

Summary:
Mitochondrial D2HGDH and L2HGDH catalyze the oxidation of D-2-HG and L-2-HG, respectively, into αKG. This contributes to cellular homeostasis in part by modulating the activity of αKG-dependent dioxygenases. Signals that control the expression/activity of D2HGDH/L2HGDH are presumed to broadly influence physiology and pathology. Using cell and mouse models, we discovered that MYC directly induces D2HGDH and L2HGDH transcription. Further, in a manner suggestive of D2HGDH, L2HGDH and αKG dependency, MYC activates TET enzymes and RNA demethylases, and promotes their nuclear localization. Consistent with these observations, in primary B cell lymphomas MYC expression positively correlated with enhancer hypomethylation and overexpression of lymphomagenic genes. Together, these data provide additional evidence for the role of mitochondria metabolism in influencing the epigenome and epitranscriptome, and imply that in specific contexts wild-type TET enzymes could demethylate and activate oncogenic enhancers.
Keywords: MYC, alpha-ketoglutarate, DNA methylation, RNA methylation, dioxygenases, metabolites
Graphical Abstract
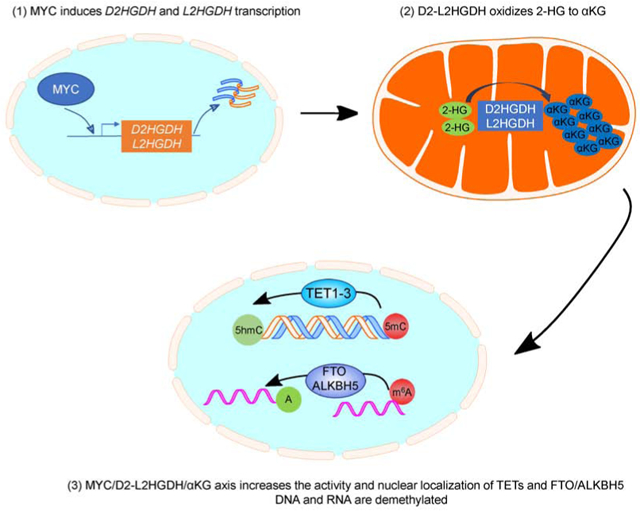
eTOC blurb:
The mitochondrial dehydrogenases D2HGDH and L2HGDH, catalyze the essential oxidation of 2-HG into αKG. Qiu et al. identify MYC as a regulator of these enzymes and show that MYC induces the activity and promotes nuclear localization of DNA and RNA demethylases in a manner suggestive of D2HGDH-/L2HGDH- and αKG dependency.
Introduction:
There is increasing recognition of mitochondria as signaling organelles. An important facet of this “adjunct” mitochondrial function is epigenetic modulation, as exemplified by the generation of acetyl-CoA and S-adenosylmethionine used in the acetylation and methylation, respectively, of DNA and histones(Chandel, 2015). The discovery of isocitrate dehydrogenase (IDH1 and IDH2) mutations in cancer further highlighted the interplay between mitochondria and epigenetics. In this instance, aberrant accumulation of the metabolite D-2-hydroglutarate (D-2-HG) competitively inhibited multiple epigenetic regulators that belong to the family alpha-ketoglutarate (αKG)-dependent dioxygenases, including TET DNA hydroxylases, histone demethylases (HDM), and RNA demethylases, resulting in a hypermethylator phenotype(Yang et al., 2012). These cancer-associated observations suggested that physiologic control of 2-HG/αKG homeostasis may play an underappreciated role in epigenetic regulation. Two mitochondrial dehydrogenases, D2HGDH and L2HGDH, catalyze the oxidation of the enantiomers D-2-HG and L-2-HG, respectively, into αKG(Ye et al., 2018). In humans, loss of either of these evolutionarily conserved enzymes leads to severe neuro-metabolic syndromes (D- or L-2-hydroxyglutaric aciduria)(Kranendijk et al., 2012), indicating that they perform an essential functional role. Interestingly, while the removal of 2-HG by D2HGDH and L2HGDH is well characterized, the generation of this “orphan” metabolite is more obscure. In mammals and other organisms, D-2-HG and L-2-HG are produced mainly by promiscuous enzymatic reactions with αKG as their apparent direct precursor(Fan et al., 2015; Struys, 2006; Struys et al., 2007; Struys et al., 2004; Struys et al., 2005). Notably, the side reactions that generate 2-HG are also influenced by environmental conditions, such as hypoxia and metabolic acidosis(Intlekofer et al., 2015; Oldham et al., 2015). Indeed, the amounts of 2-HG in distinct models (from serum of healthy volunteers, to normal tissue, primary tumors and cancer cell lines) seem to vary substantially(DiNardo et al., 2013; Gross et al., 2010; Oldham et al., 2015; Shim et al., 2014). Notably, in the few reports in which the actual concentration of 2-HG and αKG was provided side-by-side in the same cell type(Gross et al., 2010; Oldham et al., 2015), the abundance of 2-HG was sufficiently high to make it possible for it to (upon oxidation by D2HGDH and L2HGDH) meaningfully contribute to the cellular pool of αKG. Surprisingly, in spite of the essential role of D2HGDH and L2HGDH in limiting the pathogenic accumulation of 2-HG, and the broad control of epigenetic remodeling modulated by 2-HG and αKG levels, the signals that regulate the expression and activity of these mitochondrial dehydrogenases have not yet been elucidated. To start to address this knowledge gap, we characterized D2HGDH and L2HGDH promoter regions, and mapped putative binding sites that were shared by both promoters, with emphasis on those for transcriptional factors known to influence metabolism and cell growth.
Using reporter and chromatin immunoprecipitation (ChIP) assays, inducible cell lines and a transgenic mouse model we discovered that MYC transcriptionally regulates the D2HGDH and L2HGDH genes. Further, using multiple gain and loss-of-function cell models we found that MYC, in a manner suggestive of D2HGDH/L2HGDH/αKG dependency, activates TET DNA hydroxylases and RNA demethylases, thus controlling the cellular epigenome and epitranscriptome. We also found that the expression of MYC and D2HGDH/L2HGDH, and elevation of αKG levels, promoted the nuclear localization of these enzymes. Lastly, consistent with these observations, we found that in primary B cell lymphomas MYC expression positively correlated with enhancer hypomethylation and overexpression of lymphomagenic genes.
Results:
MYC directly targets and induces D2HGDH and L2HGDH transcription.
Using reporter assays, we identified DNA sequences near the annotated transcription start sites for D2HGDH and L2HGDH containing basal promoter activity (Figure 1A-B). We used the MatInspector software and the “regulation” function of the UCSC genome browser to identify putative transcription factor binding sites common to both promoter regions; canonical and non-canonical E-boxes were found in the D2HGDH (n=2) and L2HGDH (n=4) promoters. We performed ChiP followed by real-time quantitative PCR (ChIP-qPCR) and confirmed that MYC binds to specific E-boxes within these loci (Figure 1C-D), canonical E-box #2 in the D2HGDH promoter and the E-boxes #3/4 in L2HGDH (these two E-boxes were too close to allow for individual mapping of MYC binding). To test the ability of MYC to transactivate these promoters, we repeated the reporter assays in cells co-transfected with MYC or control vectors and identified a MYC-dependent reporter activity in the D2HGDH and L2HGDH promoters (Figure 1C/D). We further validated these findings, including the conservation and location of the functional E-boxes, by analyzing PhyloP and ChiP-Seq data (UCSC Encode, Figure S1A/B). To confirm the relevance of these interactions, we used the human B-cell line P493-6, which has MYC expression under the control of a tetracycline responsive element; turning MYC OFF in these cells decreased D2HGHD and L2HGDH expression while restoring MYC progressively increased their levels (Figure 1E/F). This regulation was not simply an indirect effect of MYC-driven cell cycle progression, because when MYC expression was reestablished in serum-deprived conditions, the cells did not proliferate but the expression of D2HGDH/L2HGDH was again readily induced (Figure S1C). Next, we isolated splenic mature B-cells from Eμ-Myc mice and littermate WT controls (n=6). Using qRT-PCR, we showed that the expression of D2hgdh and L2hgdh is significantly higher in mature B-cells from Eμ-Myc mice than from WT controls (Figure 1E/F). Finally, we queried the Pan-Cancer Atlas (~11,000 tumors from over 30 cancer types, from the Cancer Genome Atlas - TCGA - consortium) and found a significant correlation between MYC amplification and L2HGDH, but not D2HGDH, expression (p <2.2e-16) (Figure S1D). Together, we concluded D2HGDH and L2HGDH are direct transcriptional targets of MYC.
Figure 1. D2HGDH and L2HGDH are transcriptional targets of MYC.

a) D2HGDH and b) L2HGDH. Luciferase activity of promoter regions – arrows indicate transcriptional start sites; blue squares are putative E-boxes. c) D2HGDH and d) L2HGDH. Left panels: ChIP-qPCR of MYC binding to E-boxes in D2HGDH or L2HGDH promoters; +ctrl is the LIN28B promoter, −ctrl is a promoter region lacking a predicted E-box. Right panels: Luciferase activity of D2HGDH and L2HGDH promoters in cells co-transfected with MYC. e) D2HGDH and f) L2HGDH. Left panels: Q-RT-PCR of D2HGDH and L2HGDH mRNA in P493-6 cells; Center panel: Immunoblot of tetracycline-regulated MYC expression in P493-6 cells; Right panels: Q-RT-PCR of D2HGDH and L2HGDH mRNA in mature B-cells from Eμ-Myc mice and WT mice. Data shown are mean −/+ SD.
MYC influences TET and RNA demethylase activity and modulate DNA and RNA methylation in vitro and in vivo.
D2HGDH and L2HGDH modify 2-HG/αKG homeostasis(Ye et al., 2018), which in turn control the function of a host of enzymes belonging to the αKG-dependent dioxygenases family(Loenarz and Schofield, 2008). TET DNA hydroxylases (TET1, 2 and 3) and RNA demethylases (FTO and ALKBH5) belong to this group of enzymes, and their activity is modulated by the relative or absolute availability of αKG(Carey et al., 2015; Elkashef et al., 2017; Lin et al., 2015). We reasoned that MYC’s targeting of D2HGDH and L2HGDH, and attendant effect on 2-HG/αKG levels, may influence the epigenome and epitranscriptome. To start to address this postulate, we quantified TET and RNA demethylase activity in nuclear lysates of multiple cell models of MYC variable expression, and we used mass-spectrometry as well as antibody-based methodologies to quantify 5hmC, 5mC and m6A abundance. In P493-6 cells, turning MYC expression OFF resulted in a significant decrease in TET function, while restoring MYC expression increased TET activity. These changes were functionally relevant as they resulted in an increase in the deposition of 5hmC marks, and consequent decrease in 5mC abundance (Figure 2A). Similar results were found in HEK-293T with MYC ectopic expression and in DLBCL cell lines with shRNA-mediated knockdown of MYC (Figure S2A). Next, we isolated mature B-cells from lymphoma-free Eμ-Myc and WT mice and showed that the deregulation of MYC, increased TET activity and modulated 5hmC and 5mC levels in vivo (Figure 2B). Notably, in none of these models MYC influenced TET1-3 expression (Figure 2A/B). We also examined MYC effects towards RNA methylation. In P493-6 cells, turning MYC expression ON significantly elevated RNA demethylase activity and decreased the abundance of m6A marks (Figure 2C). A similar result was obtained when comparing B-cells from Eμ-Myc vs. WT mice (Figure 2D). In no instance, MYC modified the expression of FTO and ALKBH5, or of METTL3, METTL14 and WTAP, components of the RNA methyltransferase complex (Figure 2C-D). We confirmed that these MYC-induced changes are not simply a consequence of an increased cell proliferation, since when MYC expression is induced in cells cultured in serum deprived conditions (0.25%), an identical outcome is detected (Figure S2B). We concluded that MYC increases the activity of TET and RNA demethylases, and thus, in the tested models, it promotes DNA and RNA demethylation.
Figure 2. Regulation of demethylase activity and DNA/RNA methylation by MYC.

a) Left to right panels - TET activity in P493-6 cells following MYC suppression and re-induction, quantification of 5hmC and 5mC using mass-spectrometry or antibody-based dot-blots in MYC ON-OFF-ON P493-6 cells, and WBs of MYC, TET1, TET2 and TET3. b) Left to right panels - TET activity in mature B-cells from WT or Eμ-Myc mice (pooled nuclear lysate of 4 WT vs. 4 Eμ-Myc mice measure in a technical triplicate), quantification of 5hmC and 5mC using mass-spectrometry or antibody-based dot-blots, and WBs of TET1, TET2 and TET3. c) Left to right panels – RNA demethylase activity in P493-6 cells following MYC suppression and re-induction, quantification of m6A levels using antibody-based ELISA and dot-blots in P493-6 cells, and WBs for MYC, METL3, METTL14, WTAP, FTO and ALKBH5. d) Left to right panels - RNA demethylase activity in mature B-cells from WT or Eμ-Myc mice (pooled nuclear lysate of 3 WT vs. 3 Eμ-Myc mice measure in a technical triplicate), quantification of m6A using antibody-based ELISA and dot-blots, and WBs of FTO and ALKBH5. Data shown are mean −/+ SD. In a) and c) three sets of proteins, TET1/TET3/FTO/ALKBH5, TET2/METTL14, and METTL3/WTAP were each run in the same gel and thus share the loading control, β-actin. In b) and d) TET1/TET3 and TET2/FTO share the β-actin.
MYC modulation of TET and RNA demethylase activity is dependent on D2HGDH and L2HGDH.
We found that MYC transcriptionally activates D2HGDH and L2HGDH (Figure 1) and increase the activity of TET DNA hydroxylases and RNA demethylases (Figure 2). Here, we sought to test if these events are interconnected. To genetically isolate the contribution of all relevant players, we chose to knock-out (KO) D2HGDH or L2HGDH in the MYC-inducible P493-6 cells. In cells WT for D2HGDH and L2HGDH, turning MYC expression ON, resulted in a ~50% increase in the abundance of 5hmC marks in DNA. In contrast, in D2HGDH or L2HGDH KO cells, MYC-mediated increase in the deposition of 5hmC was limited to ~14% (ranging from 12-16%, Figure 3A, Figure S3A), a ~70% blockade on MYC-mediated increase in 5hmC abundance. Corresponding changes were detected with 5mC quantification, with KO of D2HGDH or L2HGDH limiting MYC’s ability to reduce 5mC levels (Figure 3A). In agreement with the 5hmC/5mC data, we found that MYC-mediated induction of TET activity was reduced by ~50% in D2HGDH and L2HGDH KO cells (Figure 3A). We also showed that D2HGDH and L2HGDH KO cells accumulated D-2-HG and L-2-HG, respectively (Figure S3A), and that these genetic and metabolic perturbations did not modify the inducible expression of MYC or TET1-3 levels (Figure 3A), suggesting that the changes in 5hmC/5mC result from modulation of TET activity. We next examined the role of D2HGDH and L2HGDH, downstream to MYC, in controlling RNA m6A levels and RNA demethylase activity. MYC induction in D2HGDH/L2HGDH WT cells resulted in significantly more pronounced decrease in m6A levels than in D2HGDH/L2HGDH KO cells – 22% vs. ~9% (7% to 10.5%), a ~60% blockade on MYC-associated decrease in m6A levels (Figure 3B). In addition, in cells lacking D2HGDH or L2HGDH, MYC-mediated activation of RNA demethylases was reduced by ~55% (Figure 3B). We also showed that MYC modulation or D2HGDH and L2HGDH KO did not modify the expression of FTO, ALKBH5, METTL3, METTL14 or WTAP (Figure 3B), suggesting that the MYC-D2HGDH/L2HGDH axis influences the epitranscriptome by inducing RNA demethylase activity. Expectedly, KO of D2HGDH and L2HGDH, with consequent accumulation of 2-HG was sufficient to suppress the activity of TET and RNA demethylases and to modify 5hmC and m6A levels (Figure S3B). Nonetheless, with the use of an inducible cell model, we were able to isolate MYC’s role in modulating TET and RNA demethylase activity via these mitochondrial enzymes. Still, given the accumulation of 2-HG in these cells, we cannot be entirely certain that the transcriptional activation of D2HGDH and L2HGDH by MYC accounts for all its effects on DNA and RNA methylation. Lastly, we showed that the effects of D2HGDH and L2HGDH KO on DNA and RNA methylation were not limited to the P493-6 cell model. We also generated double D2HGDH/L2HGDH KO in two MYC-expressing DLBCL cell lines and detected significant increases in 5hmC and m6A levels (Figure 3C). We concluded that MYC’s influence on the epigenome and epitranscriptome may be mediated, at least in part, by the transcriptional activation of the mitochondrial enzymes D2HGDH and L2HGDH.
Figure 3. D2HGDH and L2HGDH roles on MYC-modulated DNA/RNA methylation and demethylase activity.

a) Left to right panels – MYC-induced modulation of 5hmC and 5mC levels in WT and D2HGDH or L2HGDH KO P493-6 cells, quantification of MYC-induced TET activity in WT and D2HGDH or L2HGDH KO P493-6 cells, and WBs of D2HGDH, L2HGDH, TET1, TET2, TET3 and MYC. b) Left to right panels – MYC-induced modulation of m6A levels in WT and D2HGDH or L2HGDH KO P493-6 cells, quantification of MYC-induced RNA demethylase activity in WT and D2HGDH or L2HGDH KO P493-6 cells, and WBs of FTO, ALKBH5, METL3, WTAP and METTL14 in WT and D2HGDH or L2HGDH KO P493-6 cells. c) Left to right panels – 5hmC and m6A levels in D2HGDH/L2HGDH WT or KO clones in DLBCL cell lines OCI-Ly1 and OCI-Ly8; WBs for D2HGDH and L2HGDH are shown at the top. The percentage of MYC-induced changes in a) and b) represent the difference between the measures in MYC OFF and ON cells (full data in Figure S3). Data shown are mean −/+ SD. In a) and b), TET1/ALKBH5 and TET3/FTO were detected in the same gel and share the β-actin loading control.
Intermediary metabolism links the MYC-D2HGDH-L2HGDH axis to the control of DNA and RNA methylation.
We speculated that MYC, via modulation of D2HGDH and L2HGDH transcription, increases αKG levels and the activity of the αKG-dependent TET DNA hydroxylases and RNA demethylases. To test this concept, we quantified αKG levels in MYC-inducible D2HGDH and L2HGDH WT or KO cells. At baseline (MYC ON), D2HGDH/L2HGDH WT cells displayed higher αKG levels than D2HGDH or L2HGDH KO isogenic models (Figure 4A, Figure S4A). This observation agrees with the ability of these enzymes to oxidize 2-HG into αKG. To investigate the kinetics of the MYC/D2HGDH/L2HGDH-dependent αKG production, we suppressed and restored MYC expression in D2HGDH/L2HGDH WT or KO cells and measured αKG. In absence of MYC, and early after it was re-expressed, the levels of αKG were similar irrespectively of the genetic integrity of D2HGDH and L2HGDH. However, 24h post MYC re-expression the “baseline” status re-emerged and αKG levels were again higher in D2HGDH/L2HGDH WT cells (Figure 4A, Figure S4A). Further, in agreement with the role of these enzymes in converting 2-HG into αKG, in D2HGDH/L2HGDH WT cells the levels of 2-HG were decreased once MYC is turned ON (Figure S4B). In addition, we quantified D-2-HG and L-2-HG in D2HGDH and L2HGDH KO cells, respectively, with MYC silenced and upon its re-expression. In these KO models, we found that MYC expression increased the abundance of 2-HG, in particular that of L-2-HG (Figure S4C). These data support the concept that 2-HG levels may be sufficient for the MYC-induced, D2HGDH/L2HGDH executed, generation of αKG. However, final evidence that the 2-HG dehydrogenases can meaningfully influence the cellular αKG pool requires examination of intermediary metabolism with isotope tracing and the generation of additional KO models.
Figure 4. MYC-D2HGDH-L2HGDH axis, intermediate metabolites and TET/RNA demethylases activity.

a) Quantification of αKG in P493-6 cells (MYC ON or OFF) with WT or KO of D2HGDH and L2HGDH; MYC immunoblots are on the left, ***p<0.001, two-way ANOVA, Bonferroni's post-test. b) 5hmC and m6A levels in P493-6 cells (MYC OFF or ON; D2HGDH and L2HGDH WT or KO), with or without DMαKG rescue (5mM); WBs are on the right; p values are from two-tailed Student’s t-test c) 5hmC and 5mC (left panels) and m6A (right panel) levels in P493-6 cells (MYC OFF or ON) exposed to DMOG (1mM), octyl-D-2-HG or L-2-HG (100μM); p values are from one-way ANOVA. WBs of relevant proteins are also shown. d) Left panel: TET activity in DLBCL cell lines exposed to DMαKG (5mM) ,**p<0.01, ***p<0.001, Student’s t-test; center-panel: RNA demethylase activity of HEK-293T cells exposed to DMαKG, one-way ANOVA; right panel: RNA demethylase activity in DLBCL cell lines exposed to octyl-D-2-HG (100μM), **p<0.01, ***p<0.001, Student’s t-test. e) Fold increase in 5hmC and decrease of m6A (left and right panels) upon MYC induction in cells grown in full media (Glut+), in glutamine-free media (Glut−) or in the presence of CB-839 (1μM) (3 biological replicates for both assays, 5 and 9 technical replicates for 5hmC and m6A, respectively); p values are from two-tailed Student’s t-test. Data are mean −/+ SD. In b), the sets of proteins MYC/TET2/FTO/METLL14, TET3/METLL3/WTAP and TET1/ALKBH5 were detected in the same filter and share the β-actin loading control. In c), TET3/METLL3/METLL14, and FTO/ALKBH5 share the β-actin loading control.
We rescued DNA/RNA methylation in the D2HGDH or L2HGDH KO cells with synthetic cell-permeable αKG (Figure 4B, Figure S4D). Conversely, cell-permeable D-2-HG and L-2-HG, and dimethyloxaloylglycine (DMOG), abrogated MYC effects towards 5hmC, 5mC and m6A (Figure 4C, Figure S4E). Similarly, in additional cell models αKG increased TET and RNA demethylase activity, whereas D-2-HG suppressed them (Figure 4D), with concordant modifications on 5hmC and m6A (Figure S4F). It should be noted that the effects of these synthetic metabolites on DNA and RNA methylation are known and may be present irrespective of the mechanistic context. Once more, genetic and pharmacological modulations did not modify the total expression of TET1-3, FTO, ALKBH5, METLL3, METLL14 and WTAP (Figures 4B-C).
MYC is known to regulate other facets of intermediary metabolism that result in accumulation of αKG, notably glutaminolysis, a process in which glutamine is converted to glutamate and then αKG via the successive action of the MYC-regulated glutaminases and glutamate dehydrogenase(Hsieh et al., 2015). Thus, to test the relative contribution of this anaplerotic reaction vs. D2HGDH and L2HGDH activation on the MYC-mediated regulation of DNA and RNA methylation, we quantified 5hmC and m6A levels in conditions devoid of glutamine or in the presence of the glutaminase inhibitor CB-839. Using isogenic models, we found that inducing MYC in cells growing in glutamine-free media or in the presence of CB-839, only modestly limited (~20%) the deposition of 5hmC marks in the DNA when compared to the effects of MYC in cells grown in glutamine replete media (Figure 4E, Figure S4G). Likewise, we found that although absence of glutamine diminished the impact of MYC on reducing m6A levels, it accounted for a smaller fraction (~35%) of MYC-mediated RNA demethylation (Figure 4E, Figure S4G). Interestingly, although the effects of CB-839 on 5hmC were nearly identical to those of glutamine deprivation (21% vs. 18% block on MYC-mediated accumulation), the consequences of CB-839 exposure on m6A levels were much more pronounced than that of glutamine deprivation (68% vs. 35% block on MYC-mediated RNA demethylation) (Figure 4E). This observation does not relate to a putative CB-839-mediated change in the expression of RNA demethylases or components of the methyltransferase complex, nor does it derive from an overt off-target effect of CB-839 towards RNA demethylases (Figure S4H). Presently, we cannot exclude the possibility that an uncharacterized property of CB-839 is to promote the deposition of m6A in RNA. We concluded that the control of D2HGDH/L2HGDH by MYC, and its consequences on intermediate metabolites, plays an important part on the hydroxylation of 5-methylcytosine in DNA and the demethylation of m6A RNA. We also posit that αKG abundance is rate limiting for these dioxygenases and that increasing amounts of this co-substrate promotes TET and RNA demethylases activity.
MYC, D2HGDH/L2HGDH and intermediate metabolites modulate the subcellular localization of TETs and FTO/ALKBH5.
The existing paradigm, established in part after the discovery that IDH1/2 mutant cells accumulate D-2-HG and supported by in vitro enzymology(Dang et al., 2010; Ward et al., 2010; Xu et al., 2011), is that 2-HG (as well as fumarate and succinate(Xiao et al., 2012)) competitively inhibits the obligatory co-substrate αKG and suppress the activity of dioxygenases, including TET1-3, FTO and ALKBH5(Elkashef et al., 2017; Lin et al., 2015). Surprisingly, in our models we found that even after the cells were lysed and metabolite levels presumably equilibrated, TET and RNA demethylase activity remained distinct across comparison groups (Figures 2 to 4). This was puzzling because the expression of the relevant proteins in the whole cell lysate was consistently similar across the genetic and pharmacological models employed (Figures 2-4). Interestingly, an earlier clinical-pathological report on SDH-mutant pheochromocytomas, which accumulate succinate, showed that loss of 5hmC in these tumors was associated with nuclear exclusion of TET1(Hoekstra et al., 2015). Thus, we examined if MYC, D2-L2HGDH and their metabolic output could influence the sub-cellular localization of TET1-3, FTO and ALKBH5. In P493-6 cells, modulating MYC expression led to a marked change in the abundance of nuclear TET1-3, FTO and ALKBH5 - a decrease in their nuclear levels was detected in absence of MYC, which were restored after MYC re-expression (Figure 5A, Figure S5A). Cytoplasmic expression of TET2, TET3, ALKBH5 and FTO was readily detected (FTO was primarily found in the cytosol), and we could ascertain that in the MYC-OFF state at least a fraction of these proteins accumulated in the cytosol and that following MYC re-expression their cytoplasmic levels decreased (Figure 5A, Figure S5A). Presently, we cannot exclude the possibility that at least part of the proteins that are excluded from the nucleus relocate to insoluble cellular fractions. Importantly, as we showed earlier (Figures 2-4), the total protein level (whole cell lysate) of TET1-3, FTO and ALKBH5 did not vary between MYC OFF vs. ON states. Next, we asked if cell-permeable αKG and 2-HG could also influence this subcellular movement. In DLBCL cell lines, αKG mimicked MYC effects and promoted nuclear accumulation. Conversely, D-2-HG and/or L-2-HG decreased the levels of TETs and FTO/ALKBH5 in the nucleus in association with an increase in their cytoplasmic level (Figure 5B, Figure S5B). We showed that KO of D2HGDH or L2HGDH blunts MYC effects on 5hmC and TET activity, as well as on m6A and RNA demethylase activity (Figure 3). Concordantly, we found that in D2HGDH or L2HGDH KO cells the effects of MYC expression on the nuclear accumulation and cytoplasmic depletion of DNA hydroxylases and RNA demethylases were significantly dampened (Figure 5C, Figure S5C). We next tested the ability of ectopically expressed D2HGDH to directly mediate changes in the sub-cellular localization of αKG-dependent dioxygenases. Although the effects were, expectedly, more subtle than in the models shown in Figures 5A-C, expression of TET1-3 and FTO/ALKBH5 was reliably found to be higher in the nucleus of HEK-293 cells reconstituted with D2HGDH WT, than in isogenic models expressing an empty vector or the previously characterized loss-of-function D2HGDH mutants(Lin et al., 2015) (Figure 5D, Figure 5D). In contrast, a corresponding decrease in the cytoplasmic expression of TET1-3 and FTO/ALKBH5 was less consistently detected. The reasons for this discrepancy are not clear, but they could reflect the nature of the cell model (chronic stable ectopic expression of D2HGDH in an epithelial cell) or the overall more modest change in subcellular localization, which makes it technically difficult to consistently detect differences in the cytoplasmic fraction. Lastly, we transiently transfected HEK-293T with a FLAG-TET3 construct and analyzed immunofluorescence (IF, anti-FLAG and anti-TET3 antibodies) by confocal microscopy, following exposure of the cells to vehicle control, αKG or 2-HG. In cells exposed to control, we detected an approximately equal number of cells with nuclear-only versus nuclear and cytosolic TET3 expression. Upon exposure to aKG (5mM, 6h), there was a significant shift to nuclear-only signal, now accounting for ~80% of the cells. However, we could not detect a cytoplasmic enrichment of transiently transfected TET3 in cells exposed to 2-HG (200μM, 6h) (Figure 5E, Figure S5E). We confirmed these observations with WB analysis of the transiently transfected FLAG-TET3 construct, as well as of endogenous TET3 in HEK-293T cells (Figure S5E). Unfortunately, the endogenous TET3 protein was not detectable by IF (Figure S5E). Thus, we were unable to address the effect of 2-HG in cytoplasmatic expression of TET3 in DLBCL cell lines using IF.
Figure 5. Control of the subcellular localization of TET1-3, FTO and ALKBH5 by MYC, D2HGDH/L2HGDH, and αKG/2-HG.

a) WBs of TET1-3, FTO and ALKBH5 in nuclear and cytoplasmic fractions, and whole cell lysate (WCL) of P493-6 cells upon MYC suppression and its re-expression. Two exposures (short and long) are shown for FTO. b) WBs of TET1-3, FTO and ALKBH5 in the subcellular fractions and WCL of DLBCL cell lines exposed to DMαKG (5mM), octyl-D-2-HG or octyl-L-2-HG (100μM) for 8h. c) WBs of TET1-3, FTO and ALKBH5 in subcellular fractions and WCL of P493-6 cells (D2HGDH and L2HGDH WT or KO) upon MYC suppression and its re-expression. d) WBs of TET1-3, FTO and ALKBH5 in HEK-293 cells expressing an empty vector (MSCV), D2HGDH WT or the G131X and A208T mutants. Densitometric measurements of protein levels are shown at the bottom of WB displays – all values are relative to controls and already corrected by WCL abundance, which is shown for reference. e) immunofluorescence of HEK-293T cells transiently transfected with a FLAG-TET3 plasmid and exposed to DMαKG (5mM), octyl-D-2-HG and L-2-HG (100μM/each) or DMSO for 6h; scale bar is 10 μm. Scoring of nuclear vs. nuclear and cytoplasmic expression in the three conditions is shown on the right; p<0.001, Chi-square.
We concluded that MYC-D2HGDH/L2HGDH-αKG axis leads to accumulation of DNA hydroxylases and RNA demethylases in the nucleus. In addition, MYC silencing is associated with nuclear exclusion of TETs and FTO/ALKBH5. Conversely, the effect of 2-HG on the subcellular movement of these proteins is less clear, and it may be cell-type specific. Likewise, whereas ectopic expression of WT D2HGDH increased nuclear TET1-3 and FTO/ALKBH5 abundance, its effects on their cytoplasmic expression were not uniform. The mechanistic basis for the control of this nuclear-cytoplasmic shuttling remains to be determined, but it may involve covalent modification(s) shared by TETs and FTO/ALKBH5 (and possibly other αKG-dependent dioxygenases), which could be catalyzed by an enzyme/pathway that is stimulated by αKG. We also cannot exclude the possibility that activated TETs and FTO/ALKBH5 reside in chromatin-associated complexes and, thus, become enriched in the nucleus.
MYC expression associates with enhancer demethylation in primary DLBCL.
We showed that by modulating intermediary metabolism, the MYC-D2HGDH-L2HGDH axis increases the activity and modulate the subcellular localization of TETs (Figures 2-5). Characterization of TET KO mouse models showed that these enzymes preferentially target methylated DNA at enhancer regions, promoting their demethylation and target gene transcription(Hon et al., 2014; Lio et al., 2016; Lu et al., 2014; Rasmussen et al., 2015). To test if the positive effects of MYC on TET activity that we detected in vitro and in vivo were also present in unmodified human B cell lymphomas, we utilized the DLBCL TCGA dataset (https://portal.gdc.cancer.gov/projects/TCGA-DLBC), as it has matched methylation and expression data from primary tumors. To interrogate enhancer methylation, we mapped all probes from the Illumina 450K platform to enhancers previously characterized as active in DLBCL(Chapuy et al., 2013). To test if the global pattern of enhancer methylation associated with MYC expression in this DLBCL cohort, we calculated the percentage of unmethylated probes (beta value < 0.3) for each DLBCL sample and found that enhancer hypomethylation significantly correlated to MYC expression in this tumor type (r=0.38, p=0.009) (Figure 6A). Further validating this correlation, we found that tumors in the top and bottom quantiles of MYC expression displayed significantly higher and lower percentage of unmethylated enhancer probes, respectively (p=0.03) (Figure 6B). In this cohort of DLBCL, we also found that MYC and L2HGDH expression were highly positively correlated (r=0.6095, p=0.0000108). For each tumor in this dataset, we also applied a single sample gene set enrichment analysis (ssGSEA) to derive a “MYC activity score” and confirmed that MYC activity significantly correlated to MYC expression (r=0.46, p=0.0013), but not to TET1, TET2 or TET3 expression (Figure S6A). Expectedly, DLBCLs harboring TET2 mutation displayed significantly higher enhancer methylation than TET2 WT tumors (Figure S6B). More importantly, after excluding the potentially confounding effects of TET2-mutant DLBCLs, MYC expression remained significantly correlated with enhancer hypomethylation (r=0.33, p=0.04). We next tested if the MYC-associated enhancer demethylation was functionally relevant. Here, for each enhancer the beta values of all probes mapped to it were averaged and this aggregated value was correlated to the expression of the gene closest to, or overlapping with, the enhancer. A biologically coherent negative correlation between enhancer methylation and target transcript expression was found for multiple genes, including several products previously implicated in lymphoma pathogenesis, including, FOXP1, PIM1, ATF5, KLHL14 and BRD2 (Figure 6C and Table S1)(Baron et al., 2012; Butler and Aguiar, 2017; Dekker et al., 2016; Greenwald et al., 2004; Reddy et al., 2017; Vater et al., 2015). We concluded that DLBCLs with high MYC expression/activity display global enhancer hypomethylation and increased expression of a biologically relevant subset of the genes. Future studies should clarify the importance of these interplays to MYC lymphomagenesis. It is possible that once the MYC-driven activation of D2HGDH/L2HGDH is disrupted, MYC will lose some of its pro-growth properties. To start to address this possibility, we examined the effects of D2HGDH and/or L2HGDH KO in the MYC-driven growth of P493-6 cells. We did not find any consistent difference in the growth of D2HGDH/L2HGDH WT vs. KO cells (data not shown). These data suggest that in P493-6 cells, single KO of each of these mitochondrial dehydrogenases is not essential for the pro-growth MYC program. Presently, we cannot exclude the possibility that this outcome reflects the limitations of a model that accumulates significant amounts of 2-HG, thus making it difficult to isolate an individual gene’s role on cell growth. The generation of MYC-insensitive D2HGDH and L2HGDH alleles in lymphoma cell models should help answer this question.
Figure 6. MYC expression, enhancer methylation and activity in primary DLBCL.

a) Heat map of enhancer methylation in primary DLBCL assembled according to their MYC expression (top row). Each column is one tumor (n=46) and each row an enhancer probe (n=3000). b) comparison of the percentage of unmethylated enhancer probes in primary DLBCL mapped to the top and bottom quantiles of MYC expression. c) volcano plot depicting the relationship between enhancer methylation and target gene expression in primary DLBCL; genes with a highly significant correlation are in red, and representative examples of those with known role in lymphoma biology labeled. P value in a) is from Pearson correlation. In b) all data points are shown (min to max) and the p value is from a two-tailed Student’s t-test.
Discussion:
In this report, we showed that MYC transcriptionally activates the genes encoding the mitochondrial enzymes that perform the essential oxidation of 2-HG into αKG. Structural and functional integrity of D2HGDH and L2HGDH is critical to physiology, as demonstrated by their extensive evolutionary conservation and by the severity of 2-hydroxyglutaric acidurias, inborn errors of metabolism caused by homozygous loss of D2HGDH and/or L2HGDH(Kranendijk et al., 2012; Ye et al., 2018). MYC’s role in metabolism remodeling is broad, particularly in cancer(Hsieh et al., 2015). The data presented here add a new item to “MYC’s metabolic toolkit”, the direct control of 2-HG levels. By influencing the function and subcellular localization of DNA and RNA demethylases, and hence remodeling the epigenome and epitranscriptome, the MYC/D2-L2HGDH axis may also play a part on MYC’s role in development and stem cell reprograming. Interestingly, the relationship between MYC and D-2-HG has been suggested before in the context of breast cancer(Mishra et al., 2018). In that instance, MYC appears to modulate iron metabolism and influence ADHFE1 (alcohol dehydrogenase, Iron Containing 1) one of the mitochondrial enzymes that produces D-2-HG as a side reaction(Struys et al., 2005). We postulate that in normal cells, MYC’s transcriptional activation of D2HGDH and L2HGDH may function as a physiologic defense to the multiple enzymes that can promiscuously generate 2-HG(Fan et al., 2015; Intlekofer et al., 2015; Oldham et al., 2015; Struys et al., 2005).
A corollary of the MYC-mediated induction of D2HGDH and L2HGDH is the generation of αKG. Using KO cells, as well as pharmacological/metabolic blockade of glutaminolysis, we found that D2HGDH/L2HGDH-mediated reactions appear to contribute a meaningful fraction (but certainly not all) of the αKG accumulated downstream of MYC. The role of MYC in controlling various regulators of glutaminolysis, a significant source of αKG is well established(Hsieh et al., 2015). Our data show that a second source of αKG, 2-HG oxidation, is also regulated by MYC. It often goes unacknowledged that in primary cancer cells and cell lines the levels of 2-HG can be surprisingly high(Gross et al., 2010; Oldham et al., 2015), even in absence of IDH1/2 mutations, a finding that may reflect an environment that promotes the promiscuous generation of 2-HG (e.g., hypoxia and acidosis)(Intlekofer et al., 2015; Oldham et al., 2015). Thus, it is possible to speculate that in specific contexts, the amount of 2-HG may be high enough that upon oxidation it will influence the cellular pool of αKG. Thus, MYC influences two cellular systems that produce αKG, 2-HG oxidation and glutaminolysis, and it will be of interest to examine in future work if MYC also directly controls the transcription/function of IDHs.
Alpha-KG has pleiotropic effects in intermediary metabolism and beyond(Zdzisinska et al., 2017). However, with the recent discovery of naturally occurring competitive inhibitors to αKG (2-HG, succinate, fumarate(Xiao et al., 2012; Yang et al., 2012; Ye et al., 2018)) much emphasis has been placed on its role as an obligatory co-substrate for multiple dioxygenases. Within this context, we propose that MYC, in a manner suggestive of D2HGDH/L2HGDH/αKG dependency, may activate TET DNA hydroxylases and RNA demethylases. Remarkably, we discovered that these effects are not exclusively associated with the actual or relative availability of αKG as an enzyme co-substrate, but that it also involves a hitherto unknown influence on the subcellular location of these enzymes. We showed that the MYC/D2-L2HGDH axis and αKG promote nuclear accumulation. Conversely, the role of 2-HG in regulating the nuclear exclusion of these enzymes remains to be defined, as our findings in lymphoid vs. epithelial models were not concordant. How this modulation of subcellular movement is achieved remains to be defined, but covalent modifications previously implicated in TET nuclear localization are good candidates(Zhang et al., 2014). Lastly, although in our cell models FTO is mainly located in the cytosol, we found that its previously reported shuttling between the cytosol and nucleus(Gulati et al., 2014) is also influenced by MYC/D2-L2HGDH/αKG.
Considering the broad expression pattern of MYC, and the plethora of genes that can be simultaneously influenced by modulation of the epigenome and epitranscriptome, it is plausible that this mitochondria-centered signaling node may regulate various physiologic processes. Less intuitive, but by no means antithetical, is how to reconcile MYC oncogenicity with the increased activity of αKG-dependent dioxygenases that we uncovered. This may initially appear contrary to current thought because inhibition of dioxygenases is germane to IDH1/2-mutant tumors(Yang et al., 2012). However, recent evidence suggests that in AML, D-2-HG may in fact have anti-tumor activity(Su et al., 2018). This finding is relevant in the context of our discovery because in that report D-2-HG was found to suppress AML growth via inhibition of FTO and, downstream to it, MYC. This observation, together with our data, suggest the existence of a self-regulatory loop in which MYC, via D2HGDH/L2HGDH and αKG, activates FTO thus sustaining its own expression. Furthermore, the activation and change in subcellular location of RNA demethylases downstream to MYC and αKG that we report here, agrees with emerging evidence that FTO and ALKBH5 function as oncogenes in multiple cancer types(Deng et al., 2018), irrespective of the fact that the specificity of these two RNA demethylases towards unique chemical modification and RNA species has become a matter of recent debate(Mauer et al., 2017; Mauer et al., 2019; Wei et al., 2018).
We found that MYC, in a manner suggestive of D2HGDH/L2HGDH/αKG dependency, may activate TET DNA hydroxylases and promote their nuclear localization. In human neoplasia, TET enzymes have thus far been reported to function primarily as tumor suppressors(Huang and Rao, 2014). Thus, its activation downstream of MYC, a classical oncogene, requires rethinking of this monolithic labeling. An initial clue to the putative oncogenic role of TETs derives from their preferential targeting of enhancer sequences, promoting their demethylation and thus activation(Hon et al., 2014; Lio et al., 2016; Lu et al., 2014; Rasmussen et al., 2015). The putative influence of MYC-TET interplay on enhancer hypomethylation is noteworthy because aberrantly active enhancers (or super-enhancers) is a feature of multiple cancers, including DLBCL(Aran and Hellman, 2013; Chapuy et al., 2013; Hnisz et al., 2013). Reassuringly, when we examined the methylation pattern of DLBCL enhancers, we found that tumors with high MYC expression/activity displayed enhancer hypomethylation and active transcription of genes previously implicated in lymphoma pathogenesis. Thus, we propose that in tumors harboring MYC deregulation and wild-type TET genes, oncogenic enhancers may be activated.
Significance:
D-2-hydroxygluturate (D-2-HG) and L-2- hydroxygluturate (L-2-HG) are naturally occurring metabolites generated by fortuitous side reactions catalyzed by various enzymes. Two mitochondrial dehydrogenases, D2HGDH and L2HGDH, convert these metabolites into α-ketoglutarate (αKG). Germline loss of either enzyme leads to severe neuro-metabolic syndromes, indicating that accumulation of 2-HG is pathogenic and that D2HGDH and L2HGDH perform an essential function. Further, in cancer cells, somatically mutated IDH1 or IDH2 acquire neomorphic activity and generate D-2-HG resulting in competitive inhibition of αKG-dependent dioxygenases, including DNA and RNA demethylases. Still, despite the critical role of D2HGDH and L2HGDH in limiting the pathogenic accumulation of 2-HG, and the broad control of epigenetic remodeling modulated by 2-HG and αKG levels, the signals that regulate expression and activity of these mitochondrial dehydrogenases remains undefined. While characterizing D2HGDH and L2HGDH promoters, we identified active MYC binding sites and confirmed, in vitro and in vivo, that MYC drives the expression of these genes. Using multiple genetic models, we showed that MYC, in a manner suggestive of D2HGDH/L2HGDH dependency, modulates 2-HG/αKG homeostasis, induces the activity of the αKG-dependent TET enzymes and RNA demethylases, and thus promotes DNA and RNA demethylation. Notably, the effects of the MYC-D2HGDH/L2HGDH-αKG axis were not only due to higher levels of the co-substrate αKG, but also driven by nuclear accumulation of TET, FTO and ALKBH5, perhaps mediated by a still uncharacterized covalent modification. Consistent with the putative relevance of this interplay, in primary B-cell lymphomas MYC expression positively correlated with enhancer hypomethylation/activation, presumably due to heightened TET activity, which associated with overexpression of lymphomagenic genes. Together, these data identify MYC as a direct regulator of the essential 2-HG dehydrogenases, provide additional evidence of the multilayered control of epigenetic modifications, and reinforce the role of mitochondria metabolism in influencing the epigenome and epitranscriptome.
Lead Contact and Materials Availability
Please contact the corresponding author Ricardo Aguiar (aguiarr@uthscsa.edu) for reagents and resources included in this research. All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental Model and Subject Details
Cell lines.
The DLBCL cell lines SU-DHL8 (female), SU-DHL10 (male), OCI-Ly1 (male), OCI-Ly3 (male), OCI-Ly8 (male), OCI-Ly10 (female), Pfeiffer (male) and U2932 (female), Burkitt’s lymphoma cell line HS-Sultan (male), and P493-6 (information on sex unavailable), a human B cell line carrying a conditional MYC gene(Schuhmacher et al., 1999), were cultured at 37°C in 5%CO2 in RPMI-1640 medium (Invitrogen) containing 10% (vol/vol) fetal bovine serum (FBS), or 20% FBS (OCI-Ly3 and OCI-Ly10). HEK-293 (female,ATCC) and HEK-293T cells (female,Thermo-Fisher Scientific) were maintained in Dulbecco’s modified Eagle media (DMEM; Mediatech) with 10% FBS, as we described(Kim et al., 2009). In P493-6 cells, MYC suppression was achieved by exposing the cells to 100ng/ml of tetracycline. In most assays, samples were collected immediately before tetracycline addition to the media (MYC ON), following 36-48h of tetracycline exposure (MYC OFF), and at various time points after tetracycline is washed away (MYC ON). The identity of all cell lines was confirmed by VNTR analysis and verified online at the DSMZ and ATTC cell banks and tested for Mycoplasma contamination throughout this project. All the cell lines were preexistent in the investigator’s laboratories and/or were earlier obtained from ATCC or DSMZ cell banks.
Mice and isolation or murine mature splenic B lymphocytes.
A colony of Eμ-Myc transgenic mice (B6.Cg-Tg[IgHMyc]22Bri/J) was maintained by in-house breeding of C57BL/6 females with Eμ-Myc males. Eight to 12-week-old sex-matched WT and Eμ-Myc littermates (all without clinical evidence of lymphoma) were euthanized, and their splenic B cells purified using the mouse B-Cell Enrichment kit (StemCell Technologies). In brief, spleens were harvested from mice, single cell generated by dissociation, and mature B lymphocytes purified using the mouse B-Cell Enrichment Kit in the RoboSep Automated Cell Separator (Stem Cell Technologies). Subsequently, the purity and degree of enrichment was determined by fluorescence-activated cell sorter (FACS)-based measurement of CD19-positive cells, in pre- and post-separation aliquots. Consistently, the fraction of CD19-positive cells was >95%. as we described(Bouamar et al., 2015). High molecular weight DNA was extracted using the Gentra Puregene DNA purification kit (Qiagen), RNA isolation was completed using the Trizol method (TRIzol Reagent, Life technologies) and whole-cell lysates (WCL) were extracted in 2% Sodium Dodecyl Sulfate (SDS), 10% glycerol, 50mM Tris pH6.8, 2mM 2-Mercaptoethanol, boiled for 5-10 minutes, cooled at room-temperature and immediately thereafter loaded in the gels, as we described(Jiang and Aguiar, 2014). All animal procedures were approved by the Institute Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio, San Antonio, TX, USA.
Method Details
Compounds.
Cell lines were exposed to 1mM or 5mM of dimethyl 2-oxoglutarate (DMaKG; Sigma-Aldrich) for 8-24h, to 20-100uM of octyl-D-2-HG or octyl-L-2-HG (Toronto Research Chemicals) for 8-24h, to 1mM dimethyloxalylglycine (DMOG; Frontier Scientific) for 8-24h, and to CB-839 (Cayman Chemical) for 24h. When appropriate, dimethyl sulfoxide (DMSO; Sigma-Aldrich) was used as vehicle control. In P493-6 cells, MYC suppression was achieved by exposing the cells to 100ng/ml of tetracycline (Sigma-Aldrich).
Luciferase Reporter assays.
Two DNA fragments, 915bp and 931bp, flanking the putative transcriptional start site (TSS) of the genes D2HGDH and L2HGDH, respectively, were PCR-amplified, sequence verified and cloned into pGL3-basic vector (Promega). These constructs were co-transfected into HEK-293T cells with the pCMVβ-gal plasmid. In subsequent assays, the co-transfection also included a MYC expressing plasmid (pCDNA3-FLAG-MYC). Cells were harvested 24h after transfection, and luciferase and β-galactosidase activities measured, as we described(Ortega et al., 2015). In brief, the cells were in PBS, followed by addition of 250μl of reporter lysis buffer (Promega) and incubation for 20 minutes. The lysate was centrifuged for 30 seconds and the supernatant collected. Next, 20 μl of the cell lysate was transferred to a 1.5 ml of centrifuge tube containing 100μl of the Luciferase Assay Reagent II (LAR II) (Promega), followed by luminescence reading in luminometer. Concomitantly, 50 μl of cell lysate was incubated with 50 μl of Assay 2X Buffer (β-Galactosidase Enzyme Assay System, Promega). Incubation was performed at 37°C until a faint yellow color developed, at which time 150 μl of 1M Sodium Carbonate stop reaction was added, followed by absorbance reading . The primers sequences are listed in Supplementary Table S2.
Chromatin Immune Precipitation (ChIP)-PCR assay:
HS-Sultan cells were fixed in 1% Formaldehyde (Thermo-Fisher Scientific) for 10 minutes at room temperature and neutralized with 125mM glycine (Thermo-Fisher Scientific), followed by washing in cold PBS. The cell pellet was re-suspended in 500 ml of SDS lysis buffer (50mM Tris [pH8.0], 1%SDS, 10mM EDTA). Chromatin was sheared with samples on ice using Sonicator 3000 (Misonix Incorporated) - seven 10” bursts with 30” intervals. The sonicated prep was centrifuged at 15,000g for 10’, and the supernatant diluted 1:10 in ChIP dilution buffer (16.7 mM Tris-HCl [pH 8.1], 167 mM NaCl, 1.1% Triton X-100, 1.2 mM EDTA), and incubated for 16h at 4°C with an anti-c-MYC antibody (Santa Cruz Biotechnology) or normal mouse IgG (Santa Cruz Biotechnology) pre-bound to Dynabeads protein G (Invitrogen). The immunoprecipitation mixture beads were pulled down using a magnet, supernatant removed, followed by sequentially washing with low salt immune complex wash buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 1% Triton X-100, and 2 mM EDTA), high salt immune complex wash buffer (20 mM Tris [pH 8.0], 500 mM NaCl, 1% Triton X-100, and 2 mM EDTA), and LiCl immune complex wash buffer (10 mM Tris [pH 8.0], 0.25 M LiCl, 1% NP-40, 1% deoxycholic acid, and 1 mM EDTA), and TE (10 mM Tris [pH 8.0], 1 mM EDTA). The beads were next incubated in 0.1 M NaHCO3 and 1% SDS for 30 minutes at RT to elute chromatin DNA. Cross-link reversion was performed at 65°C for 4h and proteins removed using proteinase K in an incubation of 45°C for 1h. The naked DNA was extracted, treated with Rnase A and recovered in molecular biology grade water. Q-PCR was performed in the StepOnePlus Real-Time PCR System Thermal Cycling Block (Applied Biosystems), as we described(Bouamar et al., 2013). Primer sequences are listed in Table S1.
Generation of genetic models.
CRISPR-Cas9 technology was used to knockout (KO) the D2HGDH and/or L2HGDH genes in P493-6, OCI-Ly1 and OCI-Ly8 cell lines. In single gene KOs, the lentiCRISPR v2 puromycin was used(Sanjana et al., 2014). For double D2HGDH/L2HGDH KO, a combination of lentiCRISPR v2 puromycin and pL-CRISPR.EFS.GFP(Heckl et al., 2014) was used. lentiCRISPR v2 was a gift from Feng Zhang (Addgene plasmid # 52961); pL-CRISPR.EFS.GFP was a gift from Benjamin Ebert (Addgene plasmid # 57818). ShRNA-mediated MYC knockdown was implemented with Tet-pLKO-puro, a gift from Dmitri Wiederschain (Addgene plasmid # 21915)(Wiederschain et al., 2009); virus generation was performed, as we previously described(Kim et al., 2011). In brief, HEK-293T cells were co-transfected with the lentiCRISPR v2 (or pL-CRISPR.EFS.GFP) and the packing and envelop plasmids PAX2 and pMD2.G, respectively (Addgene #s 12260 and 12259). Cells supernatant was collected at 48h and 72h, and lymphoma cell lines transduced by spinoculation, followed by polyclonal population selection and/or clone generation by limiting dilution, as we reported(Cooney et al., 2018). D2HGDH and/or L2HGDH knockout was confirmed by western blotting and by Sanger sequencing (in clonal populations). For each gene, at least two distinct guideRNAs or shRNAs were used ( Table S2).
Protein isolation, subcellular fractionation and western blots.
Whole-cell lysates (WCL) were extracted in 2% Sodium Dodecyl Sulfate (SDS), 10% glycerol, 50mM Tris pH6.8, 2mM 2-Mercaptoethanol, boiled for 5-10 minutes, cooled at room-temperature and immediately thereafter loaded in the gels. Nuclear and cytoplasmic proteins were extracted by EpiQuik Nuclear Extraction Kit I (Epigentek), following the manufacturer’s instructions. In brief, first, the cytoplasmic fraction is removed from the nuclear pellets. Next, to extract nuclear proteins 1% NP40, 50mM Tris pH8.0,150mM NaCl, 10% glycerol, 1mM EDTA, is added to the nuclear pellet, incubated on ice for 15 minutes with vortex (5 seconds) every 3 minutes, and centrifuged for 10 minutes at 14,000 rpm at 4°C. The nuclear and cytoplasmic fractions were concentrated in Amicon Ultra Centrifugal Filters-10K (Millipore) by centrifugation, 5-15 minutes at 4°. Lastly, 10% glycerol was added to the final cytosolic extracts. All proteins were separated by SDS/polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride (PVDF) membrane, as we described(Suhasini et al., 2016). To detect TET1 and TET2, 6-8% of SDS/polyacrylamide gels were used, and transferred at 40V (4°C, for 16 hours), followed by overnight incubation with the primary antibodies. All other gel transfers were performed at 110V for 1-2h at 4°C. The following antibodies were used: c-Myc (clone 9E10; #sc-40, Santa Cruz Biotechnology), D2HGDH (#13895-1-AP, Proteintech), L2HGDH (#15707-1-AP, Proteintech), ALKBH5 (# ab69325, Abcam), FTO (c-3, #sc-271713, Santa Cruz Biotechnology), m6A (#202003, Synaptic System), TET1 (#SAB2700730, Sigma-Aldrich), TET2 (#18950, Cell Signaling Technology), TET3 (#: ABS463, EMD Millipore Corporation), METTL3, METTL14, WTAP (# 69391, # 51104 and #56501, all from Cell Signaling Technology), 5-Methylcytosine and 5-hydroxymethylcytosine (#28692 and #51660, Cell Signaling Technology), β-actin (# A2228, Sigma-Aldrich), Lamin A (#A303-433A-M, Bethyl Laboratories), Tubulin (#62204 Invitrogen). PVDF membranes were stripped with OneMinute Western Blot Stripping Buffer (#GM6001, GM Biosciences) and re-probed with relevant antibodies for loading control.
m6A RNA quantification.
m6A levels were quantified with two methodologies, as we reported(Elkashef et al., 2017). First, we used the ELISA-based EpiQuik™ m6A RNA Methylation Quantification Kit (Epigentek), according to the manufacturer guidelines. In this assay, abundance of m6A marks was quantified by absorbance and reported as relative values to the negative and positive controls, an RNA containing no m6A and an m6A oligo normalized to be 100% methylated. All assays were performed in triplicate and multiple biological replicates completed. For all models, we utilized 200ng of total RNA per well. In addition, m6A levels were measured with a dot-blot assay implemented with an anti-m6A antibody (Synaptic System), as we reported(Elkashef et al., 2017). In all applications, RNA was isolated using the Trizol method (TRIzol Reagent, Life technologies) and quantified by UV spectrophotometry.
5hmC and 5mC quantification.
High molecular weight DNA was obtained from relevant human cell lines and murine primary B cells. For the quantification of 5hmC and 5mC marks, three methods were used. In all instances, at least three independent biological replicates were performed, often utilizing more than one methodology, and including multiple technical triplicates.
Mass spectrometry:
5mC quantification was performed at Zymo Research Epigenetics Services. In brief, a selected reaction monitoring (SRM)-based mass spectrometry assay was used to quantify 5-methyl-2’-deoxycytidine (5mdC). The assay was designed to measure 5mdC concentrations as a percentage of 2’-deoxyguanosine (dG) (e.g. – [5mdC]/[dG]). The calibrated range for the analyte was 0-25% for 5mdC using a fixed 40 pmol amount of dG as an internal standard. The amount of 5mdC in the sample was calculated using the area ratio and standard curves in the range of 0-25% 5mdC/dG and reported as the percent of 5mdC. The 5hmC quantification was performed as previously described(Mariani et al., 2014). In brief, hydrolyzed genomic DNA was injected onto a HPLC Zorbax Eclipse Plus C18 RRHD column (Agilent Technologies). The analytes were separated by gradient elution using 5% methanol/0.1% formic acid (mobile phase A) and 100% methanol (mobile phase B) at a flow rate of 0.25 mL/min. Mobile phase B was increased from 0 to 3% in 5 min, to 80% in 0.5 min, kept at 80% for 2 min, then switched to initial conditions in 2.5 min. The effluent from the column was directed to the Agilent 6490 Triple Quadrupole mass spectrometer (Agilent Technologies). Calibration solutions with varying amounts of 5-hmC (0%–3%) and fixed amount of C were analyzed together with the samples. The solutions were prepared from a 200-bp DNA standard containing 57 cytosines, which are homogeneous for C and 5-hmC. Calibration plots of %5-hmC versus MRM Response ratio were constructed based on the data obtained, and %5-hmC is obtained from the ratio of [5-hmC/(5-mC+5-hmC+C)]. The %5-hmC in the samples was determined from the calibration plots.
Dot-blot assay:
DNA was extracted using the Gentra Puregene DNA purification kit (Qiagen), samples were denatured at 95°C for 10 min and placed on ice for 5 minutes before multiple dilutions were immobilized on a nitrocellulose membrane Hybond-N (Amersham Biosciences). Subsequently, the membranes were air-dried, UV crosslinked, blocked with 5% non-fat milk followed by 16h incubation with anti-5hmC and anti-5mC antibodies (Cell Signaling Technology). The membranes were then stained with methylene blue to confirm equal DNA input.
ELISA-based:
For the quantification of 5hmC and 5mC marks, 200 and 100 ng of DNA, respectively, was added to each well (96-well plate format), followed by incubation with primary (anti-5hmC or anti-5mC) and secondary antibodies and developed by colorimetric methods (Epigentek), as reported earlier(Lin et al., 2015). Abundance of 5hmC and 5mC marks was quantified by absorbance and reported as relative values to the positive controls, methylated polynucleotide containing 20% of 5hmC or 50% 5mC, respectively.
Demethylases activity.
The activity of DNA hydroxylases (TET enzymes) and m6A RNA demethylases was quantified in the nuclear protein of the relevant models. In brief, 10 million cells/sample were processed for isolation of nuclear lysate using the Nuclear Extraction Kit I (Epigentek). Next, for quantification of m6A demethylase activity the nuclear protein was incubated in wells stably coated with the m6A substrate (m6A Demethylase Activity Kit, Epigentek). The active m6A demethylases present in the lysates bind to and demethylate the m6A contained in the immobilized substrate, while the un-demethylated m6A in the substrate is recognized by an m6A antibody. The amount of un-demethylated m6A, which is inversely proportional to enzyme activity, is then colorimetrically quantified through an ELISA-like reaction. For the quantification of TET activity, the nuclear lysates is added to microplate wells stably coated with a methylated (5mC) substrate (5mC-Hydroxylase TET Activity Assay Kit, Epigentek). Active TETs bind to the substrate and convert the methylated substrate to hydroxymethylated products. The TET-converted hydroxymethylated products are recognized with a 5hmC antibody. The amount of hydroxymethylated products, which is proportional to TET activity, is colorimetrically measured by reading the absorbance in a microplate spectrophotometer.
Metabolite quantification by mass spectrometry.
The measurements of the D-2-HG, L-2-HG and αKG were performed as we described earlier(Lin et al., 2015), using liquid chromatography-tandem mass spectrometry (LC-MS/MS). In brief, 20 μl aliquot of the extracted samples were injected for chromatographic separation on an Agilent Hypersil ODS 4.0 x 250 mm, 5 μm column (Santa Clara, CA). The column oven was set at 30 °C. Solvent A was 125 mg l −1 ammonium formate, pH =3.6, solvent B was acetonitrile. The enantiomers were eluted using the programme: 0 to 0.5 min—100% A, 0.5 to 3.5 min—linear gradient to 96.5% A, 3.5 to 25 min—96.5% A and 3.5% B. The flow rate was set at 0.5 ml min −1, 50% of the post-column eluate was diverted to waste and the remainder injected into API 3000 triple–quadrupole mass spectrometer equipped with an electrospray ionization source (Applied Biosystems) and operating in negative ion mode. Turbo ionspray gas was set at 8 l min −1 the temperature at 500 °C and the ionspray voltage at −4,200V for 2-HG and at −1,700V for αKG. Product ion transitions were monitored at 363.2 to 147.2 for L-2-HG and D-2-HG, 367.1 to 151.1 for D,L-[3,3,4,4-D4]-2-hydroxyglutaric acid, 145.15 to 101.1 for αKG, and 149.2 to 105.2 for 2-ketopentanedioic acid-[D4]. Protein concentration was measured using the RC DC Protein Assay (Bio-Rad) and on SmartSpec Plus (Bio-Rad).
TET3 immunofluorescence analysis by confocal microscope:
Lipofectamine was used to transfect HEK-293T cells with an empty p3XFLAG-CMV vector (Sigma-Aldrich) or with the FLAG-tagged FH-TET3-pEF plasmid(Ko et al., 2013), a gift from Anjana Rao (Plasmid #49446, Addgene); 8h after transfection the cells were reseeded and grown for 36h in 35mm glass bottom microwell dishes (MatTek Corporation). Subsequently, the cells (three plates/condition) were exposed to DMαKG (5mM), a solution of octyl-D-2-HG and octyl-L-2-HG (100μM each) or DMSO for 6h, immediately followed by fixation in 4% paraformaldehyde (30 minutes at RT) and permeabilization in 0.1% Triton X100 (10 minutes at RT). Blocking was performed with 5% donkey serum in PBS at RT for 45 minutes. The plates were washed with PBS twice and exposed to either FLAG antibody (Sigma-Aldrich) at 1:400 dilution, or TET3 primary antibody (HPA050845, Sigma-Aldrich) at 1:25 dilution for 2h at RT. Anti-mouse (Alexa Fluor 488, Invitrogen) and anti-rabbit secondary antibodies (Alexa Fluor 568, Invitrogen) at 1:1000 dilution were added to anti-FLAG and anti-TET3 stained cells, respectively, for 45minutes. Lastly, all plates were stained with 4,6-diamidino-2-phenylindole (DAPI, 1:1000 dilution) for 10 minutes at RT, washed with PBS and stored at 4°C until images were collected. Slides were imaged with a Leica SP8 confocal microscope (Leica Microsystems, Mannheim, Germany) using a 100 × oil objective with excitations set at 355, 488, and 552 nm to detect DAPI, FLAG and TET3 signals, respectively. Images were exported as TIFF files using Leica LAS X software. Nuclear and cytosolic signals in transfected cells were identified and scored by two independent observers. Cells were sorted into the following categories: nuclear signal only, cytoplasmic and nuclear signals or exclusive cytoplasmic signal. For statistical analysis, the latter two categories were combined and compared to the nuclear only subset.
Enhancer methylation and gene expression analysis.
The DLBCL cohort studied (Schmitz et al., 2018)was from The Cancer Genome Atlas (TCGA) program and downloaded from Broad firehose (version 2016_01_28). The coordinates for DLBCL active enhancers were obtained from Chapuy et al.(Chapuy et al., 2013) and all Illumina 450K array probes mapped to these regions (n=21,856) retained; for downstream analysis, probes mapping to promoters were removed. The MYC expression value for each DLBCL in this cohort was obtained from their RNA-seq analysis. To examine the global pattern of enhancer methylation, we used median absolute deviation to select the top 3000 most variable probes from the 21,856 total. Next, we calculated the percentage of unmethylated probes (beta value < 0.3) for each sample and used Pearson correlation to examine the association between these values and MYC expression. For this analysis, we removed the two cases with the lowest MYC expression due to their exceptional profiles. The beta value cutoff 0.3 was selected based on previous reports(Zheng et al., 2016), as well as on manual examination of the beta value distribution. In an additional analysis, we divided the tumors into four quantiles according to MYC expression and calculated the percentages of unmethylated probes (amongst the 3000 most variable) in DLBCLs assigned to the top and bottom quantiles. To derive a “MYC activity score” we applied single sample gene set enrichment analysis (ssGSEA) using 17 MYC upregulated genes that are part of a reported MYC gene signature(Jung et al., 2017). We also examined the functional consequence of enhancer methylation, i.e. if methylation status correlates to the target gene expression. In brief, for each DLBCL active enhancer, we identified methylation probes and averaged their beta value to represent the enhancer methylation. Enhancers for which the methylation probes did not capture proper signal (TCGA data) or those which the closest gene was a non-coding RNAs were excluded from the downstream analysis. Subsequently, we calculated the Pearson correlation between enhancer methylation and the expression (RNA-seq data) of the gene mapping closest to, or overlapping with, each enhancer region.
Somatic copy number alterations (SCNAs) and gene expression analysis.
The GISTIC2.0 algorithm(Mermel et al., 2011), was used to define the somatic copy number alterations in the MYC locus in 10713 human tumors (TCGA sample pan-cancer dataset)(Hutter and Zenklusen, 2018). Correlation between D2HGDH or L2HGDH mRNA expression and SCNA was calculated using the Spearman test in 9890 tumors for which both expression and copy number data were available.
Quantification and Statistical Analysis
Analyses were performed using a one-way or two-way ANOVA, with Bonferroni’s multiple comparison post-hoc test, two-tailed Student’s t-test, and Chi-square test. P<0.05 was considered significant. Data analyses were performed in the Prism software (versions 5.02 and 8.02, GraphPad Software Inc) and Excel (Microsoft).
Data and Code availability:
This study did not generate any unique datasets or code
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-c-MYC (clone 9E10) | Santa Cruz Biotechnology | Cat#sc-40, ProSci Cat#51-118, RRID:AB_1947601 |
| Rabbit polyclonal anti-D2HGDH | Proteintech | Cat#13895-1-AP, RRID:AB_2089330 |
| Rabbit polyclonal anti-L2HGDH | Proteintech | Cat#15707-1-AP, RRID:AB_2133202 |
| Mouse polyclonal anti-ALKBH5 | Abcam | Cat#ab69325, RRID:AB_1267666 |
| Mouse monoclonal anti-FTO - Fatso (C-3) | Santa Cruz Biotechnology | Cat#sc-271713, RRID:AB_10707817 |
| Mouse monoclonal anti-m6A | Synaptic System | Cat#202003, RRID:AB_2279214 |
| Mouse monoclonal anti-TET1 (Clone GT1462) | Sigma-Aldrich | Cat#SAB2700730 |
| Rabbit monoclonal anti-TET2 | Cell Signaling Technology | Cat#18950, RRID:AB_2798809 |
| Rabbit polyclonal anti-TET3 | EMD Millipore Corporation | Cat#ABS463 |
| Rabbit polyclonal anti-TET3 | Sigma-Aldrich | Cat#HPA050845; RRID:AB_2681256 |
| Rabbit monoclonal anti-METTL3 (Clone D2I6O) | Cell Signaling Technology | Cat#96391; RRID:AB_2800261 |
| Rabbit monoclonal anti-METTL14 (Clone D8K8W) | Cell Signaling Technology | Cat#51104, RRID:AB_2799383 |
| Rabbit polyclonal anti-WTAP | Cell Signaling Technology | Cat#56501, RRID:AB_2799512 |
| Rabbit monoclonal anti-5-Methylcytosine (Clone D3S2Z) | Cell Signaling Technology | Cat#28692, RRID:AB_2798962 |
| Mouse monoclonal anti-5-hydroxymethylcytosine (Clone HMC31) | Cell Signaling Technology | Cat#51660, RRID:AB_2799398 |
| Mouse monoclonal anti-β-actin (clone AC-74) | Sigma-Aldrich | Cat#A2228, RRID:AB_476697 |
| Mouse monoclonal anti-FLAG (Clone M2) | Sigma-Aldrich | Cat#F3165, RRID:AB_259529 |
| Rabbit polyclonal anti-Lamin A | Bethyl Laboratories | Cat#A303-433A-M, RRID:AB_10951693 |
| Mouse monoclonal anti-Tubulin (Clone DM1A) | Thermo-Fisher | Cat# 62204, RRID:AB_1965960 |
| Normal mouse IgG | Santa Cruz Biotechnology | Cat#sc-2025, RRID:AB_737182 |
| Donkey anti-mouse IgG Alexa Fluor 488 | Thermo-Fisher | Cat#A-21202; RRID:AB_141607 |
| Donkey Anti-rabbit IgG Alexa Fluor 568 | Thermo-Fisher | Cat#A-10042, RRID:AB_2534017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dimethyl 2-oxoglutarate | Sigma-Aldrich | Cat#349631, CAS: 13192-04-6 |
| Octyl-D-2-HG | Toronto Research Chemicals | Cat#H942595, CAS:1391068-16-8 |
| Octyl-L-2-HG | Toronto Research Chemicals | Cat#H942596, CAS: 1391067-96-1 |
| Dimethyloxalylglycine (DMOG) | Frontier Scientific | Cat#D1070, CAS: 89464-63-1 |
| CB-839 | Cayman Chemical | Cat#2203 |
| Dimethyl sulfoxide | Sigma-Aldrich | Cat#D8418, CAS: 67-68-5 |
| Tetracycline | Sigma-Aldrich | Cat#T-7660, CAS: 64-75-5 |
| Glycine | Thermo-Fisher Scientific | Cat#BP381-5, CAS: 56-40-6 |
| Formaldehyde | Thermo-Fisher Scientific | Cat#28908, CAS: 50-00-0 |
| Dynabeads protein G | Invitrogen | Cat#10003D |
| OneMinute Western Blot Stripping Buffer | GM Biosciences | Cat#GM6001 |
| 4,6-diamidino-2-phenylindole - DAPI | Sigma-Aldrich | Cat#D9542, CAS: 28718-90-3 |
| Trizol | Life technologies | Cat#15596018, CAS: 108-95-2 (Phenol), CAS: 593-84-0 (Guanidine isothiocyanate), CAS: 1762-95-4 (Ammonium thiocyanate). |
| Critical Commercial Assays | ||
| 5mC-Hydroxylase TET Activity Assay Kit | Epigentek | Cat#P-3086 |
| m6A Demethylase Activity Kit | Epigentek | Cat#P-9013 |
| Methylated DNA (5mC) Quantification Kit | Epigentek | Cat#P-1034 |
| Hydroxymethylated DNA (5hmC) Quantification Kit | Epigentek | Cat#P-1036 |
| EpiQuik™ m6A RNA Methylation Quantification Kit | Epigentek | Cat#P-9005 |
| EpiQuik Nuclear Extraction Kit I | Epigentek | Cat#OP-0002 |
| Gentra Puregene DNA purification kit | Qiagen | Cat#158667 |
| Luciferase Assay Systems | Promega | Cat#E1500 |
| β-Galactosidase Enzyme Assay System | Promega | Cat#E2000 |
| Lipofectamine 2000 | Thermo-Fisher | Cat#11668-019 |
| Mouse B Cell Isolation | Stemcell Technologies Inc | Cat#19854 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| Amicon Ultra Centrifugal Filters-10K | Millipore | Cat#UFC501096 |
| Experimental Models: Cell Lines | ||
| SU-DHL8 | ATCC | CRL-2961 |
| SU-DHL10 | ATCC | CRL-2963 |
| OCI-Ly1 | DSMZ | ACC-722 |
| OCI-Ly3 | DSMZ | ACC-761 |
| OCI-Ly8 | Cellosaurus | CVCL_8803 |
| OCI-Ly10 | Cellosaurus | CVCL_8795 |
| Pfeiffer | ATCC | CRL-2632 |
| U2932 | DSMZ | ACC-633 |
| HS-Sultan | ATCC | CRL-1484 |
| P493-6 | Cellosaurus | CVCL_6783 |
| HEK-293 | ATCC | CRL-1573 |
| HEK-293FT | Invitrogen | R70007 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice | Charles River | Strain code: 027 |
| B6.Cg-Tg(IghMyc)22Bri/J | The Jackson laboratory | Stock number: 002728 |
| Oligonucleotides | ||
| Primers for PCR, see Table S2 | This paper | N/A |
| ShRNA and guide RNA, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| pGL3-basic vector | Promega | Cat# 1751 |
| pCDNA3-FLAG-MYC | gift from Dr. Yuzuru Shiio | N/A |
| pLCRISPR.EFS.GFP | Heckl et al., 2014 | Addgene plasmid #57818 |
| lentiCRISPR v2 | Sanjana et al., 2014 | Addgene plasmid #52961 |
| FH-TET3-pEF | Ko et al., 2013 | Addgene plasmid #49446 |
| Tet-pLKO-puro | Wiederschain et al., 2009 | Addgene plasmid #21915 |
| PAX2 | unpublished | Addgene plasmid #12260 |
| pMD2.G | unpublished | Addgene plasmid #12259 |
| pCMVβ-gal | Clontech | Cat# PT2004-5 |
| p3XFLAG-CMV | Sigma-Aldrich | Cat# E7658 |
| Software and Algorithms | ||
| Prism Software | GraphPad Software Inc | version 5.02 and 8.2.0 |
| UCSC genome browser | Public website | https://genome.ucsc.edu |
| The Cancer Genome Atlas (TCGA) program | Broad firehose - version 2016_01_28 | https://gdac.broadinstitute.org/ |
| GISTIC2.0 algorithm | Mermel et al. 2011 (PMID 21527027) | ftp://ftp.broadinstitute.org/pub/GISTIC2.0/all_versions |
| MatInspector | Genomatix | https://www.genomatix.de/downloads.html |
| Others | ||
| 35mm glass bottom microwell dishes | MatTek Corporation | P35GCOL-1.5-14-C |
| Nylon membrane - Hybond-N | GE Healthcare | Cat # RPN82N |
| HPLC Zorbax Eclipse Plus C18 RRHD | Agilent | Cat# 959996-902 |
| Pan-Cancer Atlas | Hutter and Zenklusen, 2018 (PMID 29625045) | https://gdc.cancer.gov/node/905/ |
| TCGA DLBCL dataset | Schmitz et al. 2018 (PMID 29641966) | http://gdac.broadinstitute.org/ |
| Enhancer coordinates | Chapuy et al. 2013 (PMID 24332044) | Table S7; https://www.sciencedirect.com/science/article/pii/S1535610813004911?via%3Dihub |
Highlights:
MYC directly induces the transcription of D2HGDH and L2HGDH
The MYC D2HGDH/L2HGDH interplay influences intermediary metabolism
The MYC-D2HGDH/L2HGDH axis activates TET and RNA demethylases
MYC-expressing B-cell lymphomas display hypomethylated and active enhancers
Acknowledgements:
We thank Dr. Dirk Eick for the P493-6 cells. Y.S. was supported by CA202485 (NCI), RP160487 and RP190385 (CPRIT), Owens Medical Research Foundation and UL1 TR001120 (NIH-CTSA). P.L.M.D. was supported by GM114102 (NIGMS), Alex’s Lemonade Stand Foundation, and National Center for Advancing Translational Sciences (UL1 TR002645). M.M. is supported by the NIH and DOD (R01GM109882, R01HL086699, R01HL142673, DOD/DHP-CDMRP PR181598P-1). R.C.T.A. was supported by RP150277, RP170146 and RP190043 (CPRIT), TRP 6524-17 (Leukemia and Lymphoma Society), and I01BX001882 (VA Merit). FACS core is supported by P30 CA054174 (NCI).
Footnotes
Declaration of interests: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aran D, and Hellman A (2013). DNA methylation of transcriptional enhancers and cancer predisposition. Cell 154, 11–13. [DOI] [PubMed] [Google Scholar]
- Baron BW, Anastasi J, Hyjek EM, Bies J, Reddy PL, Dong J, Joseph L, Thirman MJ, Wroblewski K, Wolff L, et al. (2012). PIM1 gene cooperates with human BCL6 gene to promote the development of lymphomas. Proc Natl Acad Sci U S A 109, 5735–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouamar H, Abbas S, Lin AP, Wang L, Jiang D, Holder KN, Kinney MC, Hunicke-Smith S, and Aguiar RC (2013). A capture-sequencing strategy identifies IRF8, EBF1, and APRIL as novel IGH fusion partners in B-cell lymphoma. Blood 122, 726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouamar H, Jiang D, Wang L, Lin AP, Ortega M, and Aguiar RC (2015). MicroRNA 155 Control of p53 Activity Is Context Dependent and Mediated by Aicda and Socs1. Mol Cell Biol 35, 1329–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MJ, and Aguiar RCT (2017). Biology Informs Treatment Choices in Diffuse Large B Cell Lymphoma. Trends in cancer 3, 871–882. [DOI] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, and Thompson CB (2015). Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS (2015). Evolution of Mitochondria as Signaling Organelles. Cell metabolism 22, 204–206. [DOI] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. (2013). Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney JD, Lin AP, Jiang D, Wang L, Suhasini AN, Myers J, Qiu Z, Wolfler A, Sill H, and Aguiar RCT (2018). Synergistic Targeting of the Regulatory and Catalytic Subunits of PI3Kdelta in Mature B-cell Malignancies. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. (2010). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 465, 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker JD, Park D, Shaffer AL 3rd, Kohlhammer H, Deng W, Lee BK, Ippolito GC, Georgiou G, Iyer VR, Staudt LM, et al. (2016). Subtype-specific addiction of the activated B-cell subset of diffuse large B-cell lymphoma to FOXP1. Proc Natl Acad Sci U S A 113, E577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Su R, Weng H, Huang H, Li Z, and Chen J (2018). RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell research 28, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNardo CD, Propert KJ, Loren AW, Paietta E, Sun Z, Levine RL, Straley KS, Yen K, Patel JP, Agresta S, et al. (2013). Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood 121, 4917–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkashef SM, Lin AP, Myers J, Sill H, Jiang D, Dahia PLM, and Aguiar RCT (2017). IDH Mutation, Competitive Inhibition of FTO, and RNA Methylation. Cancer cell 31, 619–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Teng X, Liu L, Mattaini KR, Looper RE, Vander Heiden MG, and Rabinowitz JD (2015). Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS chemical biology 10, 510–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald RJ, Tumang JR, Sinha A, Currier N, Cardiff RD, Rothstein TL, Faller DV, and Denis GV (2004). E mu-BRD2 transgenic mice develop B-cell lymphoma and leukemia. Blood 103, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, et al. (2010). Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. The Journal of experimental medicine 207, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati P, Avezov E, Ma M, Antrobus R, Lehner P, O'Rahilly S, and Yeo GS (2014). Fat mass and obesity-related (FTO) shuttles between the nucleus and cytoplasm. Bioscience reports 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, and Ebert BL (2014). Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nature biotechnology 32, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra AS, de Graaff MA, Briaire-de Bruijn IH, Ras C, Seifar RM, van Minderhout I, Cornelisse CJ, Hogendoorn PC, Breuning MH, Suijker J, et al. (2015). Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget 6, 38777–38788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, Yen CA, Ye Z, Mao SQ, Wang BA, et al. (2014). 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Molecular cell 56, 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AL, Walton ZE, Altman BJ, Stine ZE, and Dang CV (2015). MYC and metabolism on the path to cancer. Seminars in cell & developmental biology 43, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, and Rao A (2014). Connections between TET proteins and aberrant DNA modification in cancer. Trends in genetics : TIG 30, 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter C, and Zenklusen JC (2018). The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 173, 283–285. [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera JD, Cross JR, et al. (2015). Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell metabolism 22, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, and Aguiar RC (2014). MicroRNA-155 controls RB phosphorylation in normal and malignant B lymphocytes via the noncanonical TGF-beta1/SMAD5 signaling module. Blood 123, 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Russell AJ, Liu B, George J, Liu PY, Liu T, DeFazio A, Bowtell DD, Oberthuer A, London WB, et al. (2017). A Myc Activity Signature Predicts Poor Clinical Outcomes in Myc-Associated Cancers. Cancer research 77, 971–981. [DOI] [PubMed] [Google Scholar]
- Kim SW, Rai D, and Aguiar RC (2011). Gene set enrichment analysis unveils the mechanism for the phosphodiesterase 4B control of glucocorticoid response in B-cell lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research 17, 6723–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Rai D, McKeller MR, and Aguiar RC (2009). Rational combined targeting of phosphodiesterase 4B and SYK in DLBCL. Blood 113, 6153–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, Segal MF, Li H, Koh KP, Lahdesmaki H, et al. (2013). Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 497, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, and Jakobs C (2012). Progress in understanding 2-hydroxyglutaric acidurias. Journal of inherited metabolic disease 35, 571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AP, Abbas S, Kim SW, Ortega M, Bouamar H, Escobedo Y, Varadarajan P, Qin Y, Sudderth J, Schulz E, et al. (2015). D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2. Nature communications 6, 7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lio CW, Zhang J, Gonzalez-Avalos E, Hogan PG, Chang X, and Rao A (2016). Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loenarz C, and Schofield CJ (2008). Expanding chemical biology of 2-oxoglutarate oxygenases. Nature chemical biology 4, 152–156. [DOI] [PubMed] [Google Scholar]
- Lu F, Liu Y, Jiang L, Yamaguchi S, and Zhang Y (2014). Role of Tet proteins in enhancer activity and telomere elongation. Genes & development 28, 2103–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani CJ, Vasanthakumar A, Madzo J, Yesilkanal A, Bhagat T, Yu Y, Bhattacharyya S, Wenger RH, Cohn SL, Nanduri J, et al. (2014). TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell reports 7, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, et al. (2017). Reversible methylation of m6Am in the 5' cap controls mRNA stability. Nature 541, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ, Rentmeister A, Gross SS, Pellizzoni L, Debart F, et al. (2019). FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nature chemical biology 15, 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome biology 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Tang W, Putluri V, Dorsey TH, Jin F, Wang F, Zhu D, Amable L, Deng T, Zhang S, et al. (2018). ADHFE1 is a breast cancer oncogene and induces metabolic reprogramming. The Journal of clinical investigation 128, 323–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham WM, Clish CB, Yang Y, and Loscalzo J (2015). Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell metabolism 22, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega M, Bhatnagar H, Lin AP, Wang L, Aster JC, Sill H, and Aguiar RC (2015). A microRNA-mediated regulatory loop modulates NOTCH and MYC oncogenic signals in B- and T-cell malignancies. Leukemia 29, 968–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KD, Jia G, Johansen JV, Pedersen MT, Rapin N, Bagger FO, Porse BT, Bernard OA, Christensen J, and Helin K (2015). Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes & development 29, 910–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, Leppa S, Pasanen A, Meriranta L, Karjalainen-Lindsberg ML, et al. (2017). Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171, 481–494 e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al. (2018). Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. The New England journal of medicine 378, 1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmacher M, Staege MS, Pajic A, Polack A, Weidle UH, Bornkamm GW, Eick D, and Kohlhuber F (1999). Control of cell growth by c-Myc in the absence of cell division. Current biology : CB 9, 1255–1258. [DOI] [PubMed] [Google Scholar]
- Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, Kho EY, Ghosh AP, Kirkman R, Velu S, et al. (2014). L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer discovery 4, 1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struys EA (2006). D-2-Hydroxyglutaric aciduria: unravelling the biochemical pathway and the genetic defect. Journal of inherited metabolic disease 29, 21–29. [DOI] [PubMed] [Google Scholar]
- Struys EA, Gibson KM, and Jakobs C (2007). Novel insights into L-2-hydroxyglutaric aciduria: mass isotopomer studies reveal 2-oxoglutaric acid as the metabolic precursor of L-2-hydroxyglutaric acid. Journal of inherited metabolic disease 30, 690–693. [DOI] [PubMed] [Google Scholar]
- Struys EA, Verhoeven NM, Brunengraber H, and Jakobs C (2004). Investigations by mass isotopomer analysis of the formation of D-2-hydroxyglutarate by cultured lymphoblasts from two patients with D-2-hydroxyglutaric aciduria. FEBS letters 557, 115–120. [DOI] [PubMed] [Google Scholar]
- Struys EA, Verhoeven NM, Ten Brink HJ, Wickenhagen WV, Gibson KM, and Jakobs C (2005). Kinetic characterization of human hydroxyacid-oxoacid transhydrogenase: relevance to D-2-hydroxyglutaric and gamma-hydroxybutyric acidurias. Journal of inherited metabolic disease 28, 921–930. [DOI] [PubMed] [Google Scholar]
- Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al. (2018). R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 172, 90–105 e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhasini AN, Wang L, Holder KN, Lin AP, Bhatnagar H, Kim SW, Moritz AW, and Aguiar RCT (2016). A phosphodiesterase 4B-dependent interplay between tumor cells and the microenvironment regulates angiogenesis in B-cell lymphoma. Leukemia 30, 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vater I, Montesinos-Rongen M, Schlesner M, Haake A, Purschke F, Sprute R, Mettenmeyer N, Nazzal I, Nagel I, Gutwein J, et al. (2015). The mutational pattern of primary lymphoma of the central nervous system determined by whole-exome sequencing. Leukemia 29, 677–685. [DOI] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. (2010). The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer cell 17, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, Shi H, Cui X, Su R, Klungland A, et al. (2018). Differential m(6)A, m(6)Am, and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Molecular cell 71, 973–985 e975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer C, et al. (2009). Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8, 498–504. [DOI] [PubMed] [Google Scholar]
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, et al. (2012). Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes & development 26, 1326–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, et al. (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell 19, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ye D, Guan KL, and Xiong Y (2012). IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clinical cancer research : an official journal of the American Association for Cancer Research 18, 5562–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D, Guan KL, and Xiong Y (2018). Metabolism, Activity, and Targeting of D- and L-2-Hydroxyglutarates. Trends in cancer 4, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdzisinska B, Zurek A, and Kandefer-Szerszen M (2017). Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Archivum immunologiae et therapiae experimentalis 65, 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Liu X, Gao W, Li P, Hou J, Li J, and Wong J (2014). Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-linked beta-N-acetylglucosamine transferase (OGT). The Journal of biological chemistry 289, 5986–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, et al. (2016). Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer cell 30, 363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code