

Abstract
Purpose of Review
Proper cartilage development is critical to bone formation during endochondral ossification. This review highlights the current understanding of various aspects of glucose metabolism in chondrocytes during cartilage development.
Recent Findings
Recent studies indicate that chondrocytes transdifferentiate into osteoblasts and bone marrow stromal cells during endochondral ossification. In cartilage development, signaling molecules, including IGF2 and BMP2, tightly control glucose uptake and utilization in a stage-specific manner. Perturbation of glucose metabolism alters the course of chondrocyte maturation, suggesting a key role for glucose metabolism during endochondral ossification.
Summary
During prenatal and postnatal growth, chondrocytes experience bursts of nutrient availability and energy expenditure, which demand sophisticated control of the glucose-dependent processes of cartilage matrix production, cell proliferation, and hypertrophy. Investigating the regulation of glucose metabolism may therefore lead to a unifying mechanism for signaling events in cartilage development and provide insight into causes of skeletal growth abnormalities.
Keywords: Cartilage development, Glucose transporters, Glycolysis, Oxidative phosphorylation, Pentose phosphate pathway, Glycogen
Endochondral Ossification Is a Continuous Process of Cartilage and Bone Formation
The skeleton of vertebrates plays a critical role in supporting the body and facilitating movement. Most bones, including long bones and the irregular vertebral bones, form by the process of endochondral ossification, in which a cartilage template (or anlage) is replaced by mineralized bone tissue [1–3]. Within the anlage lie chondrocytes at different developmental stages (Fig. 1A). At the end of the bone, or the epiphysis, is the resting zone, which provides a reserve of cells for proliferation and differentiation [4, 5]. Proliferating chondrocytes form distinct columns along the long axis of the bone and express the master transcriptional regulator, Sox9, as well as the signature cartilage matrix proteins aggrecan and collagen II. As these cells exit the cell cycle, they enter the prehypertrophic phase. This is quickly followed by hypertrophy, during which chondrocytes increase in size and express another key transcription factor (Runx2) and the cartilage matrix protein collagen X.
Fig. 1.
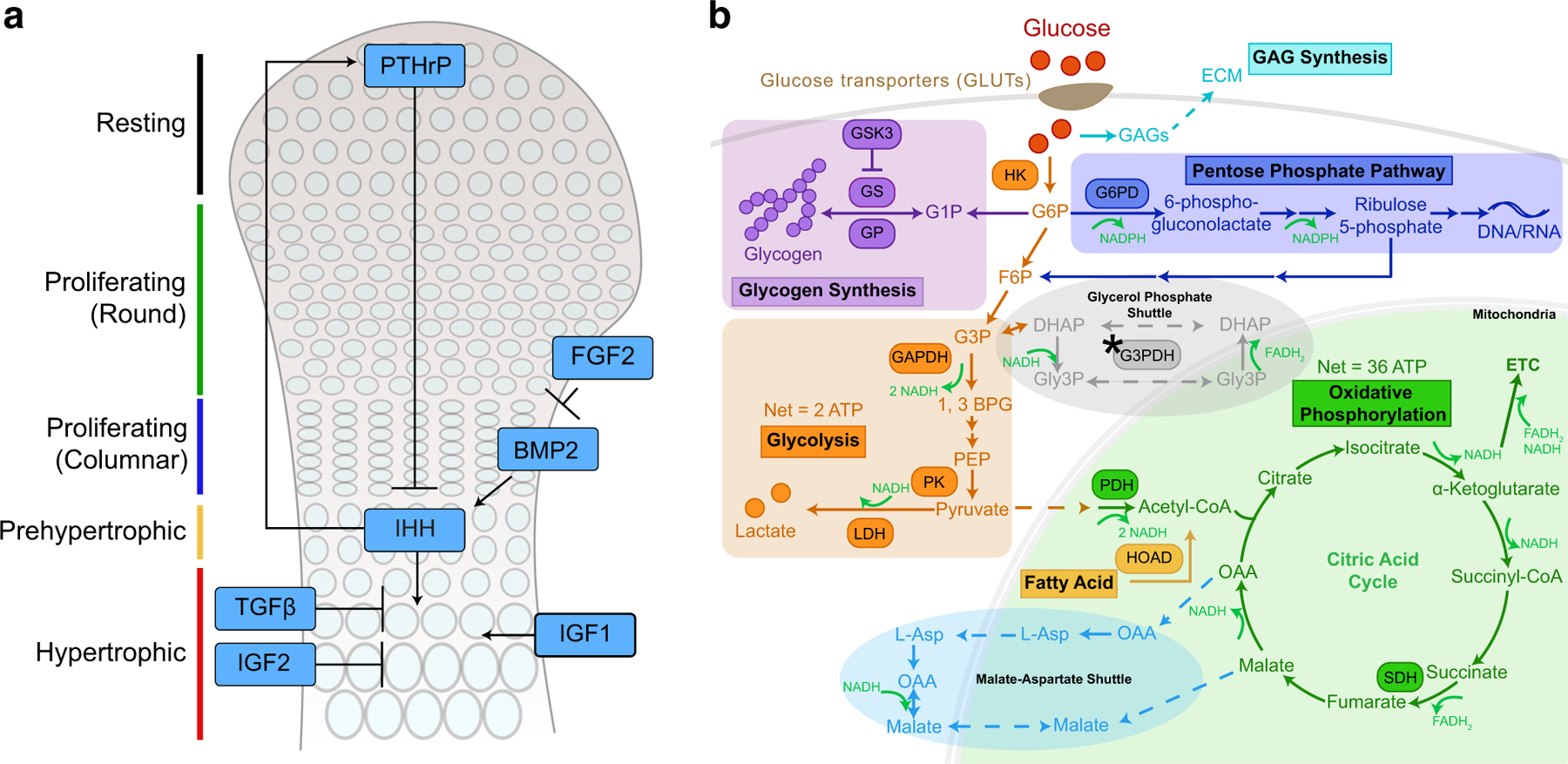
(A) Schematic diagram showing chondrocyte stages in the growth plate during development and selected signals that regulate these processes, including PTHrP, IHH, WNTs, FGFs, BMP and TGFβ, IGF1 and IGF2. Wnts have pleiotropic effects on chondrocyte development and are not illustrated here. PTHrP, parathyroid hormone-related peptide; IHH, Indian hedgehog; IGF, insulin-like growth factor; BMP2, bone morphogenetic protein 2; TGFβ, transforming growth factor β. (B) Pathways of glucose metabolism. Glucose is transported into the cells by GLUTs and converted to G6P. G6P can be utilized by multiple pathways, including glycolysis, oxidative phosphorylation, the pentose phosphate pathway, glycogen synthesis, and GAG synthesis. Two shuttles are typically used to transfer electrons associated with NADH from the cytosol to the mitochondria for the ETC to produce ATP. One is the malate-aspartate shuttle, and the other is the glycerol phosphate shuttle, although a key enzyme in the glycerol phosphate shuttle, G3PDH, is not present in chondrocytes (indicated by *). G1P, glucose 1-phosphate; G6P, glucose 6-phosphate; F6P, fructose 6-phosphate; G3P, glyceraldehyde 3-phosphate; Gly3P, glycerol 3-phosphate; DHAP, dihydroxyacetone phosphate; PEP, phosphoenolpyruvate; OAA, oxaloacetate; HK, hexokinase; GS, glycogen synthase; GSK3, glycogen synthase kinase 3; GP, glycogen phosphorylase; PFK, phosphofructokinase; GAPDH, glyderaldehyde 3-phosphate dehydrogenase; G3PDH, glycerol 3-phosphate dehydrogenase; PK, pyruvate kinase; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; SDH, succinate dehydrogenase; HOAD, β-hydroxyacyl-CoA dehydrogenase; ECM, extracellular matrix; GAG, glycosaminoglycan; ETC, electron transport chain
For many years, it was believed that chondrocytes die at the end of the hypertrophic stage [6, 7]. However, recent lineage tracing studies demonstrate that an estimated 20% to 80% of chondrocytes actually transdifferentiate into osteoblasts and osteocytes as part of a single, continuous developmental process in both the long bone and the mandibular condyle [4, 7–13]. Furthermore, these hypertrophic chondrocytes can also differentiate into marrow stromal and reticular cells, but not into bone marrow adipocytes [4]. This transdifferentiation process is additionally replicated in the fracture callus and in cartilage grafts, where bone formation in the recipient was contributed by cells derived from the donor graft [7, 11, 14]. Other studies have shown both bone morphogenetic protein receptor 1 a (BMPR1a) and β-catenin to be critical for hypertrophic chondrocyte contribution to bone formation [8, 15]. Ablation of β-catenin function in collagen X-positive hypertrophic chondrocytes in mice directly decreased the number of chondrocyte-derived osteoblasts, while increased activity increased osteoblastic cells and decreased osteoclast number [15]. It is interesting to note that hypertrophic chondrocytes during fracture healing express the well-known pluripotency genes Sox2, Oct4, and Nanog, and that conditional knockout of Sox2 prevents the transition of chondrocytes to bone cells, suggesting a progenitor-like intermediate between chondrocytes and osteoblasts [14, 16, 17]. With the transition of hypertrophic cells to osteoblasts, the cartilage of the hypertrophic zone calcifies and is subsequently destroyed, giving rise to mineralized bone matrix. This occurs first at the primary ossification center in the middle (diaphysis) of the anlage. Shortly after birth, a secondary ossification center forms at the epiphysis, thus restricting developing chondrocytes between two ossification centers.
Because of the pivotal roles of proliferation and hypertrophy in expanding the cartilage template, and the transdifferentiation of hypertrophic chondrocytes into osteoblasts and marrow stromal cells, the control of the pace of chondrocyte maturation ensures properly sized anlagen and sufficient progenitors for bone formation. Dysregulation of this process therefore leads to smaller templates and short stature, which is often associated with lower bone density and abnormal adult bone structure [18].
Multiple Signaling Pathways Regulate Cartilage Development
In developing cartilage, PTHrP and IHH function as a signaling loop, whereby chondrocytes and perichondrial cells at the end of the bone produce PTHrP to promote proliferation [19, 20]. Most recently, certain PTHrP-positive cells in the resting zone were found to be stem cells for growth plate chondrocytes, osteoblasts, and bone marrow stromal cells [4]. As chondrocytes proliferate and distance themselves from the PTHrP signal, they become prehypertrophic and produce IHH. In turn, IHH induces PTHrP expression, which feeds back into the proliferating zone to delay hypertrophy and maintain sufficient numbers of chondrocytes to lengthen the cartilage template [19, 21, 22]. IHH simultaneously promotes full hypertrophic differentiation of prehypertrophic chondrocytes in a PTHrP-independent manner [23].
In addition to PTHrP and IHH, other signaling molecules regulate bone development. BMPs, for example, directly regulate the PTHrP-IHH signaling loop by enhancing expression of IHH [24]. FGF2, on the other hand, acts upstream of IHH and negatively regulates chondrocyte proliferation and BMP activity [25]. Wnt family members also regulate the pace of cartilage development [26–29]. While the canonical Wnt pathway can inhibit the entire endochondral ossification process, the non-canonical pathways vary in that they can both promote hypertrophy and inhibit it [29–31]. Another molecule, transforming growth factor β (TGFβ), acts in a PTHrP-dependent manner to inhibit hypertrophy, but in a PTHrP-independent manner to inhibit cartilage mineralization [32]. Insulin-like growth factor 1 (IGF1) is known to mediate the effects of growth hormone and to promote chondrocyte hypertrophy in parallel to the IHH pathway, while IGF2 was found to inhibit hypertrophy [33, 34••]. The effect of IGFs may also be influenced by IGF-binding proteins [35, 36]. Additionally, biomechanical factors are known to regulate cartilage growth [37, 38]. The signals regulating chondrogenic differentiation are thus interconnected and highly complex (Fig. 1A) [39].
Despite the identification of these signaling events, it is still unclear how they control basic metabolism in chondrocytes to change cell behavior. During the long period of cartilage development from embryo to adolescence, chondrocytes experience bursts of both nutrient availability and energy expenditure, which demand sophisticated usage and control of resources. This important aspect of cartilage development is still largely elusive. The aim of this review is therefore to discuss the emerging role and regulation of glucose metabolism in growth plate development.
Overview of Glucose Metabolism
Cellular glucose metabolism initiates with the transport of glucose into cells (Fig. 1B). In the first committed step of metabolism, hexokinase uses adenosine triphosphate (ATP) to add a phosphate to glucose and form glucose 6-phosphate (G6P), which can enter a number of pathways. The vast majority of glucose will enter glycolysis to form pyruvate, producing 2 molecules of nicotinamide adenine dinucleotide (NAD+, NADH in its reduced form) and a net gain of 2 ATP. Pyruvate is either converted to lactate to regenerate NAD+ and complete the process of glycolysis or is shuttled into the mitochondria and converted into acetyl-CoA. Each acetyl-CoA that enters the citric acid (TCA) cycle produces 3 molecules of NADH and 1 molecule of flavin adenine dinucleotide (FAD, FADH2 in its reduced form). NADH and FADH2 are oxidized by the electron transport chain (ETC) to NAD+ and FAD. The complexes (complexes I to IV) of the ETC use the energy of these molecules as they are transferred to pump protons from the mitochondrial matrix into the intermembrane space, generating a proton gradient. ATP synthase (complex V) uses the energy of protons traveling down that gradient and back into the mitochondrial matrix to produce ATP [40]. Oxidative phosphorylation gives a net yield of about 36 molecules of ATP per molecule of glucose. In addition to glycolysis and oxidative phosphorylation, some glucose may be converted to glycogen, for storage, and some may enter the pentose phosphate pathway (PPP), to make nucleotides (Fig. 1B) [40].
Glycolysis is the preferred method for ATP production by cells under hypoxic conditions, and thus is traditionally called anaerobic glycolysis. However, it is widely known that certain types of cells, such as cancer cells, use glycolysis even when there is an abundance of oxygen (the “Warburg effect” or “aerobic glycolysis”) [41]. The lack of blood vessels and low oxygen tension in the tissue environment, coupled with the high energy demand for production of glucose-derived matrix glycosaminoglycans and the difference in energy requirements for proliferation and hypertrophy, constitute a unique challenge for developing cartilage in balancing various aspects of glucose metabolism.
Glucose Uptake
Because cartilage is avascular, glucose diffuses into the growth plate via capillaries derived from the epiphyseal artery, located outside the cartilage template [42]. In mammalian cells, glucose uptake is dependent on facilitative glucose transporters (GLUTs), which allow for passive diffusion across the cell membrane, and sodium-coupled glucose transporters (SGLTs), which transport Na+ and glucose across the membrane using an electrochemical gradient. There are many GLUT family members, but not all transport glucose and not all are expressed in the developing cartilage. Among the GLUTs that are expressed in the developing cartilage are GLUTs 1–4, which transport glucose, and GLUTs 5 and 9, which transport fructose and urate, respectively [43••, 44, 45]. For a comprehensive review of the GLUT family members, please refer to [44]. As no members of the SGLT family have been reported to be expressed in growing cartilage, this review will focus on GLUT family members expressed in the growth plate.
GLUT1 is considered insulin-insensitive and responsible for basal glucose uptake. In the growth plate, it appears mainly in prehypertrophic chondrocytes and the upper hypertrophic zone. GLUT2 is a low-affinity transporter, while GLUT3 is a high-affinity transporter [46, 47]. Both GLUT2 and GLUT3 are expressed in the hypertrophic zone of the rat growth plate at postnatal day 7 (P7), but disappear by P28 [43••]. GLUT4 also has a high affinity for glucose, but unlike the GLUTs previously mentioned, it is responsive to insulin. It has been widely characterized in insulin-responsive tissues such as muscle and fat, but is now also known to be present in hypertrophic chondrocytes [43••]. GLUT4 mRNA expression is interestingly different: it is more strongly expressed in proliferating chondrocytes than in hypertrophic chondrocytes [48•]. The amount of glucose consumed in each zone of cartilage has not been conclusively determined. Results from a short-term experiment with radiolabeled glucose indicated that proliferating chondrocytes took in less glucose than hypertrophic chondrocytes [43••]. Another study, however, showed that proliferating chondrocytes consumed more glucose than hypertrophic chondrocytes over time [49••]. Despite these opposite findings, studies thus far suggest that GLUT expression and glucose uptake are tightly controlled and dynamic during cartilage development (Table 1).
Table 1.
Transporters and enzymes involved in glucose metabolism in developing cartilage
| |
Protein expression |
||||
|---|---|---|---|---|---|
| Process | Protein | Function | Resting | Proliferating | Hypertrophic |
| Glucose transport | GLUT1 | Glucose transport (KM ~ 3 mM) |
+ + (upper) |
||
| GLUT2 | Glucose transport (KM ~ 17 mM) |
+ + | |||
| GLUT3 | Glucose transport (KM ~ 1.5 mM) |
+ + | |||
| GLUT4 | Glucose transport (KM ~ 6.6 mM) Insulin-responsive |
+ + | |||
| Glycolysis | LDH | Pyruvate → lactate NAD+ → NADH |
+ | + + | + + + |
| GAPDH | Glyceraldehyde 3-phosphate → 1,3-bisphosphoglycerate NAD+ → NADH |
+ | + + | + + + | |
| Citric acid cycle | SDH | Succinate → fumarate | + | + + | + + |
| Fatty acid oxidation | HOAD | Involved in converting fatty acids to acetyl-CoA NAD+ → NADH |
+ | + + | + + + |
| Pentose phosphate pathway | G6PD | G6P → 6-phosphogluconolactone NADP+ → NADPH | + | + + | + + + |
| Glycogen | GS | Glucose → glycogen | + | + (upper) |
|
| GSK | Inactivates glycogen synthase | + (GSKα) |
+ (GSKα) |
+ (GSKα and β) |
|
| Glycogen phosphorylase | Glycogen → glucose | + | + (upper) |
||
Both insulin and the IGFs play important roles in regulating glucose uptake and chondrocyte differentiation [50–52]. Using the acute diabetes mouse model of streptozotocin (STZ)-induced insulin deficiency, Maor and Karnieli showed that GLUT4, but not GLUT1, was significantly downregulated in the growth plate in this model, confirming that GLUT4 is insulin-responsive in chondrocytes [48•]. As expected, STZ-treated mice are smaller than their untreated counterparts. It is interesting to note that, although the insulin receptor (IR) is clearly expressed during proliferation, cartilage-specific knockout of this receptor did not lead to shorter bones, suggesting that insulin may also affect cartilage development indirectly [53]. Since IGF1 receptor (IGF1R) expression is induced in IR knockout mice, it is possible that some effects of insulin are mediated through the IGF1 signaling pathway [53].
IGF1 signaling functions primarily through IGF1R, and to a lesser extent, through IR [54, 55]. Knockout of IGF1R resulted in a shortened cartilage template, with a smaller hypertrophic zone and reduced GLUT4 expression, suggesting that IGF1R signaling is required for both hypertrophy and GLUT4 expression [56•]. Whether IGF1R KO affects glucose uptake in chondrocytes is not known, but this was the case in osteoblasts [50, 57]. IGF1 appears to affect glucose uptake in epiphyseal chondrocytes in a concentration-dependent way: in one study, IGF1 promoted glucose uptake only at a high concentration of 100 ng/mL, but in another study, it failed to promote glucose uptake at an even higher concentration of 1 μg/mL [58, 59]. IGF2 is also known to act through IGF1R to promote body growth, but can also bind to IR and IGF2R, though IGF2R is thought to act as a decoy receptor to sequester IGF2 [60–64].
Our laboratory has shown that the hypertrophic zone of Igf2 null bones is larger in proportion to total bone length, suggesting a very different mechanism of action than that of IGF1 [34••]. Interestingly, despite the small size of the mice, Igf2 null epiphyseal chondrocytes displayed increased glucose consumption [34••]. The results of our study and the STZ study by Maor and Karnieli imply that cartilage growth is negatively impacted by both too much and too little glucose consumption in chondrocytes. Although it is still not clear whether IGF2 regulates GLUT expression in this context, a recent study by Lee et al. revealed that BMP signaling could regulate GLUT1 expression [65••]. Additionally, Glut1 null bones exhibited growth arrest and disorganization of the proliferating and hypertrophic zones, indicating an important role for GLUT1-mediated glucose metabolism in cartilage development [65••].
As the cartilage template grows, its center becomes increasingly hypoxic. This environment induces expression of hypoxia-inducible factor 1α (HIF1α), an important regulator of cartilage development [66, 67]. HIF1α knockout studies have shown that the expression of this factor is required for Glut1 expression, thus it will be interesting to examine how HIF1α regulates glucose uptake [68]. Hypoxia may also induce expression of the glucose sensor AMP-activated kinase (AMPK) [69]. AMPK has been found to promote glucose uptake in muscle and inhibit catabolic events and osteoarthritis in articular chondrocytes [70–72]. However, it is still unclear whether AMPK is required for growth plate chondrocytes to regulate glucose uptake and whether it is controlled by IGFs or other signaling molecules in this process.
Glycolysis and Oxidative Phosphorylation
To determine whether chondrocytes use glycolysis or oxidative phosphorylation for ATP production, Rajpurohit et al. utilized the uncoupling reagent 2,4-dinitrophenol (DNP) [49••]. Uncoupling reagents separate electron transport from ATP synthesis, thus abolishing ATP production derived from oxidative phosphorylation [73]. When skin fibroblasts, a type of cell that primarily rely on oxidative phosphorylation for ATP, were treated with DNP, they exhibited the typical switch to glycolysis and lactate production [49••]. This caused an overall reduction in ATP, because glycolysis produces less ATP than oxidative phosphorylation [49••]. However, in epiphyseal chondrocytes, DNP did not increase lactate levels, a proxy measure of glycolysis, suggesting glycolysis already takes place at a high rate. Interestingly, DNP did substantially reduce ATP production, more so in embryonic chondrocytes than epiphyseal chondrocytes, suggesting that these chondrocytes still rely on oxidative phosphorylation to some extent for energy. Compared to embryonic chondrocytes, however, postnatal growth plate chondrocytes had lower mitochondrial function and were less dependent on oxidative phosphorylation for ATP production [49••].
Two shuttles facilitate transport of the electrons associated with NADH in the cytoplasm to the mitochondria, since NADH cannot diffuse across the mitochondrial membrane (Fig. 1B). The first is the malate-aspartate shuttle, in which malate is converted to oxaloacetate with electrons from NADH. Oxaloacetate crosses the mitochondrial membrane and is converted to aspartate, releasing the electrons to reform NADH [40]. In the second shuttle, the glycerol phosphate shuttle, the interconversion of glycerol-3-phosphate (Gly3P) and dihydroxyacetone phosphate (DHAP) by glycerol phosphate dehydrogenase, allows electron transport from cytoplasmic NADH into the mitochondria [40]. Brighton et al. discovered, strikingly, that glycerol phosphate dehydrogenase is inactive in chondrocytes, suggesting that chondrocytes may only depend on the malate-aspartate shuttle for this process, a notion that has yet to be verified [74].
Although epiphyseal chondrocytes as a whole rely heavily on glycolysis, they differ in glucose usage between zones of the growth plate. Using cartilage slices, Rajpurohit et al. found a lower lactate to glucose consumption ratio, and lower NADH production, in cartilage slices comprising proliferative chondrocytes than those comprising hypertrophic chondrocytes; furthermore, treatment with DNP significantly reduced ATP production in proliferating chondrocytes, but not in hypertrophic chondrocytes [49••]. These data suggest that proliferating chondrocytes partially use oxidative phosphorylation for their energy production, consistent with their higher mitochondrial membrane potential, and that hypertrophic chondrocytes are “uncoupled” from mitochondria [49••]. In fact, hypertrophic chondrocytes express higher levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and lactate dehydrogenase (LDH), enzymes used in glycolysis and lactate production, as well as β-hydroxyacyl-CoA dehydrogenase (HOAD), the enzyme that converts fatty acids into acetyl-CoA to feed into the TCA cycle (Table 1) [75, 76•]. The lower mitochondrial membrane potential in hypertrophic chondrocytes could be mediated by HIF1α, which promotes glycolysis and inhibits oxidative phosphorylation via uncoupling proteins (UCPs) [67, 77]. This may also be related to the increase in inorganic phosphate associated with increased levels of alkaline phosphatase upon hypertrophy, as inorganic phosphate has been found to reduce mitochondrial activity through the ERK pathway [49, 78–80]. Since calcium efflux from mitochondria is upregulated by low pH, heightened glycolysis may be a way to prepare for calcification after hypertrophy [81, 82]. Whether low pH is the driving force for calcification remains to be determined, but this stage is certainly critical for subsequent bone formation, as osteoblasts are now known to derive from hypertrophic chondrocytes [4, 9–12].
Is ATP generation the sole function of mitochondria in chondrocytes? A key TCA cycle enzyme, succinate dehydrogenase (SDH), which converts succinate to fumarate, is highly active; in fact, it is more active in hypertrophic chondrocytes, implicating the presence of active mitochondria even though these cells do not use them for ATP production [76•]. Mitochondrial uncoupling has been shown to reduce lactate production in hypertrophic chondrocytes, suggesting that glycolysis somehow relies on the mitochondria to achieve its full activity [49••]. The answer to the above question is not clear. Mitochondrial activity may be required for redox balance in chondrocytes, perhaps via the malate-aspartate shuttle. In articular cartilage, which is also highly glycolytic, a similar phenomenon was found. Supplementation with oxidants (electron acceptors) was sufficient to elevate lactate production even under anoxic conditions when mitochondria could not undergo respiration, suggesting mitochondria in articular chondrocytes provide electron acceptors to sustain glycolysis [83, 84]. Whether this is also the case in growth plate chondrocytes remains to be investigated.
The balance between glycolysis and oxidative phosphorylation for ATP production is thus dynamic from embryonic to postnatal growth and from the proliferation phase to the hypertrophic phase. However, very little is known about the signals that control this intricate balance in growth plate chondrocytes. Recently, our group has discovered that IGF2 is a key regulator for redox balance in epiphyseal chondrocytes. We found that Igf2 null chondrocytes exhibit increased glycolysis and oxidative phosphorylation, leading to increased reactive oxygen species (ROS) levels and ATP production [34••]. We then used an advanced two photon fluorescence imaging technique that captures the endogenous fluorescence emitted by FAD and NADH with high resolution [85]. Since FAD and NADH represent the oxidized and reduced cellular states, and FADH2 and NAD+ are not fluorescent, the optical redox ratio of FAD/(FAD+NADH) fluorescence levels reflects the redox state in cells. When there is an increase in glycolysis with respect to oxidative phosphorylation, there is less conversion of NADH to NAD+, resulting in a decreased optical redox ratio. Thus, this ratio has been used to assess the balance of glycolysis and oxidative phosphorylation [85–88]. Interestingly, two photon imaging analysis suggested that the loss of Igf2 causes a shift from oxidative phosphorylation to glycolysis, which can be restored by exogenous IGF2 treatment [34••]. Considering that Igf2 null cartilage showed premature hypertrophy, our work is consistent with the prior study demonstrating a glycolytic shift in hypertrophic chondrocytes [34••, 49••].
Little is known about the potential role of other signaling pathways on the regulation of glycolysis and oxidative phosphorylation in growth plate chondrocytes. Recent analysis using the Seahorse metabolic analyzer showed BMP2 could enhance oxygen consumption in articular chondrocytes [89]. Furthermore, glucose depletion also enhanced oxygen consumption, consistent with the “Crabtree” effect [90]. While Wnt signaling has not been shown to regulate these processes in growth plate chondrocytes, Wnt3a is known to induce a number of glycolytic proteins and stimulate glycolysis in osteoblasts [91, 92]. The importance of glucose metabolism in controlling the pace of cartilage development has been demonstrated in Igf2 knockout mice, in which a glycolysis inhibitor, 3-bromopyruvate (3-BrPA), was able to rescue premature chondrocyte hypertrophy and shortened bone phenotype in Igf2 null bones [34••]. When taken in the context of results obtained in articular chondrocytes showing that AMPK and ATP-citrate lyase are essential for regulating matrix production, our study clearly demonstrates the importance of tight control of glucose metabolism to ensure normal cartilage development [34••, 93, 94].
Pentose Phosphate Shunt
The pentose phosphate pathway (PPP) is another pathway for glucose utilization. In this pathway, G6P is converted to ribose 5-phosphate while generating reduced nicotinamide adenine dinucleotide phosphate (NADPH) (Fig. 1B) [95]. In addition, multiple sugars that are shared with glycolysis (such as glyceraldehyde 3-phosphate and fructose 6-phosphate) are generated. No ATP is consumed or produced in this process. The PPP is especially important because ribose 5-phosphate is the backbone of nucleic acids, which are required for cell proliferation and replacement. Additionally, NADPH serves as a major electron donor to balance the redox ratio and reduce oxidative stress [50, 95]. In growth plate chondrocytes, it was found that about 20% of oxygen is actually consumed by the PPP [96•, 97]. Interestingly, a higher level of glucose 6-phosphate dehydrogenase (G6PD), the first enzyme that acts on G6P in this pathway, was found in hypertrophic chondrocytes, concomitant with a lower level of G6P, but higher NADPH in these cells (Table 1) [96•]. This suggests that usage of the PPP is regulated during cartilage development. Since proliferating chondrocytes require a high level of ribose backbone for nucleic acid production, the increased PPP usage in hypertrophic chondrocytes implies that it may serve another purpose, perhaps as an energy source to compensate for the lowered oxidative phosphorylation level. Insulin and calcium supplementation were found to induce 14C incorporation into the PPP and to decrease the activity of glycolysis. This demonstrates that they regulate the balance of the PPP and glycolysis, which may directly influence chondrocyte proliferation and hypertrophy during cartilage template expansion [97]. In the case of articular chondrocytes, nitric oxide was found to attenuate the PPP and promote oxidative injury [98]. Whether the balance of the PPP versus glycolysis and oxidative phosphorylation is shifted by oxidative stress or other regulatory factors in developing cartilage remains to be determined.
Glycogen
Glucose can be stored as glycogen, a multi-branched polymer that ensures availability of glucose on-demand [99, 100]. Glycogen formation begins with conversion of G6P to glucose 1-phosphate (G1P) by phosphoglucomutase. A series of enzymes, including the glycosyltransferase glycogenin, glycogen synthase, and glycogen branching enzyme, subsequently facilitate the production of a chain of 8–12 glucose residues [99]. A key regulatory step in glycogen synthesis is the control of glycogen synthase by glycogen synthase kinase 3 (GSK3). GSK has two isoforms, GSK3α and GSK3β, that can be inactivated by a reversible phosphorylation reaction [101]. A decrease in GSK3 phosphorylation therefore leads to an increase in GSK3 activity, which subsequently decreases glycogen synthase activity and glycogen synthesis. When additional glucose is needed, glycogen is broken down in a process called glycogenolysis, facilitated by glycogen phosphorylase (to regenerate G6P for glycolysis and the PPP) and by glucose 6-phosphatase (G6Pase) to generate free glucose [100, 102]. The level of glycogen is thus balanced by the control of glycogen synthesis and glycogen breakdown (Fig. 1B).
Glycogen presence in the growth plate has been established for over a century [102–105]. Electron microscopy and histological analyses indicated that glycogen particles are present in both the proliferating and hypertrophic zones, with greater aggregation in the hypertrophic zone [56•, 103–106]. GSK3β was also found to be mainly in the prehypertrophic and hypertrophic zones, but GSK3α was uniformly expressed in all zones of the growth plate (Table 1) [107]. Mice lacking Igf1 exhibited less phosphorylated GSK3β and less glycogen in the growth plate; furthermore, Igf1R null mice showed diminished glycogen stores, suggesting that IGF1 controls GSK3β activity and glycogen levels in growth plate chondrocytes [56•, 108]. Our laboratory found that glycogen levels are also lower in Igf2 null chondrocytes, particularly in the prehypertrophic zone [34••]. This coincided with diminished expression of the prehypertrophic marker IHH, as if Igf2 null chondrocytes had prematurely entered the hypertrophic phase from the proliferating phase [34••]. Considering Igf2 null chondrocytes exhibited overactive glucose uptake, glycolysis, and oxidative phosphorylation, it appears that the lack of IGF2 sacrifices glycogen synthesis in favor of using other glucose metabolism pathways. Therefore, IGF2 plays a pivotal role in balancing the various pathways of glucose utilization [34••].
Enzymes involved in glycogen breakdown are also expressed in chondrocytes [102]. Glycogen phosphorylase and G6Pase are both strongly expressed in the proliferating zone and upper hypertrophic zone, suggesting active glycogen metabolism at these stages [102].
The roles of glycogen synthesis and utilization in the growth plate are still unclear. Inhibition of phosphorylated GSK3β increased the length of the proliferating zone and the bone as a whole, and decreased that of the hypertrophic zone [107]. However, cartilage-specific knockout of GSK3β in vivo resulted in compensatory upregulation of GSK3α and no skeletal phenotype [107]. Since GSK3β is a key component of other signaling pathways, most notably the Wnt pathway, it cannot be concluded that GSK3β affects cartilage growth solely through regulating glycogen synthesis [109]. Additional genetic manipulation of components specific to glycogen metabolism is needed to reach a more definitive conclusion.
Conclusion
Mutations and dysregulation of genes controlling growth plate development are known to cause multiple skeletal growth abnormalities. For example, IGF1 deficiency is prevalent in children with short stature [110, 111]. IGF2 mutation and insufficiency lead to growth restriction consistent with Silver-Russell Syndrome, whereas IGF2 overexpression results in Beckwith-Wiedemann Syndrome and gigantism [60, 112–115]. FGFR3 mutations cause achondroplasia, a major form of dwarfism [116]. Aside from genetic skeletal diseases, growth abnormality is also common in children with Type I diabetes and in chronic inflammatory diseases like juvenile idiopathic arthritis (JIA) and cystic fibrosis [117–119]. Insulin, IGFs, and FGFs, as well as pro-inflammatory cytokines such as IL1, IL6, and TNFα, are all known to regulate glucose metabolism and growth plate cartilage development [50–52, 58, 59, 120–125]. Although glucose metabolism has not been assessed in many growth abnormalities, the incidence of hypoglycemia in Beckwith-Wiedemann syndrome was found to reach approximately 50% [126]. It is entirely possible that dysregulation of glucose metabolism is not only casually associated with the abnormalities of cartilage growth, but is also the driving force of these changes.
The investigation of glucose metabolism was initiated decades ago, with elegant and rich biochemical characterization of multiple aspects, including glucose uptake, glycolysis, oxidative phosphorylation, the pentose phosphate pathway, and glycogen synthesis and glycogenolysis. In the current era, metabolism is increasingly recognized as a central control for countless biological processes, but how glucose metabolic pathways control cartilage development is still largely unknown, and their regulation in chondrocytes remains poorly understood. This new dimension of study may lead to a unifying mechanism explaining the actions of the elaborate web of signaling molecules, and provide insights into designing treatment options for a variety of growth abnormalities.
Footnotes
Conflict of Interest Judith M. Hollander and Li Zeng declare that they have no conflict of interest.
Human and Animal Rights All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
This article is part of the Topical Collection on Skeletal Development
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Kobayashi T, Kronenberg HM. Overview of skeletal development In: Hilton MJ, editor. Skeletal development and repair. Methods in molecular biology. Totowa, NJ: Humana Press; 2014. [Google Scholar]
- 2.Lefebvre V, Smits P. Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today. 2005;75(3):200–12. 10.1002/bdrc.20048. [DOI] [PubMed] [Google Scholar]
- 3.de Crombrugghe B, Lefebvre V, Nakashima K. Regulatory mechanisms in the pathways of cartilage and bone formation. Curr Opin Cell Biol. 2001;13:721–7. [DOI] [PubMed] [Google Scholar]
- 4.Mizuhashi K, Ono W, Matsushita Y, Sakagami N, Takahashi A, Saunders TL, et al. Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature. 2018;563:254–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abad V, Meyers JL, Weise M, Gafni RI, Barnes KM, Nilsson O, et al. The role of the resting zone in growth plate chondrogenesis. Endocrinology. 2002;143(5):1851–7. [DOI] [PubMed] [Google Scholar]
- 6.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6. [DOI] [PubMed] [Google Scholar]
- 7.Hinton RJ, Jing Y, Jing J, Feng JQ. Roles of chondrocytes in endochondral bone formation and fracture repair. J Dent Res. 2017;96(1):23–30. 10.1177/0022034516668321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jing Y, Jing J, Ye L, Liu X, Harris SE, Hinton RJ, et al. Chondrogenesis and osteogenesis are one continuous developmental and lineage defined biological process. Sci Rep. 2017;7:10020 10.1038/s41598-017-10048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Tsang KY, Tang HC, Chan D, Cheah KSE. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A. 2014;111(33): 12097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsang KY, Chan D, Cheah KSE. Fate of growth plate hypertrophic chondrocytes: death or lineage extension? Develop Growth Differ. 2015;57:179–92. [DOI] [PubMed] [Google Scholar]
- 11.Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014;10(12):e1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ono N, Ono W, Nagasawa T, Kronenberg HM. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat Cell Biol. 2014;16(12):1157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jing Y, Zhou X, Han X, Jing J, von der Mark K, Wang J, et al. Chondrocytes directly transform into bone cells in mandibular condyle growth. J Dent Res. 2015;94(12):1668–75. 10.1177/0022034515598135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bahney CS, Hu DP, Taylor AJ, Ferro F, Britz HM, Hallgrimsson B, et al. Stem cell–derived endochondral cartilage stimulates bone healing by tissue transformation. J Bone Miner Res. 2014;29(5): 1269–82. 10.1002/jbmr.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houben A, Kostanova-Poliakova D, Weissenböck M, Graf J, Teufel S, von der Mark K, et al. β-Catenin activity in late hypertrophic chondrocytes locally orchestrates osteoblastogenesis and osteoclastogenesis. Development. 2016;143:3826–38. 10.1242/dev.137489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu DP, Ferro F, Yang F, Taylor AJ, Chang W, Miclau T, et al. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development. 2017;144:221–34. 10.1242/dev.130807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park J, Gebhardt M, Golovchenko S, Perez-Branguli F, Hattori T, Hartmann C, et al. Dual pathways to endochondral osteoblasts: a novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol Open. 2015;4:608–21. 10.1242/bio.201411031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsushita M, Kito H, Mishima K, Kadono I, Sugiura H, Hasegawa S, et al. Low bone mineral density in achondroplasia and hypochondroplasia. Pediatr Int. 2016;58:705–8. [DOI] [PubMed] [Google Scholar]
- 19.Chung U-I, Lanske B, Lee K, Li E, Kronenberg HM. The parathyroid hormone/parathyroid hormone-related peptide receptor coordinates endochondral bone development by directly controlling chondrocyte differentiation. Proc Natl Acad Sci U S A. 1998;95:13030–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chagin AS, Vuppalapati KK, Kobayashi T, Guo J, Hirai T, Chen M, et al. G-protein stimulatory subunit alpha and Gq/11α G-proteins are both required to maintain quiescent stem-like chondrocytes. Nat Commun. 2014;5:3673 10.1038/ncomms4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung U-I, Schipani E, McMahon AP, Kronenberg HM. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J Clin Invest. 2001;107:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–22. [DOI] [PubMed] [Google Scholar]
- 23.Mak KK, Kronenberg HM, Chuang P-T, Mackem S, Yang Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development. 2008;135:1947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minina E, Wenzel HM, Kreschel C, Karp S, Gaffield W, McMahon AP, et al. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 2001;128:4523–34. [DOI] [PubMed] [Google Scholar]
- 25.Minina E, Kreschel C, Naski MC, Ornitz DM, Vortkamp A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev Cell. 2002;3:439–49. [DOI] [PubMed] [Google Scholar]
- 26.Witte F, Dokas J, Neuendorf F, Mundlos S, Stricker S. Comprehensive expression analysis of all Wnt genes and their major secreted antagonists during mouse limb development and cartilage differentiation. Gene Expr Patterns. 2009;9:215–23. [DOI] [PubMed] [Google Scholar]
- 27.Andrade AC, Nilsson O, Barnes KM, Baron J. Wnt gene expression in the post-natal growth plate: regulation with chondrocyte differentiation. Bone. 2007;40:1361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamamura Y, Otani T, Kanatani N, Koyama E, Kitagaki J, Komori T, et al. Developmental regulation of Wnt/β-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J Biol Chem. 2005;280(19):19185–95. [DOI] [PubMed] [Google Scholar]
- 29.Yang Y, Topol L, Lee H, Wu J. Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation. Development. 2003;130:1003–15. [DOI] [PubMed] [Google Scholar]
- 30.Church V, Nohno T, Linker C, Marcelle C, Francis-West P. Wnt regulation of chondrocyte differentiation. J Cell Sci. 2002;115: 4809–18. [DOI] [PubMed] [Google Scholar]
- 31.Dao DY, Yang X, Flick LM, Chen D, Hilton MJ, O’Keefe RJ. Axin2 regulates chondrocyte maturation and axial skeletal development. J Orthop Res. 2010;28:89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Serra R, Karaplis A, Sohn P. Parathyroid hormone–related peptide (PTHrP)-dependent and -independent effects of transforming growth factor β (TGF-β) on endochondral bone formation. J Cell Biol. 1999;145(4):783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butler AA, Le Roith D. Control of growth by the somatropic axis: growth hormone and the insulin-like growth factors have related and independent roles. Annu Rev Physiol. 2001;63:141–64. [DOI] [PubMed] [Google Scholar]
- 34.••.Uchimura T, Hollander JM, Nakamura DS, Liu Z, Rosen CJ, Georgakoudi I, et al. An essential role for IGF2 in cartilage development and glucose metabolism during postnatal long bone growth. Development. 2017;144:3533–46.This study identifies a role for IGF2 in regulating glucose metabolism in growth plate chondrocytes and a role for glucose metabolism in controlling postnatal cartilage development.
- 35.Kiepe D, Ciarmatori S, Haarmann A, Tonshoff B. Differential expression of IGF system components in proliferating vs. differentiating growth plate chondrocytes: the functional role of IGFBP-5. Am J Physiol Endocrinol Metab. 2006;290(2):E363–71. 10.1152/ajpendo.00363.2005. [DOI] [PubMed] [Google Scholar]
- 36.Yakar S, Rosen CJ, Bouxsein ML, Sun H, Mejia W, Kawashima Y, et al. Serum complexes of insulin-like growth factor-1 modulate skeletal integrity and carbohydrate metabolism. FASEB J. 2009;23:709–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ueki M, Tanaka N, Tanimoto K, Nishio C, Honda C, Lin Y-Y, et al. The effect of mechanical loading on the metabolism of growth plate chondrocytes. Ann Biomed Eng. 2008;36(5):793–800. [DOI] [PubMed] [Google Scholar]
- 38.Narváez-Tovar CA, Garzón-Alvarado DA. Computational modeling of the mechanical modulation of the growth plate by sustained loading. Theor Biol Med Model. 2012;9:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kozhemyakina E, Lassar AB, Zelzer E. A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation. Development. 2015;142(5): 817–31. 10.1242/dev.105536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dashty M A quick look at biochemistry: carbohydrate metabolism. Clin Biochem. 2013;46:1339–52. [DOI] [PubMed] [Google Scholar]
- 41.Bartrons R, Caro J. Hypoxia, glucose metabolism and the Warburg’s effect. J Bioenerg Biomembr. 2007;39:223–9. [DOI] [PubMed] [Google Scholar]
- 42.Iannotti JP. Growth plate physiology and pathology. Orthop Clin North Am. 1990;21(1):1–17. [PubMed] [Google Scholar]
- 43.••.Ohara H, Tamayama T, Maemura K, Kanbara K, Hayasaki H, Abe M, et al. Immunocytochemical demonstration of glucose transporters in epiphyseal growth plate chondrocytes of young rats in correlation with autoradiographic distribution of 2-deoxyglucose in chondrocytes of mice. Acta Histochem. 2001;103:365–78.This study demonstrates the localization of glucose transporter proteins in the epiphyseal growth plate.
- 44.Mobasheri A, Vannucci SJ, Bondy CA, Carter SD, Innes JF, Arteaga MF, et al. Glucose transport and metabolism in chondrocytes: a key to understanding chondrogenesis, skeletal development and cartilage degradation in osteoarthritis. Histol Histopathol. 2002;17:1239–67. [DOI] [PubMed] [Google Scholar]
- 45.Mobasheri A, Dobson H, Mason SL, Cullingham F, Shakibaei M, Moley JF, et al. Expression of the GLUT1 and GLUT9 facilitative glucose transporters in embryonic chondroblasts and mature chondrocytes in ovine articular cartilage. Cell Biol Int. 2005;29: 249–60. [DOI] [PubMed] [Google Scholar]
- 46.Uldry M, Ibberson M, Hosokawa M, Thorens B. GLUT2 is a high affinity glucosamine transporter. FEBS Lett. 2002;524:199–203. [DOI] [PubMed] [Google Scholar]
- 47.Colville CA, Seatter MJ, Jess TJ, Gould GW, Thomas HM. Kinetic analysis of the liver-type (GLUT2) and brain-type (GLUT3) glucose transporters in Xenopus oocytes: substrate specificities and effects of transport inhibitors. Biochem J. 1993;290:701–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.•.Maor G, Karnieli E. The insulin-sensitive glucose transporter (GLUT4) is involved in early bone growth in control and diabetic mice, but is regulated through the insulin-like growth factor I receptor. Endocrinology. 1999;140(4):1841–51.This study shows regulation of long bone development by insulin signaling and regulation of GLUT4 in the growth plate by insulin.
- 49.••.Rajpurohit R, Mansfield K, Ohyama K, Ewert D, Shapiro IM. Chondrocyte death is linked to development of a mitochondrial membrane permeability transition in the growth plate. J Cell Physiol. 1999;179:287–96.This study demonstrates the differences among zones of the growth plate in the use of glycolysis and oxidative phosphorylation.
- 50.Karner CM, Long F. Glucose metabolism in bone. Bone. 2018;115:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Torres ES, Andrade CV, Fonesca EC, Mello MA, Duarte MEL. Insulin impairs the maturation of chondrocytes in vitro. Braz J Med Biol Res. 2003;36:1185–92. [DOI] [PubMed] [Google Scholar]
- 52.Böhme K, Conscience-Egli M, Tschan T, Winterhalter KH, Bruckner P. Induction of proliferation or hypertrophy of chondrocytes in serum-free culture: the role of insulin-like growth factor-I, insulin, or thyroxine. J Cell Biol. 1992;116(4):1035–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang F, He Q, Tsang WP, Garvey WT, Chan WY, Wan C. Insulin exerts direct, IGF-1 independent actions in growth plate chondrocytes. Bone Res. 2014;2:14012 10.1038/boneres.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yakar S, Werner H, Rosen CJ. 40 years of IGF1: insulin-like growth factors: actions on the skeleton. J Mol Endocrinol. 2018;61(1):T115–T37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.White MF, Copps KD. Chapter 33 - the mechanisms of insulin action Seventh ed. Endocrinology: Adult and Pediatric. W.B. Saunders; 2016. [Google Scholar]
- 56.•.Wang J, Zhou J, Bondy CA. Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. FASEB J. 1999;13(14):1985–90.This study demonstrates the role of IGF1 in cartilage development and in regulation of GLUT4 and GSK3 in epiphyseal cartilage.
- 57.Esen E, Lee S-Y, Wice BM, Long F. PTH promotes bone anabolism by stimulating aerobic glycolysis via IGF signaling. J Bone Miner Res. 2015;30(11):1959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhaumick B, Bala RM. Differential effects of insulin-like growth factors I and II on growth, differentiation and glucoregulation in differentiating chondrocyte cells in culture. Acta Endocrinol. 1991;125:201–11. [DOI] [PubMed] [Google Scholar]
- 59.Bhaumick B, Bala RM. Parallel effects of insulin-like growth factor-II and insulin on glucose metabolism of developing mouse embryonic limb buds in culture. Biochem Biophys Res Commun. 1988;152(1):359–67. [DOI] [PubMed] [Google Scholar]
- 60.Eggenschwiler J, Ludwig T, Fisher P, Leighton PA, Tilghman SM, Efstratiadis A. Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith–Wiedemann and Simpson–Golabi–Behmel syndromes. Genes Dev. 1997;11: 3128–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Louvi A, Accili D, Efstratiadis A. Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev Biol. 1997;189:33–48. [DOI] [PubMed] [Google Scholar]
- 62.Perdue JF, LeBon TR, Kato J, Hampton B, Fujita-Yamaguchi Y. Binding specificities and transducing function of the different molecular weight forms of insulin-like growth factor-II (IGF-II) on IGF-I receptors. Endocrinology. 1991;129(6):3101–8. [DOI] [PubMed] [Google Scholar]
- 63.Pandini G, Medico E, Conte E, Sciacca L, Vigneri R, Belfiore A. Differential gene expression induced by insulin and insulin-like growth factor-II through the insulin receptor isoform A. J Biol Chem. 2003;278(43):42178–89. [DOI] [PubMed] [Google Scholar]
- 64.Zavorka ME, Connelly CM, Grosely R, MacDonald RG. Inhibition of insulin-like growth factor II (IGF-II)-dependent cell growth by multidentate pentamannosyl 6-phosphate-based ligands targeting the mannose 6-phosphate/IGF-II receptor. Oncotarget. 2016;7(38):62386–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.••.Lee S-Y, Abel ED, Long F. Glucose metabolism induced by Bmp signaling is essential for murine skeletal development. Nat Commun. 2018;9 10.1038/s41467-018-07316-5.This study identifies a critical role for BMP signaling-regulated GLUT1 in proliferation and hypertrophy of chondrocytes during skeletal development.
- 66.Watanabe H, Bohensky J, Freeman T, Srinivas V, Shapiro IM. Hypoxic induction of UCP3 in the growth plate: UCP3 suppresses chondrocyte autophagy. J Cell Physiol. 2008;216:419–25. 10.1002/jcp.21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pfander D, Cramer T, Schipani E, Johnson RS. HIF-1α controls extracellular matrix synthesis by epiphyseal chondrocytes. J Cell Sci. 2003;116(9):1819–26. [DOI] [PubMed] [Google Scholar]
- 69.Hashimoto T, Murata Y, Urushihara Y, Shiga S, Takeda K, Hosoi Y. Severe hypoxia increases expression of ATM and DNA-PKcs and itincreases their activities through Src and AMPK signaling pathways. Biochem Biophys Res Commun. 2018;505:13–9. [DOI] [PubMed] [Google Scholar]
- 70.Musi N, Goodyear LJ. AMP-activated protein kinase and muscle glucose uptake. Acta Physiol Scand. 2003;178:337–45. [DOI] [PubMed] [Google Scholar]
- 71.Yang Y, Wang Y, Kong Y, Zhang X, Zhang H, Gang Y, et al. Mechanical stress protects against osteoarthritis via regulation of the AMPK/NF-κB signaling pathway. J Cell Physiol. 2018. 10.1002/jcp.27592. [DOI] [PMC free article] [PubMed]
- 72.Chen L-Y, Wang Y, Terkeltaub R, Liu-Bryan R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthr Cartil. 2018;26: 1539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mickelson MN. Effect of uncoupling agents and respiratory inhibitors on the growth of Streptococcus agalactiae. J Bacteriol. 1974;120(2):733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brighton CT, Lackman RD, Cuckler JM. Absence of the glycerol phosphate shuttle in the various zones of the growth plate. J Bone Joint Surg. 1983;65-A(5):663–6. [PubMed] [Google Scholar]
- 75.Houten SM, Wanders RJA. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis. 2010;33:469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.•.Dunham J, Dodds RA, Nahir AM, Frost GTB, Catterall A, Bitensky L, et al. Aerobic glycolysis of bone and cartilage: the possible involvement of fatty acid oxidation. Cell Biochem Funct. 1983;1:168–72.This study demonstrates the activities of key metabolic enzymes in glucose metabolism in different zones of the growth plate.
- 77.Kim J-W, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85. 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 78.Mansfield K, Rajpurohit R, Shapiro IM. Extracellular phosphate ions cause apoptosis of terminally differentiated epiphyseal chondrocytes. J Cell Physiol. 1999;179:276–86. [DOI] [PubMed] [Google Scholar]
- 79.Orimo H The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nippon Med Sch. 2010;77:4–12. [DOI] [PubMed] [Google Scholar]
- 80.Miedlich SU, Zalutskaya A, Zhu ED, Demay MB. Phosphate-induced apoptosis of hypertrophic chondrocytes is associated with a decrease in mitochondrial membrane potential and is dependent upon Erk1/2 phosphorylation. J Biol Chem. 2010;285(24):18270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shapiro IM, Lee NH. The effect of oxygen, phosphoenolpyruvate and pH on the release of calcium from chondrocyte mitochondria. Metab Bone Dis Relat Res. 1978;1:173–7. [Google Scholar]
- 82.Matthews JL, Martin JH, Sampson HW, Kunin AS, Roan JH. Mitochondrial granules in the normal and rachitic rat epiphysis. Calcif Tissue Res. 1970;5:91–9. [DOI] [PubMed] [Google Scholar]
- 83.Lee RB, Urban JPG. Evidence for a negative Pasteur effect in articular cartilage. Biochem J. 1997;321:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee RB, Urban JPG. Functional replacement of oxygen by other oxidants in articular cartilage. Arthritis Rheum. 2002;46(12): 3190–200. [DOI] [PubMed] [Google Scholar]
- 85.Huang S, Heikal AA, Webb WW. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J. 2002;82:2811–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kunz WS, Kunz W. Contribution of different enzymes to flavoprotein fluorescence of isolated rat liver mitochondria. Biochim Biophys Acta. 1985;841:237–46. [DOI] [PubMed] [Google Scholar]
- 87.Heikal AA. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark Med. 2010;4(2):241–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Quinn KP, Sridharan GV, Hayden RS, Kaplan DL, Lee K, Georgakoudi I. Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci Rep. 2013;3: 3432 10.1038/srep03432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang C, Silverman RM, Shen J, O’Keefe RJ. Distinct metabolic programs induced by TGF-β1 and BMP2 in human articular chondrocytes with osteoarthritis. J Orthop Translat. 2018;12:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heywood HK, Knight MM, Lee DA. Both superficial and deep zone articular chondrocyte subpopulations exhibit the Crabtree effect but have different basal oxygen consumption rates. J Cell Physiol. 2010;223:630–9. [DOI] [PubMed] [Google Scholar]
- 91.Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 2013;17: 745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karner CM, Esen E, Chen J, Hsu F-F, Turk J, Long F. Wnt protein signaling reduces nuclear acetyl-CoA levels to suppress gene expression during osteoblast differentiation. J Biol Chem. 2016;291(25):13028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhao X, Petursson F, Viollet B, Lotz M, Terkeltaub R, Liu-Bryan R. Peroxisome proliferator-activated receptor γ coactivator 1α and FoxO3A mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheum. 2014;66(11):3073–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen L-Y, Lotz M, Terkeltaub R, Liu-Bryan R. Modulation of matrix metabolism by ATP-citrate lyase in articular chondrocytes. J Biol Chem. 2018;293(31):12259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stincone A, Prigione A, Cramer T, Wamelink MMC, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. 2015;90:927–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.•.Silverton SF, Matsumoto H, DeBolt K, Reginato A, Shapiro IM. Pentose phosphate shunt metabolism by cells of the chick growth cartilage. Bone. 1989;10:45–51.This study relates activity of the pentose phosphate pathway to oxygen utilization and NADPH production during chondrocyte maturation.
- 97.Hough S, Russell JE, Teitelbaum SL, Avioli LV. Regulation of epiphyseal cartilage metabolism and morphology in the chronic diabetic rat. Calcif Tissue Int. 1983;35:115–21. [DOI] [PubMed] [Google Scholar]
- 98.Clancy RM, Abramson SB, Kohne C, Rediske J. Nitric oxide attenuates cellular hexose monophosphate shunt response to oxidants in articular chondrocytes and acts to promote oxidant injury. J Cell Physiol. 1997;172:183–91. [DOI] [PubMed] [Google Scholar]
- 99.Adeva-Andany MM, González-Lucán M, Donapetry-García C, Fernández-Fernández C, Ameneiros-Rodríguez E. Glycogen metabolism in humans. BBA Clin. 2016;5:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem J. 2012;441:763–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fang X, Yu SX, Lu Y, Bast RCJ, Woodgett JR, Mills GB. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci U S A. 2000;97(22): 11960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tokunaga H, Watanabe J, Kanai K, Sakaida M, Kanamura S. Glucose 6-phosphatase and glycogen phosphorylase activities in chondrocytes in epiphyseal cartilage of growing rats. Anat Rec. 1987;219:356–62. [DOI] [PubMed] [Google Scholar]
- 103.Daimon T The presence and distribution of glycogen particles in chondrogenic cells of the tibiotarsal anlage of developing chick embryos. Calcif Tissue Res. 1977;23:45–51. [DOI] [PubMed] [Google Scholar]
- 104.Godman GC, Porter KR. Chondrogenesis, studied with the electron microscope. J Biophys Biochem Cytol. 1960;8:719–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marchand F Ueber eine Geschwulst aus quergestreiften Muskelfasern mit ungewöhnlichem Gehalte an Glykogen, nebst Bemerkungen über das Glykogen in einigen fötalen Geweben. Virchows Arch Pathol Anat Physiol Klin Med. 1885;100(1):42–65. [Google Scholar]
- 106.Hutchison MR, Bassett MH, White PC. Insulin-like growth factor-I and fibroblast growth factor, but not growth hormone, affect growth plate chondrocyte proliferation. Endocrinology. 2007;148(7):3122–30. [DOI] [PubMed] [Google Scholar]
- 107.Gillespie JR, Ulici V, Dupuis H, Higgs A, DiMattia A, Patel SC, et al. Deletion of glycogen synthase kinase-3β. Endocrinology. 2011;152(5):1755–66. [DOI] [PubMed] [Google Scholar]
- 108.Long F, Joeng KS, Xuan S, Efstratiadis A, McMahon AP. Independent regulation of skeletal growth by Ihh and IGF signaling. Dev Biol. 2006;298:327–33. [DOI] [PubMed] [Google Scholar]
- 109.Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci. 2009;35(3):161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Edouard T, Grünenwald S, Gennero I, Salles JP, Tauber M. Prevalence of IGF1 deficiency in prepubertal children with isolated short stature. Eur J Endocrinol. 2009;161:43–50. [DOI] [PubMed] [Google Scholar]
- 111.Kamboj M Short stature and growth hormone. Indian J Pediatr. 2005;72(2):149–57. [DOI] [PubMed] [Google Scholar]
- 112.Iliev DI, Kannenberg K, Weber K, Binder G. IGF-I sensitivity in silver-Russell syndrome with IGF2/H19 hypomethylation. Growth Hormon IGF Res. 2014;24:187–91. [DOI] [PubMed] [Google Scholar]
- 113.Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel H-M, et al. Paternally inherited IGF2 mutation and growth restriction. N Engl J Med. 2015;373:349–56. [DOI] [PubMed] [Google Scholar]
- 114.Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sparago A, Cerrato F, Vernucci M, Battista Ferrero G, Cirillo Silengo M, Riccio A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat Genet. 2004;36(9):958–60. [DOI] [PubMed] [Google Scholar]
- 116.Ornitz DM. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wong SC, Dobie R, Altowati MA, Werther GA, Farquharson C, Ahmed SF. Growth and the growth hormone-insulin like growth factor 1 axis in children with chronic inflammation: current evidence, gaps in knowledge, and future directions. Endocr Rev. 2016;37(1):62–110. [DOI] [PubMed] [Google Scholar]
- 118.Liem JJ, Rosenberg AM. Growth patterns in juvenile rheumatoid arthritis. Clin Exp Rheumatol. 2003;21:663–8. [PubMed] [Google Scholar]
- 119.Zhang Z, Lindstrom MJ, Lai HJ. Pubertal height velocity and associations with prepubertal and adult heights in cystic fibrosis. J Pediatr. 2013;163(2):376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Carpio LR, Bradley EW, McGee-Lawrence ME, Weivoda MM, Poston DD, Dudakovic A, et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci Signal. 2016;9(440):ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shikhman AR, Brinson DC, Valbracht J, Lotz MK. Cytokine regulation of facilitated glucose transport in human articular chondrocytes. J Immunol. 2001;167:7001–8. [DOI] [PubMed] [Google Scholar]
- 122.Mårtensson K, Chrysis D, Sävendahl L. Interleukin-1β and TNF-α act in synergy to inhibit longitudinal growth in fetal rat metatarsal bones. J Bone Miner Res. 2004;19(11):1805–12. [DOI] [PubMed] [Google Scholar]
- 123.Kiely A, McClenaghan NH, Flatt PR, Newsholme P. Pro-inflammatory cytokines increase glucose, alanine and triacylglycerol utilization but inhibit insulin secretion in a clonal pancreatic β-cell line. J Endocrinol. 2007;195:113–23. [DOI] [PubMed] [Google Scholar]
- 124.Chen L, Chen R, Wang H, Liang F. Mechanisms linking inflammation to insulin resistance. Int J Endocrinol. 2015;2015:508409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Su N, Jin M, Chen L. Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone Res. 2014;2:14003 10.1038/boneres.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Munns CFJ, Batch JA. Hyperinsulinism and Beckwith-Wiedemann syndrome. Arch Dis Child Fetal Neonatal Ed. 2001;84:F67–F9. [DOI] [PMC free article] [PubMed] [Google Scholar]