

Abstract
Smad3, a major intracellular mediator of TGFβ signaling, functions as both a positive and negative regulator in carcinogenesis. In response to TGFβ, the TGFβ receptor phosphorylates serine residues at the Smad3 C-tail. Cancer cells often contain high levels of the MAPK and CDK activities, which can lead to the Smad3 linker region becoming highly phosphorylated. Here, we report, for the first time, that mutation of the Smad3 linker phosphorylation sites markedly inhibited primary tumor growth, but significantly increased lung metastasis of breast cancer cell lines. In contrast, mutation of the Smad3 C-tail phosphorylation sites had the opposite effect. We show that mutation of the Smad3 linker phosphorylation sites greatly intensifies all TGFβ-induced responses, including growth arrest, apoptosis, reduction in the size of putative cancer stem cell population, epithelial–mesenchymal transition, and invasive activity. Moreover, all TGFβ responses were completely lost on mutation of the Smad3 C-tail phosphorylation sites. Our results demonstrate a critical role of the counterbalance between the Smad3 C-tail and linker phosphorylation in tumorigenesis and metastasis. Our findings have important implications for therapeutic intervention of breast cancer.
Introduction
TGFβ is the prototype of a large family of pleiotropic polypeptide cytokines that regulate a multitude of physiologic functions such as cell proliferation, differentiation, apoptosis, and epithelial–mesenchymal transition (EMT; refs. 1–3). Functional aberrations of the TGFβ signaling pathway underlie various forms of human cancer, fibrotic, and developmental disorders (3, 4). The ability of TGFβ signaling to suppress proliferation of epithelial, neuronal, and hematopoietic cells, from which a majority of human neoplasms originate, suggests a role of TGFβ in tumor suppression (1–3, 5). However, TGFβ has also been known to exert a multifaceted influence on gene expressions that are favorable to metastasis and cancer progression, depending on the clinical stages (1–5).
TGFβ signals are mediated through a multiplicity of pathways, including the predominant Smad signaling pathway, as well as MAPK, Rho-like GTPase, and PI3K pathways (6–8). In the canonical TGFβ pathway, Smad2 and Smad3 are phosphorylated by the type I receptor kinase at the C-terminal SXS motif, form complexes with Smad4, and then accumulate in the nucleus to regulate gene expression of target genes (6–8). Studies using mouse embryonic fibroblasts with a targeted deletion of either Smad2 or Smad3 showed that Smad3 is an essential mediator of TGFβ-induced transcriptional responses (4, 9). The Smad proteins consist of conserved N- and C-terminal domains, separated by a divergent linker region. The Smad3 linker region containing four serine/threonine phosphorylation sites at Thr179, Ser204, Ser208, and Ser213 (10–18) can be phosphorylated by several kinases, such as the MAPK family and cyclin-dependent kinase (CDK) family (10–13, 15–20). These residues except Ser213 are subject to phosphorylation in response to TGFβ (11, 12, 14). Although phosphorylation of Smad3 C-tail SXS motifs by the TGFβ type I receptor is required for the linker phosphorylation, the receptor itself does not phosphorylate the linker region (13). Interestingly, Smad linker phosphorylation has been associated with more aggressive disease in certain cancer and fibrosis models (18). Previous reports showed that mutation of the Smad3 linker phosphorylation sites increased the ability of Smad3 to activate TGFβ target genes as well as growth-inhibitory function (13, 14) and EMT (21) based on cell culture studies. However, currently, it is not clear what functional roles the Smad3 C-tail and linker phosphorylation play in tumorigenesis and metastasis.
To address this question, we abrogated Smad3 phosphorylation in breast cancer cells either at the linker or C-tail phosphorylation sites by infection with adenoviruses constitutively expressing Smad3 phosphorylation mutants. Our current study shows that aggressive breast cancer cell lines display a marked refractoriness to TGFβ, and that mutation of Smad3 linker phosphorylation sites amplifies all TGFβ responses, leading to simultaneous induction of growth arrest, apoptosis, a reduction in putative cancer stem cell population, and EMT. We have addressed the integrated output of these effects using xenograft models, and demonstrated that mutation of the Smad3 linker phosphorylation sites greatly enhances metastatic dissemination to the lung, while inhibiting growth of primary tumors. Conversely, mutation of the C-tail phosphorylation sites suppressed metastasis, while accelerating tumor growth. Our findings indicate that the dual role of TGFβ in transducing tumor-suppressive and pro-oncogenic signals is modulated by the balance between the Smad3 linker and C-tail phosphorylation. These results enable us for a deeper understanding of metastasis and tumor growth, and may provide insights for the development of novel therapies.
Materials and Methods
Cell culture
Human MCF-10A–derived breast cancer cell line MCF-10CA1a.cl1 (abbreviated as CAla; ref. 22) was from Dr. Fred Miller (Barbara Ann Karmanos Cancer Institute, Detroit, MI). Mouse breast cancer cell line 4T1 (23) and luciferase-expressing 4T1 (4T1-Luc) cells (24) were from Jeong-Seok Nam (Lee Gil Ya Cancer and Diabetes Institute, Gachon University of Medicine and Science, Incheon, Korea). In vitro experiments were performed in the medium containing 5% serum at 37°C in a CO2 incubator otherwise notified. Cells were treated with or without TGFβ1 (5 ng/mL−1).
Adenoviral infection
Adenoviral GFP, Smad3, and Smad3 mutants at C-tail or linker phosphorylation sites were kindly provided from the late Dr. Anita B. Roberts (NCI, NIH, Bethesda, MD) and Dr. Sushil G. Rane (NIDDK, NIH, Bethesda, MD). Adenovirus infection into the breast cancer cells was carried out as previously described (25).
Cell proliferation and apoptosis
After serum starvation for 4 hours, cells were cultured in serum-free condition with or without TGFβ1 for 48 hours. Cell proliferation was assayed using in situ cell proliferation kits (Roche Applied Science). For apoptotic cell detection, APO-BrdU TUNEL assay kit (Invitrogen) was used following the manufacturer’s instructions. For calculation of apoptotic cells, cells were trypsinized and combined with their medium to include the detached dead cells. Each sample was stained with trypan blue (Bio-Whittaker) and counted with TC10 automated cell counter (Bio-Rad).
Immunoblotting
Cell extracts were separated by SDS–PAGE, followed by electrotransfer to polyvinylidene difluoride membranes and probed with antibodies against E-cadherin, vimentin, fibronectin (BD Pharmingen), PAI-1, c-Myc, p21 (Santa Cruz Bio-technology), PARP, Cleaved PARP, caspase-3, cleaved caspase-3 (Cell Signaling Technology), Bcl-2, Oct4, Nanog, Sox2 (Abcam), α-tubulin and β-actin (Sigma-Aldrich). For immunoblot analyses of cell proliferation and apoptosis markers, cells were cultured with or without TGFβ1 for 48 hours in serum-free condition following serum starvation for 4 hours. For immunoblot analysis of Smad3 phosphopeptides, cells were cultured with or without TGFβ1 for 1 hour. Antibodies against the Smad3 linker regions at Thr179 (pT179), Ser204 (pS204), and Ser208 (pS208) were used as described previously (12, 13). Smad2, Smad2 C-tail phosphopeptide, and Smad3 antibodies were from Cell Signaling Technology. Smad3 C-tail phosphopeptide antibody was from Abcam. Horseradish per-oxidase-conjugated anti-mouse or rabbit antibody (Millipore) was used as a secondary antibody.
Immunofluorescence staining
Cells were cultured with or without TGFβ1 for 48 hours on Lab-TekII chamber slides (Nalge Nunc International), fixed in cold acetone, and subjected to indirect immunofluorescence with anti-E-cadherin, anti-vimentin (BD Pharmingen) or anti-Oct4 (Abcam) antibody at a 1:200 dilution overnight at 4°C. Alexa Fluor 594-conjugated goat anti-mouse IgG or goat anti-rabbit IgG was used as a secondary antibody at a 1:500 dilution for 1 hour, and cells were mounted with a medium containing DAPI (Vector Laboratories).
RT-PCR
Total RNA from cells was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The RNA was converted to cDNA using M-MLV Reverse Transcriptase (Promega). The cDNA was synthesized and RT-PCR was run with the Superscript II reverse transcriptase (Invitrogen). Synthesized cDNA was amplified by PCR using specific primers (see Supplementary Materials and Methods).
Invasion assay
Cells were cultured in the presence or absence of TGFβ1 for 24 hours. Dissociated cells (1 × 104) were plated onto the upper well of Matrigel invasion chamber (BD Biosciences) containing DMEM with 0.1% FBS. The bottom chamber contained DMEM with 10% FBS. After incubation for 48 hours, noninvasive cells were removed with a cotton swab. The cells that migrated through the membrane were fixed with ethanol and stained with 1% toluidine blue. For quantification, cells were counted under a microscope in five predetermined fields.
Mammosphere formation
Cells were cultured with or without TGFβ1 for 48 hours. Dissociated single cells (103 well−1) were plated onto 96-well ultra-low attachment plates (Corning) and grown for 10 days in serum-free DMEM as described (26). Mammospheres with diameters larger than 50 µm were counted.
Soft agar colony formation
Soft agar assay was performed as described previously (26). The colonies with diameters larger than 100 µm were counted.
Flow cytometry
CA1a cells were infected with Ad-Smad3, Ad-3SA, or Ad-EPSM for 48 hours. Dissociated single cells were subjected to FACS analysis using cell surface markers of CD44 and CD24 as previously described (27). Adenovirus-infected 4T1 cells and parental cells (5 × 105) were inoculated into Balb/c mice mammary fat pads. Tumors generated 3 weeks after inoculation were minced and incubated in DMEM with collagenase/hyaluronidase (Stem Cell Technologies) at 37°C for 20 minutes. Single cells from the dissociated tumors were analyzed by FACS using Aldefluor Kit (Stem Cell Technologies) for identification of cells expressing high levels of aldehyde dehydrogenase (ALDH; ref. 28).
Tumorigenesis and metastasis assay
All animals were maintained according to the CHA laboratory animal research center guidelines (Seoul, Korea) under approved animal study protocols. The adenovirus-infected human breast cancer cells CA1a (5 × 105) from subconfluent monolayers were injected orthotopically into 5- to 6-week-old NOD/SCID mice. For experimental metastasis assay, the adenovirus-infected CA1a (1 × 106) and mouse breast cancer cell lines 4T1 (5 × 105) were injected through the tail vein of female NOD/SCID or Balb/c mice, respectively. For tumorigenesis and metastasis assay at the same time, the adenovirus-infected 4T1 cells (1 × 105) were inoculated orthotopically into 6-week-old Balb/c mice. Five weeks after injection, mice were examined grossly at necropsy for the presence of metastases in internal organs. Microscopic quantification of metastases was performed on lung-cross sections. The total number of metastasis per lung was measured macroscopically. 4T1 cells (1 × 105) stably expressing luciferase (4T1-Luc) were injected orthotopically for bioluminescence imaging. The primary tumors and lung metastases were monitored by bioluminescence imaging after intraperitoneal injection of D-luciferin. The bioluminescence signals were detected using an IVIS-200 (Xenogen Corp.). The data were analyzed using the total photon flux emission (photons/second) in the regions of interest.
Tumor-initiating frequency of cancer cells injected into host mice in limiting dilutions
Single 4T1 cells from the primary tumor were inoculated into mammary fat pads of Balb/c mice at varying cell number ranging from 102 to 105 cells/mouse. Tumor-initiating frequency determined 3 weeks after injection was used for calculation of frequency of cancer stem cell as described previously (29).
Statistical analysis
Results were expressed as the mean ± SD. Student unpaired t test and an analysis of multiple variances by Scheffe’s method were used for statistical comparison. A P value of less than 0.05 was considered to indicate statistical significance.
Results
Mutation of the Smad3 linker phosphorylation sites markedly suppresses tumor growth while enhancing lung metastasis
To address the functional roles of linker and C-tail phosphorylation of Smad3, we used recombinant adenovirus constitutively expressing wild-type Smad3 (Smad3) or its mutant at the C-tail (3SA) or linker region (EPSM), in which all serine/threonine phosphorylation sites are replaced with alanine or valine (Fig. 1A). Adenoviral Smad3 (Ad-Smad3) and adenoviral GFP (Ad-GFP) were also included in the study. Infection of adenoviral 3SA (Ad-3SA) and adenoviral EPSM (Ad-EPSM) into high-grade metastatic breast cancer cell line, the MCF-10A-derived human breast cancer cell line, CA1a (22) specifically abrogated the Smad3 C-tail and linker phosphorylation, respectively, in the presence or absence of TGFβ1 (Fig. 1B). Although overexpression of either Smad3 or EPSM had no effect on the endogenous Smad2 C-tail phosphorylation, the dominant-negative form of Smad3, 3SA abrogated both endogenous Smad2 and Smad3 C-tail phosphorylation as reported (Fig. 1B; ref. 4). Ad-3SA and Ad-EPSM infection into another high-grade metastatic mouse breast cancer cell line 4T1 (23, 24) has also been confirmed to block the Smad3 C-tail and linker phosphorylation, respectively (Supplementary Fig. S1). Orthotopic xenografts of adenovirus-infected CA1a cells into SCID/NOD mice gave rise to distinct tumorigenesis. Ad-EPSM infection markedly suppressed tumor growth, whereas Ad-3SA infection generated the largest tumors (Fig. 1C and D). Because CA1a cells do not metastasize spontaneously to the lung when transplanted (data not shown), we injected intravenously the cells in the tail vein as previously described (22). Ad-EPSM infection gives rise to the highest number of metastatic nodules in the lung (Fig. 1E and F), contrasting with the smallest sizes of tumors generated by the orthotopic transplantation (Fig. 1C and D). Ad-3SA infection on the contrary markedly suppresses metastasis to the lung (Fig. 1E and F), while forming the largest tumors (Fig. 1C and D). The mouse breast cancer cell line 4T1 has distinct advantages over CA1a cells for metastasis study because of their biologic features such as the feasibility of syngeneic transplant into immune-competent Balb/c mice and spontaneous metastasis to the lung from the orthotopic site (23). For comparison with CA1a cells, we first injected the adenovirus-infected 4T1 cells into the tail vein of Balb/c mice. Ad-EPSM infection generated the highest number of lung metastatic nodules after intravenous injection, whereas Ad-3SA infection attained nearly perfect suppression of lung metastasis as seen in CA1a cells (Figs. 1E and F and 2A and B). For the examination of spontaneous metastasis to the lung, luciferase-expressing 4T1 cells (4T1-Luc; ref. 24) were transplanted into mammary fat pads of Balb/c mice. The primary tumors caused a gradual increase in luciferin bioluminescence after transplantation (Fig. 2C). At the 5th week, positive bioluminescent images indicative of lung metastasis became detectable at the thoracic regions of mice transplanted with Ad-GFP-, Ad-Smad3-, and Ad-EPSM–infected 4T1-Luc cells, whereas no positive image as discernible in mice transplanted with Ad-3SA–infected counterparts (Fig. 2C). Ad-EPSM infection gives rise to the smallest sizes of tumors (Fig. 2D and E). Ad-3SA infection generates larger tumors than Ad-GFP and Ad-Smad3 infection in 4T1-Luc cells, exhibiting a similar propensity as seen in after transplantation of the adenovirus-infected CA1a cells, although tumor-suppressive effects of Ad-EPSM are much higher in CA1a cells than 4T1-Luc counterparts (compare Fig. 2D and E with Fig. 1C and D). At the 5th week after the transplantation, the highest number of metastatic nodules in the lung was found in mice transplanted with Ad-EPSM–infected 4T1-Luc cells, while by far the lowest in Ad-3SA–infected counterparts (Fig. 2F and G).
Figure 1.

Mutation of the Smad3 linker phosphorylation sites greatly suppresses tumor growth of CA1a cells, but instead promotes experimental lung metastasis. A, schematic representation of phosphorylation sites of Smad3 and its mutants (3SA and EPSM). B, immunoblot analysis for the linker and C-tail phosphorylation with specific antibodies in CA1a cells infected with the recombinant adenovirus expressing Smad3 (Ad-Smad3) or its mutant (Ad-3SA or Ad-EPSM) with or without TGFβ1 for 1 hour. Ad-GFP was used as a control. Similar results were obtained from three separate experiments. C, gross appearances of tumors formed 5 weeks after orthotopic xenografts of the adenovirus-infected CA1a cells (5 × 105) into NOD/SCID mice (16 mice per group). D, tumor growth kinetics in C. Each point represents the mean tumor volume (mm3) ± SD from 16 tumors per group. *, 0.01 < P < 0.05; **, P < 0.01, compared with Ad-Smad3–infected cells. E, lung metastatic nodules of NOD/SCID mice (16 mice per group) at the ninth week after the tail-vein injection of the adenovirus-infected CA1a cells (1 × 106). Scale bar, 1.0 cm. F, number of metastatic nodules per lung. Each bar represents the number of metastatic nodules ± SD from 16 lungs per group. **, P < 0.01, compared with Ad-Smad3–infected cells.
Figure 2.
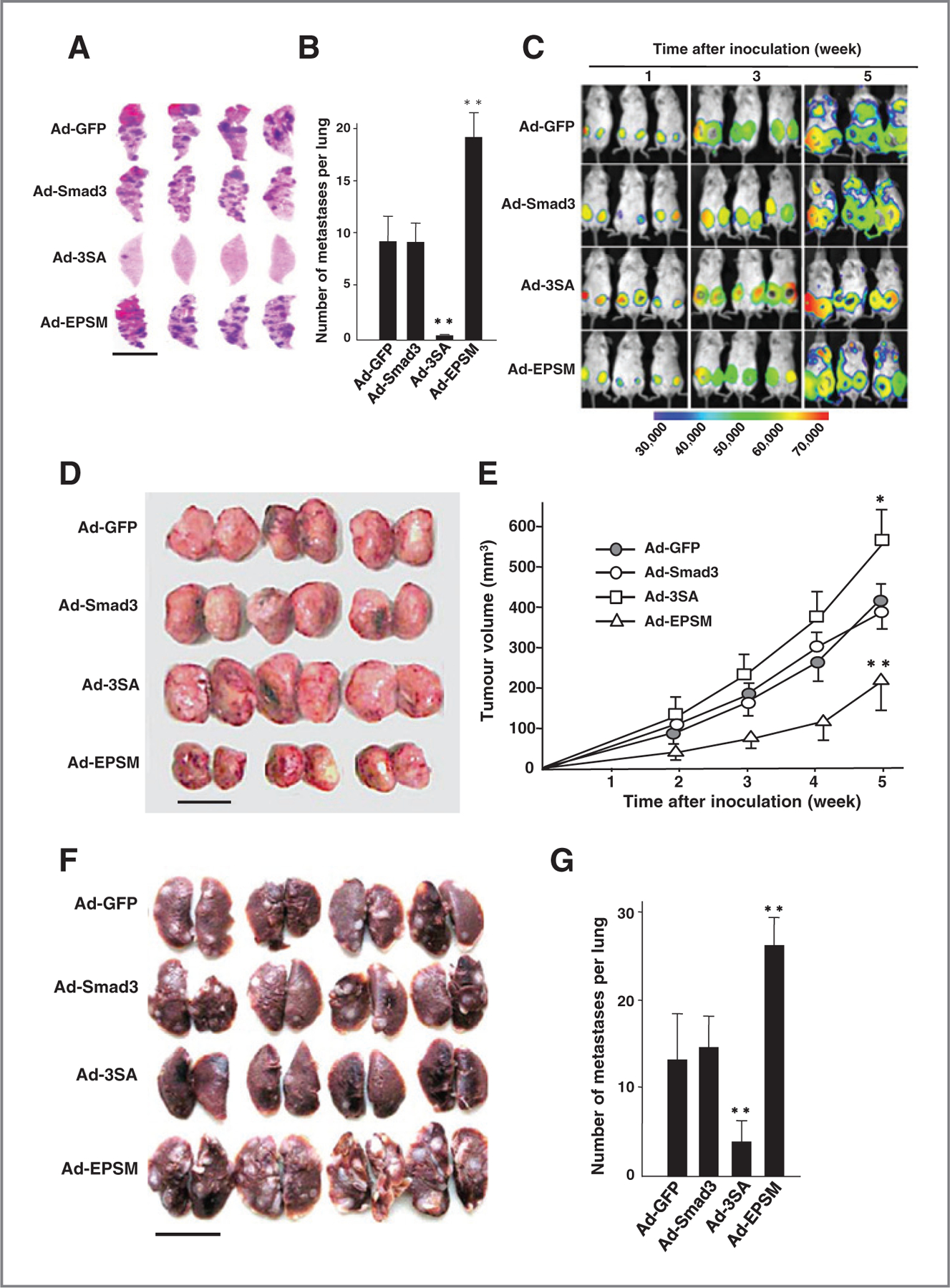
Mutation of the Smad3 linker phosphorylation sites promotes spontaneous lung metastasis of 4T1 cells, whereas mutation of the C-tail phosphorylation sites markedly suppresses it. A, lung metastasis in Balb/c mice (16 mice per group) induced by tail-vein injection of infected 4T1 cells (5 × 105) for 3 weeks. B, number of metastatic nodules per lung. Scale bar, 1.0 cm. Each bar represents the number of metastatic nodules ± SD from 16 lungs per group. C, bioluminescence imaging after orthotopic transplantation of 4T1-Luc cells (1 × 105) into Balb/c mice (16 mice per group). D, gross appearances of primary tumors formed 5 weeks after the xenograft as shown in C. Scale bar, 1.0 cm. E, tumor growth kinetics in D. The mean tumor volume (mm3) ± SD from 16 tumors per group. *, 0.01 < P < 0.05; **, P < 0.01, compared with Ad-Smad3 transfection. F, lung metastatic nodules formed 5 weeks after xenograft shown in C. Scale bar, 1.0 cm. G, number of metastatic nodules per lung as shown in F 5 weeks after the xenograft. The mean ± SD from 16 lungs per group. **, P < 0.01 compared with Ad-Smad3 transfection.
Mutation of Smad3 linker phosphorylation sites greatly suppresses cell proliferation and enhances apoptosis
We then addressed the biologic activities that might underlie the effects of modulating Smad3 activity on tumorigenesis. Tumor suppression by TGFβ involves effects on growth inhibition and apoptosis (2, 3, 5). Ad-EPSM infection markedly suppressed the expression of c-Myc in both CA1a and 4T1 cells even in the absence of TGFβ1, whereas no suppression of c-Myc was found in Ad-3SA–infected cells even in the presence of TGFβ1 (Fig. 3A and Supplementary Fig. S2A). Expression of p21WAF1 is opposite to that of c-Myc. The basal and TGFβ1-induced expression of p21WAF1 was higher in Ad-EPSM–infected cells than Ad-GFP- and Ad-Smad3–infected controls (Fig. 3A and Supplementary Fig. S2A). Their expression, however, was totally abrogated by infection with Ad-3SA even in the presence of TGFβ1. Bromodeoxyuridine (BrdUrd) incorporation was markedly suppressed by Ad-EPSM infection even in the absence of added TGFβ1, whereas no significant differences in BrdUrd incorporation were detected among the other adenovirus-infected cells (Fig. 3B and C and Supplementary Fig. S2B) . Ad-EPSM infection also conferred a TGFβ-induced growth inhibition in 4T1 cells (Supplementary Fig. S2B). For apoptosis, a marked decline of an antiapoptotic protein Bcl-2 expression was shown in Ad-EPSM–infected CA1a cells, whereas no reduction in Bcl-2 was seen in Ad-GFP-, Ad-Smad3-, and Ad-3SA–infected counterparts in the presence of TGFβ1 (Fig. 3D). Concomitant with cleavage of PARP and caspase-3, Ad-EPSM–infected CA1a cells underwent the TGFβ-induced apoptosis as shown by a positive terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining (Fig. 3E) and an increased number of apoptotic cells (Fig. 3F). Ad-EPSM–infected 4T1 cells also showed a significant albeit lower reduction in TGFβ-induced Bcl-2 expression, and cleaved PARP and caspase-3 as compared with Ad-EPSM–infected CA1a cells, resulting in a smaller increase in apoptotic cells in Ad-EPSM–infected 4T1 cells (compare Fig. 3D and F with Supplementary Fig. S2C and S2D).
Figure 3.
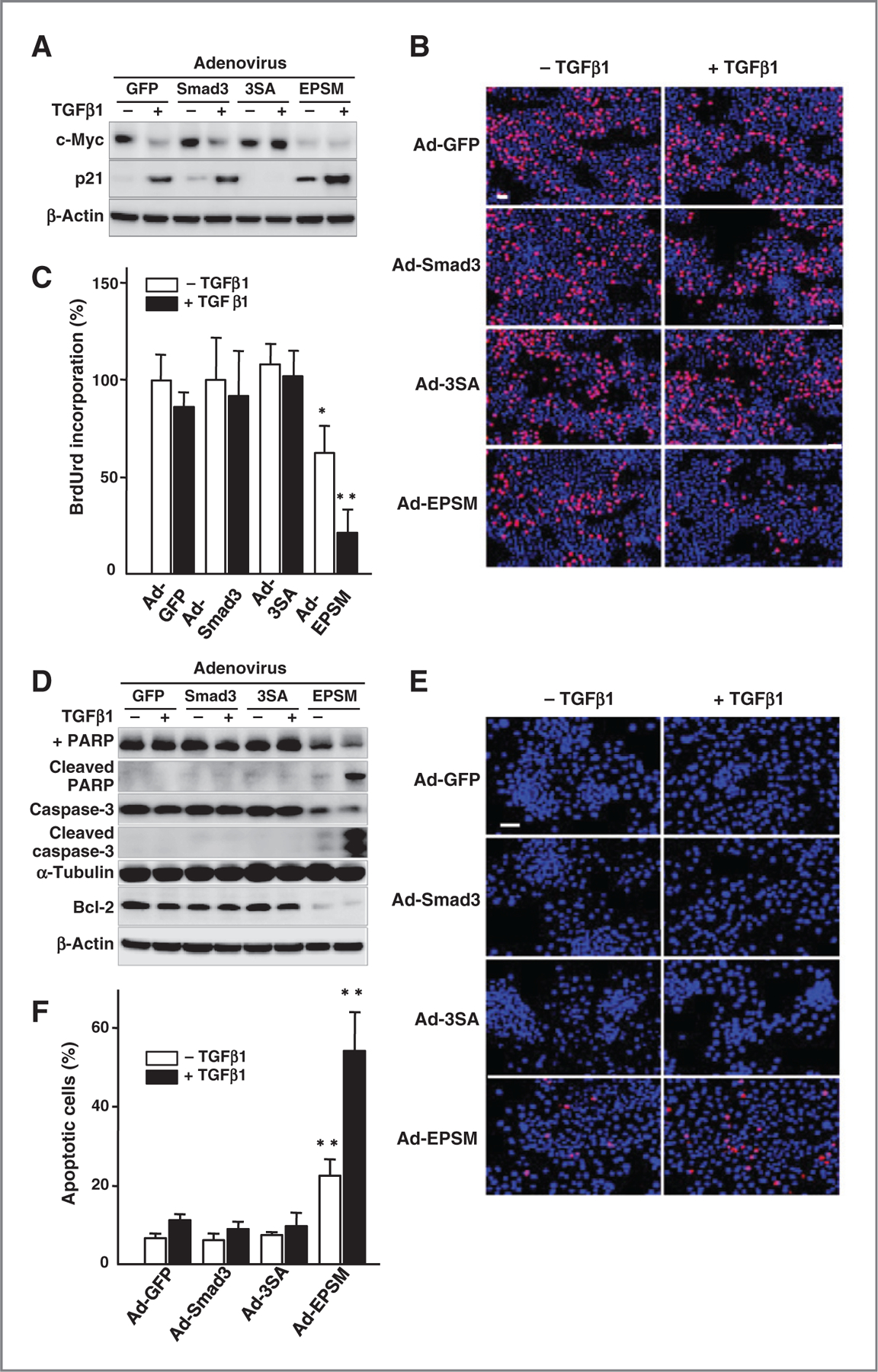
Mutation of the Smad3 linker phosphorylation sites induces a prominent TGFβ-induced growth arrest and apoptosis. A, expression of c-Myc and p21 in the adenovirus-infected CA1a cells with or without TGFβ1. B, in situ BrdUrd incorporation (red) counterstained with DAPI (blue) with or without TGFβ1. Scale bar, 50 µm. C, BrdUrd incorporation (%). The mean ± SD from nine samples per group. *, 0.01 < P < 0.05; **, P < 0.01, compared with Ad-Smad3–infected cells without TGFβ1. D, immunoblot analyses for proapoptotic (cleaved PARP and caspase-3) and antiapoptotic (Bcl-2) markers. E, TUNEL staining for apoptosis (red). Counterstained with DAPI (blue). Scale bar, 50 µm. F, apoptotic cells as estimated by trypan blue exclusion test (%). The mean ± SD from nine samples per group. **, P < 0.01, compared with Ad-Smad3–infected cells without TGFβ1. Each of the experiments was conducted four to six times with similar results.
We have further shown that the mutation of C-tail and linker phosphorylation sites suppresses and enhances the TGFβ/Smad3 transcriptional activity as determined by (CAGA)12-Luc reporter assay, respectively. Higher transcriptional activity was noted in Ad-EPSM–infected CA1a cells as compared with Ad-Smad3–infected counterparts even in the absence of TGFβ1, while its activity was totally abrogated by Ad-3SA infection (Supplementary Fig. S3A). We next attempted to determine whether basal levels of Smad3 linker phosphorylation change in the presence of an inhibitor of TGFβ type I receptor kinase (SB43152). SB43152 virtually did not alter basal levels of the linker phosphorylation, but completely suppressed the TGFβ-induced linker phosphorylation, whereas flavopiridol, although to a less extent as compared to SB43152, significantly inhibited the TGFβ-induced linker phosphorylation (Supplementary Fig. S3B). The TGFβ/Smad3 transcriptional activity was inversely correlated with levels of the linker phosphorylation (Supplementary Fig. S3B and S3C).
Mutation of Smad3 linker phosphorylation sites markedly enhances the TGFβ-induced EMT and invasive property
TGFβ is a strong inducer of EMT and enhances tumor cell invasion (1–5). Ad-GFP-, Ad-Smad3, and Ad-EPSM–infected CA1a cells shared morphologic features of the TGFβ-induced EMT as characterized by loss of epithelial characteristics leading to a fibroblastic phenotype, whereas Ad-3SA–infected cells did not display any phenotypic conversion (Fig. 4A and Supplementary Fig. S4A). Immunoblot analysis and real-time PCR (RT-PCR) further support the EMT features as revealed by reduction in an epithelial marker (E-cadherin) with increase in E-cadherin transcriptional repressors (Twist and Zeb1), and increase in mesenchymal markers (vimentin and fibronectin) together with plasminogen activator inhibitor-1 (PAI-1; Fig. 4B and Supplementary Fig. S4B). It was noticed that expression of the EMT markers such as fibronectin and vimentin, and PAI-1 were increased to a far greater extent in Ad-EPSM–infected cells as compared with Ad-Smad3- and Ad-GFP–infected counterparts (Fig. 4B and Supplementary Fig. S4B). Immunofluorescence staining depicted features of the TGFβ-induced EMT as shown by a marked reduction in E-cadherin (Fig. 4C and Supplementary Fig. S4C) and an increase in vimentin (Fig. 4D) in Ad-GFP-, Ad-Smad3-, and Ad-EPSM–infected cells but not Ad-3SA–infected counterparts. The invasive activity through Matrigel membrane was the highest in Ad-EPSM–infected cells, whereas remaining totally unchanged in Ad-3SA–infected cells in the presence or absence of TGFβ (Fig. 4E and F and Supplementary Fig. S4D and S4E).
Figure 4.
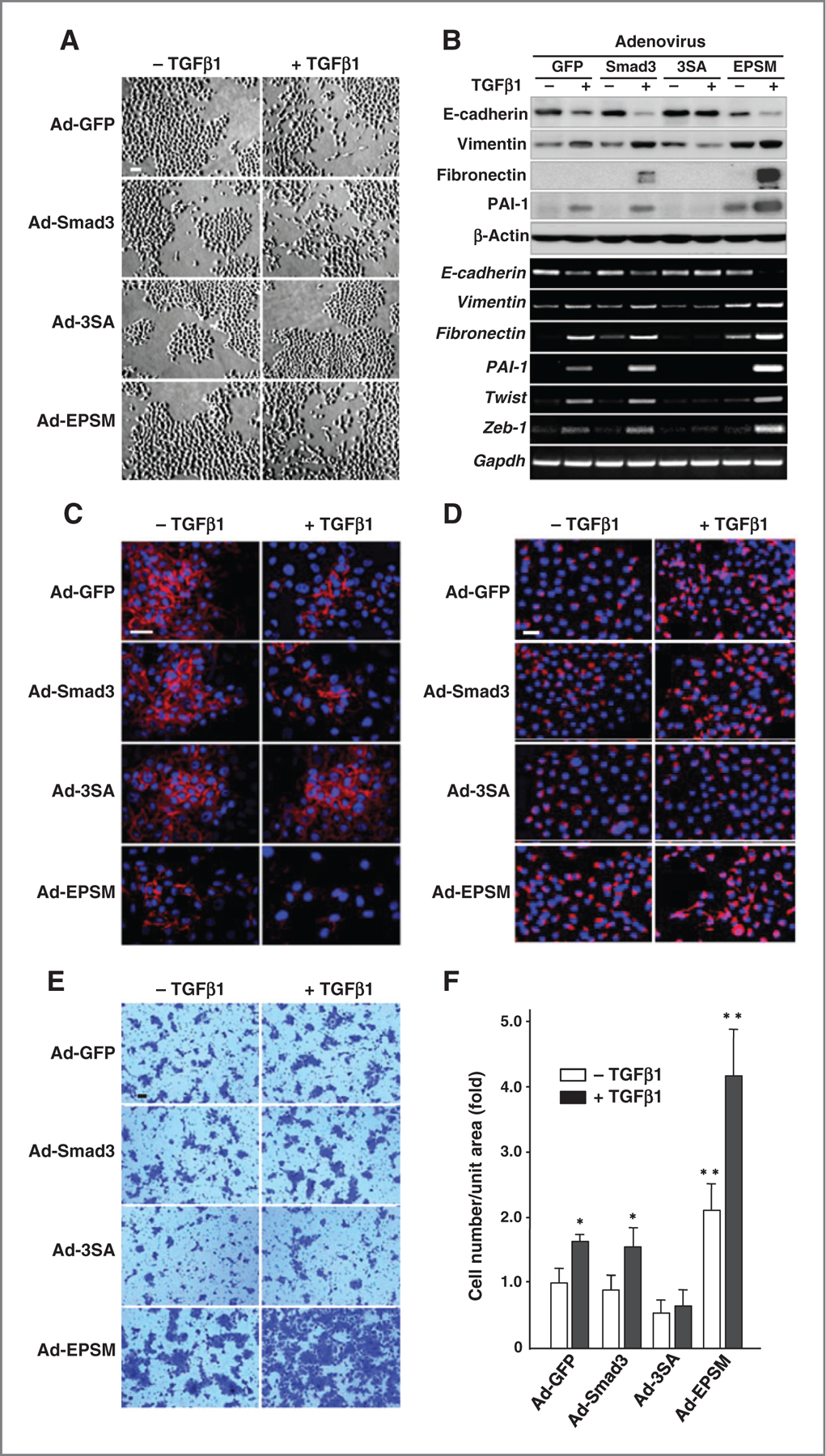
Mutation of Smad3 linker phosphorylation sites accelerates the TGFβ-induced EMT together with the invasive activity. A, phase-contrast micrographs of CA1a cells infected with Ad-GFP, Ad-Smad3, Ad-3SA, or Ad-EPSM with or without TGFβ1. Scale bar, 50 µm. B, immunoblot analyses (top) and RT-PCR (bottom) for EMT markers (E-cadherin, vimentin, and fibronectin), PAI-1, and E-cadherin suppressors (Twist and Zeb-1) in the adenovirus-infected CA1a cells with or without TGFβ1. C and D, immunofluorescence staining of E-cadherin (red; C) and vimentin (red; D) counterstained with DAPI (blue). Scale bar, 50 µm. E, invading cells through Matrigel membrane. Scale bar, 50 µm. F, invasive activities (fold) calculated from results. The mean ± SD from four experiments. *, 0.01 < P < 0.05; ** P < 0.01, compared with Ad-Smad3–infected cells without TGFβ1. Each of the experiments was conducted three to five times with similar results.
Mutation of Smad3 linker phosphorylation sites reduces putative cancer stem subpopulation along with stem cell marker expression and mammosphere formation
Recent work has suggested that TGFβ1 may regulate the cancer stem cell population (30). Ad-EPSM infection suppressed both protein and mRNA expressions of embryonic transcription factors that regulate self-renewal and pluripotency, including Oct4, Nanog, and Sox-2 (Fig. 5A). Prominent suppression of Oct4, Nanog, and Sox-2 was detected even in the absence of added TGFβ1 (Fig. 5A and Supplementary Fig. S5A). Expression of Oct4 as shown by immunofluorescence staining was suppressed as well even in the absence of TGFβ1 (Fig. 5B). Mammosphere formation together with soft agar colony formation was significantly reduced in Ad-EPSM–infected cells with and without added TGFβ1 (Fig. 5C and D and Supplementary Fig. S5B–S5E). The subpopulation of putative cancer stem cells with a cell surface marker of CD44high/CD24low was found to be the smallest in Ad-EPSM–infected CA1a cells, while no significant differences were detected among parental, Ad-Smad3- and Ad-3SA–infected counterparts (Fig. 5E and F). In the in vivo setting, analysis of dissociated 4T1 cells from primary tumors (Fig. 6A) showed that infection with Ad-EPSM but not the other constructs decreases the stem cell marker ALDH (Fig. 6B and C). Most importantly, infection with Ad-EPSM decreased the frequency of tumor-initiating cells as addressed using the gold-standard assay for cancer stem cells, the limiting dilution assay in vivo (Fig. 6D). Together, our data suggest strongly that the mutation of Smad3 linker phosphorylation sites reduces the cancer stem cell population.
Figure 5.
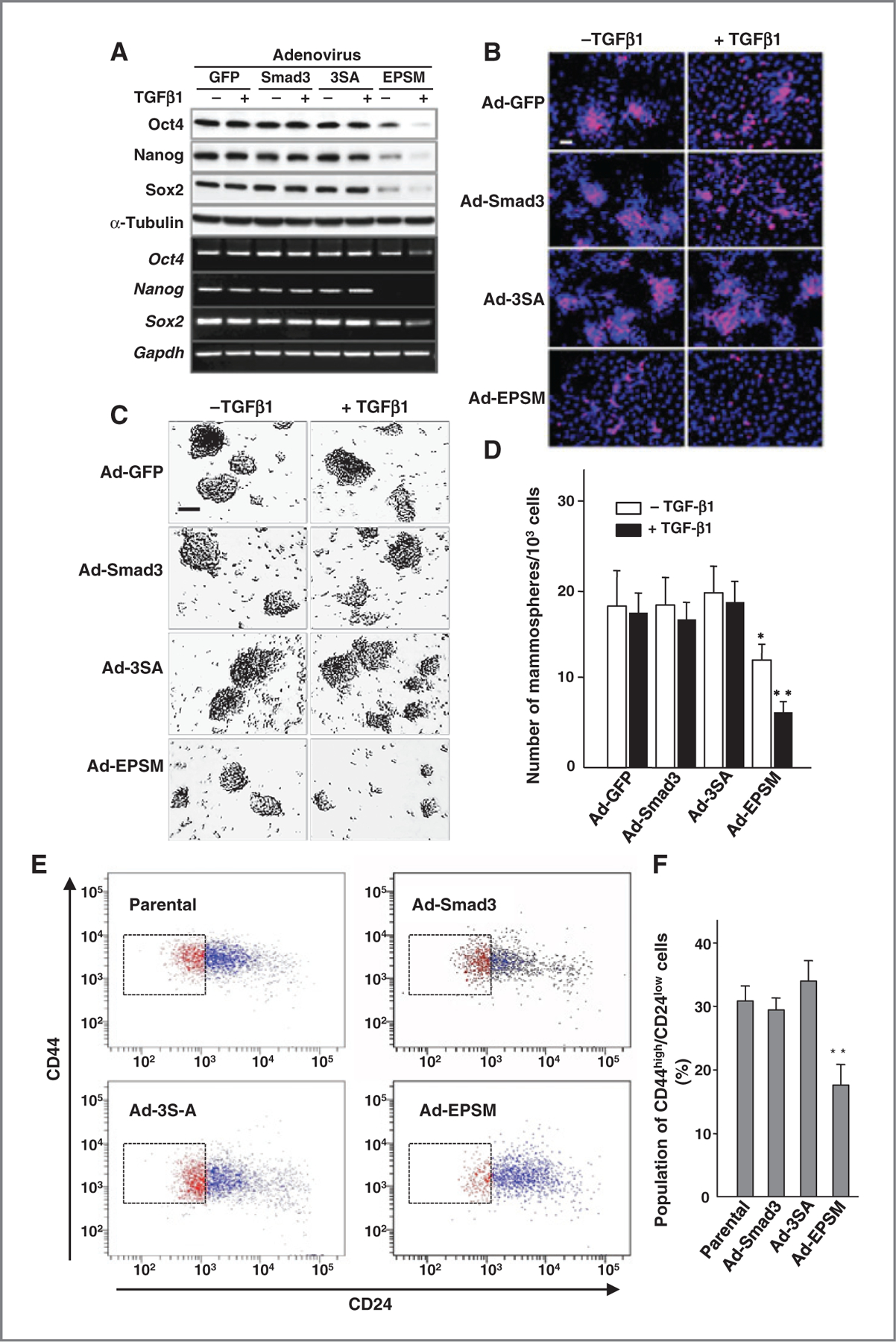
Mutation of the Smad3 linker phosphorylation sites reduces putative cancer stem cell subpopulation. A, immunoblot analysis (top) and RT-PCR (bottom) for Oct4, Nanog, and Sox-2 in the adenovirus-infected CA1a cells with or without TGFβ1. B, immunofluorescence staining of Oct4 (red) counterstained with DAPI (blue). Scale bar, 50 µm. C, representative photos of mammospheres formed from the adenovirus-infected CA1a cells treated with or without TGFβ1. Scale bar, 50 µm. D, the number of mammospheres per 1,000 seeded cells. The mean ± SD from four experiments. *, 0.01 < P < 0.05; **, P < 0.01, compared with Ad-Smad3–infected cells without TGFβ1. E, FACS analysis of cell-surface markers of CD44 and CD24 in CA1a cells infected with Ad-Smad3, Ad-3SA, or Ad-EPSM. Parental cells were used as controls. F, the putative stem cell subpopulation with CD44high/CD24low (%). Each bar represents the mean ± SD from four samples per group. **, P < 0.01, compared with Ad-Smad3–infected cells. Each of the experiments was conducted three to five times with similar results.
Figure 6.
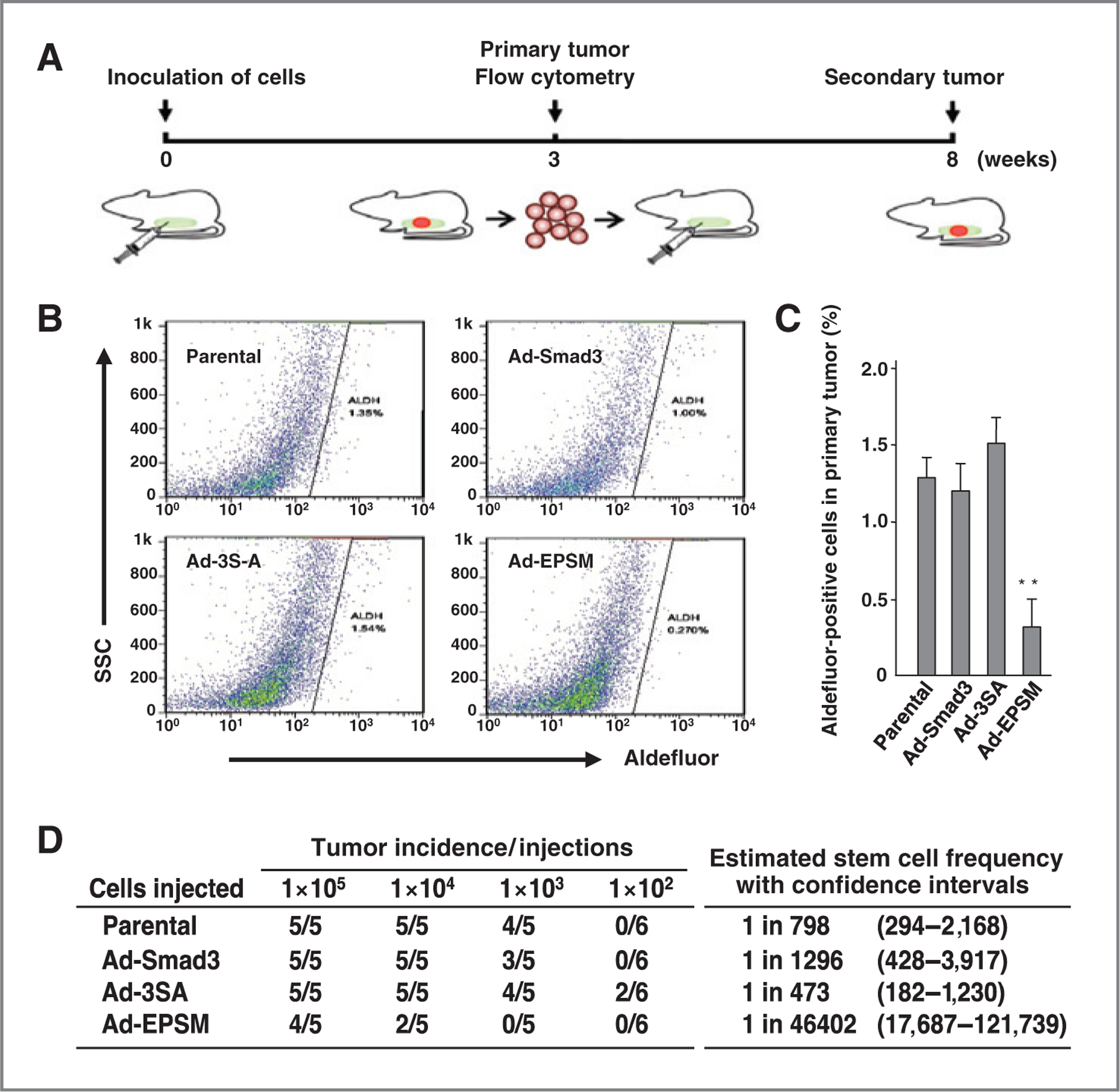
Mutation of the Smad3 linker phosphorylation sites in 4T1 cells reduces putative stem cell population and tumor initiating frequency of cells from the initial xenograft in limiting dilution. A, schematic representation of the initial and secondary xenograft. 4T1 cells (5 × 105) infected with Ad-Smad3, Ad-3SA, or Ad-EPSM were injected orthotopically into Balb/c mice (5 mice per group). Parental cells were used as controls. Three weeks after injection, single cells from the dissociated tumors were subjected to FACS analysis using the Aldefluor assay (B), and Aldefluor-positive cell population (%) was determined (C). Each bar represents the mean ± SD from four samples per group. **, P< 0.01, compared with Ad-Smad3–infected cells. D, tumor-initiating frequency of secondary tumors formed by injection of cells from the primary tumors. These experiments were conducted three times with similar results.
Discussion
We have demonstrated that mutation of the Smad3 linker phosphorylation sites intensifies all TGFβ-regulated biologic responses, regardless of whether they promote or suppress tumorigenesis. These responses included TGFβ-induced EMT, invasion, growth arrest, apoptosis, and reduction in the cancer stem cell population. Because they are fully reversed by mutation of the Smad3 C-tail phosphorylation sites, the Smad3 linker phosphorylation exerts a negative control over the TGFβ signaling pathway, and may thus contribute to the resistance to TGFβ (1, 5). Resistance of melanoma cells to TGFβ-induced growth arrest has also been attributed to their high levels of constitutive Smad3 linker phosphorylation, while neither such resistance nor phosphorylation was discernible in their normal counterparts, melanocytes (17).
Despite the prominent resistance to TGFβ conferred by the Smad3 linker phosphorylation, the cells transduced with Ad-GFP and Ad-Smad3 still retained appreciable responsiveness to TGFβ in terms of protein and mRNA expression levels for genes involved in EMT and invasiveness (E-cadherin, vimentin, fibronectin, and PAI-1; Twist and Zeb-1) and growth arrest (c-Myc and p21). These cells could still carry out the TGFβ-induced EMT program with acquisition of invasive and metastatic properties, and retained some tumor-suppressive functions when compared with Ad-3SA–infected cells. However, the Ad-GFP- and Ad-Smad3–infected cells showed no responsiveness to TGFβ in terms of expression levels of markers for apoptosis (Bcl-2, cleaved PARP, and caspase-3) and stemness (Oct4, Nanog, Sox-2, and mammosphere formation), and these cells were indeed totally insensitive to the TGFβ-induced apoptosis and reduction in stemness. Thus, endogenous levels of Smad3 linker phosphorylation in advanced metastatic cancer cell lines can selectively disrupt some TGFβ responses and not others.
Although repression of TGFβ signaling by Smad linker phosphorylation was attributed to inhibition of nuclear translocation of Smad2 and Smad3 (10), and proteasomal degradation of Smad3 through poly-ubiquitination (31, 32), mice with targeted-disruption of Smurf2 allele have now unveiled Smurf2 binding to the phosphorylated Smad3 linker regions in vivo and subsequent induction of multiple mono-ubiquitination at the MH2 domain, leading to a negative regulation of TGFβ signaling by inhibiting the formation of Smad3 complexes acting on target genes (33). This provides a molecular mechanism by which the EPSM has increased transcriptional activity. Our findings have also important implications for cancer therapy. A recent study shows that inhibition of CDK4/6 by a highly specific chemical inhibitor induces EMT and enhances invasiveness of pancreatic cancer cells, although it suppresses cell growth (34). This observation underscores the significance of our study.
A critical role of EMT in cancer metastasis has been well documented as a trans-differentiation program that is essential for cell migration and invasion (35). A previous report has shown that continual treatment with TGFβ generates cells with stem cell properties from immortalized mammary epithelial cells, suggesting a direct link between EMT and acquisition of stem cell properties (36, 37). Although EMT can generate stem cells (36–38), a variety of other factors also contribute to the population of stem cells, resulting the EPSM cells having the lowest population of stem cells in our study. In addition, EMT and stemness can be uncoupled, as recent reports have shown that abrogation of EMT or reverse EMT, mesenchymal–epithelial transition (MET), that endows cancer cells with the epithelial identity is crucial for reacquisition of stemness and colonization at distant organs after metastatic dissemination (39–41).
In conclusion, our findings suggest that neoplastic progression of the breast cancer cell lines is regulated by a balance between the Smad3 linker and C-tail phosphorylation, and each corresponding mutation has its own adverse effects, urging the need to develop combinatorial therapies with other anticancer reagents. Our report deepens the current understanding on cancer progression, and provides a therapeutic rationale for targeting Smad3 phosphorylation either at C-tail or linker phosphorylation depending on the stage of tumorigenesis and metastasis.
Supplementary Material
Acknowledgments
The authors thank the late A.B. Roberts, S.G. Rane, H.M. Lin, E.A. Kohn, and A. Kamaraju for adenoviral Smad3 mutants, and S. H. Yuspa for helpful discussion.
Grant Support
This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (NRF-2012M3A9C4048735).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflict of Interest:
No potential conflicts of interest were disclosed.
References
- 1.Roberts AB, Wakefield LM. The two faces of transforming growth factor-β in carcinogenesis. PNAS 2003;100:8621–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Millet C, Zhang YE. Roles of Smad3 in TGF-β signaling during carcinogenesis. Crit Rev Eukaryot Gene Expr 2007;17:281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Massagué J TGFβ in cancer. Cell 2008;134:215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts AB, Tian F, Byfield SD, Stuelten C, Ooshima A, Saika S, et al. Smad3 is key to TGF-β-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev 2006;17:19–27. [DOI] [PubMed] [Google Scholar]
- 5.Wakefield LM, Roberts AB. TGF-β signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev 2002;12:22–9. [DOI] [PubMed] [Google Scholar]
- 6.Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol 2005;21:659–93. [DOI] [PubMed] [Google Scholar]
- 7.Moustakas A, Heldin CH. The regulation of TGFβ signal transduction. Development 2009;136:3699–714. [DOI] [PubMed] [Google Scholar]
- 8.Massagué J TGFβ signalling in context. Nat Rev Mol Cell Biol 2012;13:616–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang YC, Piek E, Zavadil J, Liang D, Xie D, Heyer J, et al. Hierarchical model of gene regulation by transforming growth factor-β. PNAS 2003;100:10269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kretzschmar M, Doody J, Timokhina I, Massagué J. A mechanism of repression of TGFβ/Smad signaling by oncogenic Ras. Genes Dev 1999;13:804–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004;430:226–31. [DOI] [PubMed] [Google Scholar]
- 12.Matsuura I, Wang G, He D, Liu F. Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry 2005;44:12546–53. [DOI] [PubMed] [Google Scholar]
- 13.Wang G, Matsuura I, He D, Liu F. Transforming growth factor-β-inducible phosphorylation of Smad3. J Biol Chem 2009;284:9663–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Millet C, Yamashita M, Heller M, Yu L-R, Veenstra TD, Zhang YE. A negative feedback control of transforming growth factor-β signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J Biol Chem 2009;284:19808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuzaki K, Kitano C, Murata M, Sekimoto G, Yoshida K, Uemura Y, et al. Smad2 and Smad3 phosphorylated at both linker and COOH terminal regions transmit malignant TGF-β signal in later stages of human colorectal cancer. Cancer Res 2009;69:5321–30. [DOI] [PubMed] [Google Scholar]
- 16.Cooley A, Zelivianski S, Jeruss JS. Impact of cyclin E overexpression on Smad3 activity in breast cancer cell lines. Cell Cycle 2010;9:4900–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen-Solal KA, Merrigan KT, Chan JL-K, Goydos JS, Chen W, Foran D, et al. Constitutive Smad linker phosphorylation in melanoma: a mechanism of resistance to transforming growth factor-β-mediated growth inhibition. Pigment Cell Melanoma Res 2011;24:512–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuzaki K Smad phosphor-isoforms direct context dependent TGF-β signaling. Cytokine Growth Factor Rev 2013;24:385–99. [DOI] [PubMed] [Google Scholar]
- 19.Gao S, Alarćon C, Sapkota G, Rahman S, Chen P-Y, Goerner N, et al. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-β signaling. Mol Cell 2009;36:457–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wrighton KH, Lin X, Feng XH. Phospho-control of TGF-β superfamily signaling. Cell Res 2009;19:8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bae E, Kim S-J, Hong S, Liu F, Ooshima A. Smad3 linker phosphorylation attenuates Smad3 transcriptional activity and TGF-β1/Smad3-induced epithelial–mesenchymal transition in renal epithelial cells. Biochem Biophys Res Commun 2012;427:593–99. [DOI] [PubMed] [Google Scholar]
- 22.Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, et al. TGF-β switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest 2003;112:1116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heppner GH, Miller FR, Shekhar PM. Nontransgenic models of breast cancer. Breast Cancer Res 2000;2:331–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tao K, Fang M, Alroy J, Sahagian GG. Imagable 4T1 model for the study of late stage breast cancer. BMC Cancer 2008;8:228 doi: 10.1186/1471-2407-8-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin HM, Lee JH, Yadav H, Kamaraju AK, Liu E, Zhigang D, et al. Transforming growth factor-β/Smad3 signaling regulates insulin gene transcription and pancreatic islet b-cell function. J Biol Chem 2009; 284:12246–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev 2003;17:1253–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. PNAS 2003;100:3983–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ginestier C, Hur MH, Charaffe-Jauffre E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007;1:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Y, Smyth GK. ELDA: Extreme limiting dilution analysis for comparing deleted and enriched populations in stem cell and other assays. J Immunol Methods 2009;347:70–8. [DOI] [PubMed] [Google Scholar]
- 30.Caja L, Kahata K, Moustakas A. Context-dependent action of transforming growth factor β family members on normal and cancer stem cells. Curr Pharm Des 2012;18:4072–86. [DOI] [PubMed] [Google Scholar]
- 31.Alarćon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-β pathways. Cell 2009;139:757–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakano A, Koinuma D, Miyazawa K, Uchida T, Saitoh M, Kawabata M, et al. Pin1 down-regulates transforming growth factor-β (TGF-β) signaling by inducing degradation of Smad proteins. J Biol Chem 2009;284:6109–15. [DOI] [PubMed] [Google Scholar]
- 33.Tang LY, Yamashita M, Coussens NP, Tang Y, Wang X, Li C, et al. Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. EMBO J 2011;30:4777–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu F, Korc M. Cdk4/6 inhibition induces epithelial-mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther 2012;11:2138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006;7:131–42. [DOI] [PubMed] [Google Scholar]
- 36.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pirozzi G, Tirino V, Camerlingo R, Franco R, La Rocca A, Liguori E, et al. Epithelial to mesenchymal transition by TGFβ−1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS ONE 2011;6:e21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morel AP, Liévre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008;3:e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ocaña OH, Córcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012; 22:709–24. [DOI] [PubMed] [Google Scholar]
- 40.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012;22:725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139:871–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.