

ABSTRACT
Long noncoding RNA GAS5 is down-regulated in cardiomyocytes in diabetic cardiomyopathy (DCM). Here, we studied the involvement of GAS5 in DCM by analyzing its expression in DCM mouse model and cardiac muscle cell line (HL-1 cells). Compared with normal mice, GAS5 was severely down-regulated in heart tissues of DCM mice. GAS5 overexpression improved cardiac function and myocardial hypertrophy in DCM mice. In addition, the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18 were increased in heart tissues of DCM mice and high glucose-treated HL-1 cells, which was repressed by GAS5 up-regulation. GAS5 overexpression suppressed caspase-1 activity, LDH release and the levels of IL-1β, IL-18 in the high glucose-treated HL-1 cells. Moreover, GAS5 regulated AHR expression by sponging miR-34b-3p. Furthermore, GAS5 overexpression suppressed NLRP3 inflammasome activation-mediated pyroptosis by regulating miR-34b-3p/AHR axis. In summary, our study demonstrates that GAS5 acts as a competing endogenous RNA to enhance AHR expression by sponging miR-34b-3p, which consequently represses NLRP3 inflammasome activation-mediated pyroptosis to improve DCM. Thus, our data provide a novel lncRNA GAS5 that could be a valuable target for DCM treatment.
Keywords: GAS5, miR-34b-3p, AHR, NLRP3 inflammasome, pyroptosis, diabetic cardiomyopathy
Introduction
Diabetes mellitus (DM) threatens peoples’ lives and health seriously. With the rising incidence of DM, cardiovascular complications have become the main reason for the high mortality of DM patients [1]. Diabetic cardiomyopathy (DCM) is a cardiovascular complication of DM [2]. DCM causes left ventricular dysfunction or congestive heart failure, which eventually increases the incidence of heart disease [3]. DCM is characterized by reduced contractility, myocardial hypertrophy, apoptosis, and focal myocardial fibrosis [4]. In addition, there are pathological changes in the cardiac blood vessels, such as thickening of the basement membrane of capillaries, and decreasing of micro-hemangiomas and capillary density [5]. Studying the pathogenesis of DCM is of great significance for diagnosing and treating DCM and improving patients’ quality of life.
In the DM state, the increased reactive oxygen species (ROS) stimulated by high glucose (HG) is considered to be an important reason for the occurrence and development of DCM. Excessive ROS causes a boost in the expression of various inflammatory factors, such as nuclear factor κB, thioredoxin-interacting protein and inflammasomes. Previous study has confirmed that NLRP3 inflammasome is associated with DCM [6]. NLRP3 interacts with apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD) (ASC) via pyrin domain (PYD), and then ASC recruits and activates pro-caspase-1 through CARD. This interaction forms a large cytosolic protein complex, NLRP3 inflammasome [7]. The activated NLRP3 inflammasome induces the activation of caspase-1. On the one hand, the activated caspase-1 cleaves gasdermin D (GSDMD) to release active N-terminal protein, which further mediates pyroptosis [8]. On the other hand, the activated caspase-1 recruits and activates inflammatory factors such as IL-1β and IL-18, and induces inflammatory responses [9,10]. Gynostemide improves DCM by inhibiting ROS-induced NLRP3 inflammasome activation [11]. Metformin has protective effect against DCM by inhibiting NLRP3 inflammasome via AMPK/mTOR signaling pathway [12]. NLRP3 silencing inhibits NLRP3 inflammasome activation, and then ameliorates DCM in a type 2 diabetes rat model [13].
Growth arrest-specific transcript 5 (GAS5) is a long non-coding RNA (lncRNA), which participates in the development of cardiovascular disease. GAS5 overexpression attenuates cardiac fibrosis by regulating PTEN/MMP-2 signaling pathway, and improves cardiac function in coronary artery disease by inhibiting Wnt/β-catenin signaling pathway [14,15]. GAS5 knockdown inhibits hypoxia-induced myocardial apoptosis by sponging miR-142-5p [16]. However, the role of GAS5 in DCM has not been reported so far. Pant et al. have performed a microarray analysis of lncRNA profiles in DCM model in vitro, showing that GAS5 is significantly down-regulated in the cardiomyocytes [17]. Prediction analysis has revealed that there are binding sites between GAS5 and miR-34b-3p. MiR-34b-3p inhibits the expression of aryl hydrocarbon receptor (AHR). AHR is a negative regulator of NLRP3 inflammasome. The interaction between AHR and NLRP3 blocks the NF-κB binding sites and then inhibits the transcription of NLRP3, thus inhibiting the activation of NLRP3 inflammasome [18]. Therefore, we speculated that GAS5 promoted the expression of AHR by sponging miR-34b-3p, and inhibited NLRP3 inflammasome activation, pyroptosis and the expression of inflammatory factors (IL-1β and IL-18), and thus improved DCM.
Materials and methods
Animal model and treatment
Adult male C57BL/6 mice weighing 18–20 g were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The mice were housed under standardized conditions. After 4 weeks of high-sucrose/high-fat diet feeding, DCM models were established by intraperitoneal injection of streptozocin (STZ, 100 mg/kg, Cat: S0130, Sigma, St. Louis, MO, USA) in the C57BL/6 mice for 5 d. After 72 h, glucose levels greater than 16.7 mM in the tail vein as detected by a Contour glucose meter (Roche, Basel, Switzerland) were considered evidence of successful establishment of DCM models. The normal diet-fed mice were intraperitoneally injected with sodium citrate buffer with the same dosage served as control. Subsequently, DCM model mice were injected with lentivector-mediated short-hairpin GAS5 (LV-GAS5) or non-targeting plasmids (LV-NC) (RIBOBIO, Guangzhou, China) by cauda vein. All protocols were authorized by the Ethics Committee of The Second Affiliated Hospital of Nanchang University.
Echocardiographic study
The hemodynamic indexes of mice were recorded and analyzed using Ultrasound Biomicroscopy InviVue (NatureGene Corp. New Jersey, USA). The following values were obtained: left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS).
Cell culture and treatment
Cardiac muscle cell line HL-1 was obtained from ATCC (Manassas, VA, USA). HL-1 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Cat: E600008, Sangon Biotech, Shanghai, China) supplemented with 10% fetal bovine serum (FBS, Cat: E600001, Sangon Biotech) and 1% penicillin/streptomycin (Cat: B540732, Sangon Biotech). HL-1 cells were incubated with 30 mM glucose (HG) for 48 h in a humidified atmosphere at 37°C and 5% CO2. HL-1 cells were treated with 5.5 mM glucose (normal glucose, NG) as control.
Cell transfection
Lentivector-mediated short-hairpin GAS5 (LV-GAS5) or non-targeting plasmids (LV-NC) were obtained from RIBOBIO. The cDNA encoding GAS5 was amplified and then subcloned into the vector pcDNA3.1 (Invitrogen, Carlsbad, CA, USA), generating the vector pcDNA3.1-GAS5. The empty pcDNA3.1 vector served as control. The miR-34b-3p mimic, miR-34b-3p inhibitor and the corresponding negative control (NC, mimic NC or inhibitor NC) were synthesized by RIBOBIO. Small interfering RNAs specific targeting GAS5 (si-GAS5), AHR (si-AHR) and its corresponding Scramble were purchased from RIBOBIO. Plasmids were transfected into the cells using Lipofectamine 2000 Transfection Reagent (Cat: 11668019, Invitrogen) following the manufacturer’s protocol. The transfected cells were harvested after 48 to 72 h.
QRT-PCR
QRT-PCR was used to measure the expression intensity of different genes. Total RNA was extracted from heart tissues and cells using TRIzol reagent (Cat: 15596018, Invitrogen). The purity of RNA was detected using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The RNA integrity was determined by agarose gel electrophoresis. Complementary DNA was generated using PrimeScript™ RT Reagent Kit (Cat: RR037A, Takara, Tokyo, Japan). QRT-PCR was carried out using TB Green Fast qPCR Mix (Cat: RR430A, Takara) according to the instruction. The data were analyzed using the 2−∆∆CT (cycle threshold) method for quantification.
Hematoxylin-eosin (HE)staining
Fresh heart tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Five-micron sections were obtained after deparaffin and rehydration. Then, sections were stained using HE staining kit (Cat: G1121, Solarbio, Beijing, China) to observe the changes in heart tissue structure. The stained sections were then observed under the Nikon microscope (Nikon, Tokyo Metropolis, Japan).
Western blot (WB)
Total protein was extracted from heart tissues or cells using Tissue or Cell Total Protein Extraction Kit (Cat: C510003, Sangon Biotech). 1 mL lysis buffers contained 5 μL phosphatase inhibitor, 1 μL protease inhibitor, and 10 μL PMSF. The chopped heart tissues and cells were washed with PBS buffer (Cat: E607008, Sangon Biotech) for several times. Then, the chopped heart tissues were mixed with precooling lysis buffer, and homogenized by ultrasound at 4°C. The tissue homogenate was placed on ice for 10 min. Meanwhile, the cells were incubated with precooling lysis buffer at 4°C for 10 min. After that, the protein was collected by centrifugation. Equivalent protein from different samples was separated by protein electrophoresis, following by transformation onto polyvinylidene fluoride membranes (Merck Millipore, Billerica, MA, USA). The membranes were incubated with the anti-rabbit NLRP3 (1:1000, Cat: 19771-1-AP, Proteintech, Wuhan, China), caspase-1 (1:1000, Cat: 22915-1-AP, Proteintech), IL-1β (1:1000, Cat: 16806-1-AP, Proteintech), IL-18 (1:1000, Cat: 10663-1-AP, Proteintech), AHR (1:1000, Cat: 17840-1-AP, Proteintech) or Pro-caspase-1 (1:1000, Cat: ab179515, Abcam, Cambridge, MA, USA) antibodies at 4°C overnight after immersed into sealed liquid. After washed with Tris Buffered saline Tween for several times, the membranes were incubated with goat anti-mouse IgG antibody (1:5000, Cat: SA00001-1, Proteintech) labeled with horseradish peroxidase. Anti-mouse GAPDH antibody (1:20000, Cat: 1E6D9, Proteintech) was used as a reference protein for normalization. The gray levels of the protein bands were examined by Image J software.
Enzyme-linked immunosorbent assay (ELISA)
The levels of IL-1β and IL-18 in HL-1 cells were assessed using Mouse IL-1β ELISA Kit (Cat: tw040320, TW-reagent, Shanghai, China) and Mouse IL-18 ELISA Kit (Cat: tw040319, TW-reagent). The assay was performed according to the manufacturer’s instructions. The optical density values of samples were detected at 450 nm wavelength using enzyme-labeled instrument (Thermo Fisher Scientific).
Casepare-1 activity detection
Caspase-1 activity in HL-1 cells was estimated using FAM-FLICA Caspase-1 detection kit (Cat: 98, Immunochemistry Technologies, Bloomington, MN, USA). After washed with 1× PBS for several times, HL-1 cells were treated with 10 μL FLICA reagent and 300 μL DMEM containing 1% FBS for 1 h. Then, the cells were washed with 1× PBS for several times after incubation. The fluorescence intensity was measured at Ex492/Em520 nm using SpectraMax Gemini EM Microplate Reader (Molecular Devices, Sunnyvale, CA, USA).
LDH-release assay
LDH-release assay was performed to explore pyroptosis of HL-1 cells using LDH release assay kit (Cat: C0016, Beyotime, Shanghai, China). The supernatant of cells were collected by centrifugation. Then the supernatant (120 μL) was seeded into a 96-well plate and incubated with 60 μL reaction mixture (20 μL lactate, 20 μL INT and 20 μL diaphorase) at room temperature for 30 min. The absorbance was measured at 450 nm wavelength using enzyme-labeled instrument (Thermo Fisher Scientific).
Luciferase reporter assay
GAS5 or AHR containing the predicted miR-34b-3p binding sites were cloned into pGL3-GAS5-Wt (wild-type), pGL3-GAS5-Mut (mutant reporter), pGL3-AHR-Wt or pGL3-AHR-Mut vectors (RIBOBIO), respectively. The Wt (Mut) GAS5 vector or Wt (Mut) 3′ untranslated region (UTR) of AHR vector and miR-34b-3p mimic or mimic NC were co-transfected into 293 cells using Lipofectamine 2000 Transfection Reagent (Invitrogen). The luciferase activity of the cells was detected after 48 hours of transfection using the luciferase assay system (Ambion, Austin, TX, USA).
Statistical analysis
All assays were independently repeated in triplicate. The data were analyzed using SPSS 22.0 statistical software (IBM, Armonk, NY, USA). All data were exhibited as mean ± standard deviation. A two-tailed Student’s t test was used for comparison between two groups. One- or two-way ANOVA was used for comparison among multiple groups. P < 0.05 was considered statistically significant.
Results
GAS5 overexpression improves cardiac function in DCM mice
To identify the potential mechanisms by which GAS5 functioned in DCM, we established the DCM mouse model and investigated the effect of GAS5 overexpression on cardiac function of mice using echocardiography. As shown in Figure 1(a,b), LVEF and LVFS are remarkably decreased in DCM mice with respect to normal mice. GAS5 overexpression led to a boost of LVEF and LVFS in DCM mice (Figure 1(a,b)). Then, we assessed GAS5 expression in the heart tissues of DCM and normal mice, showing that GAS5 was severely down-regulated in DCM mice. GAS5 up-regulation notably enhanced the expression of GAS5 in DCM mice (Figure 1(c)). Subsequently, WB was performed to explore the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18 in heart tissues of mice. DCM mice exhibited a pronounced increase in the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18 as compared with normal mice. The expression of these proteins in DCM mice was notably inhibited by GAS5 up-regulation (Figure 1(d) and Supplementary Figure 1). Moreover, heart tissues were stained with HE to evaluate the condition of myocardial hypertrophy. DCM mice exhibited obvious myocardial hypertrophic response, which was obviously alleviated by GAS5 overexpression (Figure 1(e)). These data imply that GAS5 overexpression improves cardiac function in DCM mice.
Figure 1.
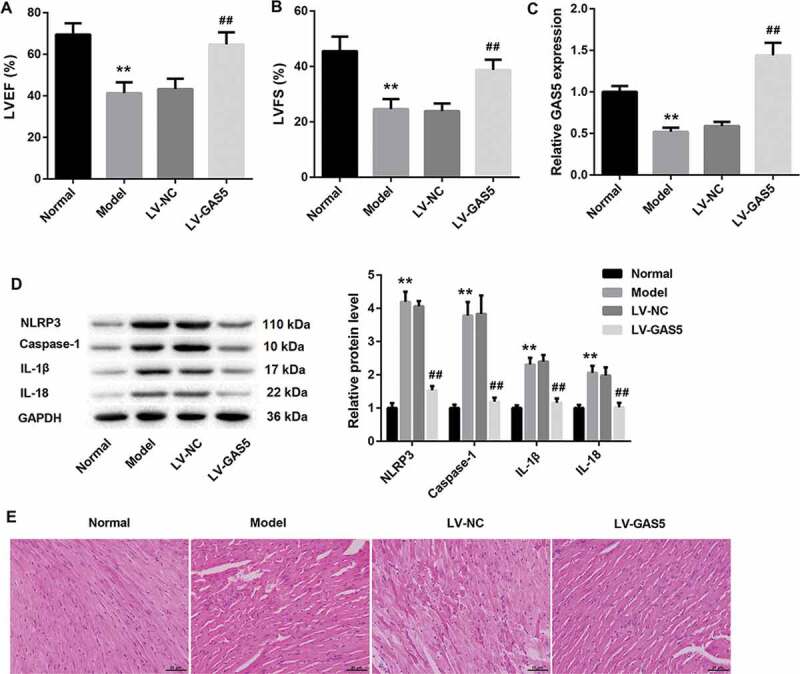
GAS5 overexpression improves cardiac function in DCM mice
DCM mouse model was established by injection of STZ. DCM mice were transfected with LV-GAS5 or LV-NC. Normal mice served as control. (a and b) The hemodynamic indexes (LVEF and LVFS) of mice were recorded and analyzed by echocardiography. (c) The expression of GAS5 in heart tissues was detected by qRT-PCR. (d) WB was performed to explore the expression of active forms of NLRP3, caspase-1, IL-1β and IL-18 in heart tissues. (e) HE staining was performed to investigate the levels of myocardial injury. (**P < 0.01, versus Normal; ##P < 0.01, versus LV-NC.)
GAS5 overexpression inhibits NLRP3 inflammasome activation-mediated pyroptosis in vitro
Next, we explore the biological function of GAS5 in HL-1 cells in vitro. HL-1 cells were transfected with LV-GAS5 to induce GAS5 overexpression, and then the modified HL-1 cells were exposed to HG. We first detected the expression of GAS5 in the HL-1 cells. The expression of GAS5 was lower in HG-induced HL-1 cells than that in the control HL-1 cells. GAS5 expression was notably increased in the HL-1 cells after transfected with LV-GAS5 (Figure 2(a)). Then, the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18 in the HL-1 cells were assessed by WB. HG-induced HL-1 cells displayed an obvious increase in the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18 with respect to the control HL-1 cells. However, GAS5 overexpression caused a pronounced decrease in the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18 in the HL-1 cells (Figure 2(b) and Supplementary Figure 1). Furthermore, we estimated the caspase-1 activity. The caspase-1 activity was enhanced in the HG-induced HL-1 cells. GAS5 overexpression significantly reduced caspase-1 activity in the HL-1 cells (Figure 2(c)). Subsequently, the levels of IL-1β and IL-18 in the HL-1 cells were examined by ELISA. As shown in Figure 2(d), the levels of IL-1β and IL-18 in the HG-induced HL-1 cells are increased as compared with the control HL-1 cells. The levels of IL-1β and IL-18 were repressed by GAS5 up-regulation in the HL-1 cells (Figure 2(d)). What’s more, the LDH release of the HL-1 cells was estimated by LDH release assay. The LDH release was enhanced in the HG-induced HL-1 cells, whereas GAS5 overexpression suppressed LDH release in the HL-1 cells (Figure 2(e)). Taken together, these data provide strong evidence that GAS5 overexpression inhibits NLRP3 inflammasome activation-mediated pyroptosis in vitro.
Figure 2.
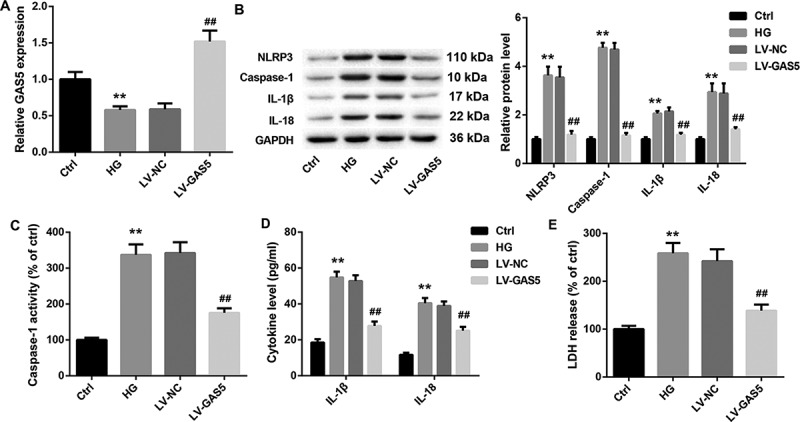
GAS5 overexpression inhibits NLRP3 inflammasome activation-mediated pyroptosis in vitro.
HL-1 cells were transfected with LV-GAS5 or LV-NC, and then the modified HL-1 cells were incubated with NG or HG. (a) The expression of GAS5 in the HL-1 cells was detected by qRT-PCR. (b) WB was performed to explore the expression of active forms of NLRP3, caspase-1, IL-1β and IL-18 in the HL-1 cells. (c) The caspase-1 activity in the HL-1 cells was estimated. (d) ELISA was performed to explore the levels of IL-1β and IL-18 in the HL-1 cells. (e) LDH release assay was performed to estimate the pyroptosis of the HL-1 cells. (**P < 0.01, versus Ctrl; ##P < 0.01, versus LV-NC.)
GAS5 overexpression represses miR-34b-3p expression and promotes AHR expression in vivo and vitro
To verify the relationship among GAS5, miR-34b-3p, and AHR, we detected the gene and protein expression of miR-34b-3p and AHR in heart tissues of DCM mice and HL-1 cells. Compared with normal mice, the gene expression of miR-34b-3p was increased and the gene and protein expression of AHR was decreased in heart tissues of DCM mice. GAS5 overexpression notably repressed miR-34b-3p expression and promoted the gene and protein expression of AHR in heart tissues of DCM mice (Figure 3(a,b)). At the same time, the gene expression of miR-34b-3p was higher in HG-induced HL-1 cells than that in the control HL-1 cells. The gene and protein expression of AHR was severely down-regulated in the HG-induced HL-1 cells. GAS5 overexpression notably repressed the expression of miR-34b-3p, while GAS5 up-regulation obviously enhanced the gene and protein expression of AHR in the HL-1 cells (Figure 3(c,d)). Taken together, these data indicate that GAS5 represses miR-34b-3p expression and promotes AHR expression in vivo and vitro.
Figure 3.
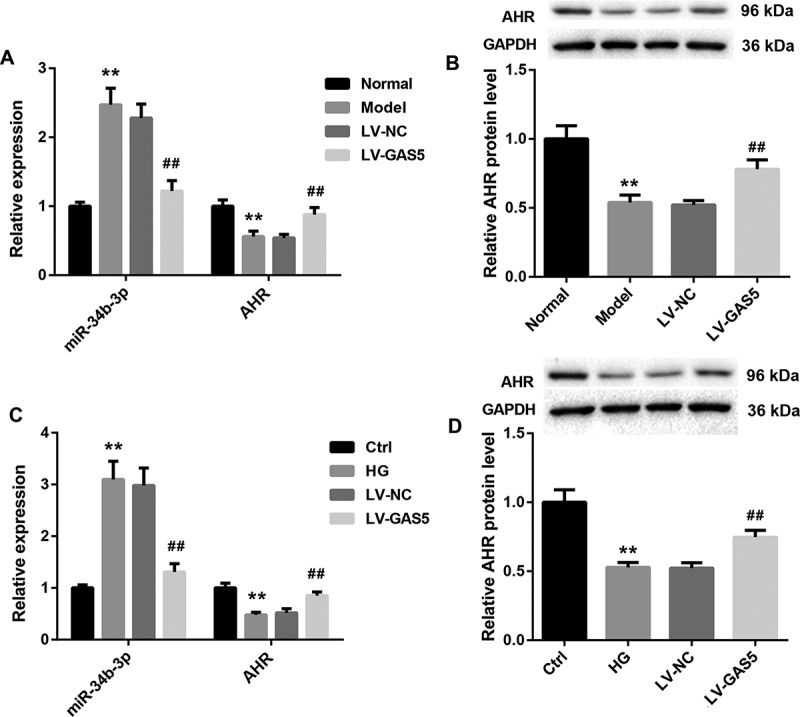
GAS5 represses miR-34b-3p expression and promotes AHR expression in vivo and vitro.
DCM mouse model was established by injection of STZ. DCM mice were transfected with LV-GAS5 or LV-NC. Normal mice served as control. (a and b) QRT-PCR and WB were performed to assess the gene or protein expression of miR-34b-3p and AHR in the heart tissues of mice. HL-1 cells were transfected with LV-GAS5 or LV-NC, and then the modified HL-1 cells were incubated with NG or HG. (c and d) QRT-PCR and WB were performed to investigate the gene or protein expression of miR-34b-3p and AHR in the HL-1 cells. (**P < 0.01, versus Normal or Ctrl; ##P < 0.01, versus LV-NC.)
GAS5 regulates AHR expression by sponging miR-34b-3p
The ability of GAS5 to regulate miR-143-3p and TGF-β1 expression is suggestive of competing endogenous RNA (ceRNA) role. To verify this hypothesis, luciferase reporter assay was performed, showing that GAS5 interacted with miR-34b-3p (Figure 4(a)). Then, HL-1 cells were transfected with pcDNA3.1-GAS5 or si-GAS5, and we measured the expression of GAS5 and miR-34b-3p in the HL-1 cells by qRT-PCR. GAS5 overexpression caused an increase of GAS5 expression in HL-1 cells, whereas GAS5 silencing obviously repressed GAS5 expression in HL-1 cells. MiR-34b-3p expression was severely down-regulated in HL-1 cells after transfected with pcDNA3.1-GAS5. GAS5 knockdown obviously enhanced the expression of miR-34b-3p in HL-1 cells (Figure 4(b)). Moreover, the relationship between miR-34b-3p and AHR was verified by luciferase reporter assay, showing that AHR was a target gene of miR-34b-3p (Figure 4(c)). Furthermore, we assessed the gene or protein expression of miR-34b-3p and AHR in HL-1 cells after transfected with miR-34b-3p mimic or miR-34b-3p inhibitor. MiR-34b-3p was highly expressed in the HL-1 cells after transfected with miR-34b-3p mimic. MiR-34b-3p knockdown repressed miR-34b-3p expression in the HL-1 cells (Figure 4(d)). MiR-34b-3p up-regulation led to a decrease in the gene and protein expression of AHR in HL-1 cells. However, miR-34b-3p down-regulation significantly promoted the gene and protein expression of AHR in HL-1 cells (Figure 4(d,e)). Therefore, these results suggest that GAS5 functions as a ceRNA for miR-34b-3p, thereby leading to the up-regulation of the activity of its endogenous target, AHR.
Figure 4.
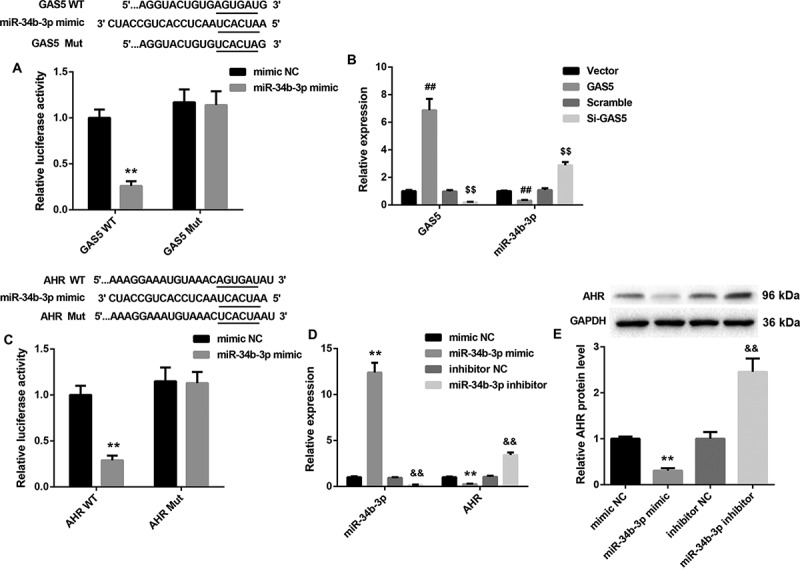
GAS5 regulates AHR expression by sponging miR-34b-3p
(a) The Wt (Mut) GAS5 vector and miR-34b-3p mimic or mimic NC were co-transfected into 293 cells. The relationship between GAS5 and miR-34b-3p was measured by luciferase reporter assay. HL-1 cells were transfected with pcDNA3.1-GAS5, si-GAS5 or its NC. (b) QRT-PCR was performed to assess the expression of GAS5 and miR-34b-3p in the HL-1 cells. (c) The Wt (Mut) 3′ UTR of AHR vector and miR-34b-3p mimic or mimic NC were co-transfected into 293 cells. The relationship between AHR and miR-34b-3p was measured by luciferase reporter assay. HL-1 cells were transfected with miR-34b-3p mimic, miR-34b-3p inhibitor or its NC. (d and e) QRT-PCR and WB were performed to investigate the gene or protein expression of miR-34b-3p and AHR in the HL-1 cells. (**P < 0.01, versus mimic NC; ##P < 0.01, versus Vector; $$P < 0.01, versus Scramble; &&P < 0.01, versus inhibitor NC.)
GAS5 overexpression inhibits NLRP3 inflammasome activation-mediated pyroptosis by inhibiting miR-34b-3p expression
We wondered if GAS5/miR-34b-3p/AHR axis could be participated in NLRP3 inflammasome activation-mediated pyroptosis in HL-1 cells. The pcDNA3.1-GAS5 or empty pcDNA3.1 vector and miR-34b-3p mimic or mimic NC were co-transfected into HL-1 cells. The expression of miR-34b-3p was inhibited by GAS5 overexpression and enhanced by miR-34b-3p up-regulation in HL-1 cells. The promoting effect of miR-34b-3p overexpression on miR-34b-3p expression was repressed by GAS5 overexpression (Figure 5(a)). Meanwhile, GAS5 overexpression caused a boost in the gene and protein expression of AHR in HL-1 cells. The gene and protein expression of AHR were severely repressed by miR-34b-3p overexpression in HL-1 cells. The influence conferred by miR-34b-3p overexpression was abolished by GAS5 overexpression (Figure 5(a,b)). Then, WB was performed to assess the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18 in the HL-1 cells. GAS5 overexpression reduced the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18 in HL-1 cells. MiR-34b-3p up-regulation enhanced the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18 in the HL-1 cells, which was abolished by GAS5 overexpression (Figure 5(c) and Supplementary Figure 1). Subsequently, we estimated caspase-1 activity and the levels of IL-1β and IL-18 in the HL-1 cells. We found that the caspase-1 activity and the levels of IL-1β and IL-18 were repressed by GAS5 overexpression in the HL-1 cells. MiR-34b-3p up-regulation notably enhanced the caspase-1 activity and the levels of IL-1β and IL-18 in the HL-1 cells, which was effectively abolished by GAS5 overexpression (Figure 5(d,e)). In addition, LDH release of HL-1 cells was estimated by LDH release assay. GAS5 overexpression inhibited LDH release, whereas miR-34b-3p overexpression promoted LDH release in the HL-1 cells. The promoting effect of miR-34b-3p overexpression on LDH release was rescued by GAS5 overexpression (Figure 5(f)). Thus, these data demonstrate that GAS5 overexpression represses NLRP3 inflammasome activation-mediated pyroptosis by inhibiting miR-34b-3p expression.
Figure 5.
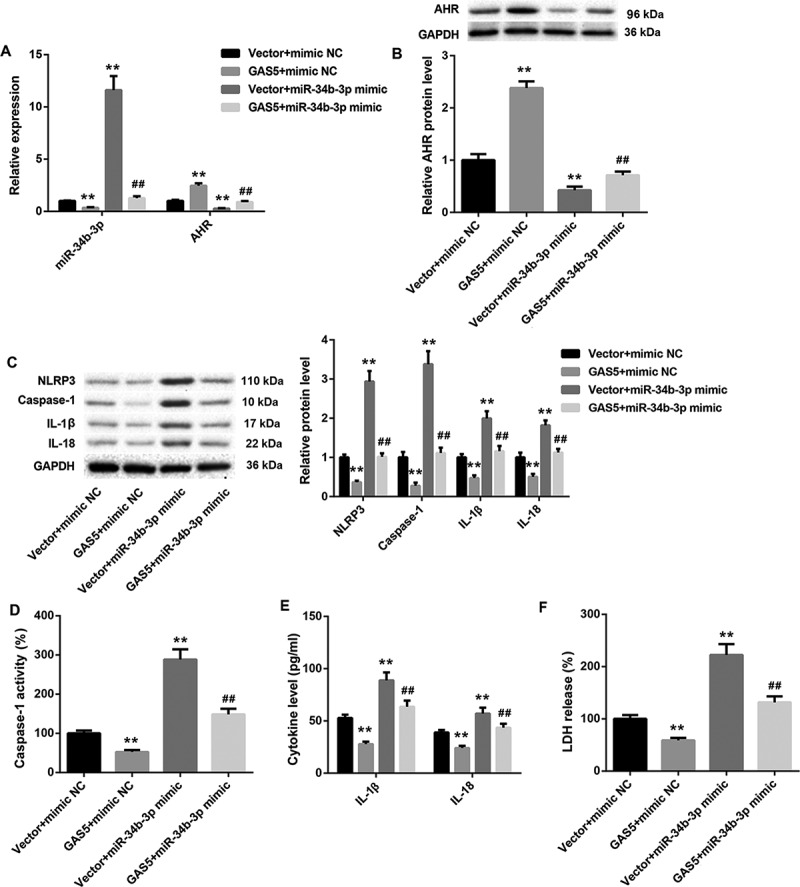
GAS5 overexpression inhibits NLRP3 inflammasome activation-mediated pyroptosis by inhibiting miR-34b-3p expression
The pcDNA3.1-GAS5 or empty pcDNA3.1 vector and miR-34b-3p mimic or mimic NC were co-transfected into HL-1 cells. (a and b) QRT-PCR and WB were performed to investigate the gene or protein expression of miR-34b-3p and AHR in the HL-1 cells. (c) WB was performed to explore the expression of active forms of NLRP3, caspase-1, IL-1β and IL-18 in the HL-1 cells. (d) The caspase-1 activity in the HL-1 cells was estimated. (e) ELISA was performed to explore the levels of IL-1β and IL-18 in the HL-1 cells. (f) LDH release assay was performed to estimate the pyroptosis of the HL-1 cells. (**P < 0.01, versus Vector+mimic NC; ##P < 0.01, versus Vector+miR-34b-3p mimic.)
GAS5 overexpression inhibits NLRP3 inflammasome activation-mediated pyroptosis by regulating miR-34b-3p/AHR axis
In order to further explore the molecular mechanism of GAS5/miR-34b-3p/AHR axis in DCM, pcDNA3.1-GAS5 or empty pcDNA3.1 vector and si-AHR or Scramble were co-transfected into HL-1 cells. As shown in Figure 6(a,b), GAS5 overexpression obviously enhances the gene and protein expression of AHR in HL-1 cells. AHR-silenced HL-1 cells displayed a pronounced decrease in the gene and protein expression of AHR. The inhibiting effect of AHR silencing on AHR expression was rescued by GAS5 overexpression (Figure 6(a,b)). Furthermore, the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β, and IL-18 was suppressed by GAS5 up-regulation and facilitated by AHR silencing in the HL-1 cells. The influence conferred by AHR silencing was abolished by GAS5 up-regulation (Figure 6(c) and Supplementary Figure 1). In addition, we estimated caspase-1 activity and the levels of IL-1β and IL-18 in the HL-1 cells. We found that GAS5 overexpression significantly suppressed the caspase-1 activity and the levels of IL-1β and IL-18 in the HL-1 cells. AHR silencing notably enhanced the caspase-1 activity and the levels of IL-1β and IL-1 in the HL-1 cells, which was effectively abolished by GAS5 overexpression (Figure 6(d,e)). Moreover, we estimated LDH release of the HL-1 cells by LDH release assay. GAS5 up-regulation notably repressed the LDH release in HL-1 cells, whereas LDH release in HL-1 cells was notably enhanced by AHR deficiency. The influence conferred by AHR silencing was abolished by GAS5 overexpression (Figure 6(f)). These data taken together demystify that GAS5 overexpression represses NLRP3 inflammasome activation-mediated pyroptosis by regulating miR-34b-3p/AHR axis.
Figure 6.
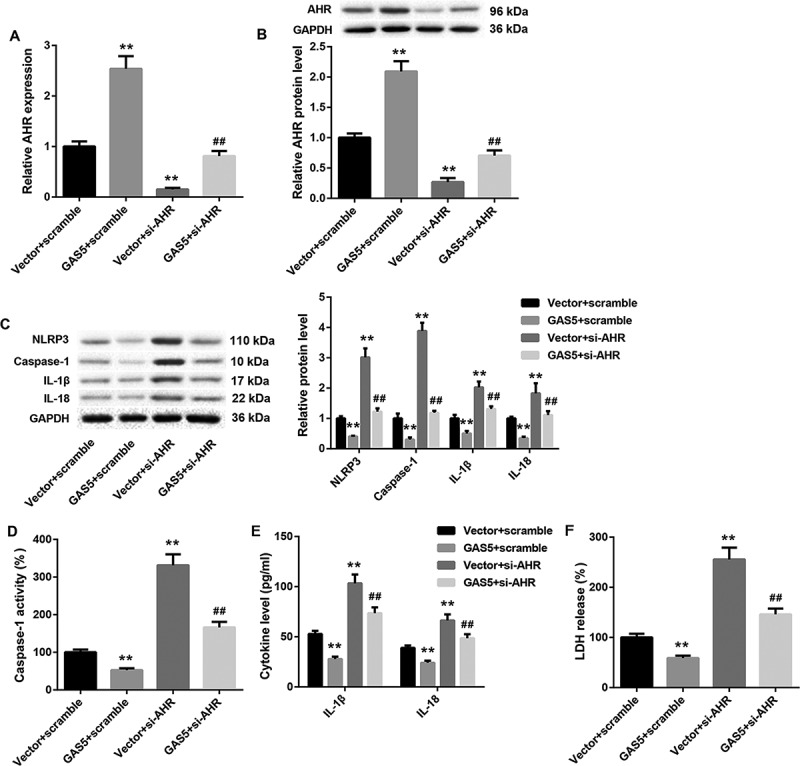
GAS5 overexpression inhibits NLRP3 inflammasome activation-mediated pyroptosis by regulating miR-34b-3p/AHR axis
The pcDNA3.1-GAS5 or empty pcDNA3.1 vector and si-AHR or Scramble were co-transfected into HL-1 cells. (a and b) QRT-PCR and WB were performed to investigate the gene or protein expression of AHR in the HL-1 cells. (c) WB was performed to explore the expression of active forms of NLRP3, caspase-1, IL-1β and IL-18 in the HL-1 cells. (d) The caspase-1 activity in the HL-1 cells was estimated. (e) ELISA was performed to explore the levels of IL-1β and IL-18 in the HL-1 cells. (f) LDH release assay was performed to estimate the pyroptosis of the HL-1 cells. (**P < 0.01, versus Vector+Scramble; ##P < 0.01, versus Vector+si-AHR.)
Discussion
Many studies have confirmed that lncRNA GAS5 is associated with various diseases. Previous study has reported that GAS5 participates in tumor-related inflammatory reaction, and GAS5 improves colorectal cancer by repressing the expression of IL-10 and vascular endothelial growth factor [19]. GAS5 is down-regulated in ovarian cancer tissues, and GAS5 overexpression represses ovarian cancer by inducing inflammasome formation and pyroptosis [20]. In addition, GAS5 is closely associated with type 2 DM and GAS5 is decreased in serum of DM patients [21]. However, the role of GAS5 in DCM has not been reported. In this study, we constructed DCM mouse model to investigate the biological role of GAS5 in DCM. Our data showed that the expression of GAS5 was significantly decreased in DCM mice. GAS5 overexpression improved cardiac function and myocardial hypertrophy in DCM mice. In addition, the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β, and IL-18 in DCM mice and HL-1 cells was inhibited by GAS5 up-regulation. Furthermore, caspase-1 activity, the levels of IL-1β and IL-18, and LDH release in the HL-1 cells were repressed by GAS5 up-regulation. Pyroptosis is an inflammation-related programmed cell death that plays a critical role in the development of DCM. Activated NLRP3 inflammasome induces pyroptosis and inflammatory reaction by activating caspase-1 [6]. Thus, our data demonstrate that GAS5 overexpression attenuates DCM by suppressing NLRP3 inflammasome activation-mediated pyroptosis in vivo and in vitro.
Existing researches have recognized the critical role played by miR-34b-3p in various cancers. Surveys such as that conducted by Feng et al. show that miR-34b-3p is down-regulated in both non-small-cell lung cancer tissues and lung cancer cell lines. MiR-34b-3p has a protective effect against non-small-cell lung cancer by inhibiting cell proliferation via regulating CDK4 expression [22]. MiR-34b-3p, as a tumor suppressor, is down-regulated in small cell lung cancer. The ectopic expression of miR-34b-3p obviously suppresses the aggressiveness of cancer cells [23]. MiR-34b-3p also has inhibiting effect on cell proliferation and migration in cervical cancer cells [24]. Our data first found that miR-34b-3p was associated with DCM. MiR-34b-3p was up-regulated and AHR was down-regulated in DCM mice and HL-1 cells. GAS5 overexpression inhibited miR-34b-3p expression and promoted AHR expression. GAS5 interacted with miR-34b-3p, and AHR was a target gene of miR-34b-3p. Therefore, theses data imply that GAS5 functions as a ceRNA for miR-34b-3p, thereby leading to the up-regulation of the activity of its endogenous target, AHR.
In the present study, we found that AHR silencing repressed the expression of AHR, whereas GAS5 overexpression enhanced AHR expression. The inhibiting effect of AHR knockdown on AHR expression was rescued by GAS5 overexpression. Furthermore, miR-34b-3p overexpression and AHR knockdown enhanced the expression of NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18, caspase-1 activity and LDH release HL-1 cells. The influence of miR-34b-3p overexpression or AHR knockdown on NLRP3 inflammasome activation-mediated pyroptosis was abolished by GAS5 overexpression in HL-1 cells. Therefore, our data elaborate that GAS5 overexpression suppresses NLRP3 inflammasome activation-mediated pyroptosis by regulating miR-34b-3p/AHR axis in HL-1 cells. Thus, the ceRNA regulatory network, GAS5/miR-34b-3p/AHR, might be an important molecular mechanism involved in the progression of DCM.
Not only in DCM, GAS5 is also associated with diabetic nephropathy. GAS5 up-regulation represses the inflammation, oxidative stress and pyroptosis of HG-induced renal tubular cells by sponging miR-452-5p [25]. GAS5 knockdown alleviates HG toxicity to HK-2 cells by regulating miR-27a expression [26]. We speculate that the difference in GAS5 expression may be caused by different concentration of HG treatment. These two studies show that miR-452-5p and miR-27a are target genes of GAS5. Our study found that there are binding sites between GAS5 miR-34b-3p. Maybe GAS5 can also regulate miR-452-5p and miR-27a to participate in the development of DCM. However, whether miR-452-5p can inhibit the expression of AHR in HL-1 cells needs further study. Zhao et al have found that GAS5 is down-regulated in HG-induced human cardiomyocyte-like AC16 cells, and GAS5 silencing alleviates inflammation by inhibiting miR-21-5p-mediated TLR4/NF-κB signaling pathway in DCM [27]. However, our data found that GAS5 was up-regulated in HG-induced HL-1 cells. It implies that GAS5 may be a cell-specific gene, which is expressed differently in different cells. GAS5 may participate in DCM by regulating inflammation.
NLRP3 inflammasome is associated with various diseases. IL-22 can repress the activation of NLRP3 inflammasome activation and the release of mature IL-1β, thereby ameliorating the acetaminophen-induced kidney injury [28]. NLRP3 inflammasome activation induces pyroptosis of cardiomyocyte, which contributes to myocardial dysfunction and the pathogenesis of non-ischemic dilated cardiomyopathy [29]. NLRP3 inflammasome activation participates in the pathogenesis of post-cardiac arrest brain injury by inducing microglial pyroptosis and inflammatory infiltration [30]. GAS5/miR-34b-3p/AHR axis may also inhibit the occurrence and development of these diseases by regulating NLRP3 inflammasome activation-mediated pyroptosis, which requires our further research.
In summary, our study demonstrates that lncRNA GAS5 is down-regulated in the heart tissues of DCM mice and may act as a ceRNA to enhance AHR expression by sponging miR-34b-3p, which consequently represses NLRP3 inflammasome activation-mediated pyroptosis to improve DCM. Our data provide a novel lncRNA GAS5 that could be a valuable target for DCM treatment.
Supplementary Material
Disclosure statement
The authors declare no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Hayat SP, Khattar R.. Diabetic cardiomyopathy: mechanisms, diagnosis and treatment. Clin Sci. 2004;107:539–557. [DOI] [PubMed] [Google Scholar]
- [2].Gulsin GS, Athithan L, McCann GP.. Diabetic cardiomyopathy: prevalence, determinants and potential treatments. Ther Adv Endocrinol Metab. 2019;10:2042018819834869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Galderisi M. Diastolic dysfunction and diabetic cardiomyopathy: evaluation by Doppler echocardiography. J Am Coll Cardiol. 2006;48:1548–1551. [DOI] [PubMed] [Google Scholar]
- [4].Cosson SKJP. Left ventricular diastolic dysfunction: an early sign of diabetic cardiomyopathy? Diabetes Metab. 2003;29:455–466. [DOI] [PubMed] [Google Scholar]
- [5].Okruhlicova L, Weismann P. Ultrastructure and histochemistry of rat myocardial capillary endothelial cells in response to diabetes and hypertension. Cell Res. 2005;15:532–538. [DOI] [PubMed] [Google Scholar]
- [6].Luo B, Huang F, Liu Y, et al. NLRP3 Inflammasome as a molecular marker in diabetic cardiomyopathy. Front Physiol. 2017;8:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. [DOI] [PubMed] [Google Scholar]
- [8].Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. [DOI] [PubMed] [Google Scholar]
- [9].He WT, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kayagaki N, Stowe I, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. [DOI] [PubMed] [Google Scholar]
- [11].Zhang H, Chen X, Zong B, et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J Cell Mol Med. 2018;22:4437–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang F, Qin Y, Wang Y, et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int J Biol Sci. 2019;15:1010–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Luo B, Li B, Wang W, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PloS One. 2014;9:e104771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu HL, Chen CH, Sun YJ. Overexpression of lncRNA GAS5 attenuates cardiac fibrosis through regulating PTEN/MMP-2 signal pathway in mice. Eur Rev Med Pharmacol Sci. 2019;23:4414–4418. [DOI] [PubMed] [Google Scholar]
- [15].Li X, Hou L, Cheng Z, et al. Overexpression of GAS5 inhibits abnormal activation of Wnt/beta-catenin signaling pathway in myocardial tissues of rats with coronary artery disease. J Cell Physiol. 2019;234:11348–11359. [DOI] [PubMed] [Google Scholar]
- [16].Du J, Yang ST, Liu J, et al. Silence of LncRNA GAS5 Protects Cardiomyocytes H9c2 against Hypoxic Injury via Sponging miR-142-5p. Mol Cells. 2019;42:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pant T, Mishra MK, Bai X, et al. Microarray analysis of long non-coding RNA and mRNA expression profiles in diabetic cardiomyopathy using human induced pluripotent stem cell-derived cardiomyocytes. Diab Vasc Dis Res. 2019;16:57–68. [DOI] [PubMed] [Google Scholar]
- [18].Huai W, Zhao R, Song H, et al. Aryl hydrocarbon receptor negatively regulates NLRP3 inflammasome activity by inhibiting NLRP3 transcription. Nat Commun. 2014;5:4738. [DOI] [PubMed] [Google Scholar]
- [19].Li Y, Li Y, Huang S. Long non-coding RNA growth arrest specific transcript 5 acts as a tumour suppressor in colorectal cancer by inhibiting interleukin-10 and vascular endothelial growth factor expression. Oncotarget. 2017;8:13690–13702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li J, Yang C, Li Y, et al. LncRNA GAS5 suppresses ovarian cancer by inducing inflammasome formation. Biosci Rep. 2017;38: BSR20171150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Carter G, Miladinovic B, Patel AA, et al. Circulating long noncoding RNA GAS5 levels are correlated to prevalence of type 2 diabetes mellitus. BBA Clin. 2015;4:102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Feng H, Ge F, Du L, et al. MiR-34b-3p represses cell proliferation, cell cycle progression and cell apoptosis in non-small-cell lung cancer (NSCLC) by targeting CDK4. J Cell Mol Med. 2019;23:5282–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mizuno K, Mataki H, Arai T, et al. The microRNA expression signature of small cell lung cancer: tumor suppressors of miR-27a-5p and miR-34b-3p and their targeted oncogenes. J Hum Genet. 2017;62:671–678. [DOI] [PubMed] [Google Scholar]
- [24].Cordova-Rivas S, Fraire-Soto I, Mercado-Casas Torres A, et al. 5p and 3p strands of miR-34 family members have differential effects in cell proliferation, migration, and invasion in cervical cancer cells. Int J Mol Sci. 2019;20:545.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xie C, Wu W, Tang A, et al. lncRNA GAS5/miR-452-5p reduces oxidative stress and pyroptosis of high-glucose-stimulated renal tubular cells. Diabetes metabolic syndrome obesity. 2019;12:2609–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lv L, Li D, Tian F, et al. Silence of lncRNA GAS5 alleviates high glucose toxicity to human renal tubular epithelial HK-2 cells through regulation of miR-27a. Artif Cells Nanomed Biotechnol. 2019;47:2205–2212. [DOI] [PubMed] [Google Scholar]
- [27].Zhao JL, Li C. Knockdown of long noncoding RNA GAS5 protects human cardiomyocyte-like AC16 cells against high glucose-induced inflammation by inhibiting miR-21-5p-mediated TLR4/NF-κB signaling. Naunyn-Schmiedeberg’s Arch Pharmacol. 2019;393:1541-1547. [DOI] [PubMed] [Google Scholar]
- [28].Shen Y, Jin X, Chen W, et al. Interleukin-22 ameliorated acetaminophen-induced kidney injury by inhibiting mitochondrial dysfunction and inflammatory responses. Appl Microbiol Biotechnol. 2020;104:5889–5898. [DOI] [PubMed] [Google Scholar]
- [29].Zeng C, Duan F, Hu J, et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020;34:101523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chang Y, Zhu J, Wang D, et al. NLRP3 inflammasome-mediated microglial pyroptosis is critically involved in the development of post-cardiac arrest brain injury. J Neuroinflammation. 2020;17:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.