cholesterol acyltransferase-deficient mice is associated with hepatic overproduction of triglycerides, increased lipogenesis, and improved glucose tolerance. J. Biol. Chem. 2004;279:7636–7642. doi: 10.1074/jbc.M309439200. [DOI] [PubMed] [Google Scholar]
- 61.Loder M.K. da Silva Xavier G. McDonald A. Rutter G.A. TCF7L2 controls insulin gene expression and insulin secretion in mature pancreatic beta-cells. Biochem. Soc. Trans. 2008;36:357–359. doi: 10.1042/BST0360357. [DOI] [PubMed] [Google Scholar]
- 62.Florez J.C. Jablonski K.A. Bayley N. Pollin T.I. de Bakker P.I. Shuldiner A.R. Knowler W.C. Nathan D.M. Altshuler D. TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N. Engl. J. Med. 2006;355:241–250. doi: 10.1056/NEJMoa062418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Waters S.T. Lewandoski M. A threshold requirement for Gbx2 levels in hindbrain development. Development. 2006;133:1991–2000. doi: 10.1242/dev.02364. [DOI] [PubMed] [Google Scholar]
- 64.Beckers S. Peeters A. Zegers D. Mertens I. Van Gaal L. Van Hul W. Association of the BDNF Val66Met variation with obesity in women. Mol. Genet. Metab. 2008;95:110–112. doi: 10.1016/j.ymgme.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 65.Beckers S. Zegers D. Van Gaal L.F. Van Hul W. The role of the leptin-melanocortin signalling pathway in the control of food intake. Crit. Rev. Eukaryot. Gene Expr. 2009;19:267–287. doi: 10.1615/critreveukargeneexpr.v19.i4.20. [DOI] [PubMed] [Google Scholar]
- 66.Mössner R. Daniel S. Albert D. Heils A. Okladnova O. Schmitt A. Lesch P. Serotonin transporter function is modulated by brain-derived neurotrophic factor (BDNF) but not nerve growth factor (NGF) Neurochem. Int. 2000;36:197. doi: 10.1016/S0197-0186(99)00122-9. [DOI] [PubMed] [Google Scholar]
- 67.Arancio O. Chao M.V. Neurotrophins, synaptic plasticity and dementia. Curr. Opin. Neurobiol. 2007;17:325–330. doi: 10.1016/j.conb.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 68.Mercader J.M. Ribases M. Gratacos M. Gonzalez J.R. Bayes M. de Cid R. Badia A. Fernandez-Aranda F. Estivill X. Altered brain-derived neurotrophic factor blood levels and gene variability are associated with anorexia and bulimia. Genes Brain Behav. 2007;6:706–716. doi: 10.1111/j.1601-183X.2007.00301.x. [DOI] [PubMed] [Google Scholar]
- 69.Zuccato C. Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]
- 70.Xiu M.H. Hui L. Dang Y.F. Hou T.D. Zhang C.X. Zheng Y.L. Chen da C. Kosten T.R. Zhang X.Y. Decreased serum BDNF levels in chronic institutionalized schizophrenia on long-term treatment with typical and atypical antipsychotics. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2009;33:1508–1512. doi: 10.1016/j.pnpbp.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 71.Maina G. Rosso G. Zanardini R. Bogetto F. Gennarelli M. Bocchio-Chiavetto L. Serum levels of brain-derived neurotrophic factor in drug-naive obsessive-compulsive patients: a case–control study. J. Affect. Disord. 2010;122:174–178. doi: 10.1016/j.jad.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 72.Kaplan A.S. Levitan R.D. Yilmaz Z. Davis C. Tharmalingam S. Kennedy J.L. A DRD4/BDNF gene-gene interaction associated with maximum BMI in women with bulimia nervosa. Int. J. Eat. Disord. 2008;41:22–28. doi: 10.1002/eat.20474. [DOI] [PubMed] [Google Scholar]
- 73.Yeo G.S. Connie Hung C.C. Rochford J. Keogh J. Gray J. Sivaramakrishnan S. O'Rahilly S. Farooqi I.S. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat. Neurosci. 2004;7:1187–1189. doi: 10.1038/nn1336. [DOI] [PubMed] [Google Scholar]
- 74.Han J.C. Liu Q.R. Jones M. Levinn R.L. Menzie C.M. Jefferson-George K.S. Adler-Wailes D.C. Sanford E.L. Lacbawan F.L. Uhl G.R. et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N. Engl. J. Med. 2008;359:918–927. doi: 10.1056/NEJMoa0801119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.