

Abstract
Objective
To determine the regional- and district-level newborn prevalence of sickle cell trait and disease, and the prevalence of haemoglobin variants and genetic modifiers of sickle cell disease, in the nine regions of north-western United Republic of Tanzania.
Methods
We repurposed dried blood spot samples from children (aged 0–24 months) born to mothers living with human immunodeficiency virus (HIV), collected as part of the HIV Early Infant Diagnosis programme, for sickle cell diagnosis. We performed isoelectric focusing to determine whether samples had normal haemoglobin, sickle cell trait, sickle cell disease or a rare haemoglobin variant. We shipped samples diagnosed as disease or variant to Cincinnati Children’s Hospital in the United States of America for deoxyribonucleic-acid-based analyses to determine the prevalence of α-thalassaemia, glucose-6-phosphate dehydrogenase (G6PD) deficiency or fetal haemoglobin genetic modifiers.
Findings
We analysed a total of 17 200 specimens during February 2017–May 2018. We observed a prevalence of sickle cell trait and disease of 20.3% (3492/17 200) and 1.2% (210/17 200), respectively. District-level trait varied from 8.6% (5/58) to 28.1% (77/274). Among confirmed sickle cell disease specimens, we noted 42.7% (61/143) had 1-gene deletion and 14.7% (21/143) had 2-gene deletion α-thalassaemia trait. We documented G6PD A– deficiency in 19.2% (14/73) of males.
Conclusion
Our calculated prevalence is twice as high as previously reported and reinforces the need for enhanced sickle cell diagnostic services. Our district-level data will inform public health policy, allowing screening and disease-modifying hydroxyurea therapy to be focused on high-prevalence areas, until universal newborn screening is available.
Résumé
Objectif
Déterminer la prévalence de la drépanocytose et du trait drépanocytaire chez les nouveau-nés, ainsi que la prévalence de variants d'hémoglobine et de modificateurs génétiques de la drépanocytose dans les neuf régions du nord-ouest de la République-Unie de Tanzanie.
Méthodes
Nous avons réutilisé des échantillons de gouttes de sang séché provenant d'enfants (âgés de 0 à 24 mois) nés de mères atteintes du virus de l'immunodéficience humaine (VIH), prélevés dans le cadre du programme de diagnostic précoce de l'infection à VIH chez le nourrisson pour détecter une éventuelle drépanocytose. Nous avons procédé à une focalisation isoélectrique pour savoir si les échantillons présentaient une hémoglobine normale, un trait drépanocytaire, une drépanocytose ou un variant d'hémoglobine rare. Ensuite, nous avons envoyé les échantillons identifiés comme porteurs de la maladie ou d'un variant à l'hôpital pour enfants de Cincinnati, aux États-Unis, afin d'analyser l'acide désoxyribonucléique et de mesurer la prévalence d'une α-thalassémie, d'un déficit en glucose-6-phosphate déshydrogénase (G6PD) ou de modificateurs génétiques de l'hémoglobine fœtale.
Résultats
Nous avons étudié 17 200 spécimens au total entre février 2017 et mai 2018. Nous avons observé une prévalence de 20,3% (3492/17 200) pour la drépanocytose, et de 1,2% (210/17 200) pour le trait drépanocytaire. D'un district à l'autre, la présence du trait variait de 8,6% (5/58) à 28,1% (77/274). Nous avons constaté que 42,7% (61/143) des spécimens chez qui une drépanocytose avait été confirmée étaient privés d'un gène, et 14,7% (21/143) de deux gènes du trait α-thalassémique. Enfin, nous avons répertorié un déficit en G6PD A– chez 19,2% (14/73) des mâles.
Conclusion
La prévalence que nous avons calculée est deux fois supérieure aux chiffres mentionnés précédemment, et souligne la nécessité d'instaurer de meilleurs services de diagnostic de la drépanocytose. Nos données réparties par district fourniront des informations en matière de politique de santé publique, afin que le dépistage et le traitement modificateur de la maladie, à base d'hydroxyurée, se concentrent sur les zones de forte prévalence jusqu'à ce qu'un dépistage universel des nouveau-nés soit disponible.
Resumen
Objetivo
Determinar la prevalencia de la enfermedad y del rasgo de las células falciformes en los recién nacidos a nivel regional y de distrito, y la prevalencia de las variantes de hemoglobina y de los modificadores genéticos de la drepanocitosis en las nueve regiones del noroeste de la República Unida de Tanzania.
Métodos
Se reutilizaron las muestras de las manchas de sangre seca de los niños (de 0 a 24 meses de edad) nacidos de madres que padecían el virus de la inmunodeficiencia humana (VIH); estas muestras se obtuvieron a través del programa de Diagnóstico precoz del VIH en niños para diagnosticar las células falciformes. Se aplicó la técnica del enfoque isoeléctrico para determinar si las muestras tenían hemoglobina, rasgos de células falciformes, drepanocitosis normales o una variante de hemoglobina poco frecuente. Se enviaron muestras diagnosticadas como enfermedad o variante al Hospital Infantil de Cincinnati (Cincinnati Children's Hospital) en los Estados Unidos de América para analizarlas a base de ácido desoxirribonucleico y así determinar la prevalencia de talasemia α, de deficiencia de glucosa-6-fosfato deshidrogenasa (G6PD, por sus siglas en inglés) o de modificadores genéticos de hemoglobina fetal.
Resultados
Se analizaron un total de 17 200 muestras entre febrero de 2017 y mayo de 2018. Se observó una prevalencia del rasgo de las células falciformes y de la drepanocitosis del 20,3 % (3492/17 200) y del 1,2 % (210/17 200), respectivamente. El rasgo a nivel de distrito varió del 8,6 % (5/58) al 28,1 % (77/274). Se observó que en las muestras de drepanocitosis confirmadas, el 42,7 % (61/143) presentaba la eliminación de un gen y el 14,7 % (21/143) la eliminación de dos genes en el rasgo de talasemia α. Se registró una deficiencia de G6PD A- en el 19,2 % (14/73) de los varones.
Conclusión
La prevalencia que se calcula aquí es el doble de la que se notificó anteriormente y refuerza la necesidad de mejorar los servicios para el diagnóstico de la drepanocitosis. Estos datos a nivel de distrito contribuirán a la política de salud pública, ya que permitirán que los cribados y la terapia con hidroxicarbamida que modifica la enfermedad se centren en las zonas de alta prevalencia, hasta que se disponga de un cribado universal de los recién nacidos.
ملخص
الغرض تحديد معدل انتشار سمات ومرض الخلايا المنجلية لدى حديثي الولادة على مستوى الأقاليم والمناطق، وانتشار متغيرات الهيموجلوبين والمعدلات الجينية لمرض فقر الدم المنجلي، في المناطق التسع الواقعة شمال غرب جمهورية تنزانيا المتحدة.
الطريقة قمنا بإعادة استخدام عينات بقع الدم المجففة من الأطفال (من حديثي الولادة إلى 24 شهرًا) المولودين لأمهات مصابات بفيروس نقص المناعة البشرية (HIV)، والتي تم جمعها كجزء من برنامج التشخيص المبكر لفيروس نقص المناعة البشرية للرضع، لتشخيص مرض الخلايا المنجلية. أجرينا تركيزًا متساوي الجهد الكهربائي لتحديد ما إذا كانت العينات تحتوي على هيموجلوبين طبيعي، أو سمة للخلايا المنجلية، أو مرض الخلايا المنجلية، أو أحد المتغيرات النادرة للهيموجلوبين. كما قمنا بشحن عينات تم تشخيصها على أنها مصابة بالمرض أو أحد المتغيرات، إلى مستشفى سينسيناتي للأطفال في الولايات المتحدة الأمريكية، لإجراء تحليلات باستخدام حمض الديوكسي ريبونيوكليك لتحديد مدى انتشار كل من ثلاسيميا ألفا، أو النقص في نازع هيدروجين الجلوكوز 6 فوسفات (G6PD)، أو المعدلات الوراثية للهيموجلوبين الجنيني.
النتائج قمنا بتحليل إجمالي 17200 عينة خلال الفترة من فبراير/شباط 2017 إلى مايو/أيار 2018. ولاحظنا انتشار سمة ومرض الخلايا المنجلية بنسبة 20.3% (3492/17200)، و1.2% (210/17200)، على الترتيب. تنوعت السمات على مستوى المناطق من 8.6% (5/58) إلى 28.1% (77/274). ولاحظنا من بين عينات مرض الخلايا المنجلية المؤكد، أن 42.7% (61/143) قد عانت من حذف جين واحد، و14.7% (21/143) عانت من حذف جينين من سمة ألفا ثلاسيميا. وكما بتوثيق النقص في G6PD A في 19.2% (14/73) من الذكور.
الاستنتاج إن معدل الانتشار الذي قمنا باحتسابه هو ضعف ما تم الإبلاغ عنه سابقًا، ويعزز الحاجة إلى خدمات محسنة لتشخيص مرض الخلايا المنجلية. ستؤدي البيانات على مستوى المقاطعات لدينا إلى توجيه سياسة الصحة العامة، مما يسمح لكل من الفحص والمعالجة باستخدام هيدروكسي اليوريا المعدل للأمراض، بالتركيز على المناطق ذات الانتشار المرتفع، حتى يتوفر فحص شامل لحديثي الولادة.
摘要
目的
确定坦桑尼亚联合共和国西北九个地区,新生儿镰状细胞性状和镰状细胞疾病的患病率,以及血红蛋白变体和镰状细胞疾病遗传修饰因子的患病率。
方法
作为 HIV 早期婴儿诊断计划的一部分,我们将携带人类免疫缺陷病毒 (HIV) 的母亲所生婴儿(0-24 个月)的干血斑样本重新用于诊断镰状细胞。我们对其实施等电聚焦,以确定样本的血红蛋白是否正常、是否具有镰状细胞性状、镰状细胞疾病或罕见的血红蛋白变体。我们将诊断出患有疾病或变异的样本运送到美国辛辛那提儿童医院,进行脱氧核糖核酸分析,以确定α地中海贫血、6-磷酸葡萄糖脱氢酶 (G6PD) 缺失症或胎儿血红蛋白遗传修饰因子的患病率。
结果
我们在 2017 年 2 月至 2018 年 5 月期间共分析了 17200 个样本。我们观察到镰状细胞性状和镰状细胞疾病的患病率分别为 20.3% (3492/17 200) 和 1.2% (210/17 200)。区级镰状细胞性状患病率则从 8.6% (5/58) 到 28.1% (77/274) 不等。在确诊的镰状细胞疾病样本中,我们发现 42.7% (61/143) 具有 1 基因缺失型 α 地中海贫血性状,14.7% (21/143) 具有 2 基因缺失型 α 地中海贫血性状。我们注意到 19.2% (14/73) 的男婴患有 G6PD A–缺乏症。
结论
我们计算得出的患病率是先前报道的两倍,这凸显了加强镰状细胞诊断服务的必要性。我们的区级数据将为公共卫生政策提供信息,从而确保在新生儿普查之前,对高发地区集中展开筛查和疾病修饰羟基脲疗法。
Резюме
Цель
Определить на национальном уровне и на уровне районов распространенность среди новорожденных носительства признака серповидно-клеточной анемии и самого этого заболевания, а также распространенность вариантов гемоглобина и генетических модификаторов серповидно-клеточной анемии в девяти регионах на северо-западе Объединенной Республики Танзания.
Методы
Для диагностики серповидно-клеточной анемии авторы использовали взятые методом высушивания пятна образцы крови детей в возрасте от 0 до 24 месяцев, чьи матери живут с вирусом иммунодефицита человека (ВИЧ); эти образцы были собраны в рамках программы ранней диагностики ВИЧ у детей. Для определения наличия в образцах нормального гемоглобина, носительства признака серповидно-клеточной анемии, собственно серповидно-клеточной анемии или редкого варианта гемоглобина использовался метод изоэлектрической фокусировки. Образцы, которые были диагностированы как соответствующие проявлениям заболевания или как содержащие вариантный гемоглобин, были направлены в Детскую больницу Цинциннати, США, для проведения ДНК-анализа с целью определения распространенности в них α-талассемии, дефицита глюкозо-6-фосфат-дегидрогеназы (Г6ФД) или наличия генетических модификаторов фетального гемоглобина.
Результаты
Авторы проанализировали 17 200 образцов в период с февраля 2017 г. по май 2018 г. Носительство признака серповидно-клеточной анемии было обнаружено в 20,3% случаев (3492 из 17 200 образцов), а серповидно-клеточная анемия — в 1,2% (210 из 17 200 образцов). Наличие признака в разных районах варьировалось от 8,6% (5 из 58 образцов) до 28,1% (77 из 274 образцов). Среди образцов с подтвержденным диагнозом серповидно-клеточной анемии в 42,7% случаев (61 из 143) наблюдался признак α-талассемии с делецией 1 гена и в 14,7% (21 из 143) — с делецией 2 генов. Дефицит Г6ФД был зафиксирован у 19,2% (14 из 73) мальчиков.
Вывод
Рассчитанная авторами распространенность заболевания вдвое превышает опубликованные ранее значения и еще раз указывает на необходимость укрепления и расширения услуг по диагностике СКА. Полученные данные по районам станут основой для выработки политики в области общественного здравоохранения, которая бы позволяла сосредоточить скрининговое обследование и лечение гидроксимочевиной (изменяющее течение заболевания) в областях с высоким распространением заболевания до тех пор, пока не станет возможным всеобщее скрининговое обследование новорожденных.
Introduction
Sickle cell disease is an inherited disorder of haemoglobin, caused by a mutation in the β-globin subunit of adult haemoglobin. In classic autosomal recessive fashion, inheritance of one abnormal and one normal allele confers sickle cell trait, a carrier state without clinical symptoms. Inheritance of two mutated alleles causes sickle cell disease, characterized by varying amounts of chronic haemolytic anaemia, recurrent debilitating pain and an array of clinical sequelae, including increased risk of infection, stroke, lung disease, splenic dysfunction and bone infarction.1
Sickle cell disease imposes a significant global burden of disease that remains underrecognized,2 especially in Africa. Approximately 400 000 infants are born each year with sickle cell disease;3–5 75% of these infants are born in the tropical regions of sub-Saharan Africa,6 home to most of the > 25 million people who live with sickle cell disease globally.7 Sickle cell disease causes substantial morbidity8 and is responsible for 5–16% of mortality in children younger than 5 years.9,10 Cumulative data from prior studies suggest that more than half of the children with sickle cell disease in sub-Saharan Africa die in early childhood,11 with substantial differences in mortality between historic rural communities12 and modern urban centres.13 Early enrolment in a comprehensive care programme that includes preventive care (immunizations and prophylactic antimicrobials) and disease-modifying therapy (hydroxyurea or prophylactic blood transfusions) can reduce symptoms and improve survival.
The World Health Organization acknowledged the global importance of addressing sickle cell disease almost 15 years ago,10,14,15 and in 2010 African leaders formally proposed sickle cell disease prevention and control strategies for the African Region.5 In response, the United Republic of Tanzania has embedded sickle cell disease targets within the national noncommunicable disease policy,16 increased advocacy,17 created a centre of excellence,18 educated health-care workers and increased research output.19 The majority of these efforts have been focused in Muhimbili National Hospital in the coastal city of Dar es Salaam, but prevalence estimates suggest that the greatest burden of sickle cell disease is in the north-western regions of the country around Lake Victoria.6,20,21
The United Republic of Tanzania does not yet have a national newborn screening programme. In the absence of recent reports, Tanzanian estimates of sickle cell disease are based on sparse data from studies performed over the past 65 years in only seven of the 30 regions of the country.22–32 Recognizing that data extracted from isolated reports can poorly reflect variation within a country,33,34 we initiated and conducted the United Republic of Tanzania Sickle Surveillance Study in the north-western regions of the country. Our primary objective was to provide contemporary regional- and district-level data on the newborn prevalence of sickle cell trait and disease to inform the national noncommunicable disease policy goals. Our study used existing public health infrastructure developed as part of the human immunodeficiency virus (HIV) Early Infant Diagnosis programme,35 while aiming to build local capacity for the accurate diagnosis of sickle cell disease. Our secondary objectives included characterization of rare haemoglobin variants and the prevalence of co-inherited genetic disorders that may affect sickle cell disease phenotypes and response to hydroxyurea treatment.
Methods
HIV Early Infant Diagnosis
In 2006, the International Center for AIDS Care and Treatment Programs, the Tanzanian Ministry of Health and Social Welfare, Bugando Medical Centre and the United States Centers for Disease Control and Prevention implemented the HIV Early Infant Diagnosis programme with the aim of preventing mother-to-child transmission of HIV.35 The country operates a decentralized testing structure with four reference laboratories. The Bugando Medical Centre, a zonal referral and teaching hospital located in the city of Mwanza, serves as the reference laboratory for the north-western catchment area, and is also the designated sickle cell centre of excellence for this area. The Bugando catchment area includes a population of approximately 17.6 million people within nine regions, who collectively represent 39.2% (17 623 047/44 928 923) of the country’s total population.36
An infant born to a mother living with HIV is brought to a Reproductive and Child Health clinic at 4–6 weeks of age for the first preventive health visit. A dried blood spot is collected from the infant, and transported to a reference laboratory for detection of HIV using a polymerase chain reaction (PCR). The dried blood spots are labelled with key demographic information including date of birth, sex, referring health facility, date of collection, date of dispatch and date of receipt at the laboratory. HIV results are communicated to the referring health facility for prompt initiation of antiretroviral medication. Dried blood spots are then stored at room temperature and made available for additional testing for sickle cell trait and disease.
Sickle cell diagnosis
We analysed all repurposed dried blood spots collected as part of the HIV Early Infant Diagnosis programme during February 2017–May 2018 by using the isoelectric focusing technique.34 Our laboratory equipment included Resolve Hemoglobin kits and JB-2 Staining System reagents (both PerkinElmer, Inc., Waltham, United States of America, USA), as well as control specimens and other consumables, all donated to the haematology section of the Bugando laboratory. Staff were trained on-site by a board-certified haematologist, and attended a 2-day seminar in Dar es Salaam, organized by the equipment manufacturer and funded jointly by the manufacturer and the United States Association of Public Health Laboratories. At the seminar, staff acquired theoretical knowledge of the isoelectric focusing technique and had the opportunity to process samples while supervised by an experienced manufacturer representative. The senior Tanzanian paediatrician leading the haematology clinic also completed a further two months of clinical and laboratory training funded and hosted by Cincinnati Children’s Hospital, USA.
All dried blood spots were analysed with a standard control specimen containing adult, fetal and sickling haemoglobin (haemoglobin A, F and S) and haemoglobin C. Isoelectric focusing results were scored independently by two Bugando Medical Centre staff for the presence and abundance of each type of haemoglobin. The results were interpreted as: normal if haemoglobin A (± haemoglobin F) was present; sickle cell disease if haemoglobin S (± haemoglobin F) was present; sickle cell trait if both haemoglobin A and S (± haemoglobin F) were present; variant if a band was present at any location other than haemoglobin A, S or C (± haemoglobin F); and uninterpretable if poor quality precluded interpretation. Dried blood spot analyses interpreted as sickle cell disease, variant or uninterpretable were repeated for confirmation and frozen for later deoxyribonucleic acid (DNA) studies. Regular teleconferences were convened with collaborators based in the Cincinnati Children’s Hospital, USA, to provide ongoing feedback on the quality of laboratory techniques and interpretation of gels; however, the Tanzanian team was responsible for all final interpretations.
DNA-based testing
Any dried blood spot diagnosed as sickle cell disease or variant was stored at −20 °C then shipped to the USA for future DNA testing. Upon arrival, we stored dried blood spots at −80 °C until testing could be performed. We extracted DNA from dried blood spots using an adapted protocol from Instagene (Bio-Rad Laboratories, Hercules, USA).37 We performed amplification of β-globin gene exons 1 and 2 using a PCR, and confirmed the presence of a haemoglobin S mutation at rs334 (c.334T > A;p.Glu6Val) using a custom TaqMan PCR probe (Applied Biosystems, Foster City, USA).37 We analysed specimens interpreted as uncommon and atypical haemoglobin variants by isoelectric focusing via the previously published algorithm37 used to investigate uncommon variants in East Africa. Uncommon variants include the α-chain variants haemoglobin G-Pest (HBA1:p.Asp75Asn) and haemoglobin Stanleyville II (HBA1:p.Asn79Lys), and the fusion variants haemoglobin P-Nilotic (β-globin gene (HBB)–δ-globin gene (HBD) fusion: β31-δ50) and haemoglobin Kenya (γ-globin gene (HBG1)–HBB fusion: Aγ81-β86).37
We detected α-thalassaemia trait resulting from the 3.7-kilobase α-globin gene deletion and glucose-6-phosphate dehydrogenase (G6PD) A– variant using DNA-based techniques,37–39 and determined the modifiers of baseline haemoglobin F production.40 We genotyped the BCL11A polymorphisms (rs1427407, rs7557939 and rs11886868) and HBS1L-MYB intergenic polymorphisms (HMIP) (rs28384513 and rs9399137) using commercially available real-time PCR assays (Applied Biosystems, Foster City, USA). To identify the XmnI single-nucleotide polymorphism at −158 basepairs to G-γ globin (rs7482144), we amplified the G-γ gene (HBG2) using PCR with G-γ-specific forward and reverse primers to ensure that the G-γ rather than an A-γ product was isolated. We then further amplified this product by performing PCR with Classic 1 forward and reverse primers. We genotyped the final product using a custom-made TaqMan PCR probe set (Applied Biosystems, Foster City, USA).
Data analysis
We entered data into an Excel database (Microsoft, Redmond, USA), treating age as a continuous variable (summarized using median and interquartile range) and treating haemoglobin type, HIV status, sex, region of origin, district of origin, G6PD status and α-thalassaemia status as categorical variables (summarized using frequencies). We calculated the prevalence of sickle cell trait and disease by dividing the number of specimens with sickle cell trait and disease by the total number of non-missing specimens with interpretable results. We determined allelic frequency by dividing the number of times that an allele was observed (once in heterozygotes, twice in homozygotes) by the total number of all alleles (twice the total number of specimens). We compared the proportions of participants living with HIV with and without sickle cell disease using a χ2 test.
Ethical considerations
Our study protocol was approved with a waiver for informed consent by the joint Catholic University of Health and Allied Sciences–Bugando Medical Centre Research and Ethics Committee, as well as the Tanzanian National Institute for Medical Research, to perform disease surveillance on de-identified archived dried blood spots previously collected by the HIV Early Infant Diagnosis programme. The study was also approved by the Cincinnati Children’s Hospital Medical Center Institutional Review Board. A formal material transfer agreement was obtained so that specimens could be shipped to the USA for genetic analysis.
Results
Sample collection and testing
Our local Bugando Medical Centre staff completed a total of 232 isoelectric focusing gels during February 2017–May 2018. After samples from children older than 24 months were excluded to obtain a more accurate prevalence for infants, the median age of infants included in the sampling population was 52 days (interquartile range, 41–93 days). Staff scored a total of 17 274 unique dried blood spot specimens from the catchment area. The quality of laboratory testing was extremely high with only 20 specimens scored as uninterpretable and 54 with missing results, meaning that we performed our primary analysis on 17 200 specimens.
Prevalence
We observed an overall prevalence of sickle cell trait and disease in our cohort of 20.3% (3492/17 200) and 1.2% (210/17 200), respectively, with a 0.1% (17/17 200) prevalence of atypical or uncommon haemoglobin variants. Our data yielded an allelic frequency of 0.114 ([3492+(2 × 210)]/(2 × 17 200)) for the sickle gene (haemoglobin S) mutation, and demonstrate perfect Hardy–Weinberg equilibrium, as expected in a stable population.41 We did not identify any haemoglobin C or other common β-globin variants. Our geospatial mapping revealed mild variation between regions, with the prevalence of sickle cell trait and disease ranging from 16.6% (146/880) to 22.5% (253/1126) and 0.5% (4/880) to 1.5% (17/1126), respectively (Table 1). Analysis of individual districts that provided more than 48 specimens (i.e. excluding Buhigwe, from which only 12 samples were provided) revealed a wider geographic variability, with sickle cell trait and disease ranging from 8.6% (5/58) to 28.1% (77/274) and from zero to 4.3% (9/208), respectively (Fig. 1 and Table 1).
Table 1. Regional and district prevalence of haemoglobin types in infants identified, north-western United Republic of Tanzania, February 2017–May 2018.
| Region or district | Total | No. (%) |
|||
|---|---|---|---|---|---|
| Normal | Sickle cell trait | Sickle cell disease | Variant | ||
| Region | |||||
| Geita | 2 436 | 1 918 (78.7) | 495 (20.3) | 23 (0.9) | 0 (0.00) |
| Kagera | 880 | 729 (82.8) | 146 (16.6) | 4 (0.5) | 1 (0.1) |
| Kigoma | 683 | 550 (80.5) | 123 (18.0) | 10 (1.5) | 0 (0.00) |
| Mara | 1 837 | 1 422 (77.4) | 388 (21.1) | 23 (1.3) | 4 (0.2) |
| Mwanza | 3 847 | 3 013 (78.3) | 778 (20.2) | 51 (1.3) | 5 (0.1) |
| Shinyanga | 2 800 | 2 158 (77.1) | 598 (21.4) | 41 (1.5) | 3 (0.1) |
| Simiyu | 1 126 | 855 (75.9) | 253 (22.5) | 17 (1.5) | 1 (0.1) |
| Singida | 1 141 | 906 (79.4) | 226 (19.8) | 9 (0.8) | 0 (0.00) |
| Tabora | 2 450 | 1 930 (78.8) | 485 (19.8) | 32 (1.3) | 3 (0.1) |
| Total | 17 200 | 13 481 (78.4) | 3492 (20.3) | 210 (1.2) | 17 (0.1) |
| District | |||||
| Bariadi | 261 | 206 (78.9) | 52 (19.9) | 2 (0.8) | 1 (0.4) |
| Biharamulo | 135 | 111 (82.2) | 24 (17.8) | 0 (0.0) | 0 (0.0) |
| Buhigwea | 12 | 11 (91.7) | 1 (8.3) | 0 (0.0) | 0 (0.0) |
| Bukoba | 174 | 142 (81.6) | 30 (17.2) | 1 (0.6) | 1 (0.6) |
| Bukombe | 394 | 294 (74.6) | 97 (24.6) | 3 (0.8) | 0 (0.0) |
| Bunda | 378 | 301 (79.6) | 73 (19.3) | 3 (0.8) | 1 (0.3) |
| Busega | 274 | 195 (71.2) | 77 (28.1) | 2 (0.7) | 0 (0.0) |
| Butiama | 208 | 161 (77.4) | 38 (18.3) | 9 (4.3) | 0 (0.0) |
| Chato | 514 | 399 (77.6) | 108 (21.0) | 7 (1.4) | 0 (0.0) |
| Geita | 1 156 | 929 (80.4) | 219 (18.9) | 8 (0.7) | 0 (0.0) |
| Igunga | 539 | 426 (79.0) | 107 (19.9) | 4 (0.7) | 2 (0.4) |
| Ikungi | 135 | 110 (81.5) | 23 (17.0) | 2 (1.5) | 0 (0.0) |
| Ilemela | 513 | 394 (76.8) | 110 (21.4) | 9 (1.8) | 0 (0.0) |
| Iramba | 291 | 230 (79.0) | 60 (20.6) | 1 (0.3) | 0(0.0) |
| Itilima | 152 | 120 (78.9) | 29 (19.1) | 3 (2.0) | 0 (0.0) |
| Kahama | 1 683 | 1281 (76.1) | 374 (22.2) | 25 (1.5) | 3 (0.2) |
| Kakonko | 49 | 39 (79.6) | 10 (20.4) | 0 (0.0) | 0 (0.0) |
| Kaliua | 374 | 284 (75.9) | 83 (22.2) | 7 (1.9) | 0 (0.0) |
| Karagwe | 95 | 81 (85.3) | 14 (14.7) | 0 (0.0) | 0 (0.0) |
| Kasulu | 159 | 120 (75.5) | 34 (21.4) | 5 (3.1) | 0 (0.0) |
| Kibondo | 162 | 129 (79.6) | 31 (19.1) | 2 (1.2) | 0 (0.0) |
| Kigoma | 207 | 171 (82.6) | 34 (16.4) | 2 (1.0) | 0 (0.0) |
| Kishapu | 424 | 333 (78.5) | 87 (20.5) | 4 (0.9) | 0 (0.0) |
| Kwimba | 282 | 230 (81.6) | 50 (17.7) | 2 (0.7) | 0 (0.0) |
| Kyerwa | 58 | 53 (91.4) | 5 (8.6) | 0 (0.0) | 0 (0.0) |
| Magu | 664 | 541 (81.5) | 116 (17.5) | 7 (1.1) | 0 (0.0) |
| Manyoni | 285 | 223 (78.2) | 60 (21.1) | 2 (0.7) | 0 (0.0) |
| Maswa | 216 | 167 (77.3) | 45 (20.8) | 4 (1.9) | 0 (0.0) |
| Mbogwe | 273 | 215 (78.8) | 53 (19.4) | 5 (1.8) | 0 (0.0) |
| Meatu | 223 | 167 (74.9) | 50 (22.4) | 6 (2.7) | 0 (0.0) |
| Missenyi | 163 | 134 (82.2) | 29 (17.8) | 0 (0.0) | 0 (0.0) |
| Misungwi | 429 | 334 (77.9) | 86 (20.0) | 6 (1.4) | 3 (0.7) |
| Mkalama | 100 | 76 (76.0) | 23 (23.0) | 1 (1.0) | 0 (0.0) |
| Muleba | 191 | 155 (81.2) | 34 (17.8) | 2 (1.0) | 0 (0.0) |
| Musoma | 461 | 352 (76.4) | 103 (22.3) | 4 (0.9) | 2 (0.4) |
| Ngara | 64 | 53 (82.8) | 10 (15.6) | 1 (1.6) | 0 (0.0) |
| Nyamagana | 1 107 | 876 (79.1) | 216 (19.5) | 13 (1.2) | 2 (0.2) |
| Nyang’hwale | 99 | 81 (81.8) | 18 (18.2) | 0 (0.0) | 0 (0.0) |
| Nzega | 459 | 366 (79.7) | 88 (19.2) | 5 (1.1) | 0 (0.0) |
| Rorya | 486 | 371 (76.3) | 108 (22.2) | 6 (1.2) | 1 (0.2) |
| Sengerema | 705 | 525 (74.5) | 169 (24.0) | 11 (1.6) | 0 (0.0) |
| Serengeti | 89 | 73 (82.0) | 15 (16.9) | 1 (1.1) | 0 (0.0) |
| Shinyanga | 692 | 543 (78.5) | 137 (19.8) | 12 (1.7) | 0 (0.0) |
| Sikonge | 233 | 182 (78.1) | 50 (21.5) | 0 (0.0) | 1 (0.4) |
| Singida | 330 | 267 (80.9) | 60 (18.2) | 3 (0.9) | 0 (0.0) |
| Tabora | 347 | 288 (83.0) | 53 (15.3) | 6 (1.7) | 0 (0.0) |
| Tarime | 215 | 164 (76.3) | 51 (23.7) | 0 (0.0) | 0 (0.0) |
| Ukerewe | 147 | 113 (76.9) | 31 (21.1) | 3 (2.0) | 0 (0.0) |
| Urambo | 211 | 165 (78.2) | 41 (19.4) | 5 (2.4) | 0 (0.0) |
| Uvinza | 94 | 80 (85.1) | 13 (13.8) | 1 (1.1) | 0 (0.0) |
| Uyui | 288 | 220 (76.4) | 63 (21.9) | 5 (1.7) | 0 (0.0) |
| Total | 17 200 | 13 481 (78.4) | 3492 (20.3) | 210 (1.2) | 17 (0.1) |
a We considered the number of samples returned from Buhigwe (12) to be too small to be statistically representative; all other districts returned ≥ 49 samples.
Note: We used isoelectric focusing to detect different haemoglobin types in infants aged 0–24 months.
Fig. 1.
District-level prevalence of sickle cell trait in infants, north-western United Republic of Tanzania, February 2017–May 2018
Note: We only included districts that provided a minimum of 49 specimens.
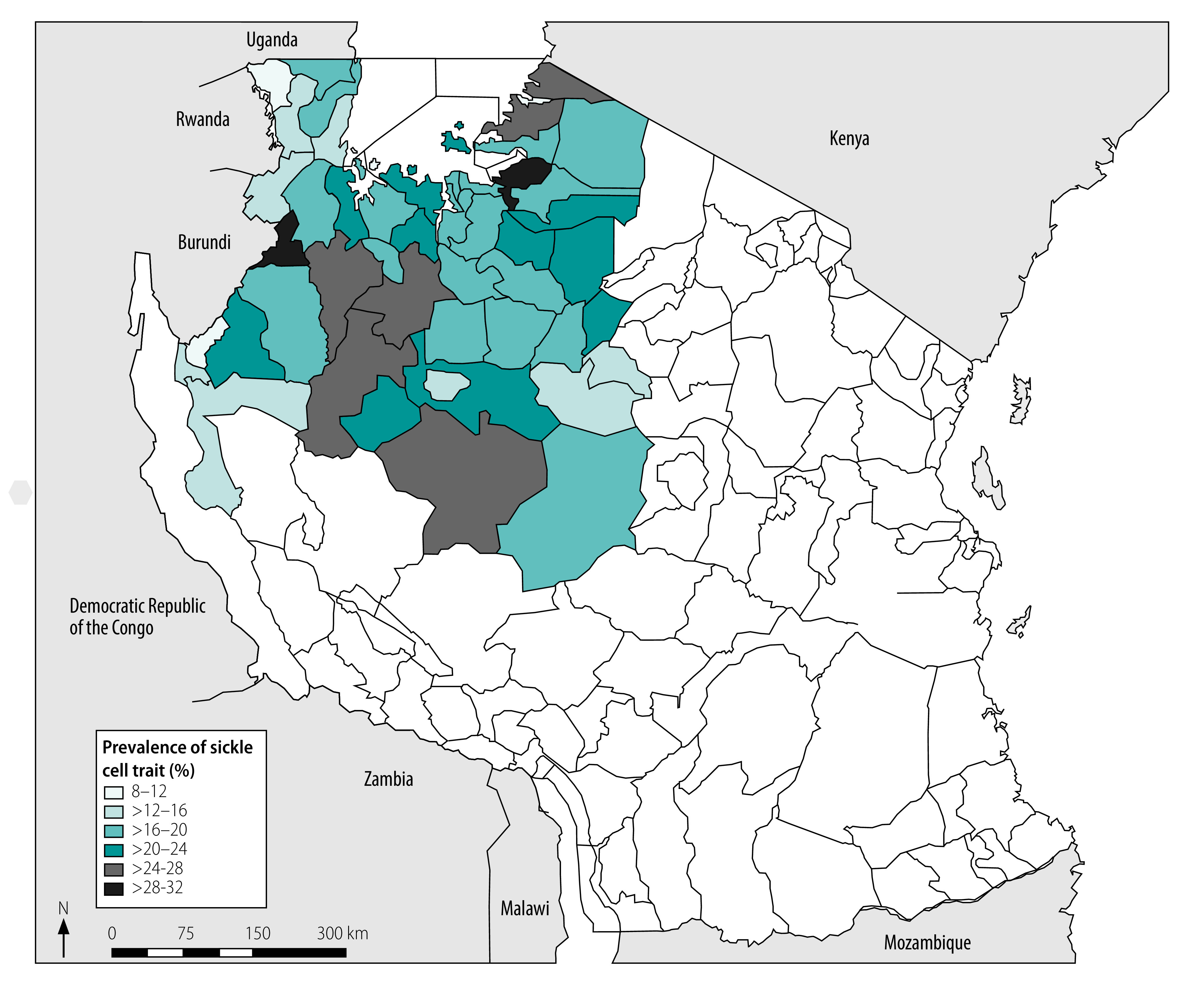
Using our regional prevalence estimates of sickle cell trait and disease, and regional census data from the 2012 Population and Housing Census for the United Republic of Tanzania,36 we calculated the estimated number of annual births with sickle cell trait or disease in each region within the study area (Table 2). We estimated that the number of births with sickle cell disease per year was 10 056 within the nine regions comprising the study area. Contributions varied by region according to their population size and sickle cell disease prevalence. We projected the lowest number of births with sickle cell disease per year for the region of Singida (526 births), and the highest for the region of Mwanza (1730 births; Table 2).
Table 2. Estimated annual region-specific numbers of infants with sickle cell trait and disease, north-western United Republic of Tanzania, February 2017–May 2018.
| Region | Populationa | Crude birth rate per 1 000 populationa | Estimated annual no. of births with sickle cell traitb | Estimated annual no. of births with sickle cell diseaseb |
|---|---|---|---|---|
| Geita | 1 739 530 | 56 | 19 775 | 877 |
| Kagera | 2 458 023 | 44 | 17 953 | 541 |
| Kigoma | 2 127 930 | 48 | 18 385 | 1 532 |
| Mara | 1 743 830 | 49 | 18 029 | 1 111 |
| Mwanza | 2 772 509 | 48 | 26 882 | 1 730 |
| Shinyanga | 1 534 808 | 44 | 14 452 | 1 013 |
| Simiyu | 1 584 157 | 52 | 18 535 | 1 236 |
| Singida | 1 370 637 | 48 | 13 027 | 526 |
| Tabora | 2 291 623 | 50 | 22 687 | 1 490 |
| Total | – | – | 169 725 | 10 056 |
Relation to HIV status
For the 16 479 samples for which HIV results were available, we analysed the co-morbidity of HIV and sickle cell disease to compare the potential effect of HIV status on mortality, as previously performed in Uganda.34 The prevalence of sickle cell disease was 1.2% among both HIV-infected (9/732) and HIV-negative (192/15 747) infants, indicating that HIV status has no effect on early mortality (Table 3).
Table 3. HIV-specific prevalence of haemoglobin types in infants, north-western United Republic of Tanzania, February 2017–May 2018.
| HIV status | Total | No. (%) |
||
|---|---|---|---|---|
| Normal | Sickle cell trait | Sickle cell disease | ||
| Negative | 15 747 | 12 388 (78.7) | 3167 (20.1) | 192 (1.2) |
| Positive |
732 |
563 (76.9) |
160 (21.9) |
9 (1.2) |
| Total | 16 479a | 12 951 (78.6) | 3327 (20.2) | 201 (1.2) |
HIV: human immunodeficiency virus.
a Only specimens for which HIV test results were available are included.
Note: We used isoelectric focusing to detect different haemoglobin types in infants aged 0–24 months.
DNA-based analysis
Of the 210 specimens that were interpreted as sickle cell disease by isoelectric focusing, 143 were genotype-confirmed and made available for further DNA-based testing. Uncommon or atypical haemoglobin variants were rare (0.1%; 17/17 200) and included four haemoglobin G-Pest (HBA1:p.Asp75Asn), two haemoglobin Kenya (HBG1–HBB fusion Aγ81-β86) and a haemoglobin P-Nilotic (HBB–HBD fusion β31-δ50). We identified 1-gene deletion α-thalassaemia trait in 42.7% (61/143) and 2-gene deletion α-thalassaemia trait in 14.7% (21/143). We detected G6PD A– deficiency in 19.2% (14/73) of males, and 25.7% (18/70) of females were heterozygous carriers (Table 4).
Table 4. Prevalence of α-thalassaemia and G6PD deficiency observed in infants, north-western United Republic of Tanzania, February 2017–May 2018.
| Genotype | Clinical effect | No. (%) |
|---|---|---|
| α-thalassaemia (n = 143) | ||
| 5 copies, αα/ααα | 1-gene duplication, unaffected | 0 (0.0) |
| 4 copies, αα/αα | 0-gene deletion, unaffected | 61 (42.7) |
| 3 copies, αα/–α3.7 | 1-gene deletion, α-thalassaemia minima | 61 (42.7) |
| 2 copies, –α3.7/–α3.7 | 2-gene deletion, α-thalassaemia trait | 21 (14.7) |
| G6PD deficiency (n = 143) | ||
| Males (n = 73) | ||
| B | Wild type, unaffected | 52 (71.2) |
| A+ | A+ variant, unaffected | 7 (9.6) |
| A– | A– variant, affected | 14 (19.2) |
| Females (n = 70) | ||
| BB | Homozygous, wild type, unaffected | 33 (47.1) |
| BA+ | Heterozygous, wild type/A+ variant, unaffected | 16 (22.9) |
| A+A+ | Homozygous, A+ variant, unaffected | 3 (4.3) |
| BA– | Heterozygous, wild type/A– variant, carrier | 15 (21.4) |
| A+A– | Heterozygous, A+ variant/A– variant, carrier | 3 (4.3) |
| A–A– | Homozygous, A– variant, affected | 0 (0.0) |
G6PD: glucose-6-phosphate dehydrogenase.
Note: We analysed dried blood spots from infants aged 0–24 months.
We provide the minor allelic frequencies for genetic modifiers that affect haemoglobin F production for the 143 samples available for genetic testing in Table 5. Three single-nucleotide polymorphisms in BCL11A had minor allelic frequencies ranging from 0.266 (rs1427407) to 0.325 (rs4671393). Our frequencies were slightly higher than those observed within the African subgroup of the 1000 Genomes Project,42 which reported values of 0.238 (rs1427407) and 0.269 (rs4671393). The frequency of two single-nucleotide polymorphisms within HMIP was more variable; the value at rs28384513 (in the HMIP-1 region) was 0.238, which is slightly higher than the 1000 Genomes Project African subgroup (0.184), while the value at rs9399137 (in the HMIP-2 region) was low (0.045) and almost identical to the 1000 Genomes Project African subgroup (0.042). Finally, the A variant of rs7482144 in HBG2 was not detected, as expected in an East African cohort.
Table 5. Prevalence of haemoglobin modifiers observed in infants, north-western United Republic of Tanzania, February 2017–May 2018.
| Chromosome | Gene | Single-nucleotide polymorphism | Allele change | Higher fetal haemoglobin allele frequency |
|---|---|---|---|---|
| 2 | BCL11A | rs1188686 | T → C | 0.322 = C |
| 2 | BCL11A | rs1427407 | G → T | 0.266 = T |
| 2 | BCL11A | rs4671393 | G → A | 0.325 = A |
| 6 | HBS1L-MYB | rs28384513 | T → G | 0.238 = G |
| 6 | HBS1L-MYB | rs9399137 | T → C | 0.045 = C |
| 11 | HBG2 | rs7482144 | G → A | 0.000 = A |
Discussion
We estimate that the annual number of live births with sickle cell disease is at least twice that previously thought to occur in the United Republic of Tanzania; we project that over 10 000 births among just 40% of the Tanzanian population are affected annually, compared with previous estimates of 86556 and 11 0229 annually affected births for the whole country. Our calculated prevalence of sickle cell trait and disease concurs with a recent pilot screening project of 919 infants conducted in Mwanza in 2014, which reported a prevalence of 1.4% (13/919) and 19.7% (181/919), respectively.20 Although we observed geographical differences between districts, our variations were not as high as those reported from neighbouring Uganda34 where specimens were collected from a roughly equivalent land mass and population. Our high prevalence and relative lack of variation between districts in the north-western part of the country may reflect the high selection pressure from malaria in a holoendemic area, and possibly lower migration rates.
Our data reinforce the urgent need to enhance sickle cell diagnostic services, the obligatory first step in the cascade of care for this neglected patient population. The sickle cell clinic at Bugando Medical Centre has approximately 600 patients currently enrolled in care, some of whom travel from neighbouring regions. However, we estimate that 1730 infants are born with sickle cell disease in Mwanza region alone each year, which indicates that many affected children in sub-Saharan Africa have not even been diagnosed with the condition. The nine regions included in this study have the highest childhood mortality in the country, ranging from 30 to 38 deaths per 1000 live births,43 and sickle cell disease contributes significantly to mortality in this age group.11–13
We also investigated the extent to which additional erythrocyte disorders are commonly co-inherited with sickle cell disease; cross-sectional analyses in the United Republic of Tanzania have reported on the impact of such disorders on sickle cell patients.44 Ongoing prospective cohort studies will help to clarify the unique interactions between specific sickle cell disease morbidities and co-inherited erythrocyte disorders, endemic arboviral and plasmodial infections, and environmental factors. Expression of haemoglobin F is another well-recognized factor affecting disease severity. Some modifiers of haemoglobin F are similar between Tanzanian and British patients,45 but genetic studies among Tanzanians have also identified unique candidate variants and pathways.46 Understanding these genetic factors and their influence on disease outcomes will become more important, especially as hydroxyurea therapy is introduced to the region.47
An important strength of our study is that we developed a sickle cell disease diagnostic service using existing infrastructure used for HIV infant diagnosis, demonstrating a potential of using this approach across sub-Saharan Africa. For example, in Uganda decision-makers have successfully expanded the use of their HIV infant diagnosis screening platform for sickle cell disease diagnosis, and have launched focused screening in 18 high-prevalence areas.48 Until the emergence of government-sponsored universal screening, local providers can implement their own strategies for optimal screening. Regardless of the location (e.g. hospitals, schools, or reproductive and child health clinics), population (e.g. all patients, mothers, newborns or children) or testing modality (isoelectric focusing, electrophoresis, point-of-care testing or high-performance liquid chromatography) used for initial screening, the HIV infant diagnosis infrastructure can be used to transport dried blood spots to a central laboratory equipped for confirmatory testing and diagnosis of sickle cell disease. Expanding testing in this way would represent a crucial step towards achieving some of the targets of the sustainable development goal 3, which are to end preventable deaths among children and reduce premature mortality from noncommunicable diseases.5,49
The testing of infants was another strength of our study. By excluding samples from children older than 24 months from our analyses, we avoided the errors of previous reports22–32 that used adults and children to develop inaccurately low estimates, or that extrapolated sparse results over large geographic areas.
Our study had two limitations. First, we analysed dried blood spots only after they had been processed by the HIV Early Infant Diagnosis laboratory. In some cases, several months had elapsed before the dried blood spots became available for sickle cell disease testing. During that time, dried blood spots were stored at suboptimal conditions for haemoglobin preservation (usually room temperature); however, our isoelectric focusing testing was robust and able to score 99.6% (17 200/17 274) of all dried blood spot specimens. Second, our study population was restricted to children born to mothers living with HIV; however, it has been reported that HIV status is unlikely to affect the genetic inheritance of the sickle allele.34
Since universal newborn screening is not immediately feasible in most countries, we recommend initially focusing screening efforts on high-prevalence districts to invest in those communities most affected by sickle cell disease, using existing public health infrastructure with minimal start-up cost and training. The cost–effectiveness of implementing a screening programme in conjunction with treatment for children who are subsequently diagnosed with sickle cell disease varies between regions and countries, especially regarding salary for personnel, but a detailed analysis in Angola provides some helpful comparisons.50
Inaccurate national and subnational estimates obscure the true burden of sickle cell disease, which is a common and important cause of death in young children in low-income countries. It is imperative that health ministries and international groups such as the Global Burden of Disease project51 receive high-quality district-level data to guide global health priorities and public health policy; our data will inform such strategies.
Acknowledgements
Emmanuela E Ambrose and Luke R Smart contributed equally to this work. Allan Anderson donated pre-owned isoelectric focusing equipment to Bugando Medical Centre and introduced laboratory staff to the technique. Jelili Ojodu and the United States Association of Public Health Laboratories donated new isoelectric focusing equipment and organized the training session in Dar es Salaam. Petri Huhtinen provided the isoelectric focusing laboratory training seminar. We thank Abel Makubi, the Director General of Bugando Medical Centre.
Competing interests:
None declared.
References
- 1.Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017. July 15;390(10091):311–23. 10.1016/S0140-6736(17)30193-9 [DOI] [PubMed] [Google Scholar]
- 2.McGann PT. Sickle cell anemia: an underappreciated and unaddressed contributor to global childhood mortality. J Pediatr. 2014. July;165(1):18–22. 10.1016/j.jpeds.2014.01.070 [DOI] [PubMed] [Google Scholar]
- 3.Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7):e1001484. 10.1371/journal.pmed.1001484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piel FB, Howes RE, Patil AP, Nyangiri OA, Gething PW, Bhatt S, et al. The distribution of haemoglobin C and its prevalence in newborns in Africa. Sci Rep. 2013;3(1):1671. 10.1038/srep01671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sickle-cell disease: a strategy for the WHO African region. Report AFR/RC60/8. Geneva: World Health Organization; 2010. Available from: https://www.afro.who.int/sites/default/files/2017-06/afr_rc60_8.pdf [cited 2020 Sep 02].
- 6.Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013. January 12;381(9861):142–51. 10.1016/S0140-6736(12)61229-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aliyu ZY, Kato GJ, Taylor J 6th, Babadoko A, Mamman AI, Gordeuk VR, et al. Sickle cell disease and pulmonary hypertension in Africa: a global perspective and review of epidemiology, pathophysiology, and management. Am J Hematol. 2008. January;83(1):63–70. 10.1002/ajh.21057 [DOI] [PubMed] [Google Scholar]
- 8.Macharia AW, Mochamah G, Uyoga S, Ndila CM, Nyutu G, Makale J, et al. The clinical epidemiology of sickle cell anemia in Africa. Am J Hematol. 2018. March;93(3):363–70. 10.1002/ajh.24986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008. June;86(6):480–7. 10.2471/BLT.06.036673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sickle-cell anaemia. Report A59/9. Geneva: World Health Organization; 2006. Available from: https://apps.who.int/iris/bitstream/handle/10665/20890/A59_9-en.pdf?sequence=1&isAllowed=y [cited 2020 Sep 02].
- 11.Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med. 2011. December;41(6)Suppl 4:S398–405. 10.1016/j.amepre.2011.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleming AF, Storey J, Molineaux L, Iroko EA, Attai ED. Abnormal haemoglobins in the Sudan savanna of Nigeria. I. Prevalence of haemoglobins and relationships between sickle cell trait, malaria and survival. Ann Trop Med Parasitol. 1979. April;73(2):161–72. 10.1080/00034983.1979.11687243 [DOI] [PubMed] [Google Scholar]
- 13.Makani J, Cox SE, Soka D, Komba AN, Oruo J, Mwamtemi H, et al. Mortality in sickle cell anemia in Africa: a prospective cohort study in Tanzania. PLoS One. 2011. February 16;6(2):e14699. 10.1371/journal.pone.0014699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Assembly of the African Union. Decisions, Declarations, and Resolution. Fifth Ordinary Session, July 4–5, 2005. Sirte, Libya. Addis Ababa: African Union; 2005. Available from: https://au.int/en/decisions-108 [cited 2020 Sep 02].
- 15.Resolution A/Res/63/237. Recognition of sickle-cell anaemia as a public health problem. In: Sixty-third session General Assembly, New York, 22 Dec 2008. New York: United Nations; 2009. Available from: https://digitallibrary.un.org/record/644334 [cited 2020 Sep 02].
- 16.Ministry of Health, Community, Development, Gender, Elderly and Children. Strategic and action plan for the prevention and control of non communicable diseases in Tanzania 2016-2020. Dar es Salaam: Government of the United Republic of Tanzania; 2016. Available from: https://www.worlddiabetesfoundation.org/sites/default/files/NCD%20Stategic%20Plan%202016%20-%202020.pdf [cited 2020 Sep 02].
- 17.Makani J, Soka D, Rwezaula S, Krag M, Mghamba J, Ramaiya K, et al. Health policy for sickle cell disease in Africa: experience from Tanzania on interventions to reduce under-five mortality. Trop Med Int Health. 2015. February;20(2):184–7. 10.1111/tmi.12428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makani J, Tluway F, Makubi A, Soka D, Nkya S, Sangeda R, et al. A ten year review of the sickle cell program in Muhimbili National Hospital, Tanzania. BMC Hematol. 2018. November 14;18:33. 10.1186/s12878-018-0125-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tluway F, Makani J. Sickle cell disease in Africa: an overview of the integrated approach to health, research, education and advocacy in Tanzania, 2004-2016. Br J Haematol. 2017. June;177(6):919–29. 10.1111/bjh.14594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ambrose EE, Makani J, Chami N, Masoza T, Kabyemera R, Peck RN, et al. High birth prevalence of sickle cell disease in Northwestern Tanzania. Pediatr Blood Cancer. 2018. January;65(1):e26735. 10.1002/pbc.26735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rwezaula SS, Magesa PM, Mgaya J, Massawe A, Newton CR, Marlow T, et al. Newborn screening for haemoglobinopathies at Muhimbili National Hospital, Dar es Salaam – Tanzania. East Afr J Public Health. 2015. March;12(1):948–56. [Google Scholar]
- 22.Allison AC. The distribution of the sickle-cell trait in East Africa and elsewhere, and its apparent relationship to the incidence of subtertian malaria. Trans R Soc Trop Med Hyg. 1954. July;48(4):312–8. 10.1016/0035-9203(54)90101-7 [DOI] [PubMed] [Google Scholar]
- 23.Allison AC. The sickle-cell and haemoglobin C genes in some African populations. Ann Hum Genet. 1956. July;21(1):67–89. 10.1111/j.1469-1809.1971.tb00266.x [DOI] [PubMed] [Google Scholar]
- 24.Chopra SA, Mbaye AH. Sickling in Zanzibar island–its incidence, regional variation, and relationship with splenomegaly rates. Trans R Soc Trop Med Hyg. 1969;63(2):270–4. 10.1016/0035-9203(69)90158-8 [DOI] [PubMed] [Google Scholar]
- 25.Enevold A, Alifrangis M, Sanchez JJ, Carneiro I, Roper C, Børsting C, et al. Associations between alpha+-thalassemia and plasmodium falciparum malarial infection in northeastern Tanzania. J Infect Dis. 2007. August 1;196(3):451–9. 10.1086/519390 [DOI] [PubMed] [Google Scholar]
- 26.Godber M, Kopeć AC, Mourant AE, Teesdale P, Tills D, Weiner JS, et al. The blood groups, serum groups, red-cell isoenzymes and haemoglobins of the Sandawe and Nyaturu of Tanzania. Ann Hum Biol. 1976. September;3(5):463–73. 10.1080/03014467600001731 [DOI] [PubMed] [Google Scholar]
- 27.Lehmann H, Mackey JP. The absence of haemoglobin C in 104 East Africans living in Dar es Salaam. Man (Lond). 1955. December;55:186 10.2307/2795332 [DOI] [Google Scholar]
- 28.Marti HR, Schoepf K, Gsell OR. Frequency of haemoglobin S and glucose-6-phosphate dehydrogenase deficiency in southern Tanzania. BMJ. 1965. June 5;1(5448):1476–7. 10.1136/bmj.1.5448.1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitchell R, Fupi F. Sickling in Tanzania. East Afr Med J. 1972. September;49(9):638–42. [PubMed] [Google Scholar]
- 30.Nhonoli AM, Kujwalile JM, Kigoni EP, Masawe AE. Correlation of glucose-6-phosphate dehydrogenase (G-6-PD) deficiency and sickle cell trait (Hb-AS). Trop Geogr Med. 1978. March;30(1):99–101. [PubMed] [Google Scholar]
- 31.Roberts DF, Papiha SS. Les polymorphisms genetiques des Sukuma (Tanzania). L’Anthropologie. 1978;82:565–74. [French]. [Google Scholar]
- 32.Stirnadel HA, Stöckle M, Felger I, Smith T, Tanner M, Beck HP. Malaria infection and morbidity in infants in relation to genetic polymorphisms in Tanzania. Trop Med Int Health. 1999. March;4(3):187–93. 10.1046/j.1365-3156.1999.43381.x [DOI] [PubMed] [Google Scholar]
- 33.Piel FB, Adamkiewicz TV, Amendah D, Williams TN, Gupta S, Grosse SD. Observed and expected frequencies of structural hemoglobin variants in newborn screening surveys in Africa and the Middle East: deviations from Hardy-Weinberg equilibrium. Genet Med. 2016. March;18(3):265–74. 10.1038/gim.2015.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ndeezi G, Kiyaga C, Hernandez AG, Munube D, Howard TA, Ssewanyana I, et al. Burden of sickle cell trait and disease in the Uganda Sickle Surveillance Study (US3): a cross-sectional study. Lancet Glob Health. 2016. March;4(3):e195–200. 10.1016/S2214-109X(15)00288-0 [DOI] [PubMed] [Google Scholar]
- 35.Nuwagaba-Biribonwoha H, Werq-Semo B, Abdallah A, Cunningham A, Gamaliel JG, Mtunga S, et al. Introducing a multi-site program for early diagnosis of HIV infection among HIV-exposed infants in Tanzania. BMC Pediatr. 2010. June 17;10(1):44. 10.1186/1471-2431-10-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Population distribution by age and sex. Dar es Salaam: National Bureau of Statistics; 2013. Available from: http://tanzania.countrystat.org/fileadmin/user_upload/countrystat_fenix/congo/docs/Population%20Distribution%20by%20Age%20and%20Sex%20Report-2012PHC.pdf [cited 2020 Sep 02].
- 37.Schaefer BA, Kiyaga C, Howard TA, Ndeezi G, Hernandez AG, Ssewanyana I, et al. Hemoglobin variants identified in the Uganda Sickle Surveillance Study. Blood Adv. 2016. November 22;1(1):93–100. 10.1182/bloodadvances.2016000950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grimholt RM, Urdal P, Klingenberg O, Piehler AP. Rapid and reliable detection of α-globin copy number variations by quantitative real-time PCR. BMC Hematol. 2014. January 24;14(1):4. 10.1186/2052-1839-14-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGann PT, Williams AM, Ellis G, McElhinney KE, Romano L, Woodall J, et al. Prevalence of inherited blood disorders and associations with malaria and anemia in Malawian children. Blood Adv. 2018. November 13;2(21):3035–44. 10.1182/bloodadvances.2018023069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheehan VA, Luo Z, Flanagan JM, Howard TA, Thompson BW, Wang WC, et al. ; BABY HUG Investigators. Genetic modifiers of sickle cell anemia in the BABY HUG cohort: influence on laboratory and clinical phenotypes. Am J Hematol. 2013. July;88(7):571–6. 10.1002/ajh.23457 [DOI] [PubMed] [Google Scholar]
- 41.Hardy GH. Mendelian proportions in a mixed population. Science. 1908. July 10;28(706):49–50. 10.1126/science.28.706.49 [DOI] [PubMed] [Google Scholar]
- 42.IGSR and the 1000 Genome Project. Cambridge: International Genome Sample Resource; 2020. Available from: https://www.internationalgenome.org/ [cited 2020 Sep 08].
- 43.Tanzania: Demographic and Health Survey and Malaria Indicator Survey 2015-16. Dar es Salaam and Rockville: Ministry of Health, Community Development, Gender, Elderly and Children [Tanzania mainland], Ministry of Health [Zanzibar], National Bureau of Statistics, Office of the Chief Government Statistician and ICF; 2016. Available from: https://dhsprogram.com/pubs/pdf/FR321/FR321.pdf [cited 2020 Sep 02].
- 44.Cox SE, Makani J, Soka D, L’Esperence VS, Kija E, Dominguez-Salas P, et al. Haptoglobin, alpha-thalassaemia and glucose-6-phosphate dehydrogenase polymorphisms and risk of abnormal transcranial Doppler among patients with sickle cell anaemia in Tanzania. Br J Haematol. 2014. June;165(5):699–706. 10.1111/bjh.12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makani J, Menzel S, Nkya S, Cox SE, Drasar E, Soka D, et al. Genetics of fetal hemoglobin in Tanzanian and British patients with sickle cell anemia. Blood. 2011. January 27;117(4):1390–2. 10.1182/blood-2010-08-302703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mtatiro SN, Singh T, Rooks H, Mgaya J, Mariki H, Soka D, et al. Genome wide association study of fetal hemoglobin in sickle cell anemia in Tanzania. PLoS One. 2014. November 5;9(11):e111464. 10.1371/journal.pone.0111464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tshilolo L, Tomlinson G, Williams TN, Santos B, Olupot-Olupot P, Lane A, et al. ; REACH Investigators. Hydroxyurea for children with sickle cell anemia in sub-Saharan Africa. N Engl J Med. 2019. January 10;380(2):121–31. 10.1056/NEJMoa1813598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kiyaga C, Hernandez AG, Ssewanyana I, Schaefer BA, McElhinney KE, Ndeezi G, et al. Sickle cell screening in Uganda: high burden, human immunodeficiency virus comorbidity, and genetic modifiers. Pediatr Blood Cancer. 2019. August;66(8):e27807. 10.1002/pbc.27807 [DOI] [PubMed] [Google Scholar]
- 49.Sustainable development goal 3. New York: United Nations; 2015. Available from: https://www.un.org/sustainabledevelopment/health/ [cited 2020 Sep 02].
- 50.McGann PT, Grosse SD, Santos B, de Oliveira V, Bernardino L, Kassebaum NJ, et al. A cost-effectiveness analysis of a pilot neonatal screening program for sickle cell anemia in the Republic of Angola. J Pediatr. 2015. December;167(6):1314–9. 10.1016/j.jpeds.2015.08.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age–sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015. January 10;385(9963):117–71. 10.1016/S0140-6736(14)61682-2 [DOI] [PMC free article] [PubMed] [Google Scholar]