

ABSTRACT
Cancer cells overexpress CD47 to subvert phagocytic elimination and evade immunogenic processing of cancer antigens. Moreover, CD47 overexpression inhibits the antibody-dependent cellular phagocytosis (ADCP) and cytotoxicity (ADCC) activities of therapeutic anticancer antibodies. Consequently, CD47-blocking antibodies have been developed to overcome the immunoevasive activities of cancer cell-expressed CD47. However, the wide-spread expression of CD47 on normal cells forms a massive “antigen sink” that potentially limits sufficient tumor accretion of these antibodies. Additionally, a generalized blockade of CD47-SIRPα interaction may ultimately lead to unintended cross-presentation of self-antigens potentially promoting autoimmunity. To address these issues, we constructed a bispecific antibody, designated bsAb CD47xEGFR-IgG1, that blocks cancer cell surface-expressed CD47 in an EGFR-directed manner. BsAb CD47xEGFR-IgG1 selectively induced phagocytic removal of EGFRpos/CD47pos cancer cells and endowed neutrophils with capacity to kill these cancer cells by trogoptosis; an alternate form of ADCC that disrupts the target cell membrane. Importantly, bsAb CD47xEGFR-IgG1 selectively enhanced phagocytosis and immunogenic processing of EGFRpos/CD47pos cancers cells ectopically expressing viral protein CMVpp65.
In conclusion, bsAb CD47xEGFR-IgG1 may be useful to reduce on-target/off-tumor effects of CD47-blocking approaches, enhance cancer cell elimination by trogoptosis, and promote adaptive anticancer immune responses.
KEYWORDS: Bispecific antibody, CD47-blockade, EGFR, ADCP, trogoptosis, cross-presentation, cancer immunotherapy
Introduction
CD47 is a multifunctional pentaspanin transmembrane glycoprotein that is expressed on virtually all normal cell types. One of its most prominent functions involves interaction with signal regulatory protein alpha (SIRPα), a cell surface glycoprotein expressed by various types of phagocytes (reviewed in1). CD47-SIRPα interaction leads to phosphorylation of the ITIM motif of SIRPα which subsequently initiates a signal transduction cascade that result in the inhibition of the phagocytic activity of, e.g. macrophages and dendritic cells (DCs).2 In this respect, CD47-SIRPα interaction has been hailed as a “Don’t eat me” immune checkpoint that serves to prevent the untimely phagocytic removal of normal healthy cells. Unfortunately, a broad variety of hematologic and solid malignancies appear to misuse the CD47-SIRPα immune checkpoint by overexpressing CD47, thereby evading phagocytic elimination and subsequent immunogenic processing of neoantigens.3–5 Moreover, CD47 overexpression by cancer cells was shown to reduce the efficacy of therapeutic anticancer antibodies by inhibiting their capacity to induce antibody-dependent cellular cytotoxicity (ADCC) and antibody dependent cellular phagocytosis (ADCP).6
Previously, it was demonstrated that CD47-blocking antibodies can be exploited to restore phagocytic elimination of CD47-overexpressing cancer cells and subsequent promote adaptive immunity toward these cancer cells.5,7 Moreover, CD47-blocking antibodies enhanced ADCP and ADCC-mediated anticancer activity of rituximab and trastuzumab.6,8,9 Currently, several CD47-blocking antibodies are being evaluated in clinical trials, alone or in combination with therapeutic anticancer antibodies (ClinicalTrials.gov identifier: NCT02953509, NCT02953782, and NCT03558139).
However, the clinical efficacy of current CD47-blocking antibodies is anticipated to be hampered by the wide-spread expression of CD47 on normal cells which may limit sufficient antibody accretion at the tumor site(s).10–13 Moreover, the lack of cancer selectivity of current monospecific CD47-blocking antibodies may result in a generalized blockade of CD47 present on normal cells which in turn may promote cross-presentation of self-antigens, thereby increasing the risk of breaking self-tolerance and inducing unpredictable immune-related adverse events.
In this respect, a bispecific antibody-based approach may be a suitable strategy to block the CD47-SIRPα immune checkpoint in a more tumor-restricted manner. Bispecific antibodies (bsAbs) can be designed to combine two independent target functionalities into one antibody-based therapeutic molecule. Moreover, bsAbs can be readily equipped with an Fc-domain Ig isotype of choice engineered to have natural, enhanced, reduced or even nullified effector activity.
The epidermal growth factor receptor (EGFR) appears to be particularly suitable tumor target for a bsAb-based approach for blocking CD47 in a more tumor-directed manner. EGFR is overexpressed and/or mutated in broad variety of malignancies and is an established target for therapeutic antibodies to selectively bind to cancer cells and inhibit EGFR-mediated oncogenic signaling.14–16
Here, we present a novel bispecific antibody (bsAb), designated CD47xEGFR-IgG1, that directs CD47-blockade toward EGFR-overexpressing cancer cells. BsAb CD47xEGFR-IgG1 has dual binding capacity for both EGFR and CD47, antagonizes their respective oncogenic activities, and is equipped with a fully functional human IgG1 Fc effector domain. Our data demonstrates that bsAb CD47xEGFR-IgG1 has multiple mutually reinforcing anticancer activities that are not available in any of the currently available conventional CD47-blocking antibodies and as such may be useful to reduce on-target/off-tumor effects, enhance cancer cell elimination by ADCC, and promote adaptive anticancer immune responses.
Materials and methods
Antibodies and reagents
The following primary fluorescently-labeled murine mAbs directed against human antigens were used: FITC/APC-labeled CD47-blocking antibody B6H12 (eBioscience), FITC-labeled nonblocking anti-CD47 antibody 2D3 (eBioscience), FITC/APC-labeled anti-EGFR (clone 528, Santa Cruz), PerCP-Cy5.5-labeled anti-CD3 (clone OKT-3, eBioscience), PE-labeled anti-CD137 (clone 4B4-1, BD Pharmingen), APC-labeled anti-CD8 (clone RPA-T8, eBioscience). Fluorescently labeled secondary antibody goat anti-human Ig PE was from Southern Biotech. Unconjugated antibodies used: CD47-blocking mAb B6H12 (Thermofisher), anti-EGFR mAb 425 (Merck), and HLA-ABC-blocking mAb W6/32 (Immunotools).
The following reagents were used: Annexin-V-FITC (Immunotools), propidium iodide (PI) (Invitrogen), and Vybrant DiD, DiO, CFSE, CellTrace Far Red cell proliferation kits (Thermofisher). Secretion of IFNγ was measured using appropriate ELISA kit (eBioscience).
Cell lines
Cell lines A2058, A375m, A431, FaDu, NCI-H292, K562, OvCAR3, DLD1, SJSA1, Pea1, SKOV3, BJAB, PR1, HEK293T, and CHO-K1 cells were obtained from the American Type Culture Collection (ATCC). Cells were cultured in RPMI-1640 or DMEM (Lonza) as indicated, supplemented with 10% fetal calf serum (FCS, Thermo Scientific) at 37°C in a humidified 5% CO2 atmosphere. CHO-K1 cells were cultured in GMEM (First Link), supplemented with 5% dialyzed FBS (Sigma Aldrich). CHO.CD47, CHO.EGFR and K562.EGFR cells stably expressing human CD47 or EGFR were generated by lipofection (Fugene-HD, Promega) of plasmid pCMV6-AC-GFP or plasmid pCMV3-C-GFPSpark, containing cDNAs encoding human CD47 (Origene) or human EGFR (Sino Biological), respectively.
Isolation of neutrophils and macrophages
Neutrophils were isolated from heparinized blood from healthy volunteers after informed consent by density gradient centrifugation followed by selective removal of remaining erythrocytes using ammonium chloride-mediated lysis method. Peripheral blood mononuclear cells (PBMCs) were obtained by density gradient centrifugation (Lymphoprep, Stemcell technologies). Monocytes were isolated from PBMCs using anti-CD14-coated magnetic beads (Miltenyi Biotec) and MACS. For generation of M0 macrophages, monocytes (2 × 105 cells/ml) were cultured in RPMI-1640/10% FCS, supplemented with 50 ng/ml GM-CSF (Immunotools) for 6 d. Macrophages were polarized toward the M1 phenotype by incubation in medium containing 20 ng/ml IFNγ (Immunotools) and 50 ng/ml LPS (Sigma Aldrich) for 24 h.
Lentiviral transduction of CMVpp65 expression in cancer cells
OvCAR3 and K562 cells were transduced using CMVpp65-encoding recombinant lentiviral particles (a kind gift from Dr. Renata Stripecke, Hannover Medical School, Germany) according to standard procedures. Ectopical expression of CMVpp65 in transduced cell lines was evaluated after 48 h by immunocytochemistry using an anti-CMVpp65 antibody cocktail (IQ Products) and an HRP-conjugated rabbit anti-mouse antibody (Dako).
CD47 knock-out (KO) A431 and FaDu cancer cells lines were generated by transfection with pSpCas9 BB-2A-GFP (PX458) plasmid (Origene) encoding sgRNA 5ʹ-ATCGAGCTAAAATATCGTGT-3ʹ17 and subsequent single-cell sorting at the UMCG Flow Cytometry facility.
Construction of bsAb CD47xEGFR-IgG
DNA fragments encoding scFv MABL and scFv 425 were generated by commercial gene synthesis service (Genscript) based on published VH and VL sequence data.18,19 For construction and production of bsAb CD47xEGFR-IgG, we used eukaryotic expression plasmid pEE14-bsAb, which contains 3 consecutive multiple cloning sites (MCS). MCS#1 and MCS#2 are interspersed by a 22 amino acid flexible linker.20 MCS#1, MCS#2 were used for directional and in-frame insertion of DNA fragments encoding scFv MABL, scFv 425, and MCS#3 for insertion of DNA fragments encoding human Fc IgG1, IgG2s21 or IgG4, respectively (Figure S1a,b). Analogously, pEE14-CD47xMock-IgG1 encoding bsAb CD47xMock-IgG1 was constructed by replacing scFv425 in pEE14-CD47xEGFR-IgG1 by scFv4-4-20 directed against fluorescein.22
Production of recombinant bsAbs
BsAbs CD47xEGFR-IgG1, -IgG2s, -IgG4, and appropriate mock control bsAbs were produced using the Expi293 expression system (Thermo Fisher Scientific). Briefly, Expi293 cells were transfected with pEE14-bsAb plasmid encoding the desired bsAb and subsequently cultured on a shaker platform (125 rpm) at 37°C, 8% CO2 for 7 d. Conditioned culture supernatant was harvested and cleared by centrifugation (3000 × g, 30 min), after which bsAbs were purified using an HiTrap protein A HP column connected to an ÄKTA Start chromatography system (GE Healthcare Life Sciences).
Assessment of dual binding activity of bsAb CD47xEGFR-IgG1
Dual binding activity of bsAb CD47xEGFR-IgG1 for EGFR and CD47 was assessed by flow cytometry using CHO.EGFR or CHO.CD47 cells versus parental CHO cells. In short, cells were incubated with increasing amounts of bsAb CD47xEGFR-IgG1 (0.01–10 µg/ml) at 4°C for 45 min after which binding was evaluated using a PE-labeled anti-human-Ig-antibody. Similarly, binding of bsAbs CD47xEGFR-IgG1 and CD47xMock-IgG1 to EGFR was assessed using a panel of EGFRposCD47pos and EGFRnegCD47pos cancer cell lines.
The capacity of bsAb CD47xEGFR-IgG1 to simultaneously bind to EGFR on one cell type and CD47 on a proximal other cell type was assessed by flow cytometry. In short, EGFRpos A431.CD47KO cancer cells (DiD-labeled) and CHO.CD47 (CSFE-labeled) were incubated with bsAb CD47xEGFR-IgG1 (1 µg/ml at 4°C for 45 min) in the presence (or not) of excess amounts (10 µg/ml) of CD47-blocking mAb B6H12 or anti-EGFR mAb 425. The amount of DiDpos/CFSEpos cell clusters was evaluated by flow cytometry.
Competitive binding assay
The overall binding strength (avidity) of bsAbs CD47xEGFR-IgG1 and CD47xMock-IgG1 for EGFRposCD47pos cancer cells was compared in a competitive binding assay.23 In short, A431 cells were incubated with an APC-labeled CD47-blocking mAb (4°C for 10 min), after which bsAb CD47xEGFR-IgG1 or CD47xMock-IgG1 was added in a concentration range from 0.01 to 10 μg/ml. After 45 min of incubation, residual binding of APC-labeled CD47-blocking antibody was quantified by flow cytometry. Where indicated, A431 cells were pre-incubated with excess amounts of mAb 425 (10 μg/ml) at 4°C for 45 min prior to addition of bsAbs.
Inhibition of EGFR-mediated cell proliferation by bsAb CD47xEGFR-IgG1
NCI-H292 cells (8 × 103/well) were seeded into an E-plate 16 (ACEA Biosciences) and treated in the continuous presence of bsAb CD47xEGFR-IgG1 or control bsAbs (2 μg/ml) at 37°C. Cell proliferation was monitored for 96 h using the xCELLigence RTCA instrument (ACEA Biosciences). Graphs show mean ± SD from duplicates.
Assessment of internalization by bsAb CD47xEGFR-IgG1
Internalization of antibody/antigen complexes was assessed by incubating EGFRpos/CD47pos NCI-H292 cells with bsAb CD47xEGFR-IgG1 or control bsAbs (2 μg/ml) in the presence of anti-human Fc saporin-6 toxin-labeled Fab (Fab-ZAP, Advanced Targeting Systems). After 72 h, apoptotic cancer cell death was evaluated by flow cytometry using annexinV/PI staining.
The capacity of bsAb CD47xEGFR-IgG1 to internalize EGFR and CD47 upon binding was assessed using a panel of solid cancer cell types by flow cytometry. In short, cancer cells were treated with bsAb CD47xEGFR-IgG1 (1 μg/ml) or appropriate controls at 37°C for 24 h, after which residual EGFR and CD47 cell surface exposure was assessed using anti-EGFR mAb 528 and anti-CD47 mAb 2D3. Of note, mAb 528 and mAb 2D3 bind to nonoverlapping epitopes on EGFR and CD47, respectively and do not interfere with the EGFR and/or CD47-binding by bsAb CD47xEGFR-IgG1.
Confocal microscopy
NCI-H292 cancer cells were seeded on PET-coated chamber slides and treated with bsAb CD47xEGFR-IgG1 (2 μg/ml) or appropriate controls at 37°C. After 24 h, cells were fixed in 4% PFA and stored in PBS at 4°C until further use. After blocking with 1% BSA (Sigma-Aldrich) for 30 minutes, the slides were incubated overnight with CD47 primary polyclonal antibody (AF4670, R&D Systems) and EGFR primary antibody (clone D38B1, Cell Signaling Technologies) at 4°C. Next, slides were stained for 1.5 h at RT with DxR 555 (1:5000 dilution, Abcam) and DxS 647 (1:250 dilution, Abcam) secondary antibodies and mounted with prolong Gold antifade with DAPI (Thermofisher). Confocal microscopy images were taken with the Confocal TCS SP8 (Leica) at the UMCG Microscopy and Imaging Center.
Assessment of bsAb-mediated ADCC
The capacity of bsAb CD47xEGFR-IgG1 and appropriate (isotype) control bsAbs to induce ADCC upon opsonizing cancer cells was assessed using an ADCC bioluminescent reporter bioassay (Promega). This assay exploits Jurkat T cells engineered to express FcγRIIIa. Binding of the Fc region of a bsAb to this FcγRIIIa activates luciferase gene transcription through the NFAT pathway. In short, EGFRpos FaDu cells and EGFRneg A375m cells (2 × 104) were seeded in a 96-wells plate and cultured for 18 h. Subsequently, Jurkat.FcγRIIIa-NFAT-luciferase cells were added at an E:T cell ratio of 1:1 and then treated for 6 h with bsAb CD47xEGFR-IgG1, -IgG2s, or -IgG4 Fc isotype variants or CD47xMock-IgG1 (all antibodies were added in 0,1 µg/ml final concentration), respectively, after which luciferase activity (RLU) was measured using a Victor V3 multilabel plate reader (Perkin Elmer). Additionally, the capacity of bsAb CD47xEGFR-IgG1 and bsAb CD47xEGFR-IgG2s to induce ADCC was assessed in a PBMC-based ADCC assay. In short, freshly-isolated PBMCs were cocultured with DiD-labeled A431 cancer target cells at an E:T cell ratio of 5:1 for 24 h in the presence (or absence) of bsAb CD47xEGFR-IgG1 or CD47xEGFR-IgG2s (both 2 µg/ml). Induction of apoptotic cancer cell death (annexin Vpos/PIpos) was assessed by flow cytometry.
Assessment of antibody-mediated trogocytosis
Antibody-dependent trogocytic uptake of cancer cell membrane parts by human neutrophils was quantified by flow cytometry. In short, cancer cells were labeled with lipophilic membrane dye DiD according to manufacturer’s protocol and cocultured at an E:T cell ratio of 1:1 with purified neutrophils in the presence of CD47xEGFR-IgG1 (1 μg/ml) or appropriate controls for 2 h. Neutrophils were gated based on their FSC/SSC characteristics after which the percentage of DiDpos neutrophils was evaluated. The flow cytometry gating strategy used for assessment of neutrophil trogocytosis was as previously described by us.19 Kinetics of the trogocytosis process were evaluated as follows; cocultures of DiDpos cancer cells and neutrophils (E:T cell ratio of 5:1) were treated with cetuximab (Merck KGaA) (1 μg/ml), bsAb CD47xEGFR-IgG2s (1 μg/ml) or anti-EpCAM-IgG124 (2 μg/ml), respectively and incubated at 37°C for 0, 10, 20, 40, 60, or 90 min.
Assessment of antibody-mediated trogoptosis
Next, we evaluated whether trogocytosis of cancer cells by neutrophils was followed by apoptotic cell death, hereafter indicated as trogoptosis as recently described in.25 In short, FaDu cells and FaDu.CD47KO (1.5 × 104/well) were seeded into an E-plate 16 and allowed to adhere for 24–48 h, after which neutrophils were added (E:T cell ratio of 10:1). Cocultures were treated with bsAb CD47xEGFR-IgG1, -IgG4 or -IgG2s (2 μg/ml) or anti-EpCAM-IgG1 (2 μg/ml), respectively. Induction of trogoptosis was monitored for up to 48 h of treatment using the xCELLigence RTCA instrument. Cell index was normalized prior to addition of neutrophils and antibodies. Graphs show mean ± SD from quadruplicates.
M1 macrophage phagocytosis assay
CellTrace Far Red-labeled M1 macrophages were coincubated with CFSE-labeled or GFP-labeled cancer cells (E:T cell ratio of 1:2) in the presence of bsAb CD47xEGFR-IgG1 (1 μg/ml) or appropriate control bsAbs at 37°C for 3 h. The capacity of M1 macrophages for ADCP of antibody-opsonized cancer cells was evaluated by flow cytometry and was indicated as the percentage of CFSEpos macrophages within the population of Far Redpos macrophages.
Generation and expansion of CMVpp65-specific CD8pos T cells
Peripheral blood lymphocytes (PBLs) were freshly isolated from PBMCs from an HLA-B*0702 CMV-positive healthy volunteer and depleted of monocytes using anti-CD14-coated magnetic beads. Subsequently, the PBLs (5 × 106/ml) were cultured in a 6-wells plate using X-VIVO15 medium (Lonza) and stimulated by adding 5–10 μl of a commercial recombinant CMVpp65 protein solution (Miltenyi Biotech) per ml of cell suspension and then cultured for 2–3 d. Next, CMVpp65-stimulated PBLs were harvested and resuspended in fresh X-VIVO15 medium supplemented with 500–1000 U/ml IL-2 (Immunotools) and cultured for 2–4 additional days. Subsequently, CMVpp65-stimulated PBLs were cultured on a feeder layer of OvCAR3.pp65 cells (HLA-B*0702pos) in X vivo 15 medium at an E:T cell ratio of 7.5:1 for 24 h. Next, CMVpp65-restimulated PBLs were harvested, washed and resuspended in X-VIVO15 medium supplemented with IL-2 and cultured for 2–4 d. Flow cytometry indicated that this stimulation protocol yielded >60% CMVpp65-dextramerepos (HLA-B*0702/TPRVTGGGAM-APC, Immudex) CD8pos T cells (Figure S5). After up to 7 stimulation rounds, CMVpp65-specific CD8pos T cells were frozen until used.
Assessment of antigen-specific T cell activation in a CMVpp65-based model system
M1 macrophages were seeded in a flat-bottom 96 wells plate (4 × 104 cells/well) using macrophage attachment medium (Promocell) and cocultured with K562.EGFR.pp65 cells (E:T cell ratio of 1:2) in the presence of bsAbs (1 μg/ml) at 37°C for 3 h. Next, K562.EGFR.pp65 cells were removed after which autologous CMVpp65-specific CD8pos T cells (see above) were added (E:T cell ratio of 1:1) in the presence or absence of HLA ABC-blocking antibodies (10 μg/ml) for 24 h. The percentage of CD137pos T cells relative to CD3pos/CD8pos T cells was evaluated by flow cytometry. The spend culture media were evaluated for the amount of T cell-secreted IFNγ by ELISA.
Results
bsAb CD47xEGFR-IgG1 simultaneously binds to CD47 and EGFR
BsAb CD47xEGFR-IgG1 dose-dependently bound to CHO.EGFR cells and not to CHO cells (Figure 1a) and binding positively correlated with the respective EGFR-expression levels of the various EGFRpos cancer cell lines evaluated (Figure S2a,b). Similarly, bsAb CD47xEGFR-IgG1 bound to CHO.CD47 cells and not to parental CHO cells (Figure 1b). Importantly, bsAb CD47xEGFR-IgG1 showed enhanced binding toward EGFRpos/CD47pos cancer cells due to the enhanced avidity effect of its dual binding capacity for both EGFR and CD47 (Figure 1c). Of note, bsAb CD47xEGFR-IgG1 has a ~ 4.5-fold higher binding capacity for K562.EGFR cells compared with K562 cells. K562 and K562.EGFR have similar CD47 expression levels; therefore, the difference in binding capacity is likely due to enhanced avidity effect of simultaneously binding to CD47 and EGFR (Figure S2c,d).
Figure 1.
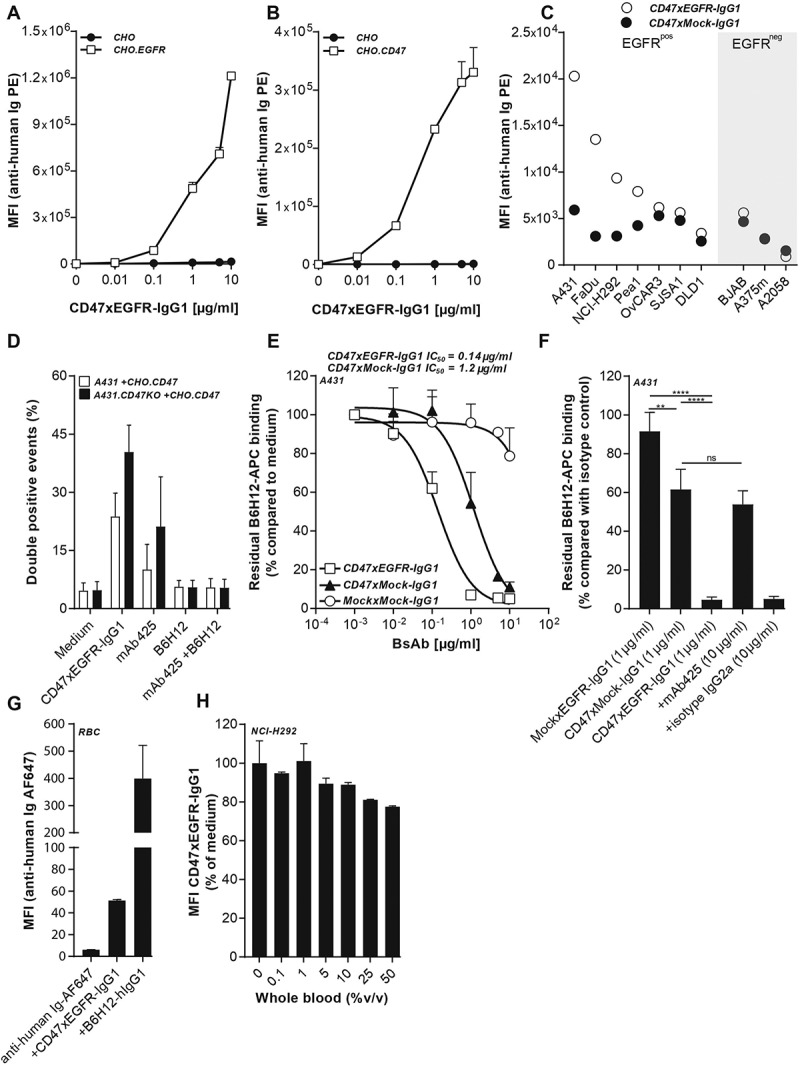
BsAb CD47xEGFR-IgG1 selectively and simultaneously binds to EGFR and CD47. Dose-dependent binding of CD47xEGFR-IgG1 to (a) CHO.EGFR or (b) CHO.CD47 cells vs. parental CHO cells. (c) Binding of CD47xEGFR-IgG1 or CD47xMock-IgG1 (1 µg/ml) to a panel of EGFRpos and EGFRneg CD47-expressing cell lines. (d) Binding of CD47xEGFR-IgG1 (1 µg/ml) to (GFP-labeled) CHO.CD47 cells and (DiD-labeled) parental A431 or A431.CD47KO cells in the presence or absence of excess CD47-blocking antibody B6H12 and/or EGFR-blocking antibody mAb 425 (both 10 µg/ml). The percentage of DiD/GFP double positive events upon incubation with indicated antibodies is shown. (e) Competitive binding assay in which APC-conjugated CD47 antibody (B6H12) competed with increasing doses (0.01–10 µg/ml) of bsAb CD47xEGFR-IgG1 (white squares), CD47xMock-IgG1 (black triangles) or MockxMock-IgG1 (white dots) for binding to A431 cells. (f) Competitive binding assay, as in E, in which A431 cells were pretreated with excess amounts of mAb 425 or isotype control IgG2a (both 10 µg/ml) prior to addition of CD47xEGFR-IgG1 (1 µg/ml), where indicated. (g) In vitro binding of CD47xEGFR-IgG1 and B6H12-hIgG1 to RBC present in whole blood. (h) Binding assay of CD47xEGFR-IgG1 (20 µg/ml) to DiO-labeled NCI-H292 cancer cells in the presence of increasing concentrations of whole blood. Of note, RBC in whole blood form an abundant “antigen-sink” for CD47-antibodies. All binding experiments were analyzed by flow cytometry. All graphs represent mean ± SD. Statistical analysis in F was performed using one-way ANOVA followed by a Tukey post-hoc test (*p < .05, **p < .01, ***p < .001, ****p < .0001, ns not significant)
Additionally, bsAb CD47xEGFR-IgG1 was able to simultaneously bind to EGFR present on one cell type and CD47 on other cell type present in close proximity. In particular, bsAb CD47xEGFR-IgG1 simultaneously bound to and cellularly bridged A431.CD47KO cancer cells (DiD-labeled) and CHO.CD47 cells (CSFE-labeled) as was evident from a marked increase in DiDpos/CFSEpos cell clusters detected by flow cytometry. Of note, DiDpos/CFSEpos cell cluster formation was strongly reduced in the presence of excess amounts of either CD47-blocking mAb B6H12 or EGFR-blocking mAb 425 (Figure 1d).
bsAb CD47xEGFR-IgG1 blocks CD47 in an EGFR-directed manner
BsAb CD47xEGFR-IgG1 and bsAb CD47xMock-IgG1 were compared for their capacity to block CD47 on EGFR-expressing cancer cells using a competitive binding assay.23 In this assay, displacement of an APC-labeled CD47-blocking mAb bound to EGFRpos/CD47pos A431 cancer cells was evaluated in the competing presence of bsAb CD47xEGFR-IgG1. The IC50 of bsAb CD47xEGFR-IgG1 for displacing APC-labeled CD47-blocking mAb from A431 cells was calculated to be 0.14 μg/ml, which is ~8.4 times lower than that of bsAb CD47xMock-IgG1 (1.20 μg/ml) (Figure 1e). Importantly, pre-incubation with an excess amount of EGFR-blocking mAb 425 essentially abrogated the capacity of bsAb CD47xEGFR-IgG1 to displace APC-labeled CD47-blocking mAb (Figure 1f). Additionally, in vitro binding (MFI) of CD47xEGFR-IgG1 to RBC present in whole blood was calculated to be ~8 times lower than B6H12-hIgG1 (Figure 1g). In an RBC competitive binding assay, binding of CD47xEGFR-IgG1 to DiO-labeled NCI-H292 cancer cells was largely preserved in the presence of increasing concentrations of whole blood (Figure 1h). Taken together, these data demonstrated that bsAb CD47xEGFR-IgG1 has strongly enhanced binding capacity toward EGFRpos/CD47pos cancer cells endowing it with capacity to block CD47 in an EGFR-directed manner.
bsAb CD47xEGFR-IgG1 inhibits cancer cell proliferation
In RTCA cell proliferation analysis, bsAb CD47xEGFR-IgG1 showed a higher capacity to inhibit proliferation of EGFRpos/CD47pos NCI-H292 cancer cells than bsAb CD47xMock-IgG1 or MockxEGFR-IgG1. After continuous treatment for 4 d with bsAb CD47xEGFR-IgG1, the proliferation of NCI-H292 cancer cells (expressed as ”cell index”) was inhibited by 49% compared to that of NCI-H292 cells treated with MockxMock-IgG1. MockxEGFR-IgG1 and CD47xMock-IgG1 inhibited the cell index by 39% and 15%, respectively (Figure 2a).
Figure 2.
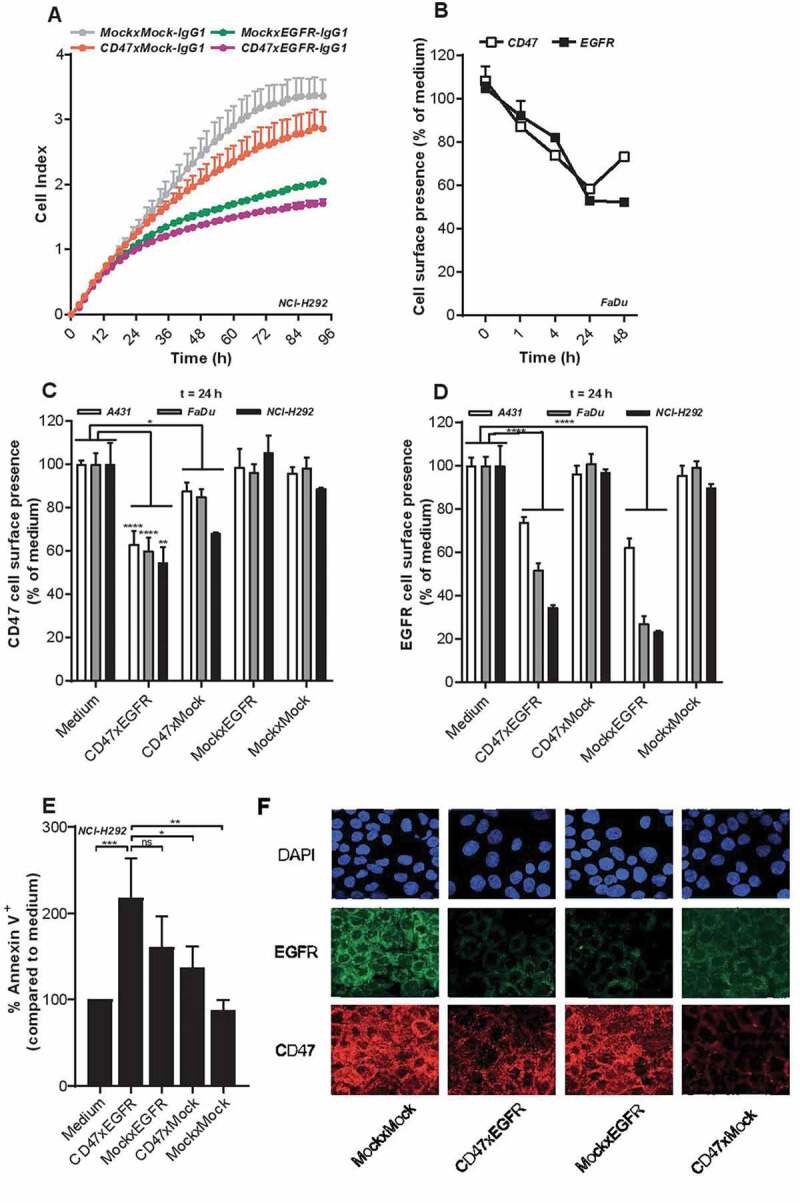
BsAb CD47xEGFR-IgG1 inhibits cancer cell proliferation and induces cointernalization of EGFR and CD47 from the cancer cell surface. (a) Cell proliferation of NCI-H292 cells was measured during 96 h of continuous treatment with bsAb CD47xEGFR-IgG1 or control antibodies (2 µg/ml) in an xCELLigence Real-Time Cell Proliferation assay. (b) FaDu cells were incubated with bsAb CD47xEGFR-IgG1 at 37°C and residual cell-surface presence of CD47 and EGFR were measured over time by flow cytometry and compared to medium control. Similar to B, residual cell-surface presence of CD47 (c) and EGFR (d) were measured on A431, NCI-H292, and FaDu cells after incubation with bsAb CD47xEGFR-IgG1 or control antibodies (1 µg/ml) at 37°C for 24 h. (e) Apoptosis of NCI-H292 cells upon internalization of EGFR and/or CD47 by indicated antibodies in the presence of Fab-ZAP, a chemical conjugate of goat anti-human monovalent antibody and saporin, was measured after 72 h. Apoptosis was measured with annexinV-staining using flow cytometry and expressed as percentage increase compared to medium control. (f) Representative confocal images of NCI-H292 cells stained with DAPI, EGFR antibody and CD47 antibody after 24 h internalization with bsAb CD47xEGFR-IgG1 or control antibodies (2 µg/ml). All graphs represent mean ± SD. Error bars in graph (a) and (b) indicate SD of duplicates. Statistical analysis in (c–e) was performed using One-way ANOVA followed by a Tukey post-hoc test (*p < .05, **p < .01, ***p < .001, ****p < .0001, ns not significant)
bsAb CD47xEGFR-IgG1 induces cointernalization and subsequent prolonged displacement of both EGFR and CD47 from the cancer cell surface
Treatment of EGFRpos/CD47pos cancer cells with bsAb CD47xEGFR-IgG1 resulted in a prolonged displacement of EGFR and CD47 molecules from the cancer cell surface for 48 h and 24 h, respectively (Figure 2b). After treatment of FaDu cancer cells with CD47xEGFR-IgG1 for 24 h, residual cell surface presence of EGFR and CD47 dropped down to ~50% and ~60%, respectively (Figure 2b–d). Moreover, a potent approximately twofold increase (p < .01) in saporin-mediated apoptotic cell death in NCI-H292 cancer cells was induced by ´piggybacking´ of the Fab-ZAP toxin on internalizing bsAb CD47xEGFR-IgG1 molecules. When using bsAbs MockxEGFR-IgG1 or CD47xMock-IgG1 a lower increase in apoptosis was observed of ~60% (ns) or ~40% (p < .05), respectively (Figure 2e). Additionally, confocal microscopy images of NCI-H292 cells treated with bsAb CD47xEGFR-IgG1 for 24 h, clearly show reduced CD47 and EGFR cell surface presence (Figure 2f). Taken together, these data demonstrated that bsAb CD47xEGFR-IgG1 strongly inhibits cancer cell proliferation and induces prolonged displacement of both EGFR and CD47 from the cancer cell surface.
bsAb CD47xEGFR-IgG1 has enhanced ADCC signaling capacity toward EGFRpos/CD47pos cancer cells
BsAb CD47xEGFR-IgG1 and the IgG4 and IgG2-silent isotype variants thereof were assessed for capacity to induce FcγIIIa (CD16)-mediated ADCC activity toward EGFRpos/CD47pos cancer cells using a Jurkat.FcγRIIIa-NFAT-luc ADCC bioassay. The ADCC-signaling capacity of bsAb CD47xEGFR-IgG1 toward EGFRpos/CD47pos cancer cells was twofold higher than that of bsAb CD47xMock-IgG1, whereas ADCC-signaling capacities toward EGFRneg/CD47pos cancer cells were essentially identical (Figure 3a). As expected, bsAbs CD47xEGFR-IgG4 and CD47xEGFR-IgG2s were devoid of ADCC-signaling activity. Moreover, bsAb CD47xEGFR-IgG1, but not bsAb CD47xEGFR-IgG2s, engaged the ADCC activity of PBMCs (5 out of 5 donors) immune effector cells toward A431 cancer target cells in a PBMC-based ADCC assay (Figure 3b). Of note, bsAbs CD47xEGFR-IgG1, IgG4, and IgG2s showed equal binding activity toward EGFRpos/CD47pos cancer cells (Figure S2e).
Figure 3.
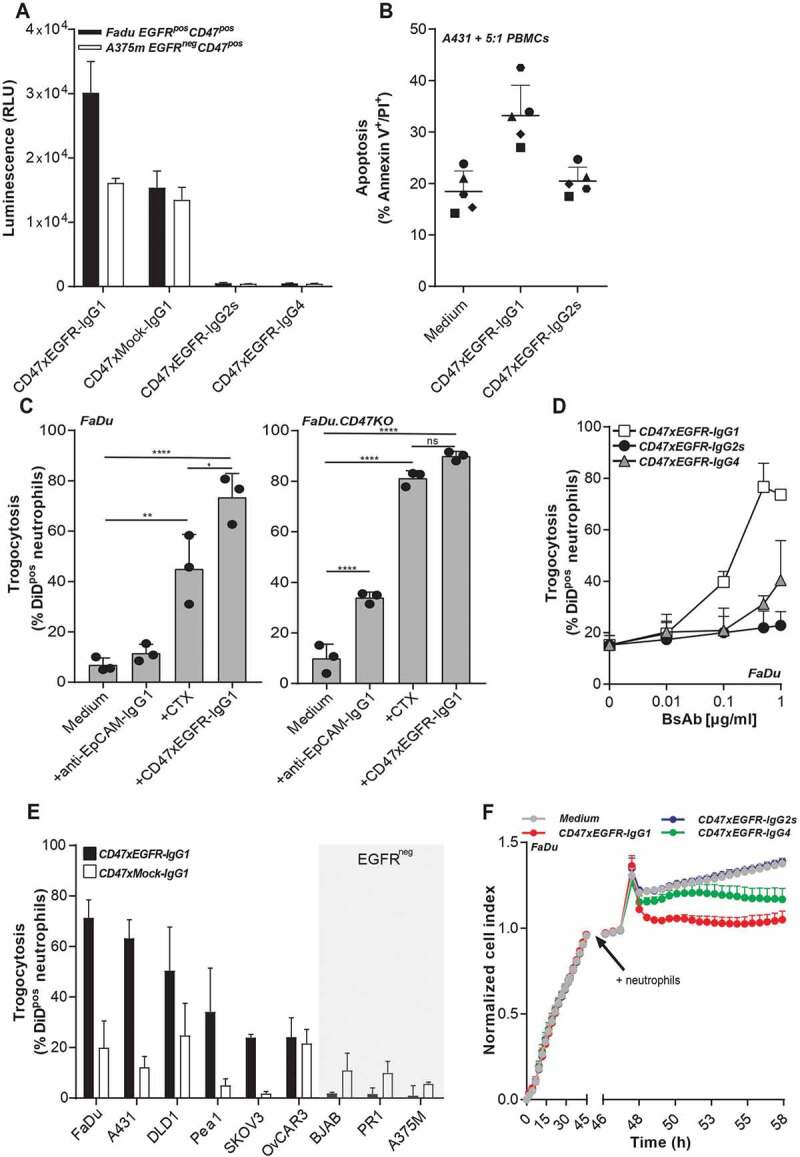
BsAb CD47xEGFR-IgG1 induces ADCC (a) ADCC reporter bioassay in which EGFRpos FaDu cells or EGFRneg A375m cells were coincubated with Jurkat. FcγRIIIa-NFAT-luc cells in the presence of bsAb CD47xEGFR-IgG1, CD47xEGFR-IgG2s, CD47xEGFR-IgG4, or CD47xMock-IgG1 (0.1 µg/ml). Luciferase activity (RLU) was measured after 6 h. (b) Evaluation of bsAb CD47xEGFR‐IgG1 and bsAb CD47xEGFR‐IgG2s for their respective capacity to engage ADCC activity of freshly isolated PBMCs from 5 healthy volunteers toward A431 cancer cells endogenously expressing EGFR and CD47. PBMCs were cocultured with DiD‐labeled A431 cancer cells at an E:T cell ratio of 5:1 for 24 h in the presence (or absence) of bsAb CD47xEGFR‐IgG1 or CD47xEGFRIgG2s (2 μg/ml each). Induction of apoptotic cancer cell death (annexin Vpos/PIpos) was assessed by flow cytometry. BsAb CD47xEGFR-IgG1 induces neutrophil-mediated trogocytosis and trogoptosis toward CD47posEGFRpos cancer cells. (c) FaDu or FaDu.CD47KO cells were DiD-labeled and incubated with neutrophils at an E:T cell ratio of 1:1 in the presence of anti-EpCAM-IgG1 (2 µg/ml), CTX or CD47xEGFR-IgG1 (both 1 µg/ml). Trogocytosis was quantified as the percentage of DiDpos neutrophils by flow cytometry after 2 h. (d) Similar as in (b), trogocytosis was measured after DiD-labeled FaDu cells were incubated with neutrophils (E:T cell ratio of 1:1) in the presence of increasing concentrations (0.01–1 µg/ml) of bsAb CD47xEGFR-IgG1, CD47xEGFR-IgG2s or CD47xEGFR-IgG4. (e) Next, neutrophil-mediated trogocytosis of a panel of EGFRpos and EGFRneg DiD-labeled CD47-expressing cell lines was assessed in the presence of bsAb CD47xEGFR-IgG1 or CD47xMock-IgG1 (both 1 µg/ml). (f) xCELLigence real-time cytotoxicity assay shows neutrophil-induced trogoptosis of FaDu cells opsonized with bsAb CD47xEGFR-IgG1, -IgG2s, or -IgG4. After adhering, FaDu cells were coincubated with IFNγ-stimulated neutrophils (E:T cell ratio of 10:1) in the presence of indicated antibodies (2 µg/ml). Cell index was normalized prior to addition of neutrophils and antibodies. All graphs represent mean ± SD. Graphs (c–e) represent data of three independent experiments with neutrophils from three different donors. Error bars in F indicate SD of quadruplicates. Statistical analysis in C was performed using one-way ANOVA followed by a Tukey post-hoc test (*p < .05, **p < .01, ***p < .001, ****p < .0001, ns not significant)
bsAb CD47xEGFR-IgG1 potentiates trogocytic activity of neutrophils toward cancer cells in an EGFR-directed manner
Cancer cell surface-expressed CD47 was recently reported to act as an inhibitory signal for neutrophil-mediated trogocytosis and trogoptosis of cancer cells25 and as such may be implicated in reducing anticancer effector activity of IgG1-isotype therapeutic antibodies such as cetuximab (CTX). In this respect, bsAb CD47xEGFR-IgG1 showed potently enhanced neutrophil-mediated trogocytic uptake of DiD-labeled cell membrane fragments from parental FaDu cancer cells which could be detected in ~75% of neutrophils. In comparison, identical treatment of FaDu cells with CTX or an anti-EpCAM-IgG1 antibody resulted in detection of FaDu cell-derived membrane fragments in only ~45% and ~10% of neutrophils, respectively (Figure 3c). Of note, when FaDu.CD47KO cancer cells were used instead, CTX and bsAb CD47xEGFR-IgG1 induced comparable neutrophil-mediated trogocytosis. As expected, IgG4- and IgG2s-isotype variants of bsAbs CD47xEGFR-IgG1 showed reduced capacity to induce trogocytic uptake by neutrophils of cell membrane fragments from parental FaDu cancer cells (40% for IgG4 and 20% for IgG2s versus 75% for bsAb CD47xEGFR-IgG1, respectively (Figure 3d).
Of note, bsAb CD47xEGFR-IgG1 potentiated the trogocytic activity of neutrophils toward 6 out of 6 different EGFRpos/CD47pos cancer cell types, whereas control bsAb CD47xMock-IgG1 showed only marginal capacity to do so (Figure 3d). BsAb CD47xEGFR-IgG1 did not enhance trogocytic activity of neutrophils toward EGFRneg/CD47pos cancer cell types (Figure 3e).
Collectively, this indicates that bsAb CD47xEGFR-IgG1 potentiates the trogocytic activity of neutrophils toward CD47pos cancer cells in an EGFR-dependent manner.
bsAb CD47xEGFR-IgG1 promotes neutrophil-mediated trogoptosis of cancer cells
Recently, it was identified that CD47-SIRPα checkpoint blockade endows neutrophils to kill antibody-opsonized cancer cells via trogoptosis, a lytic form of trogocytosis which disrupts the integrity of the cancer cell membrane.25 In RTCA cell analysis, bsAb CD47xEGFR-IgG1 (and to a lesser extend the IgG4 isotype variant thereof), promoted neutrophil-mediated trogoptosis of EGFRpos/CD47pos cancer cells, as was evident from a prominent decrease in cell index within 2 h after addition of neutrophil effector cells (Figure 3f). Of note, in this assay treatment with bsAb CD47xEGFR-IgG2s did not promote neutrophil-mediated trogoptosis of cancer cells. However, when an Fc effector function-competent anti-EpCAM-IgG1 antibody was combined with treatment with bsAb CD47xEGFR-IgG2s, neutrophil-mediated trogocytosis and trogoptosis was enhanced compared to treatment with anti-EpCAM-IgG1 antibody only, presumably due to its EGFR-directed CD47-blocking activity (Figure S3).
Taken together, these data demonstrate that bsAb CD47xEGFR-IgG1 selectively potentiates the trogocytic activity of neutrophils toward EGFRpos/CD47pos cancer cells resulting in the enhanced induction of trogoptotic cancer cell death.
bsAb CD47xEGFR-IgG1 promotes ADCP of EGFRpos/CD47pos cancer cells in an EGFR-directed manner, independent of FcR interaction
BsAb CD47xEGFR-IgG1 markedly enhanced the ADCP activity of macrophages toward EGFR-transfected/CD47pos K562.EGFR cells compared to parental EGFRneg/CD47pos K562 cells (Figure 4a,b). Similarly, bsAb CD47xEGFR-IgG1 selectively promoted macrophage-mediated ADCP activity toward various CD47pos cancer cell types with endogenous EGFR overexpression (Figures 4c and S4). In contrast, bsAb CD47xMock-IgG1 showed no enhanced capacity to induce macrophage-mediated ADCP activity toward K562.EGFR cells. Interestingly, isotype variants of bsAb CD47xEGFR-IgG1 with reduced (-IgG4) and no binding activity (-IgG2s) for human FcRs showed similar capacity to promote macrophage-mediated ADCP activity of EGFRpos/CD47pos cancer cells. This indicates that, at least in this model system, bsAb CD47xEGFR-IgG1 promotes macrophage-mediated ADCP activity toward EGFRpos/CD47pos cancer that is independent of FcR interactions.
Figure 4.
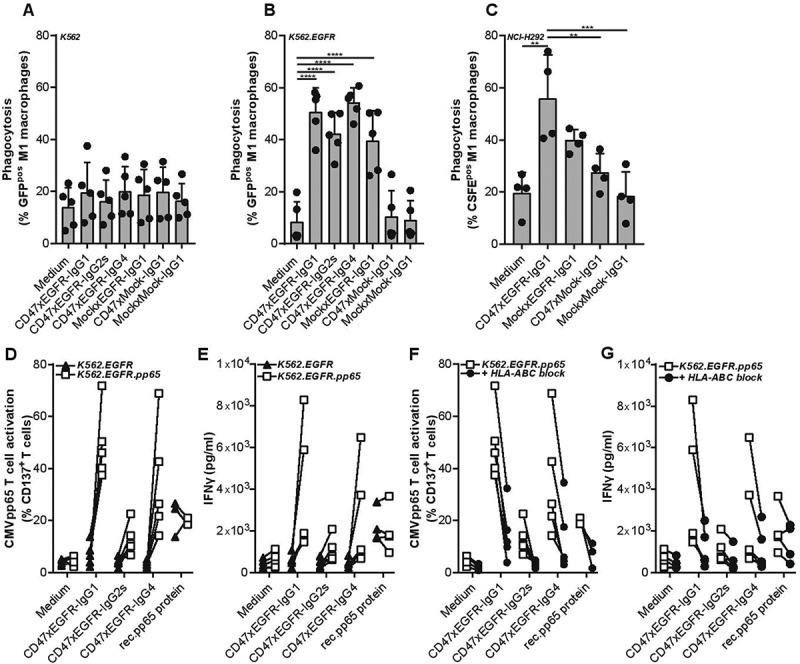
BsAb CD47xEGFR-IgG1-mediated phagocytosis of cancer cells by macrophages facilitates cross-presentation of peptides in MHC-I and subsequent activation of antigen-specific CD8+ T cells. Phagocytosis of GFP-labeled (a) K562, (b) K562.EGFR cells, or (c) CFSE-labeled NCI-H292 cells by M1 macrophages (E:T cell ratio of 1:2) in the presence of different isotypes of bsAb CD47xEGFR or control antibodies (1 µg/ml) after 3 h. (d) K562.EGFR cells or K562.EGFR.pp65 cells were coincubated with M1 macrophages from a CMV-seropositive donor (E:T cell ratio = 1:2) in the presence of CD47xEGFR-IgG1 or control antibodies (1 µg/ml) for 3 h. K562 cells and unbound antibody were washed away prior to addition of a priori expanded CMVpp65-specific autologous CD8+ T cells (E:T cell ratio of 1:1). After 24 h, the percentage of CD137pos T cells within CD8+ T cell population was measured by flow cytometry. (e) IFNγ levels in culture supernatant of D were measured by ELISA. (f) As in D, but HLA-ABC blocking antibody W6/32 (10 µg/ml) was added after addition of CMVpp65-specific autologous CD8+ T cells to block cross-presentation capacity of macrophages. (g) IFNγ levels in culture supernatant of F were measured by ELISA. Graphs (a–c) represent mean ± SD of independent experiments using at least three different donors. Statistical analysis in (a–c) were performed using One-way ANOVA followed by a Tukey post-hoc test. Graphs (d–g) represent independent experiments with macrophages from the same CMV-seropositive HLA-B7*07:02-matched donor (*p < .05, **p < .01, ***p < .001, ****p < .0001, ns not significant)
bsAb CD47xEGFR-IgG1-mediated ADCP promotes cross-presentation dependent on FcR interaction
BsAb CD47xEGFR-IgG1 was evaluated for its capacity to promote cross-presentation of cancer cell-derived peptides by macrophages to antigen-experienced CD8pos T cells. To this end, HLA-I-negative K562.EGFR.pp65 cells (ectopically expressing EGFR and CMVpp65) were treated with bsAb CD47xEGFR-IgG1 to induce macrophage-mediated ADCP activity. Subsequently, these macrophages were assessed for enhanced cross-presentation of CMVpp65-derived peptides to HLA-B7*07:02-matched CMVpp65-specific CD8+ T cells. Indeed, enhanced cross-presentation by these macrophages was evident from upregulation of CD137 and enhanced secretion of IFNγ by the CD8pos T cells (Figure 4d,e). The upregulation of CD137 and secretion of IFNγ were strongly inhibited when this experiment was performed in the presence of an excess amount of an HLA ABC-blocking antibody (Figure 4f,g). Of note, in the above setting IgG4 and IgG2s isotype variants of bsAb CD47xEGFR-IgG1 showed only marginally enhanced capacity to promote cross-presentation. This indicates that bsAb CD47xEGFR-IgG1 promotes cross-presentation in an FcR-dependent manner.
Discussion
The clinical efficacy of current monospecific CD47-blocking antibodies is potentially hampered by 'on-target/off-tumor' binding to a vast majority of normal cells that also expresses CD47.26 The lack of tumor-selectivity of conventional CD47-blocking antibodies may preclude their sufficient accretion at the tumor site(s).10–13 Moreover, blockade of CD47 on normal cells may contribute to the induction of autoimmune-related adverse events.
To address these issues, we developed a bsAb-based approach to more selectively direct CD47 blockade to cancer cells. For this purpose, we selected EGFR to serve as target antigen of our CD47-blocking bsAb approach. EGFR is a well-established cell surface target antigen with oncogenic signaling capacity that is overexpressed and/or mutated in various malignancies.14–16 Our data demonstrate that the capacity of bsAb CD47xEGFR to simultaneously bind to CD47 and EGFR resulted in a significantly enhanced overall binding strength (avidity) for EGFRpos/CD47pos cancer cells. Importantly, this binding induced rapid cointernalization of antibody-antigen complexes which resulted in prolonged displacement of both EGFR and CD47 from the cancer cell surface. This unique feature may explain the potent capacity of bsAb CD47xEGFR to selectively inhibit the proliferation of EGFRpos/CD47pos cancer cells. Although not studied here, concurrent inhibition of EGFR- and CD47-associated oncogenic signaling events by bsAb CD47xEGFR may also mediate other mutually reinforcing anticancer effects.
ADCC is an important effector mechanism of therapeutic antibodies to destroy cancer cells and can be mediated by various Fc-receptor-expressing myeloid immune effector cells, in particular by neutrophils. Recently, the mode-of-action by which neutrophils kill antibody-opsonized cancer was shown to involve trogoptosis, an alternate lytic form of ADCC that mechanically disrupts the cancer cell membrane.25 Intriguingly, it was demonstrated that neutrophil-ADCC of trastuzumab-opsonized breast cancer cells was potentiated upon cotreatment with a CD47-blocking antibody or a SIRPα-blocking antibody.9,25 Giving the combined capacities of bsAb CD47xEGFR-IgG1 to bind and block CD47/EGFR with enhanced avidity and to induce ADCC via its IgG1 Fc domain, we evaluated whether this bsAb enhanced the trogocytic activity of neutrophils in a single agent setting. Indeed, opsonization of EGFRpos/CD47pos cancer cells with bsAb CD47xEGFR-IgG1, but not its IgG4 or IgG2s isotype variants, markedly enhanced neutrophil-mediated trogocytosis resulting in rapid and selective trogoptosis of these cancer cells.
It is well established that interaction of IgG/antigen-complexes with Fc-γ receptors on phagocytes promotes shuttling of exogenous antigen into the cross-presentation pathway, thereby priming adoptive CD8pos T cell-mediated anticancer immunity (reviewed in27). In this respect, it is noteworthy that bsAb CD47xEGFR-IgG1 is equipped with a fully functional IgG1 Fc effector domain. Indeed, our results demonstrated that opsonization of CMVpp65-expressing EGFRpos/CD47pos cancer cells with bsAb CD47xEGFR-IgG1 promoted antigen cross-presentation by macrophages which was evident from their enhanced capacity to activate HLA-matched pp65-specific CD8pos T cells. Importantly, isotype variants IgG4 or IgG2s of bsAb CD47xEGFR-IgG1 only poorly induced cross-presentation.
Typically, therapeutic CD47-blocking antibodies are equipped with engineered human Fc domains with reduced or fully silenced ADCC/ADCP activity to avoid elimination of CD47-expressing normal cells.28 In the current study, we compared bsAb CD47xEGFR IgG1, IgG4 and IgG2s isotypes for their ability to induce neutrophil-mediated trogocytosis and trogoptosis, ADCC signaling and macrophage mediated cross-presentation. From this evaluation it became clear that only the bsAb CD47xEGFR-IgG1 isotype has potent activity in all these assays. Therefore, clinical CD47 antibodies equipped with (partially) silenced human Fc domains will have limited capacity to promote neutrophil-mediated trogoptosis, ADCC and APC-mediated cross-presentation of tumor antigens. This trade-off to reduce toxicity of clinical CD47-blocking antibodies toward normal cells may therefore come with a markedly reduced anti-cancer activity. However, recent clinical trials demonstrated that the efficacy of such Fc-silenced CD47-blocking antibodies can be enhanced when treatment was combined with an Fc effector-competent anticancer antibody.10 Although it is clear that more in-depth toxicity studies for bsAb CD47xEGFR-IgG1 are needed to evaluate potential toxicity toward normal cells, bsAb CD47xEGFR-IgG1 could represent a two-in-one antibody approach in which the CD47-blocking activity is selectively directed to EGFRpos cancer cell types. We uncovered that the combination of bsAb CD47xEGFR-IgG2s with an anti-EpCAM-IgG1 antibody potently enhanced neutrophil-mediated trogocytosis and trogoptosis compared to treatment with anti-EpCAM-IgG1 antibody only. Therefore, an alternative therapeutic approach could be to combine treatment of cancer cells with bsAb CD47xEGFR-IgG4 or -IgG2s with a therapeutic antibody equipped with a fully functional IgG1 Fc domain directed at an alternate tumor-associated antigen coexpressed at the cancer cell surface.
Obviously, a more detailed fit-for-purpose evaluation of bsAb CD47xEGFR-IgG1 should be evaluated in vivo in a realistic and informative animal model. In particular, a patient-derived xenograft (PDX) model using EGFR-transgenic/immune humanized NSG mice appears to be useful for such purpose. However, the results from such studies in mice should be interpreted with caution since there is a 10-fold higher binding affinity between human CD47 and mutant SIRPα known to be expressed in NOD mice29,30. Consequently, the high affinity interaction of human CD47 with murine mutant SIRPα may result in an unrealistic strong inhibition of phagocytosis in murine myeloid effector cells. Moreover, in such a PDX model, only the engrafted tumor and immune cells express human CD47, which does not fully recapitulate the massive CD47 “antigen sink” present in humans. The use of non-NOD syngeneic immunocompetent tumor models or human SIRPα transgenic Rag2nullIl2rγnull mice could allow for better evaluation of CD47-blocking antibodies.31,32 Clearly, the development of an animal model that tackles these and other issues related with the in vivo evaluation of bsAb CD47xEGFR-IgG1 are beyond the scope of our current report.
Recently, it was reported that treatment with EGFR tyrosine kinase inhibitor (TKI) gefitinib induced a marked down-regulation of CD47 expression by responsive NSCLC cells. Importantly, upon acquisition of gefitinib resistance this response was reverted, as was evident from a marked up-regulation of CD47 expression by EGFR TKI nonresponsive cancer cells. However, this selective overexpression of CD47 renders EGFR-mutant NSCLC cells more sensitive to the phagocytic clearance induced by CD47-blocking antibodies.33 In this respect, bsAb CD47xEGFR-IgG1 may provide a promising immunotherapeutic option for naïve and TKI-resistant EGFR-mutant NSCLC.
In conclusion, our data demonstrate that bsAb CD47xEGFR-IgG1 has multiple and possibly mutually reinforcing anticancer activities that are not available in any of the current conventional CD47-blocking antibodies. In particular, bsAb CD47xEGFR-IgG1 as ability to: (1) simultaneously bind to both EGFR and CD47 resulting in enhanced avidity toward EGFRpos/CD47pos cancer cells; (2) block oncogenic signaling by EGFR and possibly those mediated by CD47; (3) block CD47/SIRPα interaction in an EGFR-directed manner; (4) promote cointernalization and prolonged cell surface-displacement of both EGFR and CD47 from cancer cells; (5) enhance EGFR-directed neutrophil-mediated trogocytosis and trogoptosis of cancer cells as a single agent; (6) enhance EGFR-directed phagocytosis of cancer cells; and (7) promote cross-presentation antigens derived from EGFRpos/CD47pos cancer cells to engage the adaptive immune system.
Taken together, bsAb CD47xEGFR-IgG1 may represent novel antibody-based strategy for enhancing selectivity and therapeutic efficacy of CD47-SIRPα checkpoint inhibition approaches in EGFR-overexpressing malignancies.
Supplementary Material
Funding Statement
This work was supported by the Dutch Cancer Society [RUG2018-11464]; Dutch Cancer Society [RUG2013-6209]; Abel Tasman Talent Program [ATTP-2017]; Dutch Cancer Society [RUG2014-6986]; GSMS [MPDI-123015].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Barclay AN. Signal regulatory protein alpha (SIRPalpha)/CD47 interaction and function. Curr Opin Immunol. 2009;21(1):47–12. doi: 10.1016/j.coi.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matozaki T, Murata Y, Okazawa H, Ohnishi H. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009;19(2):72–80. doi: 10.1016/j.tcb.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, Traver D, van Rooijen N, Weissman IL. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, Park CY, Weissman IL, Majeti R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011;71(4):1374–1384. doi: 10.1158/0008-5472.CAN-10-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109(17):6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate nonhodgkin lymphoma. Cell. 2010;142(5):699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, Seita J, Inlay MA, Weiskopf K, Miyanishi M, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A. 2013;110(27):11103–11108. doi: 10.1073/pnas.1305569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsao LC, Crosby EJ, Trotter TN, Agarwal P, Hwang B-J, Acharya C, Shuptrine CW, Wang T, Wei J, Yang X, et al. CD47 blockade augmentation of trastuzumab antitumor efficacy dependent on antibody-dependent cellular phagocytosis. JCI Insight. 2019;4:24. doi: 10.1172/jci.insight.131882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao XW, van Beek EM, Schornagel K, Van der Maaden H, Van Houdt M, Otten MA, Finetti P, Van Egmond M, Matozaki T, Kraal G, et al. CD47-signal regulatory protein-α (SIRPα) interactions form a barrier for antibody-mediated tumor cell destruction. Proc Natl Acad Sci U S A. 2011;108(45):18342–18347. doi: 10.1073/pnas.1106550108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, Kline J, Roschewski M, LaCasce A, Collins GP, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N Engl J Med. 2018;379(18):1711–1721. doi: 10.1056/NEJMoa1807315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, Colevas AD, O’Rourke T, Narayanan S, Papadopoulos K, et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J Clin Oncol. 2019;37(12):946–953. doi: 10.1200/JCO.18.02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C, Wang L, Weiskopf K, Majeti R. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. MAbs. 2015;7(5):946‐956. doi: 10.1080/19420862.2015.1062192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Bommel PE, He Y, Schepel I, Hendriks MAJM, Wiersma VR, van Ginkel RJ, van Meerten T, Ammatuna E, Huls G, Samplonius DF. CD20-selective inhibition of CD47-SIRPα “don’t eat me” signaling with a bispecific antibody-derivative enhances the anticancer activity of daratumumab, alemtuzumab and obinutuzumab. Oncoimmunology. 2018;7(2):e1386361. doi: 10.1080/2162402X.2017.1386361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rokita M, Stec R, Bodnar L, Charkiewicz R, Korniluk J, Smoter M, Cichowicz M, Chyczewski L, Niklinski J, Kozlowski W, et al. Overexpression of epidermal growth factor receptor as a prognostic factor in colorectal cancer on the basis of the all red scoring system. Onco Targets Ther. 2013. July;24(6):967–976. doi: 10.2147/OTT.S42446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selvaggi G, Novello S, Torri V, Leonardo E, De Giuli P, Borasio P, Mossetti C, Ardissone F, Lausi P, Scagliotti GV. Epidermal growth factor receptor overexpression correlates with a poor prognosis in completely resected non-small-cell lung cancer. Ann Oncol. 2004. January 1;15(1):28–32. doi: 10.1093/annonc/mdh011. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida T, Okamoto I, Okabe T, Iwasa T, Satoh T, Nishio K, Fukuoka M, Nakagawa K. Matuzumab and cetuximab activate the epidermal growth factor receptor but fail to trigger downstream signaling by Akt or Erk. Int J Cancer. 2008;122(7):1530–1538. [DOI] [PubMed] [Google Scholar]
- 17.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014. August;11(8):783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kikuchi Y, Uno S, Yoshimura Y, Otabe K, Iida S, Oheda M, Fukushima N, Tsuchiya M. A bivalent single-chain fv fragment against CD47 induces apoptosis for leukemic cells. Biochem Biophys Res Commun. 2004;315(4):912–918. doi: 10.1016/j.bbrc.2004.01.128. [DOI] [PubMed] [Google Scholar]
- 19.Wiersma VR, He Y, Samplonius DF, van Ginkel RJ, Gerssen J, Eggleton P, Zhou J, Bremer E, Helfrich W. A CD47-blocking TRAIL fusion protein with dual pro-phagocytic and pro-apoptotic anticancer activity. Br J Haematol. 2014;164(2):304–307. doi: 10.1111/bjh.12617. [DOI] [PubMed] [Google Scholar]
- 20.Helfrich W, Haisma HJ, Magdolen V, Luther T, Bom VJ, Westra J, van der Hoeven R, Kroesen BJ, Molema G, de Leij L. A rapid and versatile method for harnessing scFv antibody fragments with various biological effector functions. J Immunol Methods. 2000. April 3;237(1–2):131–145. doi: 10.1016/S0022-1759(99)00220-3. [DOI] [PubMed] [Google Scholar]
- 21.Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, Scallon B, Teplyakov A, Malia TJ, Strohl WR, et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014;65(1):114–126. doi: 10.1016/j.ymeth.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 22.Midelfort KS, Hernandez HH, Lippow SM, Tidor B, Drennan CL, Wittrup KD. Substantial energetic improvement with minimal structural perturbation in a high affinity mutant antibody. J Mol Biol. 2004;343(3):685–701. doi: 10.1016/j.jmb.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 23.Koopmans I, Hendriks D, Samplonius DF, van Ginkel RJ, Heskamp S, Wierstra PJ, Bremer E, Helfrich W. A novel bispecific antibody for EGFR-directed blockade of the PD-1/PD-L1 immune checkpoint. Oncoimmunology. 2018;7(8):e1466016. doi: 10.1080/2162402X.2018.1466016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helfrich W, Kroesen BJ, Roovers RC, Westers L, Molema G, Hoogenboom HR, de Leij L. Construction and characterization of a bispecific diabody for retargeting T cells to human carcinomas. Int J Cancer. 1998;76(2):232–239. [DOI] [PubMed] [Google Scholar]
- 25.Matlung HL, Babes L, Zhao XW, van Houdt M, Treffers LW, van Rees DJ, Franke K, Schornagel K, Verkuijlen P, Janssen H. Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep. 2018;23(13):3946–3959.e6. doi: 10.1016/j.celrep.2018.05.082. [DOI] [PubMed] [Google Scholar]
- 26.Reinhold MI, Lindberg FP, Plas D, Reynolds S, Peters MG, Brown EJ. In vivo expression of alternatively spliced forms of integrin-associated protein (CD47). J Cell Sci. 1995;108:3419–3425. [DOI] [PubMed] [Google Scholar]
- 27.Baker K, Rath T, Lencer WI, Fiebiger E, Blumberg RS. Cross-presentation of IgG-containing immune complexes. Cell Mol Life Sci. 2013;70(8):1319–1334. doi: 10.1007/s00018-012-1100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, Willingham S, Howard M, Prohaska S, Volkmer J, et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS One. 2015. [Accessed 2015 September 21];10(9):e0137345. doi: 10.1371/journal.pone.0137345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwong LS, Brown MH, Barclay AN, Hatherley D. Signal-regulatory protein α from the NOD mouse binds human CD47 with an exceptionally high affinity – implications for engraftment of human cells. Immunology. 2014;143(1):61–67. doi: 10.1111/imm.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y, Ma Y, Gao P, Yao Z. Targeting CD47: the achievements and concerns of current studies on cancer immunotherapy. J Thorac Dis. 2017;9(2):E168‐E174. doi: 10.21037/jtd.2017.02.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, Xu H, Peng H, Fu Y-X, Xu MM. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21:1209–1215. doi: 10.1038/nm.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strowig T, Rongvaux A, Rathinam C, Takizawa H, Borsotti C, Philbrick W, Eynon EE, Manz MG, Flavell RA. Transgenic expression of human signal regulatory protein alpha in Rag2−/−γc−/− mice improves engraftment of human hematopoietic cells in humanized mice. Proc Natl Acad Sci U S A. 2011;108:13218–13223. doi: 10.1073/pnas.1109769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nigro A, Ricciardi L, Salvato I, Sabbatino F, Vitale M, Crescenzi MA, Montico B, Triggiani M, Pepe S, Stellato C, et al. Enhanced expression of CD47 is associated with off-target resistance to tyrosine kinase inhibitor gefitinib in NSCLC. Front Immunol. 2019;10:3135. doi: 10.3389/fimmu.2019.03135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.