

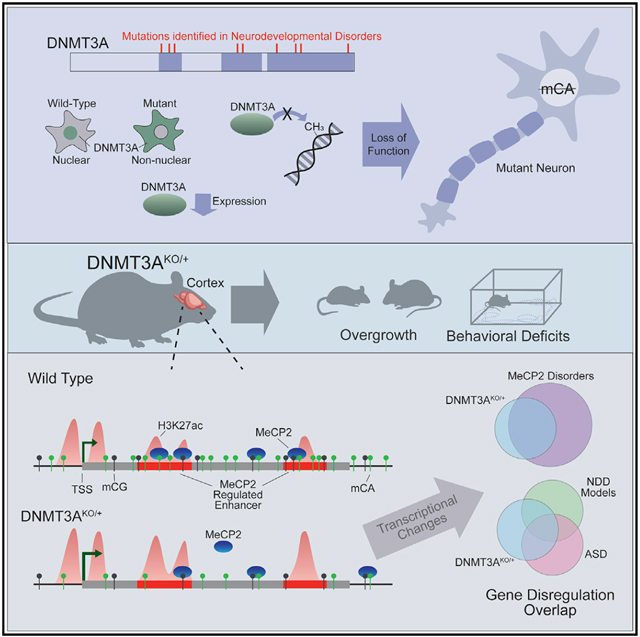
SUMMARY
Mutations in DNA methyltransferase 3A (DNMT3A) have been detected in autism and related disorders, but how these mutations disrupt nervous system function is unknown. Here, we define the effects of DNMT3A mutations associated with neurodevelopmental disease. We show that diverse mutations affect different aspects of protein activity but lead to shared deficiencies in neuronal DNA methylation. Heterozygous DNMT3A knockout mice mimicking DNMT3A disruption in disease display growth and behavioral alterations consistent with human phenotypes. Strikingly, in these mice, we detect global disruption of neuron-enriched non-CG DNA methylation, a binding site for the Rett syndrome protein MeCP2. Loss of this methylation leads to enhancer and gene dysregulation that overlaps with models of Rett syndrome and autism. These findings define the effects of DNMT3A haploinsufficiency in the brain and uncover disruption of the non-CG methylation pathway as a convergence point across neurodevelopmental disorders.
Graphical Abstract
In Brief
Christian et al. find that neurodevelopmental disease-associated DNMT3A mutations have shared neuronal DNA methylation deposition deficits. DNMT3AKO/+ mice display reduced global non-CG DNA methylation, behavior, and growth phenotypes relevant to human disease. Transcriptomic and epigenomic changes in these mice overlap models of Rett syndrome and autism, suggesting convergent pathology.
INTRODUCTION
Precise regulation of transcription is critical for nervous system development, and exome sequencing studies have revealed mutations in genes encoding epigenetic modifiers of chromatin structure as major underlying causes of neurodevelopmental disease (NDD) (McRae et al., 2017; Sanders et al., 2015). A challenge emerging from these discoveries is to define the cellular functions of the disrupted proteins during normal development and to search for shared pathways between these proteins that may be targeted for therapy development.
Gene regulation by DNA methylation has emerged as an epigenetic mechanism critical for nervous system function (Kinde et al., 2015). In addition to classical methylation of cytosines found at CG dinucleotides (mCG), neurons contain uniquely high levels of methyl-cytosine (mC) in a non-CG context, with this mark occurring primarily at CA sites (mCA) (Lister et al., 2013; Xie et al., 2012). mCA is deposited by DNA methyltransferase 3A (DNMT3A) during the early postnatal period (1–6 weeks of age in mice) until the number of non-CG methylation sites in neurons is nearly equivalent to the number of mCG sites (Lister et al., 2013; Xie et al., 2012). mCA serves as a critical binding site for methyl-CpG binding protein 2 (MeCP2) (Chen et al., 2015; Gabel et al., 2015; Guo et al., 2014). MeCP2 was initially defined by its high affinity for mCG; however, it also tightly interacts with mCA to downregulate transcription of genes with essential functions in the brain (Kinde et al., 2016; Lagger et al., 2017; Lyst and Bird, 2015). Loss of MeCP2 leads to the severe neurological disorder Rett syndrome, whereas duplication causes MeCP2 duplication syndrome, an autism spectrum disorder (ASD) (Amir et al., 1999; Van Esch et al., 2005), indicating that mCA readout is critical for nervous system function.
Exome sequencing studies have identified heterozygous mutations in DNMT3A in ASD (Feliciano et al., 2019; Iossifov et al., 2014; Sanders et al., 2015; Satterstrom et al., 2019). Separate studies have also defined heterozygous disruption of DNMT3A as the cause of Tatton-Brown Rahman syndrome (TBRS), an NDD characterized by intellectual disability (ID), overgrowth, craniofacial abnormalities, anxiety, and ASD (Tatton-Brown et al., 2014, 2018). Although some mutations in affected individuals are truncations predicted to cause complete enzyme loss, a majority of NDD-associated alleles are missense mutations, raising questions about whether loss-of-function effects are a primary mechanism in DNMT3A disorders (Tatton-Brown et al., 2014, 2018). In addition, the large percentage of missense mutations in affected individuals raises the need to establish the deleterious effects of these mutations to definitively associate mutations with NDD and end the “diagnostic odyssey” for patients (Sawyer et al., 2016). Finally, although heterozygous loss of DNMT3A has been studied in the context of hematopoietic oncogenesis (Cole et al., 2017), the effects of partial loss of DNMT3A on neuronal DNA methylation, gene expression, and brain function have not been investigated closely.
Here we examine the molecular effects of NDD-associated DNMT3A mutations and explore how heterozygous loss of DNMT3A affects the neuronal epigenome. We show that missense mutations across domains of DNMT3A disrupt different aspects of protein function, but all mutations reduce deposition of neuronal mCA. Using DNMT3A heterozygous deletion mice, we assess the effect of shared loss-of-function effects resulting from missense and deletion mutations. We detect altered growth and behavior in these mice, suggesting that haploinsufficiency contributes to pathology in DNMT3A disorders. Through integrated epigenomics analysis, we reveal a global reduction of mCA throughout the brains of DNMT3A mutants. We show that loss of mCA leads to disruption of distal regulatory enhancer activity and changes in gene expression that overlap with models of MeCP2 disorders and other NDDs. These findings define the effects of NDD-associated DNMT3A mutations and implicate disruption of mCA-mediated epigenomic regulation as a convergence site across clinically distinct NDDs.
RESULTS
Functional Analysis of NDD-Associated DNMT3A Mutations
Multiple DNMT3A mutations have been identified in individuals with ASD and TBRS. However, the myriad missense mutations identified and the phenotypic heterogeneity of affected individuals suggests that altered amino acids in different protein domains may have distinct consequences that dictate the nature and severity of disease. Furthermore, it is not clear whether missense mutations identified in patient populations are truly disruptive, raising the need for direct assessment of molecular function to ascribe mutations as potentially causative. We therefore assessed the effects of NDD-associated DNMT3A mutations on protein expression, cellular localization, and catalytic activity, looking for common effects that may be core to NDD.
We introduced alterations homologous to human disease mutations into a FLAG-tagged DNMT3A expression vector (Figure 1A; Table S1), assessing mutations within the chromatin interacting proline-tryptophan-tryptophan-proline (PWWP) domain, the auto-inhibitory ATRX-DNMT3-DNMT3L (ADD) domain, and the methyltransferase domain (Gowher and Jeltsch, 2018). Transfection into heterologous cells facilitated assessment of protein expression by western blot, cellular localization by immunocytochemistry, and catalytic activity using an in vitro methyltransferase assay. Mutations in the PWWP domain resulted in reduced DNMT3A protein and loss of nuclear localization (Figures 1B–1D; Figures S1A–S1C). When expressed at equal levels as wild-type protein, however, these mutants exhibited normal in vitro methyltransferase activity (Figures 1E and 1F). In contrast, mutations found in the catalytic domain of DNMT3A showed wild-type expression and localization but had deficits in methyltransferase activity (Figures 1B–1Fl Figures S1A–S1C). Mutations in the ADD domain of DNMT3A displayed normal protein localization and expression levels and exhibited equal or higher methylation activity compared with the wild type (Figures 1B–1F; Figures S1A–S1C).
Figure 1. NDD-Associated DNMT3A Mutations Disrupt Distinct Aspects of Protein Function.
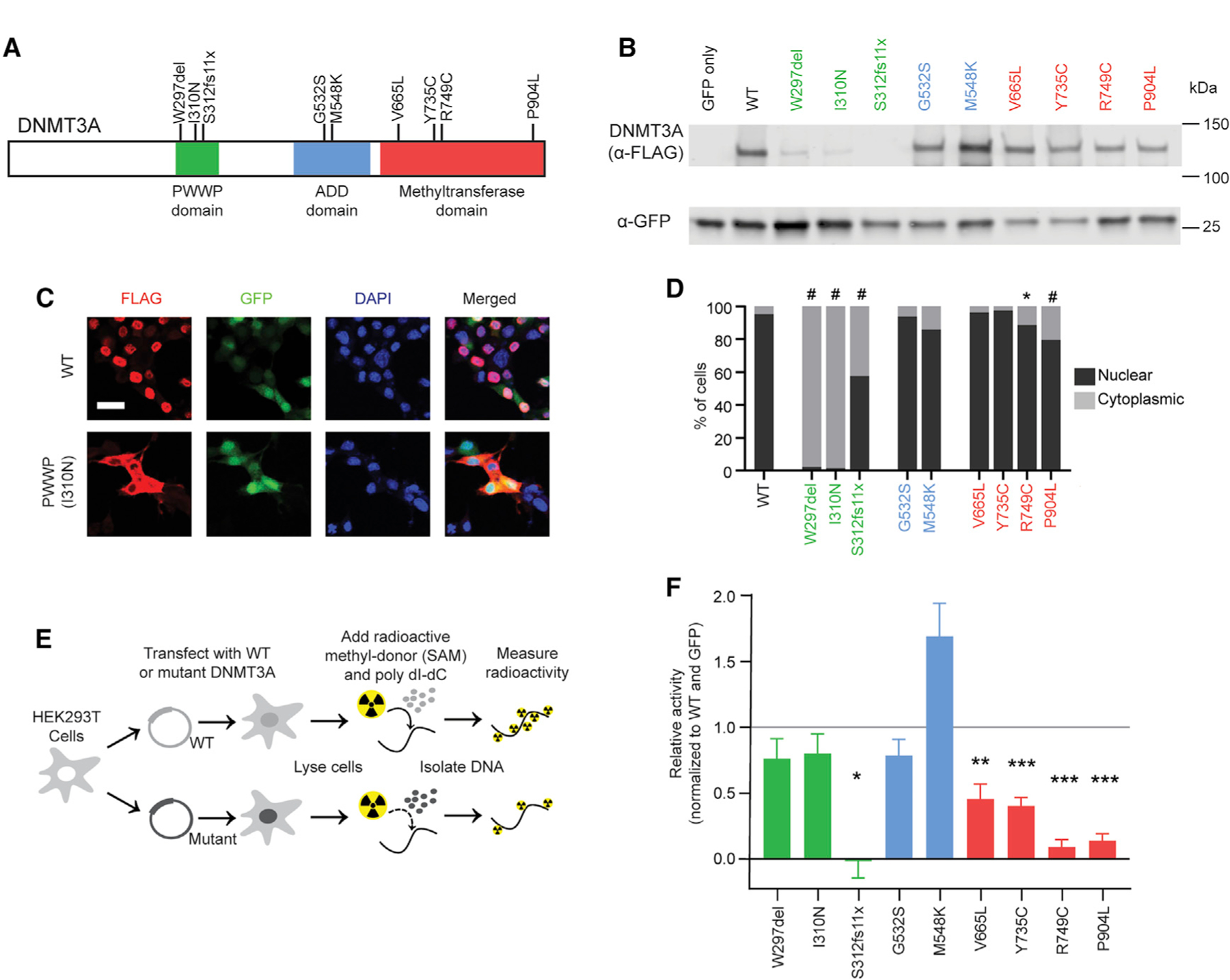
(A) Schematic of the human DNMT3A protein, showing the canonical domains and NDD-associated mutations (Sanders et al., 2015; Tatton-Brown et al., 2018) tested in this study.
(B) Example immunoblot of DNMT3A mutant protein expression in Neuro-2a cells.
(C) Example images of DNMT3A protein immunocytochemistry from the wild type and PWWP domain mutant in Neuro-2a cells. Scale bar, 20 μm.
(D) Quantification of DNMT3A mutant protein localization (n = 6–16 images; generalized linear model test with Bonferroni correction).
(E) Schematic of the in vitro methylation assay for DNMT3A mutant proteins isolated from HEK293T cells.
(F) Activity of DNMT3A mutant proteins in the in vitro methylation assay, normalized to wild-type (WT) DNMT3A (set equal to 1) and GFP-only (set equal to 0) controls (n = 4–19; one-sample t test with Bonferroni correction).
*p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.0001. Bar graphs indicate mean and SEM. See also Figure S1 and Table S1.
To further evaluate the effects of DNMT3A mutations in the context of endogenous chromatin, we tested the capacity of DNMT3A mutants to establish DNA methylation in mouse cortical neurons. For this analysis, we focused on the DNMT3A-dependent build-up of mCA in postmitotic neurons (Gabel et al., 2015; Lister et al., 2013). Cultured embryonic day 14.5 cortical neurons accumulate mCA in vitro, and this build-up is blocked by lentivirus-mediated delivery of Cre recombinase to DNMT3Aflx/flx cells at 3 days in vitro (DIV) (Figure 2A; Figures S1D and S1E). We co-transduced Cre with wild-type or mutant DNMT3A lentiviruses at equal levels (Figure S1F) to test each protein for rescue of mCA deposition. All NDD-associated mutations across domains of DNMT3A blocked mCA accumulation when tested in neurons (Figure 2B). Notably, ADD domain mutations that exhibited robust catalytic activity in vitro displayed moderate to severe deficits in mCA deposition. The ADD domain has been implicated in histone binding and autoinhibition of the protein (Guo et al., 2015); thus, the results of this neuronal assay suggest that loss of ADD function blocks DNMT3A from engaging chromatin and promoting methylation in cells. Disease mutations associated with ID (Table S1) show near-complete loss of mCA rescue, whereas mutations associated with ASD but not ID (such as V665L) have more moderate depletion of mCA. This suggests that a possible gradient of disease phenotypes may be driven by differential mutation severity. Unlike mCA, global mCG levels show no significant differences in this in vitro system, likely because of the redundant function of the DNMT1 methyltransferase in maintaining mCG sites (Figures S1G and S1H). Together our results indicate that, although NDD-associated mutations in DNMT3A affect different protein domains and alter distinct functions (e.g., localization, chromatin interaction, and catalysis), these mutations share a common outcome of reduced enzymatic activity on neuronal DNA, with multiple mutations resulting in functionally null proteins.
Figure 2. NDD-Associated DNMT3A Mutations Prevent Buildup of Neuronal CA Methylation.
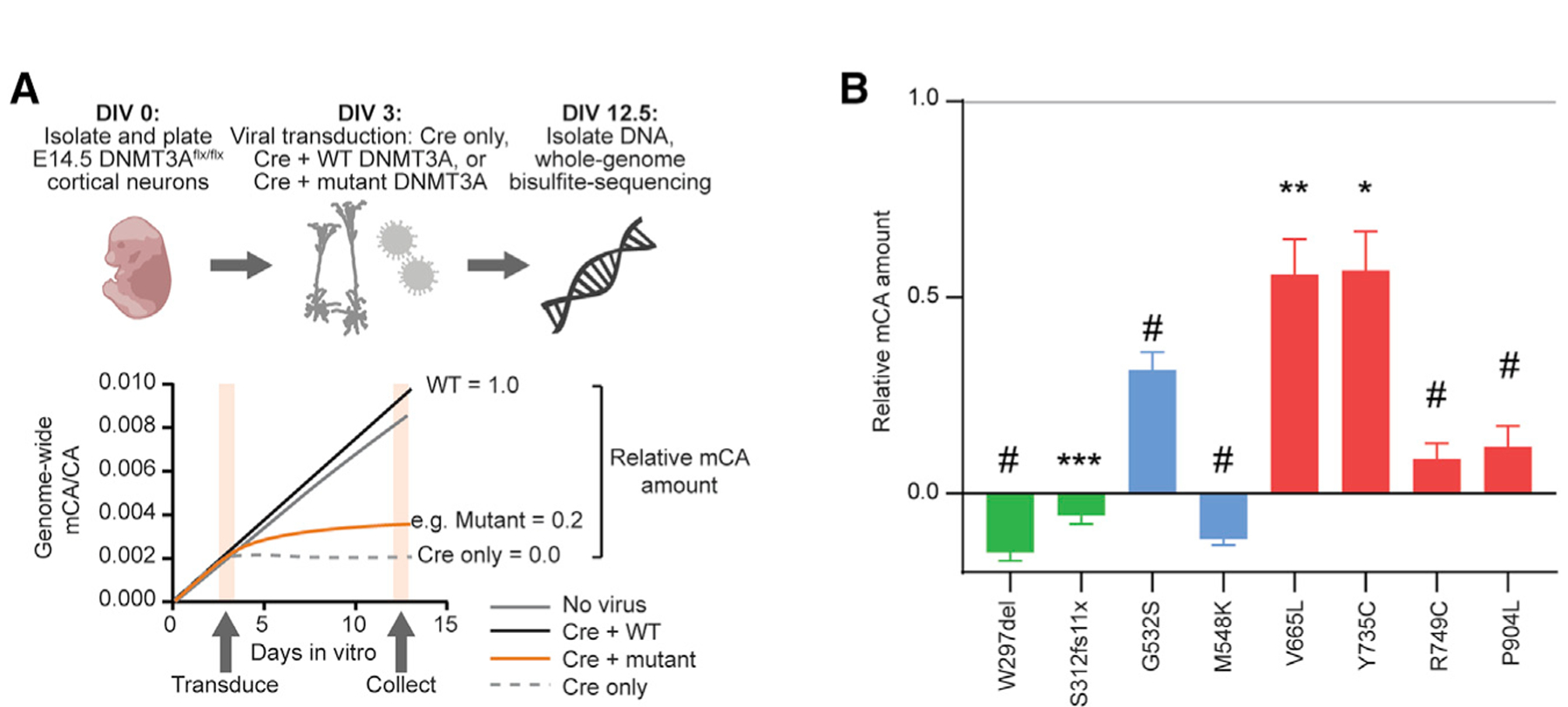
(A) Schematic of DNMT3A functional analysis in primary culture neurons. Cortical neurons are harvested from DNMT3Aflx/flx mice at embryonic day 14.5 (E14.5) and cultured. After 3 days in vitro (DIV), neurons are virally transduced with Cre recombinase and WT or mutant FLAG-tagged DNMT3A. On DIV 12.5, DNA and RNA are collected. Equal DNMT3A mRNA expression is verified by qRT-PCR (Figure S1F), and DNA is used for whole-genome bisulfite sequencing analysis.
(B) Relative mCA amount compared with Cre only (set equal to 0) and Cre+WT DNMT3A (set equal to 1) controls (n = 4–11; one-sample t test with Bonferroni correction).
*p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.0001. Bar graphs indicate mean and SEM. See also Figure S1 and Table S1.
In Vivo Growth and Behavioral Effects of Heterozygous DNMT3A Disruption
Our in vitro finding that multiple NDD-associated DNMT3A missense mutations result in severe loss of function, coupled with identification of deletion and nonsense mutations in TBRS patients, suggests that an ~50% reduction of DNMT3A activity occurs in a substantial fraction of affected individuals. Thus, studying the effects of heterozygous inactivation of DNMT3A in vivo can provide an understanding of the epigenomic and systems-level consequences of DNMT3A disruption in NDD. Previous studies have demonstrated severe developmental deficits, disruptions in movement, and lethality upon constitutive complete loss of DNMT3A (homozygous null mutation; Okano et al., 1999) or removal of DNMT3A from the brain (conditional DNMT3A deletion; Nguyen et al., 2007). However, the growth and behavioral effects of heterozygous DNMT3A inactivation have not been assessed systematically. We therefore carried out growth, behavioral, and molecular analyses of mice carrying a constitutive heterozygous deletion of exon 19 of Dnmt3a (STAR Methods; Kaneda et al., 2004). We find that this mutation leads to 50% reduction of RNA and protein expression, allowing us to study the in vivo effects of the heterozygous null mutation of DNMT3A (referred to as DNMT3AKO/+) (Figures S2A–S2C).
We first examined phenotypes relevant to overgrowth in individuals with TBRS (Tatton-Brown et al., 2018), including enlarged body size and obesity (body weight), tall stature (long-bone length), and macrocephaly (skull dimensions). DNMT3AKO/+ mice showed similar body weights as controls in the early postnatal period but were significantly heavier as adults (Figure 3A). This phenotype mimics a maturity-associated trend toward increased body weight in TBRS patients (Tatton-Brown et al., 2018). Measurements of leg bone length, a major determinant of human height (Duyar and Pelin, 2003), showed increases in the DNMT3AKO/+ mice, with significantly increased tibia length and a trend toward longer femur length (Figure 3B; Figures S3A–S3D). Morphometric analyses of the cranium and mandible indicated very subtle differences between DNMT3AKO/+ and wild-type littermates (Figures S3E–S3G). The rostrocaudal length of the interparietal bone was larger in DNMT3AKO/+ mice, whereas two facial linear distances were significantly larger in the wild type (Figure S3E). These findings uncover effects on bone length and body weight that mirror aspects of the human disorder, whereas skull development in DNMT3AKO/+ mice shows more limited effects.
Figure 3. Heterozygous Disruption of DNMT3A In Vivo Leads to Growth and Behavioral Alterations.
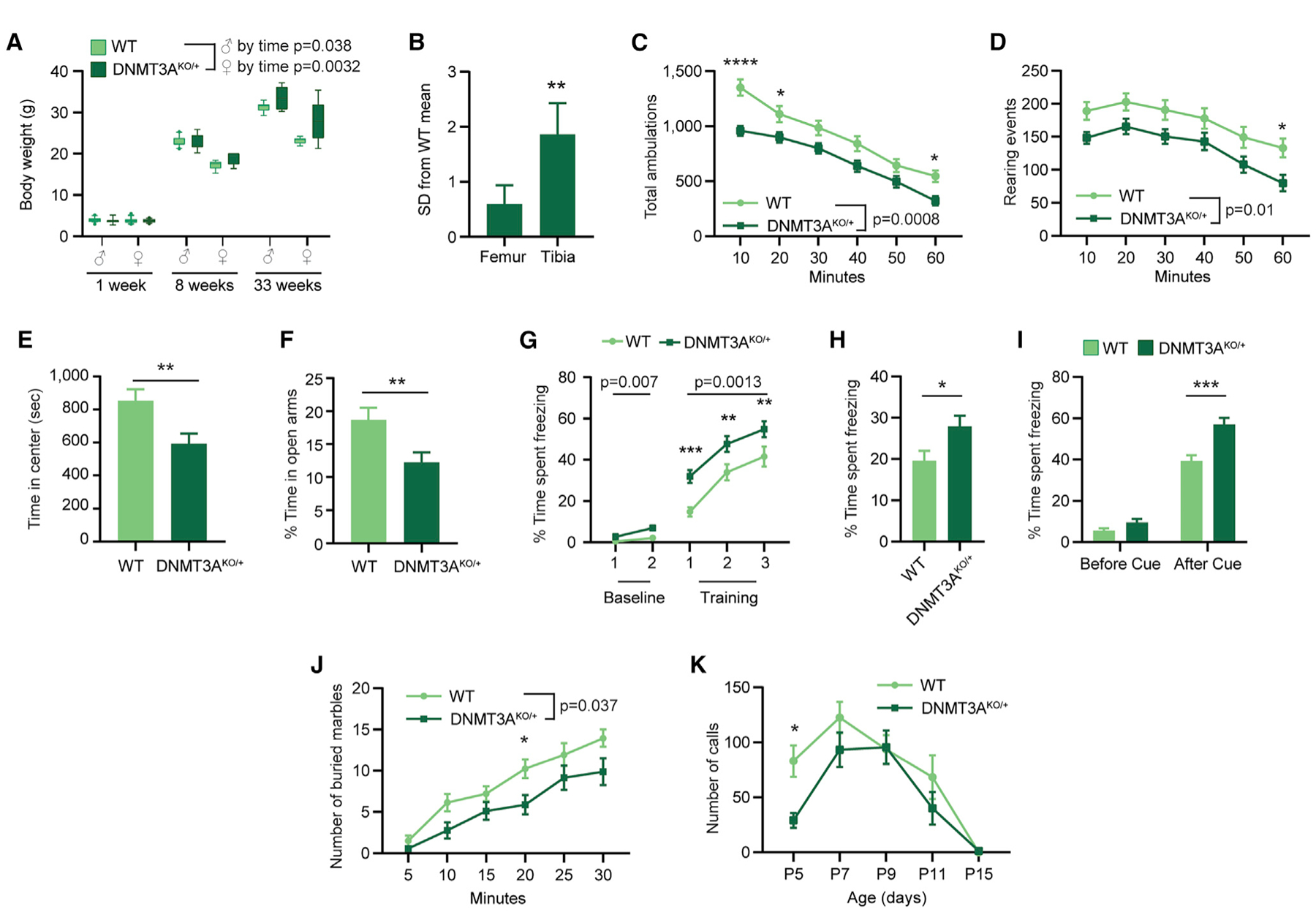
(A) Body weight of DNMT3AKO/+ and WT mice at three time points (male p = 0.038 genotype by age interaction, F(2,50) = 3.494, n = 6–18; female p = 0.0032 genotype by age interaction, F(2,48) = 6.498; female p = 0.0016 genotype effect, F(1,48) = 11.18, n = 5–17; two-way ANOVA).
(B) Bone lengths shown as standard deviations from the WT mean for DNMT3AKO/+ mice (n = 12; paired t test).
(C) Total ambulations during 1-h open-field testing (10-min bins, p = 0.0008 genotype effect, F(1,46) = 13.02, n = 21, 27; two-way repeated-measures (RM) ANOVA with Sidak’s multiple comparison test).
(D) Rearing events during 1-h open-field testing (10-min bins, p = 0.0103 effect by genotype, F(1,46) = 7.161, n = 21, 27; two-way RM ANOVA with Sidak’s multiple comparison test).
(E) Time in the center zone during open field testing (p = 0.0075, n = 21, 27; unpaired t test).
(F) Percentage of time in the open arms during elevated plus maze testing (p = 0.0069; n = 33, 39; unpaired t test).
(G–I) Percentage of time freezing in (G) conditioned fear training (baseline: p = 0.0071 genotype effect, F(1,50) = 7.897; cue: p = 0.0013 genotype effect, F(1,50) = 11.7; n = 26; two-way RM ANOVA with Sidak’s multiple comparisons test), (H) contextual fear trials (p = 0.0215, n = 26; unpaired t test), and (I) cued fear trials (before cue: p = 0.0606; after cue: p = 0.00014; n = 26; unpaired t test).
(J) Marbles buried in 30 min (5-min bins, p = 0.0374 effect by genotype, F(1,25) = 4.834, n = 14, 13; two-way RM ANOVA with Sidak’s multiple comparison test).
(K) Number of ultrasonic calls from isolated pups for 3 mine of testing at the indicated ages; p = 0.0378 genotype effect, F(1,285) = 4.355, n = 9–46; two-way ANOVA with Sidak’s multiple comparisons test.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Boxplots show 10th–90th percentiles of data. Line and bar graphs indicate mean and SEM. See also Figures S2–S5.
To examine neurological and behavioral phenotypes in DNMT3AKO/+ mice, we first assessed basic measures of sensation and motor performance, such as balance (ledge test and platform test), grip strength (inverted screen test), motor coordination (walking initiation and rotarod), and sensorimotor gating (pre-pulse inhibition). DNMT3AKO/+ mice were not significantly different in these assays (Figures S4A–S4G), indicating that heterozygous loss of DNMT3A does not grossly disrupt sensorimotor function. This allowed us to assess more complex aspects of behavior and cognition.
We next carried out assays with relevance to neuropathology in humans with DNMT3A mutations, including anxiety, autism, and ID. In open field testing, DNMT3AKO/+ mice explored the chamber less than wild-type mice, as indicated by reduced distance traveled and rearing (Figures 3C and 3D). DNMT3AKO/+ mice also displayed anxiety-like behavior, measured by less time in the center of the arena in this assay (Figure 3E). In tests of climbing behavior, DNMT3AKO/+ mice showed longer latency to climb to the bottom of a pole and to the top of mesh screens (Figures S4H–S4J), suggesting that DNMT3AKO/+ mice display differences in volitional movement. During assessment of anxiety-like behavior in the elevated plus maze, DNMT3AKO/+ mice spent less time exploring the open arms of the maze, with no change in the percentage of entries made into all arms (Figure 3F; Figure S4K). Overall, these results indicate that DNMT3AKO/+ mice display changes in exploratory behavior, suggesting altered emotionality and increased anxiety-like behaviors.
ID is a highly penetrant phenotype in TBRS. We therefore assessed learning and memory in DNMT3AKO/+ mice using contextual fear conditioning and Morris water maze tests. In foot shock-induced fear conditioning, DNMT3AKO/+ mutants showed a heightened freezing response during training and in the contextual and auditory recall phases (Figures 3G–3I; Figures S4L and S4M). Mutant mice also showed delayed extinction of freezing behavior in response to the auditory cue alone (Figure S4M). These effects do not arise from differential responses to shock pain (Figure S4N), further suggesting altered emotionality or cognition in the mutant mice. During assessment of spatial and contextual memory in the Morris water maze, DNMT3AKO/+ mice were slower to learn to find a visible platform and did not learn the location of the hidden platform over time as well as wild-type controls (Figures S4O and S4P), despite similar swimming speeds (Figures S4Q and S4R). There were no significant effects on distance traveled in target zone or platform crossings in the probe trial, although DNMT3AKO/+ mice trended toward fewer platform crossings (Figures S4S and S4T). These findings suggest that DNMT3AKO/+ mutants do not show severe deficits in learning and memory but display differences in task performance that may reflect altered emotionality or cognition.
DNMT3A has been identified as a high-confidence autism gene (Sanders et al., 2015), and autism is commonly diagnosed in TBRS patients (Tatton-Brown et al., 2018). We therefore characterized phenotypes examined in models of autism (marble burying, three-chamber social approach, and ultrasonic vocalizations). We detected a significant reduction in marble-burying activity for DNMT3AKO/+ mice, indicating alterations in repetitive digging behavior (Figure 3J; Figure S4U). Evaluation of social interaction behaviors in the three-chamber social approach assay (Yang et al., 2011) revealed that DNMT3AKO/+ mice spend reduced time investigating mice and objects, with no change in preference index for sociability or novelty (Figures S5A–S5D; Nygaard et al., 2019). We considered that testing under normal lighting and experimental conditions may induce anxiety in addition to probing sociability and that the reduced exploration of DNMT3AKO/+ mice may reflect the anxiety-like phenotypes rather than sociability changes. Indeed, when repeating the three-chamber analysis under low lighting conditions to mitigate anxiety in the mice, we detected similar exploratory behavior, sociability, and social novelty responses in wild-type and DNMT3AKO/+ mice (Figures S5E–S5H; Manno et al., 2020). To explore changes in pro-communication behaviors, we measured maternal isolation-induced ultrasonic vocalizations (Barnes et al., 2017). We detected a reduction in the number of calls in DNMT3AKO/+ mice on post-natal day 5, suggesting deficits in early pro-social behaviors or developmental delay in acquisition of this behavior (Figure 3K). These results reveal alterations in behaviors commonly assessed in mouse models of autism (Chang et al., 2017; Kazdoba et al., 2014; Takumi et al., 2020), with our findings suggesting reduced activity and exploration as well as changes in communication behaviors.
Overall, our physiological and behavioral analyses demonstrate that heterozygous deletion of DNMT3A results in growth and behavior changes in mice with relevance to the overgrowth, anxiety, communication, and memory-associated phenotypes observed in patients with DNMT3A mutations. These data support a model in which DNMT3A haploinsufficiency contributes to altered growth and behavioral circuits to drive phenotypes in NDD.
Global Disruption of DNA Methylation in the DNMT3AKO/+ Brain
We next investigated the epigenomic defects that may underlie the altered behaviors in DNMT3AKO/+ mice. We first used sparse whole-genome bisulfite sequencing to survey the effects on global methylation levels in multiple brain regions and the liver. This analysis detected limited reductions in genome-wide mCG levels in the DNMT3AKO/+ brain that were not apparent in non-neuronal tissue (liver) (Figure 4A). In contrast, mCA levels were reduced 30%–50% across all DNMT3AKO/+ brain regions examined (Figure 4B). Analysis of DNA methylation during post-natal development in the cerebral cortex, a region with high adult mCA levels, suggested that these deficits appear during accumulation of mCA (Figure 4B). Thus, global mCA levels in the brain are sensitive to heterozygous DNMT3A disruption, whereas global mCG levels are largely maintained.
Figure 4. Global DNA Methylation Levels upon Heterozygous Loss of DNMT3A.
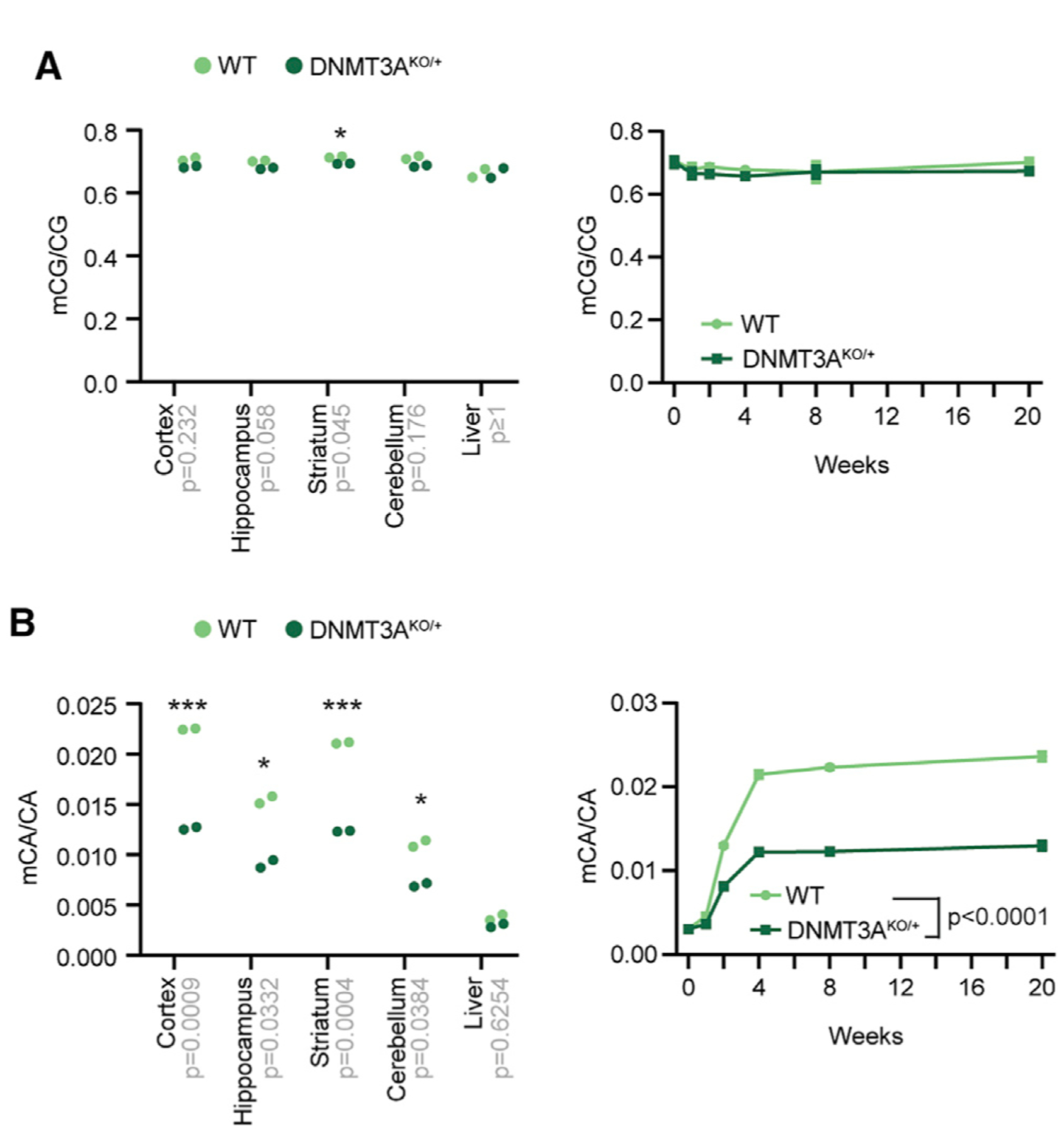
(A and B) Global mCG (A) and mCA levels (B) in DNA isolated from brain regions of 8-week-old mice (left) (unpaired t test with Bonferroni correction) and developmental time course of global mCG in the cerebral cortex (right), as measured by sparse whole-genome bisulfite sequencing (WGBS) (p < 0.0001 effect by genotype, F(1,27) = 1024; p < 0.0001 effect by age F(5,27) = 884.6; n = 3–4; two-way ANOVA). Bonferroni corrected p values are indicated on the x axis. *p < 0.05, ***p < 0.001. Line graphs indicate mean and SEM. See also Figure S6.
DNA methylation at genomic elements such as promoters, enhancers, and gene bodies can regulate transcription, and altered methylation at these regions can affect gene expression to affect brain function (Clemens et al., 2019; Nord and West, 2020; Stroud et al., 2017). We therefore performed base-pair-resolution, high-depth, whole-genome bisulfite sequencing to identify potential changes in mCG and mCA at these important regulatory sites. We focused on the cerebral cortex because this region is enriched for mCA (Figure 4B) and disrupted in ASD and MeCP2 disorders (Clemens et al., 2019; Satterstrom et al., 2019). Our analysis of mCG confirmed the subtle mCG reduction across classes of genomic elements (Figures 5A–5D). We considered, however, that CG dinucleotides at specific sites may be more sensitive to partial DNMT3A reduction. For example, in the hematopoietic system, heterozygous DNMT3A loss leads to mCG reductions in genomic regions identified as sensitive to complete loss of DNMT3A (Cole et al., 2017). We therefore evaluated CG differentially methylated regions (CG-DMRs) identified previously as becoming highly methylated in the adult cortex (Figure S6A; Lister et al., 2013), hypothesizing that these adult-specific CG-DMRs might be sensitive to a reduction in enzyme activity. Indeed, we found that these sites build up mCG during postnatal development and do not become methylated in a brain-specific DNMT3A conditional knockout mouse (DNMT3A Nestin-cKO) (Stroud et al., 2017; Figure S6B). Analysis of these adult-specific CG-DMRs in DNMT3AKO/+ mice indicated that they are particularly sensitive to partial inactivation of DNMT3A (Figures 5A, 5C, and 5D).
Figure 5. High-Resolution Analysis of DNA Methylation Changes in the DNMT3AKO/+ Cerebral Cortex.
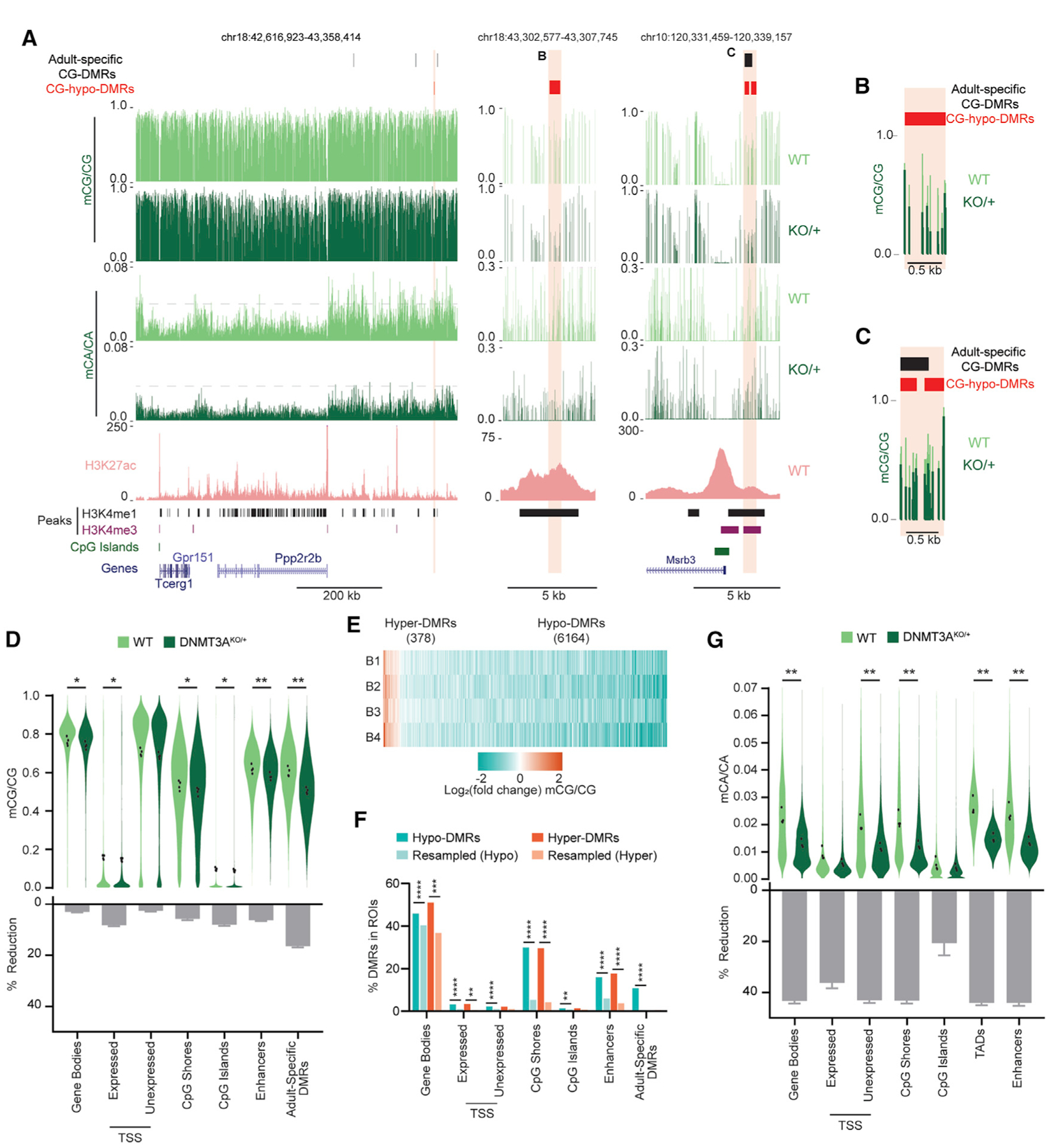
(A) Genome browser views of mCA and mCG in WT and DNMT3AKO/+ cerebral cortices, measured by WGBS. A broad view shows global reduction in mCA (left). A gray dashed line facilitates comparison of mCA levels across genotypes. Shown is magnification of DNMT3AKO/+ hypo-CG-DMRs overlapping an enhancer (center) or a CpG-island shore and an adult-specific DMR (right). A WT H3K27ac ChIP-seq signal (Clemens et al., 2019), peaks of enhancer-associated histone H3 lysine 4 methylation (H3K4me1) and promoter-associated histone H3 lysine 4 tri-methylation (H3K4me3) (Stamatoyannopoulos et al., 2012), CpG islands, and gene annotations (Haeussler et al., 2019) are also shown.
(B and C) Overlay of the mCG signal for DMRs shown in (A).
(D) Violin plot of mCG/CG levels (top) and percent reduction (bottom) in DNMT3AKO/+ versus WT across genomic regions. Dots indicate mean mCG/CG per bioreplicate (n = 4 per genotype; paired t test, Bonferroni correction).
(E) Heatmap of CG-DMRs called in the DNMT3AKO/+ cortex across bioreplicates (B1–B4).
(F) Overlap of CG-DMRs with genomic regions (Fisher’s exact test, observed versus background estimated from resampled DMRs; STAR Methods).
(G) Violin and percent reduction plots as in (D) but for mCA/CA.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Bar graphs indicate mean and SEM. See also Figure S6.
Because a substantial portion of mCG in neurons is oxidized to hydroxymethylation (hmCG), we performed oxidative bisulfite sequencing of DNMT3AKO/+ cortex DNA. This analysis revealed no evidence of widespread differential effects on oxidized or un-oxidized forms of mCG across genomic regions (Figure S6C), suggesting that hmCG is largely preserved in the DNMT3AKO/+ brain. However, future experiments will be required to uncover whether there are site-specific changes in hmCG.
To further search for local sites of altered mCG in DNMT3AKO/+, we performed de novo calling of CG-DMRs using the BSmooth algorithm (Hansen et al., 2012). We identified 6,164 hypo- and 378 hyper-CG-DMRs that met stringent size and reproducibility filters (Figures 5A–5C, and 5E; Figure S6D; Table S2; STAR Methods). Hypo-CG-DMRs significantly overlap with adult-specific CG-DMRs (Lister et al., 2013; Figure 5F; Figure S6E), further supporting the idea that DNMT3A is haploinsufficient for postnatal mCG deposition at these sites. Significant overlap of DNMT3AKO/+ hypo-CG-DMRs was also detected for putative enhancer regions, gene bodies, and promoters (Figures 5A–5C and 5F; Figure S6E). DMRs were also highly enriched for overlap with CpG island shores, regions disrupted in studies of DNMT3A mutation outside of the nervous system (Cole et al., 2017; Spencer et al., 2017; Figure 5C; Figure S6E). These findings indicate that specific mCG sites are sensitive to heterozygous DNMT3A loss and that localization of these CG-DMRs to regulatory elements could affect gene expression.
We next examined mCA in the DNMT3AKO/+ cortex at genomic elements with relevance to gene regulation. In contrast to changes in mCG levels, mCA levels were reduced 30%–50% at gene bodies, promoters, and CpG island shores (Figure 5G). CpG islands displayed less reduction of mCA, possibly because of floor effects in bisulfite sequencing (STAR Methods). Comparing mCA changes within genomic elements as a function of wild-type mCA levels revealed consistent reductions independent of the normal levels of mCA (Figure S6F). This result suggests that mCA loss in the DNMT3AKO/+ cortex does not preferentially affect specific classes of genomic elements, nor is it more severe in some regions, based on the level of mCA normally deposited there.
Recent analysis has shown that topologically associating domains (TADs) of chromatin folding affect gene regulation by influencing DNMT3A binding and subsequent mCA deposition (Clemens et al., 2019; Nothjunge et al., 2017). The average mCA level in TADs is associated with mCA levels at enhancers found in those TADs, and high-mCA enhancers within high-mCA TADs are robust targets of MeCP2 repression (Clemens et al., 2019). We therefore assessed mCA levels at TADs and enhancers genome wide. This analysis detected reductions in TAD mCA levels similar to mCA reductions at other genomic elements (Figure 5G). Enhancers also showed pervasive depletion of mCA (Figure 5G). Thus, widespread mCA loss for TADs and the enhancer elements within them occurs in DNMT3AKO/+ mice and can affect control of regulatory elements by MeCP2.
Enhancer Dysregulation Results from Methylation Deficits in DNMT3AKO/+ Mice
We next examined how disruption of DNA methylation affects epigenetic regulation and gene expression in the DNMT3AKO/+ brain. Recent analyses indicate that MeCP2 binds mCA to mediate neuron-specific gene regulation, in part by controlling the activity of enhancer elements (Boxer et al., 2020; Clemens et al., 2019). Loss of MeCP2 leads to genome-wide upregulation of the histone H3 lysine 27 acetylation (H3K27ac) active mark at enhancers that contain many mCA and mCG sites, whereas overexpression of MeCP2 leads to reciprocal downregulation of these enhancers. This altered enhancer activity in MeCP2 mutants is linked to gene dysregulation that likely contributes to nervous system dysfunction (Clemens et al., 2019). These studies suggest that reduced mCA in DNMT3AKO/+ would remove binding sites for MeCP2 in enhancers and result in enhancer dysregulation that partially phenocopies effects observed in MeCP2 mutant mice. To investigate this possibility directly, we quantified the change in mCA binding sites in DNMT3AKO/+ for enhancers found previously to be significantly repressed by MeCP2 (Clemens et al., 2019). We found that, because these MeCP2-repressed enhancers contain large numbers of mCA sites, the global 30%–50% reduction of mCA in DNMT3AKO/+ leads to a larger loss in mCA sites at these enhancers than at other enhancers genome wide (Figures 6A and 6B). Thus, MeCP2-repressed enhancers are particularly susceptible to mCA binding site loss from heterozygous DNMT3A mutation.
Figure 6. Epigenetic and Transcriptomic Dysregulation in the DNMT3AKO/+ Cerebral Cortex Overlaps with MeCP2 Mutants.
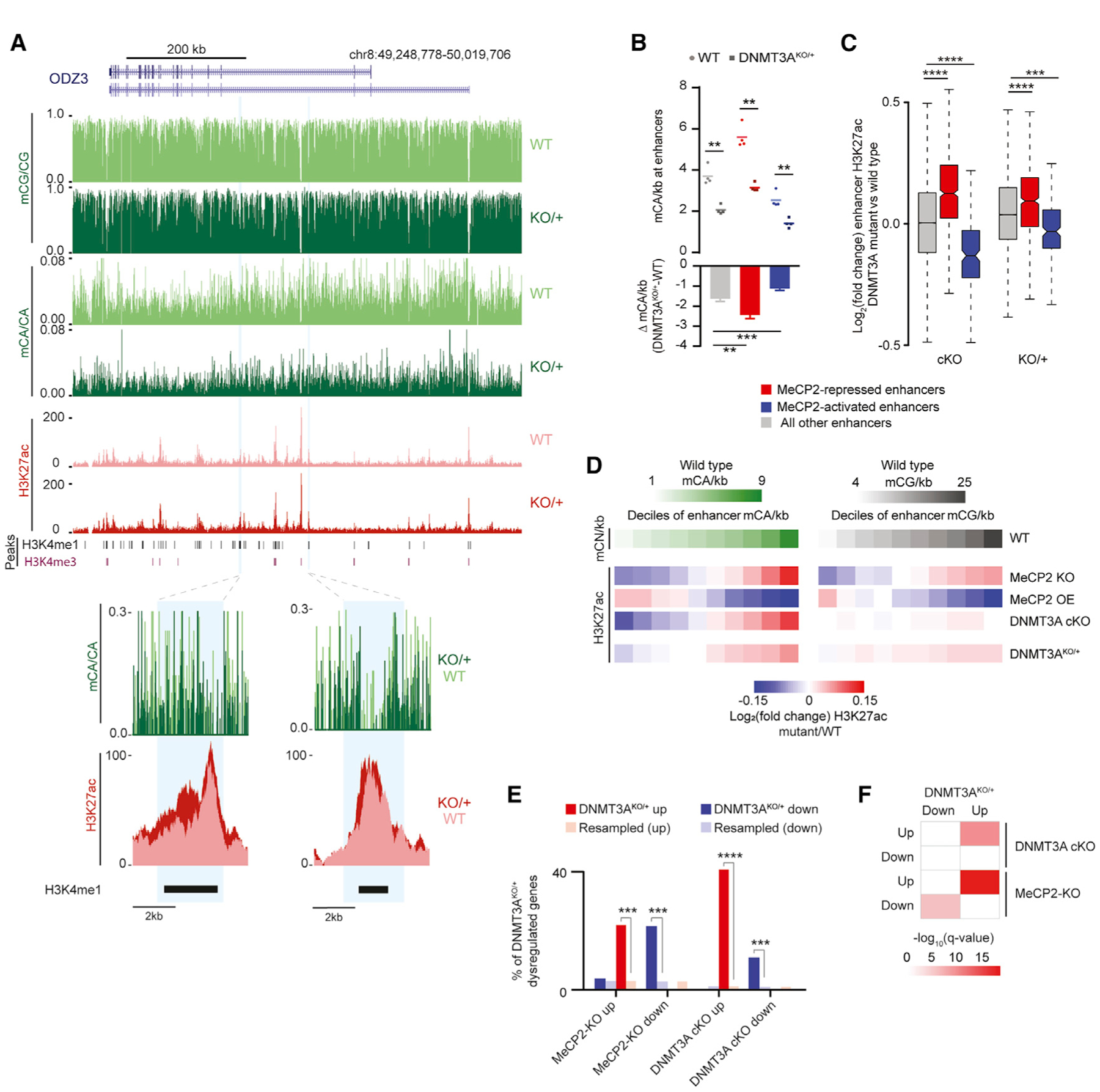
(A) Genome browser view of DNA methylation and H3K27ac ChIP-seq data from WT and DNMT3AKO/+ (top). Shown are an overlaid H3K27ac signal and mCA/CA at enhancers defined previously as dysregulated upon disruption of mCA or MeCP2 (Clemens et al., 2019) (bottom).
(B) Mean mCA sites per kilobase in WT and DNMT3AKO/+ (top) and mCA sites per kilobase lost in DNMT3AKO/+ (bottom) for enhancers significantly dysregulated in MeCP2 mutants (**p < 0.01, ***p < 0.001; n = 4; paired t test with Bonferroni correction).
(C) Boxplot of change in H3K27ac signal in DNMT3A Baf53b-cKO and DNMT3AKO/+ for enhancers defined as significantly dysregulated in MeCP2 mutants (***p < 10−8, ****p < 10−12; n = 5 bioreplicates; Wilcoxon test).
(D) Heatmap of H3K27ac changes for mutants across deciles of enhancers sorted by WT mCA or mCG sites per kilobase.
(E) Overlap of significantly dysregulated genes (padj. < 0.1) in DNMT3AKO/+ and genes dysregulated in DNMT3A Baf53b-cKO or MeCP2 mutants (***p < e−5, ****p < e−10; hypergeometric test, observed versus background estimated by resampling; STAR Methods).
(F) Significance of gene set expression changes in DNMT3AKO/+ for GAGE analysis of gene sets dysregulated in DNMT3A Baf53b-cKO or MeCP2 mutants (Clemens et al., 2019). Boxplots indicate median and quartiles. Bar graphs indicate mean and SEM.
See also Figures S6 and S7.
To determine whether mCA reductions at MeCP2-repressed enhancers affects their activity, we performed H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq) analysis of the DNMT3AKO/+ and wild-type cortex. This analysis revealed significant acetylation changes at MeCP2-repressed enhancers (Figures 6A and 6C). Consistent with these effects arising from partial loss of mCA that normally builds up post-mitotically at enhancers, we detect changes that are concordant with but smaller than effects observed upon complete mCA ablation in a neuron-specific DNMT3A cKO mouse (DNMT3A Baf53b-cKO) (Clemens et al., 2019; Figure 6C). We also find that enhancers within high-mCA TADs show significantly higher increases in acetylation compared with enhancers in other TADs (Figure S6G), further supporting a model in which high mCA levels in TADs lead to repression of enhancers in these TADs by mCA and MeCP2.
Although significantly dysregulated enhancers are detected in MeCP2 mutants, broad sub-significance-threshold effects also occur genome wide upon MeCP2 mutation, with enhancers across the genome undergoing dysregulation that is proportional to the number of mC binding sites they contain (Figure 6D; Clemens et al., 2019). Analysis of H3K27ac changes at enhancers genome wide based on the normal density of mCA sites they contain revealed broad mCA-associated derepression of enhancers in the DNMT3AKO/+ cortex that is similar to but smaller in magnitude than effects in DNMT3A Baf53b-cKO and MeCP2 knockout (KO) mice. These effects are also reciprocal to effects observed in MeCP2 overexpression (OE) mice. Consistent with limited global mCG disruption in DNMT3AKO/+, there was only a limited association between changes in enhancer activity and the normal level of mCG. This finding contrasts with MeCP2 mutants, where loss of protein binding to mCG and mCA sites leads to enhancer dysregulation associated with mCA and mCG (Clemens et al., 2019; Figure 6D).
Although there is limited evidence that a global change in mCG affects enhancers in DNMT3AKO/+, we considered that site-specific changes in mCG (DMRs) may have functional effects at these regions. Indeed, we found that enhancers overlapping hypo-CG-DMRs in DNMT3AKO/+ showed subtle but significant increases in acetylation compared with enhancers without a DMR (Figure S6H). These results suggest that site-specific enhancer mCG changes in DNMT3AKO/+ have functional effects.
Our epigenomic analyses demonstrate that loss of many of the normal mCA sites in the DNMT3AKO/+ cortex results in enhancer dysregulation that overlaps with MeCP2 mutant mice. On a more limited scale, loss of CG methylation at specific enhancers can also affect enhancer activity. These findings uncover a role of shared neuronal chromatin pathology between DNMT3A and MeCP2 disorders.
Overlapping Transcriptional Pathology between DNMT3AKO/+, MeCP2 Disorders, and ASD
The epigenetic changes we observe in DNMT3AKO/+ cerebral cortex can have direct consequences on gene expression to drive neurological dysfunction in mice. Furthermore, overlapping enhancer effects in DNMT3AKO/+ and MeCP2 mutants suggest that shared transcriptional pathology may occur upon loss of mCA in DNMT3A disorders and in MeCP2 disorders. We therefore assessed changes in gene expression in DNMT3AKO/+ mice, interrogating whether these effects overlap with those observed upon ablation of mCA in DNMT3A Baf53b-cKO and loss or OE of MeCP2 in MeCP2 mutants. RNA sequencing (RNA-seq) of the DNMT3AKO/+ cerebral cortex identified subtle mRNA and non-coding RNA changes that are consistent in magnitude with small effects observed in other heterozygous NDD models (Fazel Darbandi et al., 2018; Gompers et al., 2017; Katayama et al., 2016; Figures S7A and S7B; Tables S3 and S4).
Gene Ontology analysis of gene expression changes detected terms associated with neurodevelopment processes, such as axon guidance and recognition, that occur before and during the period when DNMT3A deposits mCA in the brain (Figure S7C). This is consistent with a role of DNMT3A in regulating transcriptional programs as neurons mature. Additionally, we see significantly reduced expression of relevant ASD genes, Shank2 and Shank3, which suggests dysfunction at the synaptic level (Table S3) (Guilmatre et al., 2014). These results suggest that genes critical for development of neural circuits are affected in DNMT3AKO/+ mice.
To determine whether transcriptional dysregulation upon disruption of DNMT3A and MeCP2 results in shared effects, we examined the overlap of significantly dysregulated genes across mutant mouse lines. We found that the genes dysregulated in the DNMT3AKO/+ overlap extensively with the gene sets dysregulated in DNMT3A Baf53b-cKO and MeCP2 mutants (Figure 6E). For example, all three models show upregulation of Shroom3, whose mutation is associated with developmental defects in humans (Deshwar et al., 2020), and latrophilin-2, a post-synaptic adhesion molecule with roles in synapse targeting and assembly in multiple brain regions (Anderson et al., 2017; Zhang et al., 2020). Shared dysregulation of genes like these in DNMT3A and MeCP2 disorders may contribute to synaptic dysfunction and disease pathology.
Although a limited gene set is detected as significantly dysregulated in DNMT3AKO/+, we considered whether genome-wide enhancer changes could lead to widespread, subtle dysregulation of gene expression below the significance threshold for detection. Such transcriptional pathology would mirror subthreshold genome-wide effects observed upon loss of neuronal mCA (DNMT3A Baf53b-cKO) and in models of Rett syndrome (MeCP2 KO) and ASD (MeCP2 OE) (Clemens et al., 2019; Gabel et al., 2015). Importantly, evidence from MeCP2 mutants suggests that pathology arises from thousands of small changes in gene expression rather than large changes in a few significant genes (Lavery and Zoghbi, 2019; Kinde et al., 2016). Disruption of the neuronal methylome in DNMT3AKO/+ could similarly lead to numerous gene expression changes that escape statistical significance but still contribute to pathology. To assess whether sub-significance level changes in DNMT3AKO/+ phenocopy the MeCP2 mutant and DNMT3A Baf53b-cKO models, we performed generally applicable gene set enrichment (GAGE) analysis (Luo et al., 2009). GAGE analysis revealed highly significant, concordant changes in gene expression in DNMT3AKO/+ for gene sets dysregulated in DNMT3A Baf53b-cKO and MeCP2 mutants (Figure 6F).
To further explore the model in which loss of methylation and enhancer dysregulation drives DNMT3AKO/+ gene expression changes, we examined enhancers associated with significantly upregulated genes. We found that these enhancers show pronounced loss of mCA sites and display significant increases in enhancer acetylation compared with other enhancers in DNMT3AKO/+ (Figures S7D and S7E). Analysis of genes containing enhancers harboring a hypo-CG-DMR also detected significant upregulation of expression compared with other genes (Figure S7F), suggesting that local changes in mCG contribute to DNMT3AKO/+ transcriptomic changes.
We next explored whether shared gene expression signatures in DNMT3AKO/+ mice extend to models of NDD without established mechanistic links to DNMT3A. GAGE analysis detected significant alterations in genes identified as dysregulated in CHD8 and PTEN mouse models of overgrowth and ASD (Gompers et al., 2017; Katayama et al., 2016; Tilot et al., 2016) as well as the SETD5 NDD model (Sessa et al., 2019; Figure 7A). These findings suggest that overlapping gene dysregulation could underlie common symptomology in individuals with mutations in distinct epigenomic regulatory genes. Extending GAGE analysis to human ASD data, we observed significant changes of gene sets dysregulated in ASD postmortem brains (Gandal et al., 2018; Voineagu et al., 2011) in the DNMT3AKO/+ cortex (Figure 7B) and also detected upregulation of genes linked to ASD from human genetics studies (Abrahams et al., 2013; Banerjee-Basu and Packer, 2010; Figure 7B). In addition, we detected overlap of DNMT3AKO/+ gene dysregulation with neurodevelopmental gene co-expression modules identified in the human brain (Parikshak et al., 2013), including M13, M16, and M17, which increases during cortical development and are enriched for ASD risk genes (Figure S7G). Modules involved in gene regulation that are expressed early in development and decrease over time (M2 and M3) are also increased in DNMT3AKO/+ (Figure S7G). Resampling analysis indicated that significant dysregulation of these mouse and human gene sets in DNMT3AKO/+ is not driven by enriched expression in the cortex (Figure S7H). These findings suggest that the DNMT3AKO/+ mouse shares overlapping transcriptional pathology with gene expression changes underlying NDD.
Figure 7. Gene Dysregulation in DNMT3AKO/+ Overlaps with Effects in Other NDDs.
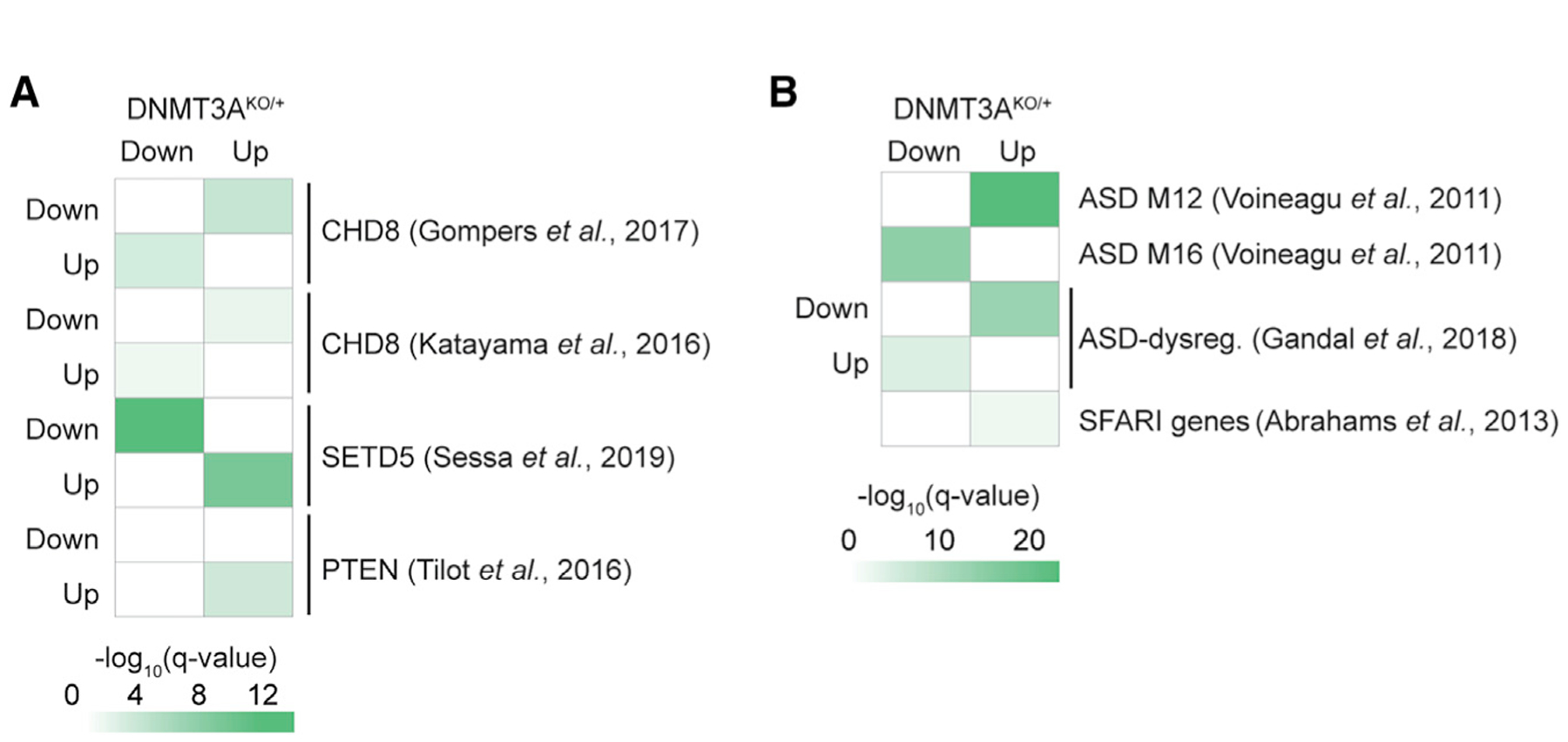
(A) GAGE analysis of expression changes in DNMT3AKO/+ for dysregulated gene sets in studies of NDD mouse models (Gompers et al., 2017; Katayama et al., 2016; Sessa et al., 2019; Tilot et al., 2016).
(B) GAGE analysis of expression changes in DNMT3AKO/+ for gene sets identified in studies of human ASD. ASD modules 12 (synaptic) and 16 (immune) were identified previously in weighted gene coexpression analysis of human ASD brains (Voineagu et al., 2011), and ASD-dysregulated genes were also identified previously (Abrahams et al., 2013; Gandal et al., 2018). See also Figure S7.
DISCUSSION
Our findings here provide an initial working model of molecular etiology in DNMT3A disorders. Diverse de novo missense mutations in affected individuals disrupt the enzyme by disabling chromatin localization in the nucleus, disrupting the ADD-regulatory domain, or impairing methyltransferase activity. Insufficient DNMT3A activity because of these missense mutations, or early truncations and deletions in other individuals, causes deficits in deposition of mCG at specific sites during development and a massive deficit in postnatal mCA accumulation throughout the brain. These changes in DNA methylation lead to alterations in epigenomic regulation, including subtle but widespread disruption of mCA-MeCP2-mediated enhancer regulation in adult neurons, resulting in gene expression changes that contribute to deficits in nervous system function.
Our study provides insights into the molecular effects of DNMT3A mutations and serves as a model to understand the functional effects of diverse de novo mutations underlying NDD. When exome sequencing studies identify heterozygous missense mutations in patients with NDD (McRae et al., 2017; Satterstrom et al., 2019), it can be unclear whether simple haploinsufficiency contributes to pathology or whether more complex functional effects occur because of individual amino acid alterations. In addition, although identification of many mutations in a gene can indicate that disruption of the gene causes NDD, a subset of observed missense mutations may not be deleterious enough to be the pathological driver in the individual in whom they are found. Thus, functional testing of these variants is necessary to determine whether they may underlie disease. In our study, the deleterious effects we observed for each mutation suggest that they are causative, but it was only by assessing multiple aspects of DNMT3A function (i.e., expression, localization, activity, and cellular mCA levels) that we detected deficits for each variant. For example, ADD domain mutations only disrupt deposition of mCA in cells, possibly because of loss of regulation that is only necessary in the endogenous chromatin context. Our findings establish loss of function effects of diverse DNMT3A mutations and underscore the importance of multidimensional analysis of de novo mutations to fully assess their role in NDD.
Our in vivo analyses revealed increased long-bone length, increased body weight, anxiety-like phenotypes, deficits in pro-social communication, and alterations in repetitive behaviors in DNMT3AKO/+ mice that mirror key features of DNMT3A disorders. In contrast, lack of severe deficits in learning and memory assays in the DNMT3AKO/+ mouse may indicate that cognitive processes are more susceptible to DNMT3A disruption in humans than in mice. However, we do detect subtly altered behavior in these assays, and the lack of strong deficits may reflect insensitivity of the methods to measure cognition in mice. Our in vivo analyses uncover effects of heterozygous DNMT3A deletion that can be probed at the molecular, cellular, and organismal levels in future studies.
We employed the DNMT3AKO/+ experimental system to assess how heterozygous DNMT3A loss affects epigenetic regulation in the brain. We detected subtle changes in mCG genome wide across brain regions, with no global mCG effects in non-neural tissue. These limited mCG effects are likely due to redundant function of the maintenance methyltransferase DNMT1, which can preserve existing mCG patterns during cell divisions (Jeltsch et al., 2018). In addition, the de novo methyltransferase DNMT3B is expressed with DNMT3A in many tissues during early development and may provide redundancy for mCG patterning (Okano et al., 1999). The focal loss of mCG we do observe in the brain (i.e., hypo-CG-DMRs) often overlaps with regulatory elements that are methylated postnatally by DNMT3A. This mCG loss has the potential to disrupt gene expression and neurological function in DNMT3AKO/+ mice. In addition, site-specific changes in mCG are likely to occur in early development and in non-neural tissues. For example, constitutive heterozygous deletion of DNMT3A disrupts mCG patterns in the blood and alters hematopoietic lineages (Cole et al., 2017). Such changes in mCG can contribute to growth and other phenotypes observed in mice and humans.
In contrast to mCG, we detect a global 30%–50% reduction in mCA in multiple brain regions of DNMT3AKO/+. These results extend findings from the hypothalamus (Sendžikaite et al., 2019), demonstrating broad sensitivity to heterozygous DNMT3A loss. The susceptibility of mCA to reduced DNMT3A is likely due to DNMT3B not being expressed in postnatal neurons (Lister et al., 2013), and DNMT1 cannot deposit mCA (Jeltsch et al., 2018), making all neuronal mCA build-up dependent on DNMT3A. In addition, DNMT3A has slow kinetics for CA methylation (Zhang et al., 2018), and deposition of mCA genome-wide must take place in a restricted time window (1–6 weeks) when the enzyme is highly expressed in neurons (Lister et al., 2013). These constraints may make enzyme levels limiting for mCA accumulation in neurons and explain the sensitivity to DNMT3A gene dosage. Notably, our findings suggest that activating the remaining DNMT3A or prolonging its high postnatal expression might rescue mCA deposition deficits. Conversely, duplication of the DNMT3A gene could result in too much mCA deposition and cause neural dysfunction akin to MeCP2 duplication disorder. Future studies can assess the feasibility of rescue approaches and explore whether DNMT3A duplication alters brain function.
Our analysis of chromatin changes downstream of altered DNA methylation has uncovered a striking point of shared molecular disruption across models of DNMT3A disorders, Rett syndrome, and MeCP2 duplication syndrome. Although the clinical profile of DNMT3A disorders is distinct from MeCP2 disorders, we show here that loss of approximately a quarter of MeCP2 binding sites in the DNMT3AKO/+ cortex results in subtle but widespread disruption of mCA-associated enhancer regulation that partially phenocopies loss of MeCP2. This enhancer dysregulation is further linked to shared gene expression changes in these models. Given the critical roles of MeCP2-regulated genes in nervous system function (Gabel et al., 2015; Lagger et al., 2017), these overlapping epigenomic and transcriptomic effects likely contribute to neurological dysfunction in DNMT3A disorders.
Overall, the deficits observed in the DNMT3AKO/+ mice are less severe than in DNMT3A cKO and MeCP2 knockout mice, which present with decreases in motor coordination, severely reduced body size, and increased mortality (Nguyen et al., 2007; Tillotson et al., 2017). These differences mirror differential effects on mCA deposition and readout in these strains; DNMT3A cKO loses all neuronal mCA and MeCP2 KO loses a major reader of mCA and mCG, whereas DNMT3AKO/+ loses only half of the mCA sites and a localized set of mCG sites. The persistence of many mCA and mCG binding sites for MeCP2 in DNMT3AKO/+ may partially explain how DNMT3A mutations in humans manifest with less severe symptomology than in Rett syndrome. Absence of DNMT3A early in prenatal development and loss of DNMT3A functions that are independent of MeCP2 (Lavery et al., 2020) are likely to drive overgrowth and other non-overlapping aspects of DNMT3A and MeCP2 disorders. Our findings show that disruption of enhancer regulation by mCA-MeCP2 likely contributes to multiple distinct disorders, defining a site of convergent molecular etiology underlying heterogeneous clinical syndromes.
Our transcriptomics analysis of changes in ASD/NDD gene sets in DNMT3A mice has detected overlap beyond MeCP2 disorders, including mouse NDD/ASD models (e.g., CHD8) and gene sets identified in human ASD. As additional NDD transcriptomics studies of mouse and human brain emerge, systematic analyses can identify shared aspects of transcriptional pathology across diverse causes of NDD. Notably, the large number of chromatin-modifying enzymes mutated in NDD suggests that shared transcriptomic effects may emerge from common chromatin pathology. Future studies may identify additional gene disruptions in which alterations in mCA and enhancer dysregulation contribute to molecular pathology, expanding the role of “methylopathies” in NDD.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for reagents and resources should be directed toward the Lead Contact, Harrison Gabel (gabelh@wustl.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
DOIs for all published gene sets used in comparison and enrichment analysis:
The accession number for the raw and aggregate data for bisulfite-seq, raw and gene-count data for RNA-seq, and raw and peak call data for ChIP-seq reported in this paper is GEO: GSE147899.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal husbandry
All animal protocols were approved by the Institutional Animal Care and Use Committee and the Animal Studies Committee of Washington University in St. Louis, and in accordance with guidelines from the National Institutes of Health (NIH). Mice were housed in a room on a 12:12 hour light/dark cycle, with controlled room temperature (20–22°C) and relative humidity (50%). Home cages measured 28.5 cm × 17.5 cm × 12 cm and were supplied with corncob bedding and standard laboratory chow and water. All mice were group-housed and adequate measures were taken to minimize animal pain or discomfort.
Transgenic animals
Male and female homozygous Dnmt3aflx/flx mice (Kaneda et al., 2004) were bred together for viral-mediated DNMT3A replacement assay culture experiments. To generate the DNMT3A heterozygous mouse model, Dnmt3aflx/flx mice were crossed to CMV:Cre (B6.C-Tg(CMV-cre)1Cgn/J) to generate Dnmt3aKO/+Cre:CMV+/− offspring. Dnmt3aKO/+Cre:CMV+/− progeny were bred to C57BL/6J to outcross the cre recombinase and generate experimental genotype (DNMT3AKO/+). Mice were genotyped with ear-DNA by PCR for Dnmt3a and Cre, and recombination was tested. Subsequent experimental animals were generated from Dnmt3aKO/+ males mated to C57BL/6J females to generate Dnmt3aKO/+ and Dnmt3a+/+ experimental and control animals for experiments. Dnmt3aKO/+ females were not used for breeding to avoid social differences in mothering from mutant dams. Mice were weighed at a variety of time points to assess growth.
METHOD DETAILS
Immunocytochemistry
Staining
Neuro-2a cells (ATCC, CCL-131) were grown on coverslips and transfected with FLAG-tagged WT or mutant mouse DNMT3A plasmids and GFP plasmid. This FLAG-tag allows for quantification of transfected DNMT3A protein only instead of endogenous DNMT3A. Coverslips were fixed with 4% paraformaldehyde in PBS for 20 minutes, permeabilized with 0.1% Triton X-100 in PBS for 10 minutes, and blocked with 1% BSA in PBS for 1 hour all at room temperature. Coverslips were incubated overnight at 4°C in anti-DDDDK tag (FLAG-tag) primary antibody (Abcam, 1:5000, ab1162). Coverslips were then washed in PBS and incubated for 1 hour at room temperature with fluorescent secondary antibody (ThermoFisher, 1:500, A-11011) and counterstained with DAPI.
Imaging
Images were captured using a Nikon A1Rsi confocal microscope with a 20× air objective. Laser settings were kept constant for each image.
Analysis/Quantification
Cells were counted using an automatic threshold in FIJI and manually classified as displaying nuclear or non-nuclear signal by a blinded observer. This was determined by evaluating the overlap of FLAG signal (DNMT3A) with DAPI signal (nucleus). For mutants that did not reach expression levels comparable to the WT or for images that had too few positive cells, cell number was counted manually. 8 separate transfections were run, with each mutant being counted over 3 or more independent experiments. Sample sizes are as follows: WT, 15 images, 880 cells; W297del, 8 images, 435 cells; I310N, 8 images, 492 cells; S312fs11x, 12 images, 321 cells; G532S, 9 images, 695 cells; M548K, 7 images, 333 cells; V665L, 6 images, 635 cells; Y735C, 16 images, 613 cells; R749C, 8 images, 667 cells; P904L, 7 images, 692 cells. While some mutants showed reduced protein expression, all proteins tested showed adequate signal assessed as nuclear or non-nuclear. Percent nuclear was assessed per image and a generalized linear model was run comparing each mutant to WT. P values for each mutant were then Bonferroni corrected. Localization to the nuclear periphery was evident for the WT FLAG-tagged protein and all nuclear-localized mutants, but no consistent differences in this localization was observed between WT and any mutants. We chose to use a generalized linear model with Bonferroni correction to allow for us to compare ratios of percent nuclear signal while accounting for experimental and bioreplicates.
Modeling of DNMT3A disease mutations
HEK293T (ATCC, ACS-4500) or Neuro-2a cells (ATCC, CCL-131) were transfected with GFP and FLAG-tagged WT or mutant mouse DNMT3A plasmids. Collected cell lysates were ruptured by 3 freeze/thaw cycles using liquid nitrogen, or sonication ~42 hours after transfection. Samples were then either used for western blotting (Neuro-2a cells), the in vitro radioactive methyltransferase assay (HEK293T cells).
qRT-PCR
RNA isolated from neuronal cortical culture or mouse brain tissue was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Dnmt3a and Actb were measured by qPCR using the Power SYBR Green PCR Master Mix and primers Actb (F:AAGGCCAACCGTGAAAAGAT, R:GTGGTACGACCAGAGGCATAC) and Dnmt3a (F:GGCCTTCTCGACTCCA-GATG, R:TTCCTCTTCTCAGCTGGCAC). Relative quantity of Actb and Dnmt3a cDNA was determined by comparing the Ct of each primer set in each sample to a standard curve and then normalizing the DNMT3A signal by the ACTB signal. We chose to compare experimental conditions to WT samples using Student’s T-Tests with Bonferroni correction, as these are two normally distributed groups (visually checked) with similar variability.
In vitro radioactive methyltransferase assay
30 μl of cell lysate from HEK293T cells was used in the methyltransferase reaction previously described (Russler-Germain et al., 2014). Lysates were incubated at 37°C for 20 hours in 5 μl reaction buffer of 20 mM HEPES, 30 mM NaCl, 0.5 mM DTT, 1 mM EDTA, 0.2 mg/ml BSA, 5 mM 3 H-labeled SAM (PerkinElmer, NET155050UC) and 500 ng/μl Poly(dI-dC) substrate (Sigma P4929). Substrate was purified (Macherey-Nagel NucleoSpin Gel and PCR Clean-up) and radioactivity measured using a scintillation counter. In instances where DNMT3A mutant showed altered protein expression, cell lysate was re-balanced to match protein expression of WT DNMT3A. The activity of endogenous DNMT3A present in the cells was accounted for by normalizing values to GFP alone. Only experimental replicates where WT DNMT3A showed a 1.5-fold increase compared to GFP alone were used for subsequent analysis. Outliers beyond 2 standard deviations above or below the mean were removed. Number of independent replicates are as follows: W297del, 18; I310N, 19; S312fs11x, 4; G532S, 10; M548K, 15; V665L, 11; Y375C, 13; R749C, 7; P904L, 14. Significance was assessed using a one-sample Student’s t test, as we are comparing groups normalized to WT and GFP back to the normalized value of 1.
Viral-mediated DNMT3A replacement assay
Functional activity of DNMT3A mutants in cortical neurons was determined by measuring methylation build-up in vitro. Timed mating of DNMT3Aflx/flx females and DNMT3Aflx/flx males was performed to collect embryonic cortical DNMT3Aflx/flx neurons at embryonic day 14.5. At E14.5, DNMT3Aflx/flx cortical neurons from both male and female embryos were rapidly isolated and plated (DIV 0). On DIV 3, neurons were either not perturbed or virally transduced with one of three conditions: 1) Cre only, 2) Cre and WT DNMT3A, or 3) Cre and mutant DNMT3A. DNA and RNA were isolated on DIV 12.5 using the AllPrep DNA/RNA Kit (QIAGEN, 80204). DNA was used for whole genome bisulfite sequencing, and RNA was used for qRT-PCR for DNMT3A. Results of pilot experiments (not shown) indicate that at this time point mCA is not saturated and sensitive to total levels of DNMT3A activity. Number of independent replicates are as follows: W297del, 5; S312fs11x, 4; G532S, 7; M548K, 6; V665L, 10; Y375C, 11; R749C, 6; P904L, 8. Buildup of methylation over development was carried out without performing any viral transduction. Significance was assessed using a one-sample Student’s t test or 1-way ANOVA.
Ultrasonic vocalization and analysis
A total of 76 DNMT3AKO/+ (n = 30, 16 male and 14 female) and litter-matched WT (n = 46, 25 male and 21 female) mice were used for ultrasonic pup vocalization (USV) recording and analyzed as previously described (Barnes et al., 2017; Dougherty et al., 2013; Holy and Guo, 2005). Dams were removed from the nest for a 10-minute acclimation, and individual pups had their body temperature measured using an infrared laser thermometer. Pups were then removed from their nest and placed in a dark, enclosed chamber. Ultrasonic vocalizations were recorded for 3 minutes with an Avisoft UltraSoundGate CM16 microphone and 416H amplifier using Avisoft Recorder software (gain = 6 dB, 16 bits, sampling rate = 250 kHz). Pups were then weighed and returned to their nest and littermates. All mice were recorded at postnatal days 5, 7, and 9, and on either day 11 or 15. Frequency sonograms were prepared and analyzed in MATLAB (frequency range = 40 kHz to 120 kHz, FFT size = 256, overlap = 50%) with individual syllables identified and counted according to previously published methods (Dougherty et al., 2013; Holy and Guo, 2005). Significance was assessed using a within-subjects repeated-measures ANOVA over time points 5–9, as these were when there was data from all experimental subjects, and these are optimal testing times where number of calls was highest.
Marble burying
A total of 27 DNMT3AKO/+ (n = 13, 8 male and 5 female) and litter-matched WT (n = 14, 7 male and 7 female) mice were used for marble burying. Marble burying is a natural murine behavior and has been used to indicate repetitive digging as well as anxiety-related behaviors. Protocol was adapted from previously published methods (Lazic, 2015; Maloney et al., 2019a). In brief, 8-week old mice were placed in a transparent enclosure (28.5 cm × 17.5 cm × 12 cm) with clean aspen bedding and 20 dark blue marbles evenly spaced in a 4 × 5 grid on top of the bedding. Animals explored freely for 30 minutes. The number of buried marbles were counted every 5 minutes by two independent blinded observers. Marbles were considered “buried” if they were at least two-thirds covered by bedding. Enclosure and marbles were cleaned thoroughly between animals. Significance was assessed using a within-subjects repeated-measures ANOVA to determine if rate of burying marbles is different between genotypes. These statistical methods are more appropriate than a simple t test at 30 minutes, as mice may have buried all marbles before this time point, and significant changes in marble burying behavior may have occurred at earlier time points in the assay.
Adult behavioral battery
A total of 72 DNMT3AKO/+ (n = 39, 18 male and 21 female) and litter-matched WT (n = 33, 15 male and 18 female) mice were used for adult behavioral testing. Mice were housed in mixed genotype home cages with 2–5 animals per cage, and all tests were performed during the light cycle. All experimenters were blinded to genotype during testing. For increased experimental rigor and reproducibility, we used three separate cohorts of mice to ensure quality and consistency in any observed phenotypes.
Testing started when mice were 3–4 months of age. The sequence of behavioral testing was designed to minimize carry-over effects across behavioral tests. Cohorts, ages, and testing order are in Table S5. Most assays were performed on cohorts 1 and 2 with cohort 3 being performed to test for reproducibility in some assays. Because of differences in testing sequences and exposure of mice to prior tests between cohorts, we examined separate cohorts individually and looked at combined cohorts (Table S5). Testing was performed by the Washington University in St. Louis Animal Behavior Core.
One-hour locomotor activity
Locomotor activity was evaluated by computerized photobeam instrumentation in transparent polystyrene enclosures (47.6 cm × 25.4 cm × 20.6 cm) as previously described (Wozniak et al., 2004). Activity variables such as ambulations and vertical rearings were measured in addition to time spent in a 33 cm × 11 cm central zone.
Sensorimotor battery
Mice were assayed in walking initiation, balance (ledge and platform tests), volitional movement (pole and inclined screens), and strength (inverted screen) as previously described (Grady et al., 2006; Wozniak et al., 2004). For the walking initiation test, mice were placed on the surface in the center of a 21 cm × 21 cm square marked with tape and the time for the mouse to leave the square was recorded. During the balance tests, the time the mouse remained on an elevated plexiglass ledge (0.75 cm wide) or small circular wooden platform (3.0 cm in diameter) was recorded. During the Pole test, mice were placed at the top of a vertical pole with nose pointing upward. The time for the mouse to turn and climb down the pole was recorded. For the inclined screen tests, a mouse was placed (oriented head-down) in the middle of an elevated mesh grid measuring 16 squares per 10 cm angled at 60° or 90°. Time for the mouse to turn 180° and climb to the top was recorded. For the inverted screen test, a mouse was placed on a similar screen and when the mouse appeared to have a secure grasp of the screen, the screen was inverted 180° and the latency for the mouse to fall was recorded. All tests had a duration of 60 s, except for the pole test which was 120 s. Two separate trials were done on subsequent days and averaged time of both trials was used for analysis. Data from the walking initiation, ledge, and platform tests were not normally distributed and therefore analyzed using Mann-Whitney U tests.
Continuous and accelerating rotarod
Motor coordination and balance were assessed using the rotarod test (Rotamex-5, Columbus Instruments, Columbus, OH) with three conditions: a stationary rod (60 s maximum), a rotating rod at constant 5 rpm (60 s maximum), and a rod with accelerating rotational speed (5 – 20 rpm, 180 s maximum) as previously described (Grady et al., 2006). This protocol is designed to minimize learning and instead measure motor coordination, so testing sessions were separated by 4 days to allow for extinction. Testing included one trial on stationary rod, and two trials on both the constant-speed rotarod and accelerating rotarod. Later time points in the constant speed rotarod test failed tests of normality, as the majority of mice stayed on the rotating rod for all 60 s. However, data were analyzed with two-way repeated-measures ANOVA.
Morris water maze
Spatial learning was assessed as previously described (Wozniak et al., 2004). Cued trials (visible platform, variable location) and place trials (submerged, hidden platform, consistent location) were conducted in which escape path latency, length, and swimming speeds were recorded. Animal tracking was done using a computerized system (ANY-maze, Stoelting). During cued trials, animals underwent 4 trials per day over 2 consecutive days with the platform being moved to a different location for each trial with few distal spatial cues available. Each trial lasted no longer than 60 s, with a 30-minute interval between each trial. Performance was analyzed across four blocks of trials (2 trials/block). After a three-day rest period, animals were tested on place trials, in which mice were required to learn the single location of a submerged platform with several salient distal spatial cues. Place trials occurred over 5 consecutive days of training, with 2 blocks of 2 consecutive trials (60 s trial maximum, 30 s inter-trial-interval after the mouse has reached the platform) with each block separated by 2 hours. Mice were released into different quadrants over different trials. Place trials were averaged over each of the five consecutive days (4 trials/block). One hour after the final block, a probe trial occurred (60 s trial maximum) in which the platform is removed, and the mouse is released from the quadrant opposite where the platform had been located. The time spent in pool quadrants, and the number of crossings over the exact platform location were recorded. DNMT3AKO/+ mice showed a small, but significant reduction in target zone time in cohort 2, though there was no difference in cohort 1. Additionally, female mice had significantly faster swimming speeds than male mice across both genotypes (Table S5).
Elevated plus maze
Anxiety-like behaviors were examined using the elevated plus maze as previously described (Boyle et al., 2006). The apparatus contains a central platform (5.5 cm × 5.5 cm) with two opposing open arms and two opposing closed arms (each 36 cm × 6.1 cm × 15 cm) constructed of black Plexiglas. Mouse position is measured using beam-breaks from pairs of photocells configured in a 16 × 16 matrix and outputs are recorded using an interface assembly (Kinder Scientific) and analyzed using software (MotoMonitor, Kinder Scientific) to determine time spent, distance traveled, and entries made into open arms, closed arms, and the center area. Test sessions were conducted in a dimly lit room with each session lasting 5 minutes and each mouse tested over 3 consecutive days. Data shown are from day 1. All mice showed a decrease in time, distance, and entries into open arms on days 2 and 3. There was no significant difference between genotypes in percent entries into open arms (Figure S4K; p = 0.137; unpaired Student’s T-Test) or total entries into arms (data not shown), indicating that both genotypes explored the maze. Percent distance traveled in open arms showed similar effects to percent time in open arms (Percent distance traveled: p = 0.027; unpaired Student’s T-Test). Analysis of these data in individual cohorts detected DNMT3AKO/+ significant effects for the percent of open arm time on the first day in cohorts 1 and 3, with no evidence of an effect in cohort 2. Individual cohorts also showed no significant difference between genotypes in percent open arm entries (Table S5) suggesting that mice explored the elevated plus maze sufficiently to detect anxiety-like behaviors.
Acoustic startle/prepulse inhibition
Sensorimotor gating was evaluated as previously described (Dougherty et al., 2013; Gallitano-Mendel et al., 2008; Hartman et al., 2001). In short, mice were presented with an acoustic startle response (120 dB auditory stimulus pulse, 40 ms broadband burst) and a pre-pulse (response to pre-pulse plus startle pulse). Stimulus onset began at 65 s, and 1ms force readings were obtained and averaged to produce an animal’s startle amplitude. 20 startle trials were presented in 20 minutes. The first 5 minutes were an acclimation period where no stimuli above the 65 dB background were presented. The session started and finished with 5 consecutive startle (120 dB pulse) trials. The middle 10 trials were interspersed with pre-pulse trials, consisting of an additional 30 presentations of 120 dB startle stimuli preceded by pre-pulse stimuli of 4, 12, or 20 dB above background (10 trials for each PPI trial type). To calculate percent pre-pulse inhibition, we used %PPI = 100 × (ASRstartle pulse alone − ASRprepulse + startle pulse)/ASRstartle pulse alone.
Conditioned fear
Fear conditioning was done as previously described (Maloney et al., 2019a, 2019b). Mice were habituated to an acrylic chamber (26 cm × 18 cm × 18 cm) containing a metal grid floor and an odorant and was illuminated by LED light which remained on for the duration of the trial. Day 1 testing lasted 5 minutes in which, after a brief baseline period with no tone, an 80 dB tone sounded for 20 s at trial time points 100, 160, and 220 s. A 1.0 mA shock (unconditioned stimulus) occurred within the last 2 s of the tone (conditioned stimulus). Baseline freezing behavior during the first 2 minutes and the freezing behavior during the last 3 minutes was quantified using image analysis (Actimetrics, Evanston, Illinois). On Day 2, testing lasted for 8 minutes in which the light was illuminated but no tones or shocks were presented. On Day 3, testing lasted for 10 minutes in which the mouse was placed in an opaque chamber with a different odorant than the original test chamber. After a brief baseline period with no tone, the 80 dB tone began at 120 s and lasted for the remainder of the trial and freezing behavior to the conditioned auditory stimulus was quantified for the remaining 8 minutes. DNMT3AKO/+ mice show elevated freezing levels during training and testing, which may reflect a baseline propensity to freeze, stronger fear conditioning, or an emotional hypersensitivity to the foot-shock. Additionally, our data suggest that the increased freezing in the DNMT3AKO/+ mice does not appear to be due to an increased pain response, as mutant mice showed similar levels of shock sensitivity. Evaluation of baseline freezing levels in individual cohorts only showed significance in one of the two cohorts tested.
3-Chamber social approach
Sociability was assayed as previously described (Moy et al., 2004; Silverman et al., 2011). Mice were tested in a rectangular all-Plexiglas apparatus (each chamber measuring 19.5 cm × 39 cm × 22cm) divided into three chambers with walls containing rectangular openings (5 cm × 8 cm) and sliding doors. The apparatus was in a room with indirect light and was cleaned between tests with Nolvasan solution. Stimulus mice were contained within a small stainless-steel withholding cage (10 cm height × 10 cm diameter; Galaxy Pencil/Utility Cup, Spectrum Diversified Designs), allowing minimal contact between mice without allowing fighting. Between tests, withholding cages were cleaned with 75% ethanol solution. A digital video camera recorded movement of the mouse within the apparatus and allowed for tracking with ANY-maze (Stoelting). Distance and time spent in each chamber and investigation zones surrounding the withholding cages were recorded. Zones were defined as 12 cm in diameter from the center of withholding cages.
The test sequence consisted of 4 consecutive 10-minute trials in which the test mouse is placed in the middle chamber and allowed to freely explore the environment. In the first trial, the mouse is placed in the middle chamber with the doors to other chambers shut. In the second trial, the mouse is placed in the middle chamber and can explore all three chambers of the task, allowing it to acclimate to the environment. Neither genotype tested showed a preference toward a side of the chamber during this habituation. For the third trial, a sex-matched novel conspecific was placed within a withholding cage with the other cage remaining empty. For the fourth trial, the same sex-matched conspecific was in one withholding cage, while a new unfamiliar sex-matched stimulus mouse was placed in the other withholding cage. The locations of stimuli mice were counterbalanced across groups for the third trial and randomized novel or familiar for the fourth trial.
Reduced anxiety 3-chamber social approach
An add-on cohort was used to investigate sociability in a low-light environment with minimal distractions and stressors. A total of 37 DNMT3AKO/+ (n = 18, 9 male and 9 female) and litter-matched WT (n = 19, 9 male and 10 female) mice 10–12 weeks of age were used. These mice had not been used for previous behavioral analysis. Two additional DNMT3A mice were removed from the cohort prior to analysis, as they appeared sickly and showed reduced movement. Social approach was carried out as described previously (Manno et al., 2020). In brief, mice were tested in a rectangular clear acrylic apparatus (60 cm × 40.5 cm) separated into three equally sized chambers divided by walls with sliding doors (6 cm × 6 cm), and with one cup placed in each of the edge chambers. The apparatus was in an isolated, quiet room with minimal sound and low-light (270 lux). Three 10-minute phases were carried out. In the first phase, the test mouse freely explored all chambers of the apparatus. In the second phase, a sex-matched conspecific mouse was added to the one of the cups in the side chambers, and the test mouse was allowed to freely explore. In the third phase, a sex-matched novel conspecific was added to the remaining empty cup and the test mouse was allowed to explore. Between experimental mice, the apparatus was cleaned with 70% ethanol. A digital video camera recorded the sessions and mouse location was measured. For analysis, only the first 5 minutes of each phase was used, as mice rapidly habituate to this task (Manno et al., 2020).
Tissue collection
Brain tissue was dissected from DNMT3AKO/+ and WT littermate mice in ice-cold PBS, flash-frozen in liquid nitrogen, and stored at − 80°C.
Western blotting
Western blotting from cell culture
Neuro-2a cells were collected and combined with 2× laemmli buffer with 5% β-mercaptoethanol. Samples were passed through a Wizard Column (Fisher, Wizard Minipreps Mini Columns, PRA7211), boiled for 5 minutes, and run on a BioRad 4%–12% acrylamide gel at 125 V for 60 minutes. Samples were then transferred to a nitrocellulose membrane, which was bisected between 37kDa and 50kDa bands. Membranes were blocked with 3% bovine serum albumin in TBS-T for 1 hour at room temperature and then the lower membrane was immunostained with anti-GFP (ThermoFisher, 1:2000, A-11122) while the upper membrane was immunostained with anti-DDDDK (Abcam, 1:1000, ab1162) for 12–16 hours at 4°C. All primary and secondary antibodies were diluted in 3% Bovine Serum Albumin in TBS-T. Membranes were next washed with TBS-T, and then incubated for 1 hour at room temperature with IR-dye secondary antibody (IRDye 800CW Donkey anti-Rabbit, LI-COR Biosciences, 1:15,000, product number: 926–32213). Blots were then washed with PBS, imaged using the LiCOR Odyssey XCL system, and quantified using Image Studio Lite software (LI-COR Biosciences). FLAG (DDDDK) and GFP levels were normalized to a standard curve, and protein levels are expressed as normalized DDDDK values divided by normalized GFP values to enable comparison of FLAG (DDDDK) levels between blots. Each blot included a standard curve and WT samples. Outliers beyond 2 standard deviations above or below the mean were removed. Number of independent replicates are as follows: WT, 29; W297del, 7; I310N, 7; S312fs11x, 12; G532S, 7; M548K, 9; V665L, 7; Y375C, 8; R749C, 6; P904L, 7. Significance was assessed using a one sample Student’s T-Test, as protein expression levels were normalized to GFP and WT, and mutant protein expression was compared to the normalized WT value of 1.
Western blotting from tissue
Brain tissue samples were homogenized with a dounce homogenizer in buffer with protease inhibitors (10mM HEPES pH 7.9, 10mM KCl, 1.5mM MgCl2, 1mM DTT, 10mM EDTA). A portion of the lysate was removed and 1% SDS was added. Samples were boiled for 10 minutes, followed by a 10-minute spin at 15,000 g. Supernatant was collected and run through a Wizard Column (Fisher, Wizard Minipreps Mini Columns, PRA7211), then diluted in LDS sample buffer with 5% β-mercaptoethanol. Samples were boiled for 5 minutes, run on an 8% acrylamide gel for 60 minutes at 125 V, and transferred to a nitrocellulose membrane. Membrane was bisected between 75kDa and 100kDa. Membranes were blocked with 3% bovine serum albumin in TBS-T for 1 hour at room temperature, and the upper membrane was immunostained with anti-DNMT3A (Abcam, 1:1000, ab13888) while the lower membrane was immunostained with anti-α-Tubulin (Abcam, 1:1000, ab52866) for 12–16 hours at 4°C. All primary and secondary antibodies were diluted in 3% Bovine Serum Albumin in TBS-T. Membranes were next washed with TBS-T, and then incubated for 1 hour at room temperature with IR-dye secondary antibody (IRDye 800CW Goat anti-Rabbit, or IRDye 800CW Goat anti-Mouse, LI-COR Biosciences, 1:15,000, product numbers: 926–32211 and 926–32210 respectively). Blots were then washed with PBS, imaged using the LiCOR Odyssey XCL system, and quantified using Image Studio Lite software (LI-COR Biosciences). DNMT3A and α-Tubulin levels were normalized to a standard curve, and protein levels are expressed as normalized DNMT3A values divided by normalized α-Tubulin values to enable comparison of DNMT3A levels between blots. For brain region analysis, sample sizes of n = 4 per genotype (2 male and 2 female pairs) were used. For time course analysis, sample sizes of n = 2 per genotype (1 male and 1 female pairs) were used for all time points except the 2-week time point in which n = 6 (3 male and 3 female pairs) was used. Significance was assessed using a Student’s T-Test for brain regions, and a two-way ANOVA considering genotype and time to determine if there was a detectable difference in protein expression over development.
Bisulfite sequencing
Whole genome bisulfite sequencing from cortical cultures
Samples were chosen for whole genome bisulfite sequencing if mutant and WT samples expressed equal amounts of Dnmt3a mRNA as measured by qRT-PCR (Figure S1F). DNA from cortical cultures was bisulfite converted and prepared for sequencing using the Tecan Ovation Ultralow Methyl-Seq Kit (Tecan, 0335–32) and the Epitect Bisulfite Kit (QIAGEN, 59824) was used for bisulfite conversion. We used alternate bisulfite conversion cycling conditions ([95°C, 5 min; 60°C, 20 min] × 4 cycles, 20°C hold) to ensure lowest possible bisulfite non-conversion rate. Libraries were PCR-amplified for 10–11 cycles. Libraries were then pooled and sequenced at a depth of 0.01–0.03× genomic coverage using an Illumina MiSeq 2×150 through the Spike-In Cooperative at Washington University in St. Louis. Significance was assessed using a one-sample Student’s t test, as we are comparing groups normalized to WT and GFP back to the normalized value of 1.
Whole genome bisulfite sequencing from tissue
DNA was isolated from tissue using the DNEasy Kit (QIAGEN). 300 ng of DNA was prepared for sequencing using the Ovation Ultralow Methyl-Seq Kit (Tecan, 0335–32) with and the Epitect Bisulfite Kit (QIAGEN, 59824) was used for bisulfite conversion. For these samples, 300 ng of DNA was fragmented for 45 s with the Covaris E220 sonicator (10% Duty Factory, 175 Peak Incidence Power, 200 cycles per burst, milliTUBE 200μL AFA Fiber). DNA was then purified using 0.7 volumes of Agencourt Beads to select for long DNA inserts for sequencing. We used alternate bisulfite conversion cycling conditions ([95°C, 5 min; 60°C, 20 min] × 4 cycles, 20°C hold) to ensure lowest possible bisulfite non-conversion rate. Libraries were PCR-amplified for 12 cycles. Libraries were then pooled and sequenced using an Illumina MiSeq 2×150 through the Spike-In Cooperative at Washington University in St. Louis. Samples for shallow-depth sequencing (Figures 4A and 4B) were sequenced at 0.01–0.03× genomic coverage. For brain region and liver methylation, n = 2 per genotype per region (one male pair, one female pair). For developmental time course methylation, n = 3–4 per genotype per time point, with at least one male and one female pair. 8-week cortex samples for deep sequencing (Figures 5 and 6A–6D; Figures S6D–S6H) were sequenced at 25–27× coverage per genotype using two male and two female WT-DNMT3AKO/+ pairs (n = 4 per genotype). Samples were sequenced either using the HiSeq 3000 or NextSeq 500 at 2×150 or 1×150, respectively. For shallow sequencing experiments, significance was assessed using either a two-sample Student’s T-Test to compare global methylation values of the cortex between two genotypes or using a two-way ANOVA to compare broad methylation changes across a variety of brain regions or time points. Genomic element comparisons were done using two-sample Student’s T-Tests with Bonferroni correction.
Oxidative bisulfite sequencing from tissue
DNA was isolated from tissue using the DNEasy Kit (QIAGEN, 69504). 450 ng of DNA was prepared for sequencing using the Ovation Ultralow Methyl-Seq Kit (Tecan, 0335–32) with TrueMethyl oxBS plugin (Tecan, 0414–32). For these samples, 450 ng of DNA was fragmented for 45 s with the Covaris E220 sonicator (10% Duty Factory, 175 Peak Incidence Power, 200 cycles per burst, milliTUBE 200μL AFA Fiber). DNA was then purified using 0.7 volumes of Agencourt Beads to select for long DNA inserts for sequencing. 2/3 of the sample (~300 ng of DNA) was used for OxBS libraries, whereas the remaining 1/3 (~150 ng of DNA) was used for bisulfite libraries. We used alternate bisulfite conversion cycling conditions ([95°C, 5 min; 60°C, 20 min] × 2 cycles; 95°C, 5 min; 60°C, 40 min; 95°C, 5 min; 60°C, 45 min; 20°C hold) to ensure lowest possible bisulfite non-conversion rate. Bisulfite and oxidative bisulfite libraries were PCR-amplified for 11 and 13 cycles respectively. Libraries were then pooled and sequenced using an Illumina MiSeq 2×150 through the Spike-In Cooperative at Washington University in St. Louis. Samples were sequenced at 0.8–2.2× genomic coverage per replicate (two male replicates per genotype). Genomic element comparisons were done using two-sample Student’s T-Tests with Bonferroni correction.
Chromatin immunoprecipitation protocol
Chromatin immunoprecipitation was performed as previously described (Clemens et al., 2019; Cohen et al., 2011). Cerebral cortex was dissected on ice in PBS from DNMT3AKO/+ and WT littermates at 8-weeks old (n = 5 pairs, 3 male, 2 female). The tissue was flash-frozen in liquid nitrogen and stored at −80°C. Chromatin was fragmented with the Covaris E220 sonicator (5% Duty Factory, 140 Peak Incidence Power, 200 cycles per burst, milliTUBE 1mL AFA Fiber). ChIP was performed with H3K27ac antibody (0.025–0.1μg; Abcam, ab4729) and libraries were generated using Ovation Ultralow Library System V2 (Tecan, 0344NB-32). Libraries were sequenced using Illumina HiSeq 3000 with the Genome Technology Access Center at Washington University in St. Louis, typically yielding 15–40 million single-end reads per sample.
RNA sequencing
Total RNA isolation was carried out as previously described (Clemens et al., 2019). In brief, cerebral cortex was dissected in ice-cold PBS from DNMT3AKO/+ and WT littermates at 8 weeks of age (n = 7 pairs, 3 male, 4 female). Cortex was lysed in RLT buffer following the RNeasy Mini Kit (QIAGEN, 74104). RNA libraries were generated from 250 ng of RNA with NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB) using a modified amplification protocol (37°C, 15 minutes; 98°C, 30 s; [98°C, 10 s; 65°C, 30 s; 72°C, 30 s] ×13; 72°C, 5 minutes; 4°C hold). RNA libraries were sequenced using Illumina HiSeq3000 1×50bp with the Genome Technology Access Center at Washington University in St. Louis, typically yielding 15–30 million single-end reads per sample.
Craniofacial morphological analyses
A total of 24 sex-matched littermate paired mice (DNMT3AKO/+ n = 12, 7 male, 5 female; WT n = 12, 7 male, 5 female) across 3 time-points (8 weeks DNMT3AKO/+ n = 4, WT n = 4; 20 weeks DNMT3AKO/+ n = 4, WT n = 4; 25 weeks DNMT3AKO/+ n = 4, WT n = 4) were fixed in 4% paraformaldehyde through intracardiac perfusion. Whole mouse heads were scanned at the Musculoskeletal Research Center at Washington University in St. Louis using a Scanco μCT40 machine. CT images had voxel dimensions of 0.018 mm and were reconstructed on a 2048×2048 pixel grid. The CT images were converted to 8bit images using ImageJ (https://imagej.nih.gov/ij/) and surface reconstructions were acquired in Avizo (https://www.thermofisher.com/fr/en/home/industrial/electron-microscopy/electron-microscopy-instruments-workflow-solutions/3d-visualization-analysis-software.html). Thirty-five three-dimensional land-marks were collected from surface reconstructions of the cranium and mandible using Stratovan Checkpoint (https://www.stratovan.com/products/checkpoint).
Generalized Procrustes Analysis in MorphoJ software was used to explore the differences and similarities of shape between the DNMT3AKO/+ mice and their WT littermates as previously described (Hill et al., 2013). To control for possible differences in size, the landmark coordinate data were natural log-transformed and analyzed with a linear regression model. Additionally, to localize differences in form to specific linear distances, landmark data were analyzed using Euclidean Distance Matrix Analysis (EDMA).
Bone length measurements
We chose to quantify long bones that may directly relate to the height phenotype seen in patients. A total of 24 sex-matched littermate paired mice (DNMT3AKO/+ n = 12, 7 male, 5 female; WT n = 12, 7 male, 5 female) across 3 time-points (8 weeks DNMT3AKO/+ n = 4, WT n = 4; 20 weeks DNMT3AKO/+ n = 4, WT n = 4; 25 weeks DNMT3AKO/+ n = 4, WT n = 4) were fixed in 4% paraformaldehyde through intracardiac perfusion. Decapitated mouse bodies were scanned at the Musculoskeletal Research Center at Washington University in St. Louis using a Faxitron Model UltraFocus100 Dual X-Ray machine. Bone lengths were measured using ImageJ. Data were taken over three age time-points: 8 weeks, 20 weeks, and 25 weeks of age for male and female mice. There was no significant difference in bone lengths based upon sex, but there was a difference based by age. To normalize for this age effect, data were expressed as DNMT3AKO/+ bone lengths normalized to the WT lengths within groups. This also mirrors how human data are presented. Left and right bones were measured and the larger was used for analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Specific bioreplicate sample numbers (n), statistical tests used, boxplot and barplot summary statistics, and p value thresholds are listed for each analysis in the corresponding figure legends. Violin plots were generated in R3.5.1 with ggplot2 using default density functions. Additional information, including software used, is included below.
Statistical analysis for behavioral tests
Behavioral data were analyzed with R v3.3.2 (including the ANOVA function from the Car package in R (Fox and Weisberg, 2011)) and plots were made using GraphPad Prism 7.03a. Normality was assessed using the Shapiro-Wilkes test and visually confirmed. Data not normally distributed were analyzed using non-parametric tests, with the exception of continuous rotarod data. No consistent genotype by sex interaction effects were observed for any tests. As expected, body weights were significantly different between males and females and we therefore presented this data separated by sex. Data were collapsed across sex for presentation across all other tests. Statistical testing was performed using planned assay-specific methods, such as using Student’s T-Tests for single parameter comparisons between genotypes, and within-subjects two-way repeated-measures ANOVA for comparisons across time points. Individual time points within repeated-measures tests were evaluated using Sidak’s multiple comparisons test. Individual cohorts were analyzed separately and in aggregate with similar trends seen across cohorts (Table S5), therefore data from all cohorts were included together.
Whole-genome bisulfite analysis
Bisulfite sequencing analysis was performed as previously described (Clemens et al., 2019). Briefly, data were adaptor-trimmed, mapped to mm9, then deduplicated and called for methylation using BS-seeker2. Methylation levels across regions were assessed using bedtools map -o sum, summing the number of reads mapping to Cs (supporting mC) and the amount of coverage in the region, then dividing those two numbers (Quinlan and Hall, 2010). Hydroxymethylation was calculated as the percent methylation found in the BS-seq minus the percent methylation found in the matching oxBS-seq. Due to count noise, this occasionally resulted in apparent negative hydroxymethylation. During bisulfite sequencing not all DNA can be efficiently bisulfite converted. Though our methods should maximize the amount of converted unmethylated C, there is still a small percentage of unmethylated cytosines that are called as methylated due to non-conversion (0.2%–0.3%). Due to this non-conversion, very lowly methylated regions (e.g., mCA at CpG islands) may not show the same percent reduction in mCA as highly methylated regions. Data were visualized using the UCSC genome browser (http://genome.ucsc.edu) (Kent et al., 2002). CpG islands were obtained from the UCSC table browser (Haeussler et al., 2019), and CpG Shores were defined as the 8kb surrounding them. Average methylation per-sample is normally distributed in all regions examined, and variance between genotypes is similar, fitting the assumptions of a 2-sample t test. Methylation levels for individual elements are not necessarily normally distributed, so non-parametric tests were used instead. This approach to mirrors multiple other studies quantifying mCA levels across the genome (Lister et al., 2013; Luo et al., 2017) where de novo calling of base-pair resolution mCA DMRs is avoided due to low statistical power because of low mCA/CA levels at any individual site, and instead focuses on quantification of mCA/CA levels in populations of pre-defined regions of known functional importance.
Differentially methylated region detection
BSmooth (Hansen et al., 2012) was used to call differentially CpG methylated regions between DNMT3AKO/+ and WT mice, using four bioreplicates. CG sites were filtered for requiring at least 2× coverage in all replicates and differentially methylated regions were called with a statistical threshold of t-stat > 2.0. These regions were further filtered for a length > 100 bp and a requirement that the smoothed per-rep methylation values were consistent. For hypomethylated regions all WT mCG/CG values needed to be greater than any KO mCG/CG value, and for hypermethylated regions all KO methylation values needed to be higher than all WT methylation values. Data fit the assumptions and requirements of BSmooth. Data were distributed evenly between chromosomes, and the overlap between DMRs and regions of interest fit a hypergeometric distribution, making a fisher’s exact test appropriate. In order to calculate an expected overlap of DMRs and genomic regions, for each chromosome, an equal number of resampled DMRs were placed, at random throughout the chromosome, as true DMRs, ensuring that the two do not overlap. From this, the overlap of genomic regions with resampled DMRs was compared to the same overlap with true DMRs.
RNA sequencing analysis
RNA sequencing analysis was performed as previously described (Clemens et al., 2019). Briefly, raw FASTQ files were trimmed with Trim Galore and rRNA sequences were filtered out with Bowtie. Remaining reads were aligned to mm9 using STAR (Dobin et al., 2013) with the default parameters. Reads mapping to multiple regions in the genome were then filtered out, and uniquely mapping reads were converted to BED files and separated into intronic and exonic reads. Finally, reads were assigned to genes using bedtools coverage -counts (Quinlan and Hall, 2010).
For gene annotation we defined a “flattened” list of longest transcript forms for each gene, generated on Ensgene annotations and obtained from the UCSC table browser. For each gene, Ensembl IDs were matched up to MGI gene names. Then, for each unique MGI gene name, the most upstream Ensgene TSS and the most downstream TES were taken as that gene’s start and stop. Based on these Ensembl gene models, we defined TSS regions and gene bodies. Differentially expressed genes were identified using a Wald test through DESeq2, running using default parameters on exonic reads from the DNMT3AKO/+ and WT.
To assess effects on other noncoding RNAs in the DNMT3AKO/+ strain, we realigned our RNA-seq data to all mouse noncoding RNAs from RNAcentral (The RNAcentral Consortium, 2019), then ran DESeq2 on the counts from each noncoding transcript.
Chromatin immunoprecipitation analysis
ChIP sequencing analysis was performed as previously described (Clemens et al., 2019). Briefly, reads were mapped to mm9 using bowtie2 and reads were extended based on library sizes and deduplicated. Bedtools coverage -counts was used to quantify ChIP signal at the transcriptional start site (TSS), gene body (GB), and transcriptional end site (TES) (Quinlan and Hall, 2010). edgeR was then used to determine differential ChIP-signal across genotypes. Data were visualized using the UCSC genome browser (http://genome.ucsc.edu) (Kent et al., 2002).
Controlled resampling
A similar resampling approach was used as previously described (Clemens et al., 2019). Briefly, for every entry in a sample set (e.g., DNMT3A-dysregulated genes), an entry in the control set (e.g., all other genes) with a similar desired characteristic (e.g., expression) was selected, generating a control set of the same size and variable distribution as the sample set.
Identification of dysregulated enhancers
Enhancer regions from Clemens et al., 2019 were used, and enhancers dysregulated in the DNMT3AKO/+ were called using the same method. Briefly, H3K27ac ChIP-seq reads were quantified in all acetyl peak regions, and edgeR was used to identify peaks with significantly different amounts of H3K27ac signal. Peak regions were then divided into promoters, enhancers, and non-identified peaks. Data fits the assumptions of BSmooth. Overlap between misregulated enhancers in different genotypes fit a hypergeometric distribution.
GAGE
Gene set enrichment analysis for the gene sets described was performed using the Generally Applicable Gene-set Enrichment (GAGE) program (Luo et al., 2009). The NDD models for comparison were chosen by searching for gene expression datasets meeting the following criteria: 1) NDD/ASD models that have at least some similar features to DNMT3A disorders (e.g., ID, ASD) 2) generated with the RNA-seq approach 3) analyzed brain tissue with enough similarity to our cortical analysis to justify a reasonable comparison. Analysis was performed directionally on the shrunken, log-normalized exonic fold changes from DESeq2 analysis of DNMT3AKO/+ versus WT RNA-seq data. For each gene set, fold changes of genes in that set were compared to a background of all expressed genes. Gene sets with an FDR q-value below 0.1 and an adjusted p value below 0.5 following expression matched resampling repeated 1,000 times were considered statistically significant. Gene sets were selected for analysis from both human and mouse studies of autism associated genes. SFARI genes (Abrahams et al., 2013) with scores of equal to or less than 3 were considered. Date accessed: 6/20/2019.
GSEA
Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005)(version 7.0, the Broad Institute of MIT and Harvard; https://www.gsea-msigdb.org/gsea/downloads.jsp) was performed on shrunken, log-normalized exonic fold changes from DESeq2 between DNMT3AKO+ and WT RNA-seq data. GSEA calculated a gene set Enrichment Score (ES) that analyzed genes were enriched in the biological signal conduction on the MsigDB (Molecular Signatures Database; https://www.gsea-msigdb.org/gsea/msigdb). Background was set to all expressed genes in this study and 1,000 permutations were set to generate a null distribution for enrichment score in the hallmark gene sets and functional annotation gene sets. The gene sets database used for enrichment analysis were ‘c5.all.v7.0.symbols.gmt’, ‘c5.bp.v7.0.symbols.gmt’, ‘c5.cc.v7.0.symbols.gmt’and ‘c5.mf.v7.0.symbols.gmt’ and FDR < 0.1 was defined as the cut-off criteria for significance.
Experimental design
Authenticated cell lines from ATCC (HEK293T, NEURO-2A) were used, and no mycoplasma contamination testing was needed. Sample sizes were chosen based upon previously published studies using similar techniques. Statistical tests and exclusion criteria (values beyond 2 standard deviations of the group mean) were similar to that of previously published studies and indicated in the appropriate methods. For all animal experiments, experimenters were blinded to genotype during data collection. No treatment conditions were used, so no samples or animals were allocated to experimental groups and no randomization was needed. Tests that assume equal variance were only run if group variances were similar, otherwise alternative tests were used.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Histone H3 (acetyl K27) | Abcam | Cat# ab4729; RRID:AB_2118291 |
| Rabbit Anti-DDDDK tag Polyclonal Antibody | Abcam | Cat# ab1162; RRID: AB_298215 |
| Mouse Anti-Dnmt3a Monoclonal Antibody, Clone 64B1446 | Abcam | Cat# ab13888; RRID: AB_300714 |
| Rabbit monoclonal alpha Tubulin antibody [EP1332Y] | Abcam | Cat# ab52866; RRID: AB_869989 |
| Goat Anti-Rabbit IgG (H+L) Antibody, Alexa Fluor 568 | ThermoFisher | Cat# A-11011; RRID: AB_143157 |
| IRDye 800CW Donkey anti-Rabbit IgG antibody | LI-COR Biosciences | Cat# 926–32213; RRID: AB_621848 |
| IRDye 800CW Goat anti-Rabbit IgG antibody | LI-COR Biosciences | Cat# 926–32211; RRID: AB_621843 |
| IRDye 800CW Goat anti-Mouse IgG antibody | LI-COR Biosciences | Cat# 926–32210; RRID: AB_621842 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat#: 74104 |
| DNeasy Kit | QIAGEN | Cat#: 69504 |
| Epitect Bisulfite Kit | QIAGEN | Cat#: 59824 |
| AllPrep DNA/RNA Kit | QIAGEN | Cat#: 80204 |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | NEB | Cat#: E7420S |
| NEBNext Mulitplex Oligos for Illumina (Index Primers Set 1) | NEB | Cat#: E7335S |
| NEBNext rRNA Depletion Kit (Human/Mouse/Rat) | NEB | Cat#: E6310L |
| Ovation Ultralow Library System V2 | NuGEN | Cat#: 0344–32 |
| Ovation Ultralow Methyl-Seq Kit | NuGEN | Cat#: 0335–32 |
| TrueMethyl oxBS plugin | NuGEN | Cat#: 0414–32 |
| High-Capacity cDNA Reverse Transcription Kit | ThermoFisher | Cat#: 4368814 |
| Power SYBR Green PCR Master Mix | ThermoFisher | Cat#: 4368577 |
| Deposited Data | ||
| RNA-sequencing data | This paper | GEO: GSE147899 |
| ChIP-sequencing data (H3K27ac) | This paper | GEO: GSE147899 |
| Bisulfite-sequencing data | This paper | GEO: GSE147899 |
| RNA, ChIP, Bisulfite-sequencing data | Clemens et al., 2019 | GEO: GSE123373 |
| Bisulfite-sequencing data | Stroud et al., 2017 | GEO: GSE104298 |
| Dixon Hi-C contact matrices | Dixon et al., 2012 | http://chromosome.sdsc.edu/mouse/hi-c/cortex.norm.tar.gz |
| Bisulfite-sequencing data | Lister et al., 2013 | GEO: GSE47966 |
| Mus musculus mm9 genome assembly | UCSC | http://hgdownload.soe.ucsc.edu/goldenPath/mm9/ |
| Ensembl gene models | UCSC | https://genome.ucsc.edu/cgi-bin/hgTables |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat#: HEK293T |
| Neuro-2a | ATCC | Cat#: CCL131 |
| Experimental Models: Organisms/Strains | ||
| Mouse: DNMT3AKO/+ | Derived from DNMT3A fl/fl strain (described in Kaneda et al., 2004; provided by M. Goodell) and CMV-Cre (IMSR Cat# JAX:006054, RRID:IMSR_JAX:006054) mice obtained from the Jackson Laboratory. After DNMT3A heterozygous deletion, Cre negative mice were backcrossed to pure C57BL6 mice (IMSR Cat# JAX:000664, RRID:IMSR_JAX:000664) obtained from the Jackson Laboratory. | JAX: 006054; RRID:IMSR_JAX:006054 JAX: 000664; RRID:IMSR_JAX:000664 |
| Mouse: Dnmt3afl/fl | Kaneda et al., 2004 | Provided by M. Goodell |
| Oligonucleotides | ||
|
Actb Forward: AAGGCCAACCGTGAAAAGAT |
IDT | N/A |
|
Actb Reverse: GTGGTACGACCAGAGGCATAC |
IDT | N/A |
|
Dnmt3a Forward: GGCCTTCTCGACTCCAGATG |
IDT | N/A |
|
Dnmt3a Reverse: TTCCTCTTCTCAGCTGGCAC |
IDT | N/A |
| Software and Algorithms | ||
| DESeq2 (v1.14.1) | Love et al., 2014 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| edgeR (v3.16.5) | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| SAMtools (v1.3) | Li and Durbin, 2009 | https://sourceforge.net/projects/samtools/files/ |
| BEDtools2 (v2.25.0) | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| Bowtie2 (v2.2.5) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| fastQC | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | |
| MACS2 (v2.1.0) | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| Trim galore | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ | |
| Tadtree | Weinreb and Raphael, 2016 | http://compbio.cs.brown.edu/projects/tadtree/ |
| BS-seeker2 | Guo et al., 2013 | https://github.com/BSSeeker/BSseeker2 |
| GREAT | McLean et al., 2010 | http://great.stanford.edupublic/html |
| BSmooth | McLean et al., 2010 | https://www.bioconductor.org/packages/release/bioc/html/bsseq.html |
| Gene Set Enrichment Analysis | Broad Institute | https://www.gsea-msigdb.org/gsea/downloads.jsp |
| Molecular Signatures Database | Broad Institute | https://www.gsea-msigdb.org/gsea/msigdb |
| ImageJ | https://imagej.nih.gov/ij/ | |
| Avizo | https://www.thermofisher.com/fr/en/home/industrial/electron-microscopy/electron-microscopy-instruments-workflow-solutions/3d-visualization-analysis-software.html | |
| Stratovan Checkpoint | https://www.stratovan.com/products/checkpoint | |
| MorphoJ | Dobin et al., 2013 | https://morphometrics.uk/MorphoJ_page.html |
| GraphPad Prism 7.03a | https://www.graphpad.com/ | |
Highlights.
Disease-associated DNMT3A mutations disrupt deposition of neuronal DNA methylation
Heterozygous DNMT3A mutant mice show disease-relevant growth and behavior phenotypes
Heterozygous DNMT3A mutation globally reduces non-CG DNA methylation in the brain
RNA and epigenomic changes in DNMT3A mutants overlap MeCP2 disorder and autism models
ACKNOWLEDGMENTS
We thank J. Hoisington-Lopez and M. Crosby at the Center for Genome Sciences for sequencing support; S. Maloney, K. McCullough, and M. Rieger for assistance with USV experiments; and J. Edwards, J. Goodman, J. Yi, A. Smith, and T. Ley for discussions. We thank our funding sources: NIH-NCRR award UL1RR024992; NIH-NIGMS award T32GM008151 and NIH-NICHD award F31HD100098 (to D.L.C.); NIH-NINDS award F31NS108574 (to A.W.C.); grants from the Klingenstein-Simons Fellowship Fund, the Mathers Foundation, the Brain and Behavior Research Foundation, and the Simons Foundation Autism Research Initiative; and NIH-NIMH R01MH117405 (to H.W.G.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108416.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, Menashe I, Wadkins T, Banerjee-Basu S, and Packer A (2013). SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, and Zoghbi HY (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet 23, 185–188. [DOI] [PubMed] [Google Scholar]
- Anderson GR, Maxeiner S, Sando R, Tsetsenis T, Malenka RC, and Südhof TC (2017). Postsynaptic adhesion GPCR latrophilin-2 mediates target recognition in entorhinal-hippocampal synapse assembly. J. Cell Biol 216, 3831–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee-Basu S, and Packer A (2010). SFARI Gene: an evolving database for the autism research community. Dis. Model. Mech 3, 133–135. [DOI] [PubMed] [Google Scholar]
- Barnes TD, Rieger MA, Dougherty JD, and Holy TE (2017). Group and Individual Variability in Mouse Pup Isolation Calls Recorded on the Same Day Show Stability. Front. Behav. Neurosci 11, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer LD, Renthal W, Greben AW, Whitwam T, Silberfeld A, Stroud H, Li E, Yang MG, Kinde B, Griffith EC, et al. (2020). MeCP2 Represses the Rate of Transcriptional Initiation of Highly Methylated Long Genes. Mol. Cell 77, 294–309.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MP, Kolber BJ, Vogt SK, Wozniak DF, and Muglia LJ (2006). Forebrain glucocorticoid receptors modulate anxiety-associated locomotor activation and adrenal responsiveness. J. Neurosci 26, 1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Cole TB, and Costa LG (2017). Behavioral Phenotyping for Autism Spectrum Disorders in Mice. Curr. Protoc. Toxicol 72, 1–, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen K, Lavery LA, Baker SA, Shaw CA, Li W, and Zoghbi HY (2015). MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. USA 112, 5509–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens AW, Wu DY, Moore JR, Christian DL, Zhao G, and Gabel HW (2019). MeCP2 Represses Enhancers through Chromosome Topology-Associated DNA Methylation. Mol. Cell 77, 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, et al. (2011). Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 72, 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole CB, Russler-Germain DA, Ketkar S, Verdoni AM, Smith AM, Bangert CV, Helton NM, Guo M, Klco JM, O’Laughlin S, et al. (2017). Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J. Clin. Invest 127, 3657–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshwar AR, Martin N, Shannon P, and Chitayat D (2020). A homozygous pathogenic variant in SHROOM3 associated with anencephaly and cleft lip and palate. Clin. Genet 98, 299–302. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, Maloney SE, Wozniak DF, Rieger MA, Sonnenblick L, Coppola G, Mahieu NG, Zhang J, Cai J, Patti GJ, et al. (2013). The Disruption of Celf6, a Gene Identified by Translational Profiling of Serotonergic Neurons, Results in Autism-Related Behaviors. J. Neurosci 33, 2732–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyar I, and Pelin C (2003). Body height estimation based on tibia length in different stature groups. Am. J. Phys. Anthropol 122, 23–27. [DOI] [PubMed] [Google Scholar]
- Fazel Darbandi S, Robinson Schwartz SE, Qi Q, Catta-Preta R, Pai EL-L, Mandell JD, Everitt A, Rubin A, Krasnoff RA, Katzman S, et al. (2018). Neonatal Tbr1 Dosage Controls Cortical Layer 6 Connectivity. Neuron 100, 831–845.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciano P, Zhou X, Astrovskaya I, Turner TN, Wang T, Brueggeman L, Barnard R, Hsieh A, Snyder LG, Muzny DM, et al. ; SPARK Consortium (2019). Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom. Med 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J, and Weisberg S (2011). An R Companion to Applied Regression (SAGE Publications; ). [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, and Greenberg ME (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallitano-Mendel A, Wozniak DF, Pehek EA, and Milbrandt J (2008). Mice lacking the immediate early gene Egr3 respond to the anti-aggressive effects of clozapine yet are relatively resistant to its sedating effects. Neuropsy-chopharmacology 33, 1266–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompers AL, Su-Feher L, Ellegood J, Copping NA, Riyadh MA, Stradleigh TW, Pride MC, Schaffler MD, Wade AA, Catta-Preta R, et al. (2017). Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci 20, 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowher H, and Jeltsch A (2018). Mammalian DNA methyltransferases: new discoveries and open questions. Biochem. Soc. Trans 46, 1191–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Wozniak DF, Ohlemiller KK, and Sanes JR (2006). Cere-bellar synaptic defects and abnormal motor behavior in mice lacking α- and β-dystrobrevin. J. Neurosci 26, 2841–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilmatre A, Huguet G, Delorme R, and Bourgeron T (2014). The emerging role of SHANK genes in neuropsychiatric disorders. Dev. Neurobiol 74, 113–122. [DOI] [PubMed] [Google Scholar]
- Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, Chen P-Y, and Pellegrini M (2013). BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics 14, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, et al. (2014). Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci 17, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Wang L, Li J, Ding Z, Xiao J, Yin X, He S, Shi P, Dong L, Li G, et al. (2015). Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 517, 640–644. [DOI] [PubMed] [Google Scholar]
- Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Hinrichs AS, Gonzalez JN, et al. (2019). The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 47 (D1), D853–D858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Langmead B, and Irizarry RA (2012). BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 13, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman RE, Wozniak DF, Nardi A, Olney JW, Sartorius L, and Holtzman DM (2001). Behavioral phenotyping of GFAP-apoE3 and -apoE4 transgenic mice: apoE4 mice show profound working memory impairments in the absence of Alzheimer’s-like neuropathology. Exp. Neurol 170, 326–344. [DOI] [PubMed] [Google Scholar]
- Hill CA, Martínez-Abadías N, Motch SM, Austin JR, Wang Y, Jabs EW, Richtsmeier JT, and Aldridge K (2013). Postnatal brain and skull growth in an Apert syndrome mouse model. Am. J. Med. Genet. A 161A, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holy TE, and Guo Z (2005). Ultrasonic songs of male mice. PLoS Biol. 3, e386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeltsch A, Broche J, and Bashtrykov P (2018). Molecular Processes Connecting DNA Methylation Patterns with DNA Methyltransferases and Histone Modifications in Mammalian Genomes. Genes (Basel) 9, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, and Sasaki H (2004). Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429, 900–903. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Nishiyama M, Shoji H, Ohkawa Y, Kawamura A, Sato T, Suyama M, Takumi T, Miyakawa T, and Nakayama KI (2016). CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 537, 675–679. [DOI] [PubMed] [Google Scholar]
- Kazdoba TM, Leach PT, Silverman JL, and Crawley JN (2014). Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res 3, 118–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde B, Gabel HW, Gilbert CS, Griffith EC, and Greenberg ME (2015). Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc. Natl. Acad. Sci. USA 112, 6800–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde B, Wu DY, Greenberg ME, and Gabel HW (2016). DNA methylation in the gene body influences MeCP2-mediated gene repression. Proc. Natl. Acad. Sci. USA 113, 15114–15119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger S, Connelly JC, Schweikert G, Webb S, Selfridge J, Ramsahoye BH, Yu M, He C, Sanguinetti G, Sowers LC, et al. (2017). MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet. 13, e1006793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery LA, and Zoghbi HY (2019). The distinct methylation landscape of maturing neurons and its role in Rett syndrome pathogenesis. Curr. Opin. Neurobiol 59, 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery LA, Ure K, Wan YW, Luo C, Trostle AJ, Wang W, Jin H, Lopez J, Lucero J, Durham MA, et al. (2020). Losing Dnmt3a dependent methylation in inhibitory neurons impairs neural function by a mechanism impacting Rett syndrome. eLife 9, e52981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazic SE (2015). Analytical strategies for the marble burying test: avoiding impossible predictions and invalid p-values. BMC Res. Notes 8, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, and Woolf PJ (2009). GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Keown CL, Kurihara L, Zhou J, He Y, Li J, Castanon R, Lucero J, Nery JR, Sandoval JP, et al. (2017). Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, and Bird A (2015). Rett syndrome: a complex disorder with simple roots. Nat. Rev. Genet 16, 261–275. [DOI] [PubMed] [Google Scholar]
- Maloney SE, Rieger MA, Al-Hasani R, Bruchas MR, Wozniak DF, and Dougherty JD (2019a). Loss of CELF6 RNA binding protein impairs cocaine conditioned place preference and contextual fear conditioning. Genes Brain Behav. 18, e12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney SE, Yuede CM, Creeley CE, Williams SL, Huffman JN, Taylor GT, Noguchi KN, and Wozniak DF (2019b). Repeated neonatal isoflurane exposures in the mouse induce apoptotic degenerative changes in the brain and relatively mild long-term behavioral deficits. Sci. Rep 9, 2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno R, Witte J, and Papouin T (2020). A modular setup to run a large line of behavioral testing in mice in a single space. Curr. Protoc. Neurosci 93, e102. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae JF, Clayton S, Fitzgerald TW, Kaplanis J, Prigmore E, Rajan D, Sifrim A, Aitken S, Akawi N, Alvi M, et al. ; Deciphering Developmental Disorders Study (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, and Crawley JN (2004). Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 3, 287–302. [DOI] [PubMed] [Google Scholar]
- Nguyen S, Meletis K, Fu D, Jhaveri S, and Jaenisch R (2007). Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev. Dyn 236, 1663–1676. [DOI] [PubMed] [Google Scholar]
- Nord AS, and West AE (2020). Neurobiological functions of transcriptional enhancers. Nat. Neurosci 23, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothjunge S, Nührenberg TG, Grüning BA, Doppler SA, Preissl S, Schwaderer M, Rommel C, Krane M, Hein L, and Gilsbach R (2017). DNA methylation signatures follow preformed chromatin compartments in cardiac myocytes. Nat. Commun 8, 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard KR, Maloney SE, and Dougherty JD (2019). Erroneous inference based on a lack of preference within one group: Autism, mice, and the social approach task. Autism Res. 12, 1171–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, and Li E (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, and Geschwind DH (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, Meyer MR, Erdmann-Gilmore P, Townsend RR, Wilson RK, and Ley TJ (2014). The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 25, 442–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. ; Autism Sequencing Consortium (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, Peng M, Collins R, Grove J, Klei L, et al. (2019). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, Bedford HM, Bernard G, Bernier FP, Brais B, et al. ; FORGE Canada Consortium; Care4Rare Canada Consortium (2016). Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin. Genet 89, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendžikaite G, Hanna CW, Stewart-Morgan KR, Ivanova E, and Kelsey G (2019). A DNMT3A PWWP mutation leads to methylation of bivalent chromatin and growth retardation in mice. Nat. Commun 10, 1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa A, Fagnocchi L, Mastrototaro G, Massimino L, Zaghi M, Indrigo M, Cattaneo S, Martini D, Gabellini C, Pucci C, et al. (2019). SETD5 Regulates Chromatin Methylation State and Preserves Global Transcriptional Fidelity during Brain Development and Neuronal Wiring. Neuron 104, 271–289.e13. [DOI] [PubMed] [Google Scholar]
- Silverman JL, Turner SM, Barkan CL, Tolu SS, Saxena R, Hung AY, Sheng M, and Crawley JN (2011). Sociability and motor functions in Shank1 mutant mice. Brain Res. 1380, 120–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer DH, Russler-Germain DA, Ketkar S, Helton NM, Lamprecht TL, Fulton RS, Fronick CC, O’Laughlin M, Heath SE, Shinawi M, et al. (2017). CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 168, 801–816.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R, Canfield T, et al. ; Mouse ENCODE Consortium (2012). An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol. 13, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Su SC, Hrvatin S, Greben AW, Renthal W, Boxer LD, Nagy MA, Hochbaum DR, Kinde B, Gabel HW, and Greenberg ME (2017). Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 171, 1151–1164.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumi T, Tamada K, Hatanaka F, Nakai N, and Bolton PF (2020). Behavioral neuroscience of autism. Neurosci. Biobehav. Rev 110, 60–76. [DOI] [PubMed] [Google Scholar]
- Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio Duarte S, Zachariou A, Hanks S, O’Brien E, Aksglaede L, et al. ; Childhood Overgrowth Consortium (2014). Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet 46, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Zachariou A, Loveday C, Renwick A, Mahamdallie S, Aksglaede L, Baralle D, Barge-Schaapveld D, Blyth M, Bouma M, et al. ; Clinical Assessment of the Utility of Sequencing and Evaluation as a Service (CAUSES) Research Study; Deciphering Developmental Disorders (DDD) Study (2018). The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res. 3, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The RNAcentral Consortium (2019). RNAcentral: a hub of information for non-coding RNA sequences. Nucleic Acids Res. 47, D221–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillotson R, Selfridge J, Koerner MV, Gadalla KKE, Guy J, De Sousa D, Hector RD, Cobb SR, and Bird A (2017). Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature 550, 398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilot AK, Bebek G, Niazi F, Altemus JB, Romigh T, Frazier TW, and Eng C (2016). Neural transcriptome of constitutional Pten dysfunction in mice and its relevance to human idiopathic autism spectrum disorder. Mol. Psychiatry 21, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gécz J, et al. (2005). Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet 77, 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, and Geschwind DH (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb C, and Raphael BJ (2016). Identification of hierarchical chromatin domains. Bioinformatics 32, 1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, and Muglia LJ (2004). Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis 17, 403–414. [DOI] [PubMed] [Google Scholar]
- Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, and Ren B (2012). Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148, 816–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Silverman JL, and Crawley JN (2011). Automated Three-Cham-bered Social Approach Task for Mice. Curr. Protoc. Neurosci 56, 8.26.1–8.26.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZM, Lu R, Wang P, Yu Y, Chen D, Gao L, Liu S, Ji D, Rothbart SB, Wang Y, et al. (2018). Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 554, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RS, Liakath-Ali K, and Südhof TC (2020). Latrophilin-2 and latrophilin-3 are redundantly essential for parallel-fiber synapse function in cerebellum. eLife 9, e54443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DOIs for all published gene sets used in comparison and enrichment analysis:
The accession number for the raw and aggregate data for bisulfite-seq, raw and gene-count data for RNA-seq, and raw and peak call data for ChIP-seq reported in this paper is GEO: GSE147899.