

Abstract
Aging and circadian rhythms are two biological processes that affect an organism, although at different time scales. Nevertheless, due to the overlap of their actions, it was speculated that both interfere or interact with each other. However, to address this question, a much deeper insight into these processes is necessary, especially at the cellular level. New methods such as single-cell RNA-sequencing (scRNA-Seq) have the potential to close this gap in our knowledge. In this review, we analyze applications of scRNA-Seq from the aging and circadian rhythm fields and highlight new findings emerging from the analysis of single cells, especially in humans or rodents. Furthermore, we judge the potential of scRNA-Seq to identify common traits of both processes. Overall, this method offers several advantages over more traditional methods analyzing gene expression and will become an important tool to unravel the link between these biological processes.
Keywords: hallmarks of aging, light resetting, melatonin, mutation rate, transcriptome
Introduction
Gene expression regulates dynamic processes, over diverse time scales. Among these processes, all occur at the cellular level and include cell division, differentiation, development, migration and many more. We are especially interested here in those processes which are also characteristic of the whole organism. Two of the most physiologically relevant ones are aging and circadian rhythms [1–3]. Both of these processes affect most of the cells, the tissues which they compose and the whole organism and have large health impacts in humans. To understand them, we need to go beyond the organism or even tissue level and study dynamics at the cell level in relation to organismal physiology. To achieve such insights at the cellular level, ideally, we should characterize phenomena within single cells. This is possible, thanks to single-cell RNA-sequencing (scRNA-Seq), which identifies the transcriptome of individual cells [4, 5]. By collecting large amounts of data from cells of different samples, it is possible to gain insight into dynamic processes such as aging. An outline of the technique is depicted (Figure 1) [6].
Figure 1.
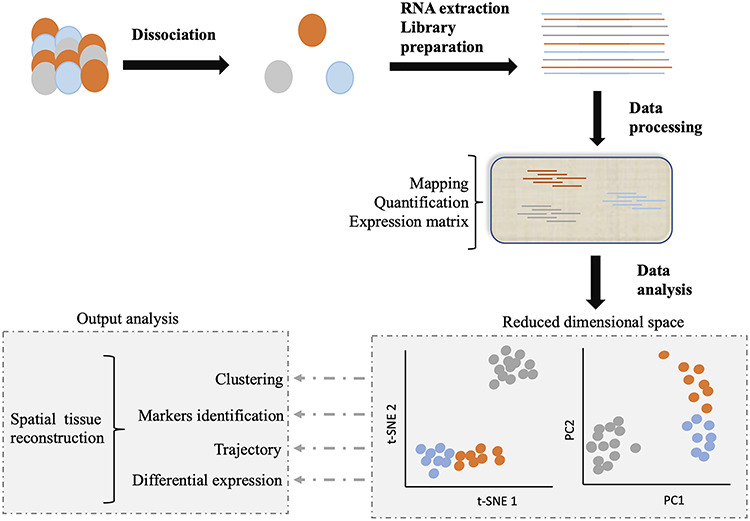
Schematic representation of scRNA-Seq. After dissociation of the sample of interest (e.g. a tissue), the mRNA content of each individually selected cell is transformed into a sequencing library. The reads of each single cell are identified by a barcode, introduced during the library construction process. Libraries are then pooled and sequenced together. For processing, the data are demultiplexed, mapped and quantified, and a count matrix is generated. Analysis of this matrix can allow to cluster per cell type, assign markers to these cell types, track cells through biological processes or detect differences in gene expression. When experimental design allows, the spatial organization of the starting tissue can be reconstructed. Image adapted from Lafzi et al. [6].
Aging is a difficult phenomenon to study because starting from primary effects, which generally follow a sequence of events, secondary effects appear more stochastic. These effects can be masked by age-related diseases and other effects, and hence, individual cells or even tissues may age at different paces [7, 8]. With time, certain metabolic or physiological functions of a body decline [9]. This functional decline renders an organism vulnerable to many adverse effects, and, consequently, aging is a process that counteracts survival. In an effort to categorize the many effects of aging observed in mammals, López-Otín et al. [10] defined nine hallmarks. The primary hallmarks comprise those causing problems to cope with different kinds of damage. These four hallmarks include an excess of mutations causing genomic instability, a shortening of the telomeres, changes of epigenetic control and problems in clearing defective proteins. Any of these hallmarks can cause substantial damage to cells. The next steps are the antagonistic hallmarks, which include processes directly affected by the damage. These hallmarks comprise three processes, i.e. problems with nutrient signaling and the involved signaling cascades, functionally impaired mitochondria and cellular senescence. All of these processes still target individual cells with local effects. Finally, the integrative hallmarks can affect the entire organ. While defective cells are replenished via stem cells, with age these stem cells become sparse. In addition, communication between different cell types gets more difficult leading to, for example, excess inflammatory responses. scRNA-Seq provides an opportunity to learn more about these hallmarks of aging, because many of them are based on transcriptional changes and—at least for the first seven hallmarks—they occur in individual cells.
On a very different time scale, circadian rhythms occur with a stable periodicity of about 1 day under constant conditions [11]. These rhythms are highly synchronized among the different tissues and drive daily changes in metabolism, physiology and behavior to bolster the performance of an organism. At their base, cell-autonomous clocks drive most of these rhythms based on transcriptional and post-translational feedback loops [12]. Such single-cell oscillations can be monitored, for example, in fibroblast cells [13]. Monitoring the circadian oscillation of a fluorescent reporter protein in individual fibroblast cells has revealed that under normal culture conditions, all cells had rhythmic activity, albeit desynchronized. The synchronization of these cells was achieved by a serum shock, which appeared to reset all the circadian oscillators to the same state, following which synchronized rhythms emerged in the culture [13]. Hence, to function properly on the organism level, a synchronizer is necessary for the synchronization of all cell-autonomous clocks.
Similar to aging processes, the transcriptional base of circadian phenomena implies that we expect scRNA-Seq to provide new insights into the circadian timing system of individual cells and their interactions. Interestingly, aging and the circadian clock affect similar pathways albeit at completely different time scales, and there might be connections between the two processes [14, 15]. For instance, in the master clock of mammals, the suprachiasmatic nuclei (SCN), the circadian accumulation of NAD+ declines with age, which causes disturbed clock outputs [16]. Stabilizing the SCN by neuron-specific expression of sirtuin-1 (Sirt1) rejuvenated the circadian clock and the organism [16]. Similarly, transplantation of young SCN tissue into aged hamsters not only improved circadian output but also increased their life span [17]. As another example, the circadian clock impacts the transcriptional organization of the metabolism [18]. This temporal organization may constitute the base for the efficiency of time-restricted feeding to revert metabolic diseases and as such potentially delay the onset of aging (see the recent meta-analysis performed by Moon et al. [19]). Hence, a better perception of the effects of the circadian clock on aging could open new avenues for therapeutic intervention [20].
In this review we focus on aging and circadian rhythms through the study of single-cell transcriptomes. New insights into transcriptional processes and their regulation have been obtained for difficult biological examples, which involve collaboration of different cell types within a tissue. Hence, scRNA-Seq offers many advantages over previously existing methods and may be the key tool to understand the interconnection of aging and circadian processes.
scRNA-Seq and aging
DNA microarrays were applied rapidly in the aging field to search for changes in gene expression. As example, changes in gene expression of actively dividing fibroblasts of different age could be associated with errors during mitosis, which may cause changes in gene expression [21]. Even earlier, a signature of aging was identified in mouse skeletal muscle, consisting of an increased stress response and reduced metabolism [22]. Applying caloric restriction to old mice brought these gene expression changes back to normal, delaying the onset of aging. Comparable conclusions were achieved after the field switched to RNA-Seq. However, both microarray and bulk RNA-Seq only provide an average view of heterogeneous cell populations, whereas many theories about aging state that aging starts in individual cells and then progresses all over the tissue.
To address this issue, scRNA-Seq appears to be the method of choice. Few studies have dissociated cells and then analyzed them to investigate aging in mammals, despite the power of this approach. Because aging is a multifactorial disease, we focus here on studies that identified the impact of aging on different cell types. For example, the pancreas has important exocrine and endocrine functions, which are mediated by different cell types. With age, the diminishing activity of the pancreas leads to severe diseases such as type II diabetes. Enge et al. [23] used scRNA-Seq to detect age-related changes in human pancreas (Figure 2).
Figure 2.
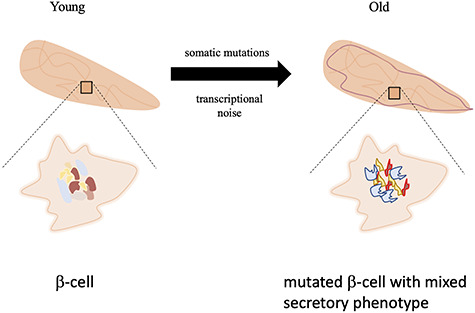
Effect of the aging process on the pancreas. The effects involve an increase of transcriptional noise and of mutation rate in the β-cells (symbolized as a scrambled genome in old cells). These effects of aging generate cells with mixed insulin and glucagon secretory phenotypes, correlating with the decline of endocrine functions of the gland.
Surprisingly, there were only few increases of typical age-related marker genes observed with progressing age, such as for Cdnk2A/p16INK4a. It has been suggested that this inhibitory regulator of the cell cycle accumulates with and consequently represents a marker for cellular senescence [24]. The increase was due to an increase of the number of cells expressing this gene and not of the expression within each cell, which would not be possible to distinguish using bulk RNA analysis. The data suggest that some cells morph faster into old cells than others in the same tissue, and this is an in vivo example for the hallmark cellular senescence [10].
However, the study also suggests that it is not easy to compare senescence data between in vitro and in vivo experiments. While in vitro the cultured cells are more or less synchronized, in vivo only a subset of cells per tissue shows the aversive signs of senescence.
With age the overall accumulation of mRNA transcripts became noisy in pancreatic α- and β-cells [23], supporting the concept that transcriptional noise increases with age [10]. Due to this increase in noise, the distinction was no longer clear between, for example, insulin- and glucagon-secreting cells. Indeed, in the pancreas of middle-aged donors, there was an increase of mixed secretors, which might partially explain the declining function of this tissue. The cause for the scattering of transcripts between individual cells is unknown but may be provoked by cellular stress and its associated pathways [23].
An increase of mutations was also observed, which is another hallmark of aging (Figure 2) [10]. Using spike-in RNA as an internal control, the authors were able to control for experimental artifacts. The subsequent comparison between the pancreas and as control brain revealed that in the pancreas, there was a specific set of mutations seemingly caused by the presence of 8-oxo-2′-deoxyguanosine incorporated into DNA of the pancreatic β-cells. This nucleotide is created upon oxidative stress and can pair erroneously with adenine or guanine. This kind of mutation is repaired by the transcription-coupled nucleotide excision pathway. Consequently, mutations caused by this nucleotide are enriched on the transcribed strand in the genome, which was found to be the case in the β-cells but not in brain cells used as control. Hence, scRNA-Seq can be used to reliably measure the mutation rate of individual cell types.
An open question remains how the increase of transcriptional noise and of mutation rate interact [23]. First, the occurrence of mutations per individual cell seems much too rare to explain a general increase in transcriptional noise. Secondly, the correlation between the mutational load and the noise in transcript accumulation with advancing age was similar. Since there was no increased correlation between the mutational load and the transcriptional noise, the authors favored the hypothesis that both processes occurred independently from each other [23]. Overall, scRNA-Seq enabled to detect an increase in variation of mRNA accumulation and in mutation rate specifically in the β-cells. Both processes together may affect changes in pancreas function with age.
As another example, the lung is strongly affected by aging, mainly because it directly interacts with the harmful outer world. In an attempt to identify age-related changes in the lung, Angelidis et al. [25] prepared suspensions of lung cells from young and aged mice (Figure 3A). The overall cell type composition of the lungs did not change. However, similar to the findings in the pancreas, in the lungs of 24-month-old mice, there was an increase of transcriptional noise in about half of the cell types. This observation provides more support for the predicted transcriptional instability with aging and also shows that this increase of noise does not occur in all cell types [10]. It would be interesting to follow up on this study to elaborate why some cells are more susceptible to transcriptional noise than others.
Figure 3.
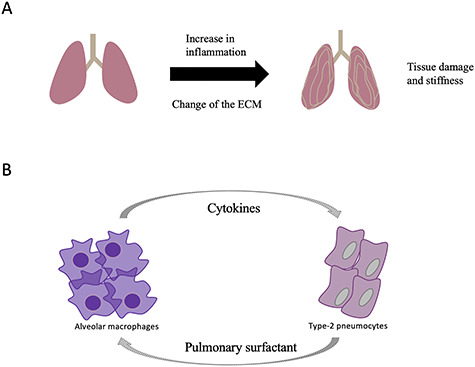
Effect of the aging process on the lung. (A) Aging causes an increase of inflammation with a subsequent change of the composition of the ECM and pulmonary surfactant. (B) Communication between the alveolar macrophages and the type-2-pneumocytes creates a vicious circle.
To validate the scRNA-Seq data, these data were compared with RNA-Seq and shotgun proteomics data from the entire lung. There was an excellent correlation of the results from both kinds of RNA-sequencing approaches, indicating that most of the cell types contributing to the tissue were found by scRNA-Seq. Combining those different methods, the authors could identify a change of the composition of the extracellular matrix (ECM) with age. This could be related to the fact that in old lungs, the elasticity and tensile strength is often reduced, which impairs proper function of the organ [26].
The main advantage, however, of the scRNA-Seq experiment was the identification of cell-specific aging events that otherwise would be masked by the other cell types. For instance, the alveolar macrophages of old mice showed a signature of immune and inflammatory responses. The type 2 pneumocytes, on the other hand, showed signatures related to interferon-γ signaling and changes in fatty acids metabolism, in particular predicting a higher production of cholesterol and triglycerides. These changes in lipids may affect the composition of the pulmonary surfactant which is secreted by these cells. It has been shown that the composition of the pulmonary surfactant changes with age [26], and it is tempting to speculate that this change could provoke local inflammation and the subsequent changes of the immune cells and cytokine signaling as found for old mice (Figure 3B). This chain of events is an example of the changing intercellular communications predicted with age [10]. Overall, this study found age-related changes within the different cell types of the lung, using a combination of scRNA-Seq and other methods.
Both examples highlight the potential of scRNA-Seq to assign changes to individual cell types and consequently to better understand the development and progression of age-related changes. These studies found that age-related changes emerge with different paces and are quite specific to each cell type. These observations are being strengthened by further examples of the application of scRNA-Seq to aging in different tissues [27–29]. Hepatocytes, which are exposed to oxidative stress like pancreatic cells, had an elevated mutation rate compared to liver stem cells [27]. This mutational load could be involved in a variety of liver diseases. Aging in fibroblasts is linked to reduced expression of genes constituting the ECM and increased expression of adipogenic-related genes [29]. Caloric restriction, one of the few means to delay aging in mammals, reverted these expression changes, demonstrating that some age-related changes are reversible at the cell level. The situation in the brain was quite complicated, because all cell types showed different aging patterns, in some cases in opposite directions [30]. For example, GABAergic and glutaminergic neurons upregulated a subset of ribosomal proteins, while dopaminergic neurons downregulated them. These differences in the expression of ribosomal genes hint towards differences in the overall protein expression. Comparing the aging gene expression signatures of the lung, kidney and spleen, Kimmel et al. [28] concluded that there were not only common age-related changes affecting protein localization and inflammatory processes but also cell type-specific changes, which were even more prominent and mixed among organs. For example, endothelial cells showed increased thyroid and type I interferon signaling compared to lymphocytes, which had changes mainly in NF-κB signaling.
scRNA-Seq and the circadian timing system
Due to the similar makeup of the circadian oscillator in all tissues, it was originally thought that all cells and tissues show the same rhythms on the gene expression level [11]. Yet DNA microarray experiments already revealed tissue-specific rhythms [31, 32]. These experiments also allowed to measure the time of peak and trough levels for genes from a particular pathway. Again, the same was found with RNA-Seq experiments. However, there were already indications that not all cells of a tissue showed circadian rhythms [33]. Research in mammals focused on a specialized center in the brain that fulfills the function of synchronizing circadian rhythms in all tissues [12]. The bipartite SCN is located above the optical chiasm to perceive light information. Specialized photosensitive cells in the retina capture the state of the external light/dark phase and transmit this information to the SCN. If the phase of the SCN is not in harmony with the external light/dark phase, then a phase shift is necessary [34]. Otherwise the circadian timing system would steadily uncouple from the environment and lose its capacity, for example, to anticipate daily recurring events. Shifting the phase by the SCN involves transcriptional changes in several of its subregions. Although we have a broad view of the changes and interactions occurring in the SCN in response to a light pulse, the cell-to-cell effects are still unknown.
Using single-cell transcriptional analysis, Park and his colleagues investigated the response of the SCN to light in the early night (Figure 4) [35]. In the SCN kept in the dark, Park et al. could identify four main clusters of neurons, three of which were already known to exist in the SCN, arginine vasopressin (AVP) and two types of vasoactive intestinal polypeptide (VIP) expressing neurons. However, a new type of neurons was identified, which had the potential to secrete the neurotransmitter pituitary adenylate cyclase-activating peptide (PACAP), based on the expression data. These neurons were interesting, because until then it was believed that PACAP—like glutamate—came solely from the optical nerves’ synapses to the SCN.
Figure 4.
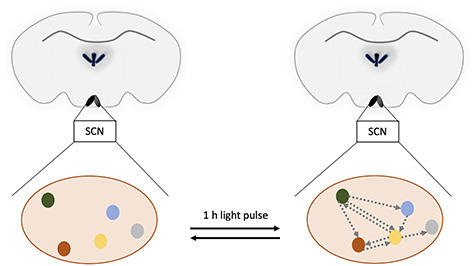
Increased communication between the subregions of the SCN after a light pulse. The different colored dots represent different subregions of the SCN expressing different neurotransmitters and receptors (red, VIP neurons; yellow, AVP neurons; green, PACAP and prokineticin 2 neurons; blue, PACAP and probably gastrin-releasing peptide neurons; gray, less defined, responsive cluster). Gray arrows symbolize the flow of neurotransmitters between the different cell clusters. Note that the figure displays only a snapshot of the situation 1 h after the light pulse and the process is probably highly dynamic.
Upon a 1-hour light pulse, the expression profiles in the SCN neurons changed (Figure 4). Thus, the authors got insight into the changes of interactions of the SCN neuronal network after a light pulse in the early dark phase. Considering autocrine and paracrine signaling pathways and the localization of these cell types within the SCN, the authors considered that a neurotransmitter network between the four neuron types became operative during the light pulse (Figure 4). Such a neurotransmitter network could on the one hand gate the incoming signal from the retina to the receiver cells in the SCN and, on the other hand, amplify it to affect other regions. This concept is interesting and invites further research on this topic.
Taken together, the single-cell analysis of the SCN neurons with and without a light pulse revealed highly organized spatial and temporal transcriptional responses. Recently, the organization of the SCN was reinvestigated by another group using scRNA-Seq [36]. They used more time points for their analysis and could identify more cell types including oligodendrocytes, astrocytes and microglia cells. Hence, the authors created a resource of cell types and their gene signatures over the circadian cycle. However, they could not reproduce all of the observations of Park et al. [35] paper, because the sequencing depth was not sufficient to detect, for example, the PACAP-expressing neurons.
One of the brain regions indirectly affected by the SCN via the sympathetic nervous system is the pineal gland (Figure 5) [37]. In this gland the information about the distribution of the light and dark phase is converted into chemical signals. To achieve this, the secretion of melatonin into the blood is restricted to the dark phase [37]. This hormonal signal affects physiological processes such as sleep and photoperiodic adaptations [38, 39]. To gain a deeper knowledge of the composition of the pineal gland, Mays et al. [40] performed scRNA-Seq on the pineal glands of rats, either in the light or dark phase (Figure 5). Among others, two distinct types of pinealocytes were identified. Although only representing 5% of the total pinealocytes, the α-pinealocytes had a metabolism shifted towards adenosine triphosphate synthesis in favor of S-adenosyl methionine production (SAM). Also, they had an increase of acetylserotonin-O-methyltransferase (ASMT), which is important for the last step of melatonin synthesis. Hence, the enzyme necessary for melatonin synthesis, ASMT, and the necessary cofactor, SAM, were enriched in these cells. The α-pinealocytes were proposed to be specialized in the output of melatonin from the pineal gland (Figure 5). As such, they would take up N-acetylserotonin, which is released by the β-pinealocytes. However, further studies are necessary to judge the contribution of these cells to overall melatonin levels.
Figure 5.
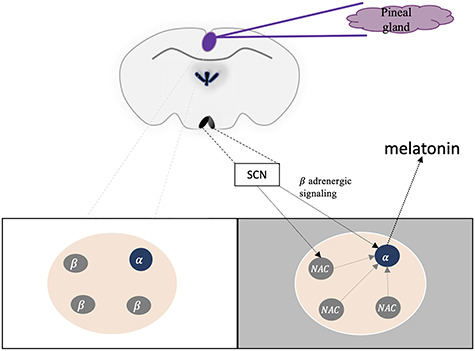
Activation of the pineal gland by the SCN. The pineal gland secretes melatonin during the night phase (gray box). Due to signals from the SCN, N-acetylserotonin (NAc) is transported from β-pinealocytes (gray circles) to α-pinealocytes (blue circle) to enhance melatonin production and secretion. Note that by contrast to the situation in Figures 2 and 3, circadian phenomena can shift freely from one condition to the other dependent on the light phase.
In the same study, diurnal changes were investigated by comparing two different time points. Most diurnal changes could be attributed to the pinealocytes, for example, changes in the expression of another enzyme involved in melatonin production, the aralkylamine N-acetyltransferase (AANAT). To understand the diurnal phenotype of the pinealocytes, they treated the rats during the light phase with isoproterenol to mimic the stimulation of the gland by adrenergic stimulation. Almost all effects of this treatment (99% of differential expression) were observed in the pinealocytes and resembled the dark phase expression signature, suggesting that the adrenergic system was mediating the effects on the pinealocytes. Hence, scRNA-Seq yielded new insights in the composition and functional organization of this gland.
Overall, the examples demonstrate that after proper design of the experiment, single-cell methods can be applied to address circadian questions. The main advantage is the identification of cell types that otherwise are masked by the bulk of the tissue. The identification of those subsets of cells allows for the construction of regulatory networks to understand the coupling of the individual cells, which is necessary for the stability of the system, for example, during aging.
Is it possible to link aging and circadian rhythms?
There exist fascinating hints that these two key dynamics at different time scales, aging and circadian processes, might interact [14, 15]. For example, with increasing age circadian oscillators in some cell types could become sloppier. This would cause further age-related changes in these affected cells and, in a kind of vicious circle, provoke more changes of the circadian oscillator. Finally, the overall synchronization of the circadian timing system—including the control of the metabolism—would decline. This sequence of events may explain the decline of the circadian system of aged mice due to a failure of the SCN [16]. Moreover, interrupting this vicious circle by rejuvenating the SCN is possible [17]. Could scRNA-Seq be a method to identify the culprits initiating this decline?
It is important to note that scRNA-Seq only provides a snapshot of gene expression of a particular cell. Unfortunately, it is not possible yet to follow the global mRNA expression profile of a cell from its birth to its death nor even a single circadian cycle. Hence, at the moment, we rely on the collection of many cells from a given tissue. The data from these cells are then clustered into distinct groups, and their differential gene expression patterns are compared (Figure 1) [6]. In essence, this represents another kind of bulk analysis. Maybe it would be possible in the future to develop algorithms capable of focusing on the aversive changes in individual cells, for example, in conjunction with the measurement of the mutation rate. An elegant means of analysis is the so-called trajectory analysis, which renders it possible to follow changes within the same cell type over multiple time points [41, 42]. Such an approach is typified here by the analyses of the transgression of fibroblasts to adipocyte-like cells with age or the circadian profile measured in the SCN [29, 36]. Such analyses would profit from the analysis of a larger number of intermediary time points. Although multiple methods exist to characterize circadian accumulation profiles in cells and tissues [43, 44], more specialized versions to analyze scRNA-Seq experiments are emerging [45]. These technological developments will certainly provide a deeper insight into the functioning of the circadian clock and potential interferences such as aging [14, 15].
As seen from our examples, scRNA-Seq has the potential to describe dynamic processes. However, this may also be a drawback of the method. The cell cycle affects the result of scRNA-Seq experiments [46], which led to the characterization of cell cycle-specific signatures in unsynchronized cells [46]. Such gene signatures can be used to clean the scRNA-Seq data and are generally filtered out prior to any further analysis [47]. As mentioned earlier, circadian rhythms occur in a cell-autonomous manner [13]. Hence, similar gene signatures may have to be introduced to correct for solely circadian phenomena. It is indeed possible to deduce the phase of a circadian oscillator from the expression of marker genes [48]. However, there is one caveat with this. Although there exists a common set of genes manifesting a core oscillator in circadian cells, the expression of circadian-regulated genes is rather tissue specific [49]; we have presented evidence that it is probably even cell type specific. Hence, for each cell type of a tissue, such a circadian signature should be established, which would be extremely laborious. A better solution would be to collect all samples at the same time point under precise experimental conditions, which is rather difficult for human samples. For example, for Enge et al. [23] paper, the samples were available probably scattered all over the day from male and female donors. Hence, much of the transcriptional change and the increase in transcriptional noise could be due to different phases of the circadian oscillator or sex-specific differences. These circumstances might also explain why only one marker of aging was detected, the increase of Cdnk2A/p16INK4a. In conclusion, the precision and consequent sensitivity of age-related research could be readily improved by considering circadian influences systematically in experimental design.
Another important application of scRNA-Seq is to understand the communication of cells within a tissue, which are important both in aging and in circadian rhythm. As seen for the SCN or the pineal gland, relevant interactions between different cell types are revealed only at the single-cell level [35, 40]. Hence, to really understand complicated regulatory systems, it is not only important to know all of the key regulatory molecules and their sites of action but also in which of the cell types of a tissue they are relevant. Again, it would be interesting to apply scRNA-Seq on the SCN of old animals. The results of Kimmel et al. [28] suggest interesting changes of gene expression in different neuronal cell types. How would such changes affect the functioning of their circadian oscillator?
The same holds true for the pineal gland. Interestingly, the synthesis of melatonin declines with age, and consequently, it is worthwhile to investigate the underlying mechanism in aged animals. Is the number of the α-pinealocytes declining, or is the transcription disturbed in these cells due to a similar reason as for the β-cells of the pancreas? It would be very interesting to distinguish these possibilities with further scRNA-Seq studies.
Overall, scRNA-Seq is a powerful method to obtain insight of regulatory processes on the single-cell level. The insights which it provides broaden our understanding of the communication and interaction of the individual cells of a tissue and provide a deeper understanding of dynamic processes which affect the whole organism.
Key Points
scRNA-Seq is useful to analyze the dynamic behavior of individual cell types.
In aging pancreatic β-cells, an increase of the mutation rate occurs due to oxidative stress.
In aging lungs, the modifying pulmonary surfactant may provoke local inflammation, which feeds back to the surfactant-producing cells.
A neurotransmitter network becomes activated in response to a light pulse in the SCN.
A subset of pinealocytes was identified, which enhances the synthesis of melatonin.
Acknowledgements
This work was supported by the Schweizer National Founds/Founds National Suisse (grant numbers 31003A_152792 to J.A.R., 31003A_173048 to M.R.-R.) and the Cantons of Fribourg and of Vaud.
Sara S. Fonseca Costa received PhD in biochemistry in the University of Fribourg, Switzerland, and is currently doing a PostDoc in the Robinson-Rechavi group at the Department of Ecology and Evolution in the University of Lausanne, Switzerland.
Marc Robinson-Rechavi is assistant then associate professor of bioinformatics at the Department of Ecology and Evolution in the University of Lausanne since 2005, and Group Leader at the Swiss Institute of Bioinformatics since 2006. He leads the group evolutionary bioinformatics.
Jürgen A. Ripperger obtained his PhD in genetics. After completing PostDoc stays in molecular biology in Geneva, Switzerland and biochemistry in Fribourg, Switzerland, he is now group leader at the Department of Biology in the University of Fribourg, Switzerland. His research focuses on the interconnection between aging and circadian rhythms.
References
- 1. Cox KH, Takahashi JS. Circadian clock genes and the transcriptional architecture of the clock mechanism. J Mol Endocrinol 2019;63:R93–R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lai RW, Lu R, Danthi PS, et al. Multi-level remodeling of transcriptional landscapes in aging and longevity. BMB Rep 2019;52:86–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nikopoulou C, Parekh S, Tessarz P. Ageing and sources of transcriptional heterogeneity. Biol Chem 2019;400:867–878. [DOI] [PubMed] [Google Scholar]
- 4. Haselgrübler T, Haider M, Ji B, et al. High-throughput, multiparameter analysis of single cells. Anal Bioanal Chem 2014;406:3279–3296. [DOI] [PubMed] [Google Scholar]
- 5. Tischler J, Surani MA. Investigating transcriptional states at single-cell-resolution. Curr Opin Biotechnol 2013;24:69–78. [DOI] [PubMed] [Google Scholar]
- 6. Lafzi A, Moutinho C, Picelli S, et al. Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies. Nat Protoc 2018;13:2742–2757. [DOI] [PubMed] [Google Scholar]
- 7. Holloszy JO. The biology of aging. Mayo Clin Proc 2000;75(Suppl):S3–S8 discussion S8-9. [PubMed] [Google Scholar]
- 8. Troen BR. The biology of aging. Mt Sinai J Med 2003;70:3–22. [PubMed] [Google Scholar]
- 9. Partridge L, Mangel M. Messages from mortality: the evolution of death rates in the old. Trends Ecol Evol 1999;14:438–442. [DOI] [PubMed] [Google Scholar]
- 10. López-Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell 2013;153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dunlap JC. Molecular bases for circadian clocks. Cell 1999;96:271–290. [DOI] [PubMed] [Google Scholar]
- 12. Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol 2010;72:517–549. [DOI] [PubMed] [Google Scholar]
- 13. Nagoshi E, Saini C, Bauer C, et al. Circadian gene expression in individual fibroblasts: cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell 2004;119:693–705. [DOI] [PubMed] [Google Scholar]
- 14. Fonseca Costa SS, Ripperger JA. Impact of the circadian clock on the aging process. Front Neurol 2015;6:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Welz PS, Benitah SA. Molecular connections between circadian clocks and aging. J Mol Biol 2019;432:3661–3679. [DOI] [PubMed] [Google Scholar]
- 16. Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 2013;153:1448–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hurd MW, Ralph MR. The significance of circadian organization for longevity in the golden hamster. J Biol Rhythms 1998;13:430–436. [DOI] [PubMed] [Google Scholar]
- 18. Bass J. Circadian topology of metabolism. Nature 2012;491:348–356. [DOI] [PubMed] [Google Scholar]
- 19. Moon S, Kang J, Kim SH, et al. Beneficial effects of time-restricted eating on metabolic diseases: a systemic review and meta-analysis. Nutrients 2020;12:E1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hui KY, Ripperger JA. Chronobiology and Aging In: Gu D, Dupre M (eds). Encyclopedia of Gerontology and Population Aging. Cham, Switzerland: Springer Nature, 2021. [Google Scholar]
- 21. Ly DH, Lockhart DJ, Lerner RA, et al. Mitotic misregulation and human aging. Science 2000;287:2486–2492. [DOI] [PubMed] [Google Scholar]
- 22. Lee CK, Klopp RG, Weindruch R, et al. Gene expression profile of aging and its retardation by caloric restriction. Science 1999;285:1390–1393. [DOI] [PubMed] [Google Scholar]
- 23. Enge M, Arda HE, Mignardi M, et al. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell 2017;171:321–330 e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hall BM, Balan V, Gleiberman AS, et al. Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 2016;8:1294–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Angelidis I, Simon LM, Fernandez IE, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun 2019;10:963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brandenberger C, Muhlfeld C. Mechanisms of lung aging. Cell Tissue Res 2017;367:469–480. [DOI] [PubMed] [Google Scholar]
- 27. Brazhnik K, Sun S, Alani O, et al. Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Sci Adv 2020;6:eaax2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kimmel JC, Penland L, Rubinstein ND, et al. Murine single-cell RNA-seq reveals cell-identity- and tissue-specific trajectories of aging. Genome Res 2019;29:2088–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Salzer MC, Lafzi A, Berenguer-Llergo A, et al. Identity noise and adipogenic traits characterize dermal fibroblast aging. Cell 2018;175:1575–1590 e1522. [DOI] [PubMed] [Google Scholar]
- 30. Ximerakis M, Lipnick SL, Innes BT, et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci 2019;22:1696–1708. [DOI] [PubMed] [Google Scholar]
- 31. Panda S, Antoch MP, Miller BH, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002;109:307–320. [DOI] [PubMed] [Google Scholar]
- 32. Storch KF, Lipan O, Leykin I, et al. Extensive and divergent circadian gene expression in liver and heart. Nature 2002;417:78–83. [DOI] [PubMed] [Google Scholar]
- 33. Gibbs JE, Beesley S, Plumb J, et al. Circadian timing in the lung; a specific role for bronchiolar epithelial cells. Endocrinology 2009;150:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Albrecht U. Timing to perfection: the biology of central and peripheral circadian clocks. Neuron 2012;74:246–260. [DOI] [PubMed] [Google Scholar]
- 35. Park J, Zhu H, O'Sullivan S, et al. Single-cell transcriptional analysis reveals novel neuronal phenotypes and interaction networks involved in the central circadian clock. Front Neurosci 2016;10:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wen S, Ma D, Zhao M, et al. Spatiotemporal single-cell analysis of gene expression in the mouse suprachiasmatic nucleus. Nat Neurosci 2020;23:456–467. [DOI] [PubMed] [Google Scholar]
- 37. Gorman MR. Temporal organization of pineal melatonin signaling in mammals. Mol Cell Endocrinol 2020;503:110687. [DOI] [PubMed] [Google Scholar]
- 38. Trivedi AK, Kumar V. Melatonin: an internal signal for daily and seasonal timing. Indian J Exp Biol 2014;52:425–437. [PubMed] [Google Scholar]
- 39. Zawilska JB. Melatonin as a chemical indicator of environmental light-dark cycle. Acta Neurobiol Exp (Wars) 1996;56:757–767. [DOI] [PubMed] [Google Scholar]
- 40. Mays JC, Kelly MC, Coon SL, et al. Single-cell RNA sequencing of the mammalian pineal gland identifies two pinealocyte subtypes and cell type-specific daily patterns of gene expression. PLoS One 2018;13:e0205883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bendall SC, Davis KL, Amir el AD, et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 2014;157:714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saelens W, Cannoodt R, Todorov H, et al. A comparison of single-cell trajectory inference methods. Nat Biotechnol 2019;37:547–554. [DOI] [PubMed] [Google Scholar]
- 43. Laloum D, Robinson-Rechavi M. Methods detecting rhythmic gene expression are biologically relevant only for strong signal. PLoS Comput Biol 2020;16:e1007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pett JP, Kondoff M, Bordyugov G, et al. Co-existing feedback loops generate tissue-specific circadian rhythms. Life Sci Alliance 2018;1:e201800078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu Z, Lou H, Xie K, et al. Reconstructing cell cycle pseudo time-series via single-cell transcriptome data. Nat Commun 2017;8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hsiao CJ, Tung PY, Blischak JD, et al. Characterizing and inferring quantitative cell cycle phase in single-cell RNA-Seq data analysis. Genome Res 2020;30:611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol 2019;15:e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ueda HR, Chen W, Minami Y, et al. Molecular-timetable methods for detection of body time and rhythm disorders from single-time-point genome-wide expression profiles. Proc Natl Acad Sci USA 2004;101:11227–11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruben MD, Wu G, Smith DF, et al. A database of tissue-specific rhythmically expressed human genes has potential applications in circadian medicine. Sci Transl Med 2018;10:eaat8806. [DOI] [PMC free article] [PubMed] [Google Scholar]