

ABSTRACT
In the human genome, there are several genes whose primary transcripts are both canonically and non-canonically spliced to generate mRNAs and RNA circles, respectively. These RNA circles are a novel class of long non-coding RNAs that became known as circular RNAs (circRNAs). Recently, a new type of circRNA was discovered and called read-through circRNAs (rt-circRNAs). They are hybrid circles that include coding exons from two adjacent and similarly oriented genes. The function of rt-circRNAs, as well as the impact of read-through transcription in our transcriptome, remains to be elucidated. Although we have just begun to scratch it, here I discussed some insights that these fascinating circRNAs are already giving us about the plasticity of RNA processing in our cells.
KEYWORDS: Non-coding RNAs, circular RNAs, read-through transcription, transcriptome, RNA processing
Introduction
There are a lot of questions about the human genome and its regulation to which we still do not have the answers for. Since the sequencing of the first whole human genome, we have been facing the fact that most of our transcripts do not code for proteins. The big and complex world of the non-coding RNAs (ncRNAs) is not easy to understand and we have just begun to scratch it, even with all remarkable advances in molecular techniques during the past few years. As said by Costa [1]: ‘Sequencing the human genome marked not the end of the field of genomics, but its beginning’.
In our genome, there are thousands of genes whose primary transcripts are both canonically and non-canonically spliced to generate mRNAs and RNA circles, respectively. These RNA circles are a recently described class of long non-coding RNAs that became known as circular RNAs (circRNAs). Interestingly, they are formed when a pre-mRNA is backspliced so its 5ʹ and 3ʹ ends are covalently closed – this process is catalysed by the canonical spliceosomal machinery and modulated by intronic base-pairing and by both cis and trans-acting factors. RNA circularization is more frequent in genes that have exons flanked by long introns harbouring repetitive sequences and reverse complement elements, allowing intronic base-pairing and facilitating the circularization process. This intronic base-pairing may occur naturally or be promoted by some RNA-binding proteins (RBPs) (Fig. 1) [2,3].
Figure 1.
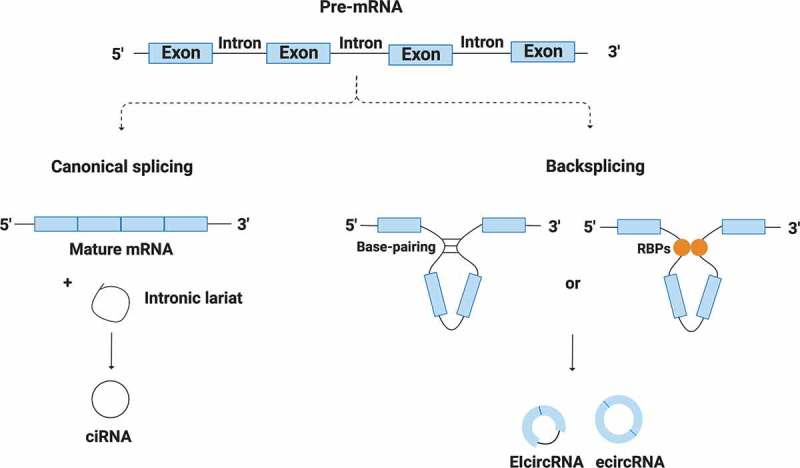
Canonical and non-canonical splicing and their products. A single gene is able to be canonically and non-canonically spliced (backsplicing) to generate mRNAs and regular RNA circles (ecircRNA, ciRNA and EIcircRNA). Circularization process is mediated by base-pairing between flanking introns and by some RBPs
As a consequence of being produced from pre-mRNA, circRNAs can be composed by exon and/or intron sequences, thus classified as exonic (ecircRNAs), intronic (ciRNAs) or exon-intron (EIciRNAs) (Fig. 1). The first class is the most common, representing 85% of all circRNAs [3–5]. According to circBase (http://www.circbase.org), a database that catalogues circRNAs in Homo sapiens and other species, there are over 90,000 circRNAs in humans being produced by more than 11,000 genes.
Besides their high diversity, most circRNAs are less abundant than the associated linear transcripts. However, there are some that can accumulate in the cell and function as microRNAs (miRNAs) or RBPs sponges, as a template for protein production, as transcriptional regulators or as immune factors [5,6]. So far, their most well-described function is the ability of sponging miRNAs – some ecircRNAs harbour miRNAs binding sites along their sequences, allowing them to retain miRNAs that have complementary sequences. When it happens, these miRNAs are prevented from regulating their target genes, leading to changes in gene expression. In fact, several reports have associated the abnormal expression of some miRNA-sponge circRNAs with numerous human diseases, including cancer and neurological diseases [2,7].
More recently, two other classes of circRNAs were described: fusion circRNAs (f-circRNAs), resulted from chromosomal translocations and deletions, and read-through circRNAs (rt-circRNAs), resulted from read-through transcription. Besides both types of circRNAs being formed by exons from different genes, they are not much alike. The first one is a chimaera between distant genes (inter-chromosomal chimaeras), while the latter is a chimaera between neighbouring genes on the same strand (intra-chromosomal chimaeras) [8,9].
Fusion circRNAs are commonly found in cancer cells, but very little is known about the rt-circRNAs – so far, they were reported in only two studies [9,10]. In this article, I will discuss some interesting points about them and will show you how informative these circRNAs are to the complexity of our transcriptome.
First of all, we need to understand what read-through (rt) transcription is. In eukaryotes, transcription usually begins from a transcription start site and ends at a regulated termination point of the same gene. However, in some cases, RNA polymerase starts transcription in the promoter of the 5ʹ gene, continues across the intergenic region and terminates beyond the annotated 3ʹ boundary, invading the adjacent gene. This process is called read-through transcription and results in the formation of hybrid transcripts that include coding exons from two adjacent and similarly oriented genes [11,12] (Fig. 2).
Figure 2.
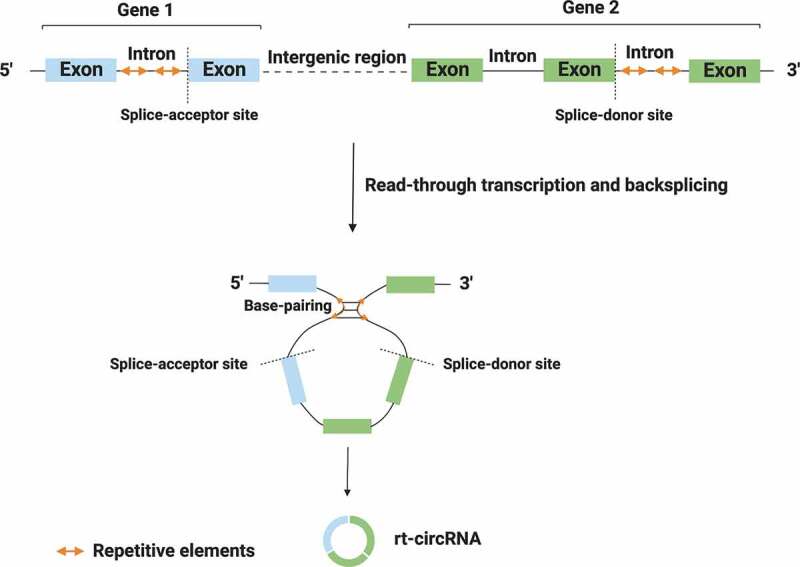
Biogenesis of rt-circRNAs. Read-through transcription results in the formation of hybrid circRNAs (rt-circRNAs) that include coding exons from two adjacent and similarly oriented genes. Circularization process is mediated by base-pairing between long introns that harbour repetitive sequences
Read-through transcription was firstly described producing only linear chimeric transcripts. Apparently, more than 20% of the human protein-coding genes are capable of producing linear read-through transcripts, but their formation mechanism and functions remain poorly studied. Surprisingly, Vo and col. [9] described 1,359 rt-circRNAs produced by our genome (complete list is available for download at: https://mioncocirc.github.io). Based on these 1,359 rt-circRNAs, I concatenated current information on this subject, answering the questions below.
Six questions about rt-circRNAs
Which and how many genes can produce rt-circRNAs?
Rt-circRNAs are formed when an acceptor site at 5ʹ end of an exon binds to a donor site at a downstream 3ʹend of an exon that belongs to the adjacent gene. Overall, there are 280 genes that produce rt-circRNAs as the 3ʹ donor site and 282 genes that serve as 5ʹ acceptor site [9].
Interestingly, there are 65 genes that can produce rt-circRNAs by being both acceptor and donor sites. For example, CYP2C19 produces six rt-circRNAs as 3ʹ donor and five as 5ʹ acceptor – these eleven rt-circRNAs are produced with two partner genes (CYP2C18 and CYP2C9). It means that a gene can produce multiple rt-circRNAs with the same or with different partner genes. Let us look at another example – YWHAE can produce three rt-circRNAs with three different partner genes (CRK, MYO1C and PITPNA), demonstrating the plasticity of our RNA processing mechanisms (Fig. 3) [9].
(2) How many rt-circRNAs can a gene produce?
Figure 3.
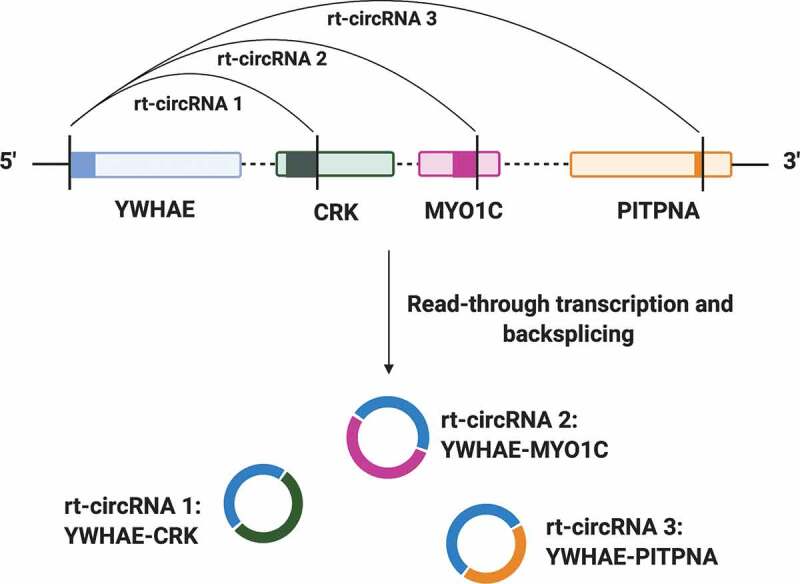
Example of the biogenesis of rt-circRNAs by read-through transcription. YWHAE produces three rt-circRNAs with three different partner genes (CRK, MYO1C and PITPNA), demonstrating the plasticity of RNA processing mechanisms. Solid coloured blocks represent exons. Vertical black lines represent splice sites
Most genes produce only one rt-circRNA. However, as mentioned before, there are some that can produce multiple rt-circRNAs with the same or with different partner genes. Of the 280 genes that produce rt-circRNAs as the 3ʹ donor site, 239 produce different isoforms with the same partner gene – in this case, the gene pair with the highest number of rt-circRNAs isoforms is YY1AP1-GON4L (18 isoforms). Considering the 282 genes that work as 5ʹ donor site, 219 produce different isoforms with the same partner gene, being the pair KLF2-KLF3 the top producer with 14 isoforms. Once again, it demonstrates the plasticity of our RNA processing mechanisms and suggests the existence of an alternative splicing for rt-circRNAs production [9].
(3) Can the same gene pair produce both linear and circular read-through RNAs?
Yes, some gene pairs are capable of producing both linear and circRNA by read-through transcription – the complete list of linear read-through RNAs in humans can be accessed at NCBI Entrez Gene database (https://www.ncbi.nlm.nih.gov/gene/). For instance, AKAP2 and PALM2 produce one linear read-through RNA (chr9: 109,640,788–110,172,512) and two rt-circRNAs (chr9:109,780,487-chr9:110,016,040; 109,942,685–110,016,040) [9]. Please notice that besides being derived from the same gene pair, they do not share the same 5ʹ and 3ʹ boundaries, suggesting that the biogenesis of linear and circular read-through transcripts is independent and presents unique splicing patterns. Further, it also indicates, once again, the existence of an alternative splicing for rt-circRNAs production.
Curiously, there are genes that can produce different isoforms of regular circRNAs (ecircRNAs, ciRNAs and EIcircRNAs) by alternative circular splicing, whereas no isoforms of mRNAs are produced by alternative splicing events [13]. Thus, a similar mechanism of RNA processing may be used for the biogenesis of multiple isoforms of rt-circRNAs, in which novel backsplicing acceptor and donor sites are adopted instead of the canonical ones.
(4) Are the rt-circRNAs tissue-specific?
Among the described 1,359 rt-circRNAs, eleven presented expression restricted to specific tissues (three for prostate and eight for liver). Interestingly, of the 18 rt-circRNAs isoforms produced by KLF2 and KLF3, only two are prostate-specific. As seen in GTEx (https://www.gtexportal.org), these two genes are expressed mostly in prostate rather than in other tissues. On the other hand, CPQ and SDC2, which present low expression in liver according to GTEx, produce a rt-circRNA that is liver-specific [9]. These findings suggest a fine-tune regulation of the rt-circRNAs’ expression across tissues.
(5) Are the rt-circRNAs related to carcinogenesis?
Vo and col. [9] analysed the expression of rt-circRNAs in several types of cancer and also in normal samples. Overall, considering the number of samples in which they were detected, the rt-circRNAs are more frequent in normal samples than in cancer. For example, the most frequent rt-circRNA in normal tissues was found in 474 samples, while the one in cancer was found in 166 samples. In addition, of the 1,359 rt-circRNAs, 817 could not be found in normal tissues.
These observations suggest that rt-circRNAs may have a more homogenous expression among the normal samples, while the rt-circRNAs found in cancer may have a more specific expression. It is also noteworthy mentioning that, of the eleven tissue-specific rt-circRNAs mentioned before, three could not be found in normal tissues, demonstrating that rt-circRNAs may present both tissue and cancer-specific expressions [9].
In order to further investigate their possible relation with carcinogenesis, I checked whether the 460 driver cancer genes described by Dietlein and col. [14] may produce rt-circRNAs. Thirty-nine of these driver genes are able to produce 67 rt-circRNAs, being none tissue-specific and 31 absent in normal tissues. The driver gene that produces the most frequent rt-circRNAs is RB1, with ITM2B as the partner gene – their rt-circRNA could be found in both normal and cancer samples.
(6) So, what do rt-circRNAs do?
So far, no functional studies have been performed to evaluate their biological roles, but let’s speculate on the matter. Liang and col. [10] found that, when canonical transcription termination is slowed or inhibited, production of circRNAs is increased because their biogenesis can occur by read-through transcription. In such cases, backsplicing becomes a preferred splicing outcome, suggesting that the read-through transcription may be a mechanism for gene regulation under specific circumstances.
Few studies showed that read-through transcription overruns and interferes with the expression of downstream genes, and that read-through transcripts repress the expression of their corresponding sense or antisense genes. Altogether, these findings suggest that read-through transcription may be a mechanism to dysregulate (or perhaps regulate) the expression of relevant genes [15,16].
Read-through circRNAs are formed by exons of different genes, suggesting that they might function as templates for translation or as sponges of miRNAs or RBPs. Previous studies showed that linear read-through transcripts may produce bifunctional fusion proteins, which contain domains of both original proteins, or alternative fusion proteins – here, the read-through event causes a frameshift, giving rise to a new protein [17,18]. In this sense, rt-circRNAs may be a mechanism for the evolution of protein complexes, being a novel source for proteins with altered domains and properties [9].
Conclusions
Several factors contributed for the delay in discovering the rt-circRNAs. Read-through transcription is still understudied, and even the linear read-through transcripts are very poorly annotated. However, advances in next-generation sequencing and in computational algorithms have allowed their identification with higher accuracy, demonstrating that they are not merely transcription or technical artefacts. Curiously, these advances have allowed the rediscovery of some fusion linear transcripts as linear read-through transcripts, suggesting that it may be more common than expected.
It is awesome (but also terrifying) to know that our transcriptome is even more complex than we ever thought. The recent findings point to the existence of a fine-tune regulation of rt-circRNAs’ biogenesis and expression, suggesting that our genes may have multiple polyadenylation and backsplicing sites that lead to transcription that extend for varied distances into the coding sequences. This process must be mediated not only by sites along the DNA sequence but also by several transcription anti-termination factors that may allow escaping of the termination signals and invasion of adjacent genes – these factors may be tissue-specific, which could explain global differences of rt-circRNAs levels across different cell types.
As said by Prakash and col. [19], I also believe that, at some point, we will have to rethink our concept of gene. We know our genome is full of overlapping and that a same DNA sequence may work in a multifunctional manner, as exon or intron for different genes, or as an intergenic region for other genes. Overall, the current concept of gene prevents us from going further and deeper in our hypotheses and, because of it, we thought that read-through circRNAs could not exist or be naturally produced. So, if our concept of gene were more flexible, we might have discovered them sooner.
Differently from fusion circRNAs, rt-circRNAs seem to be more broadly expressed across both malignant and benign samples, indicating that they may have well-defined functional roles. If they have survived evolutionary selective pressure and are conserved in different species, then this idea would be strengthened. Further research would help to clarify this possibility.
As well as the regular circRNAs, rt-circRNAs may have regulatory functions by regulating the expression of relevant genes or producing proteins. In this scenario, any disturbance in their expression may have negative effects on cellular environment, which may be associated with several diseases. Vo and col. [9] described some cancer-specific rt-circRNAs, indicating that this association is true. Hence, functional and association studies are needed to further clarify their roles and potentially establish them as biomarkers not only to cancer but also to other complex diseases.
To make it possible and reliable, it is necessary to develop accurate computational algorithms specialized in identifying and annotating rt-circRNAs. Additionally, I believe that annotation must be followed by the creation and establishment of a clear nomenclature system that could allow us to differentiate the regular circRNAs from the read-through and fusion ones.
Overall, the function of rt-circRNAs, as well as the impact of read-through transcription in our transcriptome, remains to be elucidated. Although we have just begun to scratch it, these fascinating circRNAs are already giving us novel insights about the plasticity of RNA processing in our cells. I wonder what comes next.
Acknowledgments
I thank Giovanna Cavalcante, Leandro Magalhães and Ândrea Ribeiro-dos-Santos for helpful conversations. I also thank the Federal University of Pará and the Graduate Program of Genetics and Molecular Biology for the support.
Funding Statement
This work was supported by Rede de Pesquisa em Genômica Populacional Humana (Biocomputacional—Protocol no. 3381/2013/CAPES).
Disclosure statement
No potential conflicts of interest were disclosed.
References
- [1].Costa FF. Epigenomics in cancer management. Cancer Manag Res. 2010;2:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Li X, Yang L, Chen LL.. The biogenesis, functions, and challenges of circular RNAs. Mol Cell. 2018;71(3):428–442. [DOI] [PubMed] [Google Scholar]
- [3].Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338. [DOI] [PubMed] [Google Scholar]
- [4].Vidal AF, Sandoval GT, Magalhães L, et al. Circular RNAs as a new field in gene regulation and their implications in translational research. Epigenomics. 2016;8(4):551–562. [DOI] [PubMed] [Google Scholar]
- [5].Wilusz JE. Circle the wagons: circular RNAs control innate immunity. Cell. 2019;177(4):797–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Xiao M, Ai Y, Biogenesis WJ. Functions of circular RNAs come into focus. Trends Cell Biol. 2020;30(3):226–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Guria A, Sharma P, Natesan S, et al. Circular RNAs—the road less traveled. Front Mol Biosci. 2020;6:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Guarnerio J, Bezzi M, Jeong JC, et al. Oncogenic role of fusion-circRNAs derived from cancer-associated chromosomal translocations. Cell. 2016;165(2):289–302. [DOI] [PubMed] [Google Scholar]
- [9].Vo JN, Cieslik M, Zhang Y, et al. The landscape of circular RNA in cancer. Cell. 2019;176(4):869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liang D, Tatomer DC, Luo Z, et al. The output of protein-coding genes shifts to circular RNAs when the pre-mRNA processing machinery is limiting. Mol Cell. 2017;68(5):940–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Akiva P, Toporik A, Edelheit S, et al. Transcription-mediated gene fusion in the human genome. Genome Res. 2006;16(1):30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Varley KE, Gertz J, Roberts BS, et al. Recurrent read-through fusion transcripts in breast cancer. Breast Cancer Res Treat. 2014;146(2):287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gao Y, Wang J, Zheng Y, et al. Comprehensive identification of internal structure and alter- native splicing events in circular RNAs. Nat Commun. 2016;7(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dietlein F, Weghorn D, Taylor-Weiner A, et al. Identification of cancer driver genes based on nucleotide context. Nat Genet. 2020;52(2):208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Muniz L, Deb MK, Aguirrebengoa M, et al. Control of gene expression in senescence through transcriptional read-through of convergent protein-coding genes. Cell Rep. 2017;21(9):2433–2446. [DOI] [PubMed] [Google Scholar]
- [16].Grosso AR, Leite AP, Carvalho S, et al. Pervasive transcription read-through promotes aberrant expression of oncogenes and RNA chimeras in renal carcinoma. Elife. 2015;4:e09214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Frenkel-Morgenstern M, Lacroix V, Ezkurdia I, et al. Chimeras taking shape: potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 2012;22(7):1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pintarelli G, Dassano A, Cotroneo CE, et al. Read-through transcripts in normal human lung parenchyma are down-regulated in lung adenocarcinoma. Oncotarget. 2016;7(19):27889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Prakash T, Sharma VK, Adati N, et al. Expression of conjoined genes: another mechanism for gene regulation in eukaryotes. PloS One. 2010;5(10). DOI: 10.1371/journal.pone.0013284 [DOI] [PMC free article] [PubMed] [Google Scholar]