

ABSTRACT
Ferroptosis is a newly identified form of cell death that is regulated by many metabolic pathways, including iron, lipid and amino acids. Recent two studies reveal that the mitochondria and energy stress could also mediate ferroptosis. Gao et al. report that mitochondria play an essential role in ferroptosis induced by cysteine deprivation. In addition, Lee et al. show that energy stress depressed ferroptosis partly through activation of AMP-activated protein kinase. These findings provide potential therapeutic strategies for treating ferroptosis-related diseases, such as cancer, tissue injury and neurodegenerative diseases.
KEYWORDS: Ferroptosis, mitochondria, energy stress
Ferroptosis is a recently discovered iron-dependent form of cell death, which is distinct from necrosis, apoptosis and autophagy [1]. The characteristics of ferroptosis have been defined as the accumulation of lipid peroxidation/reactive oxygen species (ROS). It is known that amino acid antiporter system XC – (cysteine glutamate transporter) and glutathione peroxidase 4 (GPX4) are required for blocking ferroptosis. For example, cysteine is imported by system XC – to synthesize glutathione which is used by GPX4 to prevent lipid accumulation [1]. Either cysteine deprivation or GPX4 inhibition is associated with the increased sensitivity to ferroptosis. Recently, ferroptosis has received increased attention across a number of diseases such as cancer, ischemia-reperfusion injury (IRI) and degenerative diseases [2]. Our previous study manifested that the process of ferroptosis has been associated with many metabolic pathways and redox biology [3]. However, whether energy metabolism plays a role in ferroptosis remains elusive. Especially, it has provoked many debates and controversies involving the role of mitochondria, a power house for energy metabolism, in the ferroptosis [4,5]. Moreover, previous reports have not yet uncovered the underlying mechanisms of the energy stress mediating ferroptosis. Two papers now respectively reported the role of mitochondria and energy stress in the ferroptosis [6,7].
Cysteine deprivation or GPX4 inhibition can induce the accumulation of lipid ROS, thus triggering ferroptosis [8]. Utilizing a selection of cellular, molecular, metabolomic and pharmacological approaches, Gao et al. demonstrated that mitochondria are a key player in ferroptosis and its membrane potential hyperpolarization is related to ferroptosis induced by cysteine deprivation [6]. Furthermore, both mitochondrial tricarboxylic acid cycle (TCA) cycle and electron transport chain (ETC) participate in the accumulation of lipid ROS, and thus initiate cysteine deprivation-induced ferroptosis. Interestingly, when GPX4 is inhibited, ferroptosis could not be suppressed either by depleting mitochondria or inhibiting ETC, suggesting mitochondria-related ferroptosis is independent of GPX4 inhibition. By contrast, cells are less sensitive to the cysteine deprivation-induced ferroptosis upon depleting fumarase, one of the enzymes involving the TCA cycle.
Lee et al. further investigated the role of energy stress in ferroptosis by inducing or mimicking energy stress and found it could potently inhibit ferroptosis induced by cystine depletion, GPX4 removal or inhibition [7]. Mechanically, energy stress depressed ferroptotic cell death partly through activation of AMP-activated protein kinase (AMPK) and cells with high basal AMPK activation are more resistant to ferroptosis. Additional experiments showed that AMPK participates in ferroptosis by AMPK-mediated phosphorylation of acetyl-CoA carboxylase 1 and 2 (ACC1/2), which are enzymes that increase the synthesis of fatty acid and prevent the oxidation of fatty acid. In addition, activation of AMPK could also inhibit the generation of polyunsaturated fatty acid and AMPK inactivation promote its synthesis in vitro. Notably, the authors demonstrated that energy stress-mediated AMPK activation had a protective effect on renal IRI via ferroptosis blockage.
Both two studies describe perturbation of energy metabolism has strong impact on cellular sensitivity to ferroptosis, suggesting modulating TCA or energy stress influences the ferroptotic cell death (shown in Figure 1). As the primary energy source in cellular functions, Gao et al. showed mitochondria also played a crucial role in ferroptosis induced by cysteine deprivation, but not GPX4 inhibition [6]. Lee et al. reported that AMPK as a key regulator in the process of energy stress has a function to block either cystine starvation or GPX4 inhibition-induced ferroptosis [7]. These findings implicate that mitochondria and AMPK-mediated ferroptosis might be mechanistically different and warrant further investigation. Nevertheless, based on the previous report [9], it is noted that mitochondria can also regulate AMPK activity and vice versa.
Figure 1.
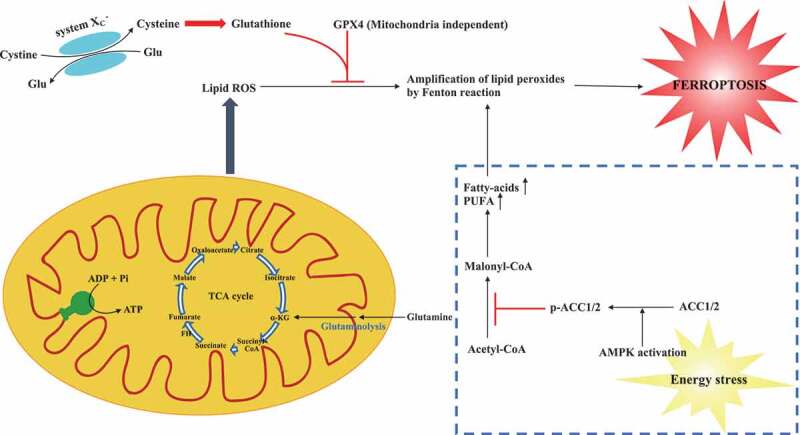
A schematic model describes the roles of energy metabolism in ferroptosis. Inhibiting system XC – (cysteine glutamate transporter) or GPX4 can trigger ferroptosis induced by the accumulation of lipid ROS. Mitochondria metabolism including TCA cycle promotes cysteine deprivation-induced ferroptosis and energy–stress-mediated AMPK activation inhibits ferroptosis. TCA cycle: tricarboxylic acid cycle; α-KG: α-ketoglutarate; ROS: reactive oxygen species; Glu: glutamate; GPX4: glutathione peroxidase 4; AMPK: AMP-activated protein kinase; AAC1/2: acetyl-CoA carboxylase 1 and 2; PUFA: polyunsaturated fatty acid
Of note, there has been intense debate about whether mitochondria play an essential role in ferroptosis. As Gao et al. shown, the role of mitochondria in the process of ferroptosis is context-dependent. The GPX4 inhibition-induced ferroptosis might be independent of mitochondria. Additionally, different cellular metabolism statuses could also affect the cellular processes such as adaptive and feedback mechanisms. Since some modalities based on ATP or metabolic activity could be influenced by mitochondrial, various cell death methods across different studies could result in considerable discrepancy. The other debate is about the functional role of AMPK in regulating ferroptosis. Song et al. concluded that AMPK-mediated BECN1 phosphorylation promoted ferroptosis by directly inhibiting the system XC −, which is opposite to the results presented by Lee et al., possibly due to context-dependent [10]. The most striking issue that emerged from these studies was the contradictory function of AMPK in cancer. It is reported that the overexpression or amplification of AMPK is highly prevalent in some cancer types [11]. Under energy stress condition, AMPK could promote tumor growth by inhibition of ferroptosis [7]. On the contrary, AMPK could also serve as a tumor suppressor mainly through suppression of biosynthesis of proteins or fatty acids [12].
Emerging evidence supports the notion that energy metabolism regulates ferroptosis. Particularly, the findings of mitochondria and energy stress modulating ferroptosis provide potential strategies for the development of prevention and treatment of ferroptosis-related diseases. A number of papers show that IRI, neurodegenerative diseases and cancer are related to ferroptosis, and targeting ferroptosis can protect from ischemic injury, degenerative diseases and cancer in mouse models [13–15]. Our previous study found that MitoTEMPO, a superoxide scavenger designed to target the mitochondria, can protect the heart from IR-induced damage via reducing the lipid peroxidation [3]. It is noted that FH, a metabolic enzyme in the TCA cycle, can suppress the cancer via promoting ferroptosis [15]. Future studies are needed to elucidate how cysteine deprivation promotes mitochondria membrane hyperpolarization and precise role of AMPK-mediated ferroptosis inhibition in the context of cancer setting. It would be interesting to investigate to what extent the ferroptosis inducers and inhibitors could be translated into clinical test for treating cancer, IRI and degenerative diseases.
Acknowledgments
This work was supported by grants from the National Key R&D Program of China (2018YFC2000400), and National Natural Science Foundation of China (81670651, and 81970573).
Funding Statement
This work was supported by the Key Technologies Research and Development Program [2018YFC2000400]; National Natural Science Foundation of China [81970573]; National Natural Science Foundation of China [81670651].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Dixon SJ, Lemberg K, Lamprecht M, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gaschler MM, Hu F, Feng H, et al. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem Biol. 2018;13(4):1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cassim S, Vučetić M, Ždralević M, et al. Warburg and beyond: the power of mitochondrial metabolism to collaborate or replace fermentative glycolysis in cancer. Cancers (Basel). 2020;12(5):1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gao M, Yi J, Zhu J, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73(2):354–363.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lee H, Zandkarimi F, Zhang Y, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dai C, Chen X, Li J, et al. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020;27(9):645–656. [DOI] [PubMed] [Google Scholar]
- [9].Trewin AJ, Berry BJ, Wojtovich AP. Exercise and mitochondrial dynamics: keeping in shape with ROS and AMPK. Antioxidants (Basel). 2018;7(1):7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Song X, Zhu S, Chen P, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc– activity. Curr Biol. 2018;28(15):2388–2399.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ross FA, MacKintosh C, Hardie DG, et al. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. Febs J. 2016;283(16):2987–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hardie DG. Molecular pathways: is AMPK a friend or a foe in cancer? Clin Cancer Res. 2015;21(17):3836–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Guiney SJ, Adlard PA, Bush AI, et al. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem Int. 2017;104:34–48. [DOI] [PubMed] [Google Scholar]
- [15].Adam J, Hatipoglu E, O’Flaherty L, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20(4):524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]