

Abstract
Maternal low-protein diet (LP) throughout gestation affects pancreatic β-cell fraction of the offspring at birth, thus increasing their susceptibility to metabolic dysfunction and type 2 diabetes in adulthood. The present study sought to strictly examine the effects of LP during the last week of gestation (LP12.5) alone as a developmental window for β-cell programming and metabolic dysfunction in adulthood. Islet morphology analysis revealed normal β-cell fraction in LP12.5 newborns. Normal glucose tolerance was observed in 6- to 8-wk-old male and female LP12.5 offspring. However, male LP12.5 offspring displayed glucose intolerance and reduced insulin sensitivity associated with β-cell dysfunction with aging. High-fat diet exposure of metabolically normal 12-wk-old male LP12.5 induced glucose intolerance due to increased body weight, insulin resistance, and insufficient β-cell mass adaptation despite higher insulin secretion. Assessment of epigenetic mechanisms through microRNAs (miRs) by a real-time PCR-based microarray in islets revealed elevation in miRs that regulate insulin secretion (miRs 342, 143), insulin resistance (miR143), and obesity (miR219). In the islets, overexpression of miR143 reduced insulin secretion in response to glucose. In contrast to the model of LP exposure throughout pregnancy, islet protein levels of mTOR and pancreatic and duodenal homeobox 1 were normal in LP12.5 islets. Collectively, these data suggest that LP diet during the last week of pregnancy is critical and sufficient to induce specific and distinct developmental programming effects of tissues that control glucose homeostasis, thus causing permanent changes in specific set of microRNAs that may contribute to the overall vulnerability of the offspring to obesity, insulin resistance, and type 2 diabetes.
Keywords: β-cell function, diabetes, fetal nutrition disorders, fetal programming, islet biology, low-protein diet
INTRODUCTION
Events in pregnancy can have far-reaching consequences for the child. Suboptimal maternal environment, such as malnourishment during pregnancy, is recognized to predispose the fetus to intrauterine growth restriction (IUGR), a condition in which the fetus in the womb fails to achieve its full genetic potential for growth and size. IUGR is now identified as a critical factor in predisposing offspring to adult-onset disorders such as cardiovascular, as well as metabolic and endocrine diseases, including type 2 diabetes (5, 23, 29). Such adverse outcomes point to the importance of optimal nutritional status during pregnancy in regulating long-term organ growth and programming of the function of key metabolic cells such as the pancreatic β-cell and insulin-sensitive tissues (11, 22, 37).
The “Barker hypothesis” proposes that specific organs, such as the pancreas, and their associated functions undergo programming during fetal development in the womb, altering the set point of physiological and metabolic responses that carry on into adulthood (6). It is likely that the degree and developmental window of insults that occur in early life can determine various aspects of metabolic programming. However, how critical “developmental windows” in utero impact on organs that determine metabolic control are not clearly defined. The majority of rodent studies have largely been conducted in the rat and involve maternal low-protein diet (LP) manipulation throughout the entire pregnancy period alone or in combination with the lactation stage. In contrast to the few studies investigating the effects of LP during the last week of gestation in the rat, the metabolic effects of this developmental window have not been investigated in the mouse. We and others have previously reported that administration of LP both in rat and mouse models impairs development of pancreatic insulin-producing β-cell and responses of peripheral tissues to insulin (2, 12, 13, 26, 31, 34, 35). Offspring of rat or mouse dams fed LP (9 versus 23% control) throughout pregnancy period show reduced β-cell mass and glucose intolerance in adult offspring (2, 12, 13, 26, 34). A longer LP exposure until weaning resulted in a significant decrease in β-cell mass early in life, as well as glucose intolerance and reduced insulin secretion in later life (31, 35). However, the effects in glucose homeostasis and β-cell mass dysfunction in the offspring of dams exposed to LP during the last week of pregnancy in the mouse remain unknown.
Fetal programming of β-cell dysfunction by low protein is complex and may involve alteration of nutrient-sensing pathways, transcriptional regulatory networks, and microRNAs (miRs) in early life, and these changes may predispose the offspring to metabolic dysfunction in adulthood. The reduction of functional β-cell mass has been associated with the downregulation of insulin and relevant transcription factors [FoxO1, pancreatic and duodenal homeobox 1 (Pdx1), Hnf4a, and MafA] as well as other essential proteins (mTOR, IGF-II) for β-cell formation, maturation, and health (2, 26, 35, 41). miRs are also likely to play a role in the mechanism of fetal programming, since they play a role in many developmental processes starting from implantation of conceptus and persisting through the prenatal and postnatal periods (21). Indeed, miRs that have been shown to regulate mouse β-cell development and function are also implicated in fetal programming of β-cell dysfunction (miR375, miR199, miR15b) by LP diet throughout pregnancy (2, 18, 35). However, miR expression in the offspring of LP12.5, rat or in the mouse, has not been explored.
A greater understanding of the critical periods of fetal programming during development is timely and significant, and may provide a window of opportunity for interventions. A typical mouse gestation period is 19 days, and the low protein exposure model during the last 7 days of pregnancy (LP12.5) would be a similar model to poor nutrient exposure during the late second and throughout the third trimester in humans. A similar phenotype is caused in humans by placental insufficiency, which is not often diagnosed before 20 wk of gestation and is associated with poor fetal growth before 32 wk of gestation. Thus the LP12.5 animal model could provide important information about the impact of poor fetal nutrient delivery during this later stage of pregnancy, a condition that can impact up to 9% of pregnancies (4).
The current studies were designed to fill a gap in our knowledge by assessing the impact of LP diet during the last week of pregnancy in mice. In contrast to LP0.5, exposure to LP during the last week of pregnancy induced glucose intolerance with aging and after diet-induced obesity. Insulin resistance with insufficient functional adaptation of the β-cell characterized this phenotype. Moreover, LP12.5 exhibited alterations in a set of islet miRNA expression that were more distinct than those observed in LP0.5, suggesting that nutritional stresses at different developmental windows result in a distinct subtype of miRNA expression. The metabolic characterization of the offspring of LP12.5 in the mouse model would provide a platform to design much needed mechanistic-oriented studies using genetically modified murine models.
MATERIALS AND METHODS
Low-protein mouse model and high-fat diet treatment.
Virgin C57B6 females and males at 8 wk of age purchased from the Jackson Laboratories were allowed to adapt to control diet (D02041001B; Research Diets, New Brunswick, NJ) for 4 wk. After acclimation to the control diet, females and males were housed together, and vaginal plugs were checked daily. On detection of a vaginal plug (designated E0.5), females were single housed and fed either low protein after 12 days [E12.5 (LP12.5), 9% protein, D02041002; Research Diets] or control 23% protein (D02041001B; Research Diets). Both control and LP diets are isocaloric but differ in the amount of protein in the form of casein. Pregnant dams delivered spontaneously on gestational day 19. Nursing dams and weanlings had access to control diet and water ad libitum until death. A cohort of 12-wk-old male LP12.5 and control mice were challenged with high-fat diet (60% kcal; Research Diet) for 20 wk. All animal care and procedures were in accordance with the Institutional Animal Care and Use Committee-approved protocols at the University of Michigan.
Primary mouse islet isolation.
Islets were isolated by collagenase digestion. The pancreas was perfused via the common duct with 1 mg/mL collagenase XI (Sigma-Aldrich, St. Louis, MO) in HBSS (Gibco Invitrogen, Grand Island, NY). Pancreatic digestion was carried out at 37°C for 10–20 min, after which cold HBSS with 10% vol/vol FBS (Gibco-Invitrogen) was added. The suspension was centrifuged at 1,000 revolutions/min for 30 s, washed three times with HBSS with 10% vol/vol FBS, resuspended in RPMI with 5 mM glucose and 10% vol/vol FBS, and poured on a 70-µm cell strainer (BD Falcon; BD Biosciences, San Diego, CA). Islets were rinsed and handpicked. Islets were allowed to recover overnight before experiments were carried out.
Pancreas section collection.
Formalin-fixed pancreatic tissues were embedded in paraffin using standard techniques. Newborn pancreata from each group (LP12.5 and control) were harvested on the day of birth. Harvested pancreas were soaked in staining dye for 10 s before incubating in 3.75% formalin (Fisher Scientific, Hampton, NH) for 5 h and then in 70% ethanol before embedding process. Newborn pancreata were sectioned from top to bottom of the pancreas (5 µm thickness), and adult tissue blocks were sectioned at 5 µm thickness, 200 µm apart as previously described (2).
β-Cell and adipose morphometric analysis.
Sections were deparaffinized, rehydrated, and incubated with blocking solution as previously described (8). Sections were incubated overnight at 4°C with antibodies against insulin (DAKO, Santa Clara, CA), and Ki-67 (Abcam, Cambridge, MA) followed by secondary antibodies conjugated to FITC or Cy5 (Jackson Immunoresearch, West Grove, PA). DAPI-containing mounting medium (Vector Laboratories, Burlingame, CA) was added to cover slips. Whole pancreas and insulin-positive cell areas from five insulin-stained sections (5 µm) separated by 50 μm (newborn pancreas) or 200 µm (adult pancreas) per animal were calculated by using Image Pro Software (Media Cybernetics) as previously described (2). β-Cell fraction in newborn is calculated as the average of insulin-positive/pancreas ratio. Adult β-cell mass is the average of five insulin-positive/pancreas ratio multiplied by the pancreas weight. Proliferating cells were identified by costaining for antigen identified by monoclonal antibody Ki-67 and insulin. Pdx1, phosphorylated S6, and TUNEL staining were done in insulin-positive cells. TUNEL staining was completed using the Millipore ApopTag Red in situ Apoptosis Detection Kit following the manufacturer’s protocol. At least 1,000–3,000 stained cells were counted from each animal. Pdx1, phosphorylated S6 (Ser240), and insulin were purchased from Abcam, Cell Signaling, and Sigma Aldrich, respectively. Validation of Pdx1 and insulin antibodies by immunofluorescence demonstrated specificity to β-cells. Ki-67 and TUNEL staining were restricted in the nucleus as expected. For immunochemical imaging, phosphorylated S6 (S24)-stained sections were incubated with secondary antibodies conjugated to biotin and then Vectastain ABC reagent (from Vectastain Elite HRP kit; Vector Laboratories). Adipose sections were then stained with DAB peroxidase substrate (Vector Laboratories) and counterstained with Mayer's hematoxylin (Sigma) before imaging on a Leica MC 120 HD microscope. The frequency distribution of adipocyte sizes across the tissue depot and the size of adipocytes per depot was estimated by counting ∼100–500 adipocytes/animal as previously described (30).
Protein and RNA isolation.
Protein and RNA samples were prepared from three to four pancreata, livers, and epididymal white adipose tissue (eWAT) from each treatment group. Samples were quickly washed with 1× PBS before incubating in RIPA lysis buffer (Cell Signaling, Danvers, MA) or with TRIzol (Sigma-Aldrich). Samples were frozen quickly with liquid nitrogen and then kept at −80°C before homogenization. Total RNA for gene expression analysis was isolated using a method with TRIzol combined with RNeasy columns (Qiagen, Valencia, CA). RNA for microRNA analysis was isolated using RNAlater and a MirVana Kit (Invitrogen, Carlsbad, CA).
Quantitative real-time PCR for gene and microRNA expression.
From isolated total RNA, cDNA synthesis was performed using random hexamers and was reverse transcribed using Superscript II (Invitrogen) according to the manufacturer’s protocol. Real-time PCR was performed on an ABI 7000 sequence detection system using Taqman gene expression assays (Applied Biosystems, Foster City, CA). Primers were purchased from Applied Biosystems. Primers used are listed in SM-1. For microRNA expression analysis, we used Taqman MicroArray Assays (Applied Biosystems).
OpenArray miR arrays.
Each TaqMan OpenArray MicroRNA Panel (Applied Biosystems) contains 754 well-characterized miR sequences from the Sanger miRBase v14. All 754 assays have been functionally validated with miR artificial templates. Full-service processing by the University of Michigan Microarray Core Facility included assessment of RNA quality by an Agilent Bioanalyzer, probe labeling from total RNA submitted, hybridization, scanning, and data analysis. Islets were harvested from male LP12.5 and control mice (8–12 wk of age), and RNA was isolated by the MirVana Kit for OpenArray microarray (n = 5).
Protein detection.
Immunoblotting was performed as described (3). Briefly, the samples were washed after treatments with PBS before adding cell lysis buffer (Cell Signaling) with protease inhibitor cocktail and PhosphoStop tablets (Roche Applied Science, Indianapolis, IN). Whole membrane blots were cut to maximize multiple protein detections. Primary antibodies against mTOR, phosphorylated S6 (Ser240) and Akt (Ser473 and T308), total Akt, Erk1/2, and vinculin were from Cell Signaling. Antibody against Pdx-1 was from Millipore. CDKAL and tubulin antibodies were purchased from Sigma-Aldrich. Secondary antibodies conjugated to HRP were from Jackson Immunoresearch.
Glucose and insulin tolerance tests.
Glucose and insulin tolerance tests were performed by intraperitoneal delivery of 2 g/kg glucose or 0.75 U/kg insulin (Novolin; Novo Nordisk, Princeton, NJ) to mice after 12 or 6 h of fasting, respectively. Blood glucose was monitored for 2 h after intraperitoneal glucose or insulin delivery. Fasting glucose and insulin were measured by obtaining blood from tail vein after overnight fasting. Glucose was measured using an AccuChek II glucometer (Roche Applied Science). Plasma insulin levels were measured using a rat insulin ELISA kit (Crystal Chem, Downers Grove, IL).
Insulin secretion analysis in vitro.
Isolated islets were cultured overnight with regular RPMI media containing 5 mM glucose and 10% vol/vol FBS. The following day, 10 islet cells were washed two times with PBS (Invitrogen) and cultured in serum-free and 2 mM glucose for 1 h before being placed in 2 or 16 mM glucose for 15 min. As a positive control for insulin secretion, we used 30 mM KCl. Insulin secretion was measured using an ELISA [Insulin (Mouse) High Range ELISA; ALPCO Immunoassays, Salem, NH].
NMR analysis of body composition.
NMR (Minispec LF90II; Bruker Biosciences, Billerica, MA) was used to investigate body composition through the University of Michigan Nutrition Obesity Research Center Animal Phenotyping Core.
Supplemental figures.
Supplemental Figs. S1–S7 are deposited at https://doi.org/10.6084/m9.figshare.12502013.
Statistical analysis.
Data were analyzed by Student’s t test or ANOVA followed by post hoc analysis where appropriate by using GraphPad Prism 8 Software (GraphPad Software, San Diego, CA). Results were considered statistically significant when the P value was <0.05.
RESULTS
Maternal low-protein diet in the LP12.5 mouse model.
To determine the developmental window of maternal LP diet programming of pancreatic β-cell mass dysfunction in the offspring, we tested the consequences of low maternal protein intake during the last week of pregnancy. In this model, LP diet was administered during the last week of pregnancy in normal wild-type virgin C57BL6 mice as shown in Fig. 1A (herein referred to as LP12.5). Females fed control and low-protein (LP12.5) diets had similar body weights at conception and comparable weight gain throughout pregnancy (Fig. 1B). No alteration in glucose levels in pregnant dams was observed at day 12.5 of pregnancy and at delivery (data not shown). After vaginal delivery on gestational day 19, nursing dams were switched to control diet ad libitum. Litter size at birth was not reduced, but the litter size on postnatal day 21 across all litters generated was reduced in the LP12.5 than the control dams (Fig. 1C). Newborn body weight was also reduced (∼5% of control) in LP12.5 compared with control (Fig. 1D), supporting the importance of maternal protein diet during the last week of pregnancy, a period of high fetal growth. No alteration in glucose, body length, insulin levels, islet area/pancreas area, or average cell/islet was observed between LP12.5 newborn and control on day 1 of life (Fig. 1, E–I), suggesting that maternal LP diet during the last week of pregnancy is not sufficient to alter β-cell fraction at birth in the mouse. Interestingly, we showed a nonsignificant trend toward increased β-cell proliferation in pancreas of LP12.5 offspring compared with control (Fig. 1J). These data indicate that LP diet during the last week of pregnancy caused smaller fetal body weight but normal β-cell fraction at birth.
Fig. 1.

Maternal low-protein (LP) diet on the last week of pregnancy and phenotype of newborn offspring. A: pregnant C57BL/6 mice were exposed to diets [control (Ctrl, 23% protein) or LP during the last week of gestation (LP12.5, isocaloric and 9% protein)] during the last week of pregnancy. After delivery, LP12.5 and Ctrl dams and offspring were introduced to Ctrl diet. B: maternal body weight during the adaptation period [days (D) 0–30], during pregnancy [embryonic day (e) 05–16.5], and after pregnancy [postnatal day (P) 1–15]. C: litter size on P21 between LP12.5 and Ctrl dams. D–F: newborn body weight (D), blood glucose (E), body length (F) of LP12.5 and Ctrl offspring. G–J: newborn circulating insulin levels (G), β-cell area (H), average number of cells per islet (I), and β-cell proliferation, as measured through Ki-67 staining (J), were assessed in LP12.5 and Ctrl offspring. n = 4 Dams were used for maternal body weight assessment (B) and litter size (C). n = 16–27 Offspring were used for D–J and from n = 4 litter. Analyzed by Student’s t test, 2 tailed and unpaired, *P < 0.05 versus Ctrl.
Adult LP12.5 males are heavier but exhibit normal glucose tolerance under regular diet.
Measurements of body weight after birth showed that body weight in LP12.5 offspring was increased during the first 4 wk (Supplemental Fig. S1A). As shown in Supplemental Fig. S1B, 6-wk-old LP12.5 male offspring were heavier compared with control, but this weight gain was transient, since LP12.5 offspring reach a similar body weight to control mice at 8 wk of age (Supplemental Fig. S1A). The changes in body weight observed in 6-wk-old male LP12.5 offspring were accompanied by normal random and fasting glucose level as well as normal islet insulin content (Supplemental Fig. S1, C–E). In contrast to males, no differences in body weight and fasting glucose were observed in 8 wk-old female LP12.5 offspring (Supplemental Fig. S1, F and H) compared with control. A tendency toward higher glucose concentrations in the fed state and lower islet insulin content was observed in female LP12.5 offspring (Supplemental Fig. S1, G–I). Assessment of body weight and some metabolic parameters in older mice showed that 48-wk-old LP12.5 males exhibited similar body weight and elevated random or nonfasted glucose levels (Supplemental Fig. S1, J and K). Comparable fasting glucose and insulin levels in the nonfasted and fasting state were observed between male LP12.5 and controls at 48 wk of age (Supplemental Fig. S1, L–N). In 48-wk-old LP12.5 females, there was a trend toward higher random or nonfasted glucose with similar body weight and fasting glucose (Supplemental Fig. S1, O–Q). Random insulin levels were significantly higher, but fasting insulin levels were only a trend toward significance (P = 0.14) (Supplemental Fig. S1, R and S). Liver and pancreas weights in 48-wk-old male LP12.5 were equivalent to control offspring (data not shown). In contrast, 51-wk-old female LP12.5 offspring displayed reduced liver and pancreas weight (data not shown). Collectively, these data show that male LP12.5 offspring show compensatory growth, and both older male and female LP12.5 mice developed mild hyperglycemia compared with controls.
Impaired glucose tolerance in aged male and female LP12.5 offspring under normal diet.
To assess glucose homeostasis in LP12.5 mice, we performed an intraperitoneal glucose tolerance test (IPGTT) in young (6 wk old) and aged (48–51 wk old) mice. Normal glucose tolerance in both male and female groups was observed between LP12.5 and control mice at 6 wk of age (Fig. 2, A and C). In contrast to the normal glucose tolerance at 6 (Fig. 2, A and C) and 24 (Supplemental Fig. S2A) wk of age, 48-wk-old male LP12.5 offspring developed significant glucose intolerance compared with control mice (Fig. 2B). Glucose intolerance was also detected in 48-wk-old LP12.5 female mice (Fig. 2D). To assess insulin sensitivity from peripheral tissues, we performed insulin tolerance tests in young and aged mice. Normal insulin sensitivity was observed in male mice at both 10 or 54 wk of age (Fig. 2, E and F). Similar to young males, LP12.5 females displayed normal insulin sensitivity at 8 wk of age (Fig. 2G). However, 51-wk-old female LP12.5 mice developed mild but significant insulin resistance (Fig. 2H). Together, these data suggest that old male and female LP12.5 mice exhibited glucose intolerance, but only females showed insulin resistance with aging.
Fig. 2.

Adult offspring administered low protein during the last week of gestation (LP12.5) exhibit glucose intolerance, but only females display insulin resistance. Intraperitoneal glucose tolerance tests were performed in 6- and 50-wk-old male [A and B, area under the curve (AUC), P = 0.026] and 6- and 48-wk-old female (C and D, AUC, P = 0.056) LP12.5 and control (Ctrl) offspring. Intraperitoneal insulin tolerance tests (ITT) were performed in 10- and 54-wk-old male (E and F, AUC, P = 0.787) and 8- and 51-wk-old female (G and H, AUC, P = 0.01) LP12.5 and Ctrl mice. *P < 0.05 vs. Ctrl. Analyzed by Student’s t test, 2 tailed and unpaired, *P < 0.05 vs. Ctrl, n = 4–9 animals in each group.
Impaired insulin secretion but increased β-cell mass in aged LP12.5 male offspring.
Because we observed normal insulin sensitivity in the setting of glucose intolerance in the aged male LP12.5 offspring, we assessed whether this phenotype was due to defects in insulin secretion. Assessment of glucose tolerance and glucose-stimulated insulin secretion (GSIS) in vivo shows that aged LP12.5 male offspring exhibited impaired glucose clearance and reduced insulin secretion after intraperitoneal glucose injection (Fig. 3, A and B). To further characterize the in vivo GSIS findings, we exposed isolated primary islets from 48-wk-old male mice to high glucose (25 mM) or KCl (30 mM) for 30 min. Islets from 48-wk-old LP12.5 males displayed lower GSIS in response to glucose but not KCl (Fig. 3C). Male LP12.5 mice (48 wk old) showed increased β-cell mass compared with controls (Fig. 3D). In contrast, normal β-cell mass was observed in 51-wk-old LP12.5 females at the time where impaired glucose tolerance was observed (Fig. 3E), which was unexpected given the insulin resistance and hyperinsulinemia seen in aged female LP12.5 offspring. However, the normal β-cell mass in female LP12.5 was supported by normal average cell size and rates of apoptosis and proliferation between female LP12.5 and control mice (Supplemental Fig. S3, A–D). Preliminary data also suggested a normal average cell size and proliferation rate in male LP12.5 and control mice (Supplemental Fig. S3, E–G). The liver and pancreas weight were reduced in aged female (Supplemental Fig. S3, H and I), but unaltered in the male (Supplemental Fig. S3, J and K). Altogether these data suggest that glucose intolerance in both males and females was not due to a defect in β-cell mass.
Fig. 3.
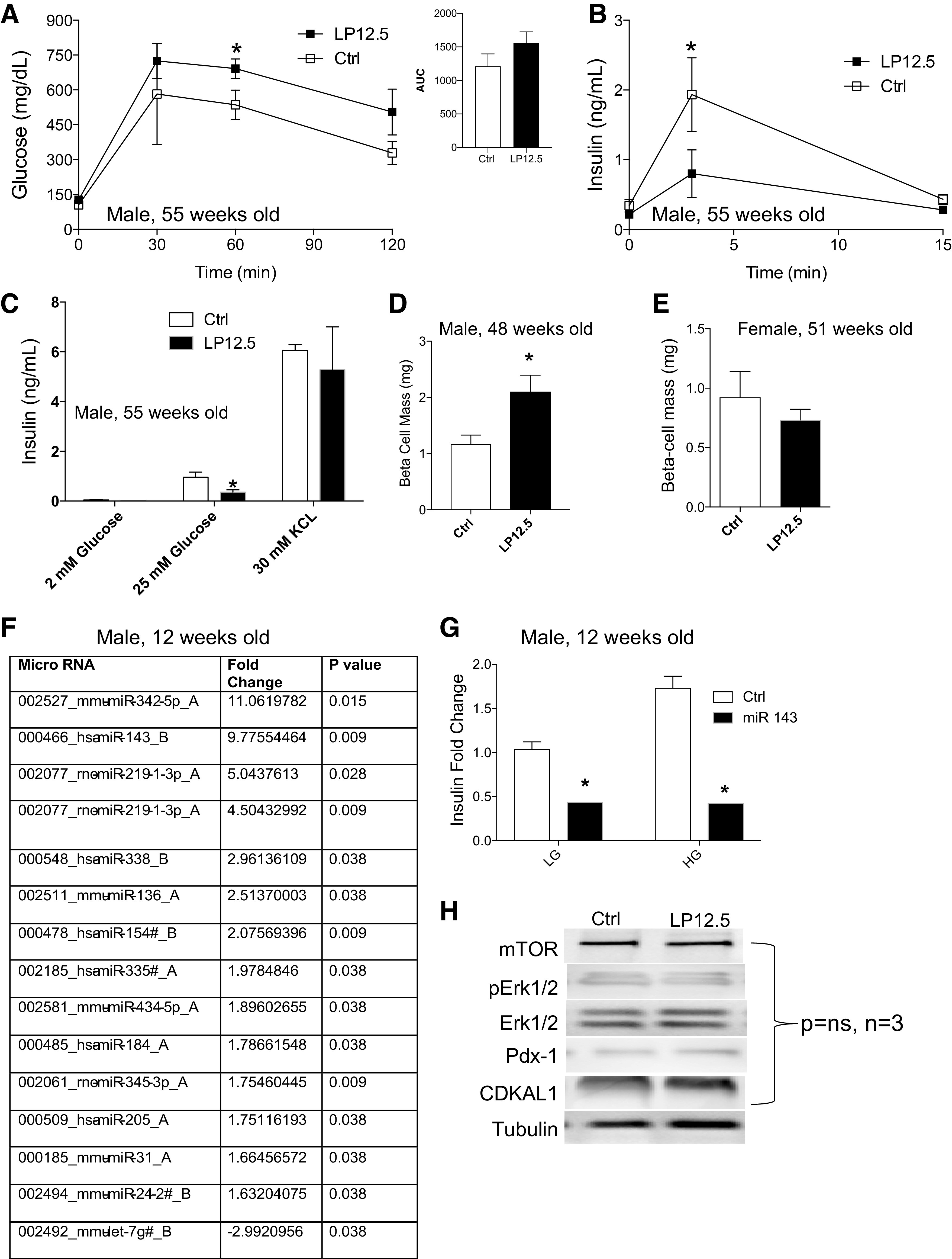
Reduced glucose-stimulated insulin secretion in aged male mice administered low protein (LP) during the last week of gestation (LP12.5). A and B: in vivo intraperitoneal glucose tolerance tests and glucose-stimulated insulin secretion (GSIS) were performed on 48-wk-old male LP12.5 and control (Ctrl) offspring [area under the curve (AUC), P = 0.2]. C: GSIS in isolated islets from adult LP12.5 and Ctrl male mice, in response to 2, 25 mM glucose and 30 mM KCl for 30 min. β-Cell mass of 48-wk-old male (D) and 51-wk-old female (E) LP12.5 and Ctrl mice. Ctrl, n = 4–5 mice. F: summary of altered microRNA level in islets of adult male LP12.5 mice. Only microRNAs passing the stringent threshold for significant change are shown. Islets were harvested from 12-wk-old LP12.5 and Ctrl mice. RNA was isolated by the MirVana Kit for OpenArray microarray. Quality of RNA was assessed by an Agilent Bioanalyzer (n = 5 mice). G: insulin secretion in primary mouse pancreatic β-cells from 12-wk-old male LP12.5 and Ctrl islets treated with or without miR143. LG, low glucose; HG, high glucose. H: representative Western blot for mTOR, pErk, total Erk, pancreatic and duodenal homeobox 1 (Pdx1), and Cdk5 regulatory-associated protein 1-like 1 (CDKAL1). Analyzed by Student’s t test, 2 tailed and unpaired, *P < 0.05 vs. Ctrl, n = 3–5.
Alteration of multiple microRNAs in LP12.5 offspring islets.
We and others have previously described a group of microRNAs (miRs) that play an important role in β-cell function in the offspring of dams exposed to low protein throughout pregnancy (LP0.5) (2, 26). Therefore, we designed unbiased studies to investigate the expression levels of miRs and elucidate the differences in miR expression and putative targets between islets of LP12.5 and LP0.5 offspring (2), by screening of 720 miRs in islets from 12-wk-old male LP12.5 and control mice using a real-time PCR-based microarray. Approximately 537 showed readable values in islets, and, of these, only 14 were altered: 13 were increased and 1 decreased (Fig. 3F). The most significantly increased miRs include miR342, -143, and -219, which have been previously implicated in playing regulatory roles in glucose metabolism, obesity (adipogenesis), and insulin resistance (20, 25, 27, 39). miR342 was also increased in LP0.5 islets and was previously shown to reduce insulin secretion in male islets from control mice (2). After miR342, miR143 was the next most upregulated miR in male LP12.5 islets. Because we have already studied the effects of miR342 in LP0.5 islets, we focused on exploring impact of miR143 on insulin secretion by transfecting mimic nucleotides into dispersed primary islet cells from control mice. We observed a decrease in insulin secretion in dispersed β-cells from control mice transfected with miR143 compared with scrambled control miR-transfected cells (Fig. 3G). microRNAs can regulate the translation of key β-cell proteins like mTOR and pancreatic and duodenal homeobox 1 (Pdx1), previously shown in islets of LP0.5 (1, 2). In addition, miR219 has binding sites on the Cdk5 regulatory-associated protein 1-like 1 (CDKAL1) (Target Scan). CDKAL1 is associated with impaired insulin response and one of the most reproducible risk genes in type 2 diabetes across different ethnic populations (14). In contrast to LP0.5 islets (2), we observed no alterations in mTOR protein, Pdx1, phosphorylated Erk, total Erk, and CDKAL1 between islets from LP12.5 and control 3 mo-old mice (Fig. 3H, P > 0.5, n = 3 mice). Pdx1 staining in insulin-positive cells was comparable in 1-yr-old male or female LP12.5 and control mice (Supplemental Fig. S4, A–C). mTOR activity, measured by downstream target phosphorylated S6, appeared unaltered in the islets of male or female LP12.5 and control mice (Supplemental Fig. S5, A and B). These data underscore the distinct developmental programming mechanisms of LP0.5 and LP12.5 in islets.
LP12.5 offspring development of glucose intolerance and insulin resistance in diet-induced obesity.
To test the responses to metabolic stress and susceptibility to diet-induced type 2 diabetes, 12-wk-old male LP12.5 and control mice were exposed to high-fat diet (HF) for 20 wk. LP12.5 mice exhibited mild but significant increase in body weight coupled with a higher rate of weight gain during HF diet exposure (Fig. 4, A and B). In contrast to comparable nonfasted glucose levels (Fig. 4C), fasting glucose was elevated in male LP12.5 contrasted to control mice (Fig. 4D). A trend toward hyperinsulinemia in the fed and fasting state was observed in male LP12.5 compared with control mice (Fig. 4, E and F).
Fig. 4.

Increased weight gain in adult male mice administered low protein during the last week of gestation (LP12.5) under high-fat (HF) diet-induced obesity. Body weight (A) and weight gain (B) over the course of HF diet in male LP12.5 and control (Ctrl) mice. Fed (C) and fasting (D) glucose over the course of HF diet in male LP12.5 and Ctrl mice. Fed (E) and fasting (F) insulin post-16-wk in HF diet of LP12.5 and Ctrl mice. Analyzed by Student’s t test, 2 tailed and unpaired, *P < 0.05 versus Ctrl, n = 6.
To investigate the glucose homeostasis of LP12.5 mice in HF diet, we performed IPGTT at 6, 10, and 14 wk post-HF diet, and we observed that LP12.5 offspring were glucose intolerant compared with controls (Fig. 5, A–C). The 2-h time point glucose level in LP12.5 was higher at 14 wk of HF diet, indicating worsening of the phenotype (Fig. 5C). Next, we assessed insulin sensitivity in these mice and noted insulin resistance in both groups, but LP12.5 offspring demonstrated more severe insulin insensitivity relative to controls (Fig. 5D). Examination of insulin secretion showed robust in vivo insulin secretion in response to HF diet when LP12.5 mice were challenged with glucose (Fig. 5E). No difference in β-cell mass was detected between LP12.5 and control mice at the end of HF exposure (Fig. 5F). The β-cell apoptosis and proliferation rates were not significantly altered in the LP12.5 compared with control mice (n = 5 mice, P = 0.15 and 0.63, respectively). These results suggest that the increase in insulin secretion observed in LP12.5 mice was still not sufficient to overcome the severe peripheral insulin resistance.
Fig. 5.
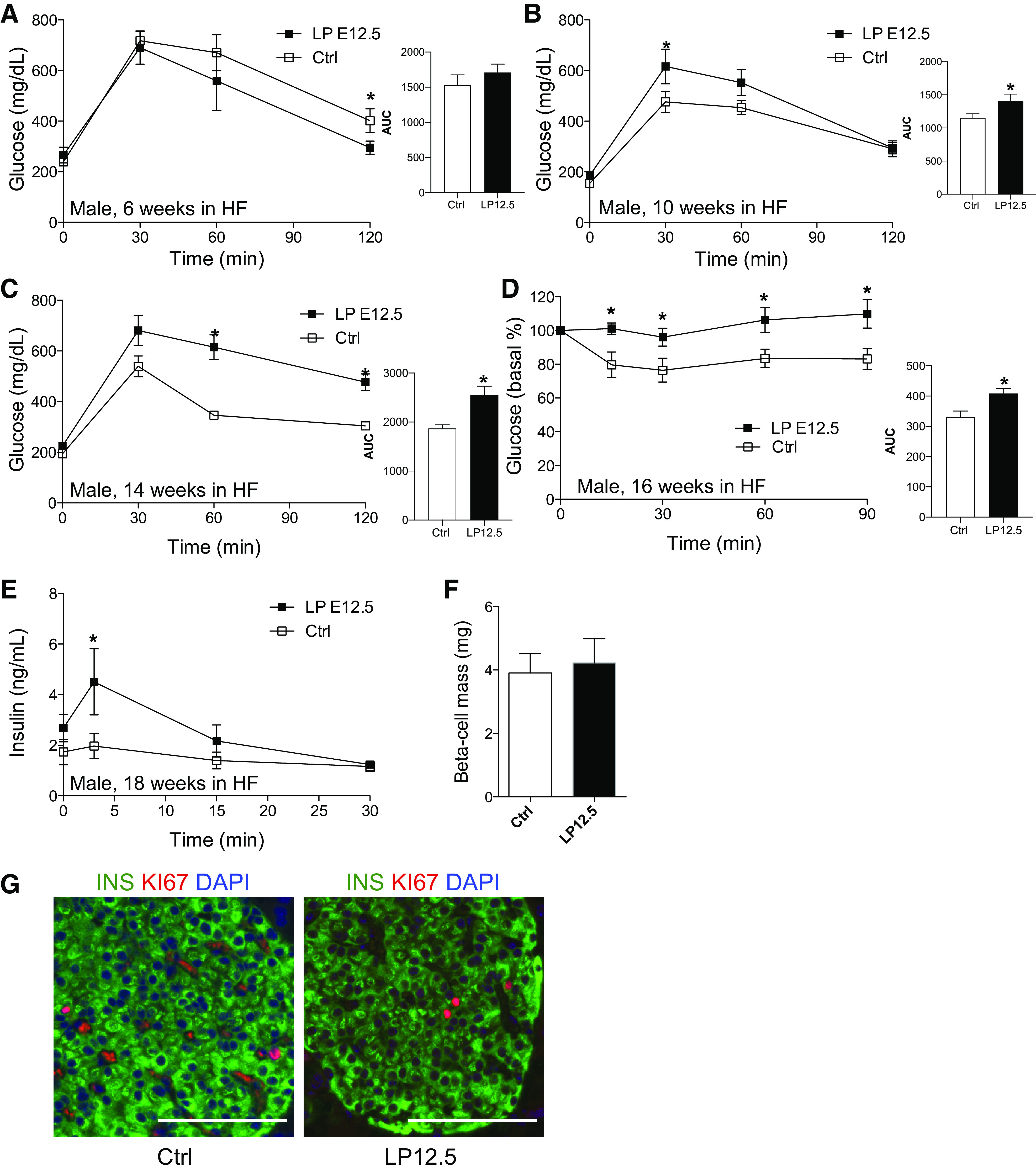
Adult male offspring administered low protein (LP) during the last week of gestation (LP12.5) exhibit glucose intolerance and insulin resistance in high-fat (HF) diet. Intraperitoneal glucose tolerance tests at 6 (A), 10 (B), and 14 (C) wk in HF [A and B: area under the curve (AUC), P = 0.05; C: AUC, P = 0.0063] and insulin tolerance tests at 16 wk in HF (D, AUC, P = 0.0238) of male LP12.5 and control (Ctrl) mice. In vivo glucose stimulated-insulin secretion (GSIS, E) and β-cell mass (F). Representative islets in male LP12.5 and Ctrl mice at 18 wk in HF diet (G, scale 100 μM). Analyzed by Student’s t test, 2-tailed and unpaired, *P < 0.05 vs. Ctrl, n = 4–6.
Increased lean but normal fat mass in obese LP12.5 offspring in high-fat diet.
To further characterize the increase in body weight observed in LP12.5 mice, we performed body composition (fat, lean, and fluid) by EchoMRI. Male LP12.5 mice were found to have no difference in fat mass or overall body fluid after 18 wk on HF diet (Supplemental Fig. S6A). No alterations were detected in epididymal, perirenal and subcutaneous adipose depot masses nor in the distribution of adipocyte sizes (Supplemental Fig. S6, B–E). Lean mass was higher in LP12.5 mice, thus implying that organs other than fat may contribute to the small but significant increase in body weight (Supplemental Fig. S6A). Tissue dissection post-HF diet showed nonsignificant differences in liver and pancreas weight in LP12.5 compared with controls (Supplemental Fig. S6, F and G). Energy expenditure measured by O2 consumption, CO2 production, and locomotor activity (Z and X) showed similar levels between LP12.5 and control mice (Supplemental Fig. S7, A–E). Moreover, food intake between LP12.5 and control mice was equivalent (Supplemental Fig. S7F).
Normal level of miR143 in LP12.5 adipose tissue.
To explore the mechanisms for enhanced weight gain and insulin resistance in LP12.5 male offspring after HF diet, we investigated the role of miR143 in adipose tissue. We focused on miR143 because this miR was the second mostly highly enriched miR identified in the male islets from LP12.5 under normal chow diet, and miR143 has been previously associated with obesity and insulin resistance. Moreover, miR143 expression in adipocytes was associated with an elevated body weight in HF diet-induced obesity in mice (27, 36) and is also increased in the liver of db/db and diet-induced obesity mice (25). miR143 expression in epididymal white adipose tissue was comparable between LP12.5 and control mice under HF diet (Fig. 6A). miR375 expression was also similar between adipose samples of male LP12.5 and control offspring after a HF diet challenge (Fig. 6B). miR143 has been shown to regulate Akt signaling (27). Validating the lack of alterations in adipocyte miR143 expression, no differences in phosphorylated Akt and phosphorylated S6 were observed between eWAT of LP12.5 offspring under HF diet exposure (Fig. 6, C–F). The phosphorylation status of S6 via immunohistochemistry demonstrated normal in the eWAT tissues was comparable between female and male LP12.5 and controls (Fig. 6G). These data suggest that miR143 expression in adipose tissue may not contribute to the phenotype of LP12.5 in HF diet.
Fig. 6.

Normal level of miR143 in adipose tissue of male offspring administered low protein (LP) during the last week of gestation (LP12.5). Quantitative PCR (qPCR) of miR143 (A) and miR375 (B) epididymal white adipose tissue (eWAT) from LP12.5 and control (Ctrl) mice fed high-fat (HF) diet. Representative Western blot (C) and quantification (relative to vinculin or total-Akt, normalized to Ctrl average) of eWAT in HF diet from LP12.5 and Ctrl mice: phospho-Akt S473 (D), phospho-Akt T308 (E), and phospho-S6 S240 (F). G: representative immunohistochemistry (IHC) images of phosphorylated S6 (S240) in eWAT adipose tissue of male (48 wk of age) and female (51 wk of age) mice. Student’s t test, 2-tailed and unpaired, n = 4 mice.
DISCUSSION
These studies extend our current knowledge by demonstrating that maternal low protein during the last week of gestation in mice is a critical developmental window for metabolic programming of the offspring. Young adult LP12.5 offspring were metabolically normal, but aged offspring developed glucose intolerance associated with β-cell dysfunction in the male. Male offspring was also more susceptible to HF diet (HFD) by a combination of increase weight with a decrease in insulin sensitivity and β-cell secretory responses. Specific upregulation of microRNAs that regulate insulin secretion may have contributed to β-cell dysfunction in aged male LP12.5 offspring. The abnormalities in glucose homeostasis (glucose and insulin intolerance) with aging were reproduced in the female offspring.
In the LP12.5 offspring, we observed normal glucose levels and glucose tolerance in young (6–8 wk of age) mice. This is in contrast to the glucose intolerance seen in the offspring of LP0.5 (LP throughout pregnancy) in the mouse (2) but in congruence with LP12.5 data in both male (15) and female (9) rats. This study is the first to demonstrate that older male and female LP12.5 offspring (48–51 wk of age) developed glucose intolerance. The mechanisms behind glucose intolerance in the male LP12.5 mice resulted in part by a defect in insulin secretion with normal insulin sensitivity. We identified increased β-cell mass in 48-wk-old male LP12.5, which may represent a compensatory mechanism in response to a primary insulin secretory defect. In contrast to males, the glucose intolerance in aged female LP12.5 mice was associated with insulin resistance (ITT and hyperinsulinemia; Fig. 2H and Supplemental Fig. 1R) and normal β-cell mass (Fig. 3E). This phenotype suggests that female LP12.5 exhibit insufficient β-cell compensation in response to insulin resistance in the peripheral tissues. The hyperinsulinemia in aged female LP12.5 may also suggest increased β-cell function as a compensatory response to insulin resistance, but insufficient to normalize glucose tolerance. The mechanisms behind the sexual dimorphism in β-cell mass and function are interesting and warrant further study to dissect the effect of sex hormones in the LP12.5 phenotype. The phenotypes observed in LP12.5 male and female mice in control chow diet support the notion that offspring of LP12.5 dams show increased vulnerability to metabolic syndrome induced by aging partially due to defects in insulin secretion (15). Altogether, our findings in mice and published data in the rat are consistent with the human insulin secretory defect associated with impaired glucose tolerance in people prenatally exposed to Dutch famine during midgestation and early gestation (9, 15, 38) (16).
To our knowledge, the assessment of glucose homeostasis in LP12.5 under metabolic stress such as HFD has not been tested in mice. In the present study, male LP12.5 offspring demonstrated increased weight gain under HFD after 4 wk but appeared to taper off and reached comparable weight gain with that observed in control mice after 12 wk of HFD. Similar responses to HFD-induced obesity have been previously observed in LP12.5 (8%) in the rat (38). Based on body composition analysis and assessment of energy homeostasis, the increase in body weight post-18 wk of HFD was caused by an increase in lean mass, suggesting that other organs except fat may have contributed to the small but significant increase in body weight in LP12.5 offspring. The changes in body composition in the LP12.5 after HFD were associated with elevations in fasting blood glucose, glucose intolerance, and insulin resistance after 10 wk of HFD. Compared with the reduced GSIS in aged male LP12.5 in normal chow, mice fed HFD displayed improved insulin secretion and normal β-cell mass, suggesting that islets from LP12.5 mice exposed to HFD are able to compensate by increasing function and not mass, but this compensatory response is inefficient to overcome the severe insulin resistance. Taken together, our observations suggest that LP12.5 exposure induces abnormalities in insulin secretion in aged male mice under normal chow diet, and these abnormalities are reversed during exposure to insulin resistance induced by HFD, but this response is not sufficient to maintain glucose homeostasis. It is possible that LP12.5 islets respond better to changes in fatty acids, and this can be tested in future studies.
microRNAs (miRs) have been linked to β-cell function, glucose metabolism, obesity, insulin resistance, and, thus, the development of type 2 diabetes (20). The two most enriched miRs in the LP12.5 islets were miR342 and -143. Previously, we showed that miR342 regulates insulin secretion in mouse islets of LP0.5 (2). In the current study, we showed that miR143 mimic is also able to reduce insulin secretion in primary mouse islets. This is likely mediated by miR143’s regulation of the insulin/Akt pathway by directly controlling the level of IGF-I/II receptor (39, 40) or Akt (27), since inhibition of Akt signaling has been shown to ameliorate insulin secretion but not mass (7). In contrast to the LP12.5, increased expression of a specific set of miRs in LP0.5 islets was associated with lower expression of mTOR, Pdx1, and Erk protein (1, 2, 26). However, in the LP12.5 islets, these proteins were not altered in young or old mice, suggesting a different mechanism between fetal programming of LP12.5 and LP0.5. This suggests that LP0.5 and LP12.5 islets share defects in insulin secretion, but the alterations in insulin secretion are regulated by different miRs or perhaps distinct mechanisms. The alterations in miR expression can also have consequences in the whole body glucose homeostasis. Hepatic miR143 overexpression has been reported to predispose animals to metabolic syndrome in the setting of diet-induced obesity (25). Mice with hepatic miR143 overexpression exhibited impaired glucose tolerance and insulin resistance as demonstrated by impairing insulin-stimulated Akt activation in the liver (25). Thus, an increased hepatic miR143 may contribute to insulin resistance in LP12.5 mice. To tease apart the contribution of hepatic miR143 in LP12.5 offspring, however, is beyond the scope of the current study and will require further investigation.
Exposure to low nutrients in different developmental windows can induce various alterations in litter size and organ growth. During pancreas development, the growing β-cell is sensitive to nutrient levels in utero, in particular amino acids (19, 32, 33). We and others have shown that exposure to low protein throughout pregnancy in LP0.5 offspring in both mouse and rat models results in reduced β-cell mass at birth (2, 34). In the current study, we show that a mouse model of maternal low-protein diet in the last week of pregnancy (LP12.5) is not sufficient to alter pancreatic β-cell area and cell size in neonates. This is in contrast to previous studies in Wistar rat (8 or 5% LP), where LP12.5 offspring show reduction in β-cell area in part due to impairment in β-cell proliferation (10, 17). Possible reasons for the discrepancy in β-cell area may arise from differences in diet formulation, total amount of LP used, and sources, as well as environmental factors (i.e., microbiome) and species (i.e., gestational day 21 in the rat versus day 19 in the mouse). The LP12.5 diet was sufficient, however, to reduce litter size (may partly be attributed to fetal survival) and fetal body weight at birth, consistent with other studies done in the rat (15, 18, 28). However, as expected with lower body weight at birth, the murine LP12.5 offspring demonstrated transient higher growth rate between 4 and 6 wk of age, at which point the LP12.5 male offspring were heavier than control offspring. This increase in weight gain was only transient, and LP12.5 offspring show comparable weight around 8 wk of age. It is possible that increased milk access due to reduced litter size by day 21 in LP12.5 offspring could play a role in the transient higher weight gain. Similar weight gain increase has been reported in the LP12.5 rat (15).
Perspectives and Significance
The current study demonstrated that maternal low-protein diet during the last week of pregnancy is associated with insulin resistance and β-cell dysfunction induced by aging or diet-induced obesity in the adult offspring. It is timely and significant to further study the mechanisms behind sex-dependent phenotypes and to identify biological targets of specific microRNAs altered in key metabolic tissues of the offspring. Collectively, these data support epidemiological results from human studies and underscore the importance of adequate maternal protein intake during the last week of pregnancy in promoting the best health outcome postnatally.
GRANTS
This work was supported by the National Institutes of Health (NIH) Grants R01-DK-084236 and DK-073716 to E. Bernal-Mizrachi; R21-DK-112144, R03-DK-114465, R01-DK-115720, and R21-HD-100840 to E. U. Alejandro; and R24-DK-092759 and RO1-DK-62876 to O. A. MacDougald. The Michigan Mouse Metabolic Phenotyping Center was supported by NIH Grant U2C-DK-110768.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.U.A. and E.B. conceived and designed research; E.U.A., S.J., B.A., P.R.L., M.G., B.G., and S.D.P. performed experiments; E.U.A., S.J., B.A., P.R.L., M.G., B.G., and S.D.P. analyzed data; E.U.A., S.J., B.A., P.R.L., M.G., B.G., S.D.P., O.A.M. and E.B. interpreted results of experiments; E.U.A., S.J., B.A., P.R.L., B.G., and S.D.P. prepared figures; E.U.A. drafted manuscript; E.U.A. and E.B. edited and revised manuscript; E.U.A., S.J., B.A., P.R.L., M.G., B.G., S.D.P., O.A.M. and E.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Taylor Wallen, Lauren See, Briana Clifton, Alicia Wong, and Tom McBrien for technical support.
REFERENCES
- 1.Abuzgaia AM, Hardy DB, Arany E. Regulation of postnatal pancreatic Pdx1 and downstream target genes after gestational exposure to protein restriction in rats. Reproduction 149: 293–303, 2015. doi: 10.1530/REP-14-0245. [DOI] [PubMed] [Google Scholar]
- 2.Alejandro EU, Gregg B, Wallen T, Kumusoglu D, Meister D, Chen A, Merrins MJ, Satin LS, Liu M, Arvan P, Bernal-Mizrachi E. Maternal diet-induced microRNAs and mTOR underlie β cell dysfunction in offspring. J Clin Invest 124: 4395–4410, 2014. doi: 10.1172/JCI74237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alejandro EU, Johnson JD. Inhibition of Raf-1 alters multiple downstream pathways to induce pancreatic beta-cell apoptosis. J Biol Chem 283: 2407–2417, 2008. doi: 10.1074/jbc.M703612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Audette MC, Kingdom JC. Screening for fetal growth restriction and placental insufficiency. Semin Fetal Neonatal Med 23: 119–125, 2018. doi: 10.1016/j.siny.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Barker DJ. The fetal origins of adult hypertension. J Hypertens Suppl 10, Suppl 7: S39–S44, 1992. doi: 10.1097/00004872-199212000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Barker DJ. The origins of the developmental origins theory. J Intern Med 261: 412–417, 2007. doi: 10.1111/j.1365-2796.2007.01809.x. [DOI] [PubMed] [Google Scholar]
- 7.Bernal-Mizrachi E, Fatrai S, Johnson JD, Ohsugi M, Otani K, Han Z, Polonsky KS, Permutt MA. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest 114: 928–936, 2004. doi: 10.1172/JCI200420016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest 108: 1631–1638, 2001. doi: 10.1172/JCI200113785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertin E, Gangnerau MN, Bailbé D, Portha B. Glucose metabolism and beta-cell mass in adult offspring of rats protein and/or energy restricted during the last week of pregnancy. Am J Physiol Endocrinol Metab 277: E11–E17, 1999. doi: 10.1152/ajpendo.1999.277.1.E11. [DOI] [PubMed] [Google Scholar]
- 10.Bertin E, Gangnerau MN, Bellon G, Bailbé D, Arbelot De Vacqueur A, Portha B. Development of β-cell mass in fetuses of rats deprived of protein and/or energy in last trimester of pregnancy. Am J Physiol Regul Integr Comp Physiol 283: R623–R630, 2002. doi: 10.1152/ajpregu.00037.2002. [DOI] [PubMed] [Google Scholar]
- 11.Boehmer BH, Limesand SW, Rozance PJ. The impact of IUGR on pancreatic islet development and β-cell function. J Endocrinol 235: R63–R76, 2017. doi: 10.1530/JOE-17-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cherif H, Reusens B, Dahri S, Remacle C. A protein-restricted diet during pregnancy alters in vitro insulin secretion from islets of fetal Wistar rats. J Nutr 131: 1555–1559, 2001. doi: 10.1093/jn/131.5.1555. [DOI] [PubMed] [Google Scholar]
- 13.Dahri S, Snoeck A, Reusens-Billen B, Remacle C, Hoet JJ. Islet function in offspring of mothers on low-protein diet during gestation. Diabetes 40, Suppl 2: 115–120, 1991. doi: 10.2337/diab.40.2.S115. [DOI] [PubMed] [Google Scholar]
- 14.Dehwah MA, Wang M, Huang QY. CDKAL1 and type 2 diabetes: a global meta-analysis. Genet Mol Res 9: 1109–1120, 2010. doi: 10.4238/vol9-2gmr802. [DOI] [PubMed] [Google Scholar]
- 15.de Oliveira JC, Gomes RM, Miranda RA, Barella LF, Malta A, Martins IP, Franco CC, Pavanello A, Torrezan R, Natali MR, Lisboa PC, Mathias PC, de Moura EG. Protein restriction during the last third of pregnancy malprograms the neuroendocrine axes to induce metabolic syndrome in adult male rat offspring. Endocrinology 157: 1799–1812, 2016. doi: 10.1210/en.2015-1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Rooij SR, Painter RC, Phillips DI, Osmond C, Michels RP, Godsland IF, Bossuyt PM, Bleker OP, Roseboom TJ. Impaired insulin secretion after prenatal exposure to the Dutch famine. Diabetes Care 29: 1897–1901, 2006. doi: 10.2337/dc06-0460. [DOI] [PubMed] [Google Scholar]
- 17.Dumortier O, Blondeau B, Duvillié B, Reusens B, Bréant B, Remacle C. Different mechanisms operating during different critical time-windows reduce rat fetal beta cell mass due to a maternal low-protein or low-energy diet. Diabetologia 50: 2495–2503, 2007. doi: 10.1007/s00125-007-0811-0. [DOI] [PubMed] [Google Scholar]
- 18.Dumortier O, Hinault C, Gautier N, Patouraux S, Casamento V, Van Obberghen E. Maternal protein restriction leads to pancreatic failure in offspring: role of misexpressed microRNA-375. Diabetes 63: 3416–3427, 2014. doi: 10.2337/db13-1431. [DOI] [PubMed] [Google Scholar]
- 19.Elghazi L, Blandino-Rosano M, Alejandro E, Cras-Méneur C, Bernal-Mizrachi E. Role of nutrients and mTOR signaling in the regulation of pancreatic progenitors development. Mol Metab 6: 560–573, 2017. doi: 10.1016/j.molmet.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, Dean NM, Freier SM, Bennett CF, Lollo B, Griffey R. MicroRNA-143 regulates adipocyte differentiation. J Biol Chem 279: 52361–52365, 2004. doi: 10.1074/jbc.C400438200. [DOI] [PubMed] [Google Scholar]
- 21.Floris I, Kraft JD, Altosaar I. Roles of microRNA across prenatal and postnatal periods. Int J Mol Sci 17: 17, 2016. doi: 10.3390/ijms17121994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gatford KL, Simmons RA. Prenatal programming of insulin secretion in intrauterine growth restriction. Clin Obstet Gynecol 56: 520–528, 2013. doi: 10.1097/GRF.0b013e31829e5b29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303: 1019–1022, 1991. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jordan SD, Krüger M, Willmes DM, Redemann N, Wunderlich FT, Brönneke HS, Merkwirth C, Kashkar H, Olkkonen VM, Böttger T, Braun T, Seibler J, Brüning JC. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol 13: 434–446, 2011. doi: 10.1038/ncb2211. [DOI] [PubMed] [Google Scholar]
- 26.King R, Hill JL, Saha B, Tong Y, Strutt BJ, Russell MA, Morgan NG, Richardson SJ, Hill DJ. Offspring of mice exposed to a low-protein diet in utero demonstrate changes in mTOR signaling in pancreatic islets of langerhans, associated with altered glucagon and insulin expression and a lower β-cell mass. Nutrients 11: 605, 2019. doi: 10.3390/nu11030605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B, Fan J, Chen N. A novel regulator of type II diabetes: microRNA-143. Trends Endocrinol Metab 29: 380–388, 2018. doi: 10.1016/j.tem.2018.03.019. [DOI] [PubMed] [Google Scholar]
- 28.Mohan R, Baumann D, Alejandro EU. Fetal undernutrition, placental insufficiency, and pancreatic β-cell development programming in utero. Am J Physiol Regul Integr Comp Physiol 315: R867–R878, 2018. doi: 10.1152/ajpregu.00072.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ozanne SE, Fernandez-Twinn D, Hales CN. Fetal growth and adult diseases. Semin Perinatol 28: 81–87, 2004. doi: 10.1053/j.semperi.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 30.Parlee SD, Lentz SI, Mori H, MacDougald OA. Quantifying size and number of adipocytes in adipose tissue. Methods Enzymol 537: 93–122, 2014. doi: 10.1016/B978-0-12-411619-1.00006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrik J, Pell JM, Arany E, McDonald TJ, Dean WL, Reik W, Hill DJ. Overexpression of insulin-like growth factor-II in transgenic mice is associated with pancreatic islet cell hyperplasia. Endocrinology 140: 2353–2363, 1999. doi: 10.1210/endo.140.5.6732. [DOI] [PubMed] [Google Scholar]
- 32.Rachdi L, Aïello V, Duvillié B, Scharfmann R. L-leucine alters pancreatic β-cell differentiation and function via the mTor signaling pathway. Diabetes 61: 409–417, 2012. doi: 10.2337/db11-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD, German MS. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 127: 3533–3542, 2000. [DOI] [PubMed] [Google Scholar]
- 34.Snoeck A, Remacle C, Reusens B, Hoet JJ. Effect of a low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol Neonate 57: 107–118, 1990. doi: 10.1159/000243170. [DOI] [PubMed] [Google Scholar]
- 35.Su Y, Jiang X, Li Y, Li F, Cheng Y, Peng Y, Song D, Hong J, Ning G, Cao Y, Wang W. Maternal low protein isocaloric diet suppresses pancreatic β-cell proliferation in mouse offspring via miR-15b. Endocrinology 157: 4782–4793, 2016. doi: 10.1210/en.2016-1167. [DOI] [PubMed] [Google Scholar]
- 36.Takanabe R, Ono K, Abe Y, Takaya T, Horie T, Wada H, Kita T, Satoh N, Shimatsu A, Hasegawa K. Up-regulated expression of microRNA-143 in association with obesity in adipose tissue of mice fed high-fat diet. Biochem Biophys Res Commun 376: 728–732, 2008. doi: 10.1016/j.bbrc.2008.09.050. [DOI] [PubMed] [Google Scholar]
- 37.Thorn SR, Rozance PJ, Brown LD, Hay WW Jr. The intrauterine growth restriction phenotype: fetal adaptations and potential implications for later life insulin resistance and diabetes. Semin Reprod Med 29: 225–236, 2011. doi: 10.1055/s-0031-1275516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venci RO, Ramos GB, Martins IP, Matiusso CCI, Saavedra LPJ, Ribeiro TA, Pavanello A, Prates KV, Tofolo LP, Moraes AMP, Fabricio GS, de Oliveira JC, Franco C, Palma-Rigo K, Mathias PCF, Malta A. Malnutrition during late pregnancy exacerbates high-fat-diet-induced metabolic dysfunction associated with lower sympathetic nerve tonus in adult rat offspring. Nutr Neurosci 23: 432–443, 2020. doi: 10.1080/1028415X.2018.1516845. [DOI] [PubMed] [Google Scholar]
- 39.Xihua L, Shengjie T, Weiwei G, Matro E, Tingting T, Lin L, Fang W, Jiaqiang Z, Fenping Z, Hong L. Circulating miR-143-3p inhibition protects against insulin resistance in Metabolic Syndrome via targeting of the insulin-like growth factor 2 receptor. Transl Res 205: 33–43, 2019. doi: 10.1016/j.trsl.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 40.Yang Z, Wang J, Pan Z, Zhang Y. miR-143-3p regulates cell proliferation and apoptosis by targeting IGF1R and IGFBP5 and regulating the Ras/p38 MAPK signaling pathway in rheumatoid arthritis. Exp Ther Med 15: 3781–3790, 2018. doi: 10.3892/etm.2018.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang L, Chen W, Dai Y, Zhu Z, Liu Q. Detection of expressional changes induced by intrauterine growth restriction in the developing rat pancreas. Exp Biol Med (Maywood) 241: 1446–1456, 2016. doi: 10.1177/1535370216638771. [DOI] [PMC free article] [PubMed] [Google Scholar]