

ABSTRACT
High mobility group box 1 (HMGB1) is involved in the induction of airway inflammation and injury in patients with chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). HMGB1 increased by transforming growth factor-β1 (TGF-β1), impairs airway epithelial barrier function in the lung. In the present study, to investigate how HMGB1 affects the barrier of normal human lung epithelial (HLE) cells, monolayer cells (2D culture) and bronchial-like spheroid cells (2.5 D Matrigel culture), which have lumen formation, were pretreated with TGF-β type I receptor kinase inhibitor EW-7197 before treatment with HMGB1. In 2D culture, treatment with HMGB1 decreased expression of angulin-1/LSR, TRIC and CLDN-1, −4, −7 and increased that of CLDN-2. Pretreatment with EW-7197 prevented the changes of all tight junction molecules induced by HMGB1. In 2.5D Matrigel culture, treatment with HMGB1 induced permeability of FITC-dextran (FD-4) into the lumen, whereas pretreatment with EW-7197 prevented the hyperpermeability of FD-4 into the lumen caused by HMGB1. In 2.5D Matrigel culture, knockdown of transcription factor p63 prevented the hyperpermeability induced by HMGB1 as well as pretreatment with EW-7197. In the 2D culture of HLE cells with HMGB1, knockdown of p63 increased the level of angulin-1/LSR and CLDN-4, while pretreatment with EW-7197 enhanced the increase of CLDN-4 induced by knockdown of p63. Immunohistochemical analysis of IPF, CLDN-2, HMGB1 and p63 revealed that their levels were higher in the regenerative epithelium of the terminal bronchial region than in normal epithelium. HMGB1 induces epithelial permeability of HLE cells via p63/TGF-β signaling in normal lung and IPF.
KEYWORDS: HMGB1, epithelial permeability, tight junctions, TGF-Β, EW-7197, TNFα-antibody, p63, 2.5D Matrigel cultures, normal human lung epithelial cells
Introduction
High mobility group box 1 (HMGB1) is one of the damage-associated molecular patterns (DAMPs) and is also a proinflammatory mediator belonging to the alarmin family1. HMGB1 is involved in the induction of airway inflammation and injury in patients with allergy, asthma, chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF) and infection of respiratory virus infections.2–4 The HMGB1 levels in sputum and serum are higher in patients with asthma, COPD, and IPF than in healthy subjects.2,5 However, the effects of HMGB1 on the epithelial barrier of normal human lung and IPF are unclear.
HMGB1 impairs airway epithelial barrier function through activation of the receptor for advanced glycation end-products (RAGE)/ERK pathway.6 The RAGE pathway is involved in lung epithelial wound repair via enhanced cell migration and cell proliferation.7 TNF-α stimulates the production and secretion of HMGB1 in cultured airway epithelial cells.8 HMGB1 expression is increased by transforming growth factor-β1 (TGF-β1) and knockdown of HMGB1 reverses TGF-β1-induced epithelial–mesenchymal transition (EMT) in human alveolar epithelial cell line A549 and normal human bronchial epithelial cell line BEAS-2B.9 EW-7197 is a TGF-β type I receptor kinase inhibitor with potential anti-inflammatory and antifibrotic properties.10 However, it is not yet known whether EW-7197 prevents the epithelial barrier disruption triggered by HMGB1 in inflammatory pulmonary diseases.
Lungs are composed of a system of branched tubes that guide the inhaled air toward alveoli and contribute to the gas exchange. The proximal and distal regions of the lung epithelial cells specialized for different functions: basal, secretory and ciliated cells in the conducting airways and type II and type I cells lining the alveoli.11 The bronchiolar and alveolar epithelium is positive for Ck7, Ck8, Ck18 and Ck19.12 The barrier function of lung epithelium depends on tight junctions. These heteromeric tight junction protein complexes form the sealing interface between adjacent epithelial cells in the bronchiolar and alveolar epithelium.13
The tricellular tight junction molecules angulin-1/lipolysis-stimulated lipoprotein receptor (LSR), and tricellulin (TRIC) and bicellular tight junction molecules occludin (OCLN) and claudins (CLDNs) form the airway epithelial barrier including lung/bronchial epithelial cells. CLDNs are major components of TJs in epithelial and endothelial cells.14 CLDN-2 is expressed in the tight junctions of leaky epithelia, where it forms cation-selective and water permeable paracellular channels.15 CLDN-2 expression is modulated by a variety of conditions.16 In the normal lung, bronchiolar epithelial cells express CLDN-1, −2, −3, −4, and −7, whereas CLDN-3, −4, and −18 are expressed in normal alveolar epithelium, mainly in type II pneumocytes.17,18 CLDN-4 has a positive correlation with alveolar fluid clearance.19 It augments alveolar epithelial barrier function and is induced in acute lung injury.20 In IPF, an increase in CLDN-2 expression in bronchiolar and alveolar epithelium and a decrease in CLDN-4 expression in type II pneumocytes are observed.21 Angulin-1/LSR contributes to the epithelial barrier.22 We previously reported that HMGB1-downregulated angulin-1/LSR induces epithelial barrier disruption via CLDN-2 in airway epithelial Calu-3 cells.23 However, the detailed roles of angulin-1/LSR remained unknown in the normal human lung and IPF.
Transcription factor p63, which is a member of the p53 family, plays a crucial role in the proliferation and differentiation of various epithelial basal cells.24 p63 regulates the epithelial barrier via various signaling pathways in the upper airway epithelium.25,26 p63 contributes to the formation and maintenance of differentiated pseudostratified bronchial epithelium and epithelial remodeling.27 In bronchioles of IPF lungs, the numbers of p63-positive cells are increased.27 Some p63-positive basal cells undergo EMT, and knockdown of p63 prevents the phenotypic switch in bronchial epithelial cells.28
In the present study, to investigate how HMGB1 affects the barrier of normal human lung epithelial (HLE) cells, monolayer cells (2D culture) and bronchial-like spheroid cells (2.5 D Matrigel culture), which have lumen formation, were derived from human lung tissues and the changes of the epithelial permeability and tight junction molecules were examined. We finally promote the development of a therapy for lung inflammation and injury in patients with IPF. For this purpose, 2.5D Matrigel culture of normal HLE cells is a useful model in vitro.
Materials and methods
Ethics statement
The protocol for the human study was reviewed and approved by the ethics committee of the Sapporo Medical University School of Medicine. Written informed consent was obtained from each patient who participated in the investigation. All experiments were carried out in accordance with the approved guidelines and the Declaration of Helsinki.
Antibodies and reagents
A rabbit polyclonal anti-lipolysis-stimulated lipoprotein receptor (LSR) antibody was obtained from Novus Biologicals (Littleton, CO, USA). Rabbit polyclonal anti-claudin (CLDN)-2 and mouse monoclonal anti-CLDN-2 (Clone 12H12) and occludin (OCLN) (Clone OC-3 F10) antibodies were obtained from Zymed Laboratories (San Francisco, CA, USA). Recombinant human HMGB1 was from Sigma-Aldrich (St. Louis, MO, USA). Mouse monoclonal anti-p63 (Clone DAK-p63) and anti-Ck7 (Clone OV-TL 12/13) antibodies were from Dako A/S (Glostrup, Denmark). Rabbit polyclonal anti-p63 and anti-HMGB1 antibodies were from Abcam (Cambridge, MA, USA). A rabbit polyclonal anti-p40 (ΔNp63) antibody was from Nichirei Biosciences Inc. (Tokyo, Japan). The TGF-β receptor type 1 inhibitor EW-7197 (N-((4-([1,2,4] triazolo [1,5-a] pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1 H-imidazol-2-yl) methyl)-2-fluoroaniline) was obtained from Cayman Chemical (Ann Arbor, MI). The anti-human-TNF-α mAb adalimumab (ADA, Humira®) was purchased from Eisai Co., Ltd. (Tokyo, Japan). Alexa 488 (green)-conjugated anti-rabbit IgG and Alexa 594 (red)-conjugated anti-mouse IgG antibodies and Alexa 594 (red)-conjugated phalloidin were from Molecular Probes, Inc. (Eugene, OR). HRP-conjugated polyclonal goat anti-rabbit IgG was from Dako A/S (Glostrup, Denmark). FITC-dextran (FD-4, MW 4.0 kDa) was obtained from Sigma-Aldrich Co. (St. Louis, MO). The detailed information of antibodies and reagents is indicated as supplemental Table 1 and 2.
Isolation and culture of primary cultured human lung epithelial (HLE) cells
Human lung tissues were obtained from patients with lung carcinoma who underwent lung resection in the Sapporo Medical University hospital. Informed consent was obtained from all patients and the study was approved by the ethics committee of Sapporo Medical University.
Macroscopic normal lung tissues surrounding lung carcinoma were minced into pieces 2 to 3 mm3 in volume and washed with PBS containing 100 U/ml penicillin and 100 μg/ml streptomycin (Lonza Walkersville, Walkersville, MD) three times. These minced tissues were suspended in 10 ml of Hanks’ balanced salt solution with 0.5 μg/ml DNase I and 0.04 mg/ml Liberase Blendzyme 3 (Roche, Basel, Switzerland) and then incubated with bubbling of mixed O2 gas containing 5% CO2 at 37°C for 10 minutes. The dissociated tissues were subsequently filtered with 300-μm mesh followed by filtration with 70-μm mesh (Cell Strainer, BD Biosciences, San Jose, CA). After centrifugation at 1000 × g for 4 minutes, isolated cells were cultured in bronchial epithelial basal medium (BEBM, Lonza Walkersville) containing 10% fetal bovine serum (FBS) (CCB, Nichirei Bioscience, Tokyo, Japan) and supplemented with BEGM Single-Quots (Lonza Walkersville; including 0.4% bovine pituitary extract, 0.1% insulin, 0.1% hydrocortisone, 0.1% gentamicin, amphotericin-B [GA-1000], 0.1% retinoic acid, 0.1% transferrin, 0.1% triiodothyronine, 0.1% epinephrine, and 0.1% human epidermal growth factor), 100 U/ml penicillin and 100 μg/ml streptomycin on 60-mm culture dishes (Corning Life Sciences, Acton, MA), coated with rat tail collagen (500 μg of dried tendon/ml of 0.1% acetic acid). Following the above protocol, tissue dissociation and cell isolation were repeated for the same sample a maximum of seven times. The cells were placed in a humidified 5% CO2:95% air incubator at 37°C. The retroviral vector BABE-hygro-hTERT (kindly provided by Dr. Robert Weinberg) was used. The viral supernatant was produced from an ecotropic packaging cell line by transfection of plasmid DNA as reported previously.21 The packaging cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% FBS and supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin. At 24 hours after plating on 60-mm dishes, HLE cells in primary culture were exposed overnight to the viral supernatant containing the retrovirus. After being washed with serum-free BEBM medium, the hTERT-transfected HLE cells were cultured in serum-free BEBM medium supplemented with the above-mentioned factors and 2.5 μg/ml Amphotericin-B. hTERT-HLE cells became confluent on the 60-mm culture dishes in 2 to 3 weeks, the first passage was done using 0.05% trypsin-EDTA (Sigma-Aldrich) in 35-mm culture dishes or 35-mm culture glass-coated dishes (Iwaki; Chiba, Japan) and was used for the experiments at days 7 to 14 after plating. Some cells treated with or without 10% FBS were treated with 10 μM EW-7197.
RNA interference studies
For RNA interference studies, small interfering RNA (siRNA) duplexes targeting the mRNA sequences of human p63 were purchased from Thermo Fisher Scientific (Waltham, MA). The sequences were as follows: siRNA-A of p63 (forward sense 5ʹ-CCCACGCAACCCAUCGUCAUCUGGA-3ʹ, reverse sense 5ʹ-UCCAGAUGACGAUGGGUUGCGUGGG-3ʹ). A scrambled siRNA sequence (BLOCK-iT Alexa Fluor fluorescent, Invitrogen) was employed as control siRNA. At one day before transfection, HLE cells were plated in medium without antibiotics such that they would be half confluent at the time of transfection. The cells were transfected with 100 nM siRNAs using Lipofectamine RNAiMAX (Invitrogen) as a carrier according to the manufacturer’s instructions for 48 h. Some cells were pretreated with 10 μM EW-7197 before treatment with 100 nM HMGB1.
2.5-dimensional (2.5D) Matrigel culture
Thirty-five-mm culture dishes or 35-mm culture glass-coated dishes were coated with 100% Matrigel (30 μl or 15 μl) at 4 C° and incubated at 37 C° for 30 min. HLE cells (5 × 104) were plated in BEGM medium with 10% Matrigel and cultured for 4 days in BEGM medium without FBS. Some cells were pretreated with 10 μM EW-7197 or 40 μg of the TNFα antibody before treatment with 100 μM HMGB1. In all experiments, 10 spheroids were examined.
Immunocytochemistry
The cells in the 35-mm glass-coated wells (Iwaki; Chiba, Japan) were fixed with cold acetone and ethanol (1:1) at −20°C for 10 min. After rinsing in PBS, the cells were incubated with anti-p63, anti-Ck7, anti-LSR and anti-OCLN antibodies (1:100) and Alexa 594-phalloidin (1:200) overnight at 4°C. Alexa Fluor 488 (green)–conjugated anti-rabbit IgG and Alexa Fluor 592 (red)–conjugated anti-mouse IgG (Invitrogen) were used as secondary antibodies. The specimens were examined and photographed with an Olympus IX 71 inverted microscope (Olympus Co.; Tokyo, Japan) and a confocal laser scanning microscope (LSM5 PASCAL; Carl Zeiss, Jena, Germany).
Fluorescein isothiocyanate (FITC) permeability assay
To assess barrier function, the permeability of Fluorescein isothiocyanate (FITC)-dextran (FD-4, MW 4.0 kDa) from the outside into the spheroid lumen was examined by using 2.5D Matrigel culture of HLE cells on 35-mm glass-coated dishes. 2.5D Matrigel culture of HLE cells was incubated in the medium with 1% FD-4 at 37 C° for 2 h. Ten spheroids of all experiments were photographed and measured by a confocal laser scanning microscope with imaging soft (LSM5 PASCAL; Carl Zeiss, Jena, Germany).
RNA isolation and reverse transcription polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted and purified using TRIzol (Invitrogen, Carlsbad, CA). One microgram of total RNA was reverse-transcribed into cDNA using a mixture of oligo (dT) and Superscript II reverse transcriptase according to the manufacturer’s recommendations (Invitrogen). The synthesis of each cDNA was performed in a total volume of 20 µl for 50 min at 42°C and terminated by incubation for 15 min at 70°C. PCR was performed in a 20-µl total mixture containing 100 pM primer pairs, 1.0 µl of the 20-µl total RT product, PCR buffer, dNTPs and Taq DNA polymerase according to the manufacturer’s recommendations (Takara, Kyoto, Japan). Amplifications were for 25–35 cycles depending on the PCR primer pair with cycle times of 15 s at 96°C, 30 s at 55°C and 60 s at 72°C. The final elongation time was 7 min at 72°C. Seven microliters of the total 20-µl PCR product was analyzed by 1% agarose gel electrophoresis with ethidium bromide staining and standardized using a GeneRuler TM 100 bp DNA ladder (Fermentas, Ontario, Canada). The PCR primers used to detect CLDN-1, −2, −4, −7, LSR, TRIC, and glucose-3-phosphate dehydrogenase (G3PDH) had the sequences shown in Supplementary Table 1.
Western blot analysis
The cells were scraped from a 35 mm dish containing 400 μl of buffer (1 mM NaHCO3 and 2 mM phenylmethylsulfonyl fluoride), collected in microcentrifuge tubes, and then sonicated for 8 s. Aliquots of 15 μl of 20 μg protein/lane for each sample were separated by electrophoresis in 5–20% SDS polyacrylamide gels (Wako, Osaka, Japan), and electrophoretically transferred to a nitrocellulose membrane (Immobilon; Millipore Co.; Bedford, UK). The membrane was incubated with anti-LSR, anti-TRIC, anti-CLDN-1, anti-CLDN-2, anti-CLDN-4, anti-CLDN-7, anti-p63 and anti-actin antibodies (1:1000) at 4°C overnight. Then it was incubated with an HRP-conjugated anti-rabbit IgG antibody at room temperature for 1 h. The immunoreactive bands were detected using an ECL Western blotting system (GE Healthcare, Little Chalfont, UK). The bands were quantitated by densitometry and the data normalized to actin.
Immunohistochemical analysis
Human lung tissues were obtained from eight patients with idiopathic pulmonary fibrosis who underwent lung resection at Sapporo Medical University. Informed consent was obtained from all patients and this study was approved by the ethics committee of the university. The tissues were embedded in paraffin after fixation with 10% formalin in PBS. Briefly, 5-μm-thick sections were dewaxed in xylene, rehydrated in ethanol, and heated with Vision BioSystems Bond Max using ER2 solution (Leica) in an autoclave for antigen retrieval. Endogenous peroxidase was blocked by incubation with 3% hydrogen peroxide in methanol for 10 min. The tissue sections were then washed twice with Tris-buffered saline (TBS) and preblocked with Block Ace for 1 h. After washing with TBS, the sections were incubated with anti-LSR (1:100), anti-CLDN-2 (1:400), anti-HMGB1 (1:400) and anti-p63 (1:400) antibodies for 1 h. They were then washed three times in TBS and incubated using Vision BioSystems Bond Polymer Refine Detection kit DS9800. After three washes in TBS, a diamino-benzidine tetrahydrochloride working solution was applied. Finally, the sections were counterstained with hematoxylin. A negative control was performed by replacing the first antibodies with normal rabbit serum.
Data analysis
Signals were quantified using Scion Image Beta 4.02 Win (Scion, Frederick, MD). Each set of results shown is representative of at least three separate experiments. Results are given as means ± SEM. Statistical analysis was tested by one-way analysis of variance (ANOVA) with the Tukey–Kramer method. Statistical significance was set at *P < .05.
Results
Characterization of 2-dimensional (2D) culture of primary cultured human lung epithelial (HLE) cells
We characterized the 2D culture of primary cultured human lung epithelial (HLE) cells isolated from lung specimens excised from lung cancer patients. Immunostaining showed that basal cell marker p63 was expressed in the nuclei and epithelial marker Ck7 was expressed in the cytoplasm (Figure 1a). RT-PCR revealed mRNAs of bicellular tight junction molecules (bTJs) CLDN-1, −2, −4, −7 and −18 and tricellular tight junction molecules (tTJs) LSR and TRIC in the cells (Figure 1b). In Western blotting, proteins of CLDN-1, −2, −4, −7, LSR and TRIC were detected in the cells (Figure 1d). In the cells without FBS, treatment with TGF-β receptor type 1 inhibitor 10 μM EW-7197 induced bTJ OCLN and LSR at the membranes in immunostaining, while OCLN was not detected in the control (Figure 1c). In Western blotting, no changes of CLDN-1, −2, −4, −7, LSR and TRIC were observed in the cells treated with 10 μM EW-7197 compared to the control (Figure 1d).
Figure 1.
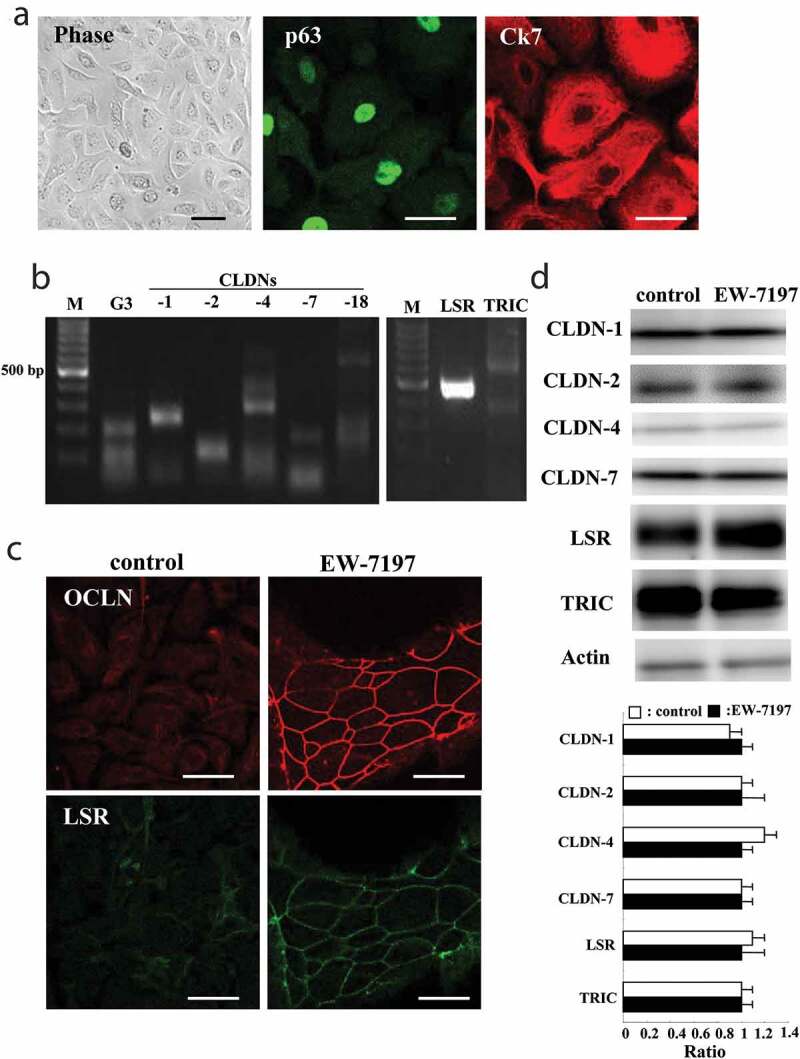
Characterization of 2D culture of HLE cells without FBS and effects of EW-7197 on tight junction molecules in 2D culture of HLE cells without FBS. (a) Phase-contrast and immunocytochemistry images for p63 and Ck7 in 2D culture of HLE cells without FBS. Scale bar, 20 μm. (b) RT-PCR for CLDN-1, −2, −4, −7, −18, LSR, TRIC and G3PDH (G3) in 2D-culture of HLE cells without FBS. M: 100 bp DNA ladder marker. (c) Immunocytochemistry for OCLN and LSR in 2D-culture of HLE cells (without FBS) treated with 10 μM EW-7197 for 24 h. (d) Western blotting for LSR, CLDN-1, −2, −4, −7, LSR, TRIC and actin in 2D culture of HLE cells (without FBS) treated with 10 μM EW-7197 for 24 h. The corresponding expression levels of D are shown as bar graphs
Effects of HMGB1 and EW-7197 on expression of bTJs and tTJs in 2D culture of HLE cells
We previously reported that HMGB1 disrupts the epithelial barrier with downregulated LSR and upregulated CLDN-2 in airway Calu-3 cells.19 To investigate whether HMGB1 affected expression of TJs in HLE cells, 2D cultures of HLE cells were treated with 100 ng/ml HMGB1 for 24 h. Western blotting showed that treatment with HMGB1 decreased expression of LSR, TRIC and CLDN-1, −4, −7 and increased that of CLDN-2 (Figure 2a). Furthermore, treatment with 10 μM EW-7197 prevented the changes of all TJs induced by HMGB1 (Figure 2a). Immunostaining showed that treatment with HMGB1 downregulated OCLN and LSR at the membranes, whereas EW-7197 prevented the changes (Figure 2b).
Figure 2.
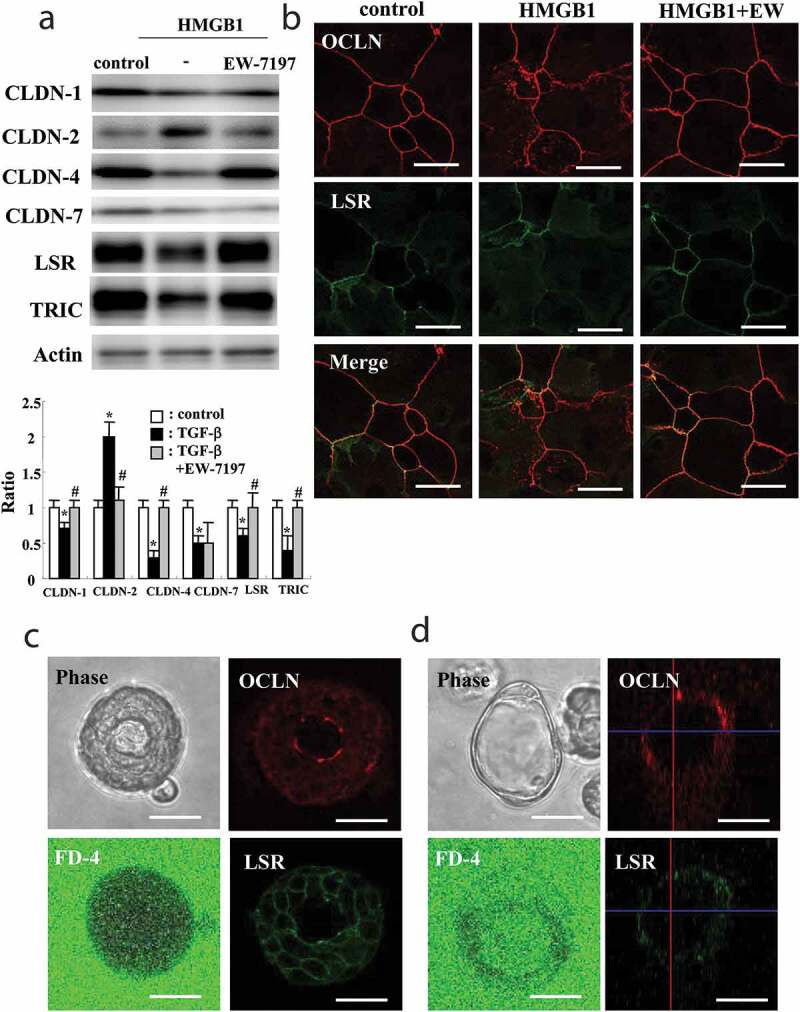
Effects of HMGB1 on tight junction molecules of 2D culture of HLE cells with 10% FBS and characterization of 2.5D Matrigel culture of HLE cells. (a) Western blotting for CLDN-1, −2, −4, −7, LSR, TRIC and actin in 2D culture of HLE cells pretreated with 10 μM EW-7197 2 h before treatment with 100 ng/ml HMGB1 for 24 h. The corresponding expression levels of A are shown as bar graphs. control vs *p < .05, TGF-β vs #p < .05. (b) Immunocytochemistry for OCLN and LSR in 2D culture of HLE cells (with 10% FBS) pretreated with 10 μM EW-7197 2 h before treatment with 100 ng/ml HMGB1 for 24 h. Scale bar, 20 μm. (c, d) Phase-contrast images and immunocytochemistry for OCLN and LSR and epithelial permeability assay using FITC-4kDa dextran (FD-4) in 2.5D Matrigel culture of HLE cells. (c) Bronchial-like spheroid cells. (d) alveolar-like spheroid cells. Scale bar, 20 μm
Characterization of 2.5-dimensional (2.5D) Matrigel culture of HLE cells
To investigate the detailed effects of HMGB1 on HLE cells, we established a 2.5D Matrigel culture system similar to the conditions in vivo. In this culture system, HLE cells with bronchial-like morphology and alveolar-like morphology were observed (Figure 2c,d). In the bronchial-like spheroid cells, FD-4 permeability was not detected, whereas in the alveolar-like cells, FD-4 was strongly observed (Figure 2c,d). Immunostaining showed that, in the bronchial-like spheroid cells, OCLN was strongly expressed at the luminal surface and LSR was expressed throughout the membranes of all cells (Figure 2c). In the alveolar-like cells, OCLN and LSR were weakly detected at the cell borders (Figure 2d).
Effects of HMGB1, EW-7197 and TNFα-antibody in 2.5D Matrigel culture of HLE cells
To investigate the effects of HMGB1, EW-7197 and the TNFα-antibody on the 2.5D Matrigel culture of HLE cells, the bronchial-like spheroid cells were used and pretreated with 10 μM EW-7197 or 40 ng/ml TNFα-antibody before treatment with 100 ng/ml HMGB1 for 24 h. Treatment with HMGB1 induced the permeability of FD-4 into the lumen of 8 spheroids/10 spheroids, whereas treatment with EW-7197 or the TNFα-antibody prevented the hyperpermeability of FD-4 into the lumen of 7 spheroids/10 spheroids or 8 spheroids/10 spheroids induced by HMGB1 (Figure 3a,c, supplemental Figure 2). Immunostaining showed that, in treatment with HMGB1, LSR was decreased at the membranes, while OCLN was detected at the luminal surface of 8 spheroids/10 spheroids (Figure 3b). Treatment with EW-7197 or the TNFα-antibody prevented the changes of TJs in 7 spheroids/10 spheroids or 8 spheroids/10 spheroids caused by HMGB1 (Figure 3d). Western blotting of the 2.5D Matrigel culture showed that treatment with EW-7197 or the TNFα-antibody decreased expression of CLDN-2 compared to the control and only HMGB1-treatment (Figure 3e,f).
Figure 3.
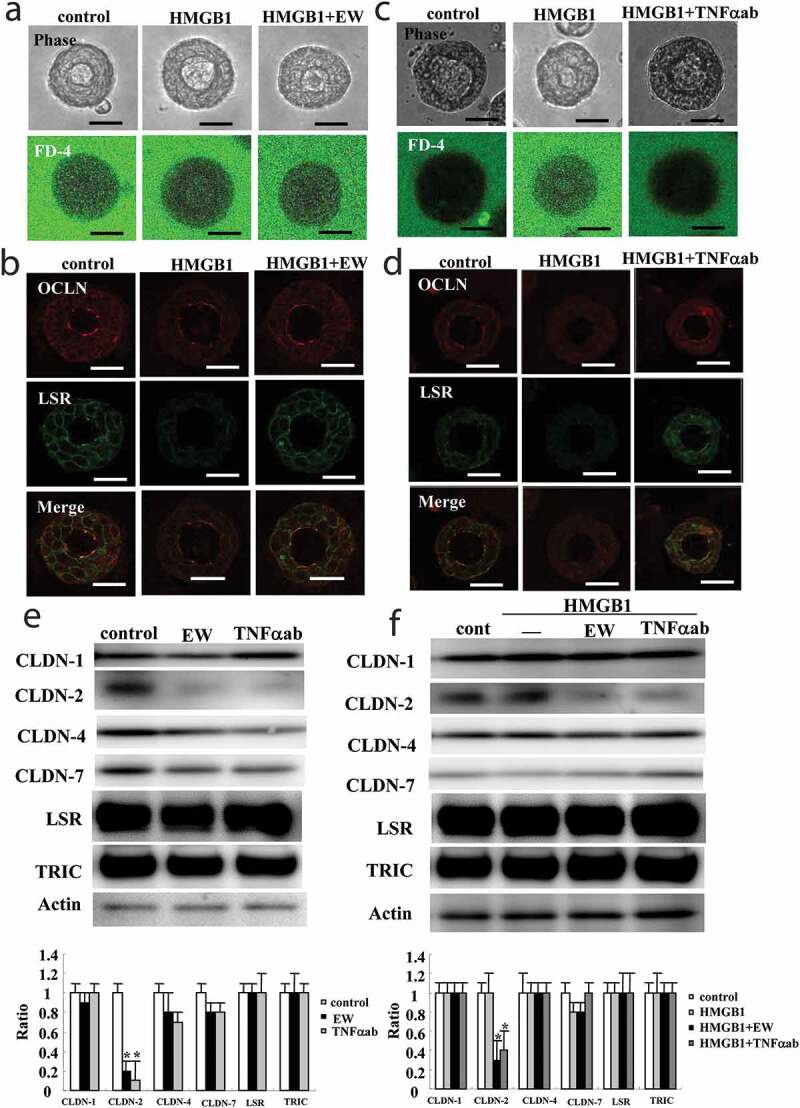
Effects of EW-7197 and TNFα-antibody on epithelial permeability and tight junction molecules treated with HMGB1 in 2.5D Matrigel culture of HLE cells. (a) Phase-contrast images and FD-4 assay of 2.5D Matrigel culture of HLE cells pretreated with 10 μM EW-7197 (EW) 2 h before treatment with 100 ng/ml HMGB1 for 24 h. Scale bar, 20 μm. (b) Immunocytochemistry for OCLN and LSR in 2.5D Matrigel culture of HLE cells pretreated with 10 μM EW-7197 (EW) 2 h before treatment with 100 ng/ml HMGB1 for 24 h. Scale bar, 20 μm. (c) Phase-contrast image and FD-4 assay in 2.5D Matrigel culture of HLE cells pretreated with 40 μM TNFα-antibody (TNFαab) 2 h before treatment with 100 ng/ml HMGB1 for 24 h. Scale bar, 20 μm. (d) Immunocytochemistry for OCLN and LSR in 2.5D Matrigel culture of HLE cells pretreated with 40 μM TNFα-antibody (TNFαab) 2 h before treatment with 100 ng/ml HMGB1 for 24 h. Scale bar, 20 μm. (e) Western blotting for CLDN-1, −2, −4, −7, LSR, TRIC and actin in 2.5D Matrigel culture of HLE cells cells pretreated with 10 μM EW-7197 2 h before treatment with 100 ng/ml HMGB1 for 24 h. (f) Western blotting for CLDN-1, −2, −4, −7, LSR, TRIC and actin in 2.5D Matrigel culture of HLE cells pretreated with 40 μM TNFα-antibody (TNFαab) 2 h before treatment with 100 ng/ml HMGB1 for 24 h. The corresponding expression levels of E and F are shown as bar graphs. Control vs *p < .05
Knockdown of p63 prevented the permeability of HLE cells induced by HMGB1
To investigate whether p63 affected the permeability of HLE cells induced by HMGB1, the 2.5D Matrigel culture of HLE cells was pretransfected with siRNA of p63 and Scrambled siRNA as the control for 48 h before treatment with 100 ng/ml HMGB1 or 100 ng/ml HMGB1 + 10 μM EW-7197 for 24 h. Knockdown of p63 prevented the hyperpermeability induced by treatment with HMGB1, like treatment with EW-7197 (Figure 4a, supplemental Figure 3), while knockdown of p63 did not affect the permeability. In Western blotting of the 2D culture of HLE cells, knockdown of p63 was found to increase expression of LSR and CLDN-4 (Figure 4b). Furthermore, EW-7197 enhanced the increase of CLDN-4 induced by knockdown of p63 (Figure 4b). In Scrambled siRNA controls, no change was observed.
Figure 4.
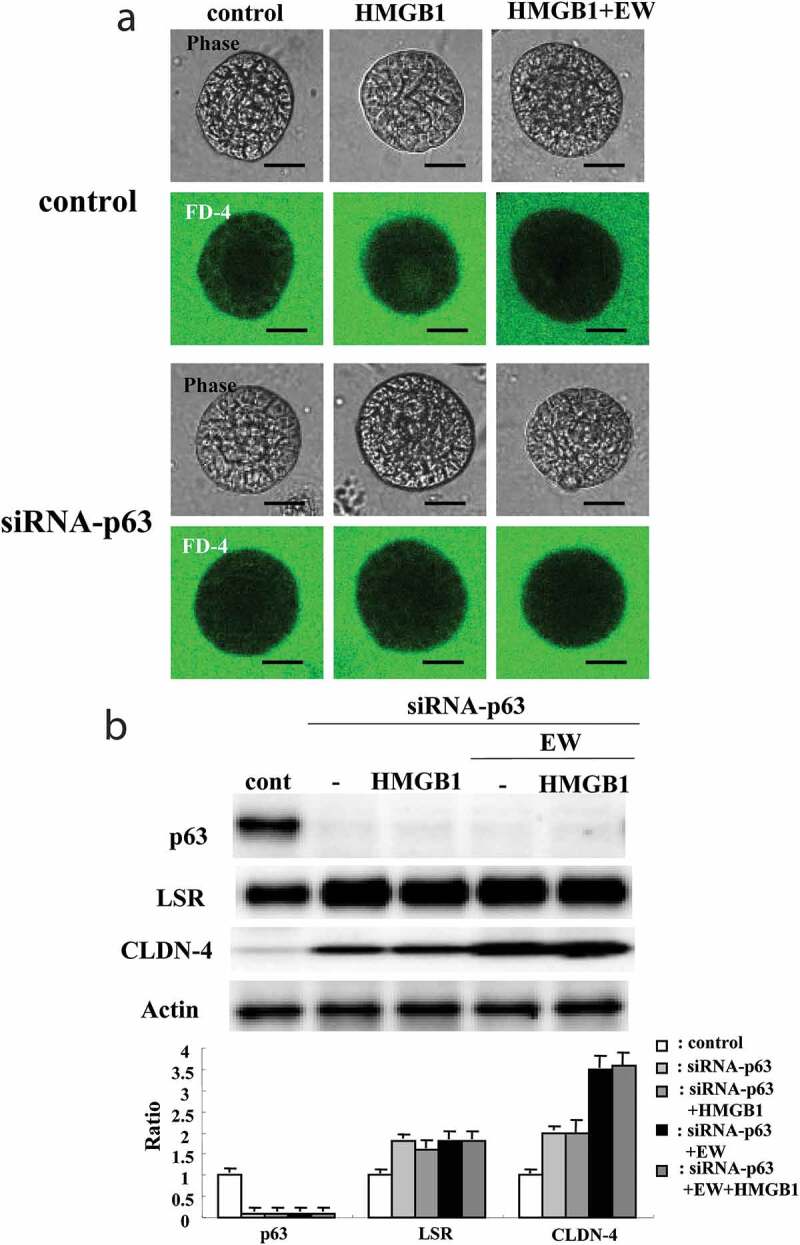
Effects of p63 knockdown on epithelial permeability and tight junction molecules in 2.5D Matrigel culture of HLE cells. (a) Phase-contrast images and FD-4 assay in 2.5D Matrigel culture of HLE cells transfected with siRNA of p63 for 48 h and then pretreated with 10 μM EW-7197 2 h before treatment with 100 ng/ml HMGB1 for 24 h. Scale bar, 20 μm. (b) Western blotting for p63, LSR, CLDN-4 and actin in 2.5D Matrigel culture of HLE cells transfected with siRNA of p63 for 48 h and then pretreated with 10 μM EW-7197 2 h before treatment with 100 ng/ml HMGB1 for 24 h. The corresponding expression levels of B are shown as bar graphs
Expression patterns of LSR, CLDN-2, HMGB1 and p63 in the peripheral bronchial epithelium of idiopathic pulmonary fibrosis (IPF)
We performed immunohistochemical analysis for LSR, CLDN-2, HMGB1 and p63 in the normal peripheral bronchial epithelium and that of idiopathic pulmonary fibrosis (IPF). The immunohistochemical results for IPF showed that, in the regenerative epithelium of the terminal bronchial region, the expression of CLDN-2, HMGB1 and p63 was higher than that of normal epithelium, whereas LSR was not detected. In degenerative epithelium where p63 was not detected, LSR, CLDN-2, HMGB1 levels were similar to those in the normal epithelium (Figure 5).
Figure 5.
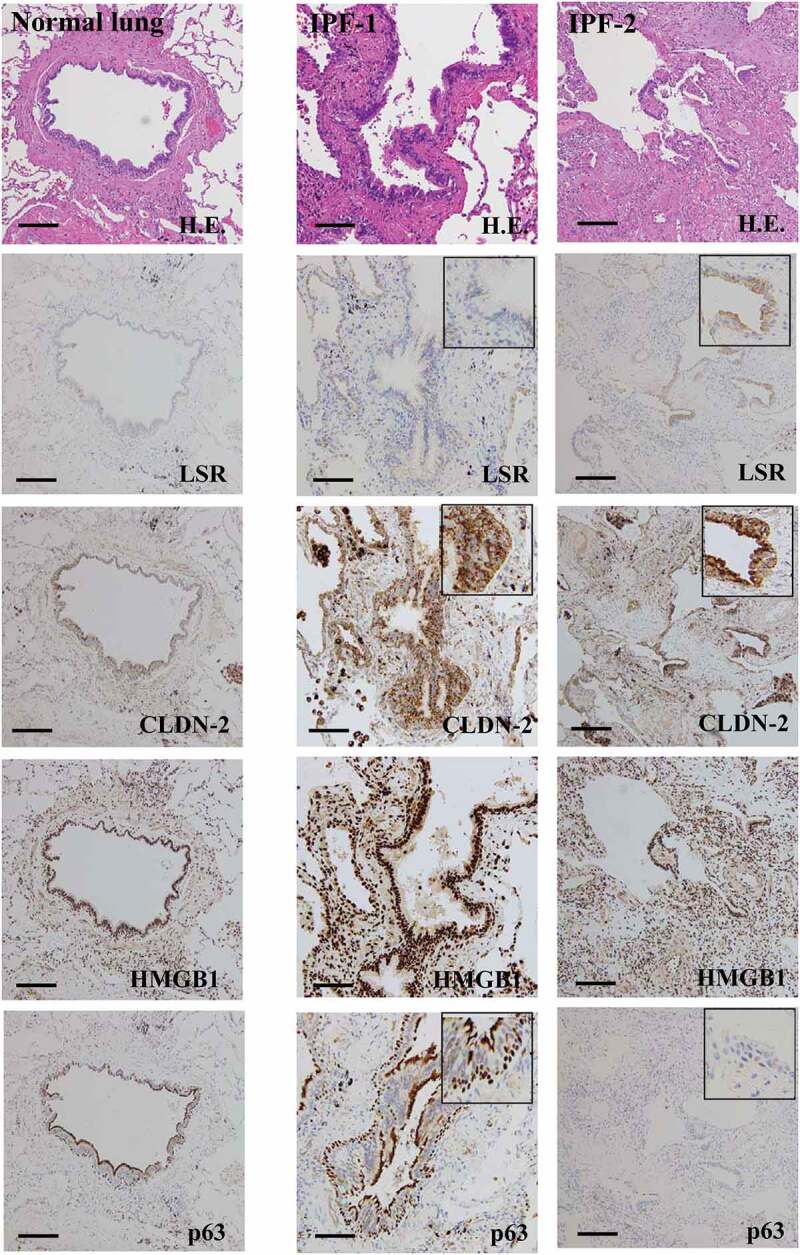
Expression and distribution of LSR, CLDN-2, HMGB1 and p63 in human normal lung tissues and IPF lung tissues. Hematoxylin-eosin (H.E.) staining and immunohistochemical staining for p63, CLDN-2 and LSR in terminal bronchial tissues of human normal lung and terminal bronchial tissues of IPF lung. Bar: 50 μm
Discussion
In the present study, HMGB1 induced epithelial permeability of normal HLE cells with downregulation of LSR, TRIC and CLDN-1, −4, −7 and upregulation of CLDN-2 in 2D and 2.5D cultures. The TGF-β type I receptor kinase inhibitor EW-7197 prevented the changes of hyperpermeability and expression of tight junction molecules induced by HMGB1. The knockdown of p63 prevented the hyperpermeability by HMGB1 and enhanced expression of LSR and CLDN-4. In the immunohistochemical analysis of IPF, CLDN-2, HMGB1 and p63 were more highly expressed in the regenerative epithelium of the terminal bronchial region than in the normal epithelium and degenerative epithelium, where p63 was not detected, and LSR, CLDN-2 and HMGB1 levels were observed to be similar to the normal condition.
HMGB1 is a proinflammatory mediator belonging to the alarmin family.29 The HMGB1 level in sputum or serum is higher in patients with asthma, COPD, and IPF than in healthy subjects.2–4 In airway epithelial cell line Calu-3, HMGB1-downregulated angulin-1/LSR induces epithelial barrier disruption via the upregulation of CLDN-2 expressed in the tight junctions of leaky epithelia.23 The knockdown of angulin-1/LSR by the siRNA decreases the epithelial barrier with the upregulation of CLDN-2 in Calu-3 cells.23 Loss or downregulation of angulin-1/LSR disrupts the barriers with the relocalization of the tTJ molecule tricellulin in various cell types.30,31 HMGB1-mediated barrier hyperpermeability is caused by the downregulation of ZO-1, OCLN and CLDN-1 in primary nasal epithelial cells.32 In the present study, HMGB1 induced epithelial permeability of normal HLE cells with downregulation of LSR, TRIC and CLDN-1, −4, −7 and upregulation of CLDN-2. These findings suggested that in normal HLE cells, HMGB1 disrupted the epithelial barrier and induced the permeability via downregulation of the tight type of tight junction molecules LSR, TRIC, CLDN-1, −4, −7 and upregulation of the leaky type tight junction molecule CLDN-2 in lung/bronchial epithelial cells. Furthermore, in bronchial-like spheroids in 2.5D Matrigel culture of normal HLE cells, HMGB1 markedly decreased LSR expression at the membranes with the induction of the permeability of FD-4 into the lumen.
EW-7197 is a transforming growth factor-β type I receptor kinase inhibitor with potential anti-inflammatory and antifibrotic properties.7 It prevents ulcerative colitis-associated fibrosis and inflammation33 and prevents changes in the distribution of angulin-1/LSR and the epithelial barrier function by TGF-β in a pancreatic cancer cell line.34 It also prevents the epithelial barrier disruption due to downregulated angulin-1/LSR and upregulated CLDN-2 induced by HMGB1 in Calu-3 cells.23 In the present study, EW-7197 induced expression of OCLN and LSR at the membranes without a change of the protein level in normal HLE cells without FBS. EW-7197 prevented the changes of hyperpermeability and tight junction molecules induced by HMGB1 in normal HLE cells with FBS. TGF-β1 alters the epithelial barrier with the modification of CLDNs in various cells.35,36 In the present study of normal HLE cells without FBS, treatment with TGF-β1 decreased expression of CLDN-1, −2, −7 and EW-7197 prevented the changes of all tight junction molecules (Supplemental Figure 1). These results indicated that in normal HLE cells, HMGB1 affected epithelial barrier and tight junction molecules via TGF-β signaling and that EW-7197 could prevent the epithelial barrier disruption induced by HMGB1.
HMGB1 enhances the release of TNFα and IL-1β in macrophages via TLR4.37 In the present experiment, an anti-TNFα antibody as well as EW-7197 prevented the hyperpermeability induced by HMGB1 in 2.5D Matrigel culture of normal HLE cells. In this culture system, treatment with the anti-TNFα antibody or EW-7197 both also downregulated the expression of CLDN-2.
Transcription factor p63 plays a crucial role in the proliferation and differentiation of various epithelial basal cells.25,26 p63 contributes to the formation and maintenance of differentiated pseudostratified bronchial epithelium and epithelial remodeling.22 In bronchioles of IPF lungs, the numbers of p63-positive cells are increased.27 Some p63-positive basal cells undergo EMT in bronchial epithelial cells.28 p63 is also upregulated in the airway epithelium of chronic rhinosinusitis (CRS) and nasal polyps.25
On the other hand, knockdown of p63 prevents EMT in bronchial epithelial cells.27 Furthermore, knockdown of p63 induces CLDN-1 and −4 with Sp1 activity and enhanced epithelial barrier function in normal human nasal epithelial cells.25 It is thought that p63 negatively regulates the epithelial tight junctional barrier of the airway epithelium.25 In the present study, knockdown of transcription factor p63 prevented the hyperpermeability by HMGB1 and increased expression of angulin-1/LSR and CLDN-4 in normal HLE cells. Furthermore, in immunohistochemical analysis of IPF, levels of CLDN-2, HMGB1 and p63 were found to be higher in the regenerative epithelium of the terminal bronchial region than in the normal epithelium and degenerative epithelium, where p63 was not detected, LSR, and CLDN-2 and HMGB1 levels were observed to be similar to those in the normal epithelium. These findings suggested that HMGB1 in part contributed to the epithelial permeability and tight junction molecules via p63 in normal lung and IPF.
HMGB1 is a non-histone nuclear protein that binds to DNA in the nucleus and indirectly regulates the activities of various transcription factors.38 It is possible that HMGB1 may regulate transcription factor p63 in lung/bronchial epithelial cells, although that was unclear in the present study.
In conclusion, our findings indicated that HMGB1 affected the epithelial barrier and tight junction molecules via p63/TGF-β signaling in normal HLE cells. Treatment with EW-7197 or knockdown of p63 prevented the epithelial barrier disruption induced by HMGB1 in 2.5D Matrigel culture of normal HLE cells. p63/TGF-β signaling is an attractive target for the treatment of inflammatory lung diseases. Our findings may lead to the development of a therapy for lung inflammation and injury in patients with IPF, although further study is necessary. 2.5D Matrigel culture of normal HLE cells is similar to peripheral bronchial structures in vivo and might be a useful in vitro model for studying IPF and other lung disease.
Supplementary Material
Funding Statement
This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology of Japan [T. Kojima:19K07464; W. Arai:20K17186] and by a grant-in-aid from GlaxoSmithKline Japan [T. Konno].
Authors contributions statements
Y.K., T Kohno, A.W., H.C., H.T. and T. Kojima designed and coordinated the study and wrote the main manuscript text. T. Konno, M.T., M.M., and Y.S. analyzed the data. W.A., Y.S. and A.W. contributed reagents/materials. All authors reviewed the manuscript.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Yang H, Wang H, Chavan SS, Andersson U.. High mobility group box protein 1 (HMGB1): the prototypical endogenous danger molecule. Mol Med. 2015;21:1–12. PMID: 26605648. doi: 10.2119/molmed.2015.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hou C, Zhao H, Liu L, Li W, Zhou X, Lv Y, Shen X, Liang Z, Cai S, Zou F. High mobility group protein B1 (HMGB1) in asthma: comparison of patients with chronic obstructive pulmonary disease and healthy controls. Mol Med. 2011;17:807–815. PMID: 21380479. doi: 10.2119/molmed.2010.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hosakote YM, Brasier AR, Casola A, Garofalo RP, Kurosky A. Respiratory syncytial virus infection triggers epithelial HMGB1 release as a damage-associated molecular pattern promoting a monocytic inflammatory response. J Virol. 2016;90:9618–9631. PMID: 27535058. doi: 10.1128/JVI.01279-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamada N, Maeyama T, Kawaguchi T, Yoshimi M, Fukumoto J, Yamada M, Yamada S, Kuwano K, Nakanishi Y. The role of high mobility group box1 in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39:440–447. PMID: 18441281. doi: 10.1165/rcmb.2007-0330OC. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi K, Iwamoto H, Sakamoto S, Horimasu Y, Masuda T, Miyamoto S, Nakashima T, Ohshimo S, Fujitaka K, Hamada H, et al. Serum high-mobility group box 1 is associated with the onset and severity of acute exacerbation of idiopathic pulmonary fibrosis. Respirology. 2020;25:275–280. PMID: 31270920. doi: 10.1111/resp.13634. [DOI] [PubMed] [Google Scholar]
- 6.Huang W, Zhao H, Dong H, Wu Y, Yao L, Zou F, Cai S. High-mobility group box 1 impairs airway epithelial barrier function through the activation of the RAGE/ERK pathway. Int J Mol Med. 2016;37:1189–1198. PMID: 27035254. doi: 10.3892/ijmm.2016.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhai R, Blondonnet R, Ebrahimi E, Belville C, Audard J, Gross C, Choltus H, Henrioux F, Constantin JM, Pereira B, et al. The receptor for advanced glycation end-products enhances lung epithelial wound repair: an in vitro study. Exp Cell Res. 2020;112030. PMID: 32330509. doi: 10.1016/j.yexcr.2020.11203. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu S, Kouzaki H, Kato T, Tojima I, Shimizu T. HMGB1-TLR4 signaling contributes to the secretion of interleukin 6 and interleukin 8 by nasal epithelial cells. Am J Rhinol Allergy. 2016. May;30(3):167–172. doi: 10.2500/ajra.2016.30.4300.PMID:27216346. [DOI] [PubMed] [Google Scholar]
- 9.Gui Y, Sun J, You W, Wei Y, Tian H, Jiang S. Glycyrrhizin suppresses epithelial-mesenchymal transition by inhibiting high-mobility group box1 via the TGF-β1/Smad2/3 pathway in lung epithelial cells. PeerJ. 2020;8:e8514. PMID: 32117622. doi: 10.1111/jcmm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park SA, Kim MJ, Park SY, Kim JS, Lee SJ, Woo HA, Kim DK, Nam JS, Sheen YY. EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-β/Smad and ROS signaling. Cell Mol Life Sci. 2015;72:2023–2039. PMID: 25487606. doi: 10.1007/s00018-014-1798-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barkauskas CE, Chung MI, Fioret B, Gao X, Katsura H. Hogan BL.Lung organoids: current uses and future promise. Development. 2017;144:986–997. PMID: 28292845. doi: 10.1242/dev.140103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlage WK, Bülles H, Friedrichs D, Kuhn M, Teredesai A. Cytokeratin expression patterns in the rat respiratory tract as markers of epithelial differentiation in inhalation toxicology. II. Changes in cytokeratin expression patterns following 8-day exposure to room-aged cigarette sidestream smoke. Toxicol Pathol. 1998;26:324–343. doi: 10.1177/019262339802600307. [DOI] [PubMed] [Google Scholar]
- 13.Wittekindt OH. Tight junctions in pulmonary epithelia during lung inflammation. Pflugers Arch. 2017. January;469(1):135–147. PMID: 27921210. doi: 10.1007/s00424-016-1917-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochim Biophys Acta. 2008;1778:631–645. PMID: 18036336. doi: 10.1111/j.1749-6632.2000.tb05235.x. [DOI] [PubMed] [Google Scholar]
- 15.Amasheh S, Meiri N, Gitter AH, Schöneberg T, Mankertz J, Schulzke JD, Fromm M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci. 2002;115:4969–4976. PMID: 12432083. doi: 10.1242/jcs.00165. [DOI] [PubMed] [Google Scholar]
- 16.Venugopal S, Anwer S, Szászi K. Claudin-2: roles beyond permeability functions. Int J Mol Sci. 2019;20:E5655. PMID: 31726679. doi: 10.3390/ijms20225655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaarteenaho-Wiik R, Soini Y. Claudin-1, −2, −3, −4, −5, and −7 in usual interstitial pneumonia and sarcoidosis. J Histochem Cytochem Histochem Soc. 2009;57:187–195. PMID: 18955738. doi: 10.1369/jhc.2008.951566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Overgaard CE, Mitchell LA, Koval M. Roles for claudins in alveolar epithelial barrier function. Ann N Y Acad Sci. 2012;1257:167–174. PMID: 22671603. doi: 10.1111/j.1749-6632.2012.06545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rokkam D, Lafemina MJ, Lee JW, Matthay MA, Frank JA. Claudin-4 levels are associated with intact alveolar fluid clearance in human lungs. Am J Pathol Am Soc Invest Pathol. 2011;179:1081–1087. PMID: 21741940. doi: 10.1016/j.ajpath.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wray C, Mao Y, Pan J, Chandrasena A, Piasta F, Frank JA. Claudin-4 augments alveolar epithelial barrier function and is induced in acute lung injury. Am J Physiol - Lung Cell Mol Physiol. 2009;297:L219–L227. PMID: 19447895. doi: 10.1152/ajplung.00043.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou J, Li Y, Yu J, Dong L, Husain AN, Shen L, Weber CR. Idiopathic pulmonary fibrosis is associated with tight junction protein alterations. Biochim Biophys Acta Biomembr. 2020;1862:183205. PMID: 32001212. doi: 10.1016/j.bbamem.2020.183205. [DOI] [PubMed] [Google Scholar]
- 22.Higashi T, Tokuda S, Kitajiri SI, Masuda S, Nakamura H, Oda Y, Furuse M. Analysis of the “angulin” proteins LSR, ILDR1 and ILDR2 - tricellulin recruitment, epithelial barrier function and implication in deafness pathogenesis. J Cell Sci. 2013;126:966–977. PMID: 23239027. doi: 10.1242/jcs.116442. [DOI] [PubMed] [Google Scholar]
- 23.Kodera Y, Chiba H, Konno T, Kohno T, Takahashi H, Kojima T. HMGB1-downregulated angulin-1/LSR induces epithelial barrier disruption via claudin-2 and cellular metabolism via AMPK in airway epithelial Calu-3 cells. Biochem Biophys Res Commun. 2020;527:553–560. PMID: 32423802. doi: 10.1016/j.bbrc.2020.04.113. [DOI] [PubMed] [Google Scholar]
- 24.Candi E, Terrinoni A, Rufini A, Chikh A, Lena AM, Suzuki Y, Sayan BS, Knight RA, Melino G. p63 is upstream of IKKα in epidermal development. J Cell Sci. 2006;119:4617–4622. PMID: 17093266. doi: 10.1242/jcs.03265. [DOI] [PubMed] [Google Scholar]
- 25.Kaneko Y, Kohno T, Kakuki T, Takano KI, Ogasawara N, Miyata R, Kikuchi S, Konno T, Ohkuni T, Yajima R, et al. The role of transcriptional factor p63 in regulation of epithelial barrier and ciliogenesis of human nasal epithelial cells. Sci Rep. 2017;7:10935. PMID: 28883651. doi: 10.1038/s41598-017-11481-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaneko Y, Konno T, Kohno T, Kakuki T, Miyata R, Ohkuni T, Kakiuchi A, Yajima R, Ohwada K, Kurose M, et al. Induction of airway progenitor cells via p63 and KLF11 by Rho-kinase inhibitor Y27632 in hTERT-human nasal epithelial cells. Am J Transl Res. 2019;11:599–611. PMID: 30899365. [PMC free article] [PubMed] [Google Scholar]
- 27.Chilosi M, Poletti V, Murer B, Lestani M, Cancellieri A, Montagna L, Piccoli P, Cangi G, Semenzato G, Doglioni C. Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: the role of ΔN-p63. Lab Invest. 2002;82:1335–1345. PMID: 12379768. doi: 10.1097/01.lab.0000032380.82232.67. [DOI] [PubMed] [Google Scholar]
- 28.Jonsdottir HR, Arason AJ, Palsson R, Franzdottir SR, Gudbjartsson T, Isaksson HJ, Gudmundsson G, Gudjonsson T, Magnusson MK. Basal cells of the human airways acquire mesenchymal traits in idiopathic pulmonary fibrosis and in culture. Lab Invest. 2015;95:1418–1428. PMID: 26390052. doi: 10.1038/labinvest.2015.114. [DOI] [PubMed] [Google Scholar]
- 29.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. PMID: 20192808. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 30.Shimada H, Satohisa S, Kohno T, Takahashi S, Hatakeyama T, Konno T, Tsujiwaki M, Saito T, Kojima T. The roles of tricellular tight junction protein lipolysis-stimulated lipoprotein receptor in malignancy of human endometrial cancer cells. Oncotarget. 2016;7:27735–27752. PMID: 27036040. doi: 10.18632/oncotarget.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Czulkies BA, Mastroianni J, Lutz L, Lang S, Schwan C, Schmidt G, Lassmann S, Zeiser R, Aktories K, Papatheodorou P. Loss of LSR affects epithelial barrier integrity and tumor xenograft growth of CaCo-2 cells. Oncotarget. 2017;8:37009–37022. PMID: 27391068. doi: 10.18632/oncotarget.10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng J, Wei X, Zhan JB, Jiang HY. High mobility group box1 contributes to hypoxia-induced barrier dysfunction of nasal epithelial cells. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2017;31:1178–1181. PMID: 29798353. doi: 10.13201/j..1001-1781.2017.15.009. [DOI] [PubMed] [Google Scholar]
- 33.Binabaj MM, Asgharzadeh F, Avan A, Rahmani F, Soleimani A, Parizadeh MR, Ferns GA, Ryzhikov M, Khazaei M, Hassanian SM. EW-7197 prevents ulcerative colitis-associated fibrosis and inflammation. J Cell Physiol. 2019;234:11654–11661. PMID: 30478959. doi: 10.1002/jcp.27823. [DOI] [PubMed] [Google Scholar]
- 34.Kyuno T, Kyuno D, Kohno T, Konno T, Kikuchi S, Arimoto C, Yamaguchi H, Imamura M, Kimura Y, Kondoh M, et al. Tricellular tight junction protein LSR/angulin-1 contributes to the epithelial barrier and malignancy in human pancreatic cancer cell line. Histochem Cell Biol. 2020;153:5–16. PMID: 31650247. doi: 10.1007/s00418-019-01821-4. [DOI] [PubMed] [Google Scholar]
- 35.Li F, Pascal LE, Wang K, Zhou Y, Balasubramani GK, O’Malley KJ, Dhir R, He K, Stolz D, DeFranco DB, et al. Transforming growth factor beta 1 impairs benign prostatic luminal epithelial cell monolayer barrier function. Am J Clin Exp Urol. 2020;8:9–17. PMID: 32211449. [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen N, Fernando SD, Biette KA, Hammer JA, Capocelli KE, Kitzenberg DA, Glover LE, Colgan SP, Furuta GT, Masterson JC. TGF-β1 alters esophageal epithelial barrier function by attenuation of claudin-7 in eosinophilic esophagitis. Mucosal Immunol. 2018;11:415–426. PMID: 28832026. doi: 10.1038/mi.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El Gazzar M. HMGB1 modulates inflammatory responses in LPS-activated macrophages. Inflamm Res. 2007;56:162–167. PMID: 17522814. doi: 10.1007/s00011-006-6112-0. [DOI] [PubMed] [Google Scholar]
- 38.Ranzato E, Martinotti S, Patrone M. Emerging roles for HMGB1 protein in immunity, inflammation, and cancer. Immuno Targets Ther. 2015;4:101–109. PMID: 27471716. doi: 10.2147/ITT.S58064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.