

ABSTRACT
Hippo pathway is a chain of kinases consists of a series of protein kinases and transcription factors. Meanwhile, oxidative stress is a condition of elevated concentrations of reactive oxygen species (ROS) that cause molecular damage to vital structures and functions. Both of them are key regulators in cell proliferation, survival, and development. These processes are strictly regulated by highly coordinated mechanisms, including c-Jun n-terminal kinase (JNK) pathway, mTOR pathway and a number of extrinsic and intrinsic factors. Recently, emerging evidence suggests that Hippo pathway is involved in the responses to cellular stresses, including mechanic stress, DNA damage, and oxidative stress, to mediate biological process, such as apoptosis, pyroptosis, and metastasis. But the exact mechanism remains to be further explored. Therefore, the purpose of this review is to summarize recent findings and discuss how Hippo pathway, oxidative stress, and the crosstalk between them regulate some biological process which determines cell fate.
KEYWORDS: Hippo pathway, redox, crosstalk, biological process, oxidative stress
1. Introduction
Oxidative stress, the imbalance between oxidation and anti-oxidation in the body, induces varieties of biological effects mainly through the accumulation of reactive oxygen species (ROS). ROS is a group of highly active oxygen-free radicals and their derivatives [1,2]. Although deleterious at high concentrations, ROS plays an extremely important role in cell survival, apoptosis, autophagy, and other processes as essential signaling messengers under physiologic conditions [3]. The redox hypothesis emphasizes the dual effects of reactive oxygen species. This hypothesis suggests that the primary effect of redox state changes is the change in cellular signaling, not simply oxidative damage [4].
Hippo pathway is first defined in drosophila by genetic mosaic screening and mutation of Hippo leads to a strong overgrowth phenotype [5]. It is proved that Hippo pathway also controls cell proliferation, differentiation, and apoptosis [6]. In mammals, when the Hippo pathway is activated, STE20-like protein kinase 1/2 (MST1/2) co-phosphorylates with Salvador family WW domain-containing protein 1 (SAV1), then large tumor suppressor 1/2 (LATS1/2) and MOB kinase activator 1A/B (MOB1A/B) constitute a kinase cascade that phosphorylates Yes-associated protein (YAP) in conjunction with TEA domain family members1-4 (TEAD1-4) and they mediate major physiological functions of the Hippo pathway.
ROS and Hippo pathways interact directly or indirectly through a variety of intermediate factors and they mediate different biological effects. Further study of molecular mechanisms involved in the initiation and progression of this connection needs to be clear elucidated. For this reason, an accumulating quantity of recent studies focuses on the relationship between ROS and Hippo pathway. Therefore, the main scope of this review is discussing the function of Hippo/ROS in biological process, as well as the crosstalk between Hippo pathway and oxidative stress which induces biological effects such as proliferation, autophagy, apoptosis, and so on.
2. The role of redox signaling in biological effects
Oxidative stress refers to the imbalance between oxidation and anti-oxidation in vivo, leading to inflammatory infiltration of neutrophils and destruction of cell structure [7]. It is a process of negative effects of free radicals in vivo, which is considered to be an important factor leading to aging [8].
Reactive oxygen species are key components of oxidative stress, including not only oxygen radicals such as superoxide anion (O2-) and hydroxyl radical (HO•), but also atoms or small molecules without unpaired electrons, such as hydrogen peroxide (H2O2) [9]. High concentrations of ROS can cause oxidative damage associated with many diseases [10] and even carcinogenesis, but appropriate levels of ROS have been shown to be necessary for normal cell survival [11]. ROS are produced from a variety of sources, including mitochondrial respiratory chain, enzymatic activation of cytochrome p450 enzyme and NADPH oxidase [12,13]. The resulting ROS can activate various signaling proteins such as NF-κB, PI3K, MAPK, and p53 and play corresponding biological roles [14–16].
2.1. Apoptosis
In 1991, Pearce [9,17] found that catalase could reduce the production of hydrogen peroxide to inhibit apoptosis. Subsequent studies found that during the process of cell apoptosis, the effect of ROS mainly depended on its dose [18]. At low doses, cells could gradually adapt to ROS and become more tolerant to subsequent redox exposure [19]. High doses of reactive oxygen species can enhance cell anti-oxidation function, and YAP1p and Msn2/4 transcription factor [19] are the most active transcription factor which can reduce the gene expression and delay the cell division cycle [17,20]. With the accumulation of ROS to higher doses, ROS may promote the release of cytochrome c by oxidizing mitochondrial pores to induce apoptosis [21,22].
The role of drug-generated ROS in apoptosis has been demonstrated by ROS scavenging agents [23]. However, the sequential relationship between cell apoptosis and ROS generation has not been fully clarified at present. Whether the cell damage caused by ROS first leads to apoptosis, or whether the cell damage leads to the increase of ROS production, still remains to be further studied.
2.2. Autophagy
Autophagy is the process that misfolded proteins and damaged organelles in cells move into vesicles and fuse with lysosomes to form autophagy lysosomes, which are eventually degraded. Reactive oxygen species produced by mitochondria and NADPH oxidases have been proved to activate autophagy and regulate cell survival, death, and several human diseases even cancers. ROS regulates autophagy through a variety of internal regulatory mechanisms, including ROS-FOXO3-lLC3/BNIP3-autophagy [24], ROS-Nrf2-p62-autophagy [25,26], ROS-HIF1-BNIP3/NIX-autophagy, ROS-tigar-autophagy [27] and other molecular signaling pathways. It has been found that a variety of drugs with ROS regulatory ability can become effective tools for the treatment of cancer by mediating autophagy. For example, ROS-modulating natural product such as juglanin, a flavonol derived from polygonum aviculare, may induce ROS-mediated autophagy in breast cancinogenesis [28]. Coadministration of resveratrol and temozolamide can reduce tumor volumes and suppress ROS/MAPK-mediated autophagy and subsequently induce apoptosis [29]. The content of ROS in the microenvironment plays an important role in cell survival and may provide a new thought for treating cancer.
2.3. Other effects
Pyroptosis is a programmed cell death induced by the dissolution of inflammasomes, and studies have shown that iron-activated ROS can lyse gasdermin D (GSDMD) through the Tom20-Bax-caspase-1 pathway. The N-terminal pore forming region (PFD) of GSDMD oligomerized to form nonselective pores on the membrane, resulting in cell swelling and membrane rupture [30]. In addition, melatonin attenuates smoking-induced apoptosis of endothelial cells by inhibiting the ROS/NLRP3 axis [31].
Cell migration is also regulated by ROS [32]. For example, Luteolin, a naturally occurring flavonoid, induces apoptosis of breast cancer cells and suppresses metastasis of breast cancer cells to lung in vivo [33]. Indomethacin, a non-steroidal anti-inflammatory drug (NSAID), may induce the generation of ROS [34] and suppresses the focal complex formation that inhibits colon cancer cell migration by attenuating calcium influx [35].
In addition, Luigi Donato et al. [36,37] explored the specific mechanism of ROS causing retinal dystrophy and compared the transcriptome of human RPE cells exposed to the oxidant N-retinylidene-N-retinylethanolamine (A2E), to found changes in gene expression in 10 different pathways which include regulation and/or alterations of angiogenesis, extracellular matrix integrity, isoprenoid-mediated reactions, physiological or pathological autophagy and cell death induction, etc. And these pathways are closely related to the Hippo pathway. Furthermore, by the similar experimental method, they also identified 22 Oxidative Glyoxalase 1 (GLO1) related genes that change their expression, which are involved in a complex network of biochemical mechanisms that might be associated with retinitis pigmentosa onset and progression [38]. GLO1 is a ubiquitous cellular enzyme for methylglyoxal (MG), a cytotoxic byproduct of glycolysis whose excess can produce oxidative stress and cause tissue damage.
Furthermore, the retinaldehyde-binding protein 1 gene, RLBP1, is the most frequently involved in retinitis punctata albescens (RPA), characterized by progressive retinal degeneration due to alteration in visual cycle and consequent deposit of photopigments in retinal pigment epithelium [39]. Relevant studies have shown that oxidative stress changes both intracellular RLBP1 and autophagy levels, involved in the pathological process of inflammatory neurodegenerative diseases [40]. To sum up, oxidative stress is also inextricably related to retinal pigment degeneration and other neurodegenerative diseases.
3. The role of Hippo pathways in biological effects
Hippo pathway is a large protein network which is highly conserved, including MST1/2 (Drosophila Hippo), SAV1 (Drosophila Salvador), LATS1/2 (Drosophila Warts), MOB1A/B (Drosophila Mats), MAP4K4 (Drosophila Misshapen), YAP (Drosophila Yorkie), and TTEAD1-4 (Drosophila Scalloped) [41,42]. The Hippo pathway can respond to various upstream stimuli from the cellular micro-environment, including mechanical signal, cell stress, polarity, and adhesion. These signals activate LATS1/2 protein and MOB1A/B complex, thus promoting the kinase cascade activation. Then, YAP/TAZ is phosphorylated and transferred out of the nucleus through the interaction with the 14-3-3 protein or protease degradation, eventually stops TEAD-mediated transcription and affects cell function [43]. Hippo pathway has emerged as an exciting and potent regulator of organ development, homeostasis, and regeneration. A growing body of work has advanced our understanding of the Hippo pathway’s involvement in mechanisms of biological effects.
3.1. Apoptosis
There is a complex dual relationship between YAP and apoptosis. YAP has been proven to promote apoptosis and thus reduce tumor growth in vivo [44,45]. In fact, Yuan et al. [46] have pointed out that as a potential tumor suppressor gene for breast cancer, the deletion of YAP promotes the migration and invasion of cells in vitro, and they have also proved the deletion of YAP expression in 63% invasive ductal breast cancer, but they have not described the specific correlation between the reduction of YAP expression and breast cancer. Subsequent studies have shown that YAP promotes apoptosis in a p73-dependent manner [47]. Nuclear YAP has been shown to interact with p73 to enhance apoptosis in response to DNA damage and tumor formation. In addition, Keshet R et al. reported that when blood cell DNA was damaged, tyrosine kinase c-Abl entered the nucleus and phosphorylated YAP. Phosphorylated YAP binds to p73 and promotes transcription of pro-apoptotic genes such as Bax [48], DR5 [49], and PUMA [50].
However, on the contrary, most clinical trials have proved that YAP has a pro-cancer effect. YAP is overexpressed in lung tumors [51] and pancreatic tumors [52,53], which is consistent with the results of the study. Over-expression of YAP can significantly inhibit the apoptosis of liver [54], pancreas [55], and lung cancer cells [45], and thus accelerate tumor growth and lead to the decreased survival rate of patients. The dual role of YAP in apoptosis mainly depends on the transcriptional partner. In the nucleus, YAP binds to the TEAD family of transcription factors to initiate anti-apoptotic expression while interacting with p73 to enhance the transcription of pro-apoptotic genes.
3.2. Autophagy
The anti-cancer effect of YAP is also related to autophagy [56]. The study found that in ESCs, the increase of autophagy level and the decrease of YAP expression occurred simultaneously, suggesting that there may be a negative regulatory relationship between YAP and autophagy [26]. In the study of autophagy response in triple-negative breast cancer (TNBC) [57], the association between autophagy response and YAP at the cellular level was further confirmed. The study found that YAP was significantly up-regulated in TNBC, and when the TNBC autophagy response was initiated, YAP target gene ankyrin repeat domain 1 (ANKRD1) expression was significantly increased, and YAP was transferred into the nucleus. The relationship between autophagy and YAP activation was demonstrated at the single-cell level. In addition, Wilkinson et al. [58] reported that phosphorylation of LC3 by STK3/STK4 is an essential step in autophagy, and STK3/STK4 is a mammalian homolog of Hippo that antagonizes YAP activity, suggesting a possible relationship between the YAP pathway and autophagy. But the specific mechanism has not been fully clarified.
3.3. Metastasis
We found that abnormal YAP activity can promote metastasis of breast cancer and melanoma in a manner that is highly dependent on the interaction between the YAP domain and the transcription factor TEAD family [59]. YAP plays a role in promoting metastasis at both primary and metastatic sites, which is critical for YAP-mediated tumor growth and metastasis [59]. Compared with primary tumors, the expression of TAZ in BC metastatic tumors tends to be higher [60]. Similarly, in specific pancreatic cancer patients, the expression of YAP in metastasis is higher than that in primary tumors [61], suggesting that YAP may be closely associated with tumor metastasis. But the drawback at this stage is that, on the one hand, most experiments do not distinguish between nucleolus and cytoplasmic YAP. On the other hand, the role of the Hippo pathway in cancer metastasis is largely derived from in vitro studies. While in vitro studies may provide clues about how a pathway regulates metastasis, it is unclear that how reliable these experiments are in reflecting what happens in vivo.
4. Crosstalk between redox and Hippo pathway
The Hippo pathway is an evolutionary-conserved regulator of organ size and tumorigenesis that controls cell proliferation and death. There is an increasing evidence that oxidative stress can modulate various aspects of the typical or atypical Hippo pathways and produce different functional outcomes. In addition, the Hippo pathway also regulates intracellular redox homeostasis.
4.1. Regulation of MST1/2 by oxidative stress
A variety of upstream factors stimulate the activation of MST1/2 (Figure 1). Some enzymes are modified through a more specific mechanism involving the thioredoxin [62]. Studies have shown that the specific mechanism of oxidative stress-inducing MST1/2 is realized by thioredosin-1 (Trx1) [63]. Trx1 can physically associate with the SARAH domain of MST1, inhibit the homodimerization and autophosphorylation of MST1, thus prevent the activation of MST1. But H2O2 can eliminate this interaction, then lead to MST1 activation. Therefore, Trx-1 may act as a molecular switch to turn off oxidative stress-induced activation of MST1 [64].In addition, amyotrophic lateral sclerosis (ALS) associated SOD1 (G93A) mutants induced the dissociation of Mat1 with the REDOX protein Trx-1 and promoted the activation of MST1 in spinal neurons in a reactive oxygen associated manner [65].
Figure 1.
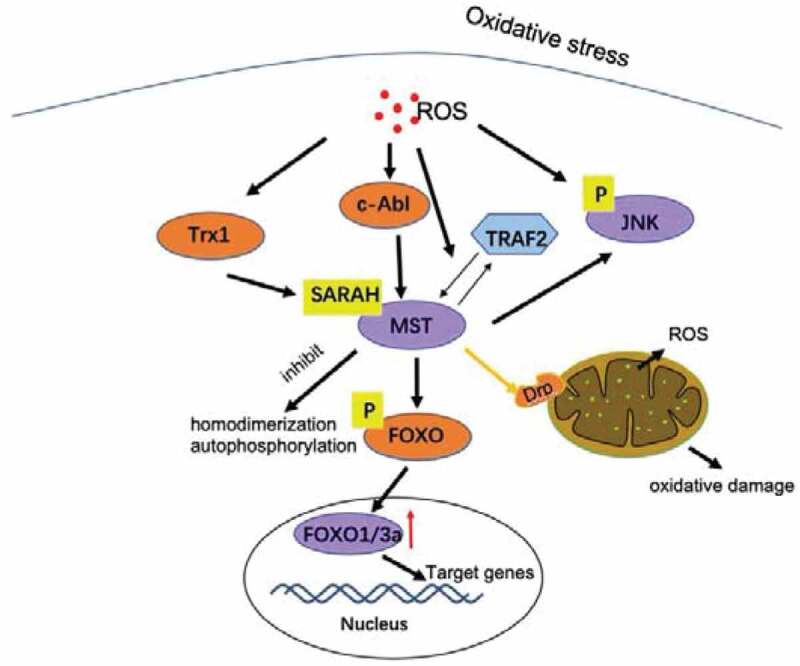
Crosstalk between ROS and MST. Oxidative stress induces Trx1, which physically binds to the SARAH domain of MST1 and inhibits homologous dimerization and autophosphorylation of MST1, thereby preventing MST1 activation [64]. Oxidative stress causes MST1 to be activated by c-Abl, promoting FOXOs forkhead phosphorylation, and this modification promotes FOXOs nuclear entry and transcriptional activation [68,69]. ROS also induces the physical interaction between TRAF2 and MST1, promotes homodimerization and activation of MST1 [73]. In addition, after transfection with MST1 adenovirus, Drp1 expression increased, promoting mitochondrial division. Active mitochondrial division mediates mitochondrial damage, which is manifested by increased mitochondrial oxidative stress, decreased mitochondrial energy production, and decreased mitochondrial respiratory complex function [75]
Meanwhile, MST1/2 can also maintain redox homeostasis of cells and prevent senescence and death of phagocytes by sensing excessive ROS and regulating the stability of the key antioxidant transcription factor Nrf2 [66]. We found that Nrf2 expression increased after MST1 deletion, while Nrf2 silencing eliminated the protective effect of MST1 deletion on survival of nasal epithelial cells and mitochondrial homeostasis. Nrf2 overexpression also protects nasal epithelial cells from TNF-induced inflammatory damage [67].
Meanwhile, MST1 has been shown to be activated by c-Abl-dependent tyrosine phosphorylation and to promote the role of FOXOs in response to oxidative stress, leading to cell death in primary-cultured neurons and rat hippocampal neurons [68]. These results indicate that c-Abl-MST-FOXO signaling cascade is an important pathway in the process of oxidative stress in cells [69]. A similar H2O2–Abl–MST1–FOXO3 pathway was identified in astrocytes [70].
Concomitant with Tyr433 phosphorylation, MST1-catalyzed phosphorylation of the FOXO3 forkhead domain is enhanced which promotes the nuclear entry and transcriptional activation of FOXO1. In fact, we found that H2O2 induces the phosphorylation and activation of MST1/2, then it promotes the accumulation of FOXO3a nuclear communication [71], which is consistent with previous reports that activated MST1/2 directly phosphorylates FOXO3a at S207 and affects nuclear accumulation [72]. In addition, oxidative stress inhibits HER2 or PI3K-mediated tumor metastasis through the MST2-FOXO3a-ΔNp63 pathway.
We found that H2O2 can also induce the physical interaction between TRAF2 and MST1, which promotes homodimerization and activation of MST1. MST1 requires TRAF2-mediated stimulation of JNK and p38 kinase induced by H2O2 to promote apoptosis [73]. In addition, significant activation of exogenous pathways was also observed through Fas expression and caspase-8 activation [74]. Meanwhile, after transfection with adenovirus loaded MST1, the expression of Drp1 increased, and promoted mitochondrial division. Active mitochondrial division mediates mitochondrial damage, which is manifested by increased mitochondrial oxidative stress, decreased mitochondrial energy production, and decreased mitochondrial respiratory complex function [75].
Furthermore, Hippo activities can also regulate the protein phosphatase 2A (PP2A), Striatin-interacting phosphatase and kinase (STRIPAK) complex and lead to a common vascular disease called cerebral cavernous malformation (CCM), which can be caused by mutation of three CCM genes (CCM1, CCM2, and CCM3) [76,77]. Among them, the ability of CCM3 to promote either cell survival or death may be determined in part by the kinase with which it interacts. For example, CCM3 enhances the phosphorylation of the ERM family by MST4 to protect cells from death induced by oxidative stress [78]. In contrast, CCM3 interacts with YSK1 to promote apoptosis under oxidative stress [79]. In addition, the depletion of YSK1 and either MST3 or MST4 in human endothelial cells replicates the lumen formation defects seen with CCM3 knockdown. Further studies have found that CCM1 limits the accumulation of intracellular ROS and prevent oxidative stress-mediated cellular and DNA damage [80]. Therefore, we suspect that Hippo and the STRIPAK complexes can lead to CCM by altering hypoxia, inflammation, or oxidative-stress conditions [81].
4.2. The relationship between YAP/TAZ and ROS
YAP is the main downstream target of Hippo pathway and also mediates ROS triggered signaling pathway. The expression and activity of YAP could be inhibited by pro-oxidation conditions. In addition, YAP itself can affect the redox state of cells through a variety of mechanisms (Figure 2).
Figure 2.
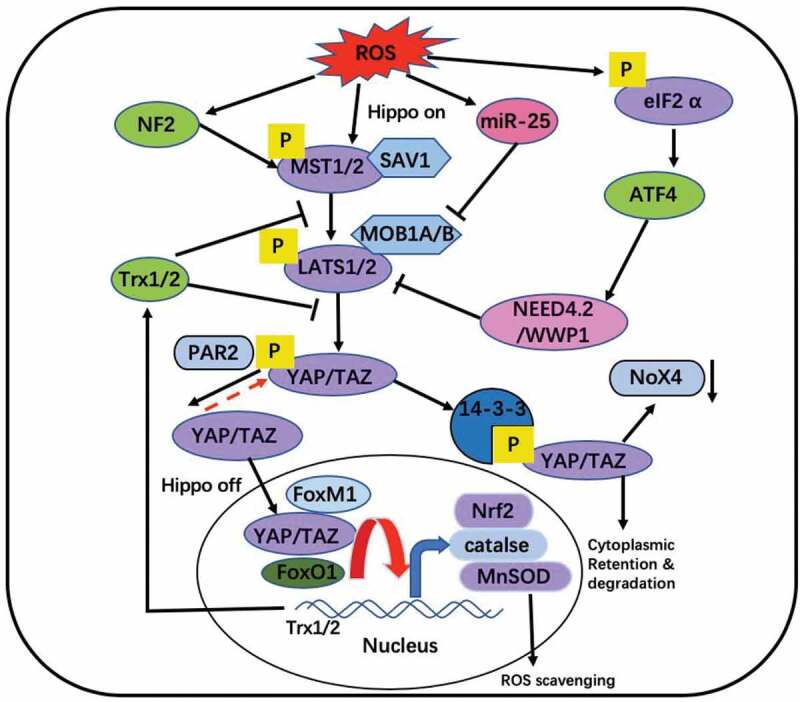
Interaction between ROS and Hippo pathway. ROS can activate Hippo pathway. YAP and FOXO1 forms functional complexes on antioxidant gene promoters such as catalase and MnSOD [82,83]. YAP induced redox proteins Trx1/2, which mediated YAP up-regulation (through MST1), thereby controlling redox status [85]. YAP can combine FoxM1 to promote the expression of Nrf2, a kind of antioxidant factor [87]. Proteinase-activated receptor 2 (PAR2) signals induce dephosphorylation of YAP [89]. Down-regulation of TAZ/YAP can inhibit TGF-induced Nox4 (NADPH oxidase) protein [91]. Oxidative stress induces the overexpression of miR-25 which inhibits LATS to promote expression of YAP [98]. ROS also phosphorylates eIF2 to activate ATF-NEED4.2 chain to inactivate Hippo pathway [99]
There is abundant evidence that YAP can inhibit oxidative stress in cells. In cardiomyocytes, YAP-FOXO1 (a transcription factor) axis forms functional complexes on antioxidant gene promoters such as catalase and MnSOD [82,83] and stimulates their transcription. In hepatic ischemia reperfusion injury (IRI), the activation of YAP also promotes induction of antioxidant genes to protect the liver from IRI injury [84]. In pancreatic cells, YAP over-expression up-regulates small molecule redox proteins Trx1/2 [85]. In human angiosarcoma, endothelial cells with low CD31 expression are more resistant to oxidative stress and DNA damage due to enhanced YAP signaling [86]. Meanwhile, BSO treatment silenced Nrf2, exhausted GSH, and inhibited the expression of YAP. Either Nrf2 or YAP silencing increased the drug sensitivity of bladder cancer cells [87]. YAP can also cross-link with other pathways to play an antioxidant role. Overexpression of Mitofusin 2 (Mfn2) in cultured mouse neuroblastoma N2a cells can improve the expression of YAP to reduce oxidative stress and reactive oxygen species excess [88]. Proteinase-activated receptor 2 (PAR2) signals induce dephosphorylation of YAP and significantly reduce the production of ROS in colon epithelium [89]. The activation of YAP leads to excessive accumulation of ROS by down-regulating the antioxidant enzyme GPX2 in a manner related to the blocking of p63 [90]. Down-regulation of MRTF and TAZ/YAP can inhibit TGF-induced Nox4 (NADPH oxidase) protein and ROS production during TGF-induced fibroblast/myofibroblast transformation [91].
The expression and activity of YAP can be directly or indirectly inhibited by pro-oxidation conditions. In both gastric and breast cancer [92,93], curcumin analog WZ35 that induced ROS down-regulates YAP. 4-hydroxynonenal (HNE), a pro-oxidant that can consume GSH, inhibit the expression of YAP and its target gene in bladder cancer cells through redox-dependent mechanism [94]. The transcriptional level of YAP is regulated by GABP in the liver. Oxidative stress inactivates GABP and down-regulates YAP, which leads to weakened oxidative-stress defense [95]. Controversially, some studies have shown that oxidizing conditions can sometimes promote the expression of YAP. The protein levels of YAP in glioma cells are elevated when treated with Chaetocin, a histone methyltransferase inhibitor that is known to induce ROS generation [96,97]. Oxidative stress induces the overexpression of miR-25, which promotes the proliferation and metastasis of lung cancer cells by targeting the LATS2/YAP signaling pathway [98]. Rajesh et al. [99] found that exposure to oxidative stress-induced phosphorylation of the translation initiation factor eIF2 protein, which promoted mRNA translation of activated transcription factor 4 (ATF4) and promoted LATS1 to inactivate YAP thereby inducing apoptosis.
In addition, the correlation of Hippo pathway components in influencing mitochondrial function and ROS production provides a further understanding of the interaction between cellular redox environment and Hippo signaling [100]. This interaction is well documented in a study using drosophila epithelium, in which cellular ROS plays a mediating role in oncogenic Ras driven JNK-mediated Hippo signal inactivation in the presence of functional mutations in the mitochondrial respiratory chain complex [101]. In general, YAP maintains mitochondrial potential, normalizes mitochondrial respiratory function and reduces ROS overproduction [102]. By shutting down the ROCK1/F-actin signaling pathway, Yap attenuates mitochondrial fission mediated by brain hypoxia-reoxygenation injury [102]. In rectal cancer cells, YAP silencing drives JNK phosphorylation, which induces Drp1 activation and translocation to the mitochondrial surface, triggering mitochondrial division [103].
5. Interaction between Hippo-YAP signal and redox mediates biological process
5.1. Apoptosis
In recent years, there has been a number of evidence that the Hippo pathway is related to oxidative stress or ROS activated signaling pathway and multiple pathological processes. And redox can regulate various aspects of typical and/or atypical Hippo pathway to affect apoptosis (Figure 3).
Figure 3.
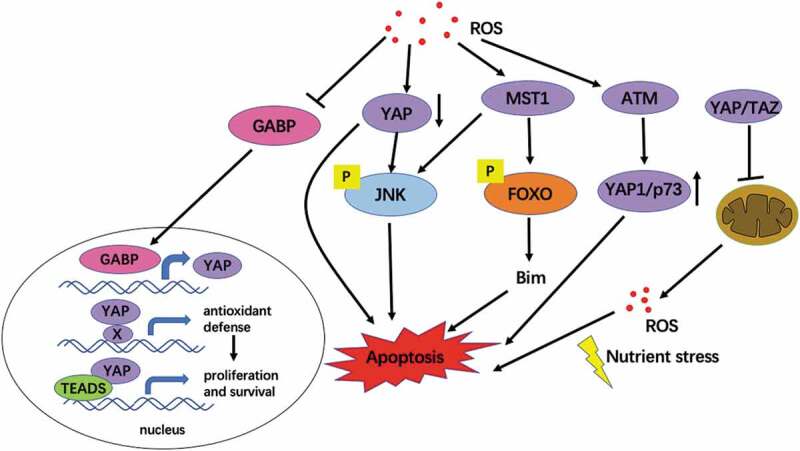
The interaction between ROS and Hippo pathway mediates cell survival. Oxidative stress inhibits the expression of YAP and induces apoptosis. Upon the loss of Hippo signaling, GABP translocates to the nucleus, where it activates the expression of a set of genes, including YAP. YAP is essential for several cellular and tissue responses against oxidative stress. Overactive YAP can also cooperate with TEADs to promote organ growth and cell survival [95]. MST1 was activated under oxidative stress, and the activated MST1 kinase phosphorylated the S207 site of FOXO3, leading to transcriptional activation of Bim protein, thus triggering neuronal apoptosis [65]. Chaetocin-induced ROS induce YAP1 expression independent of the typical Hippo pathway and activation of ATM and JNK. YAP1 interacts with p73 and JNK to induce apoptosis in an ATM – dependent manner [96]. YAP/TAZ inhibits mitochondrial function and ROS production and ultimately induces apoptosis [67,108]
MST1 is the first Hippo pathway component that has been studied to play an important role in ROS-induced cell death and ROS defense [72]. As it is mentioned above, oxidative stress, the upstream activator of MST, induces Trx1 binds to the SARAH domain of MST1 and inhibits the homodimerization of MST1 [64]. Among them, Lehtinen et al [68] found that MST1 was activated under oxidative stress, and the activated MST1 kinase phosphorylated the S207 site of FOXO3, led to its nuclear localization and transcriptional activation of BH3-only Bcl-2 protein, thus triggered neuronal apoptosis. After cisplatin treatment, the ROS reactive protein peroxiredoxin-1 is specifically associated with MST1, which induces apoptosis in U2OS cells [104]. Other studies have shown that NF2 can also promote I/R damage by simultaneously activating MST1 and inhibit YAP through oxidative stress, thereby regulating Hippo signals in adult hearts and promoting apoptosis [105]. Redox-mediated MST1-dependent FOXO3 phosphorylation has been shown to be a natural T-cell survival mechanism that activates transcription of antioxidant genes such as MnSOD and catalase [106]. It was found that H2O2 induced the physical interaction between TRAF2 and MST1, promoted homodimerization and activation of MST1, and activated JNK and p38 kinase, then caused cell apoptosis [107].
YAP is the main downstream target of the Hippo pathway and a key regulator that regulates cell proliferation and apoptosis. It is known that overexpression of YAP can resist apoptosis. In mouse hearts, YAP over-expression protects cardiomyocytes from H2O2-induced cell death [82]. In liver cells, YAP activation protects cells from hepatic ischemia reperfusion injury (IRI) by inducing regeneration and antioxidant genes, while reducing oxidative stress, necrosis, apoptosis, and innate inflammatory response [84]. As it is mentioned above, oxidative stress reduces the expression of YAP by inactivating GABP, which leads to weakened oxidative-stress defense [95] and G1/S cell cycle arrest and increased cell death. In colonic epithelial cells, PAR2 signal transduction stabilizes YAP protein and significantly reduces ROS production and sensitivity to nitric oxide-induced apoptosis [89]. Chaetocin-induced ROS induces YAP1 expression independent of the typical Hippo pathway and activation of ATM and JNK. YAP1 interacts with p73 and p300 to induce apoptosis in an ATM – dependent manner [96]. In NF2-mutant cells, Consumption of YAP/TAZ inhibits glycolysis and increases mitochondrial respiration and the accumulation of ROS, leading to oxidative stress and cell death [105].
The conserved Hippo signaling pathway also plays a role in mitochondrial regulation in drosophila and human cells [108], which indirectly affects apoptosis. Mitophagy activation can reduce oxidative stress injury, as well as mitochondrial calcium overload, and block mitochondrial pro-apoptotic factor leakage. In gastric cancer cells, the absence of YAP reduced the expression of SIRT1 and inhibited the Mfn2-mediated activity of mitophagy, which led to mitochondrial apoptosis and cellular oxidative stress [109]. In microglia BV-2, mitotic acid 5 (MA-5) regulates mitophagy through the MAPK-ERK-YAP signaling pathway and BNIP3 to reduce mitochondrial apoptosis and neutralize the overproduction of ROS [110].The absence of YAP promotes mitochondrial dysfunction, manifested by increased mitochondrial oxidative stress, pro-apoptotic factor leakage, and caspase-9 activity, which mediates the apoptosis of lung cancer cells [111].
In addition to the above studies, Hippo pathway further regulates apoptosis by regulating mitochondrial homeostasis (Figure 4). Mitochondrial division mediates oxidative stress, reduces mitochondrial respiratory complex function, increases mitochondrial permeability, and promotes the leakage of pro-apoptotic molecule cyt-c into the nucleus and cytoplasm, leading to caspase-9-related mitochondrial apoptosis [112]. The Hippo pathway interacts with mitochondrial homeostasis to mediate injury and apoptosis of cardiomyocytes, neurons, and so on. Tian et al [113] found that MST1 promotes mitochondrial division by up-regulating Drp1 and JNK pathway, enhancing oxidative stress and inflammatory signals, thus promoting neuronal dysfunction and cell death. Ji et al [114]found that IL-2/TAZ regulates mitochondrial fission by activating the JNK/F-actin pathway, thus inducing the apoptosis of HCC cells, and inhibiting TAZ can promote the induction of IL-2 on the apoptosis of HCC cells. Moreover, YAP overexpression can limit mitochondrial division and thus promote cell survival. Jun et al [103] found that knockout of YAP can promote JNK phosphorylation, thereby inducing Drp1 activation and translocation to the mitochondrial surface, leading to mitochondrial division. Wei et al [115] found that melatonin, by activating the Hippo-YAP pathway, enhances OPA1-related mitochondrial fusion to inhibits neuronal apoptosis.
Figure 4.
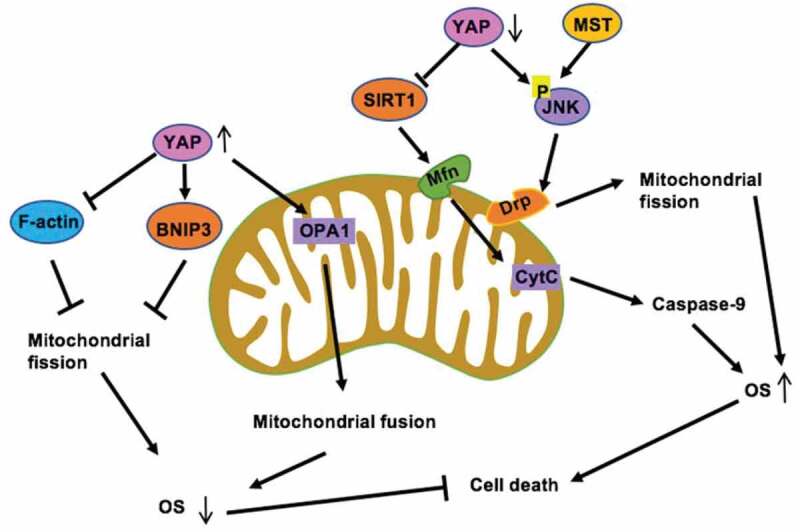
YAP and MST sustain cell viability and survival via mitophagy. On the one hand, YAP activates SIRT1 which enhances the mitophagy receptor Mfn2 expression, strongly maintaining mitophagy activity [109], on the other hand, YAP can phosphorylate JNK and activate Drp, induce Mitochondrial fission [103]. Mitophagy blocks the caspase-9 apoptotic pathway, abates cellular oxidative and prevents mitochondrial function. Meanwhile, overexpressed YAP activates OPA1 to enhance mitochondrial fusion [115] and also enhances BNIP3 [110] to reduce mitochondrial apoptosis
Nowadays, a group of lncRNAs have been delineated to directly or indirectly target the core components of Hippo cascade, such as YAP, TAZ, LATS1/2, and MST1 [116]. By contrast, Hippo can also modulate certain lncRNAs by affecting their transcriptional activity and form a reciprocal feedback loop. In addition, Luigi Donato et al. [37] further found that lncRNAs could be involved in several biochemical pathways related to compromised response to oxidative stress, leading to retinal cell death.
5.2. Aging
Human beings are facing emerging degenerative diseases, in large part, as a consequence of increased life expectancy. The Hippo-ROS pathway is also involved in some important mechanisms that control the aging process [72,117]. The study confirmed that down-regulation of CST-1, a homolog of Hippo kinase in Drosophila, accelerated aging and shortened lifespan. Meng et al [118] reported that attenuation of redox-stress response capacity (RRC) is a substantial feature of senescence. Specifically, RRC contains three main activities: the ability to produce ROS or RNS, the ability to regulate antioxidants, and the ability to maintain protein homeostasis. When MST1/2 is lost, the oxidative stress of macrophages increases, respiratory burst decreases and the ability to clear bacteria decreases [119]. Therefore, we suspect that MST1/2 defect will lead to premature aging of macrophages. Further research found that Kinase MST1 and MST2 induce ROS and maintain redox balance and through adjusting the stability of transcription factor Nrf2, ROS can recruit MST1/2 from cytoplasm to phagocytic membrane or mitochondrial membrane, and activate MST1/2 to phosphorylate kelch like ECH associated protein 1 (Keap1) and prevent Keap1 polymerization, thus blocking Nrf2 ubiquitination and degradation, and then protect cells against oxidative damage [120]. Therefore, the loss of MST1/2 can lead to oxidative damage, senescence and death of phagocytes [121,122].
In addition, transmission electron microscopy (TEMs) showed that there are more lipofuscin granules in the macrophages of MST1/2 double knockout mice (DKO mice), which is a recognized marker of aging [123].
5.3. Phagocytosis
Phagocytes are special immune cells that devour harmful microorganisms and destroy them in the phagocytes [124]. The destruction process is mainly dependent on the production of large amounts of ROS, which have long been thought to be produced entirely mechanically by the phagocytic membrane NADPH oxidase (NOX) [125,126]. But maximal phagocytic ROS generation and bactericidal activity require mitochondrial ROS production [127,128].
The kinases MST1 and MST2 play an important role in the optimal ROS production and bactericidal activity of phagocytes by activating small GTPase Rac and mitochondrial transport, as well as by assembling the TRAF6-ECSIT complex [124]. The high incidence of bacterial infections in the MST1 and MST2 double-deficient mice prompted us to investigate the role of MST1 and MST2 in the innate immune system, which provides immediate defense against infection by activating phagocytes.
5.4. Pyroptosis
Recent studies have shown that ROS can induce apoptosis and inhibit cancer progression [129,130], but whether the Hippo pathway or its core components are involved remains to be clarified. Cui et al [131] found that MST expression was decreased in Pancreatic ductal adenocarcinoma (PDAC), while MST1 recovery promoted PDAC cell death and inhibited PDAC proliferation, migration, invasion, and cell globular formation. Further studies have shown that MST1-induced pyroptosis is not related to the Hippo pathway, but is mediated by ROS, and overexpression of MST1 increases the levels of caspase-1, caspase-3, and caspase-7. Activation of caspase-3 and caspase-7 is mainly involved in apoptosis, while caspase-1 mainly mediates pyroptosis.
5.5. metastasis
The role of ROS in cancer development is complex. For example, a moderate ROS level has been shown to promote cell proliferation and migration, therefore contributing to tumor development [132], while excessive ROS can cause oxidative damage to lipids, proteins, and DNA, eventually leading to apoptosis and senescence to prevent tumor development [133,134]. However, the role of ROS in tumor metastasis remains largely unclear. Study reported that ROS can limit distant metastasis and only cells with increased antioxidant capacity can metastasize [135]. Furthermore, we already find that oxidative stress can inhibit tumor metastasis via activation of Hippo kinase MST1/2, which leads to the nuclear accumulation and activation of FOXO3a, causing up-regulation of ΔNp63α expression and suppression of cell migration [71].
A recent study reported that WZ35 may play a critical role in ROS-mediated regulation of cell migration and tumor metastasis. WZ35 is a new-discovered curcumin derivative, which can activate YAP, and then phosphorylate JNK. The activated JNK can induce apoptosis by phosphorylation of 14-3-3 protein, thereby releasing pro-apoptotic proteins such as BAX and FOXO [136]. To inhibit the migration and metastasis of breast cancer cells [93]. Being consistent with this finding, Zou et al. has demonstrated that ROS generation is the upstream regulator of WZ35-induced apoptosis and metastasis inhibition in gastric cancer [137].
5.6. autophagy
We found that WZ35 can also effectively inhibited the growth of hepatocellular carcinoma cells in vitro and in vivo by promoting apoptosis [138]. YAP is significantly overexpressed in liver cancer tissues, WZ35 can down-regulate YAP and promote autophagy by mediating the generation of ROS. Meanwhile, YAP upregulation can also induce multi-drug resistance in HCC cells via the Rac1-ROS-mTOR pathway, thus inhibiting autophagy-related cell death [139]. Furthermore, in vitro, MST1 up-regulation induces mitochondrial damage, including reduced mitochondrial potential, ROS overload, cell-c release, and activation of the caspase-9 apoptotic pathway. In vitro, MST1 up-regulation induces mitochondrial damage, including reduced mitochondrial potential, ROS overload, cell-c release, and activation of the caspase-9 apoptotic pathway. Further studies have found that RASSF1A inhibits PI3K-AKT-mTOR through the Hippo pathway regulating component MST1, thus enhancing autophagy initiation, and recruits autophagosomes on RASSF1A-stabilized acetylated microtubules through MAP1S, thus promoting autophagy maturation. RASSF1A deletion leads to a blockade of autophagy flux. Therefore, RASSF1A may inhibit HCC and improve survival by activating autophagy fluxes [140].
6. Concluding remarks
In this review article, we discussed the crosstalk between the Hippo pathway and oxidative stress, and their critical function in the various aspects of biological process, with special attention to apoptosis. Despite some studies highlighting the key components of the Hippo pathway has been proved to participate in ROS-mediated cell death or ROS scavenge in a variety of species and organs as well as the subcellular organelles, our knowledge regarding many of its crosstalk between oxidative stress is still pretty rudimentary, many questions still await answers before the full gamut of ROS/Hippo pathway effects is realized.
However, the crosstalk between cellular redox milieu and Hippo components has already provided a potential nodal point for direct and/or indirect intervention of cancer. In other words, in addition to inhibiting cancer by regulating cell growth and damage, redox may also play an important role in promoting the Hippo-driven execution of cancer cells. In addition, we hypothesized that the novel cancer suppressor drug could work effectively by activating Hippo through oxidative stress, because the core kinases involved in Hippo signaling, such as MST and YAP, suggests the tumor-inhibiting function need to identify “inducers” rather than “inhibitors.” But at present, studies on the interaction between Hippo and oxidative stress to mediate biological effects (even cancer) are limited, especially the specific mechanism of how Hippo core kinases are activated by ROS remains to be further studied.
Considering the evidence that various Hippo components interact with oxidative stress process, and the deregulation of Hippo signaling is strongly associated with the development of cancer, we believe that the crosstalk between cellular redox milieu and Hippo components provides an attractive potential target for cancer therapy. As mentioned above, the Hippo pathway is frequently deregulated in many human cancers, suggesting that YAP/TAZ and MST1/2 are the vital tumor driving components, which interplay with other cellular signaling networks to influence the balance of ROS. Notably, YAP/TAZ is critical in the hepatocellular carcinoma, breast cancer, and non-small cell lung carcinoma whereas MST1 is important in pancreatic carcinoma and colorectal carcinoma. These conclusions will provide many novel ideas for the treatment of cancer. For example, the strategies to inhibit YAP/TAZ transcriptional activity and/or promoting their cytoplasmic sequestration could have potential implications. Digitoxin increases YAP nuclear sequestration and boosts YAP activity, then YAP activation promotes ROS accumulation through down-regulation of GPX2, and finally suppresses lung squamous cell carcinoma progression. In addition, YAP up-regulation can also endow hepatocellular carcinoma cells with multi-drug resistance via the RAC1-ROS-mTOR pathway, which means the blockade of YAP may serve as a promising novel therapeutic strategy for overcoming chemoresistance in cancer. In addition, the crosstalk between cellular redox milieu and Hippo components provides a potential nodal point for direct and/or indirect intervention. In this respect, regulating the ROS level, as a novel therapeutic strategy, may also play a significant role in promoting the Hippo-driven execution of cancer cells. In fact, Yu et al. has found that curcumin can induce ROS-dependent Mst1-FOXO3 activation, which triggers JNK-dependent apoptosis in malignant melanoma cells. ROS activation can also inhibit tumor metastasis via the MST2-FOXO3a-ΔNp63α pathway. Meanwhile, MST1 suppressed the progression of PDAC cells partly through ROS-induced pyroptosis. Furthermore, recent studies reported that the anti-cancer activity of WZ35 is stronger than curcumin both in vitro and in vivo. WZ35 inhibits glycolysis through the ROS-YAP-JNK pathway, which may be a potential therapeutic for the treatment of gastric and breast cancer.
These observations demonstrated that MST1/2 and YAP would be valuable biomarkers and promising molecular targets for many human cancers, which could be combined with other therapies. We demonstrated that the blockade of YAP/TAZ facilitated the pro-apoptotic effects of sorafenib via augmenting mitochondrial fragmentation in a manner dependent on JNK signaling pathway and TAZ protein. In addition, the combination of Curcumin and PTX could not only inhibit gastric and breast cancer via ROS-YAP-JNK pathway, but also enhance anti-tumor activity of PTX against different cancers and reduce adverse effects of PTX. But up to now, there are few studies on the use of targets in the Hippo and ROS pathways for cancer treatment, and further studies on the optimal therapeutic regimen and toxic effects are needed.
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China (No.81701007), the Fundamental Research Funds for the Central Universities (No.2018SCUH0006), Research Funding for Talents Developing, West China Hospital of Stomatology Sichuan University (No.RCDWJS2020-6), and Basic and Applied Basic Research Projects of West China Hospital of Stomatology of Sichuan University (No.RD-02-201902).
Disclosure statement
The authors declare no potential conflict of interests.
References
- [1].Rahal A, Kumar A,Singh, V, et al. Oxidative stress, prooxidants, and antioxidants: the interplay. Biomed Res Int. 2014;1(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Boveris A. Determination of the production of superoxide radicals and hydrogen-peroxide in mitochondria. Methods Enzymol. 1984;105(5):429–435. [DOI] [PubMed] [Google Scholar]
- [3].Halliwell B, Gutteridge JMC.. Lipid-peroxidation,oxygen radicals,cell-damage,and antioxidant therapy. Lancet. 1984;1(8391):1396–1397. [DOI] [PubMed] [Google Scholar]
- [4].Johnston AD, Ebert PR. The redox system in c. Elegans, a phylogenetic approach. J Toxicol. 2012;2:e546915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Justice RW, Zilian O, Woods DF, et al. The drosophila tumor-suppressor gene warts encodes a homolog of human myotonic-dystrophy kinase and is required for the control of cell-shape and proliferation. Genes Dev. 1995;9(5):534–546. [DOI] [PubMed] [Google Scholar]
- [6].Harvey KF, Pfleger CM, Hariharan IK. The drosophila mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114(4):457–467. [DOI] [PubMed] [Google Scholar]
- [7].Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84. [DOI] [PubMed] [Google Scholar]
- [8].Jones DP. Redox theory of aging. Redox Biol. 2015;5:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pradedova EV, Nimaeva OD, Salyaev RK, et al. Redox processes in biological systems. Russ J Plant Physiol. 2017;64(6):822–832. [Google Scholar]
- [10].Lo Conte M, Carroll KS. The redox biochemistry of protein sulfenylation and sulfinylation. J Biol Chem. 2013;288(37):26480–26488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yun SB, Oh H, Rhee SG, et al. Regulation of reactive oxygen species generation in cell signaling. Mol Cells. 2011;32(6):491–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45(7–8):466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Loschen G, Azzi A, Flohé L. Mitochondrial H2O2 formation: relationship with energy conservation. FEBS Lett. 1973;33(1):84–88. [DOI] [PubMed] [Google Scholar]
- [14].Burdo RH, Rice-Evans C. Free radicals and the regulation of mammalian cell proliferation. Free Rad Res Comm. 1989;6(6):345–358. [DOI] [PubMed] [Google Scholar]
- [15].Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10(2):248–253. [DOI] [PubMed] [Google Scholar]
- [16].Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192(1):1–15. [DOI] [PubMed] [Google Scholar]
- [17].Lee JW, Romeo A, Kosman DJ. Transcriptional remodeling and g(1) arrest in dioxygen stress in saccharomyces cerevisiae. J Biol Chem. 1996;271(40):24885–24893. [DOI] [PubMed] [Google Scholar]
- [18].Perrone GG, Tan S-X, Dawes IW. Reactive oxygen species and yeast apoptosis. Biochimica Et Biophysica Acta-Molecular Cell Research. 2008;1783(7):1354–1368. [DOI] [PubMed] [Google Scholar]
- [19].Collinson LP, Dawes IW. Inducibility of the response of yeast-cells yeast cells to peroxide stress. J Gen Microbiol. 1992;138(2):329–335. [DOI] [PubMed] [Google Scholar]
- [20].Flattery-O’Brien JA, Dawes IW. Hydrogen Peroxide Causes RAD9-dependent Cell Cycle arrest in g(2) in Saccharomyces cerevisiae whereas Menadione Causes g(1) Arrest Independent of rad RAD9 Function. J Biol Chem. 1998;273(15):8564–8571. [DOI] [PubMed] [Google Scholar]
- [21].Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ros) in apoptosis induction. Apoptosis. 2000;5(5):415–418. [DOI] [PubMed] [Google Scholar]
- [22].Castello PR, David PS, McClure T, et al. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 2006;3(4):277–287. [DOI] [PubMed] [Google Scholar]
- [23].Wedi B, Straede J, Wieland B, et al. Eosinophil apoptosis is mediated by stimulators of cellular oxidative metabolisms and inhibited by antioxidants: involvement of a thiol-sensitive redox regulation in eosinophil cell death. Blood. 1999;94(7):2365–2373. [PubMed] [Google Scholar]
- [24].Hamacher-Brady A, Brady NR, Logue SE, et al. Response to myocardial ischemia/reperfusion injury involves bnip3 and autophagy. Cell Death Differ. 2007;14(1):146–157. [DOI] [PubMed] [Google Scholar]
- [25].Moscat J, Diaz-Meco MT. P62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tao Jiang BHT, Harder M, Rojo de la Vega M, et al. P62 links autophagy and nrf2 signaling. Free Radic Biol Med. 2015;88:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ros levels by tigar controls autophagy. The EMBO Journal. 2009;28(19):3015–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sun Z-L, Dong J-L, Wu J. Juglanin induces apoptosis and autophagy in human breast cancer progression via ros/jnk promotion. Biomed Pharmacother. 2017;85:303–312. [DOI] [PubMed] [Google Scholar]
- [29].Lin C-J, Lee -C-C, Shih Y-L, et al. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic Biol Med. 2012;52(2):377–391. [DOI] [PubMed] [Google Scholar]
- [30].Zhou B, Zhang J-Y, Liu X-S, et al. Tom20 senses iron-activated ros signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28(12):1171–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang X, Bian Y, Zhang R, et al. Melatonin alleviates cigarette smoke-induced endothelial cell pyroptosis through inhibiting ros/nlrp3 axis. Biochem Biophys Res Commun. 2019;519(2):402–408. [DOI] [PubMed] [Google Scholar]
- [32].Tochhawng L, Deng S, Pervaiz S, et al. Redox regulation of cancer cell migration and invasion. Mitochondrion. 2013;13(3):246–253. [DOI] [PubMed] [Google Scholar]
- [33].Cook MT, Liang Y, Besch-Williford C, et al. Luteolin inhibits lung metastasis, cell migration, and viability of triple-negative breast cancer cells. Breast Cancer. 2017;9:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tomita T, Sadakata H, Tamura M, et al. Indomethacin-induced generation of reactive oxygen species leads to epithelial cell injury before the formation of intestinal lesions in mice. J Physiol Pharmacol. 2014;65(3):435–440. [PubMed] [Google Scholar]
- [35].Guo Y-C, Chang C-M, Hsu W-L, et al. Indomethacin inhibits cancer cell migration via attenuation of cellular calcium mobilization. Molecules. 2013;18(6):6584–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Donato L, D’Angelo R, Alibrandi S, et al. Effects of a2e-induced oxidative stress on retinal epithelial cells: new insights on differential gene response and retinal dystrophies. Antioxidants. 2020;9(4):307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Donato L, Scimone C, Alibrandi S, et al. Transcriptome analyses of lncrnas in a2e-stressed retinal epithelial cells unveil advanced links between metabolic impairments related to oxidative stress and retinitis pigmentosa. Antioxidants. 2020;9(4):318–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Donato L, Scimone C, Alibrandi S, et al. Discovery of glo1 new related genes and pathways by rna-seq on a2e-stressed retinal epithelial cells could improve knowledge on retinitis pigmentosa. Antioxidants. 2020;9(5):416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Scimone C, Donato L, Esposito T, et al. A novel rlbp1 gene geographical area-related mutation present in a young patient with retinitis punctata albescens. Hum Genomics. 2017;11(1):18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ieong C, Ma J, Lai W. Ralbp1 regulates oral cancer cells via akt and is a novel target of mir-148a-3p and mir-148b-3p. J Oral Pathol Med. 2019;48(10):919–928. [DOI] [PubMed] [Google Scholar]
- [41].Hong W, Guan K-L. The yap and taz transcription co-activators: key downstream effectors of the mammalian hippo pathway. Semin Cell Dev Biol. 2012;23(7):785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hansen CG, Moroishi T, Guan K-L. Yap and taz: A nexus for hippo signaling and beyond. Trends Cell Biol. 2015;25(9):499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Varelas X. The hippo pathway effectors taz and yap in development, homeostasis and disease. Development. 2014;141(8):1614–1626. [DOI] [PubMed] [Google Scholar]
- [44].Xu Z, Chen J, Shao L, et al. Promyelocytic leukemia protein enhances apoptosis of gastric cancer cells through yes-associated protein. Tumor Biol. 2015;36(10):8047–8054. [DOI] [PubMed] [Google Scholar]
- [45].Sun L, Zhang Q, Ren H, et al. Overexpression of yes-associated protein contributes to apoptosis of lung cancer. Int J Clin Exp Pathol. 2016;9(2):540–547. [Google Scholar]
- [46].Yuan M, Tomlinson V, Lara R, et al. Yes-associated protein (yap) functions as a tumor suppressor in breast. Cell Death Differ. 2008;15(11):1752–1759. [DOI] [PubMed] [Google Scholar]
- [47].Jia L, Gu W, Zhang Y, et al. Activated yes-associated protein accelerates cell cycle, inhibits apoptosis, and delays senescence in human periodontal ligament stem cells. Int J Med Sci. 2018;15(11):1241–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Strano S, Monti O, Pediconi N, et al. The transcriptional coactivator yes-associated protein drives p73 gene-target specificity in response to DNA damage. Mol Cell. 2005;18(4):447–459. [DOI] [PubMed] [Google Scholar]
- [49].Xiao Q, Qian Z, Zhang W, et al. Depletion of cabyr-a/b sensitizes lung cancer cells to trail-induced apoptosis through yap/p73-mediated dr5 upregulation. Oncotarget. 2016;7(8):9514–9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Matallanas D, Romano D, Yee K, et al. RASSF1A Elicits apoptosis through an MST2 Pathway Directing Proapoptotic Transcription by the p73 Tumor Suppressor Protein. Mol Cell. 2007;27(6):962–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lau AN, Curtis SJ, Fillmore CM, et al. Tumor-propagating cells and YAP/taz activity contribute to lung tumor progression and metastasis. The EMBO Journal. 2014;33(13):1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rozengurt E, Sinnett-Smith J, Eibl G. Yes-associated protein (yap) in pancreatic cancer: at the epicenter of a targetable signaling network associated with patient survival. Signal Transduct Target Ther. 2018;3(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Maugeri-Saccà M, De Maria RJP. The hippo pathway in normal development and cancer. pharmthera. 2018;186:60–72. [DOI] [PubMed] [Google Scholar]
- [54].Wang X, Wu B, Zhong Z. Downregulation of yap inhibits proliferation, invasion and increases cisplatin sensitivity in human hepatocellular carcinoma cells. Oncol Lett. 2018;16(1):585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhou X, Su J, Feng S, et al. Antitumor activity of curcumin is involved in down-regulation of yap/taz expression in pancreatic cancer cells. Oncotarget. 2016;7(48):79062–79074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Pei T, Huang X, Long Y, et al. Increased expression of yap is associated with decreased cell autophagy in the eutopic endometrial stromal cells of endometriosis. Mol Cell Endocrinol. 2019;491:e328–329. [DOI] [PubMed] [Google Scholar]
- [57].Chen W, Bai Y, Patel C, et al. Autophagy promotes triple negative breast cancer metastasis via yap nuclear localization. Biochem Biophys Res Commun. 2019;520(2):263–268. [DOI] [PubMed] [Google Scholar]
- [58].Wilkinson DS, Jariwala J, Anderson E, et al. Phosphorylation of lc3 by the hippo kinases stk3/stk4 is essential for autophagy. Mol Cell. 2015;57(1):55–68. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lamar JM, Stern P, Liu H, et al. The hippo pathway target, yap, promotes metastasis through its tead-interaction domain. Proc Natl Acad Sci U S A. 2012;109(37):2441–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bartucci M, Dattilo R, Moriconi C, et al. Taz is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene. 2015;34(6):681–690. [DOI] [PubMed] [Google Scholar]
- [61].Yang S, Zhang L, Purohit V, et al. Active yap promotes pancreatic cancer cell motility, invasion and tumorigenesis in a mitotic phosphorylation-dependent manner through lpar3. Oncotarget. 2015;6(34):36019–36031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nishiyama A, Masutani H, Nakamura H, et al. Redox regulation by thioredoxin and thioredoxin-binding proteins. IUBMB Life. 2001;52(121):29–33. [DOI] [PubMed] [Google Scholar]
- [63].Graves JD, Gotoh Y, Draves KE, et al. Caspase-mediated activation and induction of apoptosis by the mammalian ste20-like kinase mst1. The EMBO Journal. 1998;17(8):2224–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chae JS, Gil Hwang S, Lim D-S, et al. Thioredoxin-1 functions as a molecular switch regulating the oxidative stress-induced activation of mst1. Free Radic Biol Med. 2012;53(12):2335–2343. [DOI] [PubMed] [Google Scholar]
- [65].Sanphui P, Biswas SC. Foxo3a is activated and executes neuron death via bim in response to beta-amyloid. Cell Death Dis. 2013;4:e625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chen L. Non-canonical hippo signaling regulates immune responses. Adv Immunol. 2019;144:87–119. [DOI] [PubMed] [Google Scholar]
- [67].Song HG, Wang M, Xin T. Mst1 contributes to nasal epithelium inflammation via augmenting oxidative stress and mitochondrial dysfunction in a manner dependent on nrf2 inhibition. J Cell Physiol. 2019;234(12):23774–23784. [DOI] [PubMed] [Google Scholar]
- [68].Xiao L, Chen D, Hu P, et al. The c-abl-mst1 signaling pathway mediates oxidative stress-induced neuronal cell death. J Neurosci. 2011;31(26):9611–9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Liu W, Wu J, Xiao L, et al. Regulation of neuronal cell death by c-abl-hippo/mst2 signaling pathway. Plos One. 2012;7(5):e36562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lee S-J, Seo B-R, Choi E-J, et al. The role of reciprocal activation of cabl and mst1 in the oxidative death of cultured astrocytes. Glia. 2014;62(4):639–648. . [DOI] [PubMed] [Google Scholar]
- [71].Wang Y, Li J, Gao Y, et al. Hippo kinases regulate cell junctions to inhibit tumor metastasis in response to oxidative stress. Redox Biol. 2019;26:e101233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lehtinen MK, Yuan Z, Boag PR, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125(5):987–1001. . [DOI] [PubMed] [Google Scholar]
- [73].Roh K-H, Choi E-J. TRAF2 functions as an activator switch in the reactive oxygen species-induced stimulation of MST1. Free Radic Biol Med. 2016;91:105–113. [DOI] [PubMed] [Google Scholar]
- [74].Ahmed K, Matsuya Y, Nemoto H, et al. Mechanism of apoptosis induced by a newly synthesized derivative of macrosphelides with a thiazole side chain. Chem Biol Interact. 2009;177(3):218–226. [DOI] [PubMed] [Google Scholar]
- [75].Ma C, Fan L, Wang J, et al. Hippo/MST1 overexpression induces mitochondrial death in head and neck squamous cell carcinoma via activating β-catenin/Drp1 pathway. Cell Stress Chaperones. 2019;24(4):807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Rinaldi C, Bramanti P, Scimone C, et al. Relevance of ccm gene polymorphisms for clinical management of sporadic cerebral cavernous malformations. J Neurol Sci. 2017;380(1):31–37. [DOI] [PubMed] [Google Scholar]
- [77].Scimone C, Donato L, Katsarou Z, et al. Two novel KRIT1 and CCM2 mutations in patients affected by cerebral cavernous malformations: new information on ccm2 penetrance. Front Neurol. 2018;9:e00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Fidalgo M, Guerrero A, Fraile M, et al. Adaptor protein cerebral cavernous malformation 3 (CCM3) mediates phosphorylation of the cytoskeletal proteins ezrin/radixin/moesin by mammalian ste20-4 to protect cells from oxidative stress. J Biol Chem. 2012;287(14):11556–11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Chen S, Fang Y, Xu S, et al. Mammalian sterile20-like kinases: signalings and roles in central nervous system. Aging Dis. 2018;9(3):537–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Goitre L, Villoria-Recio M, Cutano V, et al. KRIT1 and reactive oxygen species: A novel molecular pathway involved in cerebral cavernous malformations. Free Radic Biol Med. 2012;53(1):S58. [Google Scholar]
- [81].Hwang J, Pallas DC. STRIPAK complexes: structure, biological function, and involvement in human diseases. Int J Biochem cell Biol. 2014;47:118–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Del Re DP, Yang Y, Nakano N, et al. Yes-associated protein isoform 1 (YAP1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem. 2013;288(6):3977–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Shao D, Zhai P, Del Re DP, et al. A functional interaction between hippo-yap signalling and foxo1 mediates the oxidative stress response. Nat Commun. 2014;5(1):3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Liu Y, Lu T, Zhang C, et al. Activation of yap attenuates hepatic damage and fibrosis in liver ischemia-reperfusion injury. J Hepatol. 2019;71(4):719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Yuan T, Rafizadeh S, Azizi Z, et al. Proproliferative and antiapoptotic action of exogenously introduced YAP in pancreatic beta β cells. Jci Insight. 2016;1125(185):e86326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Stroebel P, Truemper L, Wulf G, et al. Cd31 expression determines redox status and chemoresistance in human angiosarcomas. Virchows Arch. 2018;473:S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ciamporcero E, Daga M, Pizzimenti S, et al. Crosstalk between nrf2 and yap contributes to maintaining the antioxidant potential and chemoresistance in bladder cancer. Free Radic Biol Med. 2018;115:447–457. [DOI] [PubMed] [Google Scholar]
- [88].Hou S, Wang L, Zhang G. Mitofusin-2 regulates inflammation-mediated mouse neuroblastoma n2a cells dysfunction and endoplasmic reticulum stress via the yap-hippo pathway. J Physiol Sci. 2019;69(5):697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].He L, Ma Y, Li W, et al. Protease-activated receptor 2 signaling modulates susceptibility of colonic epithelium to injury through stabilization of yap in vivo. Cell Death Dis. 2018;9(10):949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Huang H, Zhang W, Pan Y, et al. YAP Suppresses Lung Squamous Cell Carcinoma Progression via Deregulation of the DNp63–GPX2 Axis and ROS Accumulation. Cancer Res. 2017;77(21):5769–5781. [DOI] [PubMed] [Google Scholar]
- [91].Rozycki M, Bialik JF, Speight P, et al. Myocardin-related transcription factor regulates nox4 protein expression: linking cytoskeletal organization to redox state. J Biol Chem. 2016;291(1):227–243. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Chen T, Zhao L, Chen S, et al. The curcumin analogue wz35 affects glycolysis inhibition of gastric cancer cells through ros-yap-jnk pathway. Food Chem Toxicol. 2020;137:e111131. [DOI] [PubMed] [Google Scholar]
- [93].Wang L, Wang C, Tao Z, et al. Curcumin derivative wz35 inhibits tumor cell growth via ros-yap-jnk signaling pathway in breast cancer. J Exp Clin Cancer Res. 2019;38(1):460. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Cucci MA, Compagnone A, Daga M, et al. Post-translational inhibition of yap oncogene expression by 4-hydroxynonenal in bladder cancer cells. Free Radic Biol Med. 2019;141:205–219. [DOI] [PubMed] [Google Scholar]
- [95].Wu H, Xiao Y, Zhang S, et al. The ets transcription factor gabp is a component of the hippo pathway essential for growth and antioxidant defense. Cell Rep. 2013;3(5):1663–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Dixit D, Ghildiyal R, Anto NP, et al. Chaetocin-induced ROS-mediated apoptosis involves atm-yap1 ATM–YAP1 axis and JNK-dependent inhibition of glucose metabolism. Cell Death Dis. 2014;5(5):e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Mao B, Gao Y, Bai Y, et al. Hippo signaling in stress response and homeostasis maintenance. Acta Biochim Biophys Sin (Shanghai). 2015;47(1):2–9. [DOI] [PubMed] [Google Scholar]
- [98].Wu T, Hu H, Zhang T, et al. Mir-25 promotes cell proliferation, migration, and invasion of non-small-cell lung cancer by targeting the lats2/yap signaling pathway. Oxid Med Cell Longev. 2019;23(12):3945–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Rajesh K, Krishnamoorthy J, Gupta J, et al. The eif2α serine 51 phosphorylation-atf4 arm promotes hippo signaling and cell death under oxidative stress. Oncotarget. 2016;7(32):44–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ashraf A, Pervaiz S. Hippo circuitry and the redox modulation of hippo components in cancer cell fate decisions. Int J Biochem Cell Biol. 2015;69:20–28. [DOI] [PubMed] [Google Scholar]
- [101].Ohsawa S, Sato Y, Enomoto M, et al. Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in drosophila. Nature. 2012;490(7421):547. [DOI] [PubMed] [Google Scholar]
- [102].Geng C, Wei J, Wu C. Yap-hippo pathway regulates cerebral hypoxia-reoxygenation injury in neuroblastoma n2a cells via inhibiting rock1/f-actin/mitochondrial fission pathways. Acta Neurol Belg. 2018. DOI: 10.1007/s13760-13018-10944-13766. [DOI] [PubMed] [Google Scholar]
- [103].Li H, He F, Zhao X, et al. Yap inhibits the apoptosis and migration of human rectal cancer cells via suppression of jnk-drp1-mitochondrial fission-htra2/omi pathways. Cell Physiol Biochem. 2017;44(5):2073–2089. [DOI] [PubMed] [Google Scholar]
- [104].Morinaka A, Funato Y, Uesugi K, et al. Oligomeric peroxiredoxin-i is an essential intermediate for p53 to activate mst1 kinase and apoptosis. Oncogene. 2011;30(40):4208–4218. [DOI] [PubMed] [Google Scholar]
- [105].Matsuda T, Zhai P, Sciarretta S, et al. NF2 Activates Hippo Signaling and Promotes Ischemia/Reperfusion Injury in the Heart. Circ Res. 2016;119(5):596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Choi J, Oh S, Lee D, et al. Mst1-FoxO Signaling Protects naive Naïve T Lymphocytes from Cellular Oxidative Stress in Mice. Plos One. 2009;4(11):e8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Shang X, Li J, Yu R, et al. Sepsis-related myocardial injury is associated with mst1 upregulation, mitochondrial dysfunction and the drp1/f-actin signaling pathway. J Mol Histol. 2019;50(2):91–103. . [DOI] [PubMed] [Google Scholar]
- [108].Nagaraj R, Gururaja-Rao S, Jones KT, et al. Control of mitochondrial structure and function by the yorkie/yap oncogenic pathway. Genes Dev. 2012;26(18):2027–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Yan H, Qiu C, Sun W, et al. Yap regulates gastric cancer survival and migration via sirt1/mfn2/mitophagy. Oncol Rep. 2018;39(4):1671–1681. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [110].Lei Q, Tan J, Yi S, et al. Mitochonic acid 5 activates the MAPK–ERK–yap signaling pathways to protect mouse microglial BV-2 cells against tnf alpha-induced TNFα-induced apoptosis via increased Bnip3-related mitophagy. Cell Mol Biol Lett. 2018;23(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Zho J, Zhang S, Li Z, et al. Yap-hippo promotes a549 lung cancer cell death via modulating mief1-related mitochondrial stress and activating jnk pathway. Biomed Pharmacother. 2019;113:e108754. [DOI] [PubMed] [Google Scholar]
- [112].Lu C, Chen X, Wang Q. et al. TNFα promotes glioblastoma A172 cell mitochondrial apoptosis via augmenting mitochondrial fission and repression of MAPK–ERK–YAP signaling pathways. Onco Targets Ther. 2018;11:7213–7227. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [113].Tian H, Wang K, Jin M, et al. Proinflammation effect of mst1 promotes bv-2 cell death via augmenting drp1-mediated mitochondrial fragmentation and activating the jnk pathway. J Cell Physiol. 2020;235(2):1504–1514. [DOI] [PubMed] [Google Scholar]
- [114].Ji K, Lin K, Wang Y, et al. Taz inhibition promotes il-2-induced apoptosis of hepatocellular carcinoma cells by activating the jnk/f-actin/mitochondrial fission pathway. Cancer Cell Int. 2018;18(1):117–126. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wei N, Pu Y, Yang Z, et al. Therapeutic effects of melatonin on cerebral ischemia reperfusion injury: role of yap-opa1 signaling pathway and mitochondrial fusion. Biomed Pharmacother. 2019;110:203–212. [DOI] [PubMed] [Google Scholar]
- [116].Tu C, Yang K, Wan L, et al. The crosstalk between lncrnas and the hippo signalling pathway in cancer progression. Cell Prolif. 2020;12(1):e12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Yuan Z, Lehtinen MK, Merlo P, et al. Regulation of neuronal cell death by mst1-foxo1 signaling. J Biol Chem. 2009;284(17):11285–11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Meng J, Lv Z, Qiao X, et al. The decay of redox-stress response capacity is a substantive characteristic of aging: revising the redox theory of aging. Redox Biol. 2017;11:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Geng J, Sun X, Wang P, et al. Kinases mst1 and mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat Immunol. 2015;16(11):1142–1152. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wang P, Geng J, Gao J, et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the mst-nrf2 axis. Nat Commun. 2019;10(1):755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Avruch J, Zhou D, Fitamant J, et al. Protein kinases of the hippo pathway: regulation and substrates. Semin Cell Dev Biol. 2012;23(7):770–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Rawat SJ, Creasy CL, Peterson JR, et al. The tumor suppressor Mmst1 promotes changes in the cellular redox state by phosphorylation and inactivation of peroxiredoxin-1 protein. J Biol Chem. 2013;288(12):8762–8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Vida C, de Toda IM, Cruces J, et al. Role of macrophages in age-related oxidative stress and lipofuscin accumulation in mice. Redox Biol. 2017;12:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Geng J, Sun X, Wang P, et al. The kinases mst1 and mst2 positively regulate phagocyte ros induction and bactericidal activity. Nat Immunol. 2015;16(11):1142–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Lambeth JD. Nox enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–189. [DOI] [PubMed] [Google Scholar]
- [126].Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. [DOI] [PubMed] [Google Scholar]
- [127].West AP, Brodsky IE, Rahner C, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Emre Y, Hurtaud C, Nübel T, et al. Mitochondria contribute to lps-induced mapk activation via uncoupling protein UCP2 in macrophages. Biochem J. 2007;402(2):271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Pizato N, Luzete BC, Kiffer LFMV, et al. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triple-negative breast cancer cells. Sci Rep. 2018;8(1):1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Wu X, Zhang H, Qi W, et al. Nicotine promotes atherosclerosis via ROS-nlrp3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018;9(2):171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Cui J, Zhou Z, Yang H, et al. Mst1 suppresses pancreatic cancer progression via ros-induced pyroptosis. Mol Cancer Res. 2019;17(6):1316–1325. [DOI] [PubMed] [Google Scholar]
- [132].Gorrini C, Harris IS, Mak TW, et al. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931–947. [DOI] [PubMed] [Google Scholar]
- [133].Liao Y-J, Bai H-Y, Li Z-H, et al. Longikaurin a, a natural ent-kaurane, induces g2/m phase arrest via downregulation of skp2 and apoptosis induction through ros/JNK/c-jun pathway in hepatocellular carcinoma cells. Cell Death Dis. 2014;5(3):e1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Schieber MS, Chandel N. Ros links glucose metabolism to breast cancer stem cell and emt phenotype. Cancer Cell. 2013;23(3):265–267. [DOI] [PubMed] [Google Scholar]
- [135].Piskounova E, Agathocleous M, Murphy MM, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527(7577):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Nikolaou K, Tsagaratou A, Eftychi C, et al. Inactivation of the deubiquitinase cyld in hepatocytes causes apoptosis, inflammation, fibrosis, and cancer. Cancer Cell. 2012;21(6):738–750. [DOI] [PubMed] [Google Scholar]
- [137].Zou P, Zhang J, Xia Y, et al. Ros generation mediates the anti-cancer effects of wz35 via activating jnk and er stress apoptotic pathways in gastric cancer. Oncotarget. 2015;6(8):5860–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Wang L, Zhu Z, Han L, et al. A curcumin derivative, WZ35, suppresses hepatocellular cancer cell growth via downregulating YAP-mediated autophagy. Food Funct. 2019;10(6):3748–3757. [DOI] [PubMed] [Google Scholar]
- [139].Zhou Y, Wang Y, Zhou W, et al. Yap promotes multi-drug resistance and inhibits autophagy-related cell death in hepatocellular carcinoma via the RAC1--mtor pathway. Cancer Cell Int. 2019;19(1):179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Li WJ,Yue F, Dai Y, et al. Suppressor of hepatocellular carcinoma RASSF1a activates autophagy initiation and maturation. Cell Death Differ. 2019;26(8):1379–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]