

Abstract
Nucleoside, nucleotide, and base analogs have been in the clinic for decades to treat both viral pathogens and neoplasms. More than 20% of patients on anticancer chemotherapy have been treated with one or more of these analogs. This review focuses on the chemical synthesis and biology of anticancer nucleoside, nucleotide, and base analogs that are FDA-approved and in clinical development since 2000. We highlight the cellular biology and clinical biology of analogs, drug resistance mechanisms, and compound specificity towards different cancer types. Furthermore, we explore analog syntheses as well as improved and scale-up syntheses. We conclude with a discussion on what might lie ahead for medicinal chemists, biologists, and physicians as they try to improve analog efficacy through prodrug strategies and drug combinations.
Graphical Abstract

1. INTRODUCTION AND MOTIVATION
Cancer is the second leading cause of death in the United States, accounting for 1 in 4 deaths annually. Importantly, cancer incidence continues to increase worldwide.1 Cancer encompasses a broad range of diseases in which host cells escape their normal cell cycle regulation. This phenomenon can be linked to several factors, including gender, ethnicity, age of onset, and lifestyle,2 but cancer can also be caused by cellular transformation linked to viral infection, chemical exposure, or radiation exposure, or the cause can be unknown (spontaneous) in nature.3–5 Advances in oncology treatments have increased the number of cancer survivors from 3 million in 1971 to more than 13 million in 2012.6 This has led to a 39% improvement in 5-year survival rates for different cancers and stages of disease as indicated by the 2010 data, and rates should continue to improve with recent advances in new therapies.1,7
The primary standard-of-care treatment for cancer includes surgery, radiation, chemotherapy, hormone therapy, immuno-therapy, and targeted therapy.1 Cutting-edge therapies may include one or more of these procedures listed above depending upon the type and stage of the cancer being treated. Sometimes surgery and radiation may be a second tier treatment, with chemotherapy being used first to reduce the tumor burden. Chemotherapies are divided into several main drug classes: alkylating agents,8 antimetabolites, anti-tumor antibiotics,9 topoisomerase inhibitors,10 mitotic inhibitors,11,12 and corticosteroids.13 Recently, antibodies directed against programmed cell death 1 (PD-1) protein, PD-1 ligands 1 and 2, and cytotoxic T-lymphocyte-associated protein 4 (CLTA-4) have been found to activate the immune system to attack tumors, and they are therefore used with varying success to treat some types of cancer.14–16
Chemotherapeutic nucleoside, nucleotide, and base analogs, herein referred to as “nucleoside analogs”, are antimetabolites. They are chemically modified analogs of natural nucleosides, nucleotides, and bases, which are endogenous metabolites involved in many essential cellular processes, such as DNA and RNA synthesis, and purinergic signaling. Nucleoside analogs have been a cornerstone of anticancer and antiviral chemotherapy for decades. Currently, there are 15 FDA-approved nucleoside analogs used to treat various cancers, and they account for a large percentage of the current cancer chemotherapeutic arsenal.17 Whereas chemotherapeutic nucleoside analogs alone seldom lead to a cure for cancer, they do provide a valuable treatment option for cancer patients. In addition, many other nucleoside analogs are currently being investigated in clinical trials as monotherapy or combination therapy for multiple cancers. These studies attempt to increase the potency or bioavailability of these agents while diminishing associated dose-limiting toxicity.
In this review, we focus on both the biology and chemical syntheses of the current FDA-approved anticancer nucleoside analogs (Figure 1), investigational anticancer nucleoside analogs in development since 2000 or analogs used outside of the United States (Figure 2), and nucleoside analogs in development since 2000 that have stalled in clinical trials (Figure 3). Additionally, we present an overview of the cellular metabolism, the biochemical mode of action, and the types of cancers treated with these agents, as well as focusing on current methodologies for their chemical synthesis.
Figure 1.

FDA-approved anticancer nucleoside analogs.
Figure 2.


Various nucleoside analogs in clinical trials or used outside of the U.S.
Figure 3.

Various nucleoside analogs that have stalled in clinical trials.
2. GENERAL BIOLOGICAL ACTIVITY OF ANTICANCER NUCLEOSIDE ANALOGS
The two main routes for administering nucleoside analogs are by intravenously infusion and oral formulation. Once inside the body, the analogs can face a major bottleneck in cellular uptake. Both protein facilitated diffusion using either concentrative nucleoside transporters (CNT or SLC28; three members) and/or equilibrative nucleoside transporters (ENT or SLC29; four members) are involved in the cellular uptake for the majority of agents.18,19 Passive diffusion is limited in regard to cellular uptake for parent nucleoside analogs, with troxacitabine 48 being the exception, requiring very high extracellular concentrations in order to occur.20 Certain nucleoside analog prodrugs, such as NUC-1031 32, NUC-3373 37, or elacytarabine 51, however, can enter the cell independent of transporters.
Nucleoside analogs are prodrugs and require cellular metabolism to be converted to their active metabolites. This is accomplished by the cellular salvage pathway—a group of enzymes that can phosphorylate nucleoside and nucleotide substrates. After transport across the plasma membrane, nucleoside analogs undergo an initial phosphorylation—or if a base analog, ribosylation followed by phosphorylation—to generate nucleoside-5′-monophosphate forms,21 which is often the rate-limiting step within the cell in the process to generate active metabolites (Figure 4).22 The cell primarily uses four kinases for nucleoside-5′-monophosphate phosphorylation: 2′-deoxycytidine kinase, thymidine kinase 1, thymidine kinase 2, and deoxyguananine kinase.23 Each kinase has multiple substrates and can have feedback inhibition to regulate their activities.24,25 The initial phosphorylation is often performed by 2′-deoxycytidine kinase. Cancer cells usually express 2′-deoxycytidine kinase at a 3- to 5-fold higher protein level than most normal cells, affording some degree of selectivity.26 Phosphoramidate prodrugs such as NUC-1031 32 or NUC-3373 37, however, enter the cell as masked monophosphates and pass through a different enzymatic pathway in order to be converted to their nucleoside-5′-monophosphate forms.
Figure 4.

General biological mechanism of action of anticancer nucleoside analogs.
Nucleoside-5′-monophosphate analogs are converted to nucleoside-5′-diphosphate and nucleoside-5′-triphosphate forms by various cellular kinases. Nucleoside-5′-triphosphate analogs are substrates for DNA polymerases and can be incorporated into DNA during replication or DNA excision repair synthesis, which gives rise to stalled replication forks and chain termination. These events activate various DNA damage sensors, which stimulate DNA repair, halt cell progression, and often lead to apoptosis.27,28 Since most cancer cells replicate their genome more frequently than a majority of normal adult cells, which are quiescent and not actively synthesizing their DNA, this phenomenon allows for a degree of cancer cell selectivity.17 Moreover, certain nucleoside-5′-triphosphate analogs can be incorporated into RNA, leading to transcriptional termination, and messenger RNA (mRNA) and ribosomal RNA (rRNA) instability.29,30
Nucleoside and nucleotide (mono-, di-, or triphosphates) derivatives can also inhibit key cellular enzymes, providing a secondary mode of action that inhibits cell growth. Such enzymes include ribonucleotide reductase,31 which removes the 2′-OH group from the ribose sugar in order to generate de novo 2′-deoxyribonucleoside diphosphate,32 purine nucleoside phosphorylase,33,34 which is involved in purine metabolism catalyzing the reversible phosphorolysis of purine nucleosides,35 and thymidylate synthase,36 discussed below (see Section 5.1.1.1). A few newer nucleoside analogs, however, exert their anticancer activities as adenosine receptor antagonists, or by inhibiting cellular enzymes that are not involved in nucleic acid synthesis. These cellular enzymes include NEDD8-activating enzyme, which is involved in the ubiquitin-proteasome degradation system and has an important role in cellular processes associated with cancer cell growth,37 and DOT1L (histone-lysine N-methyltransferase, H3 lysine-79 specific), which is involved in post-translational gene modification.38 Furthermore, the enzymatic activity of DOT1L represents an oncogenic driver of MLL-r leukemia.39
Enzymes that dephosphorylate nucleotides are also present in the cell. 5′-Nucleotidases (5′-NTs) can dephosphorylate noncyclic nucleoside-5′-monophosphates to nucleosides and inorganic phosphates, leading to the regulation of cellular nucleotide and nucleoside levels.40,41 There are seven intracellular 5′-NTs: cN-IA, cN-IB, cN-II, cN-III, cN-II-like, cdN, and mdN in the cell,42,43 and they have been reported to modulated antiviral and anticancer nucleotide levels.41 Their cellular functions involve cell–cell communication and signal transduction,44 as well as nucleic acid repair.45 Other enzymes involved in nucleotide degradation include eN (CD73), an ecto-5′-nucleotidase that converts AMP to adenosine plus phosphate,43 and ectonucleoside triphosphate diphosphohydrolase-1 (CD39), which can hydrolyze nucleoside-5′-triphosphates into nucleoside-5′-monophosphate and nucleoside-5′-diphosphate products. Tumor expression of these two enzymes, in an animal model, appears to enhance tumor growth by negatively regulating the immune response.46,47 Furthermore, alkaline phosphatases are also present and can be targets for anticancer treatment, but they might have a lesser impact on antimetabolite metabolism.48 Recently, a sterile alpha motif and histidine/aspartic acid domain containing protein 1 (SAMHD1) was discovered to be a dGTP/GTP-dependent deoxynucleotide triphosphohydrolase,49,50 generating 2′-deoxynucleosides and inorganic triphosphates from the cellular canonical 2′-deoxyribonucleoside-5′-triphosphates. Clofarabine-5′-triphosphate has been shown to be a substrate for SAMHD1.51 Furthermore, the SAMHD1 gene was shown to be mutated in CLL cancer,52 and the protein level downregulated in lung cancer.53
Cancer chemotherapy is limited in effectiveness by off target toxicity and drug resistance, with the latter leading to a decreased ability to deliver an adequate concentration of drug to cancer cells. Drug resistance can arise from pre-existing intrinsic properties of cancer cells or by acquired traits, which cancer cells develop in response to limited drug exposure.54 Cancer cells can acquire resistance to nucleoside analogs by several different mechanisms. First, many studies using cell lines exposed to nucleoside analogs have shown transporter mutations, which generally do not occur in cancer patients. Although a decrease in nucleoside transport in patients was detected, it was dependent on cell differentiation state: myeloblasts versus lymphoblasts.55,56 For example, high expression of hENT1 was a potential co-determinant of poor clinical response to 5-FU 2 in cells ex vivo from colorectal cancer patients,57 or using RNA1 to downregulate hENT1 in the pancreatic cell line.58 In contrast, high hENT1 expression was reported as a biomarker for survival in patients with pancreatic cancer treated with gemcitabine.59 Second, modulation of phosphorylating enzymes, such as 2′-deoxycytidine kinase, has been reported when using cancer cell lines.60 In patients, however, resistance to nucleoside analogs by phosphorylating enzymes has been reported to be linked to decreases in mRNA levels and a decrease in enzyme activity.61,62 Third, resistance to nucleoside analogs can also occur by augmentation of nucleotide excision repair machinery.63
An increase in cellular nucleopside analog efflux provides another mechanism of resistance.64 Cellular nucleoside analog efflux is performed by several ATP-Binding Cassette (ABC) transporters, such as ABCB1 (MDR1), ABCC4 (MRP4), ABCC5 (MRP5), and ABCC11 (MRP8).65,66 Furthermore, metabolic inactivation of nucleoside analogs can occur by the actions of cellular enzymes such as adenosine deaminase, cytidine deaminase, and purine nucleoside phosphorylase.67–69
3. GENERAL SYNTHETIC APPROACHES TO ANTICANCER NUCLEOSIDE ANALOGS
Syntheses highlighted in this review use three general approaches to access anticancer nucleoside analogs: divergent, convergent, and trans-glycosylation (Figure 5).70 The divergent approach begins with an intact nucleoside that is subsequently modified at either the sugar or base. A major advantage of this method is that the stereochemistry, especially of the key glycosidic bond (β-anomer), is already fixed. A drawback of this approach is the presence of two or three hydroxyl groups, which are relatively similar in chemical reactivity, and often require multiple protection/deprotection steps.
Figure 5.
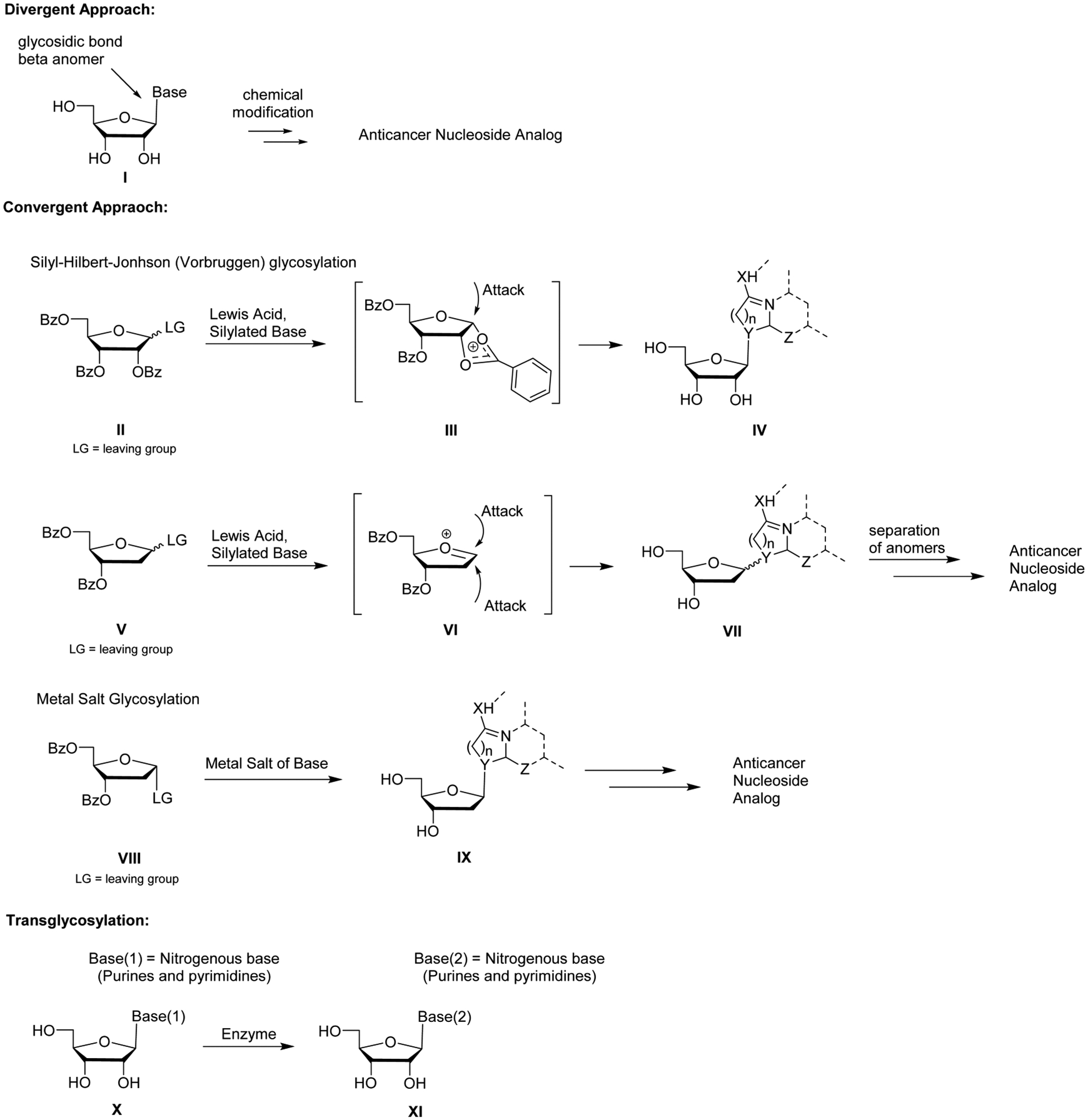
General synthetic approaches to anticancer nucleoside analogs.
The convergent approach couples a nitrogenous base with a modified sugar in a glycosylation reaction. This method allows for more synthetic diversity, and it is more widely used, although the stereochemistry of the glycosylation can be an issue. The reaction is most often performed under Silyl–Hilbert–Johnson (Vorbrüggen) conditions or metal salt coupling conditions.
Benzoyl-protected furanose sugars are often used in glycosylations performed under Vorbrüggen conditions, because the stereochemistry of the glycosylation reaction is almost completely selective. Anchimeric assistance of the 2′-O-benzoyl group, protected furanose sugar II, leads to intermediate III, which effectively has its α-face blocked. This allows for selective formation of the protected β-anomer IV. Difficulties arise when dealing with 2′-deoxy furanose sugars. The absence of a 2′-O-benzoyl group precludes formation of an intermediate with a sterically hindered α-face, often leading to an anomeric mixture of products VII, and many times requires laborious chromatographic separation.
The metal salt glycosylation generally couples a furanose sugar possessing a leaving group (often halogens) with α-stereochemistry and a salt of a nitrogenous base. Conditions are sought to make the reaction predominantly (or exclusively if possible) SN2 in nature, in an attempt to elude anomerization of the leaving group or avoid the formation of an oxonium ion intermediate similar to VI, favoring a SN1-like reaction.
Enzyme-mediated base exchange is an addition method for nucleoside synthesis. Purine nucleoside phosphorylase or uridine phosphorylase can be used to replace one nucleoside base with another in a trans-glycosylation reaction. Overall, enzymes can selectively generate the desired β-anomer nucleoside.
4. PURINE AND ACYCLIC ANALOGS
4.1. Thiopurines
4.1.1. Thiopurines Biology.
In the early 1950s, Elion’s group, while at the Wellcome Research Laboratories, discovered that hypoxanthine and guanine substituted with sulfur at the 6 position led to purine synthesis inhibition, which in turn decreased growth of Lactobacillus casei.71 Subsequently, 6-mercaptopurine (6-MP, 1) and 6-thioguanine (6-TG, 3) were demonstrated to have potent anticancer activities against a wide variety of rodent tumors. The more promising 6-MP 1 was rapidly advanced into clinical trials; in 1953 it received FDA approval for treating pediatric acute lymphocytic leukemia (ALL), a rapid-developing cancer of the blood and bone marrow in which immature lymphocytes are overproduced.
Currently, 6-MP 1 and 6-TG 3 have various clinical applications. They can effectively treat hematologic cancers (childhood and adult leukemias).72 6-TG 3 is used to treat acute myelogenous leukemia, a cancer involving abnormal white blood cells of myeloid linage that rapidly divide and interfere with normal white blood cell production, whereas 6-MP 1 is used in combination with other chemotherapeutic agents and remains part of the standard of care for acute lymphocytic leukemia.73 These compounds are also being used to treat autoimmune disorders, such as rheumatoid arthritis, psoriasis, and ulcerative colitis,74–76 and they have also been employed to prevent solid organ transplant rejection.30,77,78
Thiobases require further metabolism by cellular enzymes to reach their active forms (Figure 6).79 6-MP 1 is converted by hypoxanthine/guanine phosphoribosyl transferase to 6-thioinosine-5′-monophosphate III and then converted to 6-thio-guanosine-5′-monophosphate VI, which is also the product of 6-TG 3 and hypoxanthine/guanine phosphoribosyl transferase. Further phosphorylation leads to 6-thio-guanosine-5′-triphosphate VIII, which can be incorporated into RNA.29 However, Nelson et al. have reported that 6-MP 1 activity was not directed toward specifically inhibiting RNA synthesis, suggesting that 6-MP-5′-triphosphate VIII incorporation into RNA was not a major mechanism for its anticancer activity in vitro.80
Figure 6.

Metabolism of 6-MP 1 and 6-TG 3.
A major active metabolite of both 6-MP 1 and 6-TG 3 is 6-thio-2′-deoxyguanosine-5′-triphosphate XI, which is produced from ribonucleotide reductase-mediated deoxygenation of 6-TG-5′-diphosphate VII, followed by phosphorylation.17 6-Thio-2′-deoxyguanosine-5′-triphosphate XI can be incorporated into DNA. DNA polymerases continue polymerization after 6-thio-2′-deoxyguanosine-5′-monophosphate incorporation, resulting in multiple thiobase derivatives embedded in the newly synthesized DNA strand. The reactive thiol group of the incorporated active metabolite is susceptible to nonenzymatic methylation, leading to incorporated 6-methylthioguanine-5′-monophosphate IX, which preferentially base-pairs with a thymine during DNA replication.81 The 6-methylthioguanine:-thymine base-pairings resemble DNA replication errors, which are recognized by cellular mismatch repair enzymes. It is hypothesized that 6-methylthioguanine:thymine base-pairing leads to a futile cycle of repair, which ultimately generates cytotoxic DNA damage.81
Another major active metabolite of 6-MP 1 is methylthioino-sine-5′-monophosphate IV, which is formed by the methylation of thioinosine-5′-monophosphate III by thiopurine S-methyltransferase.82 Metabolite IV inhibits phosphoribosylpyrophosphate (PRPP) amidotransferase, the first enzyme in the de novo purine biosynthesis pathway.83 PRPP amidotransferase inhibition leads to a decrease in purine nucleotide pools, which can disrupt DNA synthesis and repair, ultimately leading to cell death in vitro.84 6-Thioguanine-5′-monophosphate VI can also be methylated by S-methyltransferase to form methylthioguano-sine-5′-monophosphate IX. However, this metabolite is a weak inhibitor of PRPP amidotransferase.
Two enzymes involved in the detoxification of 6-MP 1 and 6-TG 3 are thiopurine methyltransferase and xanthine oxidase. The former directly methylates 6-MP 1 and 6-TG 3, to metabolites II and V, respectively. Xanthine oxidase catabolizes 6-MP 1 to the 6-thiouric acid derivative I. However, because xanthine oxidase expression is low in hematopoietic cells, thiopurine S-methylation is considered to be the predominant pathway of detoxification of 6-MP 1 and 6-TG 3.79 In addition, six different variant alleles have been identified from clinical samples that lead to lower thiopurine methyltransferase activity.79
Thiopurine resistance can arise due to the action of ATP-Binding Cassette (ABC) transporters. 6-MP-5′-monophosphate III and 6-TG-5′-monophosphate IV are substrates for ABC transporters: MRP4 and MRP5, for active transport out of cancer cells.85,86 Additional mechanisms of resistance identified using cell lines are the up regulation of P-glycoprotein87 and decreased hypoxanthine-guanine phosphoribosyltransferase activity.88 A genomic approach revealed that overexpression of the ATM/p53/p21 pathway, overexpression of TNFRSF10D, and overexpression of CCNG2 may also contribute to thiopurine resistance in cell lines.89 Besides the natural mutations in thiopurine S-methyltransferase gene,79 mutations in NT5C2 transporter gene led to an increase in nucleotidase activity and resistance to 6-mercaptopurine 1 and 6-thioguanine 3 in ALL patients.90,91 Over 170 clinical trials evaluating thiopurines as a cancer treatment are listed at ClinicalTrials.gov.
4.1.2. Thiopurines Synthesis.
Elion and Hitchings reported the preparation of 6-MP 1 in 1952 (Scheme 1).92 For their studies, they developed a multigram-scale synthesis of hypoxanthine 57 starting from 4-amino-6-hydroxy-5-nitrosopyrimidine-2-thiol 54.93 The heterocycle 54 was reduced, dethiolated, and cyclized, forming 57, which was subsequently treated with P2S5, furnishing the desired 6-MP 1 Conversion of hypoxanthine 57 to 6-MP 1 using P2S5 has also been used in a recent process synthesis, which was done in toluene on a 200-g-scale with a yield of 93%.94 Additionally, this transformation has been effected on a gram-scale using the crystalline and storable P4S10-pyridine complex reagent 58.95
Scheme 1. Synthesis of 6-Mercaptopurine 1a.

aReagents and conditions: (a) Na2S2O4, H2O, 60–70 °C, 80–85%; (b) Raney Ni, Na2CO3, H2O, reflux, 2 h then H2SO4, 88%; (c) HCO2H, reflux, 2 h then aq NaOH, AcOH, 93%; (d) P2S5, tetralin, 190–200 °C, 12 h, 40%; or 58, DMSO2, 165–170 °C, 85%.
Elion and Hitchings synthesized 6-thioguanine (6-TG, 3) in 1954 (Scheme 2).96 Guanine 59 was converted to 6-TG 3 by treatment with P2S5 in refluxing pyridine. Diacylated guanine 60 has also been used to produce 3. Sulfuration with P2S5, followed by deprotection, furnished the product on 100-g-scale (Scheme 2).97 In addition, the sulfuration of 60 has been effected by treatment with Lawesson’s reagent.98
Scheme 2. Synthesis of 6-Thioguanine 3a.
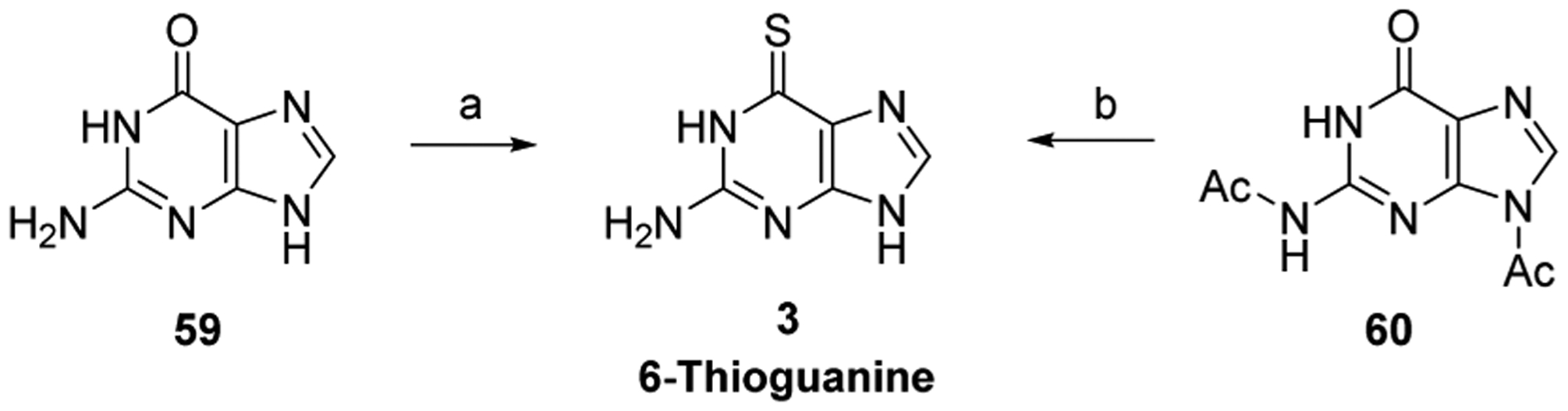
aReagents and conditions: (a) P2S5, pyr, reflux, 2.5 h, 32%; (b) P2S5, pyridine hydrochloride, pyr, 110 °C, 4 h, then HCl, H2O, pH 4, recrystallization, 75%.
4.2. GS-9219
4.2.1. GS-9219 Biology.
Several acyclic nucleotide derivatives, such as 9-(2-phosphonylmethoxyethyl)guanine (PMEG), 9-(2-phosphonylmethoxyethyl)-2,6-diaminopurine (PMEDAP), and 9-(2-phosphonylmethoxyethyl)adenine (PMEA), display antiproliferative activities in cancer cells (Figure 7).99 The derivatives bear a phosphonate group, which is a bioisostere of a phosphate, but metabolically and chemically more stable.100 The acyclic nucleotide-5′-diphosphates are structural analogs of naturally occurring 2′-deoxyribonucleoside-5′-triphosphates and can be used as substrates for cellular DNA polymerases, acting as absolute chain terminators.101
Figure 7.
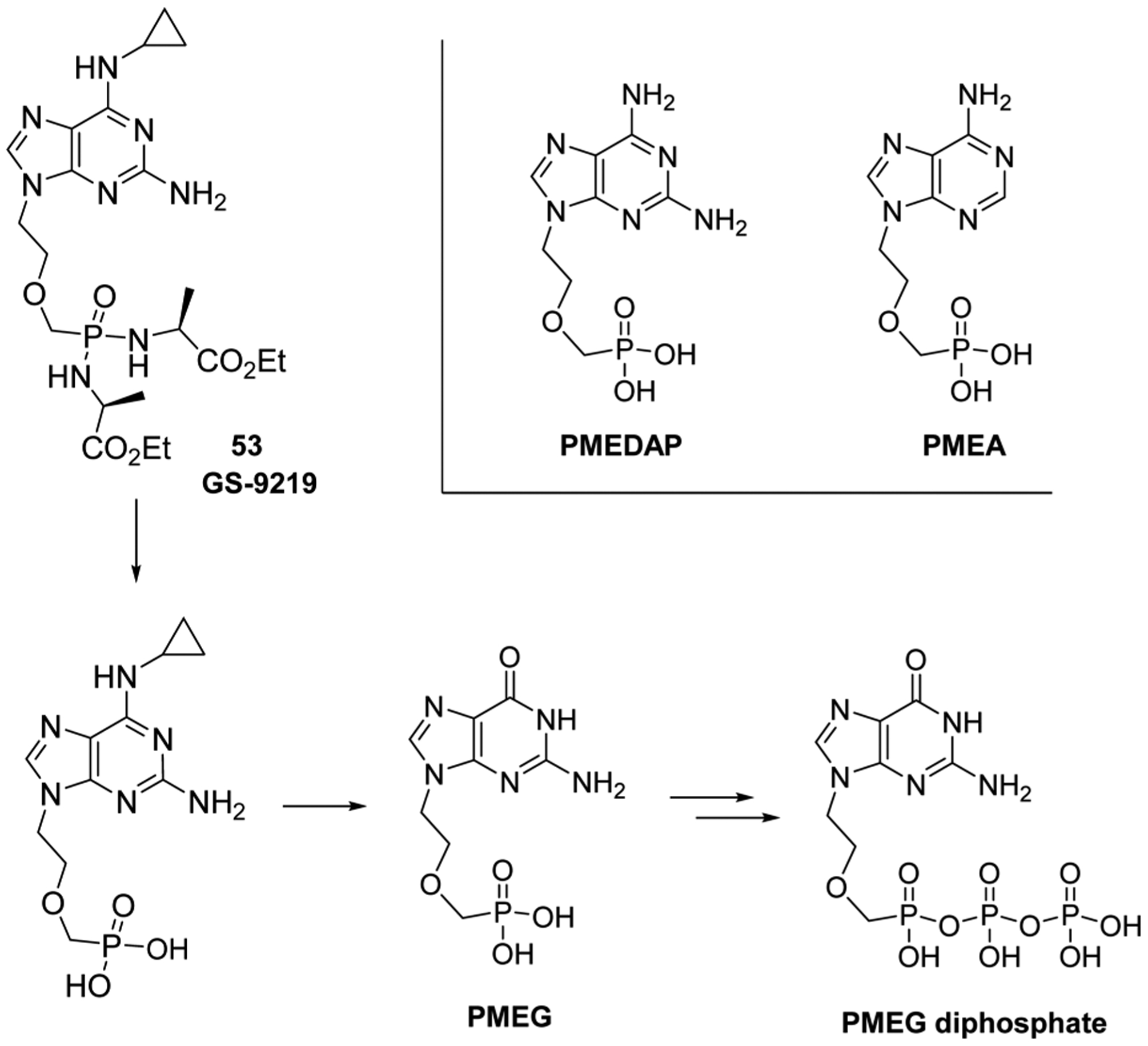
Structures of PMEG, PMEDAP, and PMEA, and proposed cellular metabolism of GS-9219 53.
PMEG has been shown to be the most cytotoxic among the acyclic derivatives studied.99 Within the cell, PMEG is converted to its diphosphate form, which is the active metabolite. The PMEG-5′-diphosphate potently inhibits DNA polymerases α, δ, and ε (DNA chromosomal replication enzymes), leading to inhibition of DNA synthesis and repair.102 The 3′ → 5′ exonuclease activity of DNA polymerase can remove PMEG incorporated during DNA synthesis.99 However, if not excised from the growing DNA strand, the PMEG-5′-monophosphate, which lacks a 3′-OH moiety, acts as an absolute DNA synthesis chain terminator.
Unfortunately, PMEG displays poor cellular permeability properties, in addition to kidney and gastrointestinal tract associated toxicities.102 Therefore, researchers at Gilead developed GS-9219 (53, VDC-1101), a double prodrug of PMEG, in an attempt to preferentially target lymphoid tissues.103 The N6-cyclopropyl moiety was installed to increase specific intracellular activation as well as decrease plasma exposure to the toxic PMEG. The phosphorodiamidate prodrug was installed to increase lymphoid cell and tissue loading efficacy. Such prodrugs are also often employed to increase compound solubility, cellular penetration, and selective tissue accumulation.104 GS-9219 53 showed potent antiproliferative activity against activated lymphocytes and hematopoietic cancer cells in vitro and in canines.103 It was proposed that GS-9219 53 is intracellularly converted to PMEG-5′-diphosphate in three steps: enzymatic hydrolysis of the amino acid of the prodrug (involving cathepsin A), deamination of the cyclopropyl amino moiety (involving N6-methyl-AMP-aminohydrolase), and phosphorylation of PMEG (Figure 7).105
Mutations in human adenosine deaminase-like proteins (H286R and S180N) have recently been found to promote resistance to GS-9219 53 and to 9-(2-phosphonylmethoxyethyl)-N6-cyclopropyl-2,6-diaminopurine, an intermediate in the conversion pathway of GS-9219 53 to PMEG.106 Furthermore, point mutations in human guanylate kinase (S35N and V168F) were shown to promote GS-9219 resistance in cell lines.107
Unfortunately, GS-9219 53 has stalled in human clinical trials for treatment of non-Hodgkin’s lymphoma, chronic lymphocytic lymphoma, and multiple myeloma due to an unacceptable safety profile (ClinicalTrials.gov identifier: NCT00499239—terminated—no results posted). However, the agent was FDA-approved for canine lymphoma in June 2013,108,109 and an additional Phase II study was done evaluating the effectiveness in canine cutaneous T-cell lymphoma.110 GS-343074 and GS-424044 are two additional prodrugs synthesized after GS-9219 53 and are being evaluated as anticancer prodrugs, particularly for canine cancers. No structures have been revealed for these two compounds.
4.2.2. GS-9219 Synthesis.
In 2005, Gilead disclosed their discovery synthesis of GS-9219 53 (Scheme 3).111 2-Amino-6-chloropurine 61 was selectively N9-alkylated, and subsequent nucleophilic aromatic substitution with cyclopropylamine furnished alkyl diester 63. TMSBr-mediated deprotection led to the phosphonic acid derivative 64, which was sufficiently pure to require no chromatography after the reaction. Bis-amidate formation was effected by treatment of acid 64 with 2,2′-dithiodipyridine, triphenylphosphine, and D-alanine ethyl ester HCl.
Scheme 3. Synthesis of GS-9219 53a.

aReagents and conditions: (a) HO(CH2)2OCH2PO(O-iPr)2, DIAD, PPh3, DMF, −15 °C to rt, 4 h, 63%; (b) cyclopropylamine, CH3CN, 100 °C, 4 h, 90%; (c) TMSBr, CH3CN, rt, overnight, 90%; (d) d-alanine ethyl ester HCl, 2,2′-dithiodipyridine, PPh3, Et3N, pyr, 60 °C, overnight, 50%.
Recently, Jansa et al. reported an improved route to bisamidate prodrugs.112 These researchers surmised that the intermediate 65 would react directly with 2,2′-dithiodipyridine, triphenylphosphine, and the amino acid to produce the bisamidate (Scheme 4). This would obviate the need for the laborious purification step often needed to isolate and purity the intermediate diacid. The approach was successful and was used to synthesize GS-9219 53.113 TMS bromide-mediated deprotection of the phosphonate diester 63113 formed the bis(TMS)ester intermediate 65, maintained by strictly avoiding any contact with air. The bis(TMS)ester intermediate was then directly converted to GS-9219 53.
Scheme 4. Synthesis of GS-9219 53a.
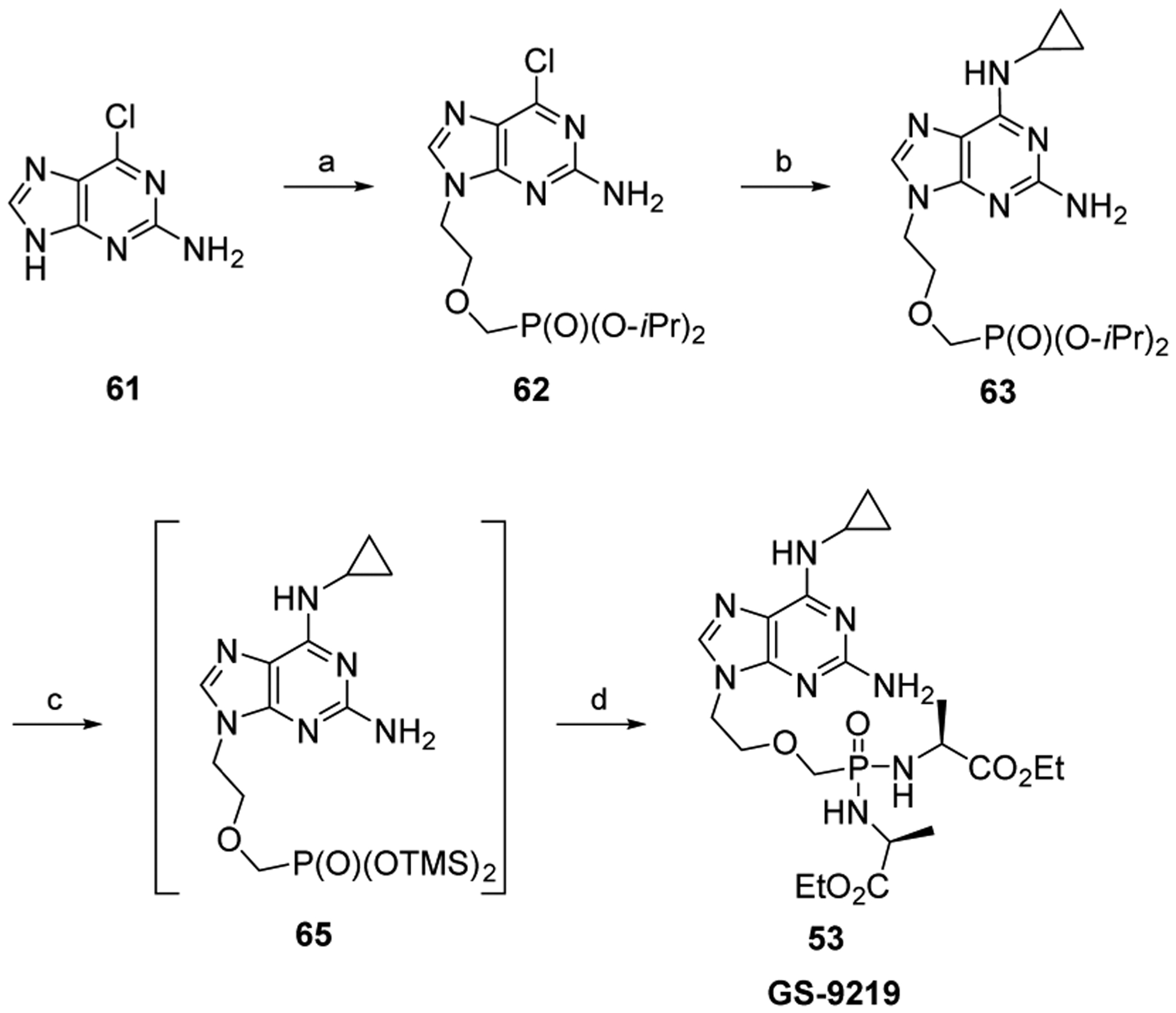
aReagents and conditions: (a) Cl(CH2)2OCH2P(O)(O-iPr)2, Cs2CO3, DMF, 80 °C, 8 h, 56%; (b) cyclopropylamine, EtOH, reflux, 3 h, 65%; (c) TMSBr, CH3CN, rt, overnight; (d) d-alanine ethyl ester, aldrithiol-2, PPh3, Et3N, pyr, 50 °C, 3 h, 92–98% (over two steps).
5. PYRIMIDINE ANALOGS
5.1. Fluorinated Pyrimidines
5.1.1. Fluorouracil (5-FU), Prodrugs, and Combinations Biologies.
5.1.1.1. Fluorouracil (5-FU) Biology.
In 1954, Rutman and colleagues observed that rat liver tumor cells utilized more uracil 18 as compared to normal liver cells.114 Thymidylate synthase can catalyze the conversion of deoxyuridine-5′-monophosphate to thymidine-5′-monophosphate using 5,10-methylenetetrahydrofolate as a methyl source. These observations led Heidelberger and colleagues to synthesize fluorouracil (5-FU, 2). Their hypothesis was that the 5-FU 2 would be converted to 5-fluorouracil-5′-monophosphate, and this product would selectively inhibit thymidylate synthase by forming a complex that could not breakdown due to prevention of elimination by the 5-fluorine (Figure 8).36,115 Furthermore, they surmised that this would selectively target tumor cells, since 5-FU-5′-monophosphate would be found at higher concentrations in these cells. Moreover, the inhibition of thymidylate synthase and subsequent decrease of thymidine would be deleterious for cancer cells growth.
Figure 8.

Complex of 5,10-methylenetetrahydrofolate and thymidylate synthase with 5-FU-5′-monophosphate.
These researchers subsequently validated their hypothesis by showing that 5-FU 2 inhibited the production of thymine in vitro,36 and soon thereafter reported that the agent inhibited tumor growth in animals.116 Other reports have shown that the main mechanism of action for 5-FU 2 was the inhibition of synthesis,117 although RNA metabolism was also influenced.118
5-FU 2 is used as a palliative treatment for colorectal, head and neck, and breast cancers.119 Treatment can also include a biomodulator, such as leucovorin, which increases folate cofactors and the efficacy of 5-FU 2.120,121 Although introduced over 50 years ago, 5-FU 2 is still extensively studied, with over 2000 clinical trials listed at ClinicalTrials.gov.
Mechanisms of action for 5-FU 2 are complex, and an overview of its metabolism is shown in Figure 9. The agent is administered by intravenous infusion and is rapidly eliminated from the plasma, having a half-life of 8–20 min.122 5-FU 2 enters the cell by facilitated transport,123 and subsequent ribosylation and phosphorylation occur by orotate phosphoribosyltransferase or in two steps via uridine phosphorylase and uridine kinase.119,124 Nucleotide kinases convert 5-FU-5′-monophosphate IV to the active metabolite 5-FU-5′-triphosphate VIII, which may be incorporated into many species of RNA. This leads to several levels of RNA disruption, including rRNA maturation inhibition101,125 and disruption of post-translational modifications of tRNAs,126 which can lead to cell death.
Figure 9.
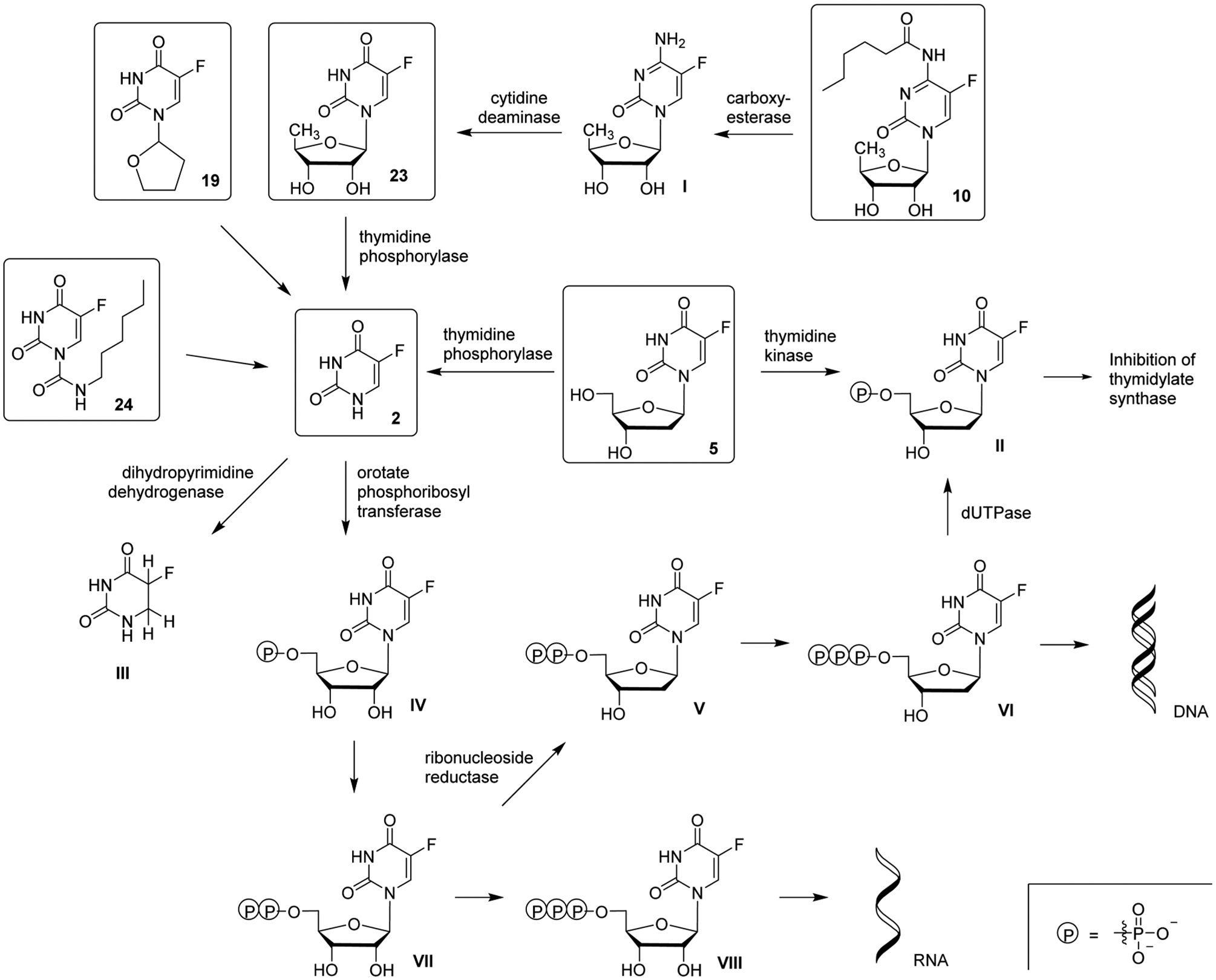
Metabolism of 5-FU 2 and derivatives.
5-FU-5′-diphosphate VII is also a substrate of ribonucleotide reductase, resulting in 2′-deoxyribose-5-FU-5′-diphosphate generation. It is further phosphorylated to 2′-deoxyribose-5-FU-5′-triphosphate VI (an additional active metabolite), which then can be incorporated into DNA by cellular polymerases. 2′-Deoxyribose-5-FU-5′-monophosphate embedded into DNA does not act as a chain terminator or delayed chain terminator as compared to other antimetabolites. Instead, uracil glycosylase can excise the embedded 2′-deoxyribose-5-FU-5′-monophosphate from the DNA, or the activation of homologous recombination can occur.127 This apyrimidinic site is recognized by apurinic/apyrimidinic endonuclease 1 and can promote a DNA single-strand break.17 DNA repair enzymes recognize this break in the DNA, and a futile cycle of excision, repair, and nucleotide misincorporation ensues, which eventually causes cell death.
2′-Deoxyribose-5-FU-5′-triphosphate VI is converted by dUTPase to 2′-deoxyribose-5-FU-5′-monophosphate II, which then inhibits the enzyme thymidylate synthase. Inhibition of thymidylate synthase leads to a decrease in dTMP cellular concentration and an increase in dUMP concentration. As a result, less dTTP is available for DNA polymerases during DNA synthesis. This can promote the misincorporation of dUTP into newly synthesized DNA, which may result in the futile cycle mentioned above. Incorporation of dUTP into DNA has been shown to be mutagenic and promote genomic instability.128–130
There are several major obstacles for the effective use of 5-FU 2 as a chemotherapeutic. First, up to 80% of administered 5-FU 2 is catabolyzed by dihydropyrimidine dehydrogenase, which is abundantly expressed in the liver and acts as an innate resistance mechanism. Dihydropyrimidine dehydrogenase converts 5-FU 2 to dihydrofluorouracil III and is the rate-limiting enzyme in the catabolic pathway.122 Secondly, 2′deoxy-5-FU-5′-monophosphate and 5-FU-5′-monophosphate metabolites (II and IV) can be exported from cancer cells via the ABC transporters MRP5 and MRP8, limiting the therapeutic impact of this compound in vitro.131,132
Several mechanisms for 5-FU 2 resistance have been reported. The miRNA-320a has been shown to downregulate PDCD4 (programmed cell death protein 4) gene expression, thereby promoting 5-FU 2 resistance in vitro.133 Also, miRNA-21 induces resistance by regulating PTEN and PDCD4 levels in vitro, which correlate to cell survival and cell death pathways, respectively.134 Overexpression of nicotinamide N-methyltransferase is common in many cancer cell lines and has been linked to 5-FU 2 resistance by regulating nitric oxide production in vitro. Reducing reactive oxygen species leads to inactivation of the ASK1-p38 mitogen-activated protein kinase (MAPK) pathway, leading to a reduction in 5-FU 2-induced apoptosis in vitro.135 Furthermore, putrescine released by macrophage has been shown to promote 5-FU 2 resistance in vitro, which might be important for regulating 5-FU 2 concentration in the microenvironment of solid tumors in vivo.136 Overall, 5-FU 2 resistance appears to be a multifactorial event which includes transport mechanisms, metabolism, molecular mechanisms, protection from apoptosis, and resistance via cell cycle kinetics.
In patients, 5-FU 2 resistance has been linked to thymidylate synthase methylenetetrahydrofolate reductase, dihydropyrimidine dehydrogenase, and thymidine phosphorylase, which influence 5-FU 2 metabolism.137,138 There have been over 1500 clinical trials performed that evaluated 5-FU 2 therapy listed at ClinicalTrials.gov.
5.1.1.2. Doxifluridine Biology.
5-FU 2 is administered intravenously due to the high level of dihydropyrimidine dehydrogenase in the gut wall, which rapidly degrades the agent. Researchers at Roche used prodrug strategies to develop an orally available prodrug that overcomes degradation in the gut leading to enhanced absorption and an improved pharmacokinetic profile.139 This work led to the development of doxifluridine (5′-deoxy-5-fluorouridine) 23, an orally available prodrug which is converted to 5-FU 2 in tumors by either thymidine phosphorylase or pyrimidine-nucleoside phosphorylase.140,141
High expression levels of pyrimidine-nucleoside phosphorylase have been reported for esophageal squamous cell carcinoma and colorectal cancer patients.142,143 Furthermore, thymidine phosphorylase is expressed at relatively high levels in tumor tissues such as esophageal, breast, cervical, pancreatic, and hepatic,144,145 which increase drug specificity. However, high thymidine phosphorylase expression is also found in the human intestinal tract. Therefore, doxifluridine treatment can result in dose-limiting toxicity (diarrhea) in some individuals.146 In addition, the most frequent adverse effects for doxifluridine 23 were neurotoxicity and mucositis, whereas leukopenia and nausea were reported for 5-FU 2 in patients.147 Several clinical trials have been done using doxifluridine 23 as a monotherapy or in combination therapy with mitomycin-C and cisplatin (administered intravenously).148
5.1.1.3. Capecitabine Biology.
As mentioned above, doxifluridine 23 treatment had gastrointestinal toxicity in patients. This led Roche to develop capecitabine 10 (5′-deoxy-5-fluoro-N-[(pentyloxy)carbonyl]cytidine), a third-generation prodrug of 5-FU 2 that was FDA-approved in 1998.149,150 Capecitabine 10 is almost 100% orally bioavailable and is not a substrate for thymidine phosphorylase.151 After passage through the intestinal mucosa, carboxylesterases in the liver cleave the N4-pentyl carbamate. Next, cytidine deaminase, in either the liver or tumor, catalyses the formation of doxifluridine 23, which is subsequently converted by thymidine phosphorylase to 5-FU 2.152 Capecitabine 10 was highly effective in HCT116 human cancer xenograft models and showed better selective tumor delivery as compared to either doxifluridine 23 or 5-FU 2.153 In addition, it was found that radiation therapy stimulated thymidine phosphorylase activity, and combination therapy with capecitabine 10 enhanced tumor cell selectivity.154
Drug resistance to capecitabine 10 appears to involve the interplay between dihydropyrimidine dehydrogenase and thymidine phosphorylase, which are involved in the conversion of capecitabine 10 to 5-FU 2 and the degradation of 5-FU 2 within tumors.155 In addition to these resistance mechanisms, drug efflux is associated with gene polymorphisms of the ABCB1 transporter in patients.66
Currently, capecitabine 10 is FDA-approved for treatment of metastatic colorectal and breast cancers. Moreover, several clinical trials are underway using capecitabine 10 in combination therapy. A Phase II study for capecitabine 10 and bendamustine (DNA alkylating agent) in women with pretreated locally advanced or metastatic Her2-negative breast cancer (MBC-6) is being conducted (ClinicalTrials.gov identifier: NCT01891227—active status—not recruiting patients). A Phase III study is being conducted that examines capecitabine 10 maintenance therapy following treatment with capecitabine 10 in combination with docetaxel (an antimitotic taxane) in treatment of mBC (CAMELLIA study) (ClinicalTrials.gov identifier: NCT01917279—recruiting patients). Capecitabine 10 is also being evaluated as a follow up therapy for efficacy of capecitabine 10 metronomic chemotherapy to triple-negative breast cancer (SYSUCC-001 study) (ClinicalTrials.gov identifier: NCT01112826—recruiting patients). It is also under examination for maintenance therapy: a Phase II study of maintenance capecitabine 10 to treat resectable colorectal cancer (CAMCO study) (ClinicalTrials.gov identifier: NCT01880658—recruiting patients).
5.1.1.4. NUC-3373 Biology.
In 2011 a novel 5-FU 2 prodrug NUC-3373 37 (aka. NUC-3073 and FUDR) was reported having potent anticancer activity in vitro.156,157 NUC-3373 37, a phosphoramidate prodrug of floxuridine 5, was shown to be resistant to thymidine phosphorylase activity, and showed (in comparison to 5) a significantly decreased loss of potency in CEM cells deficient in hENT1 transporters.157 The authors also proposed a mechanism for the prodrug decomposition sequence including enzyme-mediated carboxyl ester hydrolysis, spontaneous cyclization with concomitant loss of the aryloxy leaving group, water-mediated opening of the mixed anhydride, and phosphoramidase-mediated P–N bond cleavage, resulting in 2′-deoxyribose-5-FU-5′-monophosphate II (Figure 10).
Figure 10.

Proposed mechanism for phosphoramidate decomposition of NUC-3373 37.
NUC-3373 37 is a mixture of phosphoramidate diastereomers, and it has been shown to have many impressive prodrug attributes, overcoming the major 5-FU 2 resistance mechanisms. The agent retains activity in thymidine kinase deficient cells, is resistant to thymidine phosphorylase activity, and maintains activity in hENT1 deficient cells by entering independently of nucleoside transporters. It is resistant to dihydropyrimidine dehydrogenase catabolism, shows lower toxicity than 5-FU 2 in a variety of cell lines, and achieves high intracellular levels of 2′-deoxyribose-5-FU-5′-monophosphate II. Furthermore, the agent rapidly distributes to tissues following bolus infusion, shows low plasma levels of degradation metabolites, and reduces tumor weigh in HT29 colorectal xenografts significantly more than 5-FU 2.157 This impressive profile has led to NUC-3373 37 entering a Phase I clinical trial in late 2015 for treatment of advanced solid tumors (ClinicalTrials.gov identifier: NCT02723240—recruiting patients).
5.1.1.5. Floxuridine and 5′-Fluoro-2′-deoxycytidine with Tetrahydrouridine Biologies.
The National Cancer Institute was an early developer of floxuridine 5 (5-fluoro-2′-deoxyuridine, FdUrd), which has been reported to be 10–100 times more effective at inhibiting tumor cell proliferation in vitro than 5-FU 2.158 Floxuridine 5 gained FDA approval in 1970, and was marketed by Roche.159 It is approved, but not widely used, for colon and colorectal cancers that have metastasized to the liver, kidney, and stomach.160
Floxuridine 5 is a hydrophilic compound, and is likely transported into cells by pyrimidine nucleoside transporters.161 Thymidine kinase converts floxuridine 5 to floxuridine-5′-monophosphate II, which, as mentioned above, inhibits thymidylate synthase (Figure 9).
In the intestine, a significant amount of floxuridine 5 is converted to 5-FU 2 by thymidine phosphorylase. Therefore, hepatic arterial infusion has been employed to bypass the intestine and deliver more intact floxuridine 5 to the liver.162 Furthermore, the agent exhibits two properties associated with drugs that are best fit for hepatic arterial infusion: a high hepatic extraction and a short plasma half-life.160 These properties can lead to increased hepatic levels of floxuridine 5 and decreased systemic levels of the compound, which can decrease overall toxicity. This procedure has shown improvements for floxuridine 5 treatment for colorectal cancers.163–165
Several mechanisms of floxuridine resistance have been reported. Studies have shown a decrease in orotate phosphoribosyltransferase activity and thymidine kinase activity in vitro, as well as an increase of thymidylate synthase mRNA expression in vitro.166 In patients, an increase in thymidylate synthase levels has been correlated with floxuridine resistance.167
Several clinical trials that include floxuridine 5 are currently underway. These include hepatic arterial infusion with floxuridine 5 and the anti-inflammatory and immunosuppressive steroid derivative dexamethasone (ClinicalTrials.gov identifier: NCT01862315—recruiting patients). Another trial examines hepatic arterial infusion with floxuridine 5 and dexamethasone in combination with gemcitabine hydrochloride 9 or the antiangiogenic monoclonal antibody bevacizumab (ClinicalTrials. gov identifiers: NCT01938729—recruiting patients and NCT00200200—active status—not yet recruiting patients). Recent results from a Phase I trial using floxuridine 5 with modified oxaliplatin, 5-fluorouracil 2, and leucovorin (m-FOLFOX6) were reported.164
A major problem for anticancer cytidine analogs (gemcitabine, cytarabine 4, azacytidine 11, decitabine 14, capecitabine 10, and 5-fluoro-2′-deoxycytidine 38) is that they are substrates for cytidine deaminase and can be rapidly converted to uridine analogs, which are often inactive metabolites.168 For example, the liver expresses high levels of cytidine deaminase that provide a microenvironment to protect cancer cells from decitabine 14 treatment.169 In attempts to overcome the problem of deamination, cytidine deaminase inhibitors, tetrahydrouridine (39, NSC-112907) and (4R)-2′-deoxy-2′,2′-difluoro-3,4,5,6-tetrahydrouridine (Figure 11), are being investigated with nucleoside analogs as potential combination therapies.
Figure 11.

Structures of tetrahydrouridine 39 and (4R)-2′-deoxy-2′,2′-difluoro-3,4,5,6-tetrahydrouridine.
Tetrahydrouridine 39 has been investigated since the 1970s and been evaluated in cell lines and mice with cytarabine 4, gemcitabine, azacytidine (11, and decitabine 14.170–174 How ever, clinical data has shown that patients with the cytidine deaminase gene G208A polymorphism had several adverse reactions to gemcitabine treatment.175,176 In addition, the cytidine deaminase gene K27Q polymorphism promotes greater catalytic activity toward deoxycytine and cytarabine 4.177 Collectively, these data suggest prescreening patients for polymorphisms would be helpful in guiding treatment options.
Currently 5-fluoro-2′-deoxycytidine (FdCyd, 38) is being studied in combination with tetrahydrouridine 39 in mice.168,174 Coadministration of 38 with tetrahydrouridine 39 led to higher levels of FdCyd-5′-monophosphate, produced by 2′-deoxycyti-dine kinase in mice.178 Once in the cell, FdCyd 38 can be converted to FdUrd by the action of cytidine deaminase, and subsequently, phosphorylation leads to FdUrd-5′-monophosphate, which inhibits thymidylate synthase. Furthermore, FdCyd-5′-monophosphate can be phosphorylated to FdCyd-5′-triphosphate and incorporated into DNA. Once incorporated into DNA, FdCyd 38 has been shown to inhibit DNA methyltransferase with comparable activity to azacytidine 11 (see Section 5.2.1).179 Therefore, FdCyd 38 exposure can lead to the following direct and indirect cytotoxic activities in cells: (1) FdCyd-5′-monophosphate inhibition of DNA methylation, (2) 2′-deoxyribose-5-FU-5′-monophosphate II inhibition of thymidylate synthase, and (3) 2′-deoxy-5-FU-5′-triphosphate VI and 5-FU-5′-triphosphate VIII incorporation into DNA and RNA, respectively.180
Both preclinical and clinical trials are currently underway using FdCyd 38 with tetrahydrouridine 39. In 2016, a preclinical evaluation of FdCyd 38 and tetrahydrouridine 39 in pediatric brain tumors was reported,181 and a methods development report was published evaluating a combination treatment of tetrahydrouridine 39 and FdCyd 38 from human serum using HPLC-MS analysis.182 In 2015, Newman et al. reported a Phase I trial that examined the toxicity, plasma exposures, peak response, and dosing for the two compounds.183 Additional clinical studies are underway for patients with advanced non-small cell lung cancer, breast cancer, bladder cancer, head and neck cancer, and solid tumors (ClinicalTrials.gov identifiers: NCT00978250—recruiting patients and NCT01534598—recruiting patients). Recently, the oral and intravenous pharmacokinetics of FdCyd 38 and tetrahydrouridine 39 were reported for cynomolgus monkeys and humans trial.184
(4R)-2′-Deoxy-2′,2′-difluoro-3,4,5,6-tetrahydrouridine is a newer cytidine deaminase inhibitor being evaluated in preclinical studies.185 The agent has an IC50 of 0.4 μM for cytidine deaminase and exhibited enhanced acid stability at its N-glycosyl bond as compared to tetrahydrouridine 39. Rhesus monkeys cotreated with (4R)-2′-deoxy-2′,2′-difluoro-3,4,5,6-tetrahydrouridine and decitabine 14 had higher detectable levels of 14 in the serum.
5.1.1.6. Tegafur-Uracil, TS-1, Carmofur, and Flucytosine Biologies.
Tegafur 19, TS-1, carmofur 24, and flucytosine 31 are other examples of prodrugs of 5-FU 2 (Figure 9). The tegafururacil combination, TS-1 (a triple drug combo including tegafur 19, chloro dihydropyridine 20, and potassium oxonate 21), and carmofur (24, mifurol), are used globally in cancer treatments.186 In 1983, tegafur–uracil (18 + 19) was approved for use in Japan. The orally bioavailable combination is given in a 4:1 mol ratio of tegafur 19 and uracil 18. Tegafur 19 is metabolized to 5-FU 2, which can be converted to the inactive dihydrofluorouracil mainly by dihydropyrimidine dehydrogenase in the liver.187 The coadministration of uracil 18 inhibits this enzyme, thus helping maintain high levels of 5-FU 2 in the liver and in the circulation. Tegafur-uracil (18 + 19) is approved in Japan and Taiwan for various advanced gastrointestinal cancers. Patients are still being recruited for a Phase II clinical trial using metronomic therapy with tegafur–uracil (18 + 19) to treat head and neck cancer (ClinicalTrials.gov identifier: NCT00855881—recruiting patients). While a Phase II study in combination with sorafenib (kinase inhibitor) for advanced hepatocellular carcinoma has been terminated in Jan 2015, with no results posted (ClinicalTrials.gov identifier: NCT01539018—terminated in 2015—no results posted).
TS-1 includes tegafur 19, chlorodihydropyridine (gimeracil) 20, and potassium oxonate 21 used in a respective molar ratio of 1:0.4:1.188 TS-1 (19 + 20 + 21) is an orally administered triple combination drug regiment, which was approved in Japan in 1999. Chlorodihydropyridine 20 is a more potent inhibitor of dihydropyrimidine dehydrogenase than uracil 18.188,189 Potassium oxonate 21 may decrease gastrointestinal toxicity by selectively inhibiting 5-FU 2 activity within the intestinal lumen. TS-1 (19 + 20 + 21) is approved for use and is currently undergoing multiple clinical trials. Some of these studies include coadministration with cisplatin (DNA alkylating agent) for treatment of advanced non-small-cell lung cell cancer (ClinicalTrials.gov identifier: NCT01874678—completed status—with no results), a Phase II study for combination treatment of advanced hepatocellular carcinoma with the DNA alkylating agent oxaliplatin (ClinicalTrials.gov identifier: NCT01429961–unknown status–no updates for 2 years), and a Phase II study for monotherapy treatment of advanced metastatic breast cancer (ClinicalTrials.gov identifier: NCT01492543—unknown status—no updates for 2 years). In addition, Phase I clinical studies using combination therapy of TS-1 (19 + 20 + 21) and TAS-114, a first-in-class dual inhibitor of dUTPase (deoxyuridine-5′-triphosphate nucleotide hydro-lase) and dihydropyrimidine dehydrogenase,190 is being conducted (ClinicalTrials.gov identifier: NCT01610479—active status—not recruiting).
Product marketing for carmofur 24 started in 1981. Carmofur 24 is a lipophilic-masked analog of 5-FU 2 that can be administered orally. The carbamoyl moiety of the drug is removed in vivo to release 5-FU 2. Carmofur 24 has been used to treat colorectal cancer in China, Japan, and Finland for many years.186 However, the agent has been shown to induce delayed leukoencephalopathy, characterized by progressive damage to white matter in the brain with stroke-like symptoms.191–193 There are currently no open clinical trials for this agent. A trial treating patients with stage II hepatocellular carcinoma was suspended prematurely, because 56% of the treated patients had unacceptable side effects and offered no survival advantage for certain cancers in stage 1 and 2 of the disease.194 This may be a reason why carmofur 24 was never pursued for FDA-approval in the US.
Flucytosine 31 is another prodrug of 5-FU 2 in clinical trials. It is converted to 5-FU 2 by cytosine deaminase, which is not encoded by the human genome. Therefore, flucytosine 31 is used in combination with gene therapy of cancer cells in an attempt to have drug-specific targeting.195 Engineered mesenchymal stromal cells have been examined to convert flucytosine 31 into its active compound,196 and modified neural stem cells are currently being tested for the same purpose (ClinicalTrials.gov identifiers: NCT02015819—recruiting status, and NCT01172964—completed status—no results posted). Flucytosine 31 is currently in a Phase I/II clinical trial in combination with maltose and APS001F, a recombinant anaerobic bacteria Bifidobacterium engineered to express the cytosine deaminase gene, for treatment of solid tumors (ClinicalTrials.gov identifier: NCT01562626—recruiting status).
The progression of fluoropyrimidine analogs shows the importance of a biological observation–chemical modification interplay. The intravenously-administered 5-FU 2 and floxuridine 5 was followed by the orally-bioavailable carmofur 24 and flucytosine 31. The next step in the progression involved drug combinations TS-1 (19 + 20 + 21), tegafur–uracil (18 +19), and the recently FDA-approved TAS-102 (15 and 16), in order to decrease 5-FU 2 degradation. Further chemical modifications led to double (doxifluridine 23) and triple (capecitabine 10) prodrugs, which showed improvement in toxicity and pharmacokinetics profiles. Currently, NUC-3373 37 appears to overcome both innate and drug-derived resistance mechanisms. Newer analogs could be used as combination therapy with DNA damaging agents such as mitomycin-C and cisplatin, and immunotherapies—antiCTLA-4 and anti-PD-1, to improve patient survival rates.
5.1.2. Fluorouracil (5-FU), Fluorouracil Prodrugs, and Combinations Syntheses.
5.1.2.1. Fluorouracil (5-FU) Synthesis.
In 1957, a synthesis of 5-FU 2 was reported in which the compound was prepared from pseudourea salts and α-fluoro-β-keto ester enolates.197–199 Twenty years later, Robins et al. reported a two-step synthesis of 5-FU 2 from uracil 18 (Scheme 5).200 Trifluoromethyl hypofluorite was used to introduce a fluorine atom into the uracil ring, and the resulting fluorinated adduct 66 underwent base-catalyzed elimination of MeOH, generating 5-fluoro-pyrimidine 2. An alternative synthesis used the cheap and readily available ortic acid (vitamin B13, 67) as the starting material.201 Fluorination of 67 with fluorine gas and trifluoroacetic acid furnished acid 68, which in turn was decarboxylated to produce 5-FU 2 on a gram-scale.
Scheme 5. Synthesis of Fluorouracil 2a.

aReagents and conditions: (a) CF3OF/CCl3F, MeOH, −78 °C, 5 min; (b) Et3N, MeOH, H2O, rt, 11 min, 90% (over two steps); (c) F2 (g), TFA, 0 °C to 10 °C, 3 h, 80%; (d) triethyleneglycol dimethyl ether, 130 °C to 200 °C, 20 min, 90%.
Baasner et al. reported a 100-g-scale preparation of 5-FU 2.202 This procedure used selective fluorine/chlorine exchange and chlorine hydrogenolysis reactions (Scheme 6). Tetrafluoropyr-imidine 72, which can be prepared in four steps from urea 69 and diethylmalonate 70 via 2,4,5-trichloropyrimidine 71, was treated with HCl gas forming the dichlorinated intermediate 73. Dechlorination by selective palladium-catalyzed hydrogenolysis was followed by hydrolysis with aqueous sodium hydroxide to complete the synthesis.
Scheme 6. Synthesis of Fluorouracil 2a.

aReagents and conditions: (a) Na, MeOH; (b) POCl3, PhNMe2; (c) proton sponge hydrofluoride; (d) NaF, F2, CFCl3; (e) HCl (g), 160 °C, 4 h, 38%; (f) Pd/C, H2, Et3N, EtOAc, 3.5 h, 82%; (g) aq NaOH, 80 °C, 4 h, 93%.
5.1.2.2. Doxifluridine and Capecitabine Syntheses.
The first synthesis of capecitabine 10 was reported in the mid-1990s (Scheme 7).150,203 Compound 79 is a key intermediate and is synthesized in six steps from d-ribose through a cyclization, mesylation, iodidation, reductive dehalogenation, hydrolysis, and cyclization/acetylation sequence. 5-Fluorocytosine, prepared by treating cytosine with trifluoromethyl hypofluorite in MeOH,204 was glycosylated with protected sugar 79 in good yield. N4-Amino acylation followed by selective deprotection of the 2′,3′-acetyl groups by treatment with aqueous sodium methoxide afforded crude capecitabine 10. Finally, pure capecitabine 10 was precipitated as a white solid from ethyl acetate/hexanes on a 100 gram-scale.
Scheme 7. Synthesis of Capecitabine 10a.

aReagents and conditions: (a) HCl, MeOH/Me2CO, 78%; (b) MsCl, pyr; (c) NaI, DMF; (d) Pd/C, H2, 27%; (e) aq HCl, 100 °C, 97%; (f) Ac2O, pyr, 64%; (g) 5-fluorocytosine, HMDS, SnCl4, DCM, 76%; (h) n-pentyl chloroformate, pyr; (i) aq NaOH, MeOH.
Two large-scale process syntheses of capecitabine 10 were reported by Hoffman-La Roche205 and Gore et al.206 Both syntheses use 5′-deoxy-5-fluorocytidine 85 as a key intermediate (Scheme 8). 5-Fluorocytidine 81, synthesized by treating protected cytidine with CF3 OF/CCl3 F,200 was protected, tosylated, and subsequently converted to 5′-iodo 83.207 Reductive dehalogenation followed by deprotection produced intermediate 85.208 This same sequence can also be employed to prepare doxifluridine 23, although using 5-fluorouridine as the starting material.
Scheme 8. Synthesis of Doxifluridine 23 and Capecitabine 10a.

aReagents and conditions: (a) 2,2-dimethoxypropane, TsOH, Me2CO, rt, 2 h, 95%; (b) methyltriphenoxyphosphonium iodide, DMF, rt, 2.5 h, then MeOH, rt, 1 h, 74%; (c) Et3N, H2, Pd/C, MeOH, rt, 1.5 h, 93%; (d) TFA, 40 min, 78%; (e) CH3(CH2)4OCOCl, pyr, DCM, −5 °C to rt, overnight, 92%; (f) NaOH, MeOH, −10 °C, 15 min, then concd HCl, 87%; (g) 87, THF, reflux, 66%.
The Hoffmann-La Roche synthesis converted 85 to the trisacylated intermediate 86, using 3 equiv of n-pentyl chloroformate in cold dichloromethane and pyridine. Selective removal of 2′,3′-ester groups was accomplished by treatment with aqueous sodium hydroxide in cold methanol, and precipitation from cold ethyl acetate afforded pure capecitabine 10.
The Gore et al. approach converted 85 directly to the product 10 by selective N4 acylation. In refluxing THF, intermediate 85 was reacted with pentyloxycarbonyl-1-hydroxybenzotriazole 87, which can be easily made from pentyl chloroformate and HOBt. This was followed by aqueous workup and precipitation to afford 10. Other acylating agents, such as pentyloxycarbonyl-imidazole, pentyloxycarbonyl-4-nitrophenoxy, and pentyloxycarbonyl-pentafluorophenoxy were also used, and produced similar yields.
The Jamison group used continuous flow chemistry to prepare capecitabine 10.209 The reaction exhibited impressive yields, “green” conditions, and very short reaction times (Scheme 9). The glycosylation reaction between triacetate intermediate 88 and silylated pyrimidine 90, while under BrØnsted acid-catalyzed conditions, was completed in 20 min. This procedure does not require aqueous workup. Mixing conditions were carefully monitored to form the N4-carbamate intermediate. Deprotection with a mixture of NaOH/MeOH/H2O occurred at room temperature in only 2 min. The overall process produced milligrams of capecitabine 10 in less than 1 h and a 72% yield from starting materials 88 and 90.
Scheme 9.

Flow Synthesis of Capecitabine 10
In the same report,209 doxifluridine 23 was also synthesized using protected sugar 88, which was condensed with silylated 5-FU 2 in a glycosylation reaction. Deprotection of the resulting intermediate with sodium methoxide furnished the product. This one-flow, two-step process produced doxifluridine 23 in 89% yield in 10 min.
5.1.2.3. NUC-3373 Synthesis.
In 2011, McGuigan and co-workers reported the synthesis of NUC-3373 37.156 The agent was prepared by reaction of phosphochloridate 93 (produced using l-alanine benzyl ester salt and 1-naphthyl-dichlorophosphate 92) with floxuridine 5 in THF in the presence of t-BuMgCl (Scheme 10). The synthesis produces milligrams of the final product and requires column chromatography to produce the purified mixture of diastereomers. Furthermore, no large-scale syntheses of NUC-3373 37 have been reported.
Scheme 10. Synthesis of NUC-3373 37a.

aReagents and conditions: (a) l-alanine benzyl ester salt, Et3N, DCM, −78 °C, 1–3 h, 74%; (b) t-BuMgCl, THF, rt, 18 h, 8%.
5.1.2.4. Floxuridine, 5-Fluoro-2′-deoxycytidine, and Tetrahydrouridine Syntheses.
In the late 1950s, floxuridine 5 was originally prepared enzymatically and later converted to 5-fluoro-2′-deoxycytidine 38 via chemical synthesis.210–212 Robin’s method (see Sections 5.1.2.1) was used to prepare both compounds in only two steps from 2′-deoxyuridine 94a and 2′-deoxycytidine 94b, with the key ring fluorination and sugar deprotection occurring in one pot (Scheme 11). This approach has the advantage of starting from a deoxynucleoside, which already has the needed stereochemistry fixed.
Scheme 11. Synthesis of Floxuridine 5, 5-Fluoro-2′-deoxycytidine 38, and Tetrahydrouridine 39a.

aReagents and conditions: (a) Ac2O, DMAP, rt, 24 h, 88% (for 95a); Ac2O, pyr, rt, 4.5 h, 99% (for 95b); (b) CF3OF, CCl3F, CHCl3, −30 °C, evaporation, then Et3N, MeOH, H2O, rt, (55% for 5),(69% for 38); (c) Rh/Al, H2 (g), H2O, overnight; (d) H2O, pH 6, 92% (over two steps); (e) Rh/Al, H2 (g), NaOH, H2O, 24 h, 45% of 99, 35% of 39; (f) NaBH4, H2O, freezer, overnight.
Surprisingly few syntheses of tetrahydrouridine 39 have been reported. In 1967, Hanze reported two approaches to the compound (Scheme 11).213 Cytidine 96 was “overreduced” to tetrahydrocytidine 97 in one step using rhodium on aluminum in water, and subsequent hydrolysis afforded tetrahydrouridine 39. Uridine 98 was reduced to 5,6-dihydrouridine 99 also using rhodium and aluminum, and subsequent treatment with sodium borohydride furnished the target compound. The syntheses suffered from complex reaction mixtures, and no yields were reported.
Aoyama reported a gram-scale synthesis of floxuridine 5,214 in which the key step was the condensation of 5-fluoro-2,4-bis(trimethylsilyloxy)pyrimidine 102 with 3,5-bis(O-p-chorobenzoyl)-2-deoxy-α-D-ribofuranosyl chloride 101. Chlorosugar 101 was made as the pure α anomer in three steps from 2′-deoxyribose (Scheme 12).215,216 The condensation was performed in the presence of p-nitrophenol, which led, interestingly, to the stereoselective production of the desired β-anomer 103, whereas the combination of p-nitrophenol and pyridine as catalysts led to formation of the α-anomer. Recrystallization from acetic acid gave the pure β-anomer, which was deprotected with methanolic ammonia, concluding the synthesis.
Scheme 12. Synthesis of Floxuridine 5a.

aReagents and conditions: (a) AcCl, MeOH, 25 °C, 45 min, then pyr; (b) pyr, DMAP, 4-ClBzCl, 0 °C, 1 h then 25 °C, 12 h; (c) HOAc, HCl (g), 0 °C, 7–10 min, 82% (α-anomer crystallizes from the solvent); (d) p-nitrophenol, CHCl3, 30 °C, 12 h, 92%; (e) NH3/MeOH, 30 °C, 16 h, 81%.
5.1.2.5. Tegafur, Carmofur, and Flucytosine Syntheses.
In 1976, an original synthesis of tegafur 19 was reported that coupled 2-acetoxytetrahydrofuran with silylated 5-FU 2 in the presence of sodium iodide in 95% yield.70,217 Five years later, Miyashita et al. reported an alternative preparation (Scheme 13).218 The synthesis started with a one-pot condensation of urea 69, triethyl orthformate 104, and methyl malonate 105 under solvent-free conditions. The resulting methyl ureidomethylenemalonate 106 was then cyclized in methanolic sodium methoxide to give 5-methoxycarbonyluracil 107. Condensation of 107 with 2,3-dihydrofuran followed by ring fluorination and subsequent hydrolysis concluded the synthesis of 19.
Scheme 13. Synthesis of Tegafur 19 and Carmofur 24a.

aReagents and conditions: (a) 130 °C, 4 h, 69%; (b) NaOMe, MeOH, reflux, 10 min, 84%; (c) 2,3-dihydrofuran, pyr, 180 °C; (d) F2, AcOH, rt; (e) 1 N NaOH, rt, 1 h, 62% (over two steps); (f) aq HCHO, 55 °C, 6 h, 82%; (g) KBrO3, 80 °C, then H2N(CH2)5CH3, DCC, MeCN, 0 °C to rt, 6 h, 40%.
Carmofur 24 has been synthesized by treating 5-FU 2 with phosgene and hexylamine.219,220 An alternative approach also utilized 5-FU 2 as starting material but used aqueous form-aldehyde to effect N-formylation. This was followed by oxidation with potassium bromate and finally condensation with hexylamine to conclude the synthesis of 24 (Scheme 13).221
Flucytosine 31 was synthesized by reacting cytidine 110 with CF3OF/CCl3F in methanol for 5 min followed by addition of triethylamine in water (Scheme 14).200
Scheme 14. Synthesis of Flucytosine 31a.

aReagents and conditions: (a) CF3OF, CCl3F, MeOH, −78 °C to rt; (b) Et3N, MeOH, H2O, rt, 8 h, 85% (over two steps).
5.1.2.6. Gimeracil and Oteracil Syntheses.
In 1953, Kolder and Hertog reported a synthesis of the TS-1 additive gimeracil 20, which was completed in seven steps using 4-nitropyridine N-oxide as starting material.222 Later, Yano et al. reported an alternative gram-scale synthesis (Scheme 15).223 The one-pot, three component condensation of malononitrile 111, 1,1,1-trimethoxyethane, and 1,1-dimethyoxytrimethylamine generated the dicyano intermediate 112, which was into 2(1H)-pyridinone 113.224 Selective chlorination of 113 was followed by acid-mediated demethylation, hydrolysis, and decarboxylation, to afford gimeracil 20. Interestingly, Xu et al. found that treatment of intermediate 113 with sulfuryl chloride resulted in dichloro 115 formation, which could still be converted to gimeracil 20 by treatment with sulfuric acid.225
Scheme 15. Synthesis of Gimeracil 20a.

aReagents and conditions: (a) CH3C(OCH3)3, MeOH, then (CH3)2NHCH(OCH3)2, reflux, 92%; (b) aq AcOH, 130 °C, 2 h, 95%; (c) SO2Cl2, HOAc, 50 °C, 0.5 h, 91%; (d) 40% H2SO4, 130 °C, 4 h, 91%; (e) SO2Cl2, HOAc, 50 °C, 45 min, 86%; (f) 75% H2 SO4, 140 °C, 3 h, then NaOH, then pH 4–4.5, 89%.
Poje et al. reported a two-step, gram-scale preparation of the TS-1 additive oteracil 21 (Scheme 16).226 Iodine-mediated-oxidation of uric acid 116 produced dehydroallantoin 117 as the major product, and subsequent treatment with potassium hydroxide resulted in the rearranged product oteracil 21.227
Scheme 16. Synthesis of Oteracil 21a.

aReagents and conditions: (a) LiOH, I2, H2O, 5 °C, 5 min, then AcOH, 75%; (b) aq KOH, 20 min, rt, 82%.
5.1.3. Trifluorothymidine and Tipiracil Hydrochloride (TAS-102) Biology.
In 1964, Heidelberger and co-workers synthesized trifluorothymidine (TFT, 15) and demonstrated that it had promising anticancer activity.228–231 Initial animal studies showed that TFT 15 was degraded to trifluorothymidine and catabolite 5-carboxyuracil, in the liver, spleen, and intestines.230 The agent showed reductions in some tumor sizes during a clinical trial but, like other antimetabolites, exhibited a plasma half-life of 15 min in cancer patients.232 Research on TFT 15 was discontinued due to inadequate information on the pharmacokinetics and toxicity profile.233,234
TFT 15 is a substrate for thymidine kinase, generating TFT-5′-monophosphate (an active intracellular metabolite). Similar to 5-FU-5′-monophosphate II, TFT-5′-monophosphate inhibits thymidylate synthase (Figure 8).235 In contrast to 5-FU-5′-monophosphate II, TFT-5′-monophosphate does not form a ternary complex with thymidylate synthase but inhibits it by binding to the active site of the enzyme.236 Furthermore, TFT-5′-monophosphate is a reversible inhibitor of thymidylate synthase, and removal of the agent results in rapid recovery of enzyme activity, whereas inhibition caused by formation of the 5-FU-5′-monophosphate II ternary complex has prolonged effects.237 TFT-5′-monophosphate can be further phosphorylated to its triphosphate form (another active metabolite) and then incorporated into DNA to promote single-strand breaks.238 DNA-incorporated TFT-5′-monophosphate is resistant to DNA glycosylase,239 and incorporated TFT-5′-monophosphate can promote DNA instability and double-stranded breaks.240,241
The major drawback of TFT 15 monotherapy is its rapid degradation within the body, primarily by the action of thymidine phosphorylase, providing a natural TFT resistance mechanism.242 However, coadministration of TFT 15 and a thymidine phosphorylase inhibitor improved the pharmacokinetic profile in animal models and antitumor activity in cell lines and animal models.243 Currently, Taiho Pharmaceuticals is developing TAS-102 (15 + 16) as a combination therapy that uses TFT 15 with tipiracil hydrochloride 16, a thymidine phosphorylase inhibitor with an IC of 35 nM,243 in a respective molar ratio of 2:1. This combination was shown to greatly decreased the biodegradation of TFT 15 in vitro.240 Interestingly, tipiracil hydrochloride 16 has also been shown to have antiangiogenic activity.228
TAS-102 (15 + 16) was FDA-approved in Sept 2015 for treatment in patients with colorectal cancer,244 and it is also currently being evaluated in clinical trials for the treatment of advanced solid tumors and metastatic refractory colorectal cancer233 (ClinicalTrials.gov identifier: NCT01607957—active status, not recruiting). A study was initiated evaluating TAS-102 (15 + 16) plus nivolumab in patients with microsatellite stable refractory metastatic colorectal cancer ClinicalTrials.gov identifier: NCT02860546—recruiting patients). In addition, TFT 15 has been pursued as an antiviral agent (registered as Viroptic) for use against the herpes simplex virus.245,246
5.1.4. Trifluorothymidine and Tipiracil Hydrochloride (TAS-102) Syntheses.
Heidelberger et al. reported the first synthesis of TFT 15 (Scheme 17), featuring an enzyme-mediated transglycosylation of 5-trifluoromethyluracil 126 with thymidine.229 The trifluoro base 126 was prepared in an eight-step sequence. The synthesis began with treatment of trifluoroacetone 118 with hydrogen cyanide to produce cyanohydrin 119, and subsequent acetylation and ester pyrolysis afforded alkene 120. Treatment of 121 with anhydrous HBr and urea provided ureidoamide 123, which was then refluxed in acid to generate 5,6-dihydro-5-trifluoromethyluracil 124. This intermediate was subjected to a bromination–dehydrobromination sequence, furnishing 5-trifluoromethyluracil 126.
Scheme 17. Synthesis of Trifluorothymidine 15a.

aReagents and conditions: (a) NaCN, H2SO4, H2O, 10 °C, 3 h, 99%; (b) Ac2O, concd H2SO4, reflux, 1 h, 78%; (c) 500 °C, 4 h, 64%; (d) HBr (g), MeOH, 0 °C, 36 h, 82%; (e) (NH2)2CO, H2O, 100 °C, 30 min, 28%; (f) aq HCl, reflux, 1 h, 58%; (g) Br2/AcOH, reflux, 3 h, 85%; (h) DMF, 140 °C, 75 min, 80%; (i) thymidine, bactotryptone, NaCl, Escherichia coli B, 0.067 M phosphate buffer (pH 6.7), 37 °C, 3.5 h, 14%.
Komatsu et al. have reported a more modern and enzyme-free preparation of TFT 15.247 The preparation featured a green glycosylation reaction performed on the 100-g-scale using the TMS-protected analog of thymine 127 and the corresponding chlorosugar 101, made in two steps from 2′-deoxyribose. Conditions were sought to increase the rate of glycosylation (e.g., 101α to 128) and decrease the rate of chlorosugar anomerization (e.g., 101α to 101β), thereby increasing the yield of the β-glycosylation product 128 (Scheme 18).248
Scheme 18. Synthesis of Trifluorothymidine 15a.

aReagents and conditions: (a) AcCl, MeOH, 25 °C then pyr; (b) pyr, DMAP, 4-ClBzCl, 0 °C, 1 h, then 25 °C, 12 h; (c) HOAc, HCl (g), 0 °C, 7–10 min, 82% (over three steps); (d) 127, anisole, 50 °C, 3.5 h, 85% (β/α = 85.7:14.3); (e) NaOMe, MeOH, 4 °C, 3.5 h, then AcOBu, 97% (β/α = 99.92:0.08).
Nonpolar solvents were used to decrease chlorosugar anomerization, and temperature and reactant concentrations were explored empirically. Optimized large-scale conditions included condensation of silylated trifluorothymidine with an equimolar amount of chlorosugar 101 in minimal amounts of anisole (96% w/w of 101) at 50 °C, producing an 85:15 β/α anomeric mixture of 128/129 in 71% yield. Deprotection with NaOMe at 4 °C, precipitation and filtration, and finally washing with AcOBu (to remove residual methyl 4-chlorobenzoate) produced pure trifluorothymidine 15.
A concise 100-g-scale synthesis of tipiracil hydrochloride 16 has been reported (Scheme 19).249 Amination of 134 with methanolic ammonia led to the cyclized intermediate 2-iminopyrrolidine 135.250 In parallel, 6-chloromethyl uridine, which can be prepared in four steps from urea 69 and ethyl 2-acetoacetate 130 via intermediate 131,251 was chlorinated at the 5-position to furnish the dichloromethyl uracil derivative 133.252 Finally, condensation of 133 with 135 produced tipiracil hydrochloride 16.249
Scheme 19. Synthesis of Tipiracil Hydrochloride 16a.

aReagents and conditions: (a) SO2Cl2, AcOH, 50 °C, 2.5 h, 83%; (b) NH3/MeOH, 120 °C, 10 h, 83%; (c) NaOEt, DMF, rt, 16 h, then aq HCl, 38%.
5.2. Azanucleosides
5.2.1. Decitabine, Azacytidine, CP-4200, and SGI-110 Biologies.
The azanucleosides, decitabine (Dacogen, 14), and azacytidine (Videza, 11), were first developed as cytostatic agents nearly half a century ago.253,254 These cytostatic agents were later found to inhibit DNA methylation in human cell lines and were developed as epigenetic drugs.
Decitabine 14 is a 2′-deoxy-5-azanucleoside analog of cytidine that enters the cell by ENT-1.255 It is subsequently converted to decitabine-5′-monophosphate by 2′-deoxycytidine kinase. Further phosphorylation leads to the active metabolite decitabine-5′-triphosphate, which is a substrate for DNA polymerase α and is incorporated into the DNA.256 The incorporated decitabine-5′-monophosphate cannot be methylated, which can influence epigenetic gene regulation.257 DNA methylation usually involves the transfer of a methyl group from S-adenosyl-l-methionine, catalyzed by DNA methyl transferases (DNMTs), to the 5-position of a cytosine base within a cytosine-phosphate-guanosine dinucleotide.258,259 Furthermore, DNA hypermethylation often silences tumor suppressor genes in hematopoietic malignancies,260 and inhibition of DNA methylation at regions of decitabine-5′-monophosphate incorporation in the DNA most likely restores the expression of some of these genes.261–264
The ability of decitabine 14 to inhibit DNA methylation is often attributed to the formation of a complex between the azacytosine-guanine dinucleotide and DNMT1.265 This maypossibly occur via the covalent trapping paradigm (Figure 12).266 A covalent bond is formed between DNMT1 and the C6-position of the azacytosine base. Methylation of the nitrogen at position 5 of the base then occurs; however, β-elimination cannot occur due to the lack of a hydrogen atom at this position. As a consequence, DNMT1 remains covalently bound to the aza-base and is unable to continue its methylation activity. The covalent complex also triggers DNA damage signaling to result in DNMT1 degradation.267 A recent study, however, indicated that decitabine 14 could induce degradation of methyltransferases without the formation of the covalent complex, suggesting that the agent may work by additional mechanisms as well.268
Figure 12.

Covalent trapping paradigm: mechanism-based DNMT1 degradation by decitabine 14.
Decitabine 14 suffers from some chemical stability issues. The triazine ring of the drug is prone to hydrolytic opening and deformylation, whereas the sugar is susceptible to anomerization. Several groups have reported a half-life of decitabine 14 ranging from 3.5 to 21 h at physiological pH and temperature,269 whereas the in vivo half-life of decitabine 14 has been reported to be 15–20 min.270 Once the prodrug is metabolized to release decitabine 14 in the liver or cells, the high levels of cytidine deaminase protein will generate an inactive nucleoside byproduct,169 which in turn limits the intracellular concentration and toxicity of decitabine 14. Finally if patients have reduced levels of 2′-deoxycytidine kinase activity, this may also contribute to natural decitabine resistance, as observed with decitabine.271
At low concentrations, decitabine 14 has the ability to inhibit DNA methyltransferase 1, but it is cytotoxic at high concentrations in vitro.272,273 Optimal treatment with decitabine 14 has been tested using continuous treatment at a low concentration in patients.274 However, the agent has poor oral bioavailability (currently administered by intravenous infusion) and is also a substrate for cytidine deaminase, which provides a natural resistance mechanism. Additionally, ex vivo studies suggest that reduction in phosphorylation by 2′-deoxycytidine kinase may also contribute to decitabine resistance.271 Attempts to overcome these problems include the preparation of a decitabine mesylate salt,275 designed to increase the oral bioavailability, as well as coadministration of oral decitabine 14 with oral tetrahydrouridine 39 in patients.276
Azacytidine 11, a riboside analog of decitabine 14, is given by injection and enters cells by the uridine/cytidine transport system.277 After phosphorylation by uridine-cytidine kinase,278,279 the active metabolite azacytidine-5′-triphosphate is incorporated into RNA to disrupt RNA metabolism and protein synthesis.279 Azacytidine-5′-diphosphate can also be reduced by ribonucleotide reductase to form 2′-deoxyazacytidine-5′-diphosphate and then follows the mechanism of action of decitabine 14.
After years of clinical research and dosage refinement, conditions were finally produced to effectively treat myelodysplastic syndrome, with azacytidine 11 and decitabine 14 being FDA-approved in 2004 and 2006, respectively.280,281 Myelodysplastic syndrome, also known as preleukemia, is a group of related diseases originating in the bone marrow in which hematopoietic stem cells produce ineffective myeloid cells.282 A genetic polymorphism in the cytidine deaminase gene (79A>C) and promoter depletion (–31delC) can lead to a rapid-deaminator phenotype and an increase in mRNA expression, leading to increased toxicity in patients treated with azacytidine 11.283,284 Sorting patients based on cytidine deaminase genotypes remains difficult due to genotype to phenotype relationships, whereas developing a functional cytidine deaminase assay would be beneficial.285
Azacytidine 11 and decitabine 14 treatments are being evaluated in numerous clinical studies. An oral formulation of azacytidine (CC-486, Celgene) is currently in Phase I clinical trials (alone and in combination) for treatment of refractory solid tumors and Japanese myelodysplastic syndrome (ClinicalTrials. gov identifiers: NCT01478685—completed—no results posted, and NCT01908387—terminated status—no results posted). A Phase I/II Study of azacitidine 11 and with capecitabine 10 and oxaliplatin (DNA alkylating agent) is underway (ClinicalTrials. gov identifier: NCT01193517–active status–not recruiting). A Phase II study is examining the kinase inhibitor sorafenib with azacitidine 11 for primary response and secondary toxicity profile (ClinicalTrials.gov identifier: NCT02196857–actively recruiting patients). For decitabine 14 and tetrahydrouridine 39 clinical trials, one is currently recruiting patients— adjuvant oral decitabine 14 and tetrahydrouridine 39 with or without celecoxib in people undergoing pulmonary metastasectomy (ClinicalTrials.gov identifiers: NCT02839694–recruiting patients). Four additional studies have yet to start recruiting patients with refractory/relapsed lymphoid malignancies, pancreatic cancer, and non-small cell lung cancer using different combination treatments (ClinicalTrials.gov identifiers: NCT02846935–not yet recruiting patients, NCT02847000–not yet recruiting patients, NCT02795923–not yet recruiting patients, and NCT02664181–not yet recruiting patients).
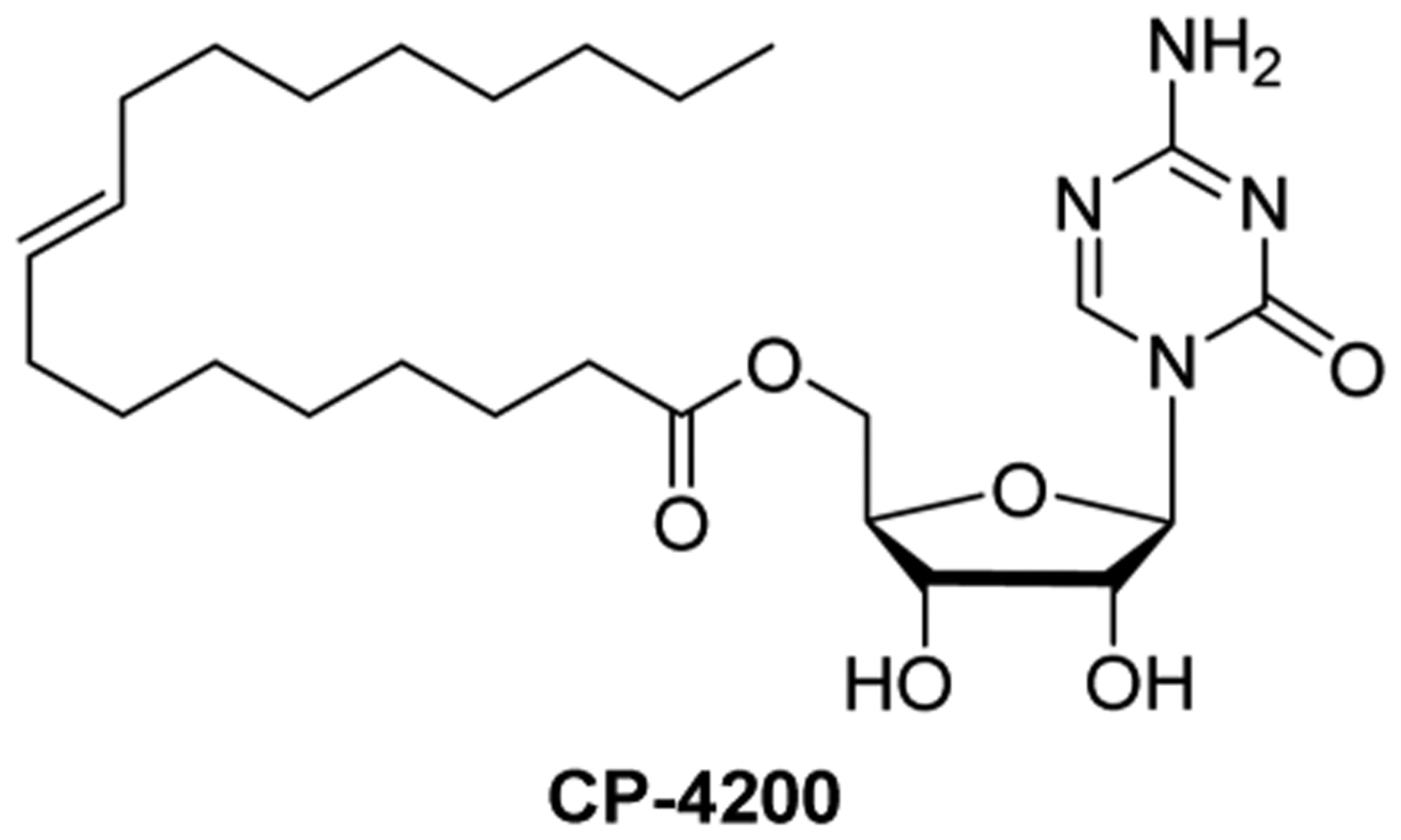
Clavis Pharma is developing CP-4200, an elaidic acid derivative of azacytidine, which has strong epigenetic modulatory potency in human cancer cell lines (Figure 13).286 The aim of this drug is to circumvent therapy resistance in patients with MDS/AML due to decrease in drug uptake by hENT1, because the agent enters the cell by a hENT1-independent mechanism.287 CP-4200 has not yet moved past pre-clinical development. CP-4200 might have an acceptable bioavailability profile for oral application.
Figure 13.
Chemical structure of CP-4200.
Potential hurdles for CP-4200 include the following. First, upon prodrug metabolism to release azacytidine 11 in the liver or tumor cells, high levels of cytidine deaminase activity will generate an inactive nucleoside byproduct.288 Second, patients with reduced levels of 2′-deoxycytidine kinase activity may have some level of natural azacytidine resistance, as observed with decitabine 14.271 Third, two other elaidic acid derivative prodrugs: elacytarabine (51, Phase I trial) and CP-4126 (49, two Phase III trials) have not been successful in clinical trials.
Astex Pharmaceuticals has developed the dinucleotide SGI-110 22, a second-generation prodrug of decitabine 14, which couples the agent with deoxyguanosine via a phosphodiester bond.289 It was synthesized in order to increase the in vivo exposure and efficacy of decitabine 14 by protecting it from cytidine deaminase activity.290,291 SGI-110 22 is being evaluated as a hypomethylation agent with an epigenetic mechanism of action in tumors.292,293 In a Phase I/II dose-escalating study, SGI-110 22 showed a favorable pharmacokinetic profile.294 The dinucleotide was efficiently converted to decitabine 14 in vivo. Moreover, it exhibited a longer apparent half-life, lower Cmax, and prolonged plasma exposures of decitabine 14 than equivalent amounts of IV-administered decitabine 14. Furthermore, SGI-110 22 exhibited a greater stability, and lower doses of the agent achieved equal or better methylation inhibition than decitabine 14 monotherapy. SGI-110 22 is currently in several Phase I/II studies (as monotherapy and combination therapy) for treatment of ovarian cancer, metastatic colorectal cancer, myelodysplastic syndrome, acute myelogenous leukemia, and advanced hepatocellular carcinoma (ClinicalTrials.gov identifiers: NCT01696032–active status–not recruiting patients, NCT01896856–recruiting patients, NCT01261312–active status–not recruiting patients, NCT01752933–completed in 2015–no results posted, and NCT01966289–recruiting patients).
5.2.2. Azacytidine, Decitabine, and SGI-110 Syntheses.
In 1964, Piskala et al. reported the first synthesis of azacytidine 11 (Scheme 20).295 Chlorination of the 1′-position of tetraacetate riboside 136 followed by treatment with silver isocyanate produced 1-glycosyl isocyanate 137. Conversion of 137 to isobiuret 138 using 2-methylisourea was followed by cyclization with triethyl orthoformate and deprotection with methanolic ammonia to give azacytidine 11.
Scheme 20. Synthesis of Azacytidine 11a.

aReagents and conditions: (a) HCl (g), Ac2O, Et2O; (b) AgNCO, 60%; (c) 2-methylisourea, 68%; (d) HC(OC2H5)3; (e) NH3/MeOH, 40%.
In 2006, Ionescu and Blumbergs reported a large-scale synthesis of azacytidine (11, Scheme 21).296 Cyanoguanidine 139 was hydrolyzed and subsequently cyclized to produce the highly water-sensitive triazine 141. The glycosylation reaction between silylated triazine 142 and 1,2,3,5-tetra-O-acetyl-β-d-ribofuranose was performed in cold dichloromethane, with CF3SO3H added at 0 °C, followed by reaction warming to room temperature. Cooled solutions were used during the quenching/extraction step, which also minimized decomposition of the triazine base. Deprotection with a sodium methoxide/methanol solution, followed by recrystallization from DMSO/MeOH afforded azacytidine 11 on kilogram-scale. The last three steps, from glycosylation to recrystallization, were also performed in one pot starting with 5 g of the triazine, producing azacytidine 11 in 41% yield.
Scheme 21. Synthesis of Azacytidine 11a.

aReagents and conditions: (a) HCO2H, 80 °C, 15 min, 85%; (b) (CH3)2NCH(OCH3)2, NaOMe, MeOH, 40–50 °C, 4 h, 90%; (c) HMDS, (NH4)2SO4, CH3CN, reflux, 4–8 h; (d) 1,2,3,5-tetra-O-acetyl-β-d-ribofuranose, CF3SO3H, EtOAc, 45 °C, 30 min, then HCl, DCM, rt, 45 min; (e) n-BuNH2, MeOH, 65 °C, 1 h, 49% (over three steps).
An additional process synthesis of azacytidine 11 using triflic acid in the glycosylation was reported.297 Detailed empirical studies of the reaction found that a 1:1.2:0.95 ratio (silylated triazine 142:triflic acid:1,2,3,5-tetra-O-acetyl-β-d-ribofuranose) produced the highest yield. Exposure times to water or acid during the workup were minimized in order to protect the triazine base of intermediate 143. Further empirical studies found optimized conditions for acetyl group deprotection of 143, which involved n-butylamine in refluxing methanol. Recrystallization of the crude azacytidine 11 from DMSO/toluene gave the pure product in 99.9% purity on a kilogram-scale.
In 1964, the first synthesis of decitabine 14 was reported, which used a multistep sequence similar to the preparation of azacytidine 11 shown in Scheme 20. However, no yield was reported, as the α/β-anomers were not separated.298 Several years later, Winkley and Robins reported a synthesis of pure β decitabine (14, Scheme 22).299 Triacetyl protected sugar 144 was converted to 3′,5′-di-O-acetyl-2′-deoxy-d-ribofuranosyl chloride and then treated with the silylated 5-azacytosine 142, forming 145 as a mixture of diastereomers. Deprotection with ethanolic ammonia followed by α/β anomer separation, involving fractional crystallization followed by preparative layer chromatography on silica gel, gave decitabine 14 in 7% yield. Unfortunately, the undesired α-anomer was the predominant product, forming in 52% yield.
Scheme 22. Early Synthesis of Decitabine 14a.

aReagents and conditions: (a) AcCl, HCl, Et2O, 0 °C; (b) bis(trimethylsilyl)-5-azacytosine 142, CH3CN, 10%; (c) NH3/EtOH; (d) anomer separation, 7%.
Two difficulties for large-scale synthesis of decitabine 14 are the lack of stability of the triazine base and the absence of anomeric selectivity during the glycosylation step. Indeed, when conjugated to a carbohydrate, 5-azacytosine is sensitive to hydrolytic decomposition under acidic, basic, and neutral conditions.300 Furthermore, coupling of the silylated triazine with a 2′-deoxyribosugar generally leads to a 1:1 mixture of α/β anomers.
Henschke et al. recently performed detailed studies under various conditions of the glycosylation step in attempts to improve anomeric selectivity (Scheme 23).301 Solvent choice, reaction temperature, and ratio of the silylated triazine to the protected ribosugar were all found to affect anomeric selectivity, producing glycosylation products ranging from an anomeric ratio of 1:1 to 1:3 (α:β). Optimal large-scale conditions included a 1:1 molar ratio of the protected sugar to silylated triazine 142 with the addition of 1.05 equiv of TMSOTf in cold DCM. Kilograms of protected decitabine 147 were produced in a 1:2.7 α/β ratio in up to 55% yield. Furthermore, it was discovered that immediate quenching of the reaction with an organic amine (usually a primary amine) was needed in order to preserve the α/β-ratio. Since they had observed isomerization of the β-anomer under different conditions, the authors hypothesized that prolonged reaction standing led to the resultant decrease of the β-anomer. Moreover, the decrease in the resultant was a result of its anomerization to the α-product and not due to decomposition of the triazine base of protected decitabine. The synthesis of decitabine 14 was concluded by deprotection of a finely milled 147; high quality of this intermediate was required for reaction success, in methanol with or without sodium methoxide. Subsequent filtration and recrystallization from DMSO and methanol afforded kilograms of API-grade decitabine 14.
Scheme 23. Kilogram-Scale Synthesis of Decitabine 14a.

aReagents and conditions: (a) bis(trimethylsilyl)-5-azacytosine 142, TMSOTf, DCM, cold, 55%; (b) NaOMe/MeOH, 65%.
Kolla et al. have recently reported a process synthesis of decitabine (14, Scheme 24).302 Bis-acetyl protected sugar 149, formed by treating 2′-deoxy-α/β-d-ribose 100 with acetyl chloride in methanol followed by acetyl chloride in pyridine/DCM, was condensed with silylated 5-azacytosine in the presence of TMSOTf, giving a 1:1 α/β mixture of acylated 150. Deprotection with methanolic ammonia treatment followed by crystallization afforded decitabine 14 in an HPLC purity of 90–99%. The resulting solid was dissolved in DMSO, filtered, washed with MeOH/EtOAc, and suction dried to produce crystalline decitabine 14 at 99.8% purity on a gram-scale.
Scheme 24. Synthesis of Decitabine 14a.

aReagents and conditions: (a) AcCl/MeOH, 99%; (b) AcCl/pyr/DCM, 78%; (c) 5-azacytosine, HMDS, NH4SO4, TMSOTf, 60% (β/α = 1:1); (d) NH3/MeOH, 34%.
Recently, Redkar reported an approach to SGI-110 22 which increased the scale of compound production from 500 mg to 1 kg (Scheme 25).303 Decitabine 14 was selectively protected at the N4- and 5′-positions by treatment with DMF dimethyl acetal and then vinyl acetate in the presence of Lipozyme RM IM. Conversion to the 3′-phosphoramidite 151 was followed by coupling with 3′,N2-protected guanosine 152, and subsequent oxidation by tert-butyl hydrogen peroxide furnished dinucleotide 154. Deprotection in methanolic ammonia followed by hydrolysis produced SGI-110 22, which precipitated as a sodium salt.
Scheme 25. Synthesis of SGI-110 22a.

aReagents and conditions: (a) DMF, dimethylacetal; (b) vinyl acetate, Lipozyme RM IM, CH3CN, dioxane; (c) [(iPr)2N]POCH2CH2CN, DCM; (d) DCM; (e) tBuOOH; (f) NH3/MeOH; (g) NaOAc, H2O, EtOH.
5.3. Ribosugar-Modified Cytidine Analogs
5.3.1. Gemcitabine and Gemcitabine Prodrugs Biologies.
5.3.1.1. Gemcitabine Biology.
Hertel and colleagues at Eli Lilly originally synthesized gemcitabine (2′,2′-difluoro-2′-deoxycytidine) hydrochloride 9 as an antiviral nucleoside analog agent.304 However, it was also discovered to be very effective at inhibiting cancer proliferation in cell culture.304 Gemcitabine was originally screened against hematological cancers, but was also surprisingly shown to have activity in solid tumors. The agent is the only FDA-approved cytidine analog to be effective in solid tumors.305,306
Gemcitabine is imported into the cell by hENT-1,307 and to a lesser extent by hCNT-1 and hCNT-3.308–310 The agent is initially phosphorylated by 2′-deoxycytidine kinase, which is the rate-limiting step in its activation. Subsequent phosphorylations lead to the active metabolites: gemcitabine-5′-triphosphate and gemcitabine-5′-diphosphate. Gemcitabine-5′-triphosphate inhibits DNA synthesis by being incorporated into the newly synthesized DNA strand. Once incorporated into DNA, one additional nucleotide is inserted past gemcitabine-5′-monophosphate before chain elongation is inhibited.311,312 This “masked chain termination” cannot be detected by exonucleases and eventually causes cell cycle arrest primarily in the S phase, resulting in apoptosis.313 Alternatively, gemcitabine-5′-triphosphate can also be incorporated into DNA without chain elongation termination,17,314 and is recognized by a DNA-dependent protein kinase/p53 protein complex, which coincides with cellular apoptosis.27 Gemcitabine was also shown to have anticancer activity by being incorporated into RNA.315 Furthermore, synergistic interaction of gemcitabine and sphingomyelin can induce ceramide-mediated apoptosis through Fas-induced cytotoxicity.316
Gemcitabine has self-potentiation mechanisms.317,318 Gemcitabine-5′-diphosphate is a major active metabolite that inhibits ribonucleotide reductase, leading to one self-potentiation mechanism. Blocking ribonucleotide reductase activity decreases the natural deoxyribonucleotide pools, promoting more gemcitabine-5′-triphosphate to be incorporated into genomic DNA by DNA polymerase.31,319,320 Furthermore, inhibition of ribonucleotide reductase leads to an indirect self-potentiation mechanism involving the regulation of 2′-deoxycytidine kinase. The enzyme is inhibited by a high dCTP concentration by a negative feedback loop regulation, and gemcitabine-induced ribonucleotide reductase inhibition decreases the dCTP and dATP concentrations.321 This in turn leads to an increase in the activity of 2′-deoxycytidine kinase and more gemcitabine-5′-monophosphe accumulation in the cell. Gemcitabine-5′-triphosphate induces another direct potentiation mechanism involving the inhibition of deoxycytidylate, an enzyme that deaminates deoxycytidine-5′-monophosphate as well as gemcitabine-5′-monophosphate, preventing deamination of gemcitabine-5′-monophosphate into 2′,2′-difluoro-2′-deoxyuridine-5′-monophosphate, an inactive metabolite.322
There are several mechanisms of resistance for gemcitabine reported from tissue culture studies using cell lines, and some (but not all) of the proteins and pathways being discovered include the following. The deaminated byproduct of gemcitabine, 2′,2′-difluoro-5′-deoxyuridine, is a competitive inhibitor for its uptake by the hENT transporter,323 and mutations in nucleotide transporters and 2′-deoxycytidine kinase prevent the uptake and initial phosphorylation of gemcitabine in the cell.60,324 Intracellularly, gemcitabine-5′-monophosphate can be deaminated to form 2′,2′-difluoro-2′-deoxycytidine-5′-monophosphate, and high gene expression of NT5C3 has been correlated with lower levels of phosphorylated gemcitabine metabolites.325 Furthermore, upregulation of ABCC5 and hENT1 mRNA or protein was detected in gemcitabine resistance cell lines but has not been directly linked to promoting drug resistance in patients.326 Additional studies have shown that histone methyltransferase G9a,327 p38MAPK,328 and Nutlin-3329 might be correlated with gemcitabine resistance. Furthermore, gemcitabine resistance can arise from changes in high expression of drug efflux pumps and in nucleotide metabolism enzymes, inactivation of the apoptosis pathway, changes in miRNA expression, and modulation of Hedgehog, Wnt, and Notch pathways.330
In patients, gemcitabine deamination appears to be a major mechanism limiting drug efficacy. Gemcitabine can be deaminated at the N4 position of the cytidine base in the serum to the inactive 2′,2′-difluoro-5′-deoxyuridine metabolite by cytidine deaminase,331 or gemcitabine-5′-monophoshate can be deaminated by deoxycytdine deaminase in the cell. This metabolism is responsible for the very short half-life of the drug, thus limiting availability at the tumor.332 Changes in ribonucleotide reductase subunit protein levels or polymorphisms, which regulate dNTP cellular concentrations, have been demonstrated.333–335 Furthermore, collective cell line data suggest that modulation of mRNA expression, cell survival pathways, and tumor suppressor genes all might contribute to gemcitabine resistance. These mechanisms for gemcitabine resistance will need to be validated in the future using ex vivo models to determine their importance in patient populations. Since gemcitabine resistance is polygenic, combination treatments with alkylating agents, topoisomerase inhibitors, mitotic inhibitors, and biologicals (such as anti-PD-1 and anti-CTLA-4) appear to be a logical choice for new investigatory treatments.
Gemcitabine hydrochloride 9 is currently being used to treat non-small cell lung, pancreatic, breast, bladder, and ovarian cancers.336–338 The agent is also being used to treat pancreatic cancer; however, treatment with gemcitabine hydrochloride 9 monotherapy showed only a modest benefit.339,340 Currently, various clinical trials studying using gemcitabine hydrochloride 9 in combination therapy are underway. Some of these trials include: gemcitabine-cisplatin (DNA alkylating agent) combination therapy for advanced non-small-cell lung cancer,341 Hodgkin’s lymphoma, and non-Hodgkin’s lymphoma,342 gemcitabine-erlotinib (tyrosine kinase inhibitor) regimen to treat locally advanced and metastatic pancreatic cancers,343 and gemcitabine-oxaliplatin (DNA alkylating agent) treatment for biliary adenocarcinoma,344 pancreatic cancer,345 hepatocellular cancer,346 testicular cancer,347 and epithelial ovarian cancer.348 Additional combination treatments of gemcitabine with necitumumab (a monoclonal antibody which binds the epidermal growth factor receptor) and radiation treatments are under investigation (ClinicalTrials.gov identifiers: NCT01606748–active status–not recruiting patients and NCT02254681–recruiting patients).
5.3.1.2. LY2334737 Biology.
As an N4-valproic acid prodrug, LY2334737 52 was designed to improve the oral bioavailability of gemcitabine hydrochloride 9, primarily by blocking the site of deamination and decreasing first-pass metabolism.349,350 The prodrug passes through the intestinal epithelium and enters the systemic circulation largely intact,350 thereby reducing gut exposure to gemcitabine. LY2334737 52 is hydrolyzed by slow systemic cleavage via the actions of carboxyesterase 2, liberating gemcitabine and valproic acid, and leads to prolonged gemcitabine exposure in vitro.351 Preclinical studies showed that low-dosing of LY2334737 52 was efficient for human colon and lung tumor xenograft models.352 Moreover, elevated carboxyesterase 2 activity and ENT1 expression may enhance LY2334737 tumor response.
In a clinical study to determine the maximum tolerated dose and dose-limiting toxicities of LY2334737 52, stable disease was achieved in 22 of the 65 patients with advanced solid tumors.353 In 2012, a Phase I clinical trial was conducted in persons with solid and metastatic tumors (ClinicalTrials.gov identifier: NCT01648764–completed in 2014–no study results were posted). However, in 2013 Eli Lilly discontinued the development of LY2334737 52,354 due to results from a Japanese study that showed hepatic toxicities in patients.355
5.3.1.3. CP-4126 Biology.
CP-4126 (49, CO-1.01) is an elaidic acid ester derivative of gemcitabine developed by Clavis Pharma. The lipophilic side chain was installed to increase the ability of gemcitabine to diffuse through the plasma membrane. This modification makes CP-4126 49 a transporter-independent analog as compared to gemcitabine, which enters the cell primarily by hENT1.356 Once in the cell, cleavage to gemcitabine is believed to occur via the action of carboxyesterases, and subsequent phosphorylation to gemcibine-5′-monophosphate is performed by deoxycytidne kinase.357 Furthermore, resistance studies showed that CP-4126 49 and gemcitabine can arise by down regulation of 2′-deoxycytidine kinase in vitro.324
CP-4126 49 has been assessed in several Phase I/II clinical trials for safety and efficacy as monotherapy or combination therapy,357 involving non-small-cell lung cancer, metastatic pancreatic adenocarcinoma, and other advance solid tumors. The results demonstrated no marked difference in overall survivals of individuals treated with CP-4126 49 versus gemcitabine hydrochloride 9 having pancreatic ductal adenocarcinoma and a low hENT1 protein expression level.358 Two Phase I clinical trials have been completed (ClinicalTrials.gov identifiers: NCT01392976–completed in 2013–results were published). For one clinical trial, CP-4126 49 was poorly absorbed and was subject to metabolism before systemic exposure. Overall, this led to further testing of CP-4126 49 in patients by intravenous administration.359 Another Phase I clinical trial had CP-4126 49 given intravenously and reported positive results (ClinicalTrials.gov identifiers: NCT00778128–completed in 2010–results were published).360 Another Phase I trial testing CP-4126 49 and cisplatin was stopped (ClinicalTrials.gov identifiers: NCT01641575–terminated in 2013–no results posted). As a result of these studies, Clavis suspended development of CP-4126 49.
We have not covered all the potential prodrug combinations or technologies under development for gemcitabine. Other unique modifications of the agent include mesoporous silica nanoparticles,361 amino acid monoester prodrugs,362 Hoechst conjugates,363 and further base modifications.364
5.3.1.4. NUC-1031 Biology.
The ProTide concept has been widely used in the development of antiviral and anticancer nucleoside-5′-monophosphate prodrugs.104,365 Protides were designed as protected nucleotide-5′-monophosphate isomers to increase cellular penetration as well as bypass the often rate-limiting monophosphorylation of the nucleoside analog.104 Furthermore, they are also believed to improve the delivery of the parent compound to the liver.366 NUC-1031 (Acelarin, 32) is a 5′-phosphoramidate prodrug of gemcitabine.367 This agent is able to overcome gemcitabine resistance mechanisms: decreased uptake by cellular transporters, decreased activity of 2′-deoxycytidine kinase (by mutation), and cytosine deamination.368 However, it would still be sensitive to gemcitabine-5′-monophosphate deamination by deoxycytidine deaminase. Like gemcitabine, NUC-1031 32, is administered by intravenous injection.
A Phase I study in people with advanced solid tumors has been launched in 2012.368,369 The agent could be given at four times the maximum dose of gemcitabine, and over half the patients achieved stable disease, with a few patients having a reduction in tumor size. In addition, administration of NUC-1031 32 produced a 13-fold increase in intracellular gemcitabine concentration as compared to gemcitabine intravenous injection. An additional study funded by NuCana BioMed Limited examining combination treatment of NUC-1031 32 with carboplatin (DNA alkylating agent) for ovarian cancer is planned (ClinicalTrials.gov identifiers: NCT01621854–completed June 2015–no results posted, and NCT02303912–recruiting patients).
5.3.2. Gemcitabine and Gemcitabine Prodrugs Syntheses.
5.3.2.1. Gemcitabine Syntheses.
Hertel and co-workers reported the discovery synthesis of gemcitabine hydrochloride 9 (Scheme 26).370 It commenced with a Reformatsky reaction involving ethyl bromodifluoroacetate and aldehyde 155, which is easily prepared by isopropylidene protection and subsequent oxidative cleavage of d-mannitol, forming 156 as a mixture of diastereomers (3:1 anti/syn) which were separated by HPLC. Acid-mediated deprotection and cyclization were followed by 3′,5′-O-silylation, affording the protected lactone 157. Reduction, mesylation, and glycosylation under Vorbrüggen conditions produced protected sugar 158 as a 4:1 mixture of α/β anomers. Deprotection allowed for reverse phase HPLC separation of the anomers. The unfortunate α/β anomeric ratio produced under these conditions gave gemcitabine hydrochloride 9 in only 10% yield from intermediate 158.
Scheme 26. Synthesis of Gemcitabine Hydrochloride 9a.
aReagents and conditions: (a) BrCF2CO2Et, Zn, THF, Et2O, 87% (3:1 anti/syn); (b) Dowex 50, MeOH, H2O; (c) TBDMSOTf, lutidine, DCM, 86% (over two steps); (d) DIBAL-H; (e) MsCl, Et3N, DCM, 71% (over two steps); (f) 2,4-bis(trimethylsilyl)cytosine, TMSOTf, DCE, reflux; (g) AG 50W-X8 resin, MeOH; (h) reverse phase HPLC, 10% (over two steps).
In 2010, Chang et al. reported a large-scale synthesis of gemcitabine 163 using 2,2-fluoro-α-ribofuranosyl bromide 162 as a key intermediate to increase the β-anomeric selectivity in the glycosylation reaction (Scheme 27).371 The stereoisomers of 156 were protected with p-phenylbenzyl chloride and subjected to ester hydrolysis under basic conditions. Fortunately, the reaction volume reduction led to selective precipitation of the desired potassium erythro-pentonate 159, and filtration furnished the pure diastereomer, representing a marked increase in efficiency when compared to the laborious diastereomeric separations of other syntheses (e.g., HPLC purification in the Hertel method). Deprotection, cyclization, and 5′-O-benzoylation of intermediate 159 were followed by lactone reduction and phosphorylation of the crude anomeric mixture, directly furnishing intermediate 161 (1:10.8 α/β ratio). Subsequent bromination led to a 10.8:1 α/β mixture, and recrystallization from isopropanol afforded the pure α-anomer 162, with the phenylbenzoyl protecting group being necessary for recrystallization.
Scheme 27. Synthesis of Gemcitabine 163a.

aReagents and conditions: (a) PhBzCl, Et3N, DCM; (b) K2CO3, H2O, THF, MeOH, 67%; (c) HCl, CH3CN, reflux; (d) BzCl/Pyr, EtOAc, rt, 72%; (e) LiAl(O-tBu)3H, THF; (f) ClP(O)(OPh)2, Et3N, PhMe, rt, 77%; (g) HBr/HOAc, rt, 82%; (h) 2,4-bis(trimethylsilyl)cytosine, octane/heptane, Ph2O, 140–150 °C; (i) NH3/MeOH, H2O, rt, 65%.
In order to maximize the SN2-displacement of α-bromide 162, the glycosylation conditions were optimized. Therefore, non-polar solvents were used, and in situ generated TMSBr was removed via continuous distillation, in an attempt to decrease anomerization of 162 or formation of the oxocarbenium ion. Indeed, this led to an improved β/α anomeric mixture of 5.5:1. Deprotection with methanolic ammonia, followed by isolation of the pure β-diastereomer by simple extraction, evaporation, and crystallization from water, produced the hemi- or dihydrate of gemcitabine, depending on workup conditions.
Jiang et al. also recently reported a process preparation for gemcitabine hydrochloride 9 on a 100-g-scale (Scheme 28).372 A diastereomeric mixture of difluoro lactone 164 was acylated with cinnamoyl chloride, and when the solution was cooled, stereospecific crystallization afforded the desired lactone 165. Reduction and tosylation gave a 1:1 mixture of anomeric tosylates 166, which was found to crystallize in ethyl acetate/petroleum ether. Subsequent glycosylation of the anomeric mixture 166 with N-acetyl cytosine in the presence of TMSOTf, followed by direct treatment of the reaction mixture with sodium carbonate, allowed for the precipitation of the undesired α-nucleoside analogue. Deprotection of β-isomer 167 with methanolic ammonia at room temperature gave crude gemcitabine 163, which was acidified with HCl and crystallized in acetone/H2O, affording the target salt in 99.9% purity.
Scheme 28. Synthesis of Gemcitabine Hydrochloride 9 and LY2334737 52a.

aReagents and conditions: (a) Dowex 50W-X12 resin, MeOH, H2O, rt, 4 days, 94%; (b) (E)-cinnamoyl chloride, pyr, EtOAc, 30 °C, 3 h, 43%; (c) LiAl(O-tBu)3H, THF, −10 °C, 2 h, then TsCl, Et3N, PhMe, −10 °C, 5 h, 62%; (d) N-acetyl cytosine, (TMS)2NH, TMSOTf, DCE, reflux, 12 h, then 5% NaHCO3, 47%; (e) NH3/MeOH, rt, overnight, then HCl, Me2CO, rt, 12 h, 80%; (f) 2-propylpentanoic acid, EDC, HOBt, NMM, DMF, DMSO, 55 °C, 17 h, 95%.
5.3.2.2. LY2334737, CP-4126, and NUC-1301 Syntheses.
The gemcitabine prodrug LY2334737 52 was formed by directly coupling valproic acid with gemcitabine hydrochloride 9, using peptide coupling conditions (EDC/HOBt/NMM).350 Prodrug LY2334737 52 is not crystalline; therefore, it was purified as a p-toluenesulfonic acid (pTSA) salt co-crystal (Scheme 28).
CP-4126 49 was formed by direct esterification of the 5′-OH of gemcitabine 163 using elaidic acid chloride as reagent (Scheme 29).373 NUC-1301 32 was prepared by reaction of phosphochloridate 169, which was produced by treating l-alanine benzyl ester hydrochloric salt 168 with phenyl-dichlorophosphate 92, with gemcitabine 163 in THF/pyridine in the presence of N-methyl imidazole.374
Scheme 29. Synthesis of CP-4126 49 and NUC-1031 32a.
aReagents and conditions: (a) Et3N, DCM, −80 °C, 1 h, 98%, (b) NMI, pyr, THF, −80 °C to rt, 2 h, 16%; (c) HCl (g), DMF, elaidic acid chloride, rt, 12 h, 30%.
An improved synthesis for NUC-1031 32 has recently been reported, which affords the agent on a 20-gram-scale.375 Boc protection of the 3′-OH and N4-amino group of gemcitabine 163 was followed by reaction with phenyl-dichlorophosphate 92 and L-alanine benzyl ester hydrochloride 168, which installed the protide. Subsequent Boc deprotection completed the synthesis.
5.3.3. Sapacitabine Biology.
In the early 1990s, Matsuda et al. published the synthesis and anticancer activity of 2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine (CNDAC) along with the rationale for the structure of the agent.376 Since a cyanoethyl group beta to a phosphate diester of a nucleoside was reported to undergo β-elimination under alkaline conditions,377 it was hypothesized that a 2′-β-C-cyano nucleoside when incorporated into DNA could lead to β-elimination of the 3′-phosphodiester moiety and produce DNA strand breaks (Figure 14). This mechanism of anticancer activity would be similar to that of radiation therapy, which was shown to produce similar DNA strand breaks that were believed to result in tumor cell death.378,379
Figure 14.
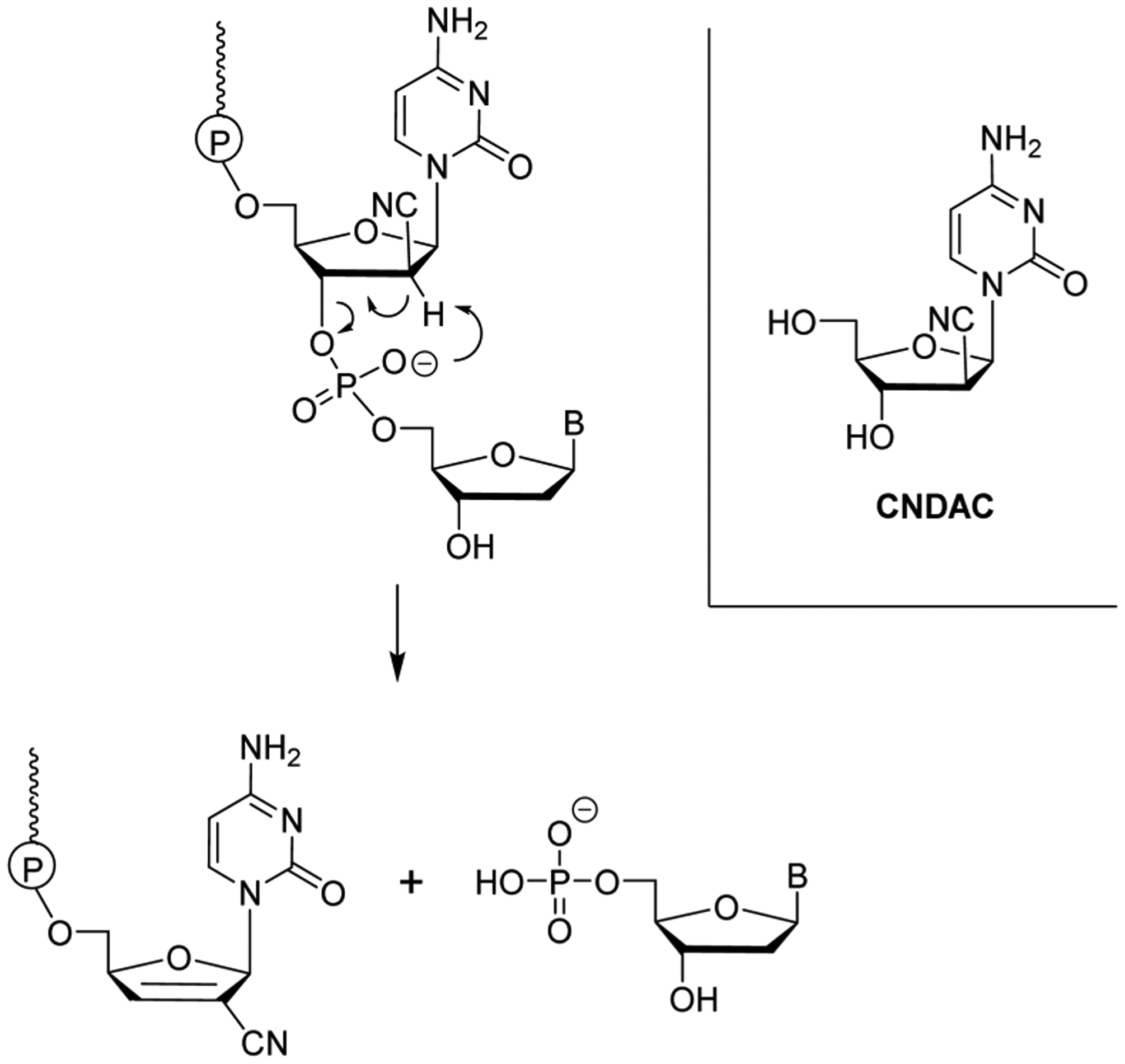
CNDAC structure and hypothesized mechanism of DNA strand break with incorporated CNDAC.
Sapacitabine 25 (Cyclacel Pharmaceuticals) is an N4-palmitoyl derivatized analog of CNDAC and is orally bioavailable. It is primarily metabolized in the plasma, gut, and/or liver to CNDAC, and eventually transported into cells.380 CNDAC is phosphorylated by 2′-deoxycytidine kinase and further phosphorylated by other cellular kinases to generate the triphosphate form.381 DNA polymerases incorporate the CNDAC-5′-triphosphate into the growing DNA strands, and further elongation of the chain is sluggish due to the steric effects of CNDAC analog.26 Upon incorporation of CNDAC-5′-monophosphate into DNA, β-elimination of the 3′-phosphodiester moiety can result in a severed DNA strand that is terminated with 2′-C-cyano-2′,3′-didehydro-2′,3′-dideoxycytidine-5′-monophosphate (Figure 14).382 This represents a unique anticancer mechanism of a nucleoside derivative, and results in cell cycle arrest at the G2 phase, which is different from the majority of other anticancer nucleoside analogs that cause arrest in the S phase.383
Sapacitabine 25 is a weak substrate for cytidine deaminase, whereas CNDAC is a substrate and is converted into the inactive uracil derivative: 2′-C-cyano-2′-deoxy-1-β-d-arabino-(pentofuranosyl)uridine. In addition, unlike other nucleoside analogs in their corresponding di- or triphosphates forms, CNDAC-5′-diphosphate does not effectively inhibit ribonucleotide reductase, and CNDAC-5′-triphosphate does not act as a feedback inhibitor of 2′-deoxycytidine kinase.26
Cells deficient in nucleotide excision repair (NER) of endonuclease XPF are 4- to 5-fold more sensitive to CNDAC exposure,384 suggesting that the NER pathway is used to excise the agent from the DNA strand. If DNA single-strand breaks caused by CNDAC exposure are not repaired, they become more lethal DNA double-stranded breaks during the following round of DNA replication.385 These CNDAC-induced DNA double-stranded breaks can occur when the replication fork encounters a DNA single-stranded break or after collapse of a stalled replication fork,386 which are repaired primarily by homologous recombination. Cells with defects or mutations in the homologous recombination pathway (ATM, RAD51, Brac2, Xrcc3) are up to 100-fold more sensitive to CNDAC exposure.387 In addition to DNA incorporation, CNDAC exposure was also shown to inhibit RNA synthesis to a limited extent in vitro.26
Sapacitabine 25 is in several clinical trials (Phase I–III) either as a monotherapy or in combination with other drugs for treatment of acute myelogenous leukemia, chronic lymphocytic leukemia, non-small cell lung cancer, and some solid tumors.349,387,388 However, it failed to achieve end point efficacy in a Phase III study using oral sapacitabine 25 in elderly patients with newly diagnosed acute myelogenous leukemia (ClinicalTrials.gov identifier: NCT01303796–unknown, >2 years since reported). In 2013, Cyclacel Pharmaceuticals announced that sapacitabine 25 had activity against 75% of primary ovarian cancer samples isolated from individuals. DFP-10917 (26, intravenously administered hydrochloride salt form of CNDAC) is in Phase I/II studies to treat relapsed or refractory acute leukemia (ClinicalTrials.gov identifier: NCT01702155–recruiting patients).
5.3.4. Sapacitabine Synthesis.
The discovery synthesis of CNDAC is shown in Scheme 30.376,389 N4-Acylation of cytidine 96 followed by 3′,5′-TIPS protection and 2′-OH oxidation provided ketosugar 170. Treatment of 170 with sodium cyanide followed by reaction with phenyl chlorothionocarbonate produced an epimeric mixture of 2′-cyanohydrins 171. Subsequent deoxygenation under standard Barton conditions produced 172 diastereoselectively, presumably due to steric hindrance on the β-face. Silyl deprotection followed by N4-acetyl removal with HCl in MeOH afforded DFP-10917 (26, the hydrochloride salt of CNDAC). Interestingly, attempted deacetylation of 172 with NH3/MeOH produced mainly cytosine, adding further evidence for the increased acidity of the 2′-α of 26.
Scheme 30. Discovery Synthesis of DFP-10917 26 and Process Synthesis of Sapacitabine 25a.

aReagents and conditions: (a) 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane, pyr; (b) Ac2O, EtOH; (c) CrO3/pyr/Ac2O, DCM; (d) NaCN, NaHCO3, Et2O, H2O; (e) PhOC(S)Cl, DMAP, CH3CN; (f) Bu3SnH, AIBN, PhMe (57% over 3 steps); (g) Bu4NF, CH3CO2H, THF then HCl/MeOH, 49% (over two steps); (h) Et3N, MeOH, DCM, 30 °C, 10 min, 97%; (i) palmitic anhydride, dioxane, H2O, 85 °C, 89%.
Soon after the discovery synthesis of CNDAC hydrochloride (DFP-10917, 26), a process synthesis for sapacitabine 25 was reported.390 The N4-palmitoyl group was installed in the first step, and the rest of the synthesis followed a route similar to that of Scheme 30, with a few variations in reagent choice (i.e., pyridinium dichromate for the 2′-oxidation) producing sapacitabine 25 in nine steps in 20% overall yield.
More recently, Westwood et al. reported a kilogram preparation of sapacitabine 25.391,392 The synthesis followed a similar route as the two above-mentioned preparations with some variation in reagent use and synthetic order. Deoxygenation was performed using the economical lauroyl peroxide with collidine on a substrate similar to intermediate 171, except with a less-bulky ethyl xanthate ester. Additionally, 3′,5′-OH silyl protection came before N4-acylation, generating a solid intermediate that was easy to purify and use in subsequent steps. The final step of the preparation involved N4-acylation with palmitic anhydride, producing sapacitabine 25 on a kilogram-scale in nine steps and 37% overall yield.
5.3.5. TAS-106 Biology.
Many antineoplastic agents exert their cytotoxic effects during the S phase (DNA synthesis) of the cell cycle. This poses a problem for more slow growing solid tumors, which have decreased DNA replication rates as compared to leukemias, making the incorporation of 2′-deoxynucleoside-5′-triphosphate analogs very inefficient. However, RNA synthesis occurs throughout the cell cycle except in the M phase. Therefore, a drug that targets both DNA and RNA was hypothesized to offer a broader interval in the cell cycle to exert anticancer effects.26
Drawing on the observation that CNDAC-5′-triphosphate (see Section 5.3.3) was shown to have some RNA synthesis inhibition, TAS-106 46 (ECyd; [1-(3-C-ethynyl-β-d-ribopentofuranosyl)cytosine]) was developed by Taiho Pharmaceuticals in hopes that the TAS-106–5′-triphosphate analog would inhibit RNA polymerase.382,393 Indeed, the main mechanism of action of TAS-106 46 was shown to be inhibition of RNA polymerases I, II, and III, which essentially blocks RNA synthesis to cause apoptosis.394 In addition, TAS-106 46 was shown to inhibit DNA repair proteins (BRAC2 and Rad51) in the homologous repair pathway, suggesting it might also act via some of the mechanisms of sapacitabine 25.395
TAS-106 46 is phosphorylated to the monophosphate form by uridine/cytidine kinase, which is preferentially expressed in tumor cells versus normal cells.396 It is subsequently converted to the TAS-106–5′-diphosphate and TAS-106–5′-triphosphate forms by uridine/cytidine monophosphate kinase and nucleoside diphosphate kinase, respectively.397 TAS-106–5′-triphosphate remains in cells for a prolonged period of time even after short-term exposure to TAS-106 46, producing significant exposure of cells to the active form of the drug in vitro.398 TAS-106 46 is a poor substrate for cytidine deaminase, although the deaminated uridine analog of the agent also exhibited potent cytotoxicity in vitro.399,400 TAS-106–5′-diphosphate was anticipated to also block activity of the enzyme due to the precedent that other 2′-deoxyribonucleoside anticancer agents are ribonucleotide reductase inhibitors. However, contrary to the original hypothesis, TAS-106–5′-diphosphate was not found to inhibit ribonucleotide reductase. Additional investigational studies have examined duplex drug linking of 5FdU(5′−5′)TAS-106 against gastric adenocarcinoma cell lines401 and 5-FdU(3′−5′)TAS-106 exposure to hepatoblastoma cells lines.402 Both studies showed positive cytotoxic results in vitro, but further studies are needed to examine the full potential of using these duplex analogs.
TAS-106 46 therapy has produced modest results in clinical trials. In a Phase II clinical trial for salvage metastatic or recurrent head and neck squamous cell cancers and nasopharyngeal cancer, TAS-106 46 treatment was reasonably well tolerated for platinum-failure patients, but had myelosuppression as its main toxicity.403 However, the agent showed no anticancer efficacy (ClinicalTrials.gov identifier: NCT00737360–terminated in 2012–limited number of patients). TAS-106 46 treatment in combination with carboplatin, a DNA alkylating agent, recently underwent a Phase I dose-escalating study in subjects with solid tumors.404 The combination was well tolerated, and a few individuals experienced stabilized disease, in which the cancer did not progress (>4 months). There are no current clinical trials underway for TAS-106 46.
5.3.6. TAS-106 Synthesis.
The discovery synthesis of TAS-106 46 is shown in Scheme 31.399 The chosen starting material was 1,2-O-isopropylidene-α-d-xylofuranose 173, which was 5′-OH protected and then 3′-oxidized, forming intermediate 174.405 Stereoselective addition of lithiated (trimethyl)acetylene to 174 gave the tertiary alcohol 175 as the desired β-adduct. Subsequent desilylatation, selective 5′-OH benzoylation, isopropylidene deprotection, and 2′-OH benzoylation generated 178. Glycosylation of 178 with cytosine under Vorbrüggen conditions followed by deprotection furnished the desired TAS-106 46 in good overall yield.
Scheme 31. Synthesis of TAS-106 46a.
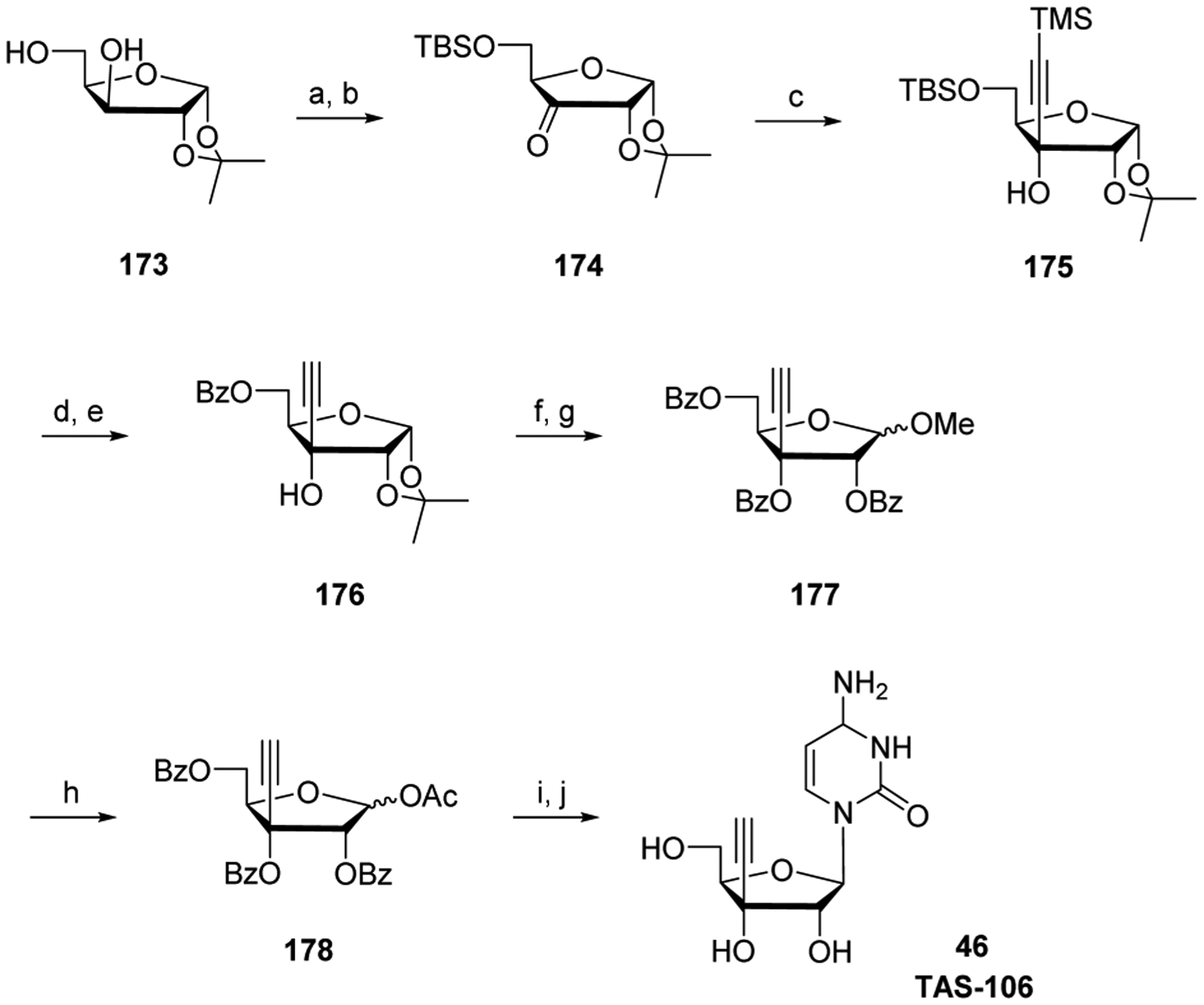
aReagents and conditions: (a) TBDMSCl, pyr, rt, 1.5 h, 100%; (b) CrO3, Ac2O, pyr, rt, 1.5 h, 77%; (c) trimethylsilylacetylene, n-BuLi, THF, −78 °C, 99%; (d) n-Bu4NF, THF, rt; (e) BzCl, pyr, rt, 93% (over two steps); (f) 20% HCl, MeOH, rt; (g) BzCl, DMAP, pyr, 100 °C, 87% (over two steps); (h) concd H2SO4, AcOH, Ac2O, rt, 93%; (i) 2,4-bis(trimethylsilyl)cytosine, SnCl4, CH3CN, 0 °C to rt, 18 h, 81%; (j) NH3/MeOH, rt, 2 days, 90%.
In preparation for clinical trials, a large-scale synthesis of TAS-106 46 was developed (Scheme 32).406 Most of the intermediates were crystalline and required no chromatography. Selective 5′-OH protection of 1,2-O-isopropylidine-D-xylofuranose 173 with p-chlorobenzoyl chloride was followed by TEMPO-mediated oxidation of the 3′-OH group. Stereoselective introduction of a TMS-ethynyl group followed by protecting group manipulation set the stage for the key glycosylation reaction. Multiple leaving groups at the anomeric position were screened, and the isobutyryloxy leaving group proved to be most effective, affording the protected intermediate 184 in good yield and stereoselectivity. Final deprotection and crystallization from aqueous methanol furnished 128 g of crystallized TAS-106 46.
Scheme 32. Synthesis of TAS-106 46a.

aReagents and conditions: (a) 4-ClBzCl, Et3N, DCM, 0 °C, 2 h, 74%; (b) TEMPO, DCM NaClO4, NaHCO3, H2O, 0 °C, 30 min, then 2-propanol, rt, 10 min, 88%; (c) trimethylsilylacetylene, EtMgBr, THF, 4 °C, 30 min, 25%; (d) HCO2H, H2O, reflux, 45 min, 97%; (e) i-BuCl, Et3N, DMAP, rt, 5 h, 87%; (f) 2,4-bis(trimethylsilyl)cytosine, SnCl4, MeCN, 30 °C, 3 h, 81%; (g) DBU, MeOH, 30 °C, 3 h, 68%.
5.3.7. Tezacitabine Biology.
Tezacitabine 45 [MDL 101,731; (E)-2′-deoxy-2′-(fluoromethylene)cytidine] was designed by Hoechst Marion Roussel Inc. as a mechanism-based irreversible ribonucleotide reductase inhibitor.407 Such inhibitors are modified at the 2′-position, and often form covalent complexes with the enzyme. Tezacitabine 45 employs a 2′-fluorovinyl moiety, which was proposed to aid in the ribonucleotide reductase-catalyzed abstraction of the 3′-hydrogen atom from tezacitabine-5′-diphosphate, by resonance stabilization of the resultant 3′-radical (Figure 15).408 The design was successful, as tezacitabine-5′-diphosphate was indeed found to be an irreversible inhibitor of ribonucleotide reductase.409 Furthermore, tezacitabine-5′-triphosphate was found to be a DNA polymerase chain terminator.410
Figure 15.

Proposed mechanism of catalytic reduction of nucleoside-5′-diphosphates by ribonucleotide reductase (upper), and proposed mechanism of ribonucleotide reductase inhibition by tezacitabine-5′-diphosphate (lower).
Although a cytidine nucleoside analog, tezacitabine 45, is relatively resistant to cytidine deaminase activity,411 upon entering the cell, the agent is phosphorylated by 2′-deoxycytidine kinase and then by other endogenous kinases to the active metabolites: tezacitabine-5′-diphosphate and tezacitabine-5′-triphosphate. Tezacitabine-5′-triphosphate is a substrate for DNA polymerase α, being incorporated into the growing DNA strand in the place of cytidine, and it prevents further chain elongation by DNA polymerases.412
In tissue culture tumor cell lines and mouse tumor models, tezacitabine 45 has been examined as a candidate for treatment of colorectal and hematological solid cancers, wherein hydroxyurea and cytarabine 4 have limited antitumor activities.413,414 Furthermore, the radiosensitizing effects of tezacitabine 45 in the presence of zidovudine (a nucleoside analog inhibitor of reverse-transcriptase) were tested in the colon cancer cell line WiDr.415 In addition, tezacitabine 45 treatment was also shown to have antiangiogenic activity in xenograft models.416
A phase I clinical study showed that tezacitabine 45 treatment caused myelotoxicity in 53% of the patient and 83% had febrile episodes.412 An additional Phase I study showed that tezacitabine 45 plus 5-FU 2 treatment had activity in patients with advanced esophageal and other gastrointestinal carcinomas.417 However, tezacitabine 45 treatment failed to have significant activity as a stand-alone therapy.417 In 2004 Chiron reported that it had discontinued development of tezacitabine 45 after it failed to meet expectations in a Phase II trial (ClinicalTrials.gov identifier: NCT00051688–terminated in 2004).
5.3.8. Tezacitabine Synthesis.
McCarthy et al. reported a synthesis of tezacitabine 45,418 which proceeded in seven steps from uridine in 10% overall yield, requiring five column chromatography purifications. Two years later, an improved process was published that used cytidine as starting material, allowing for the preparation of multigram quantities of the compound in five steps with a 29% overall yield (Scheme 33).419 A one-pot protection of the 3′-OH and 5′-OH of cytidine 96 by treatment with TIPSCl and of the N4-amino group by the addition of N,N-dimethylformamide dimethyl acetal gave intermediate 185. This derivative underwent swern oxidation to provide the keto intermediate 186, which was worked up by a nonaqueous silica gel plug filtration in order to avoid ketone hydration. A Horner–Wadsworth–Emmons reaction between 186 and the in situ-generated diethyl-1-fluoro-1-(phenylsulfonyl)-methylphosphonate produced a separable mixture of fluorovinyl sulfones (Z/E ratio 10:1, respectively). Deprotection of the N4-amino group with methanolic ammonia yielded the corresponding α-fluorovinyl sulfone 187, which readily crystallized from hexanes. Radical-mediated sulfone/stannane interchange provided 188 with retention of configuration, and treatment of the intermediate with cesium fluoride in refluxing methanol allowed for the simultaneously removal of the TIPS and tributyltin groups.
Scheme 33. Synthesis of Tezacitabine 45a.

aReagents and conditions: (a) TIPSiCl, pyr, 34 °C, 18 h; (b) (CH3)2NCH(OCH3)2, 37 °C, 4 h, 85% (over two steps); (c) (COCl)2, DMSO, DCM, −75 °C, 0.5 h, then Et3N, −75 °C to rt, 1 h, 90%; (d) PhSO2CH2F, ClP(O)(OEt)2, 1 M LiHMDS, −60 °C to rt, 3 h; (e) NH3/MeOH, rt, overnight, 66% (over two steps); (f) Bu3SnH, AIBN, cyclohexane, reflux, 18 h, 83%; (g) CsF, MeOH, reflux, 24 h, 69%.
5.3.9. Troxacitabine Biology.
Troxacitabine 48, β-l-dioxolane-cytidine (Troxaty, SGX Pharmaceuticals), was originally studied as an anti-HIV agent, and later it was shown to have potential as an anticancer agent.420 Moreover, this agent is the first L-nucleoside studied for cancer.421
Troxacitabine 48 enters cancer cells by passive diffusion, possibly due to the lack of 3′-OH making it not a good substrate for facilitated transporters.422 The agent is not a substrate for cytidine deaminase.423 However, it is recognized by 2′-deoxycytidine kinase, suggesting more chiral specificity from the former enzyme and less from the latter.424
Troxacitabine-5′-monophosphate is further phosphorylated to the di- and triphosphate forms by cellular enzymes. In contrast to a majority of other anticancer nucleoside analogs, troxacitabine-5′-diphosphate is the predominant intracellular metabolite, and a large pool accumulates intracellularly.311,425 Phosphoglycerate kinase converts troxacitabine-5′-diphosphate to troxacitabine-5′-triphosphate, the major intracellular active metabolite of troxacitabine 48.426
Troxacitabine-5′-triphosphate is incorporated into newly synthesized DNA, being a substrate for DNA polymerases α, β, δ, γ, and ε, leading to polymerase inhibition by an absolute DNA chain termination mechanism.427,428 The incorporated troxacitabine-5′-monophosphate is not readily excised from the DNA chain by proof reading 3′ → 5′ exonuclease activities associated with DNA polymerases, due to the chiral specificity of the enzymes. However, removal of the incorporated troxacitabine-5′-monophosphate from DNA can occur by the APE-1 excision repair mechanism.420 Resistance to troxacitabine 48 exposure can arise by decreased expression of 2′-deoxycytidine kinase in vitro.429
Troxacitabine 48 initially showed promising anticancer activity against solid tumors and leukemias.424,427,430–432 Tumor cells with catalytically inactive p53 have increased sensitivity to troxacitabine 48 exposure.429 In addition, pancreatic cell lines were shown to have increased sensitivity to troxacitabine prodrugs bearing N4 single carbon chain amides to increase drug lipophilicity.433 However, no additional information is available addressing preclinical efficacy study models for this compound.
Unfortunately, despite the promising preclinical results, troxacitabine 48 therapy had limited success in clinical trials for leukemias and neoplasms (administered by intravenous infusion)434,435 and ended in the termination of multiple trials (ClinicalTrials.gov identifiers: NCT00129948–terminated in 2006, NCT00104468–terminated in 2007, and NCT00104286–terminated in 2006). In 2007, results were reported for a Phase II study evaluating troxacitabine 48 therapy in relapsed or refractory lymphoproliferative neoplasms or multiple myeloma.436 All the patients had one adverse event, with 62% with one serious adverse event. Overall, troxacitabine 48 treatment had limited benefit in patients with these advanced disorders.436 Furthermore, the agent also exhibited significant toxicity issues, indicating that further development of the agent is unlikely.
5.3.10. Troxacitabine Synthesis.
An initial diastereoselective synthesis of troxacitabine 48 used l-ascorbic acid as starting material (Scheme 34).437 Acid 189 was condensed with benzyloxyacetaldehyde dimethyl acetal in the presence of tosylic acid, affording dioxolane 190 as a mixture of diastereomers. Oxidative degradation followed by benzyltriethylammonium chloride-catalyzed Wolfe oxidation produced a mixture of carboxylic acid products 192, which could be separated with flash chromatography. Oxidative decarboxylation followed by glycosylation, separation of the α and β isomers, and deprotection furnished troxacitabine 48.
Scheme 34. Synthesis of Troxacitabine 48a.

aReagents and conditions: (a) PhCH2OCH2CH(OMe)2, TsOH, CH3CN, 95%; (b) H2O2, K2CO3, EtOH; (c) RuCl3 hydrate, NaOCl, DCE/MeCN/H2O, BnEt3NCI; (d) H+, 51% (over three steps); (e) Pb(OAc)4, MeCN/DCM, pyr, 80%; (f) silylated N-acetylcytosine, TMSOTf, DCM, rt; (g) deprotection, 30%.
Kim et al. reported a gram-scale synthesis (Scheme 35)438 starting from 2,3:5,6-di-O-isopropylidene-L-gulofuranose 194, which is made by protecting and reducing L-gulono-6,3-lactone. Acid-mediated rearrangement of 194 to pyranose 195 followed by oxidative cleavage and reduction produced dioxolane 196. Protecting group manipulation, oxidation, and Pb(OAc)4-mediated decarboxylation furnished intermediate 198. Glycosylation of benzoyl-protected cytosine with 198 followed by deprotection with methanolic ammonia afforded troxacitabine 48.
Scheme 35. Synthesis of Troxacitabine 48a.

aReagents and conditions: (a) aq HCl, reflux, 20 h, 60%; (b) NaIO4, MeOH, H2O, then NaBH4; (c) TsOH, Me2CO, 6 h, 62%; (d) BzCl, pyr, DCM, rt, 2 h; (e) TsOH, MeOH, rt, 2 h, 83% (over two steps); (f) NaIO4, RuO2·H2O, CH3CN:CCl4:H2O (1:1:1.5 v/v), rt, 5 h; (g) Pb(OAc)4, pyr, THF, rt, 45 min, 63% (over two steps); (h) N4-Bzcytosine, HMDS, NH4SO4, DCE, reflux, 2.5 h, then TMSOTf, DCE, rt, 1.5 h; (i) NH3/MeOH, 0 °C, rt, 72 h, 38% (over two steps).
5.3.11. Thiarabine Biology.
Thiarabine 47 was first prepared in the 1970s and showed significant anticancer activity. However, difficulty in its synthetic preparation limited further research.439,440 Two decades later, Southern Research Institute drew upon this finding as well as the report that 4-thionucleo-sides showed resistance to purine nucleoside phosphorylase cleavage441 to continue the development of this next-generation anticancer nucleoside analog.442,443
Thiarabine 47 (T-ara C; 4′-thio-arabinofuranosylcytosine; OSI-7836) is phosphorylated to the monophosphate form via 2′-deoxycytidine kinase, although the phosphorylation rate of thiarabine 47 is 1% as compared to cytarabine 4.444 The thiarabine-5′-monophosphate exhibits reduced susceptibility to deaminase activity yet is still a good substrate for uridine/cytidine monophosphate kinase, leading to thiarabine-5′-diphosphate.445,446
Thiarabine-5′-diphosphate is further converted to the triphosphate form (the active metabolite of thiarabine 47). Thiarabine-5′-triphosphate accumulates slowly in the cell but was shown to have long intracellular retention,447 and yet was still efficiently degraded to the nucleoside when using cellular extracts.446
Thiarabine-5′-triphosphate is a substrate for the nuclear DNA polymerase and is incorporated into DNA, inhibiting DNA synthesis and generating DNA lesions,444,448 with the primary mechanism of action being disruption of DNA polymerase activity.449 In addition, HCT 116 colon carcinoma cells exposed to thiarabine 47 led to cleavage of caspase 3 and PARP that promoted cell death via apoptosis.447,450 Thiarabine 47 was also reported to have antiangiogenic properties.442
Preclinical studies for thiarabine 47 were promising, having broad spectrum activities against human solid tumors and leukemia/lymphoma xenografts in mice.17,446 In addition, cross-resistance in multidrug resistant cell lines did not occur for thiarabine 47, suggesting that it potentially could be used in combination drug therapy.451,452 Previously, a Phase I/II clinical trial showed positive results when using clofarabine 12 and cytarabine 4, having a proposed mechanism of increased cytarabine-5′-triphosphate concentration in cells.453 These results led to a combination therapy of clofarabine 12 and thiarabine 47 study, which showed synergistic activity of 12 and 47 leading to delayed tumor growth in mice.17
A few clinical trials have been reported for thiarabine 47. In 2006, two Phase I trials were reported for thiarabine 47.454,455 Study results indicated excessive fatigue reversible grade-3 lymphopenia. Changes in the schedule and duration of thiarabine 47 did not improve its tolerability. Development of the agent has stalled, although some have called for continued clinical evaluation of the agent for hematological and/or solid tumors.449
5.3.12. Thiarabine Synthesis.
The first synthesis of thiarabine 47 was accomplished by Whistler et al. (Scheme 36)439,456–459 through a long and arduous sequence. The protected glucanofuranose 199, made from glucose in two steps (isopropylidene installation followed by benzylation), was transformed into the protected tosyl sugar 200 in six steps after protecting group manipulation and tosylation. Subsequent treatment of 200 with sodium methoxide, sodium benzyloxide, and tosyl chloride furnished 201, which was further converted to intermediate 202 by exposure to potassium thioacetate. Isopropylidene removal followed by oxidative cleavage gave the ring-opened aldehyde 203, and subsequent cyclization, deprotection/protection, and bromination furnished 204 as a mixture of diastereomers. The glycosylation step afforded an anomeric mixture, with the desired β-adduct crystallizing from solution, and deprotection concluded the synthesis of thiarabine 47.
Scheme 36. Synthesis of Thiarabine 47a.

aReagents and conditions: (a) Na, ether, reflux, 16 h, then BnBr, 70 °C, 16 h; (b) Ac2O, H2O, 40 °C, 16 h; (c) AcCl, pyr, 25 °C, 16 h, 78% (over three steps); (d) Na, MeOH, 16 h; (e) BzCl, pyr, −15–25 °C, 20 h; (f) TsCl, pyr, CHCl3, 40 °C, 36 h, 92% (over three steps); (g) NaOMe, CHCl3, MeOH, −15 to 0 °C, 2 h, 87%; (h) NaOBn, BnOH, 40 °C; (i) TsCl; (j) KSC(O)CH3, DMF, 115 °C, 73%; (k) HOAc, H2O, 70 °C, 36 h, 78%; (l) NaIO4, EtOH, H2O, 30 °C, 0.5 h, 97%; (m) aq HCl, MeOH, reflux, 3 h, 41%; (n) Na, NH3, ~100%; (o) BnCl; (p) HBr/HOAc; (q) bis(trimethylsilyl)-N-acetylcytosine, 130 °C, 3 h, 20%; (r) NH3/MeOH, 0 °C, 2 days, 89%.
Secrist et al. developed an improved gram-scale synthesis of thiarabine 47 in order to access sufficient quantities for biological study (Scheme 37).441,443 l-Xylose 205 was converted into the benzyl-protected dithioacetal 208 by selective methoxylation, O-benzylation, and dithioacetalization. Treatment of 208 with a triphenylphosphine-iodine-imidazole reagent resulted in a single inversion cyclization at C-4, affording the desired 4-thiosugar 209 in the d-arabino configuration.441 Coupling of 209 with silylated uracil led to a separable anomeric mixture of 210; as compared to making the cytosine analog, this reaction has several advantages, including a higher β:α ratio, higher yield, and easier purification process. 210β was converted to 212 via C-4 carbonyl activation and amination. Subsequent benzyl deprotection gave thiarabine 47.443
Scheme 37. Synthesis of Thiarabine 47a.

aReagents and conditions: (a) HCl, MeOH, rt, 5 h, 95%; (b) BnBr, NaH, TBAI, THF, rt, 3 days, 87%; (c) BnSH, SnCl4, DCM, rt, overnight, 57%; (d) Ph3P, imidazole, I2, PhMe, CH3CN, 90 °C, 24 h, 83%; (e) uracil, BSA, NBS, MeCN, 50–55 °C, 18 h, column purification (α:β = 1:1.15), 36%; (f) TPSCl, Et3N, DMAP, 5 °C to rt, overnight; (g) NH4OH, MeCN, rt, overnight, 50%.
5.3.13. RX-3117 Biology.
RX-3117 29, a fluorocyclopentenyl-cytosine nucleoside analog, was shown to have anticancer activity460,461 with an IC50 range of 0.4 to >30 μM for 59 cell lines.462 The agent was also shown to inhibit DNA and RNA synthesis and induce apoptotic cell death of tumor cells.349 Interestingly, preclinical studies using nude mice implanted with various tumors, including colon, lung, renal, and pancreatic, indicated that the ribose form and not the 2′-deoxyribose form was the active compound.463
Biological data were recently published on the metabolism and mechanism of action of RX-3117 29.462 Cellular uptake of the agent occurs by hENT, and the agent is a poor substrate for cytidine deaminase, preventing its degradation within the cell.462 Furthermore, uridine-cytidine kinase generates RX-3117–5′-monophosphate, which is subsequently phosphorylated to the di- and triphosphate forms by cellular kinases. RX-3117–5′-trisphophate is incorporated into RNA and inhibits RNA synthesis at high concentrations, but at IC50 values it does not affect RNA structural integrity. RX-3117–5′-diphosphate is a substrate for ribonucleotide reductase, and the 2′-deoxy-RX-3117–5′-triphosphate can be incorporated into DNA, potentially providing another mechanism of cancer cell growth inhibition. The 2′-deoxy-RX-3117–5′-triphosphate also downregulates DNA methyltransferase-1 protein (at IC50 values for specific cell line) and Cdc2 protein expression, which are both involved in the cell cycle division process.462
Preclinical and clinical studies of RX-3117 29 showed potent efficacy in xenograph models.464 In 2012, Rexahn Pharmaceuticals reported the completion of a Phase I clinical trial for RX-3117 29 in Europe and indicated that the drug had 56% orally bioavailable, had a plasma half-life of 14 h, and was well tolerated in cancer patients. Currently, a Phase Ib trial is ongoing to test the dose-finding and safety of RX-3117 29 as an oral monotherapy to treat advanced malignancies (ClinicalTrials.gov identifier: NCT02030067–recruiting patients).
5.3.14. RX-3117 Synthesis.
A synthesis of RX-3117 29 is illustrated in Scheme 38.463 Protection of d-ribose 213 followed by Wittig olefination, Swern oxidation, and vinyl Grignard addition afforded diene 217. Silyl ether deprotection and selective primary alcohol benzylation, followed by ring-closing metathesis and oxidative rearrangement via a [3,3]-sigmatropic shift yielded benzylated cyclopentenone 221. Subsequent iodination of the double bond, lactone reduction, and protection of the resulting lactol provided TBDPS ether 224. Electrophilic vinyl fluorination followed by desilylation gave fluorocyclopentenol 225, which was coupled with N3-benzoyluracil under Mitsunobu conditions, providing protected nucleoside analogue 226. Removal of the N3-benzoyl group by methanolic ammonia and the 5′-benzyl group by BBr3 generated 227. Finally, triacetatyl intermediate 227 was converted to the cytidine analog RX-3117 29 by a sequence involving formation of a 4-triazole intermediate and nucleophilic displacement with ammonia.
Scheme 38. Synthesis of RX-3117 29a.

aReagents and conditions: (a) concd H2SO4, Me2CO, then TBDPSCl, imidazole, 99%; (b) Ph3PCH3Br, t-BuOK, THF, 93%; (c) (COCl)2, DMSO, DCM, −78 °C, 1 h, then Et3N, rt, 96%; (d) CH2=CHMgBr, THF, −78 °C, 84%; (e) n-Bu4NF, THF, 97%; (f) Bu2Sn(O), PhMe, 15 h, then TBAI, BnBr, 50 °C, 16 h, 76%; (g) Grubbs’ catalyst (2nd generation), PhMe, 80 °C, 90%; (h) PDC, DMF, rt, 18 h, 59%; (i) I2, pyr, THF, 55%; (j) NaBH4, CeCl3, MeOH, 93%; (k) TBDPSCl, imidazole, DMF, 97%; (l) NFSI, n-BuLi, THF, −78 °C; (m) n-Bu4NF, THF, 63% (over two steps); (n) N3-benzoyluracil, DEAD, Ph3P, THF, 87%; (o) NH3/MeOH; (p) BBr3, DCM, −78 °C, 50%; (q) Ac2O, pyr, then POCl3, Et3N, 1,2,4-triazole; (r) NH4OH, dioxane; (s) NH3/MeOH, 40% (over three steps).
An alternative and slightly improved approach to RX-3117 29 uses a trityl protecting group at the 5′-position of intermediate 214, which avoids a deprotection/reprotection step, decreasing the total number of steps of the synthesis by two.465 However, both processes include long and laborious syntheses, producing only small quantities of the final product in low yield. A large-scale synthesis is most likely needed for significant clinical treatment of RX-3117 29.
5.4. Cytarabine and Cytarabine Prodrugs
5.4.1. Cytarabine Biology.
In the 1940s, Werner Bergmann isolated two nucleosides from the marine sponge Cryptotethya crypta, which he termed spongothymidine and spongouridine. These spongonucleosides were determined to have a 2′-OH in the arabino configuration. In the late 1950s, various unnatural nucleosides (including spongonucleosides) were prepared with the hypothesis that they would kill cancer cells by interfering with DNA replication, and indeed, many were found to inhibit DNA synthesis in vitro. In the 1960s, Seymour Cohen reported promising cytotoxic effects of a new spongonucleoside in cancer cells: cytarabine 4 (aka spongocytidine, cytosine arabinoside, or ara-C).466 The anticancer agent rapidly progressed through rodent models and clinical trials to gain FDA approval as a cancer therapy.
Cytarabine 4 is transported into the cell primarily by hENT-1.467,468 Cytarabine 4 is monophosphorylated by 2′-deoxycyti-dine kinase, which is the rate-limiting step in the formation of the active form: cytarabine-5′-triphosphate.469 Cytarabine-5′-tri-phosphate competes with dCTP for incorporation into DNA and is a weak competitive inhibitor of DNA polymerases. Cytarabine-5′-triphosphate is also a substrate for DNA polymerase α and is incorporated into both the leading and lagging strands of DNA. This incorporation leads to delayed chain termination, allowing several additional nucleotides to be incorporated before the DNA polymerase stops extending and falls off the replication fork. Cell cycle arrest occurs with the DNA repair mechanism deployed. If the embedded cytarabine-5′-monophosphate is not removed, the cell will enter apoptosis.470–472
Various factors decrease the effectiveness of cytarabine 4. The agent is deaminated by cytidine deaminase, and cytarabine-5′-monophosphate can be deaminated by deoxycytidylate deaminase, leading to the inactive uridine analogs.473 In addition, cytarabine-5′-triphosphate is a feedback inhibitor of 2′- deoxycytidine kinase, limiting the formation of cytarabine-5′-monophosphate.474
Resistance to cytarabine 4 exposure is afforded by several different mechanisms, including nucleotide transporter and 2′-deoxycytidine kinase gene mutations and decreased expression levels,475 and changes in cytosolic nucleotidase II,476 cytidine deaminase, and deoxycytidylate deaminase activities.477 Recently, a correlation was reported between increased expression of the ABC transporter MRP8 and a poor treatment response of individuals with acute myelogenous leukemia treated with cytarabine 4.478
Cytarabine 4 has been FDA-approved since 1969 for the treatment of acute myeloid leukemia,479 with the exception of acute promyelocytic leukemia.480 It is usually given in combination with a topoisomerase II inhibitor such as daunorubicin, idarubicin, or mitoxantrone.481–483 Several clinical trials are underway using cytarabine 4 alone or in combination treatment studies. (ClinicalTrials.gov identifiers: NCT01191541–recruiting patients, NCT01289457–recruiting patients, and NCT01756677–recruiting patients).
5.4.2. Elacytarabine Biology.
Elacytarabine (51, CP-4055), an oleic acid ester of cytarabine 4, was designed to enter cancer cells independent of nucleoside transporters, which would also overcome the low expression level of hENT-1 at the plasma membrane or loss of function mutation in hENT-1.484,485 The lipophilic prodrug diffuses through the phospholipid bilayer, being retained in a membrane fraction within the cell.486 After the action of an unidentified esterase, cytarabine 4 is released into the cytosol.349 In comparison to cytarabine 4, elacytarabine 51 exposure led to an increased intracellular level of active metabolite, which was also retained longer in cells after drug removal.486
Elacytarabine 51 showed promising results in both preclinical and clinical studies.487–489 However, in April 2013, despite the initial success, Clavis Pharma announced that elacytarabine 51 did not perform better than the regular standard of care therapy in a pivotal Phase III trial354 (ClinicalTrials.gov identifier: NCT01147939–completed in 2013). Moreover, patients with relapsed/refractory acute myeloid leukemia had no meaningful benefit from elacytarabine 51 when directly compared to seven other treatment regiments.490 As a result, Clavis has suspended all further developmental work on elacytarabine 51.
5.4.3. MB 07133 Biology.
Ligand Pharmaceuticals Inc. developed MB 07133 34, a phosphoramidate prodrug of cytarabine 4. In 2003, MB 07133 34 entered a phase I/II clinical trial as an intravenous infusion agent for the treatment of unresectable hepatocellular carcinoma (ClinicalTrials.gov identifier: NCT00073736–completed–no results posted). MB 07133 34 was well tolerated with evidence of disease stabilization. In 2011, Ligand Pharmaceuticals granted the license to Chiva Pharmaceuticals to develop MB 07133 34 in China.
5.4.4. Cytarabine, Elacytarabine, and MB 07133 Syntheses.
In 1959, cytarabine 4 and 3-β-d-arabinofuranosyluracil were originally synthesized by the Dekker group (Scheme 39).491 Cytidine 93 was treated with polyphosphoryic acid and purified by gradient elution chromatography to afford the diphosphate intermediate 228 as a crystalline cyclohexylammonium salt. Subsequent enzymatic dephosphorylation with prostatic phosphatase furnished cytarabine 4. In order to verify the hypothesis that the transformation proceeded via a cyclonucleoside-5′-diphosphate intermediate, O2-2′-cyclocyti-dine 229 was isolated by ion-exchange fractionation. The intermediate could further be converted to cytarabine 4 by enzyme-mediated dephosphorylation and hydrolysis.
Scheme 39. Synthesis of Cytarabine 4a.

aReagents and conditions: (a) polyphosphoric acid; (b) prostatic phosphatase; (c) OH–.
Two different process syntheses of cytarabine 4 used 1-β-d-arabinfuranosyl uracil 231 as a key intermediate, which can be synthesized on a large-scale in two steps from uridine 98 via 2,2′-anhydro-uridine 230 (Scheme 40).492 One process treated 231 with HMDS under high pressure and high temperature, leading to the intermediate 232, which upon treatment with MeOH provided cytarabine 4 on a 50-g-scale.493 The other process converted 231 to the O-acyl-substituted, 1,2,4-triazole derivative 233, and subsequent deprotection produced cytarabine 4 on a kilogram-scale.494
Scheme 40. Synthesis of Cytarabine 4a.
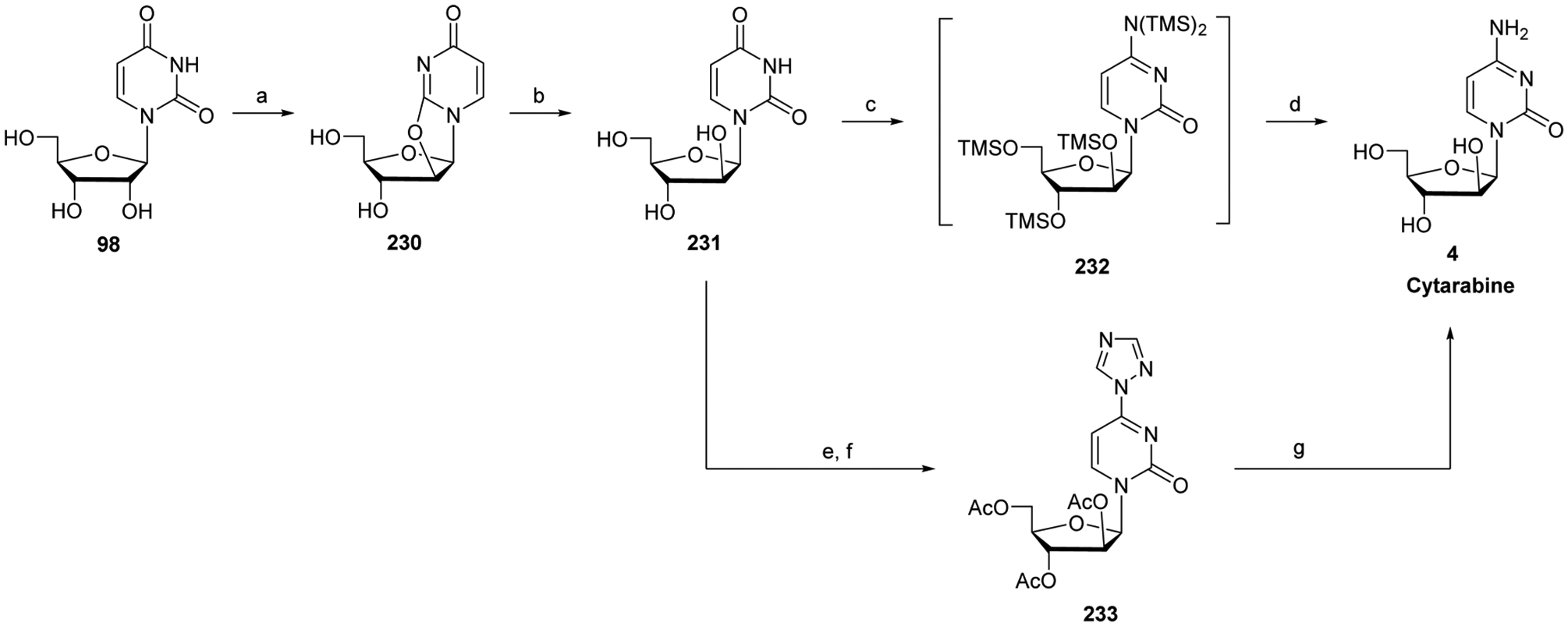
aReagents and conditions: (a) (PhCO)2O, DMF, 100 °C, then K2CO3, 137 °C, 1.5 h, 78%; (b) aq HCl, 80 °C, 2 h, 68%; (c) HMDS, high pressure, 150 °C, 80 h; (d) MeOH, 5 °C, 1 h, 87% (over two steps); (e) Ac2O, pyr, rt, 3 h, 91%; (f) POCl3, 1H-1,2,4-triazole, Et3N, CHCl3, <8 °C then rt, overnight, 84%; (g) NH4OH, NH3, dioxane, H2O, rt, overnight, 70%.
Elacytarabine 51 can be prepared in one step by enzyme-mediated esterification of the 5′-OH of cytarabine 4 with oleic anhydride in DMF,495 whereas the cyclic protide MB 07133 34 has been synthesized from cytarabine 4 in four steps (Scheme 41).496,497 Selective 2′,3′-silyl protection followed by treatment with N,N-dimethylformamide dimethyl acetal generated intermediate 236.496 In parallel, trans-p-nitrophenylphosphate 239 was prepared in three steps from methyl 3-hydroxy-3-(pyridin-4-yl)-propanoate 237 via esterification (with l-N,N-dimethyl phenylalanine) and subsequent diastereomer separation, reduction with LiAlH4, and coupling with p-nitrophenyl phosphorodichloridate. Condensation of protected nucleoside 236 with p-nitrophenyl phosphorochloridate 239 followed by TBS deprotection furnished MB 07133 34.497
Scheme 41. Synthesis of Elacytarabine 51 and MB 07133 34a.

aReagents and conditions: (a) oleic anhydride, DMF, Mucor miehei lipase (MML), 60 °C, 48 h, 88%; (b) TBSCl, imidazole, DMF, rt, overnight, 85%; (c) 50% TFA, THF, −10 °C, 18 h, 65%; (d) DMF-dimethyl acetal, pyr, rt, 16 h, 90%;(e) EDC, DMAP, l-N,N-dimethyl-phenylalanine, DCM, rt, 16 h, 83%; (f) LiAlH4, ether, −20 °C, 1 h, 66%; (g) 4-nitrophenyl phosphorodichloridate, THF, pyr, 0 °C to rt, 3.5 h, then sodium 4-nitrophenoxide, 40 °C, 4 h, 63%; (h) t-BuMgCl, THF, rt, 16 h, 78%; (i) 70% TFA, 60 °C, 16 h, 83%.
6. PURINE ANALOGS
6.1. Deoxypurines
6.1.1. Cordycepin Biology.
In 1951, cordycepin 27 was isolated from the fungus Cordyceps militaris498 and exhibited anticancer, anti-inflammatory, and antifungal activities.499,500 It is currently thought to be the active ingredient in various traditional Chinese medicines.501 The mechanism of cellular uptake of this agent has not been fully elucidated. However, the 3′-OH has been shown to be required for efficient uptake of nucleosides and nucleoside analogs by hENTs, suggesting that cordycepin 27 does not efficiently use this mechanism for uptake.502 Once in the cell, the agent is converted to the cordycepin-5′-monophosphate by adenosine kinase,503 and then to the corresponding di- and triphosphate forms by other cellular kinases. Cordycepin-5′-triphosphate is incorporated into the poly A tail of the growing mRNA chain, presumably causing chain termination due to its lack of a 3′-OH.504 Furthermore, cordycepin 27 can inhibit ribose-phosphate pyrophosphokinase and 5-phosphoribosyl-1-pyrophosphate amidotransferase in vitro.505,506
Cordycepin 27 exposure was shown to cause DNA double-stranded break in breast cancer cells.507 A possible explanation for this observation is the inhibitory effect this agent has on poly(ADP-ribose) polymerase, an enzyme involved in the repair of DNA single-strand breaks.28 Unrepaired DNA single-strand breaks could lead to more extensive DNA damage, such as double-stranded breaks. In addition, cordycepin 27 is a substrate for adenosine deaminase, which can lead to significant degradation in vivo. Other mechanisms of action for cordycepin 27 are binding A3 adenosine and DR3 receptors, activating caspase-8/−3 pathways for cell death, suppressing cyclin D1 and c-myc expression cell signal pathways, inhibiting cAMP formation, and decreasing glycogen synthase kinase-3 β/β-catenin activation.508–511
In 2008, Oncovista initiated a Phase I/II clinical trial of cordycepin 27 in combination with pentostatin 6 (adenosine deaminase inhibitor; see Section 6.1.5) for the treatment of refractory TdT-positive leukemia (ClinicalTrials.gov identifier: NCT00709215–unknown status >2 years without an update). Currently, no study updates have been reported. Earlier in 2000, a Phase I trial was conducted using cordycepin 27 plus pentostatin 6 in treating patients with refractory acute lymphocytic or chronic myelogenous leukemia (ClinicalTrials. gov identifier: NCT00003005–completed in 2001–no results reported). Development of cordycepin 27 seemed to have stalled, until Rainforest Pharmacal announced plans to reactivate the IND in 2016 and start a Phase I/II trial in TdT-positive refractory leukemia patients in 2017. However, issues with deamination will have to be addressed before cordycepin 27 can progress significantly in clinical trials.
6.1.2. Cordycepin Synthesis.
In 1961, Lee et al. reported a pioneering synthesis of cordycepin 27, which involved desulfurization of 3′-ethylthioadenosine by treatment with Raney nickel in refluxing 2-methoxyethanol.512 Later, Hansske and Robins used 2-acetoxyisobutyryl bromide (Mattock’s bromide) to synthesize cordycepin (Scheme 42).513 Thus, reaction of adenosine 240 with 2-acetoxyisobutyryl bromide afforded the corresponding trans-bromoacetates (241a and 241b). Cordycepin 27 was finally obtained after treatment with acidic resin to provide epoxide 242 and regioselective reduction with lithium triethylborohydride.
Scheme 42. Synthesis of Cordycepin 27a.

aReagents and conditions: (a) 2-acetoxyisobutyryl bromide, CH3CN, H2O, rt, 1 h; (b) Amberlite IRA-400 (OH–) resin, MeOH, 92% (over two steps); (c) 1 M LiEt3BH, THF, Me2SO, 0 °C to rt, overnight, then 5% HOAc/H2O, Et3B, 98%.
Aman et al. used a similar approach for a process synthesis of cordycepin 27 (Scheme 43).514 Initially, the authors thought to use the epoxide 242 as a key intermediate, but they had difficulty controlling the regioselectivity of the subsequent reduction. They noticed, however, that trans-bromoacetate 243b fortuitously precipitated selectively from solution. This intermediate was isolated and subsequently deprotected at the 2′- and 5′-positions in order to increase compound solubility and reactivity (decreased steric hindrance) in the final reduction reaction. The deprotection required a delicate balance of reaction temperature, acid concentration, and methanol concentration in order to fully hydrolyze 243b to 244, without leading to compound decomposition via depurination. Depurination also plagued the final dehalogenation step, and conditions had to be optimized. Finally, hydrogenation in the presence of 4–5 molar equivalent of sodium acetate in a 1:1 solvent mixture of ethanol and water effectively debrominated the hydrochloride salt of 244. Ethyl acetate was used to extract crude cordycepin 27 from the reaction mixture, which had a high salt load. Subsequent addition of methanol to the organic solution followed by cooled agitation precipitated cordycepin 27. The overall process required no chromatography and produced the product cordycepin 27 on a 100-g-scale at 99% purity.
Scheme 43. Synthesis of Cordycepin 27a.
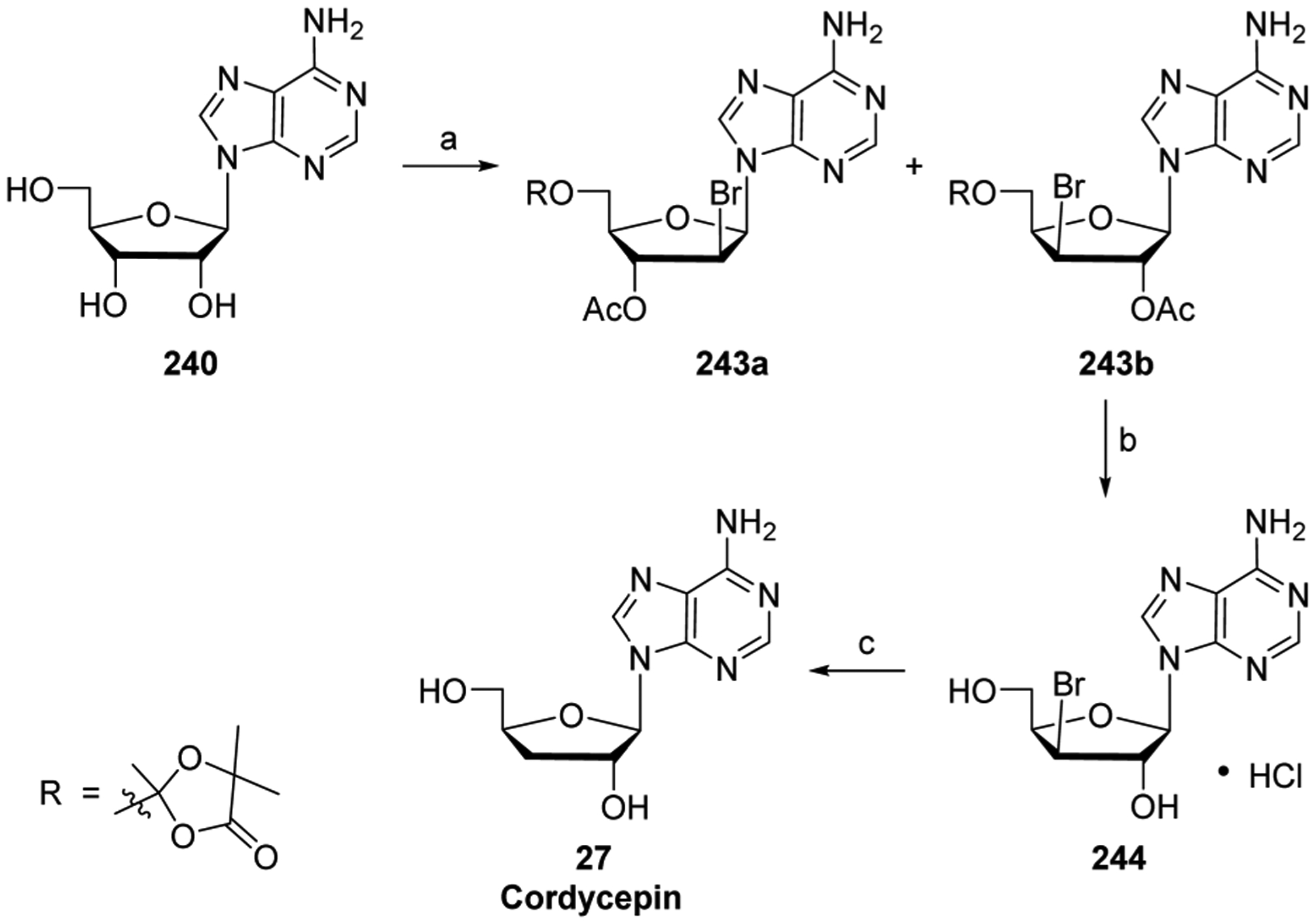
aReagents and conditions: (a) 2-acetoxyisobutyryl bromide, CH3CN, EtOAc, <35 °C, 6 h, 52% for 243b; (b) AcBr, MeOH, concd HCl, 20 °C, rt, 2–4 days, 70%; (c) AcONa, EtOH, H2O then Pd/C, H2 (50 psi), rt to 50 °C, 60%.
6.1.3. Cladribine Biology.
Inherited adenosine deaminase deficiencies were reported to cause lymphopenia, an immunodeficiency in humans.515 Furthermore, the accumulation of deoxyadenosine nucleotides had been implicated in the pathogenesis of this disorder. An increase in dATP levels was shown to kill lymphocytes.516 Experimentally, a high dADP level in lymphoid cells led to negative feedback inhibition of ribonucleotide reductase, and the subsequent decrease in dNTP pools resulted in DNA synthesis inhibition and cell death.517 It is now known that inhibition of adenosine deaminase causes an increase in both dATP and dADP levels.
Carson et al. reasoned that deoxyadenosine analogs, which are resistant to adenosine deaminase activity, may be phosphorylated into their triphosphate forms in the cell. Moreover, a deoxyadenosine analog that inhibits adenosine deaminase activity might be successful for the treatment of lymphocytic malignancies.518 In collaboration with John Montgomery, Carson studied 2-chloro-2′-deoxyadenosine (cladribine, 8), which was shown to have significant cytotoxicity in malignant T lymphocytes.518 The presence of the C2 chlorine in the purine base was believed to make the agent resistant to adenosine deaminase activity.519 Furthermore, cladribine 8 was shown to be converted to the cladribine-5′-triphosphate in vitro, and to exert significant cytotoxicity both in vitro and in vivo.519–521
Cladribine 8 is transported by both hCNT and hENT into cells.522 It is phosphorylated by cytosolic 2′-deoxycytidine kinase516 and mitochondrial deoxyguanosine kinase to generate cladribine-5′-monophosphate. This phosphorylation is the rate-limiting step in the activation of the parent nucleoside analog.523 Subsequent phosphorylation generates cladribine-5′-diphosphate and cladribine-5′-triphosphate forms; both are active metabolites of cladribine 8. Cladribine-5′-triphosphate is readily incorporated into DNA.524 Chain extension by DNA polymerase continues past a single DNA incorporated cladribine-5′-monophosphate, but elongation is terminated when the polymerase encounters three successive incorporated cladribine molecules in the growing DNA strand.17 In addition, cladribine-5′-triphosphate inhibits ribonucleotide reductase activity.525 Collectively, these actions can promote cell death by dNTP imbalance and DNA double-stranded breaks.526,527 Furthermore, cladribine 8 was shown to interfere with methyltransferases and may promote hypomethylation in vitro.528–530 Other effects of cladribine 8 include the induction of caspase-3 activation, thereby promoting cell apoptosis in vitro.531 Interestingly, the agent has also been shown to impede mononuclear cells from crossing the blood brain barrier and to inhibit T cells and B cells in the central nervous system.532
Resistance to cladribine 8 exposure can arise from compound metabolism and detoxification as well as increased egress of the agent from cancer cells in vitro.533 Interestingly, a recent report examining metabolism data for cladribine 8 treatment in animals and humans indicated the formation of 2-chlorodeoxyinosine and 2-chlorohypoxanthine (inactive metabolites), suggesting a sensitivity to adenosine deaminase degradation.534 Furthermore, cladribine 8 was shown to be a substrate for the ABCG2 transporter, leading to cell egress.535
Cladribine 8 gained FDA approval in 2004 for use as a first-line monotherapy for hairy cell leukemia536–538 and has a favorable toxicity profile in comparison to other lymphocytic drugs.539 It has been used in combination treatment for chronic lymphocytic leukemia35 and abnormal mast cells debulking.540 Cladribine 8 has also been reported to kill immature and mature monocyte-derived dendritic cells.537 In addition, the agent was shown to be effective for treatment of Langerhans cell histiocytosis.541 Cladribine 8 therapy has also been investigated for relapsingremitting multiple sclerosis, but has not gained FDA approval for this disease due to lack of safety information.542
6.1.4. Cladribine Synthesis.
Interestingly, the first reported preparation of cladribine 8 was as an intermediate in a synthesis of 2′-deoxyguanosine.543,544 In 1972, Christensen et al. reported a nonstereoselective fusion glycosylation synthesis of cladribine 8 from diprotected sugar 245, made by treatment of 2′-deoxyribose in methanol with a catalytic amount of acid followed by p-toluoyl protection (Scheme 44).545 The anomeric mixture of glycosylated product 246 was resolved by silica gel chromatography, and the β-anomer was treated with methanolic ammonia, to give cladribine 8.
Scheme 44. Synthesis of Cladribine 8a.

aReagents and conditions: (a) 2,6-dichloropurine, melt, then dichloroacetic acid, and fusion under vacuum; (b) NH3/NaOMe.
In 2006, Robins reported a new synthesis of cladribine 8 that focused on improving the yield of the glycosylation step between N6-imidazolyl purine 248 and α−1′-chlorosugar 247, which is formed by treating 2′-deoxyribose with HCl in methanol followed by tosylation and stereoselective crystallization of the α-anomer (Scheme 45).546,547 The modified purine base exhibited improved solubility in organic solvents as well as better glycosylation regioselectivity, due to steric hindrance of the N7 position of the base, leading to exclusive formation of the N9 adduct. A binary solvent system was employed in the glycosylation in attempts to increase the solubility of the modified purine base as well as improve the stereoselectivity of the reaction. The rational for this system drew on a previously reported observation that preferential solvation of reactants can lead to different free energies of activation, and therefore alter the ratios of two competitive pathways.548 Indeed, a judicious solvent choice of cold dichloromethane and acetonitrile (1:1 mixture) allowed for the selective formation of the desired β-nucleoside in 95% yield. Furthermore, the use of cold solvent led to decreased anomerization of chlorosugar 247. The synthesis was concluded by converting the imidazoyl group to an imidazolium group by treatment with in situ-generated benzyl iodide, and the resulting salt was heated in methanolic ammonia to give, after recrystallization, pure cladribine 8, in >90% from chlorosugar 247.
Scheme 45. Synthesis of Cladribine 8a.

aReagents and conditions: (a) NaH, CH3CN, rt, 8 h; (b) then 247, 0 °C to rt, 22 h, 100% (over two steps); (c) BnI, CH3CN, 60 °C, 1.5 h; (d) NH3/MeOH, 60 °C, 11 h, >90% (over two steps).
More recently, a kilogram-scale synthesis has been reported for cladribine 8 (Scheme 46).549 The researchers chose to explore Vorbrüggen conditions for the glycosylation. Generally, when this reaction involves 2′-deoxy lactones, it usually gives a product ratio of roughly 50% α/β nucleosides. This was indeed found to be the case when the glycosylation of the 4-chlorobenzyl protected sugar 249 and silylated 2-chloroadenine was performed in solvents such as tetrahydrofuran and dichloromethane. However, when solvents such as acetonitrile or lithium hexamethyldisilazane were employed, the desired β-anomer fortuitously precipitated from solution. It was hypothesized that a dynamic equilibrium existed in the reaction, involving glycosylation and retroglycosylation of the α-anomer along with concomitant precipitation of the β-anomer, driving the equilibrium to the desired β-nucleoside product 253. The reaction was complete within 1 h; however, an aging step at 60 °C was used to further increase the yield. Furthermore, only a true catalytic amount of the Lewis acid trimethylsilyl trifluoromethanesulfonate was needed, which greatly simplified the purification of the product by obviating the need for a catalyst quench and aqueous workup. Isolation was accomplished by direct filtration. Deprotection with 10–20 mol % of sodium methoxide in MeOH led to in situ crystallization of high purity cladribine 8, in 99.8–99.9% HPLC purity with an overall yield of 43%.
Scheme 46. Synthesis of Cladribine 8a.

aReagents and conditions: (a) 25% NaOMe/MeOH.
6.1.5. Pentostatin Biology.
Pentostatin (6, [(R)-3-(2-deoxy-β-D-erythro-pento-furanosyl)-3,6,7,8-tetrahydro-imidazo-[4,5-d][1,3-diazepin-8-ol]) is an antibiotic isolated from a culture broth of Streptomyces antibioticus.550 It is an irreversible, stoichiometric inhibitor of mammalian adenosine deaminase, with a Ki in the nanomolar range.551,552 Importantly, pentostatin 6 directly inhibits this enzyme in order to truly mimic adenosine deaminase deficiencies, which is in contrast to cladribine 8, which was designed as an adenosine deaminase-resistant antimetabolite (see Section 6.1.3.). Pentostatin 6 is unique among anticancer nucleoside analogs in that it is active without metabolism (cellular phosphorylation). It has been proposed that the tetrahedral carbon at position 8 of the diazepine ring of pentostatin 6 mimics the purported transition state tetrahedral carbon in the deamination of adenosine to inosine.553 In addition to this effect, pentostatin 6 also targets cellular methyltransferases, hindering the ability of the cell to methylate both DNA and mRNA.554–556
Pentostatin 6 is used to treat hairy cell leukemia and is effective against lymphoid malignancies with high adenosine deaminase activities.557,558 The agent has also been used to prevent and to treat both acute and chronic graft-versus-host disease.559 Pentostatin 6 is currently being tested in several clinical trials. An additional Phase I/II clinical trial uses pentostatin 6 and cyclophosphamide (DNA-alkylating agent) cotreatment for low-intensity stem cell transplantation with multiple lymphocyte infusions and to treat advanced kidney cancer (ClinicalTrials.gov identifier: NCT00923845–active status–primary objective of trial was not achieved). A Phase II trial combines pentostatin 6, cyclophosphamide (DNA alkylating agent), and rituximab (monoclonal antibody against B cell protein CD20) followed by lenalidomide (antiangiogenic/immunomodulatory agent) treatment for relapsed or refractory B cell chronic lymphocytic leukemia (ClinicalTrials.gov identifier: NCT00074282–unknown status–no updates in 2 years). Finally, a Phase I trial involves the combination of pentostatin 6, bendamustine (DNA-alkylating agent), and ofatumumab (monoclonal antibody against B cell protein CD20) for the treatment of chronic lymphocytic leukemia (ClinicalTrials.gov identifier: NCT01352312–active status–not recruiting patients).
6.1.6. Pentostatin Synthesis.
Parke-Davis developed a large-scale industrial production of pentostatin 6 using fermentation cultures of Streptomyces antibioticus NRRL 3238. To date, this approach is the major form of production of the compound, although a few other syntheses have been reported.560
In 1982, the first synthesis of pentostatin 6 was reported by chemists at Warner-Lambert/Parke-Davis (Scheme 47).561,562 Knoevenagel condensation of azole 254 with benzaldehyde produced styrylimidazole 255, and subsequent benzylation formed regioisomers 256 and 257. The mixture was subjected to ozonolysis, and 258 was separated from its isomeric product by differential precipitation. Carbon homologation of 258 was followed by nitro reduction, ring closure, and debenzylation, producing heterocycle 261. Due to the instability of heterocycle 261, glycosylation conditions had to be optimized. The use of tin(IV) chloride as Lewis acid and BSTFA as silylating agent were found to produce the desired product in a 1:1 mixture of α/β diastereomers. The mixture can be separated by preparative HPLC on a small-scale and by fractional crystallization on a larger-scale. Deprotection of the β-anomer 262 in the presence of sodium methoxide followed by ketone reduction produced a separable 1:1 mixture of pentostatin 6 and its stereoisomer. It is noteworthy that Zhang et al. improved the conclusion of the synthesis by using a chiral ruthenium catalyst for asymmetric transfer hydrogenation of ketone 262.563 This reaction was completely diastereoselective and gave a yield of 80%.
Scheme 47. Synthesis of Pentostatin 6a.

aReagents and conditions: (a) PhCHO, piperidine, 95 °C, 21 h, 70%; (b) BnCl, K2CO3, DMF, 75 °C, 6 h, 96%; (c) O3, DCM, −78 °C, 8 h, then HCO2H, H2O2, 0 °C to rt, overnight, 67%; (d) CDI, THF, reflux, 1 h, then t-BuOK, CH3NO2, THF, 0 °C, 45 min, 68%; (e) SnCl2, concd HCl, 60 °C, 2.5 h, then H2S, rt, 75%; (f) H2, Pd/C, MeOH, H2O, rt, 16 h, 96%; (g) HC(OEt)3, Me2SO, 65 °C, 15 min, 81%; (h) N,N-bis(trimethylsilyl)trifluoroacetamide, pyr, CH3CN, rt, overnight; (i) 2-deoxy-3,4-di-O-p-toluoyl-D-pentofuranosyl chloride, SnCl4, DCE, CH3CN, −50 °C, 45 min; (j) NaHCO3, H2O, rt, 0.5 h, 23% (over three steps); (k) NaOMe, MeOH, rt, 1 h; (l) NaBH4, H2O, MeOH, 25 °C, 30 min, 26% (over two steps).
Phiasivongsa and Redkar reported a ring expansion of protected 2′-deoxyinosine 263 using diazomethane.564 This was followed by deprotection and reduction to furnish a diastereomeric mixture of pentostatin 6, which was separated by following the Warner-Lambert/Parke-Davis procedure in Scheme 48.564 Unfortunately, no yields were reported.
Scheme 48. Synthesis of Pentostatin 6a.
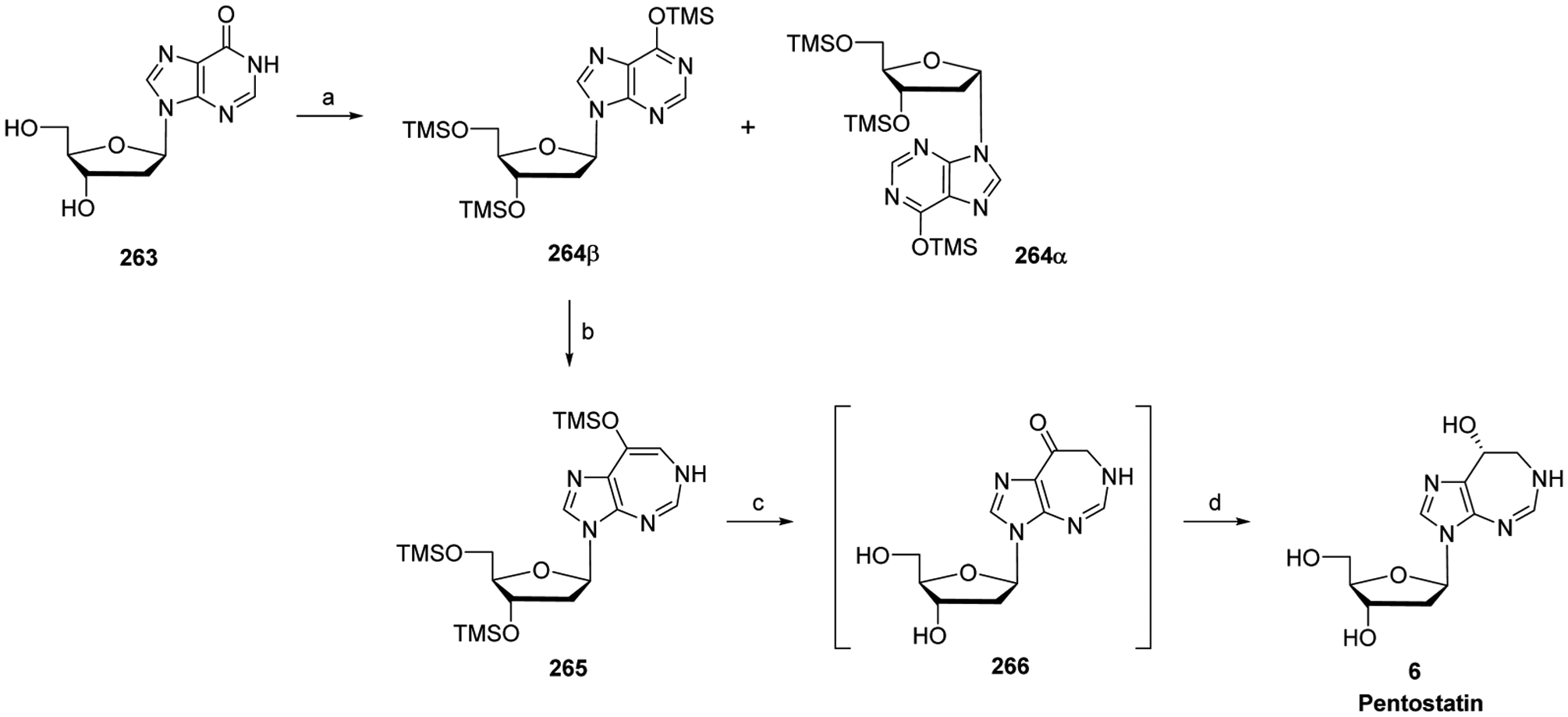
aReagents and conditions: (a) (Me3Si)2NC(O)CF3, pyr, MeCN, 12 h; (b) BF3·Et2O, DCM, 0 °C, 30 min; (c) TBAF, THF, 0 °C, 2 h; (d) NaBH4, MeOH, H2O, rt, 1 h.
6.1.7. Clofarabine Biology.
Clofarabine 12 is a second-generation purine nucleoside analog designed to overcome the limitations of fludarabine-5′-monophosphate 7 (see Section 6.2.3) and cladribine 8 (see Section 6.1.3),565 which are both susceptible to glycosidic bond cleavage.521 Indeed, the introduction of a C2′-fluorine in the arabino configuration in clofarabine 12 significantly increased the stability of the glycosidic bond in acidic solution and toward phosphorolytic cleavage.566,567 Chlorine substitution instead of fluorine at the 2-position of the adenine base was chosen in order to avoid production of the 2-fluoroadenine, a precursor to the toxic 2-fluoro-adenosine-5′-triphosphate, should the glycosidic bond be cleaved.568
Clofarabine 12 is usually administered intravenously; however, the increased stability of the glycosidic bond also allows for oral administration.566,569,570 Clofarabine 12 enters the cell via hENT1, hENT2, and hCNT2, and to a much lesser extent by passive diffusion.429 2′-Deoxycytidine kinase generates clofarabine-5′-monophosphate;571 however, the enzyme has a lower Km for clofarabine 12 than cladribine 8.572 Moreover, clofarabine resistance arises from decreased 2′-deoxycytidine kinase activity in vitro.572 Formation of clofarabine-5′-diphosphosphate is generated by purine nucleotide monophosphate kinase, and is the rate-limiting step in the activation of the agent.573 This is in contrast to many other nucleoside analogs, in which the rate-limiting step is the generation of clofarabine-5′-monophosphate.
Clofarabine-5′-diphosphate serves as an intracellular reservoir for the active-metabolite clofarabine-5′-triphosphate, which is retained in the cell longer than clofarabine-5′-triphosphate in cells from CLL and AML patients ex vivo.572 Clofarabine-5′-triphosphate has several mechanisms of action in the cell. It inhibits ribonucleotide reductase (presumably by allosteric inhibition), which results in a decrease in dCTP and dATP concentrations.520 The reduction in the amount of dCTP likely leads to DNA synthesis inhibition, and a decrease in the concentrations of dATP leads to greater DNA incorporation of clofarabine-5′-triphosphate (self-potentiation).568 A low intracellular concentration of clofarabine-5′-triphosphate can be incorporated by DNA polymerases α and ε574 into DNA and can promote polymerase arrest at the replication fork, inhibiting DNA repair and inducing strand breaks in vitro.573,574 This DNA damage leads to the induction of cytochrome c-mediated apoptosis in vitro.575 Studies have shown that clofarabine-5′-triphosphate can also be incorporated into RNA in cell lines.573
Clofarabine 12 is an FDA-approved drug for the treatment of relapsed or refractory pediatric acute lymphoblastic leukemias.35,576 It is being evaluated for the treatment of acute myeloid leukemia in both children and the elderly.577 The agent was examined for treatment of non-Hodgkin’s lymphomas, myelodysplastic syndrome, and solid tumors.520 Oral clofarabine 12 has been studied in relapse/refractory non-Hodgkin’s lymphomas578 and in high-risk myelodysplastic syndrome patients.579 Currently clofarabine 12 in combination treatments is being investigated in over a dozen clinical trials for chronic lymphocytic leukemia, acute myelogenous leukemia, myelodysplastic syndrome, and mixed phenotype acute leukemia with nucleoside analogs such as cytarabine 4, gemcitabine, and fludarabine-5′-monophosphate 7 and with other chemotherapies such as but not limited to busulfan, idarubicin, etoposide, and mitoxantrone.
6.1.8. Clofarabine Synthesis.
In the early 1990s, Montgomery et al. utilized differentially protected sugar 272 in order to access various nucleoside analogs, including clofarabine 12 (Scheme 49).566,580,581 The synthesis began with the stereoselective fluorination of diprotected tosylate 267, which can be formed by isopropylidene protection and tosylation of α-d-glucofuranose, followed by selective deprotection, leading to fluorinated sugar 268. Benzoylation of the 5′-OH followed by acid-mediated deprotection afforded lactol 269. Oxidative cleavage and subsequent rearrangement produced 270, which was eventually converted to bromosugar 272. Condensation of 2,6-dichloropurine with 272 occurred in refluxing dichloroethane, and chromatographic separation of the anomeric isomers produced nucleoside derivative 273. Three days of treatment of 273 in a steel bomb with ethanolic ammonia afforded a mixture of clofarabine 12 and the partially deprotected 5′-O-benzoyl derivative. Treatment of the mixture with LiOH in MeCN/H2O removed the residual protecting group, and pure clofarabine 12 was produced by three crystallizations from water.
Scheme 49. Synthesis of Clofarabine 12a.

aReagents and conditions: (a) KF, acetamide, 210 °C, 62%; (b) MeOH–0.7% H2SO4 (1:1 v/v); (c) BzCl in DCM, pyr, −15 °C, 80% (over two steps); (d) Amberlite IR-120 (H+), H2O/dioxane, 80 °C, 78%; (e) KIO4, H2O; (f) NaOMe, MeOH; (g) Ac2O, pyr, 80% (over three steps); (h) HBr/HOAc, DCM; (i) 2,6-dichloropurine, DCE, 100 °C, 32% (β-anomer); then NH3/EtOH, steel bomb, 3 days, solvent switch to MeCN/H2O, LiOH, 42%.
Twelve years later, Bauta et al. reported an improved and scalable process synthesis of clofarabine 12 (Scheme 50).582 Studies were performed on bromosugar 279,583 with the desire to increase β-anomer formation in the glycosylation step. Compound 279 was easily prepared by first conversion of 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose 274 to protected sugar 276584 via formation of the benzoxonium ion intermediate and solvolysis. Subsequent installation of the imidazolylsulfonate leaving group, stereoselective deoxyfluorination using excess KHF2, and finally bromination under acidic conditions completed the synthesis of 279.583 Empirical studies of the glycosylation step of 2-chloroadenine found potassium tert-butoxide to be the optimum base, with the potassium counterion being particularly important for both yield and β-selectivity. Solvent choice was guided by the precedent that low dielectric constants of a solvent favor the formation of the β-anomer in glycosylations involving bromosugar 279. It is believed that this effect arises from suppression of the formation of the oxonium ion intermediate 280 and subsequent SN1-type condensation, which would lead to both α- and β-glycosylation products. Nonpolar solvents would therefore be needed in order to favor a SN2 reaction pathway. However, solubility in such solvents would also be an issue, particularly with the 2-chloroadenine base. In order to find a balance between the two conflicting requirements of anomeric selectivity and substrate solubility, a 1:2:1 mixture of MeCN/tert-amyl alcohol/DCE was eventually chosen after considerable experimentation. In addition, the additive CaH2 was found to increase overall yield and anomeric ratio, due in part to removal of trace amounts of water from the solvent.
Scheme 50. Synthesis of Clofarabine 12a.

aReagents and conditions: (a) 0.5 M HBr, DCM, 0 °C; (b) H2O, 81% (over two steps); (c) NaH, DMF, 0 °C, 0.5 h, then N,N′-sulfuryldiimidazole, −40 °C to rt, 2 h, 85%; (d) KHF2, 2,3-butanediol, HF (50% in H2O), 160 °C, 1 h, 63%; (e) HBr/HOAc, DCM, rt, 16 h, 98%; (f) 2-chloroadenine, CH3CN, t-amyl alcohol, tert-BuOK, CaH2, 50 °C, 40 min, then 279, DCE, 50 °C, 19 h, 50% (β/α = 80:1); (g) NaOMe, MeOH, 33 °C, 7 h, 64%.
A number of challenges were overcome to separate protected clofarabine 281 from the heterogeneous reaction mixture. Excess 2-chloroadenine was filtered off. A subsequent solvent exchange to n-butyl acetate dissolved the mixture to allow for heptane-induced precipitation of crude 281, which after two subsequent pH-maintained methanol slurries was isolated in a >50:1 β/α ratio. Finally, deprotection with a catalytic amount of sodium methoxide in methanol followed by recrystallization formed drug-pure clofarabine 12 on a 25-g-scale.
6.2. Arabinose Purine Analogs
6.2.1. Nelarabine Biology.
Arabinosylguanine (9-β-d-arabinofuranosylguanine; ara-G) was discovered in the 1960s, but was not significantly developed at that time. In the 1970s, studies on the rare autosomal disorder purine nucleoside phosphorylase deficiency rekindled interest in ara-G.393,585 A deficiency in purine nucleoside phosphorylase, an enzyme that catalyzes the phosphate-mediated deribosylation of certain purine nucleosides, results in a profound dGTP-mediated imbalance in the endogenous nucleotide pools that leads to T cell lymphopenia without concomitant decreases in B cell populations.586 This sensitivity of human T cells (especially immature T cells) to purine nucleoside phosphorylase deficiency is hypothesized to arise from their relatively high ratio of kinase activity versus nucleotidase activity (in contrast to other cell types) which results in a high accumulation of intracellular dGTP.587 Deoxyguanosine, but not ara-G, is a substrate for purine nucleoside phosphorylase.588,589
Nelarabine (13, 2-amino-9-β-d-arabinosyl-6-methoxy-9H-guanine; 506U78) is a prodrug of ara-G and has a 10-fold higher solubility as compared to ara-G.590,591 Nelarabine 13 is converted to ara-G in the serum by adenosine deaminase.592 Ara-G is phosphorylated by either cytosolic 2′-deoxycytidine kinase or mitochondrial deoxyguanidine kinase into ara-G-5′-monophosphate593 and, subsequently, into ara-G-5′-diphosphate and ara-G-5′-triphosphate by cellular kinases. The active metabolite ara-G-5′-triphosphate is a substrate for DNA polymerases and is incorporated into DNA. This incorporation leads to inhibition of the DNA polymerases, causing inhibition of DNA replication and resulting in cell death via apoptosis.594 Moreover, it was believed that ara-G-5′-triphosphate would accumulate to high intracellular levels and would promote cytotoxic effects similar to those created by high dGTP concentrations. When T cell lymphoma cell lines were exposed to nelarabine, this resulted in FasL-mediated cytotoxicity, whereas myeloid and B cells did not accumulate ara-G-5′-triphosphate, but were stopped in the S phase of the cell cycle.595 High concentrations of ara-G-5′-triphosphate accumulated in malignant T cells from human bone marrow when tested in a rodent model.596
In 2005, nelarabine 13 was approved by the FDA for the treatment of relapse T cell acute lymphocytic leukemia and relapse T cell lymphoblastic lymphoma.597 Additional clinical trials are being conducted studying pharmacokinetic and pharmacodynamics properties in addition to efficacy in combination with etoposide (topoisomerase inhibitor) and cyclophosphamide (DNA-alkylating agent) treatment in lymphomas (ClinicalTrials.gov identifiers: NCT01094860–recruiting patients and NCT00981799–terminated status for Phase I/II in 2015).
6.2.2. Nelarabine Synthesis.
An early synthesis of nelarabine 13, shown in Scheme 51,598 goes through a key enzyme catalyzed trans-glycosylation between 1-β-d-arabinosyl uracil 231 and 2-amino-6-methoxy-9H-purine 282. The reaction uses uridine phosphorylase and purine nucleoside phosphorylase and is completely regio- and stereoselective, furnishing only the N9, β-anomer nelarabine 13 (Scheme 51).
Scheme 51. Synthesis of Nelarabine 13a.

aReagents and conditions: (a) 10 mM K3PO4, n-PrOH, H2O, pH 6.75, uridine phosphorylase, purine nucleoside phosphorylase, 37 °C, 26 days, 17%.
A more recent process synthesis for nelarabine 13 was reported by Zong et al. (Scheme 52).599 Glycosylation of 2-amino-6-chloropurine 61 with 2,3,5 tri-O-benzyl-arabifuranyl 283β (see Section 6.2.4 for preparation) followed by methoxy displacement of the chlorine at position 6 of the purine base furnished benzylated intermediate 285.600,601 Debenzylation by catalytic transfer hydrogenation, using ammonium formate as the in situ hydrogen source, afforded nelarabine 13 on a 30-g-scale at 99.8% purity after HPLC.
Scheme 52. Synthesis of Nelarabine 13a.

aReagents and conditions: (a) NaH, MeCN; (b) MeONa, MeOH; (c) Pd/C, HCOONH4, MeOH, reflux, 90 min, 91%.
6.2.3. Fludarabine and Fludarabine-5′-monophosphate Biologies.
Arabinofuranosyladenine (ara-A; vidarabine) has been around since the 1960s and was originally studied for its antiviral effects.602 The agent was also tested for anticancer effects, but showed limited success due to its rapid degradation by adenosine deaminase. In order to avoid adenosine deaminase-mediated deactivation, a fluorine was installed at the 2-position of the purine base of ara-A, producing fludarabine 291.603 This agent, however, had limited solubility and difficulties with its formulation. Therefore, the fludarabine-5′-monophosphate prodrug 7 (Fludara or fludarabine phosphate) was subsequently explored in clinical trials, which led to its FDA approval in 1991. Fludarabine-5′-monophosphate 7 is a commonly used drug against chronic lymphoid leukemia and hairy cell leukemia.538
The negatively charged fludarabine-5′-monophosphate 7 is dephosphorylated in the plasma by ecto-5′-nucleotidase, and fludarabine 291 is transported into cells by nucleoside transporters hENT1, hENT2, and hCNT3.429,592 Although fludarabine-5′-monophosphate is adenosine deaminase resistant,604 the drug is susceptible to glycosidic bond cleavage, which results in the formation of 2-fluoroadenine, a precursor to the toxic 2-fluoroadenosine-5′-triphosphate (see section 6.1.7).605 After entering the cell, fludarabine is initially phosphorylated by cellular 2′-deoxycytidine kinase, although it is a relatively poor substrate for the enzyme.606,607 Deoxyguanosine kinase has also been reported to phosphorylate fludarabine.608 Fludarabine-5′-monophosphate 7 is then phosphorylated to the fludarabine-5′-diphosphate and fludarabine-5′-triphosphate by adenylate kinase and nucleoside diphosphate kinase, respectively, in the cell.
Fludarabine-5′-triphosphate is the active cytotoxic agent. It is a ribonucleotide reductase inhibitor, which leads to a decrease in cellular dNTP concentrations, and therefore negatively influences DNA synthesis. As the concentrations of dNTP diminish during cell division, this results in more fludarabine-5′-triphosphate being incorporated into the host DNA (self-potentiation).572,592 Furthermore, the incorporated fludarabine-5′-monophosphate prevents further DNA chain elongation by restricting the addition of more dNTPs, leading to effective chain termination.603 Moreover, fludarabine-5′-triphosphate can inhibit DNA polymerase, DNA primase, and DNA ligase, causing DNA strand breaks, which may lead to apoptosis.592,609 Fludarabine-5′-triphosphate was also shown to be incorporated into RNA and can inhibit RNA synthesis.312,610 In addition, fludarabine 291 exposure can also suppress the excision repair enzyme ERCC1 and led to cytotoxic synergy with SJG-136 (DNA minor groove binding agent) in chronic lymphocytic leukemia cells.611 However, the side effects of fludarabine-5′-monophosphate 7 treatment are becoming an increasing concern, because secondary myelodysplastic syndrome and leukemia have been reported in at least 3% of persons treated with fludarabine-5′-monophosphate 7 (Fludara).609
Several different mechanisms have been reported that lead to fludarabine resistance in vivo. Changes in the expressions of miRNA-29a, miRNA-181a, and miRNA-221 have been reported.612 The amplification of MYC gene and its transcript level miRNAs can promote fludarabine resistance.613 Differential overexpression of sulfatase was correlated to fludarabine resistance.613
Fludarabine-5′-monophosphate 7, cyclophosphamide (DNA-alkylating agent), and rituximab (monoclonal antibody against B cell protein CD20) combination therapy is generally considered the standard treatment for younger fit patients with chronic lymphocytic leukemia.614 A combination treatment of fludarabine-5′-monophosphate 7, bendamustine (DNA-alkylating agent), and rituximab for chronic lymphocytic leukemia is being investigated, in addition to the fludarabine-5′-monophosphate 7, clofarabine 12, SAHA (vorinostat, histone deacetylase inhibitor), and busulfan (antineoplastic alkylating agent) combination for acute leukemia (ClinicalTrials.gov identifiers: NCT01096992–active status–not recruiting patients yet, and NCT02083250–recruiting patients).
6.2.4. Fludarabine and Fludarabine-5′-Monophosphate Syntheses.
In 1969, Montgomery and Hewson reported the discovery synthesis of fludarabine 291 (Scheme 53).615 Key chlorosugar 283 was prepared from d-ribose by first cyclization in methanolic sulfuric acid to methyl-β-d-ribofuranoside, followed by 3′,5′-O-benzylation, and subsequent hydrolysis, p-nitrobenzoylation, and chlorination of the anomeric position.616,617 The glycosylation step between chlorosugar 283 and 2,6-dichloropurine 286 gave a column-chromatography-separable mixture of α/β diastereomers, with the desired β-anomer 287 formed in only 11%. Treatment with sodium azide followed by catalytic reduction furnished the diaminopurine intermediate 289. Due to solubility issues of 289, a mixed solvent system was employed for the modified Balz–Schiemann reaction, which introduced the N2-fluorine. Debenzylation concluded the synthesis, producing fludarabine 291.
Scheme 53. Synthesis of Fludarabine 291 and Fludarabine-5′-monophosphate 7a.

aReagents and conditions: (a) Hg(CN)2, CaSO4, MeNO2, reflux, 3 h, 11%; (b) NaN3, EtOH, H2O, 1 h, 98%; (c) Pd/C, H2, EtOH, rt, 6 h, 75%; (d) HBF4, NaNO2, CHCl3, −10 °C, 40 min, 36%; (e) Na, NH3, 34%; (f) POCl3, PO(OEt)3, 0 °C, 3.5 h, 96%.
Montgomery later reported a gram-scale preparation of fludarabine 291, which utilized the same basic route, although the glycosylation reaction was performed using a silylated or acylated derivative of 2,6-diaminopurine.618 Soon thereafter, he reported a gram-scale preparation of fludarabine-5′-monophosphate 7 by treatment of fludarabine 291 with phosphoryl chloride and triethyl phosphate (Scheme 53).619
Blumbergs et al. developed a kilogram-scale preparation of fludarabine-5′-monophosphate 7, which followed a similar route used by Montgomery (Scheme 54).620,621 Yields were improved, allowing the synthesis of the drug in five steps from chlorosugar 283α (12% yield). Improvements included the use of 2,6-di(methoxyacetamido)purine 293 during the glycosylation, which increased base solubility and greatly reduced the volume of solvent (ethylene dichloride) required for the reaction. Furthermore, chlorosugar 283α, which was produced almost exclusively as the α-anomer by treating the p-nitrophenol intermediate 294 with hydrogen chloride in dichloromethane,617 was used in the glycosylation step, which allowed for higher yield of the desired β-anomer of 295.
Scheme 54. Synthesis of Fludarabine 291 and Fludarabine-5′-monophosphate 7a.

aReagents and conditions: (a) methoxyacetic anhydride, pyr, 88 °C, 1 h, then methyl ethyl ketone, overnight, 90%; (b) HCl (g), DCM, 4–6 °C, 2 h, quantitative; (c) DIPEA, DCE, reflux, overnight, quantitative; (d) NaOMe, MeOH, 68%; (e) POCl3, TMP, 0 °C, 18 h, 56%.
Another highlight of the synthesis involved the use of hydrochloric acid in the debenzylation step to avoid the partial-defluorination problem associated with Montgomery’s catalytic hydrogenation, and to simplify the purification procedure. Furthermore, the phosphorylation step yield was significantly increased when using scrupulously dried nucleoside as the starting material. Recently, another publication effected the debenzylation by transfer hydrogenation, using ammonia formate as an in situ hydrogen donor, to produce kilograms of fludarabine 291 at 99.8% purity in 90–95% yield.622
Farina et al. prepared fludarabine 291 through enzymatic transglycosylation involving Enterobacter aerogenes, 2-fluoroade-nine, and 9-β-d-arabinofuranosyl-uracil (Scheme 55).623 The crude product was treated with acetic anhydride, forming 2′,3′,5′-tri-O-acetyl-9-β-d-arabinofuranosyl-2-fluoroadenine, which was hydrolyzed and recrystallized to the pure fludarabine 291. Subsequent phosphorylation afforded fludarabine-5′-monophosphate 7.
Scheme 55. Synthesis of Fludarabine 291 and Fludarabine-5′-monophosphate 7a.
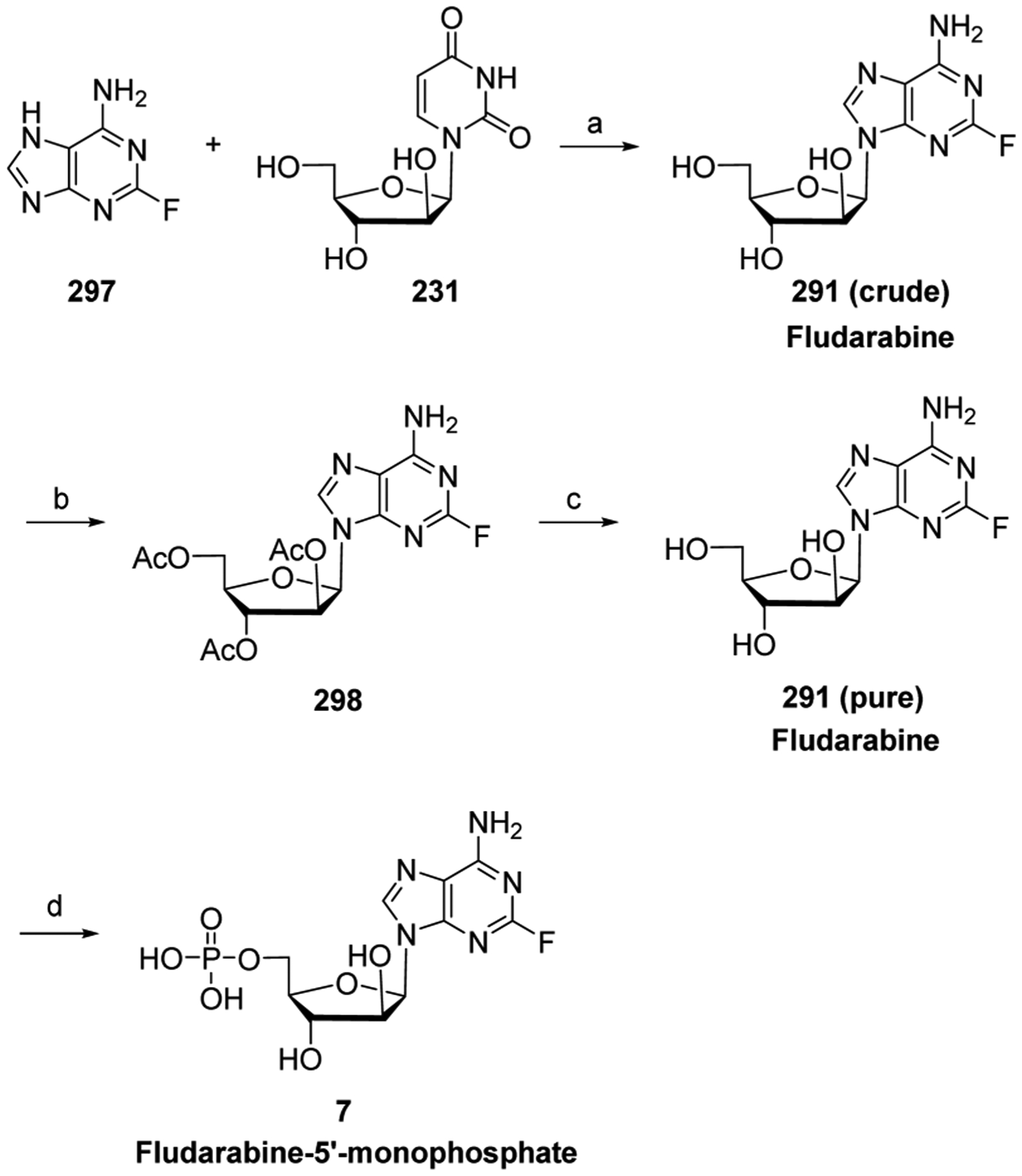
aReagents and conditions: (a) Enterobacter aerogenes, K2HPO4, H2O, 60 °C, 24 h, 35%; (b) Ac2O, 95 °C, 9 h, 87%; (c) NH4OH, H2O, MeOH, rt, 19 h, 74%.
6.3. Base Modified Purine Nucleosides
6.3.1. 8-Chloro-adenosine and Tocladesine Biologies.
In the 1980s, 8-chloro-adenosine 28 and tocladesine (50; 8-chloroadenosine 3′,5′-monophosphate; 8-Cl-cAMP) were reported to have potent anticancer activities.624–627 Tocladesine 50 is converted extracellularly to 8-chloro-adenosine 28,628 which is then transported by hCNT1 and hCNT2 into the cell.629 Once in the cell, this agent is converted via adenosine kinase to 8-chloro-adenosine-5′-monophosphate, and ultimately to the active metabolite 8-chloro-adenosine-5′-triphosphate by cellular kinases. 8-Chloro-adenosine-5′-triphosphate accumulates to almost millimolar concentration in the cell.630 8-Chloroadenosine is a poor substrate for adenosine deaminase, and therefore, deamination of the agent is minimal.603
The 8-chloro-adenosine-5′-triphosphate affects cellular energetics by decreasing the levels of intracellular ATP, leading to RNA synthesis inhibition. Furthermore, RNA polymerase II incorporates 8-chloro-adenosine-5′-triphosphate into mRNA, and inhibits polyadenylation of full-length mRNA transcripts.631,632 A decrease in these short-lived mRNA transcripts can also lead to a reduction in protein levels, which promote growth and survival of tumor cells. 8-Chloro-adenosine 28 exposure has been found to deplete the cyclin E level in breast cancer cell lines,624 decrease the level of the receptor tyrosine kinase MET in multiple myeloma cell lines,633 and depress the level of Mcl-1 in chronic lymphocytic leukemia cells.630 Collectively, all of these mechanisms promote apoptosis.
Although many effects of 8-chloro-adenosine 28 are RNA-directed, 8-chloro-adenosine-5′-triphosphate has also been found to inhibit topoisomerase II and induce DNA double-stranded breaks in human myelocytic leukemia K562 cells.634 8-Chloro-adenosine 28 exposure also inhibits the rate of DNA synthesis and decreases dATP pools in mantle cell lymphoma cell lines.635 Furthermore, decrease in cancer cell proliferation by 8-chloro-adenosine 28 exposure has been linked to AMP-activated protein kinase and p38 mitogen-activated protein kinase inhibitions in vitro.636,637
Glucose-6-phosphate dehydrogenase has been linked to 8-chloro-adenosine 28 resistance in multiple myeloma and chronic lymphocytic leukemia cell lines.638 Also, a report indicated that the agent influences glucose consumption, leading to autophagy activation in multiple myeloma cells.639
In 2008, the M.D. Anderson Cancer Center sponsored a Phase I clinical trial of a 8-chloro-adenosine 28 therapy to study dosing tolerance in subjects with chronic lymphocytic leukemia (ClinicalTrials.gov identifier: NCT00714103–active status–not recruiting patients yet). In June 2015, the City of Hope Medical Center began a Phase I/II clinical trial evaluating 8-chloro-adenosine 28 in treating patients with relapsed or refractory acute myeloid leukemia (ClinicalTrials.gov identifier: NCT00004902–currently recruiting participants). Tocladesine 50 was evaluated in two clinical trials. In 2000, it was evaluated in a Phase II clinical trial for the treatment of persons with recurrent or refractory multiple myeloma (ClinicalTrials.gov identifier: NCT00004902–completed status). In 2001 the agent was studied in a Phase I trial for the treatment of progressive metastatic colorectal cancer (ClinicalTrials.gov identifier NCT00021268–unknown status–greater than 2 years since updated). No trial results have been reported.
6.3.2. 8-Chloroadenosine and Tocladesine Syntheses.
Synthesis of 8-chloroadenosine 28 has been achieved in one step from adenosine 240 by using either tetrabutylammonium iodotetrachloride as the chlorinating reagent640 or m-CPBA and acid (HCl or acetyl chloride) in aprotic solvents (Scheme 56).641,642 Additionally, the synthesis of 8-chloroadenosine 28 has been achieved by direct coupling of 8-chloroadenosine 28 with 2,3,5-tri-O-benzoyl-d-ribofuranosyl bromide. However, the major product of the reaction is the N3 glycosylated product and not the desired N9 product.643 Tocladesine 50 can be prepared by treatment of 8-chloroadenosine 28 with phosphoryl chloride in triethyl phosphate.642
Scheme 56. Synthesis of 8-Chloroadenosine 28 and Tocladesine 50a.
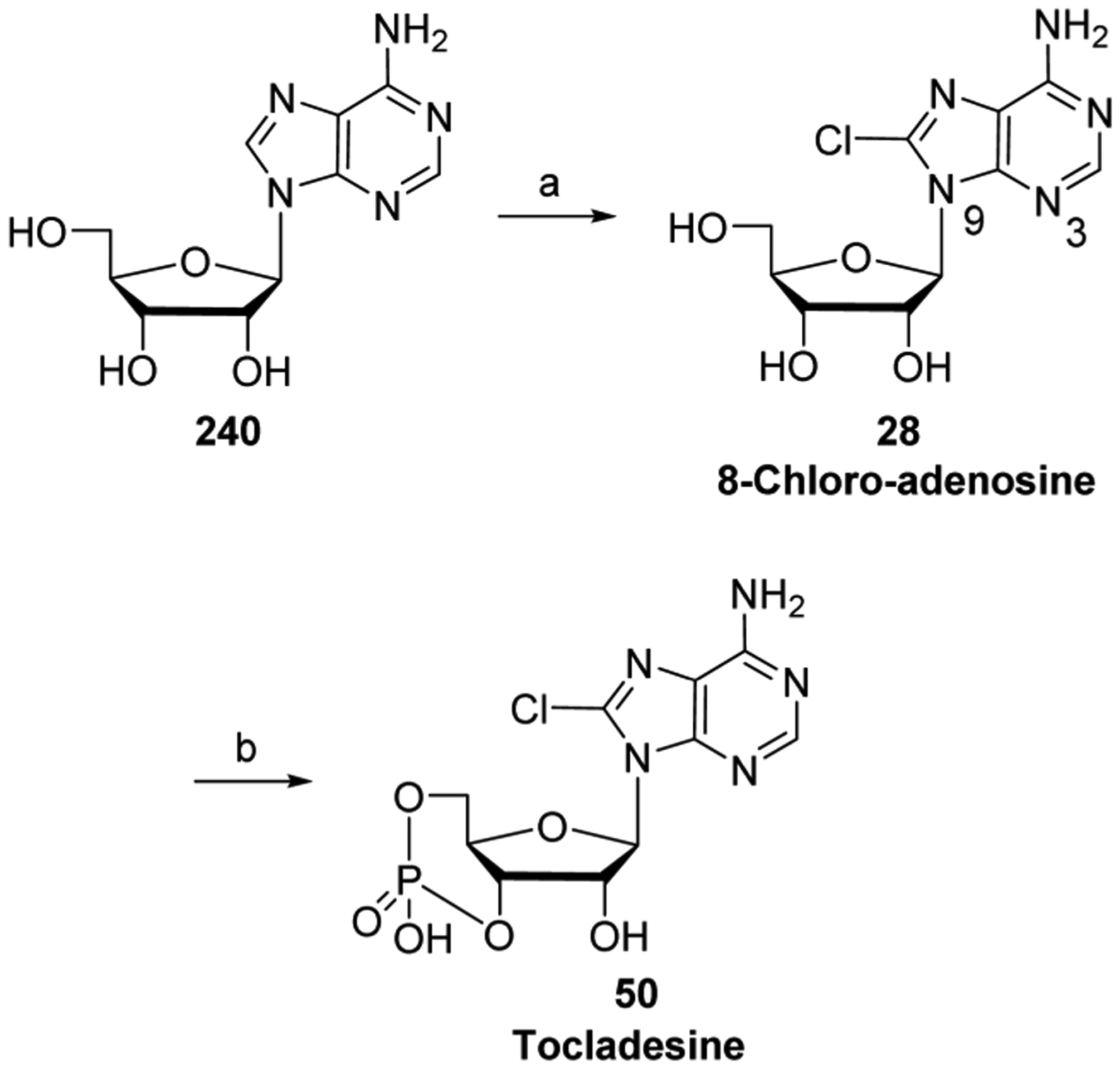
aReagents and conditions: (a) AcCl, m-CPBA, DMF, rt, 20 min, 40%; (b) POCl3, PO(OEt)3, 0 °C, 6 h, then aq NaOH, 2 h, 23%.
6.3.3. Forodesine Hydrochloride Biology.
Forodesine hydrochloride (36, (1S)-1-(9-deazaguanin-9-yl)-1,4-dideoxy-1–4-imino-d-ribitol; BCX-1777; Immucillin H) was developed as a purine nucleoside phosphorylase inhibitor (see Section 6.2.1). Transition state analysis of the purine nucleoside phosphorylasecatalyzed phosphorolysis of inosine was used as a model system in the design of the agent (Figure 16).33,34 The protonated nitrogen of the iminoribitol ring of forodesine exhibits a similar charge distribution to the ribosyl moiety oxocarbenium ion of the transition state. Furthermore, the 9-deazapurine of forodesine can be protonated at N7, which provides additional interaction with a residue of the purine nucleoside phosphorylase active site.644 In addition, instead of a natural glycosidic bond, forodesine has a carbon–carbon bond which is not readily cleaved by purine nucleoside phosphorylase.630
Figure 16.

Transition state model for the phosphorolysis of inosine by purine nucleoside phosphorylase and the protonated form of forodesine.
Forodesine was shown to be a potent purine nucleoside phosphorylase inhibitor. Complete inhibition of the homotrimeric complex of the enzyme at a concentration as low as 72 pM has been reported, with the agent exhibiting a high equilibrium binding constant for and a very slow dissociation rate from the enzyme.337,630 Inhibition occurs at a 1:1 molar ratio of inhibitor to purine nucleoside phosphorylase homotrimeric enzyme complex, with just one of the three substrate sites needing to be occupied by the agent to block enzyme activity.34
Forodesine hydrochloride 36 is administered by intravenous infusion. Like pentostatin 6 (see Section 6.1.5), the agent is active without metabolism, meaning it does not require phosphorylation. It blocks purine nucleoside phosphorylase, causing a high plasma concentration of deoxyguanosine, and it is not incorporated into DNA.645 Deoxyguanosine is further converted to dGMP, dGDP, and dGTP by cellular kinases. High cellular levels of dGDP inhibit the ribonucleotide reductase complex and lead to imbalances of the nucleotide pool concentrations, resulting in p53-based apoptosis.591 Because of this, forodesine works best in malignant cells containing high deoxyguanosine activity.646
Forodesine hydrochloride 36 has undergone clinical trials for the treatment of T-cell malignancies, including T-cell acute lymphocytic leukemia.35,647 Additionally, the agent is being examined for treatment of chronic lymphocytic leukemia B cells, which may require an additional nucleoside treatment for effectiveness.35,648 Forodesine hydrochloride 36 did not inhibit the growth of colon cancer cell lines or normal human nonstimulated T cells, indicating some specificity of the drug toward normal versus malignant tissues.33 In 2007 a phase IIb clinical trial of forodesine hydrochloride 36 in patients with leukemia/lymphoma was terminated due to manufacturing issues (ClinicalTrials.gov identifier: NCT00419081–terminated), indicating a need to improve the synthesis process in order to produce sufficient quantities of the agent for further clinical trials. In January 2013, Mundipharma began a Phase I/II clinical trial evaluating forodesine hydrochloride 36 in recurrent/refractory peripheral T cell lymphoma subjects in Japan (ClinicalTrials.gov identifier NCT01776411–active status–not recruiting patients).
6.3.4. Forodesine Synthesis.
The discovery synthesis of forodesine involved formation of the key iminoribitol intermediate 306 (see Scheme 57) followed by a 16-step linear construction of the base, producing only milligram quantities of the compound.649 Therefore, a different route was designed, which could produce the required kilogram quantities of the agent needed for drug development. This new synthesis involved the coupling of imine 307 and brominated 9-deazahypoxanthine 313 (Scheme 57). One metric ton of 306 was prepared from d-ribose 213 by first bromine oxidation, protection, lactone epimerization,650 and Fleet chemistry (five steps).651 The same quantity of 9-deazahypoxanthine 310 was produced by condensing (ethoxymethylene)cyanoacetate 308 with diethyl aminomalonate followed by cyclization and acid-mediated decarboxylation.652 Three more steps were required to produce the bromo hypoxanthine derivative 312: chlorination, protection and methoxy displacement, and finally bromination. At this stage, the condensation of lithiated heterocycle 313 (generated in-situ from 312) with imine 307 afforded the β-anomer of 314 exclusively, due to steric hindrance of the α-face by the isopropylidene protecting group. In order to avoid a hard-to-remove tarry byproduct, 314 was not converted directly to the product, but instead passed through a process of protection and sequential deprotection, eventually furnishing pure forodesine hydrochloride 36.653
Scheme 57. Synthesis of Forodesine Hydrochloride 36a.

aReagents and conditions: (a) Br2, K2CO3, H2O, 5 °C to rt, overnight; (b) concd H2SO4, Me2CO, reflux, 4 h, then 30 °C, 8 h, 55% (over two steps); (c) MsCl, Et3N, DCM, −20 °C, 1 h, then rt, 8 h; (d) aq KOH, <30 °C, 4 h, 59% (over two steps); (e) TBDMSCl, imidazole, DMF, 20 °C, 4 h, 91%; (f) LiBH4, THF, −30 °C to rt, 24 h, 75%; (g) (MeSO2)2O, pyr, 0 °C to rt, 18 h, 88%; (h) NaN3, DMF, 100 °C, 2 h, 42%; (i) H2, Pd, NaOAc, dioxane, 20 °C, 72 h, 94%; (j) NCS, pentane, rt, 1 h, then lithium tetramethylpiperidide (LiTMP), THF, −78 °C, 85%; (k) H2NCH(CO2C2H5)2, NaOMe, MeOH, reflux, 4 h, 68%; (l) formamidine acetate, C2H5OH, reflux, 27 h, 61%; (m) aq KOH, reflux, 40 h, 74%; (n) POCl3, reflux, 2 h; (o) BOMCl, NaH, THF, 0 °C to rt, 1 h, then NaH, MeOH, 1 h; (p) NBS, DCM, 52% (over three steps); (q) n-BuLi, −78 °C; (r) 307, −78 °C to 0 °C; (s) (Boc)2O, DCM, rt, 1 h, 76% (over three steps); (t) H2, Pd/C, 18 h, then NH4OH, H2O, 1 h; (u) concd HCl, MeOH, reflux, 1.5 h, 85% (over two steps).
In order to avoid some of the cumbersome manipulations from the above-mentioned route, a different approach to forodesine hydrochloride 36 explored a novel route to prepare the aza-sugar core (Scheme 58).654 Lactam 317, which is prepared by esterification and reduction of l-pyroglutamic acid, was transformed into unsaturated bicycle 318 by treatment with benzaldehyde and catalytic acid followed by treatment with Meyer’s reagent (methyl phenolsulfonate).655 Osmium tetr-oxide-catalyzed dihydroxylation of compound 318 gave the desired diol stereochemistry, and subsequent acetal protection furnished the intermediate 319. The coupling of lithiated 9-deazahypoxanthine to lactam 319 led to the ring-opened product 320. Treatment of the intermediate 320 with BBr3 gave the cyclized product 321, which was then reduced with BH3·Me2S and deprotected under acidic condition to give forodesine hydrochloride 36 as a 4:1 β:α mixture.
Scheme 58. Synthesis of Forodesine Hydrochloride 36a.
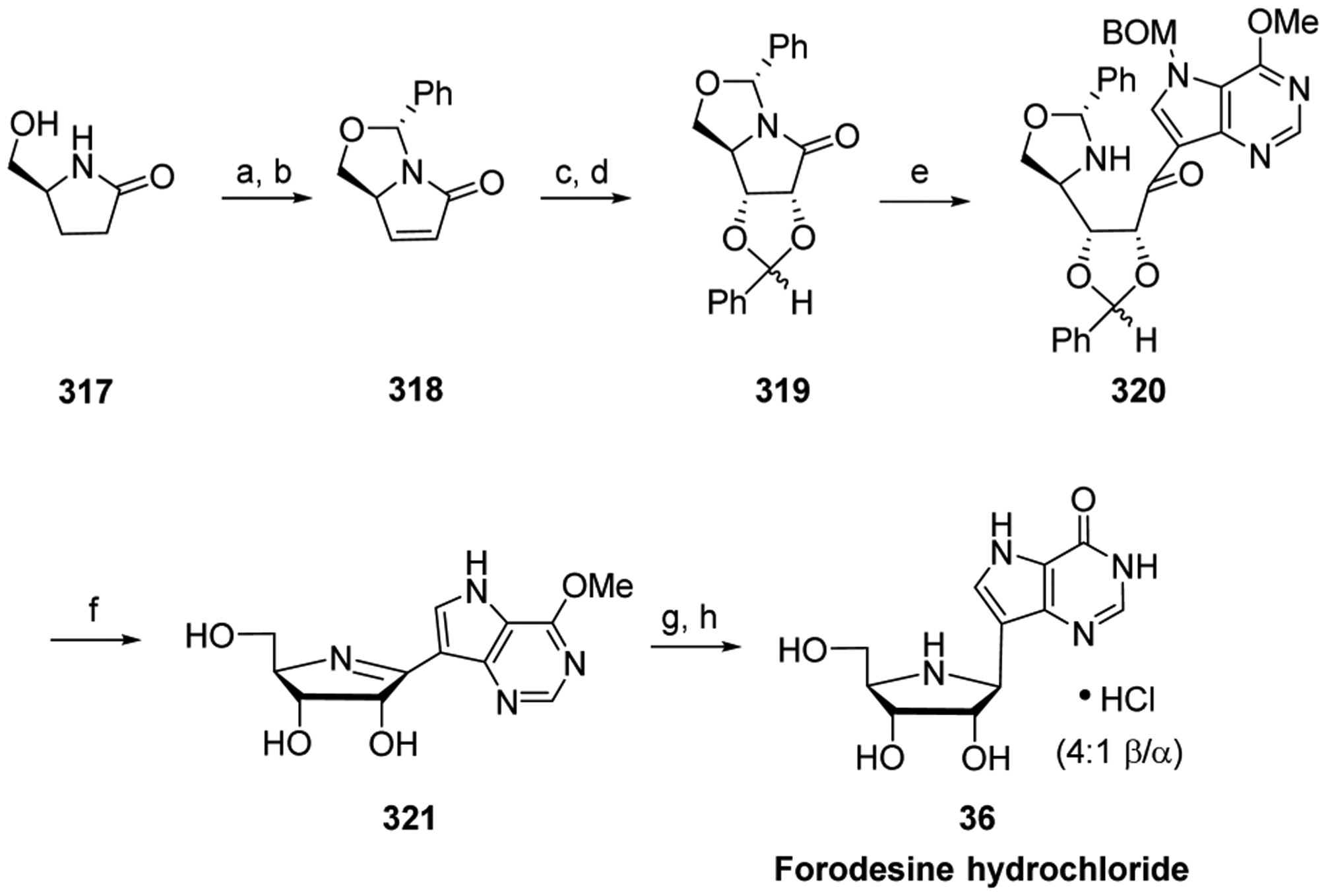
aReagents and conditions: (a) PhCHO, TsOH; (b) PhSO2Me, 59%; (c) OsO4, Me CO; (d) DMP/H+ 2, 63%; (e) lithiated 9-deazahypoxanthine, anisole, Et2O, −20 °C, 55%; (f) BBr3, DCM; (g) BH3·Me2S, DMF; (h) MeOH/H+, 90%.
In 2016, a 50-g-scale synthesis of forodesine hydrochloride 36 was reported.656 Differences from the original preparation (Scheme 57) include coupling of imine 307 with lithiated heterocycle 313 under milder condition, and the use of concentrated HCl instead of hydrogen for BOM removal in 315.
7. ENZYME INHIBITOR AND CELL SURFACE RECEPTOR ANTAGONIST NUCLEOSIDE ANALOGS
7.1. Triciribine and Triciribine-5′-Monophosphate Biologies
In 1971, Schram and Townsend synthesized triciribine 30, a tricyclic derivative of a purine nucleoside as a potential anticancer drug,657 which has also been studied as an antiretroviral agent against HIV-1 and HIV-2.658 Triciribine 30, however, is poorly soluble, and this suboptimal property led to the development of the highly soluble prodrug triciribine-5′-monophosphate 33.
Interestingly, triciribine 30 does not work by blocking host DNA or viral polymerases. It inhibits the phosphorylation of Akt1, Akt2, and Akt3,659 and appears to influence the PI3K/Akt/mTOR cell survival pathway. The PI3K/Akt/mTOR pathway regulates a variety of cellular pathways, including cell proliferation and survival.660,661 Akt is dysregulated in a variety of malignancies.662 Initially, triciribine 30 was reported to inhibit de novo purine nucleotide and DNA synthesis in cell lines.425,663 However, this would not explain the antiretroviral activity in HIV-1 and HIV-2 infected cell lines, because the reverse transcription step for proviral DNA synthesis of the viral life cycle was completed. Moreover, viral replication was no longer influenced by cellular dNTP concentrations or nucleoside reverse transcriptase inhibitors, which are chain terminators. Based on current data, the antiviral and anticancer activities of triciribine metabolite appear to be associated with Akt inhibition (Figure 17).664 In addition to breast cancer, triciribine 30 may be effective when used as a combination therapy in bladder cancer patients prescreened for alterations in the Akt pathway.665,666
Figure 17.

Proposed biological mechanisms of actions of triciribine 30 as an Akt inhibitor (see Section 7.1, Triciribine Biology); MLN4924 44 as a Cullins inhibitor (see Section 7.3, MLN4924 Biology); and acadesine 41 as an AMPK activator (see Section 8.5, Acadesine Biology).
Triciribine-5′-monophosphate 33 is currently being evaluated in solid tumors, such as breast cancer.662,667,668 A Phase I/II study is currently underway in patients with metastatic and locally advanced breast cancer that uses triciribine-5′-monophosphate monohydrate with paclitaxel (antimitotic taxane), which is followed by dose-dense doxorubicin (DNA intercalating agent) and the DNA alkylating-agent cyclophosphamide (ClinicalTrials.gov identifier: NCT01697293–recruiting patients). The H. Lee Moffitt Cancer Center and Research Institute recently began a Phase I/II clinical trial for the treatment of ovarian cancer with a combination of triciribine 30 and the DNA alkylating agent carboplatin (ClinicalTrials.gov identifier: NCT01690468–recruiting patients).
7.2. Triciribine and Triciribine-5′-Monophosphate Syntheses
Triciribine 30657 and triciribine-5′-monophosphate 33669 were originally prepared from the intermediate 322 by Townsend and Schram (Scheme 59). Aryl chloride displacement of 322 followed by tricycle formation led to triciribine 30, and subsequent phosphorylation produced triciribine-5′-monophosphate 33.
Scheme 59. Early Synthesis of Triciribine 30 and Triciribine-5′-monophosphate 33a.

aReagents and conditions: (a) CH3NHNH2, EtOH, rt, 3 min; (b) H2O, reflux, 16 h; (c) POCl3, PO(OMe)3, 0 °C, 16 h, 21%;
More recently Shen et al. developed a new approach to prepare triciribine 30 from commercially available intermediates in only 4 steps (Scheme 60).659 Thus, reaction of 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose with 6-chloro-7-iodo-7-deazapurine 324 under classical glycosylation conditions gave intermediate 325, which was then selectively cyanated by treatment with tributyltin cyanide in the presence of catalytic Pd(0) to give compound 326. Aryl chloride displacement with methyl hydrazine followed by deprotection and ring closure with sodium methoxide in refluxing methanol afforded triciribine 30 in a 37% overall yield.
Scheme 60. Synthesis of Triciribine 30a.
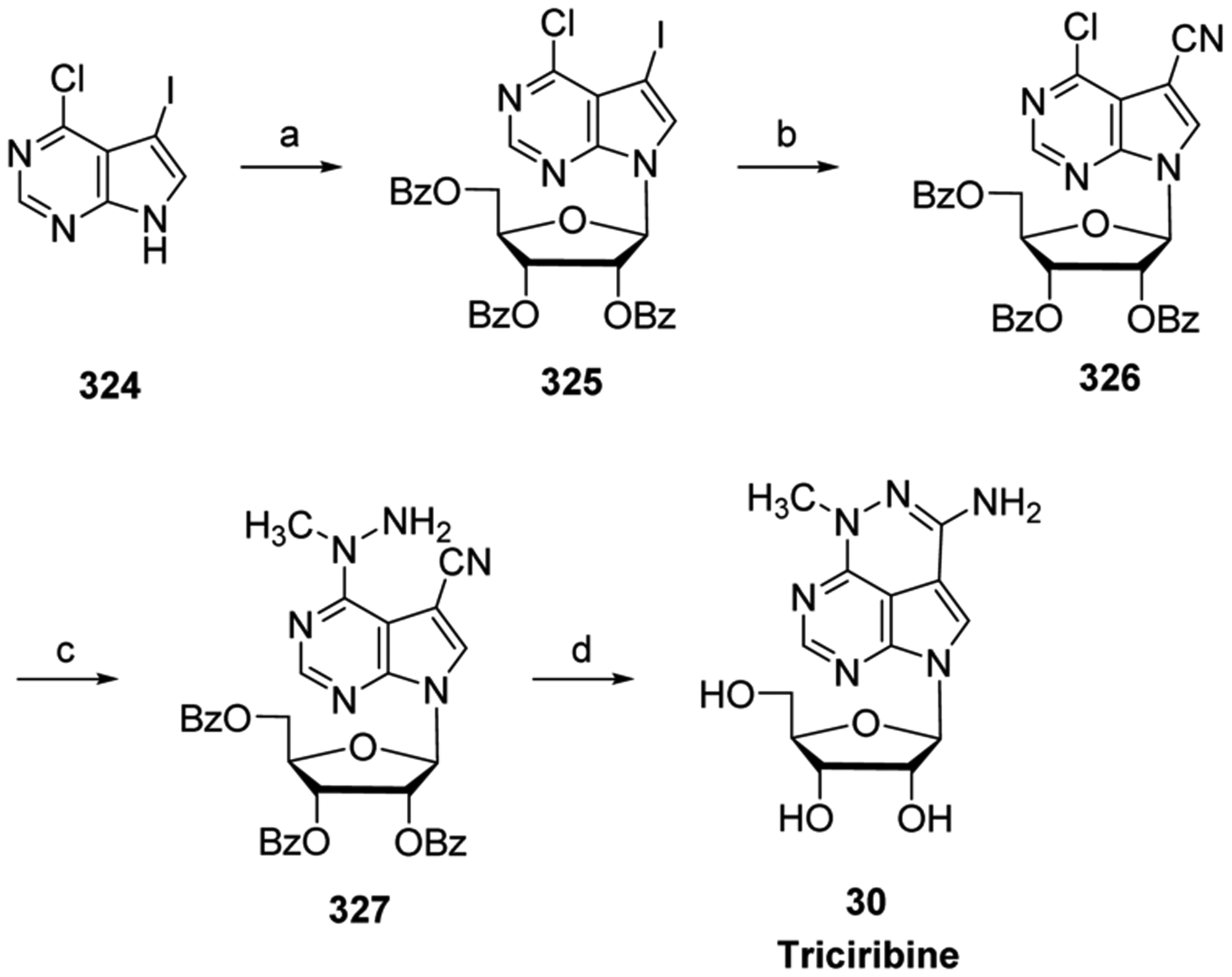
aReagents and conditions: (a) BSA, 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose, TMSOTf, CH3CN, 80 °C, 80%; (b) Bu3SnCN, Pd(PPh3)4, DMF, 95 °C, 6 h, 72%; (c) H2NNHCH3, C2H5OH, CHCl3, rt, 3.5 h, 92%; (d) NaOMe, MeOH, rt, 1 h, then reflux, 18 h, 74%.
7.3. MLN4924 Biology
Whereas the function of most FDA-approved nucleoside analogs involves their incorporation into DNA or RNA after phosphorylation, MLN4924 (44; Pevonedistat; TAK-924) suppresses tumor development mainly through blocking the proteasomal degradation pathway. MLN4924 44 is a structural analog of adenosine-5′-monophosphate. MLN4924 44 tightly binds to NEDD8-activating enzyme (NAE), which is an ubiquitin-like protein involved in the ubiquitin-proteasome degradation system (Figure 18).37 An initial high throughput screening of adenosine-5′-monophosphate analogs identified N6-benzyl adenosine as an inhibitor of NAE, and further structural elaboration lead to MLN4924 44.37
Figure 18.
Structures of MLN4924 44 and adenosine-5′-monophosphate.
Functionally, MLN4924 44 was shown to selectively inhibit NAE, and did not inhibit transfer RNA synthetases or other ATP-consuming enzymes.37 NAE is the enzyme that initiates the important pathway to maintain protein homeostasis, by forming a tight binding adduct with NEDD8.670,671 This is a multistep enzymatic process in which Cullin-RING E3 ligases (CRLs) add the NEDD8 polypeptide onto target proteins.672 MLN4924 44 was shown to block polypeptide addition to target proteins. Furthermore, in cancer cells MLN4924 44 blocks the important homeostatic balance between protein synthesis and degradation, which is essential for cell survival, cell division, and regulating both innate and adaptive immune responses.673–675 As a result, CRLs are deactivated and consequently multiple known substrates of CRLs accumulate inside the target cell (e.g., DEPTOR and HIF-1α), leading to cell cycle arrest and apoptosis of the cancer cells (Figure 17).37,670,676
Cells in the S phase are more sensitive to exposure to MLN4924 44, a unique feature of the agent.37 A recent study using the A375 human melanoma-derived cell line indicated that MLN4924 44 induced cell cycle arrest might largely be triggered by a p21-mediated intra-S phase checkpoint.676 Parallel to this study, an in vivo study with diffuse large B cell lymphoma models indicated that MLN4924 44 might also suppress tumor growth via its inhibition of the NF-κB pathway.677 Studies in mice demonstrated good toleration for MLN4924 44, as well as potent antiproliferative activity against human tumor cell xenografts derived from human colorectal and lung carcinomas.37 However, HLC116 cells, a human colorectal carcinoma cell line, exposed to MLN4924 44 developed a point mutation at A171T in the UBS3 subunit of NAE, which greatly decreased sensitivity to the drug.678 Another study attempted to address a similar issue and found that both HIF1-REDD1-TSC1-mTORC1 and DEPTOR might play important roles in MLN4924-induced autophagy, which is a survival strategy in cancer cells in response to chemotherapy-mediated DNA damage.679 This study suggested that cells exposed to an autophagy inhibitor may have a marked improvement in MLN4924 44 response.679
MLN4924 hydrochloride affords better solubility than the parent compound, MLN4924 44. However, clinical studies indicate that MLN4924 44 pevonedistat is being administered. Currently the safety and efficacy of MLN4924 44 therapy are being assessed in several Phase I clinical studies involving solid tumors, melanoma, acute myeloid leukemia, refractory or relapsed/refractory multiply myeloma, non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, and cisplatin resistant ovarian cancers.680,681 Current studies also include investigating fluconazole and itraconazole (antifungals often administered with cancer chemotherapeutics) effects on MLN4924 44 safety and efficacy in solid tumors. Phase I trials using MLN4924 44 in combination therapy with docetaxel (antimitotic taxane), gemcitabine hydrochloride 9, or a combination of carboplatin (DNA alkylating agent) and paclitaxel (antimitotic taxane) in solid tumors are underway (ClinicalTrials.gov identifiers: NCT02122770–recruiting patients, and NCT01862328–active status–not recruiting patients).
7.4. MLN4924 Synthesis
A preparation of MLN4924 44 is summarized in Scheme 61.682,683 Starting from diene 217, ring closing metathesis led to the tertiary β-alcohol product 328, which had the required stereochemistry that allowed oxidative rearrangement to cyclopentenone 329.682 Stereoselective catalytic hydrogenation, due to hydrogen incorporation from the convex face of the intermediate, followed by Luche reduction with CeCl3, provided 330 as a single diastereomer. Regioselective cleavage of the acetonide group of 330 was followed by treatment with SOCl2 and RuCl3/NaIO4 to furnish the cyclic sulfate 331. Subsequent condensation with N6-indanyl-7-deazaadenine in the presence of NaH afforded the nucleoside analogue 332. Standard radical-mediated deoxygenation and O-TBDPS deprotection provided the intermediate 333, which was then reacted with chlorosulfonamide and trifluoroacetic acid to furnish MLN4924 44.683
Scheme 61. Synthesis of MLN4924 44a.

aReagents and conditions: (a) Grubbs’ catalyst (2nd generation), DCM, rt, 2 days, 95%; (b) PDC, DMF, rt, 2 days, 84%; (c) H2, Pd/C, MeOH, rt, overnight, 100%; (d) NaBH4, CeCl3·H2O, MeOH, 0 °C to rt, 0.5 h, 98%; (e) Me3Al, DCM, 0 °C to rt, 2 days, 62%; (f) SOCl2, Et3N, 0 °C, 10 min, then RuCl3·3H2O, NaIO4, CCl4, CH3CN, H2O, 87%; (g) N6-indanyl-7-deazaadenine, NaH, 18-crown-6, THF, 80 °C, overnight, then concd HCl, 80 °C, 2 h, 65%; (h) PhOC(S)Cl, DMAP, DCM, rt, overnight, then Bu3SnH, AIBN, PhMe, 110 °C, 1 h, 82%; (i) HF-pyridine, THF, pyr, rt, 1 h, 99%; (j) NH2SO2Cl, Et3N, CH3CN, 0 °C to rt, 1 h, 92%; (k) TFA, rt, 2 h, 90%.
In 2007, Langston et al. published an alternative synthesis of MLN4924 44 (Scheme 62).684 The starting diol 334,685 formed in a four step sequence from cyclopentadiene and glycolic acid involving a hetero-Diels–Alder and oxidation/reduction reactions, was oxidized and treated with p-anisaldehyde to produce the cyclic epoxide 335. Condensation with pyrrolopyrimidine 337 was followed by deoxygenation, deprotection, selective 5′-OH silylation, and 3′-OH acetylation. Finally, desilylation, sulfamate ester formation, and deacetylation led to MLN4924 44.
Scheme 62. Synthesis of MLN4924 44a.
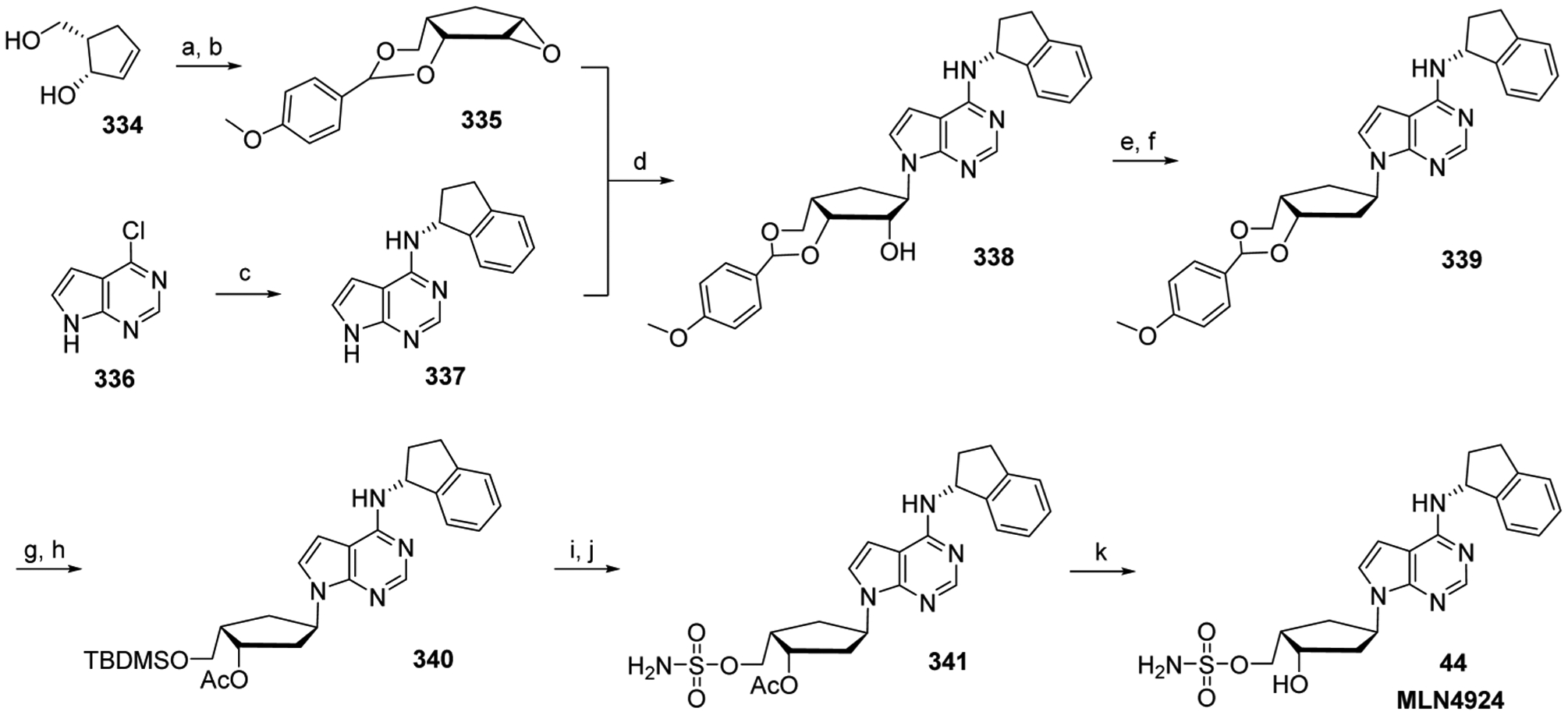
aReagents and conditions: (a) m-CPBA, DCM, 4 h, 76%; (b) 1-(dimethoxymethyl)-4-methoxybenzene, PPTS, rt, overnight, 78%; (c) (S)-(+)-1-aminoindan, DIPEA, 1-butanol, reflux, 60 h, 80%; (d) NaH, DMF, 110 °C, 2 h, 69%; (e) DMAP, PhOC(S)Cl, DCM, rt, 1 h, 99%; (f) AIBN, Bu3SnH, PhMe, reflux, 30 min, 79%; (g) AcOH, THF, H2O, rt, 60 h, 98%; (h) 1H-imidazole, DMAP, TBDMSCl, DMF, rt, 2 h, then DMAP, Ac2O, pyr, rt, overnight, 86%; (i) HF-pyridine, THF, pyr, rt, 1 h, 80%; (j) chlorosulfonamide, Et3N, MeCN, rt, 1 h, 94%; (k) NH3/MeOH, rt, 5 days, 90%.
In 2015, a 100-g-scale process synthesis of MLN4924 hydrochloride 350 was reported (Scheme 63).686 The key intermediate amino-diol 346 can be prepared from commercially available carboxylic acid 342 in four steps: bromolactonization, reductive ring opening, Boc deprotection, and hydrogenation. Subsequent coupling of 346 with acetaldehyde and (S)-(+)-1-aminoindane afforded diol 348. Selective sulfamoylation of diol 348 was carried out using a Burgess-type reagent 349, which was produced on a 100-kg-scale by reaction of chlorosulfonyl isocyanate with tert-butanol and subsequent treatment with DABCO. MLN4924 44 was then separated from the disulfamoylated byproduct by stepwise hydrolysis with increasing concentration of HCl, requiring no chromatography. Finally, salt formation and crystallization from ethanol furnished MLN4924 hydrochloride 350.
Scheme 63. Synthesis of MLN4924 Hydrochloride 350a.

aReagents and conditions: (a) Br2, py, DME, H2O, 0 °C, 68%; (b) LiBH4, THF, H2O, −5 °C; (c) 8.84 M HBr, i-PrOH, 55 °C; (d) H2, Pd/C, DIPEA, MeOH, 25 °C, 80% (over three steps); (e) 2-(4,6-dichloropyrimidin-5-yl)acetaldehyde, Et3N, i-PrOH, 75 °C, 78%; (f) (S)-(+)-1-aminoindane hydrochloride, DIPEA, 2-butanol, 130 °C, 81%; (g) 349, MeCN, 53 °C, 5 h, then HCl, 59%; (h) 1.25 M HCl, EtOH, crystallization, 93%.
7.5. EPZ-004777 and EPZ-5676 Biologies
Histone methyl transferases (HMT) use S-adenosyl-L-methionine as a cofactor to methylate histone amino acid side chains, leading to post-translational gene modification. An important cellular HMT is DOT1-like histone H3K79 methyltransferase (DOT1L histone methyltransferase). It targets residue Lys79 of histone 3 (H3K79) for methylation, as illustrated in Figure 19.687 The DOT1L histone methyltransferase is unique among HMTs for several reasons, making it an attractive therapeutic target.688 The enzyme has recently been implicated as a therapeutic target for acute leukemias bearing rearrangements in the mixed lineage leukemia (MLL) gene.689,690
Figure 19.

DOT1L-catalyzed methylation of H3K79.
A translocation in the MLL gene occurs in 5–10% of acute leukemias and is especially common in childhood acute leukemias.691,692 These cancers are particularly aggressive and have a relatively poor prognosis with a five-year survival of <40%.693 In MLL-rearranged leukemia cells, DOT1L histone methyltransferase has been shown to physically bind to a complex of the rearranged MLL protein fused with sequences from genes such as AF4, AF9, AF10, and ENL. This interaction recruits DOT1L, leads to hypermethylation of H3K79 and aberrant expression of leukemogenic genes such as Hoxa9, Hoxa7, and Meis1.559 Furthermore, preclinical models have shown that MLL-rearranged leukemias depend on abnormal DOT1L histone methyltransferase methylation of H3K79.39
Epizyme Inc. recently developed a potent and selective inhibitor of DOT1L histone methyltransferase. The compound, EPZ-004777 351 (Figure 19) is a 7-deaza-5′-amino-alkylurea substituted analogue of adenosine. The agent inhibits S-adenosyl methionine in the enzyme-binding pocket. EPZ-004777 binding leads to a conformational change in DOT1L histone methyltransferase, which opens a hydrophobic pocket past the amino acid site and produces additional interaction surfaces.694 Consequently, 351 selectively binds to DOT1L histone methyltransferase with high affinity. Furthermore, the agent was shown to selectively inhibit H3K79 methylation in vitro and inhibit the proliferation of MLL-rearranged cells. It also blocked MLL fusion target gene expression and led to prolonged survival in a mouse xenograft model of MLL leukemia.695 However, the poor pharmacokinetic properties of EPZ-004777 351 led to weak in vivo activity and precluded clinical development.
To overcome the suboptimal pharmacokinetic profile of 351, Epizyme synthesized additional analogs. In 2013, the company reported promising preclinical data for EPZ-5676 43, which exhibited improved drug-like properties and potency.696 Structural modifications were made to the 5′-side chain in order to decrease the number of hydrogen bonds and conformational freedom. EPZ-5676 43 also occupies the S-adenosyl methionine binding pocket of DOT1L histone methyltransferase, and it was found to be a potent and selective inhibitor of the enzyme with a Ki of <0.08 nM, having >37,000-fold binding selectivity against 15 other human methyltransferases.696
EPZ-5676 43 demonstrated selective inhibition of MLL-rearranged versus non-MLL-rearranged leukemia cells. In particular, an IC50 of 3.5 nM for the agent was demonstrated in the MV4–11 cell line following 14 days of exposure, with antiproliferative activity being most evident after 7 days.696 This delayed onset could be explained by a progressive series mechanism involving a decrease of H3K79 methylation, lowered MLL-fusion target gene expression, and inhibition of the expression of leukemogenic genes.696 EPZ-5676 43 produces a concentration-dependent decrease of both cellular methylated H3K79 levels as well as mRNA transcript levels of the MLL-fusion target genes HOXA9 and MEIS1. Continuous intravenous infusion of EPZ-5676 43 (70 mg/kg) for 21 days produced a complete regression of tumors in a rat model of MLL-rearranged leukemia. Little or no tumor regrowth was detected 30 days after treatment cessation. Evaluation of subcutaneous administration of EPZ-5676 43 has also been reported.697
However, EPZ-5676 43 still exhibits suboptimal pharmacokinetics with a short plasma half-life and with fecal excretion being the main route of excretion.698,699 A carbocyclic analogue of EPZ-5676 43 was synthesized 352 in an attempt to produce increased metabolic stability, and this compound (352; see Figure 19) did indeed show increased stability in human plasma and liver microsomes while maintaining a selective DOT1L histone methyltransferase Ki of 1.1 nM.559
In September 2012, Epizyme began a phase I clinical trial of EPZ-5676 43 for subjects with MLL-rearranged leukemia. Currently there are two ongoing Phase I clinical studies examining the agent for dose escalation and the first-in-human study for malignancies with rearrangement of the MLL gene (ClinicalTrials.gov identifiers: NCT02141828–competed in June 2016–no results posted, and NCT01684150–competed in February 2016–no results posted).
7.6. EPZ-5676 Synthesis
The synthesis of EPZ-5676 43 is shown in Scheme 64.700,701 Amine 353 is prepared from adenosine in four steps: 2′,3′-O-isopropylidine protection, installation of a 5′-O leaving group, 5′-azide displacement, and azide reduction. Reductive amination involving 5′-amino 353 and methyl 3-oxocyclobuganecarboxylate produced a cis/trans mixture (separated using chiral HPLC) furnishing intermediate 354. Subsequent N-alkylation, combined reduction, and Wittig olefination, alkene reduction, and saponification furnished intermediate 357. A two-step process was used to form the benzoimidazole ring of 359, without isolation of the intermediate amide 358. Finally, acid-mediated deprotection completed the synthesis of EPZ-5676 43.
Scheme 64. Synthesis of EPZ-5676 43a.

aReagents and conditions: (a) methyl 3-oxocyclobutanecarboxylate, Ti(O-iPr)4, MeOH, 45 °C, 2 h, then NaCNBH4, rt, overnight, 41%; (b) 2-iodopropane, K2CO3, CH3CN, 95 °C, overnight, 86%; (c) DIBAL-H, DCM, −78 °C to rt, then ethyl 2-(diethoxyphosphoryl)acetate, DBU, LiCl, rt, 1 h, 83%; (d) Pd/C, MeOH, rt, overnight, 78%; (e) LiOH·H2O, THF, MeOH, rt, overnight, 28%; (f) 4-(tert-butyl)benzene-1,2-diamine, EDC, HOBt, Et3N, rt, overnight, 50%; (g) AcOH, 65 °C, overnight, 98%; (h) HCl/MeOH, rt, 2 h, then HPLC, 51%.
In 2014, a 700-g-scale process of EPZ-5676 trihydrate was reported (Scheme 65).702 Cyclobutanone 366 can be prepared from 360 on kilogram-scale in a six-step sequence: protection, [2+2] cyloaddition, dechlorination, deprotection, acyl substitution, and iron-catalyzed reduction followed by condensation. 353 was subsequently converted to isopropyl amine 367, which was further converted to 368 as a mixture of diastereomers. Deprotection, followed by two crystallizations from MeCN/H2O, afforded pure EPZ-5676 43.
Scheme 65. Synthesis of EPZ-5676 43a.

aReagents and conditions: (a) BnBr, K2CO3, TBAI, Me2CO, rt, 2 d, 92%; (b) Zn(Cu), ClCOCCl3, CH3OCH2CH2OCH3, Et2O, 50 °C, 3 days, 96%; (c) Zn, AcOH, 50 °C, 2 h, 93%; (d) H2, Pd/C, (CH3)2CHCOOCH3, PhMe, rt, 20 h, then DCHA, 20 °C, 18 h, 85%; (e) dioxane, DMF, (COCl)2, 20 °C, 18 h, then 4-tert-butyl-2-nitroaniline, dioxane, 20–40 °C, 5 h, then 20 °C, 18 h, 89%; (f) Fe, AcOH, 75 °C then rt, overnight, 95%; (g) STAB, Me2CO, MeOH, AcOH, 20 °C, 2 h, 95%; (h) STAB, MeCN, 55 °C, 16 h, 83%; (i) HCl, MeOH, 45 °C, 9 h then rt, overnight, 93%; (i) MeCN/H2O, crystallization, 65%.
7.7. CF102 Biology
In 2007, preclinical studies developed by Can-Fite Biopharma showed that CF102 (2-chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyl-uronamide) 42 had potent anticancer activity in hepatocellular carcinoma,703 activity against hepatitis B virus, and anti-inflammatory activity (demonstrated in pre-clinical animal models of liver inflammation).704
CF102 42 is as antagonist for the A3 adenosine receptor. Hepatocellular carcinoma cells treated with CF102 had Wnt and NF-κB signaling pathways deregulation, which promoted apoptosis.703 Preclinical studies in rats with N1S1 hepatocellular carcinoma tumors, which highly express the A3 adenosine receptor, showed that CF102 42 encouraged tumor apoptosis.703 Furthermore, A3 adenosine receptor expression appears to be inflammation induced.705,706 The A3 adenosine receptor is also highly expressed on PBMCs in hepatocellular carcinoma patients and patients with rheumatoid arthritis, psoriasis, dry eye syndrome, and glaucoma, in addition to hepatocellular carcinoma and hepatitis.707,708
Currently, a Phase I/II study for dose escalation using CF102 42 was reported in patients with unresectable hepatocellular carcinoma.709 Overall study results showed that CF102 42 was well tolerated and had favorable pharmacokinetic characteristics. A Phase II study examining the safety and efficacy of CF102 42 is being planned (ClinicalTrials.gov identifier: NCT02128958–recruiting patients).
7.8. CF102 Synthesis
Kim et al. reported the discovery synthesis of CF102 42 in 1994 (Scheme 66).710 Protecting group manipulation of 1-O-methyl-β-D-riboside 370 produced the intermediate 371. Subsequent oxidation and esterification of the 5′-OH followed by methyl amide introduction, 2′,3′-O-benzoylation, and 1′-acetylation produced intermediate 374. Condensation of intermediate 374 with silylated N6-(3-iodobenzyl)-2-chloroadenine 376, made by reacting 2,6-dichloropurine 375 with 3-iodobenzylamine, followed by debenzoylation produced CF102 42 in milligram quantities in an overall yield of 1.7%.
Scheme 66. Synthesis of CF102 42a.

aReagents and conditions: (a) TBDPSCl, DMAP, DMF, rt, 18 h, 55%;(b) BzCl, py-DCM, 0 °C to rt, 17 h, 99%; (c) n-Bu4NF, THF, rt, 2 h, 89%; (d) RuO2, NaIO4, CHCl3, CH3CN, H2O, rt, 2.5 h, 90%; (e) EDC, DMAP, MeOH rt, 3 h, 72%; (f) MeNH2, THF, 50 °C, 15 h; (g) BzCl, py-DCM, rt, 3 h, 72% (over two steps); (h) Ac2O, H2SO4, AcOH, rt, 15 h, 34%; (i) 3-iodobenzylamine·HCl, Et3N, EtOH, rt, 5 d, 60%; (j) HMDS, (NH4)2SO4, reflux, 4 h; (k) TMSOTf, DCE, reflux, 62 h, 33%; (l) NH3/MeOH, rt, 16 h, 69%.
Hou et al. recently reported an alternative preparation of CF102 42 starting from the commercially available 1,2,3,5-tetra-O-acetyl-β-d-ribofuranose 136β, which is easily made from D-ribose 213 (Scheme 67).711 The synthesis performed the glycosylation before constructing the 5′-methyl amide and furnished the target compound on a gram-scale in 27% overall yield.
Scheme 67. Synthesis of CF102 42a.

aReagents and conditions: (a) BSA, TMSOTf, CH3CN, 2,6-dichloropurine, 50 °C, 18 h, 88%; (b) 3-iodobenzyl amine, Et3N, EtOH, rt, 2 d, 85%; (c) NaOMe, MeOH, DCM, rt, 1 h, 92%; (d) 2,2-dimethoxypropane, TsOH, DMF, rt, 12 h, 95%; (e) PDC, DMF, rt, 18 h, 70%; (f) MeNH2·HCl, HOBt, EDAC, DIEA, DCM, DMF, rt, 12 h, 71%; (g) 80% HCO2H, rt, 12 h, 83%.
8. ANTIVIRAL NUCLEOSIDE ANALOGS BEING EXPLORED FOR CANCER TREATMENT
8.1. Valacyclovir Hydrochloride and Valganciclovir Hydrochloride Biologies
Valacyclovir hydrochloride 17 and valganciclovir hydrochloride 35 are l-valyl ester prodrugs of acyclovir and ganciclovir, respectively. They were FDA-approved in 2009 and 2010, for the treatments of cytomegalovirus and herpes infections, respectively. After oral administration, the parent drug is released in vivo.712 Importantly the monophosphorylated agents occur by the viral-specific thymidine kinases, making them less toxic to uninfected cells. The monophosphorylated agents are further converted to their active triphosphate forms to block viral DNA synthesis.713 Since the viral thymidine kinase does not exist in human cells, gene therapy involving an adenoviral vector (disabled virus) designed to express the herpes simplex virus thymidine kinase gene in combination with valacyclovir hydrochloride 17 or valganciclovir hydrochloride 35 may lead to targeted cancer treatment (see Section 5.1.1.6).714 Furthermore, bystander killing of cancer cells has been proposed to be one mechanism of action for ganciclovir and acyclovir.715,716
Several clinical studies are evaluating valacyclovir hydrochloride 17. A Phase I trial is underway using combination therapy with adenovirus expressing viral-specific thymidine kinase (AD-VTK) and radiation for pediatric brain tumors (ClinicalTrials.gov identifier: NCT00634231–active status–not recruiting patients). Another Phase I trial is using intrapleural AD-VTK therapy with valacyclovir hydrochloride 17 in patients with malignant pleural effusion (ClinicalTrials.gov identifier: NCT01997190–active status–not recruiting patients). A Phase I/II trial of the 17 in combination with HSV-TK and brachytherapy for recurrent prostate cancer is ongoing (ClinicalTrials.gov identifier: NCT01913106–recruiting patients), as well as a Phase III trial with AD-VTK and valacyclovir hydrochloride 17 combination therapy for localized prostate cancer (ClinicalTrials.gov identifier: NCT01436968–recruiting patients). Currently, valganciclovir hydrochloride 35 is being used in a Phase IV trial for chronic lymphocytic leukemia, but the action of the agent is to inhibit cytomegalovirus (CMV) replication in an attempt to stop chronic lymphocytic leukemia propagation (ClinicalTrials.gov identifier: NCT01552369–recruiting patients). The agent has also been examined as an add-on treatment for controlling CMV in patients being treated for the malignant brain tumor glioblastoma. Overall, results showed that the addition of valacyclovir hydrochloride 17 to the standard treatment improved patient survival,717 which has encouraged a discourse on the possibility that CMV infection influences the development of brain tumors.718,719
8.2. Valacyclovir and Valganciclovir Syntheses
Valganciclovir hydrochloride 35 has been prepared almost exclusively from its parent compound ganciclovir 387, following the chemistry described in Scheme 68.720 Epichlorohydrin 383 was converted to dibenzyl-protected 384, which was subsequently subjected to chloromethylation by treatment with paraformaldehyde and HCl. Exposure of the resulting intermediate to potassium acetate in acetone afforded 385. Acid catalyzed condensation of 385 with diacetylguanine furnished a mixture of N7:N9 regioisomers, and the desired N9 isomer was crystallized from toluene. Debenzylation followed by deacetylation completed the synthesis of ganciclovir 387, on a 250-g-scale.
Scheme 68. Synthesis of Ganciclovir 387a.
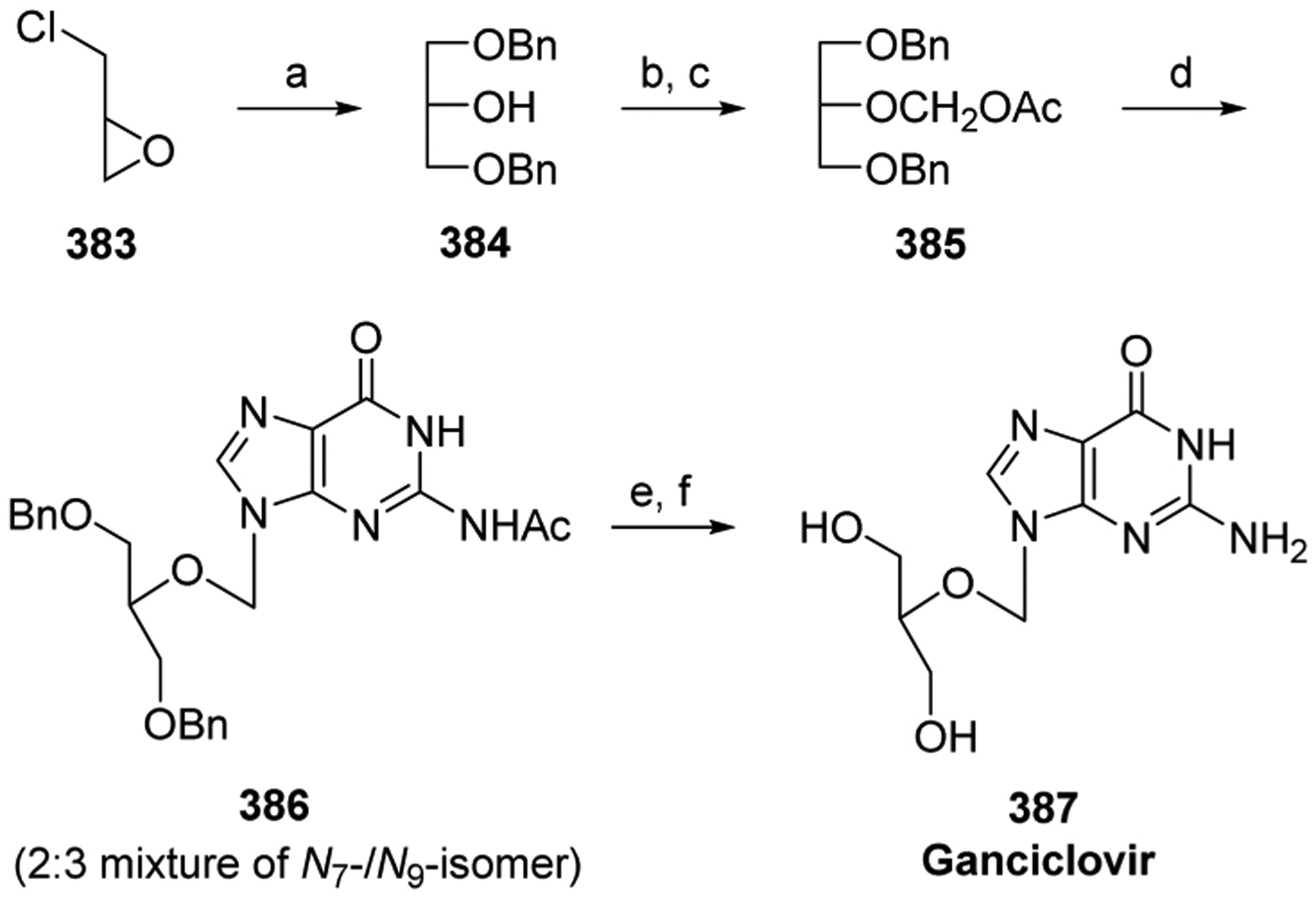
aReagents and conditions: (a) BnOH, aq NaOH, rt, 16 h, 63%; (b) HCl (g), (CH2O)n, DCM, 0 °C, 16 h; (c) KOAc, Me2CO, rt, 16 h, ~100%; (d) Diacetylguanine, TsOH, sulfolane, 95 °C, 5 days, 31% (N9-isomer); (e) 20% Pd(OH)2/C, cyclohexene–EtOH, reflux, 32 h; (f) NH4OH–MeOH (1:1 v/v), rt, 16 h, 86% (2 steps).
In 2004, a 100-g-scale process synthesis of triacetyl-protected ganciclovir 389 was reported that significantly increased the yield of the base condensation step.721 The reaction coupled diacetylguanine 60 and protected glycerol (388 in the presence of TsOH (Scheme 69). This step occurs under thermodynamic conditions, with the desired N9 isomer being the thermodynamic product. Recycling of the undesired N7 isomer was possible, allowing for increased yields of the desired N9 isomer (overall 70%). Compound 389 can be deprotected to furnish ganciclovir 387.
Scheme 69. Synthesis of 389a.

aReagents and conditions: (a) TsOH, DMF, 95–100 °C, 40 h; (b) Crystallization, 70% (N9-isomer).
In 1998, Arzeno et al. reported a preparation of valganciclovir hydrochloride 35 from ganciclovir 387 (Scheme 70).722 Treatment of 387 with trityl chloride and DMAP in DMF followed by coupling with Boc-L-valine furnished the intermediate 390. Subsequent treatment with a TFA/TFE mixture, in order to remove the trityl groups, and hydrogenolysis of the resulting Boc-protected intermediate with palladium on carbon in a methanol/HCl solution afforded kilograms of valganciclovir hydrochloride 35 with an HPLC purity of 98.4%.
Scheme 70. Synthesis of Valganciclovir Hydrochloride 35a.

aReagents and conditions: (a) TrCl, DMAP, Et3N, DMF, 50 °C, overnight; (b) Boc-l-valine, DCC, DMAP, Et3N, DCM, rt, 16.5 h; (c) TFA, TFE, 20 °C, 5 h; (d) H2, Pd/C, then HCl/MeOH, 34% (over four steps).
Khanduri et al. reported a preparation of valganciclovir hydrochloride 35 directly from ganciclovir 387 (Scheme 71).723 Treatment of ganciclovir 387 with Cbz-l-valine, DCC, and DMAP in DMSO led to a mixture of ganciclovir 387, mono-l-valyl ester 391, and bis-l-valyl ester 392. Addition of water and ethyl acetate caused 387 and 391 to precipitate, while 392 was removed in the organic layer. Ganciclovir 387 was removed from the 387/391 mixture by treatment with TFA followed by extraction with dichloromethane. Mono-l-valyl ester 391 was then separated from the organic layer by recrystallization. Subsequent hydrogenolysis of 391 in acidic solution produced valganciclovir hydrochloride 35, which was purified by recrystallization to give the target compound on a 60-g-scale in greater than 99% HPLC purity. Both ganciclovir 387 and bis-l-valyl ester 392 were recovered and recycled.
Scheme 71.

Preparation of Valganciclovir Hydrochloride 35
The discovery synthesis of valacyclovir hydrochloride 17 was reported in 1989 (Scheme 72). Acyclovir 393, which is prepared in ways similar to those of ganciclovir 387, was condensed directly with Cbz-L-valine.724 Palladium-catalyzed deprotection of the intermediate followed by treatment with aqueous HCl produced 17. An alternative preparation used Boc-l-valine instead of Cbz-l-valine, furnishing the compound on a 50-g-scale.725
Scheme 72. Synthesis of Valacyclovir Hydrochloride 17a.

aReagents and conditions: (a) Cbz-L-val-OH, DCC, DMAP, DMF, 60 °C to rt, 12 h; (b) Pd/C, H2, MeOH, THF, aq HCl, 55% (over two steps); (c) SO2Cl2, imidazole, MeCN; (d) l-valine, CuSO4, K2CO3, MeCN; (e) Acyclovir, DCC, DMAP, DMF, 10–15 °C, 45 min, 82%; (f) Raney Ni, EtOH, 50–60 °C then aq HCl, 82%.
An alternative route utilized azido-intermediate 395 to avoid protection–deprotection steps that increase risks of racemization (Scheme 72).726 Sodium azide 394 was treated with sulfuryl chloride and imidazole followed by L-valine and copper sulfate, producing the intermediate 395. Condensation of 395 with acyclovir followed by reduction with Rainey nickel furnished valacyclovir hydrochloride 17 in three steps.
8.3. Ribavirin Biology
Ribavirin (1-β-d-ribofuranosyl-1,2,4-triazole-3-carboxamide) 40 was first discovered as an effective antiviral agent against influenza A and B viruses in the 1970s using cell lines and and in vivo models.727,728 It was FDA-approved 15 years ago for the combination treatment of hepatitis C virus.729 The agent’s antiviral activity is primarily attributed to ribavirin-5′-triphosphate, which can inhibit RNA polymerase.730 Ribavirin-5′-monophosphate is also a potent inhibitor of inosine monophosphate dehydrogenase (IMPDH),731 an enzyme involved in the rate-limiting step of purine de novo synthesis. It is an important factor in maintaining guanine nucleoside pools, and a virus is highly dependent on this process. IMPDH Type I is ubiquitously expressed in normal cells, whereas a high level of IMPDH Type II is also associated with many hematological cancers.732 In addition, ribavirin 40 has a negative impact on eukaryotic translation initiation factor eIF4E activity, which is dysfunctional in 30% of cancers.733–735 High levels of eIF4E have been correlated with clinical progression, increased angiogenesis, and poor prognosis in breast cancer,736,737 and ribavirin 40 was shown to suppress growth in breast cell lines.738
Ribavirin 40 is currently being evaluated in a Phase I/II study for malignant solid tumors (ClinicalTrials.gov identifier: NCT01309490–unknown status–no updates for over 2 years) and in a Phase I trial using combination therapy for head and neck cancer and squamous cell cancer (ClinicalTrials.gov identifier: NCT01721525–active status–not recruiting patients). Ribavirin 40 is also being examined in combination with an inhibitor of hedgehog proteins, which regulate cell growth, differentiation, and survival, with or without azacytidine 11 in acute myelogenous leukemia patients (ClinicalTrials.gov identifier: NCT02073838–recruiting patients). The pharmacodynamic effects of ribavirin 40 therapy are being investigated for patients with tonsil and/or base of tongue squamous cell carcinoma (ClinicalTrials.gov identifier: NCT01268579–active status–not recruiting patients). Ribavirin 40 is being evaluated in combination with decitabine 14 in high-risk acute myelogenous leukemia patients (ClinicalTrials.gov identifier: NCT02109744–recruiting patients). Furthermore, a Phase I/II study of ribavirin 40 given as monotherapy in solid tumor cancer patients is ongoing (ClinicalTrials.gov identifier: NCT01309490–unknown status–no updates for over 2 years).
8.4. Ribavirin Synthesis
Witkowski et al. reported the discovery synthesis of ribavirin 40 in 1972 (Scheme 73).727,739 The compound was prepared by acid-catalyzed fusion of acylated sugar 136β with 1,2,4-triazole-3-carboxylate 397, which was prepared by reacting aminoguanidine bicarbonate with oxalic acid and subsequent esterification of the resulting 1,2,4-triazole-3-carboxylic acid. Treatment of methyl ester 398 with methanolic ammonia effected concomitant deprotection and amide formation, completing the synthesis.
Scheme 73. Synthesis of Ribavirin 40a.

aReagents and conditions: (a) SnCl4, DCM, 15–20 ° C, then reflux, 2 h, 70%; (b) NaOMe/MeOH, 10 °C, 3 h then NH3/MeOH, 20 °C, 4 h, 79%.
In 2007, Banfi et al. reported a large-scale synthesis, which allowed for the preparation of ribavirin 40 on a 400-g-scale (Scheme 74).740 Improvements over other preparations that allowed for scale up include the following: (1) a glycosylation involving direct coupling of triazole 397, thereby avoiding the need for silylating reagents, (2) intermediates pure enough to be used directly in the following stages without difficult purifications, and (3) relatively mild reaction conditions.
Scheme 74. Large-Scale Synthesis of Ribavirin 40a.

aReagents and conditions: (a) TfOH, 135 °C, 2 h, vacuum, 92%; (b) NH3/MeOH, 20 °C, 40 h, 83%.
Another recent preparation makes use of cheap nucleosides as starting materials.741 Inosine, adenosine, and guanosine were cleaved to the tetra-acylated sugar 136β and the corresponding acetylated purine base by treatment with acetic acid and TFA as catalyst. The glycosylation between 136β and triazole 397 was catalyzed by TfOH, and after deprotection, ribavirin 40 was produced on a 30-g-scale.
Ribavirin 40 can also be synthesized enzymatically. It has been formed on gram-scale. A transglycosylation involving purine nucleoside phosphorylase, 7-methylguanosine and 1,2,4-triazole-3-carboxamide has been used to produce grams of the agent.742 Furthermore, purine nucleoside phosphorylase has been used in conjunction with phosphopentomutase to couple ribose-5′-monophosphate 399 with the triazole 400, forming ribavirin 40 (Scheme 75).743
Scheme 75. Synthesis of Ribavirin 40a.
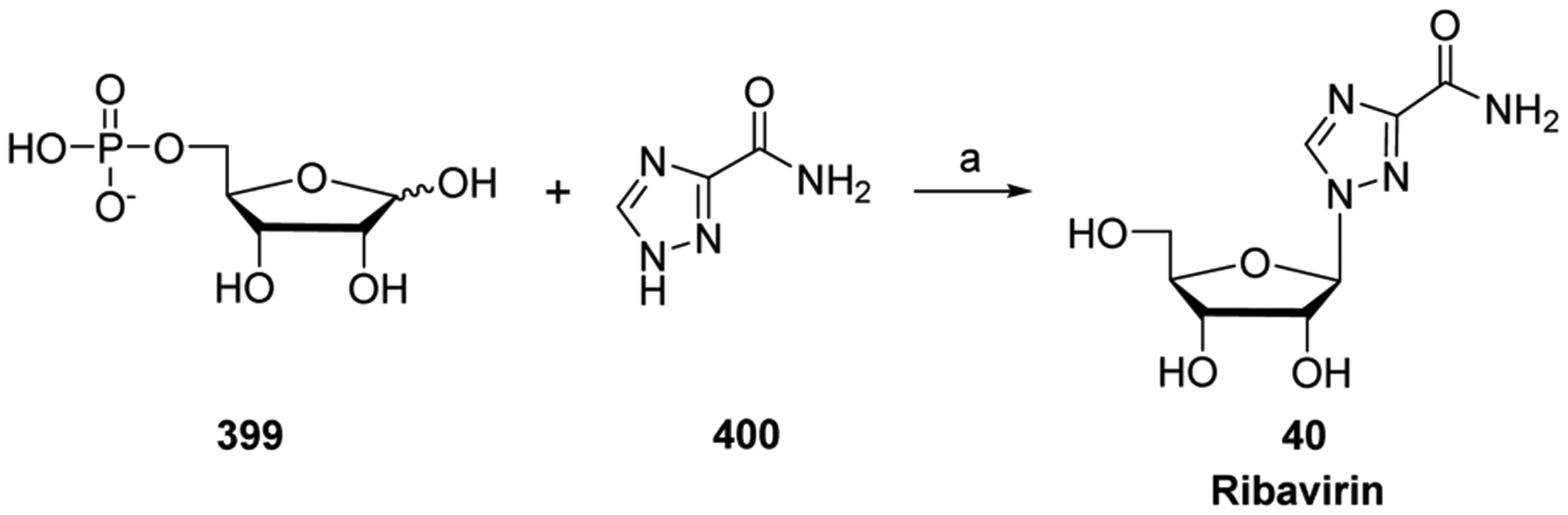
aReagents and conditions: (a) Phosphopentomutase, purine nucleoside phosphorylase, tris-buffer, 100%.
8.5. Acadesine Biology
Acadesine (AICA-Ribonucleotide (AICAR); 5-amino-1-β-d-ribofuranosyl-imidazole-4-carboxamide, 41) was previously under investigation for preventing ischemia-reperfusion injury in persons undergoing coronary artery bypass graft surgery.744 The agent was found to exert its cardio-protective effect via regulation of adenosine monophosphate activated protein kinase (AMPK) activity.745 Acadesine-5′-monophosphate is a highly potent activator of AMPK, and it can bind to the allosteric site of the enzyme as an AMP mimic. In a recent study, this activation was found to exert anti-proliferative and apoptotic effects on acute lymphocytic leukemia cells by regulating different signaling pathways (Figure 17).746,747 For this reason, cell-specific cytotoxicity of acadesine 41 might ascertain its future usage in anti-cancer therapy.
Preclinical trials with using acadesine 41 and rituximab showed synergistic anticancer activity in tissue culture and animal models.748 Acadesine 41 therapy is currently being evaluated in a Phase I/II study for myelodysplastic syndromes and acute myelogenous leukemia (ClinicalTrials.gov identifier: NCT01813838–trial status suspended in 2015) and a Phase I/II open label dose escalation study for safety and tolerability in B cell chronic lymphocytic leukemia patients (ClinicalTrials.gov identifier: NCT00559624–completed in 2010). Clinical trial results indicated that acadesine 41 had a manageable toxicity profile and might be a valuable agent for the treatment of relapsed/refractory CLL patients.749
8.6. Acadesine Synthesis
Acadesine 41 is now produced industrially from the fermentation broth of glucose,750–752 but Baddiley et al. reported the first synthesis of the agent in 1959 (Scheme 76).753 The key intermediate 403 was synthesized from d-ribose 213 in a five-step sequence involving cyclic acetal formation and benzoylation, followed by anomeric bromination, acetylation, and chlorination.754 In the presence of silver chloride, protected chlorosugar 403 was condensed with amide 405, formed from 4-nitro-5-styrylglyoxaline 404 in three steps via a carboxylic acid and a methylester intermediate.755 The resulting 1:1 mixture of regioisomers 406a and 406b was separated using descending paper chromatography. Hydrogenation of 406b using Adams’ catalyst afforded acadesine 41.
Scheme 76. Synthesis of Acadesine 41a.

aReagents and conditions: (a) cat. HCl, MeOH; (b) BzCl, pyr, DCM; (c) HBr/HOAc; (d) Ac2O, pyr, 48 h, 57% (from D-ribose); (e) HCl/ether, rt, 7 days; (f) AgCl, xylene, reflux, 1 h; (g) NH3/MeOH, 90 h, 19% (406a:406b = 1:1); (h) H2 (1 atm), PtO2, H2O, 1 h, 12%.
Kohyama et al. reported a gram-scale synthesis of acadesine 41, which features a hydrolysis of N-MEM protected inosine 408 by treatment with aqueous ammonia in methanol (Scheme 77).756 The resulting deprotected triol was subjected to aqueous alkaline hydrolysis, furnishing the ring-opened product acadesine 41 in four steps from inosine.
Scheme 77. Synthesis of Acadesine 41a.

aReagents and conditions: (a) MEMCl, DIPEA, DCM, 0 °C, 65 min, 86%; (b) NH4OH, MeOH, rt, 1 h; (c) aq NaOH, reflux, 1 h, 73%.
9. CONCLUSIONS
Nucleoside analogs are used to treat a high percentage of clinical cancers, usually in combination with other agents. They interact with various biological targets, such as cellular kinases, ribonucleotide reductase, and cellular polymerases in order to produce cytotoxic effects. Overall, a majority of cancers treated with nucleoside analogs are haematological. Newer nucleoside analogs, however, such as RX-3117 29, triciribine-5′-monphosphate 33, NUC-1031 32, and NUC-3373 37 are exhibiting promising activity against solid tumors in patients, but further testing is needed. With the exceptions of 6-MP 1 and cytarabine 4, nucleoside analogs usually do not produce cures as standalone agents.
As is the case with most classes of compounds explored in the clinic, attrition rates for nucleoside analogs remains high. Continuing efforts are directed toward improving the selectivity and targeting of anticancer nucleoside analog therapies. Nucleoside analogs interact with various biological targets, such as cell membrane transporters for cellular uptake, kinases to promote their phosphorylation, as well as DNA and RNA polymerases in order to produce their cytotoxic effects. Therefore, the tendency to increase chemical modifications to the parent nucleoside analog in hopes to design a better derivative should be considered with caution. Major efforts to improve analog biological efficacy currently focus on developing new prodrug strategies and using antimetabolites in combination treatments.
Prodrug strategies have emerged as promising approaches in the search for new anti-cancer nucleoside analogs. Next generation nucleoside analogs have been developed in an attempt to overcome cellular uptake deficiencies (e.g., elacytarabine 51, SGI-110 22, and sapacitabine 25) as well as overcome sensitivity to cytidine deamination and adenosine deamination (e.g., FdCyd 38, MB 07133 34, fludarabine-5′-monophosphate 7, clofarabine 12). Compared to the parent compound, nucleoside analog prodrug candidates generally offer enhanced efficacy and less toxicity, with the most successful being capecitabine 10.
Nucleoside analog prodrugs that have progressed to Phase III trials include elacytarabine 51, SGI-110 22, and sapacitabine 25. Elacytarabine 51 and CP-4126 49 have stalled in development, with the former failing to achieve end point efficacy in a Phase III clinical trial involving elderly patients with acute myeloid leukemia,490 indicating that 5′-elaidic acid ester conjugates are not successful clinical candidates. Elacytarabine 51 is still under clinical investigation, but questions remain about its clinical prospects as a monotherapy. The N4-alkylester moiety was not a successful approach for LY2333727 52, but it may be for sapacitabine 25, which is still being investigated in a Phase III clinical trial as a monotherapy. Furthermore, in June 2016, Cyclacel Pharmaceuticals announced positive results from a Phase I clinical trial involving sapacitabine 25 with seliciclib (CDK inhibitor) treatment for advanced solid tumors. SGI-110 22 is also currently being evaluated in a Phase III clinical trial in patients with AML. In addition, in May 2016 BioCryst Pharmaceuticals submitted regulatory paperwork to obtain forodesine hydrochloride 36 approval in Japan.
The phosphoramidate prodrug strategy has been successful in HCV treatment.757 Currently, ProTide analogs NUC-3373 37 and NUC-1031 32 are still being evaluated in Phase I clinical trials. Furthermore, the phosphoramidate prodrugs of many of the nucleoside analogs in this review (e.g., cladribine 8, fludarabine-5′-monophosphate 7, clofarabine 12, thiarabine 47, troxacitabine 48, etc.) have been claimed in patents, but no interesting biological effects have been reported.433,494 Beyond prodrugs, parent nucleoside analogs such as L-nucleosides seem to have an uncertain future, given the poor performance of troxacitabine 48, which produced multiple terminated clinical trials.
There are several other prodrug strategies being evaluated in preclinical studies. SGI-110 22 employs a duplex linked drug of decitabine(3′−5′)deoxyguanosine. Additional duplex drug linking of 5FdU(5′−5′)TAS-106401 and 5-FdU(3′−5′)TAS-106402 has been investigated in vitro. They may provide a means of delivering two nucleoside analogs at once to allow targeting of different cellular pathways. Preliminary data of the duplex drug linking approach seem promising, but further evidence is needed to show synergy and clinical efficacy. Other unique ways to modify nucleoside analogs being tested in vitro are mesoporous silica nanoparticles,361 5′-mono-amino acid monoester nucleoside prodrugs,362 and Hoechst nucleoside conjugates.363
A better understanding of cellular degradation pathways may greatly aid in maintaining high intracellular triphosphate forms of the anticancer nucleosides. SAMHD1 hydrolyzes dNTPs into 2′-deoxynucleoside and inorganic triphosphates.49,50 The tight catalytic pocket of the enzyme excludes ribonucleoside-5′-triphosphates.758 Clofarabine-5′-triphosphate was shown to be a substrate for SAMHD1 in a biochemical assay.51 Therefore, we postulate that nucleoside analog triphosphates such as decitabine-5′-triphosphate, cladribine-5′-triphosphate, floxuri-dine-5′-triphosphate ara-C-5′-triphosphate, ara-A-5′-triphosphate, ara-G-5′-triphosphate, fludarabine-5′-triphosphate, cladribine-5′-triphosphate, and clofarabine-5′-triphosphate may be sensitive to dNTP hydrolysis activity by SAMHD1 both in vitro and in vivo. Cotreatment of these nucleoside analogs with an effective inhibitor of SAMHD1, which still has not been developed, may increase the intracellular antimetabolite concentrations. Moreover, decreasing SAMHD1 protein levels have been shown to promote dNTP imbalance both in vitro and in vivo,759,760 and inhibition of the dNTP hydrolytic activity of the enzyme might lead to higher mutation rates in cancer cells. An alternative approach has been suggested to increase SAMHD1 expression in cancer cells. Methylation of the promoter of SAMHD1 has been shown to decrease mRNA and protein levels in certain cancers,53,761,762 and gene dosage of SAMHD1 has been shown to influence cellular dNTP levels in a mouse model.763 The current concept is to increase the SAMHD1 protein level within cells in order to decrease cellular dNTPs, which, in turn, might inhibit cell proliferation.764,765
Continued investment in basic synthetic organic chemistry provides the ability to access novel nucleoside analogs on both discovery and production scales. For example, the Jamison group’s syntheses of capecitabine 10 and doxifluridine 23 (see section 5.1.2.2) using continuous flow chemistry show great potential for the synthesis of nucleoside, nucleotide, and base analogs. Flow chemistry is widely used in academia and currently is attracting great interest in the pharmaceutical industry (e.g., establishment of the MIT-Novartis Center for Continuous Manufacture).766 This burgeoning technology offers great potential for both microscale synthetic drug discovery efforts and macroscale production of active pharmaceutical agents. On the laboratory scale, the microreactor allows for rigorously controlled reaction conditions, leading to better heat and mass transfer, while also requiring much smaller reaction volumes.767 This allows for much greater efficiency in comparison to batch systems—a reduction in reaction times (minutes versus hours), and a significant drop in waste (particularly solvent) production. On the macroscale, the significantly reduced reaction time has the potential to produce larger quantities of drug per unit time, in comparison to batch. Furthermore, the continuous flow process joins well with other enabling technologies (microwave irradiation, electrochemistry, 3D-printing, etc.), potentially leading to fully automated processes.768 Although questions remain about anomeric selectivity in glycosylations involving 2′-deoxysugars as well as phosphoramidate prodrug synthesis, application of this technology to nucleoside analog drug discovery and drug production seems promising.
The recent FDA approval of TAS-102 (15 + 16) illustrates that possibilities still exist for anticancer nucleoside analogs. The wealth of information available for these analogs produced from decades of research offers a strong foundation for future pursuits, including prodrug and combination strategies. In addition, further development of novel nucleoside analog chemical entities as well as continued refinement of biological applications of current nucleoside analogs will continue to contribute positively to the fight against cancer.
ACKNOWLEDGMENTS
We would like to thank Megan Browning for her initial contributions to this manuscript and Judy Mathew for her critical evaluation. This work was supported in part by NIH Grant 5P30-AI-50409 (CFAR).
Biographies
Jadd Shelton received his B.S. in Biology (Chemistry minor) from Brigham Young University—Idaho in 2008, where he worked in the lab of Jason Hunt, studying mink glycogen metabolism as a result of hormonal treatment. Also as an undergraduate, and NIH-funded Idaho INBRE fellow in the laboratory of Ron Strohmeyer at Northwest Nazarene University, he researched pharmacologic manipulation of microglial cell inflammatory gene expression related to Alzheimer’s disease. In 2012, he received his Ph.D. in Chemistry from Brigham Young University, working in the lab of Matt Peterson, where his research focused on the synthesis of anticancer nucleoside analogs. From 2012 to 2015, Jadd worked as a postdoctoral researcher in the laboratory of Raymond F. Schinazi at Emory University, synthesizing and studying nucleoside analogs for antiviral applications. He currently works as an R&D chemist for Autoliv Inc. His research interests include the design and synthesis of energetic materials for pyrotechnic formulations.
Xiao Lu received his B.S. degree in Pharmaceutical Sciences in 2005 and his M.S. degree in Chemical Biology in 2008 from Peking University. He then earned his Ph.D. degree in Pharmaceutical and Biological Sciences from The University of Georgia in 2012. After finishing his doctoral studies, he joined Prof. Schinazi’s Laboratory of Biochemical Pharmacology (LOBP) as a postdoctoral fellow at Emory University, working on medicinal chemistry for viral infectious diseases. He is currently a postdoctoral researcher at The National Center for Advancing Translational Sciences (NCATS), where he is contributing to developing drugs that block malaria transmission.
Joseph A. Hollenbaugh received his doctorate from the Department of Cellular and Molecular Biology at University of Vermont in 2006. He was trained as a cellular immunologist under the mentorship Richard Dutton (Trudeau Institute in Saranac Lake, NY) and examined CD8+ T cell responses to EG7OVA tumor in C57BL/6 mice. He did postdoctoral training in immunology and virology in the laboratories of David Topham and Baek Kim (University of Rochester). He is currently an Instructor in the Pediatrics Department, in the Laboratory of Biochemical Pharmacology at Emory University School of Medicine. His research interests include viral biology, cancer biology, and nucleoside biosynthesis regulation at the cellular level. His recent focus has been on the newly identified cellular protein SAMHD1, which degrades nucleoside triphosphates, in order to understand their biological implications in diseases and to develop a nucleoside inhibitor for potential therapeutic applications.
Jong Hyun Cho was born in Jangyumyeon, Gimhae-si, South Korea, in 1967. In 2002, he received his Ph.D. in Organic Chemistry from Seoul National University, working on biologically active peptide mimetics under the direction of B. M. Kim. He then joined David C. K. Chu’s group at the Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of Georgia, where he studied nucleoside chemistry and medicinal chemistry from 2003 to 2006. He is currently working as an instructor research fellow for Raymond F. Schinazi at Emory University. His research interests include synthetic approaches to nucleosides and peptidomimetics with antiviral activities against DNA and RNA viruses.
Franck Amblard was born in Châteauroux, France. He studied chemistry at the University of Orléans (France), where he received his Ph.D. in 2004 under the guidance of Luigi A. Agrofoglio, working on the synthesis of new nucleoside analogs using metathesis and palladium-catalyzed reactions. In 2005, he moved to the USA to join Raymond F. Schinazi’s research group at Emory University (Atlanta, GA) and worked, as a postdoctoral fellow, on new nucleosides and nucleotides prodrugs. He is now Assistant Professor at the Department of Pediatrics, Emory University School of Medicine. His main research interests include the study of nucleosides analogs and small molecules as potential antiviral agents.
Raymond F. Schinazi is the Frances Winship Walters Professor of Pediatrics, Director of the Laboratory of Biochemical Pharmacology at Emory University, and Adjunct Professor at Georgia State University. He also serves as Director of the HIV Cure Scientific Working Group within the NIH-sponsored Emory University Center for AIDS Research. Dr. Schinazi is a world leader in the area of nucleoside chemistry and biology, having published more than 500 papers and having 100 US patents issued. He is the founder of several biotechnology companies focusing on antiviral drug discovery and development, including Pharmasset, Inc., Triangle Pharmaceuticals, Idenix Pharmaceuticals, and Cocrystal Pharmaceuticals, Inc. He is best known for his innovative and pioneering work on stavudine, lamivudine, emtricitabine, telbivudine, and sofosbuvir, all of which have been approved by the United States Food and Drug Administration and are widely used clinically. More than 94% of HIV-infected individuals take at least one of the antiretroviral agents he invented as a fixed dose combination.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Siegel R; Jemal A Cancer facts & figures 2014; American Cancer Society: 2014; pp 1–68. [Google Scholar]
- (2).Rasool S; Kadla SA; Rasool V; Ganai BA A comparative overview of general risk factors associated with the incidence of colorectal cancer. Tumor Biol 2013, 34 (5), 2469–2476. [DOI] [PubMed] [Google Scholar]
- (3).Saeki N; Ono H; Sakamoto H; Yoshida T Genetic factors related to gastric cancer susceptibility identified using a genome-wide association study. Cancer Sci 2013, 104 (1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zimet GD; Buffler P Prevention of human papillomavirus-related diseases: Impediments to progress. Prev. Med 2013, 57 (5), 407–408. [DOI] [PubMed] [Google Scholar]
- (5).Yanik EL; Tamburro K; Eron JJ; Damania B; Napravnik S; Dittmer DP Recent cancer incidence trends in an observational clinical cohort of HIV-infected patients in the US, 2000 to 2011. Infect. Agents Cancer 2013, 8 (1), 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).PhRMA’s Communications & Public Affairs Department. Medicines in development: Cancer 2014 report; Pharmaceutical Research and Manufacturers of America: 2014; pp 1–104. [Google Scholar]
- (7).Siegel RL; Miller KD; Jemal A Cancer statistics, 2016. Ca-Cancer J. Clin 2016, 66 (1), 7–30. [DOI] [PubMed] [Google Scholar]
- (8).Fortin S; Berube G Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discovery 2013, 8 (8), 1029–1047. [DOI] [PubMed] [Google Scholar]
- (9).Povirk LF; Goldberg IH A role of oxidative DNA sugar damage in mutagenesis by neocarzinostatin and bleomycin. Biochimie 1987, 69 (8), 815–823. [DOI] [PubMed] [Google Scholar]
- (10).Chen T; Sun Y; Ji P; Kopetz S; Zhang W Topoisomerase IIα in chromosome instability and personalized cancer therapy. Oncogene 2015, 34 (31), 4019–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Casaluce F; Sgambato A; Maione P; Ciardiello F; Gridelli C Emerging mitotic inhibitors for non-small cell carcinoma. Expert Opin. Emerging Drugs 2013, 18 (1), 97–107. [DOI] [PubMed] [Google Scholar]
- (12).Deep G; Agarwal R New combination therapies with cell-cycle agents. Curr. Opin. Investig. Drugs 2008, 9 (6), 591–604. [PMC free article] [PubMed] [Google Scholar]
- (13).Paulsen O; Aass N; Kaasa S; Dale O Do corticosteroids provide analgesic effects in cancer patients? A systematic literature review. J. Pain Symptom Manage 2013, 46 (1), 96–105. [DOI] [PubMed] [Google Scholar]
- (14).Antonioli L; Yegutkin GG; Pacher P; Blandizzi C; Hasko G Anti-CD73 in cancer immunotherapy: awakening new opportunities. Trends Cancer 2016, 2 (2), 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Voena C; Chiarle R Advances in cancer immunology and cancer immunotherapy. Discovery Med 2016, 21 (114), 125–133. [PubMed] [Google Scholar]
- (16).Carvajal RD; Yang J; Asmar R Clinical utility of nivolumab in the treatment of advanced melanoma. Ther. Clin. Risk Manage 2016, 12, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Parker WB Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev 2009, 109 (7), 2880–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang J; Visser F; King KM; Baldwin SA; Young JD; Cass CE The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev 2007, 26 (1), 85–110. [DOI] [PubMed] [Google Scholar]
- (19).Pastor-Anglada M; Casado FJ In Deoxynucleoside Analogs in Cancer Therapy; Humana Press: Totowa, NJ, 2006; pp 1–28. [Google Scholar]
- (20).Gourdeau H; Clarke ML; Ouellet F; Mowles D; Selner M; Richard A; Lee N; Mackey JR; Young JD; Jolivet J; et al. Mechanisms of uptake and resistance to troxacitabine, a novel deoxycytidine nucleoside analogue, in human leukemic and solid tumor cell lines. Cancer Res 2001, 61 (19), 7217–7224. [PubMed] [Google Scholar]
- (21).Van Rompay AR; Johansson M; Karlsson A Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol. Ther 2000, 87 (2–3), 189–198. [DOI] [PubMed] [Google Scholar]
- (22).Smal C; Vertommen D; Bertrand L; Ntamashimikiro S; Rider MH; Van Den Neste E; Bontemps F Identification of in vivo phosphorylation sites on human 2′-deoxycytidine kinase. Role of Ser-74 in the control of enzyme activity. J. Biol. Chem 2006, 281 (8), 4887–4893. [DOI] [PubMed] [Google Scholar]
- (23).Arner ES; Eriksson S Mammalian deoxyribonucleoside kinases. Pharmacol. Ther 1995, 67 (2), 155–186. [DOI] [PubMed] [Google Scholar]
- (24).Saada-Reisch A Deoxyribonucleoside kinases in mitochondrial DNA depletion. Nucleosides, Nucleotides Nucleic Acids 2004, 23 (8–9), 1205–1215. [DOI] [PubMed] [Google Scholar]
- (25).Knecht W; Petersen GE; Munch-Petersen B; Piskur J Deoxyribonucleoside kinases belonging to the thymidine kinase 2 (TK2)-like group vary significantly in substrate specificity, kinetics and feed-back regulation. J. Mol. Biol 2002, 315 (4), 529–540. [DOI] [PubMed] [Google Scholar]
- (26).Matsuda A; Sasaki T Antitumor activity of sugar-modified cytosine nucleosides. Cancer Sci 2004, 95 (2), 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Achanta G; Pelicano H; Feng L; Plunkett W; Huang P Interaction of p53 and DNA-PK in response to nucleoside analogues: potential role as a sensor complex for DNA damage. Cancer Res 2001, 61 (24), 8723–8729. [PubMed] [Google Scholar]
- (28).Jackson SP; Bartek J The DNA-damage response in human biology and disease. Nature 2009, 461 (7267), 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Melvin WT; Milne HB; Slater AA; Allen HJ; Keir HM Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J. Biochem 1978, 92 (2), 373–379. [DOI] [PubMed] [Google Scholar]
- (30).Sahasranaman S; Howard D; Roy S Clinical pharmacology and pharmacogenetics of thiopurines. Eur. J. Clin. Pharmacol 2008, 64 (8), 753–767. [DOI] [PubMed] [Google Scholar]
- (31).Perez MA; Cerqueira NM; Fernandes PA; Ramos MJ Ribonucleotide reductase: a mechanistic portrait of substrate analogues inhibitors. Curr. Med. Chem 2010, 17 (26), 2854–2872. [DOI] [PubMed] [Google Scholar]
- (32).Hofer A; Crona M; Logan DT; Sjoberg BM DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol 2012, 47 (1), 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kicska GA; Long L; Horig H; Fairchild C; Tyler PC; Furneaux RH; Schramm VL; Kaufman HL Immucillin H, a powerful transition-state analog inhibitor of purine nucleoside phosphorylase, selectively inhibits human T lymphocytes. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (8), 4593–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Miles RW; Tyler PC; Furneaux RH; Bagdassarian CK; Schramm VL One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry 1998, 37 (24), 8615–8621. [DOI] [PubMed] [Google Scholar]
- (35).Robak P; Robak T Older and new purine nucleoside analogs for patients with acute leukemias. Cancer Treat. Rev 2013, 39 (8), 851–861. [DOI] [PubMed] [Google Scholar]
- (36).Heidelberger C; Chaudhuri NK; Danneberg P; Mooren D; Griesbach L; Duschinsky R; Schnitzer RJ; Pleven E; Scheiner J Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179 (4561), 663–666. [DOI] [PubMed] [Google Scholar]
- (37).Soucy TA; Smith PG; Milhollen MA; Berger AJ; Gavin JM; Adhikari S; Brownell JE; Burke KE; Cardin DP; Critchley S; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458 (7239), 732–736. [DOI] [PubMed] [Google Scholar]
- (38).Tatum D; Li S Evidence that the histone methyltransferase Dot1 mediates global genomic repair by methylating histone H3 on lysine 79. J. Biol. Chem 2011, 286 (20), 17530–17535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bernt KM; Zhu N; Sinha AU; Vempati S; Faber J; Krivtsov AV; Feng Z; Punt N; Daigle A; Bullinger L; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20 (1), 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hunsucker SA; Mitchell BS; Spychala J The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol. Ther 2005, 107 (1), 1–30. [DOI] [PubMed] [Google Scholar]
- (41).Eriksson S Is the expression of deoxynucleoside kinases and 5′-nucleotidases in animal tissues related to the biological effects of nucleoside analogs? Curr. Med. Chem 2013, 20 (34), 4241–4248. [DOI] [PubMed] [Google Scholar]
- (42).Buschmann J; Moritz B; Jeske M; Lilie H; Schierhorn A; Wahle E Identification of Drosophila and human 7-methyl GMP-specific nucleotidases. J. Biol. Chem 2013, 288 (4), 2441–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bianchi V; Spychala J Mammalian 5′-nucleotidases. J. Biol. Chem 2003, 278 (47), 46195–46198. [DOI] [PubMed] [Google Scholar]
- (44).Chernogorova P; Zeiser R Ectonucleotidases in solid organ and allogeneic hematopoietic cell transplantation. J. Biomed. Biotechnol 2012, 2012, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Reichard P Interactions between deoxyribonucleotide and DNA synthesis. Annu. Rev. Biochem 1988, 57, 349–374. [DOI] [PubMed] [Google Scholar]
- (46).Stagg J; Smyth MJ Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010, 29 (39), 5346–5358. [DOI] [PubMed] [Google Scholar]
- (47).Gaudreau PO; Allard B; Turcotte M; Stagg J CD73-adenosine reduces immune responses and survival in ovarian cancer patients. Oncoimmunology 2016, 5 (5), e1127496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Stella J; Bavaresco L; Braganhol E; Rockenbach L; Farias PF; Wink MR; Azambuja AA; Barrios CH; Morrone FB; Oliveira Battastini AM Differential ectonucleotidase expression in human bladder cancer cell lines. Urol Oncol 2010, 28 (3), 260–267. [DOI] [PubMed] [Google Scholar]
- (49).Powell RD; Holland PJ; Hollis T; Perrino FW Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem 2011, 286 (51), 43596–43600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Goldstone DC; Ennis-Adeniran V; Hedden JJ; Groom HC; Rice GI; Christodoulou E; Walker PA; Kelly G; Haire LF; Yap MW; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480 (7377), 379–382. [DOI] [PubMed] [Google Scholar]
- (51).Arnold LH; Kunzelmann S; Webb MR; Taylor IA A continuous enzyme-coupled assay for triphosphohydrolase activity of HIV-1 restriction factor SAMHD1. Antimicrob. Agents Chemother 2015, 59 (1), 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Rossi D SAMHD1: a new gene for CLL. Blood 2014, 123 (7), 951–952. [DOI] [PubMed] [Google Scholar]
- (53).Wang JL; Lu FZ; Shen XY; Wu Y; Zhao LT SAMHD1 is down regulated in lung cancer by methylation and inhibits tumor cell proliferation. Biochem. Biophys. Res. Commun 2014, 455 (3–4), 229–233. [DOI] [PubMed] [Google Scholar]
- (54).Holohan C; Van Schaeybroeck S; Longley DB; Johnston PG Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13 (10), 714–726. [DOI] [PubMed] [Google Scholar]
- (55).Wiley JS; Jones SP; Sawyer WH; Paterson AR Cytosine arabinoside influx and nucleoside transport sites in acute leukemia. J. Clin. Invest 1982, 69 (2), 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Jamieson GP; Brocklebank AM; Snook MB; Sawyer WH; Buolamwini JK; Paterson AR; Wiley JS Flow cytometric quantitation of nucleoside transporter sites on human leukemic cells. Cytometry 1993, 14 (1), 32–38. [DOI] [PubMed] [Google Scholar]
- (57).Phua LC; Mal M; Koh PK; Cheah PY; Chan EC; Ho HK Investigating the role of nucleoside transporters in the resistance of colorectal cancer to 5-fluorouracil therapy. Cancer Chemother. Pharmacol 2013, 71 (3), 817–823. [DOI] [PubMed] [Google Scholar]
- (58).Hu X; Chen W; Xu J Downregulation of human equilibrative nucleoside transporter 1 by RNAi enhances 5-fluorouracil response in pancreatic cancer. Hepatogastroenterology 2010, 57 (104), 1567–1572. [PubMed] [Google Scholar]
- (59).Mohelnikova-Duchonova B; Melichar B Human equilibrative nucleoside transporter 1 (hENT1): do we really have a new predictive biomarker of chemotherapy outcome in pancreatic cancer patients? Pancreatology 2013, 13 (6), 558–563. [DOI] [PubMed] [Google Scholar]
- (60).Saiki Y; Yoshino Y; Fujimura H; Manabe T; Kudo Y; Shimada M; Mano N; Nakano T; Lee Y; Shimizu S; et al. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem. Biophys. Res. Commun 2012, 421 (1), 98–104. [DOI] [PubMed] [Google Scholar]
- (61).Flasshove M; Strumberg D; Ayscue L; Mitchell BS; Tirier C; Heit W; Seeber S; Schutte J Structural analysis of the 2′-deoxycytidine kinase gene in patients with acute myeloid leukemia and resistance to cytosine arabinoside. Leukemia 1994, 8 (5), 780–785. [PubMed] [Google Scholar]
- (62).Veuger MJ; Honders MW; Willemze R; Barge RM 2′-deoxycytidine kinase expression and activity in patients with resistant versus sensitive acute myeloid leukemia. Eur. J. Haematol 2002, 69 (3), 171–178. [DOI] [PubMed] [Google Scholar]
- (63).Ferry KV; Hamilton TC; Johnson SW Increased nucleotide excision repair in cisplatin-resistant ovarian cancer cells: role of ERCC1-XPF. Biochem. Pharmacol 2000, 60 (9), 1305–1313. [DOI] [PubMed] [Google Scholar]
- (64).Nambaru PK; Hubner T; Kock K; Mews S; Grube M; Payen L; Guitton J; Sendler M; Jedlitschky G; Rimmbach C; et al. Drug efflux transporter multidrug resistance-associated protein 5 affects sensitivity of pancreatic cancer cell lines to the nucleoside anticancer drug 5-fluorouracil. Drug Metab. Dispos 2011, 39 (1), 132–139. [DOI] [PubMed] [Google Scholar]
- (65).Fukuda Y; Schuetz JD ABC transporters and their role in nucleoside and nucleotide drug resistance. Biochem. Pharmacol 2012, 83 (8), 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Gonzalez-Haba E; Garcia MI; Cortejoso L; Lopez-Lillo C; Barrueco N; Garcia-Alfonso P; Alvarez S; Jimenez JL; Martin ML; Munoz-Fernandez MA; et al. ABCB1 gene polymorphisms are associated with adverse reactions in fluoropyrimidine-treated colorectal cancer patients. Pharmacogenomics 2010, 11 (12), 1715–1723. [DOI] [PubMed] [Google Scholar]
- (67).Steuart CD; Burke PJ Cytidine deaminase and the development of resistance to arabinosyl cytosine. Nature: New biology 1971, 233 (38), 109–110. [DOI] [PubMed] [Google Scholar]
- (68).Abraham A; Varatharajan S; Abbas S; Zhang W; Shaji RV; Ahmed R; George B; Srivastava A; Chandy M; Mathews V; et al. Cytidine deaminase genetic variants influence RNA expression and cytarabine cytotoxicity in acute myeloid leukemia. Pharmacogenomics 2012, 13 (3), 269–282. [DOI] [PubMed] [Google Scholar]
- (69).Damaraju VL; Damaraju S; Young JD; Baldwin SA; Mackey J; Sawyer MB; Cass CE Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22 (47), 7524–7536. [DOI] [PubMed] [Google Scholar]
- (70).Vorbrüggen H; Ruh-Pohlenz C Organic Reactions; American Chemical Society: 1999; pp 1–630. [Google Scholar]
- (71).Elion GB The purine path to chemotherapy. Science 1989, 244 (4900), 41–47. [DOI] [PubMed] [Google Scholar]
- (72).Levinsen M; Rotevatn EO; Rosthoj S; Nersting J; Abrahamsson J; Appell ML; Bergan S; Bechensteen AG; Harila-Saari A; Heyman M; et al. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: Influence on cure rates and risk of second cancer. Pediatr. Blood Cancer 2014, 61 (5), 797–802. [DOI] [PubMed] [Google Scholar]
- (73).Vora A; Mitchell CD; Lennard L; Eden TO; Kinsey SE; Lilleyman J; Richards SM; Medical Research C; National Cancer Research Network Childhood Leukaemia Working, P.. Toxicity and efficacy of 6-thioguanine versus 6-mercaptopurine in childhood lymphoblastic leukaemia: a randomised trial. Lancet 2006, 368 (9544), 1339–1348. [DOI] [PubMed] [Google Scholar]
- (74).Lemann M Treatment of chronic inflammatory bowel diseases. Bull. Acad. Natl. Med 2007, 191 (6), 1125–1141. [PubMed] [Google Scholar]
- (75).Jabin D; Kumar S; Gow PJ Outcome of patients on azathioprine: a need for a better pre-treatment assessment and dosing guideline. N. Z. Med. J 2010, 123 (1324), 67–73. [PubMed] [Google Scholar]
- (76).Chaparro M; Ordas I; Cabre E; Garcia-Sanchez V; Bastida G; Penalva M; Gomollon F; Garcia-Planella E; Merino O; Gutierrez A; et al. Safety of thiopurine therapy in inflammatory bowel disease: Long-term follow-up study of 3931 patients. Inflamm. Bowel Dis 2013, 19 (7), 1404–1410. [DOI] [PubMed] [Google Scholar]
- (77).Kudo M; Saito Y; Sasaki T; Akasaki H; Yamaguchi Y; Uehara M; Fujikawa K; Ishikawa M; Hirasawa N; Hiratsuka M Genetic variations in the HGPRT, ITPA, IMPDH1, IMPDH2, and GMPS genes in Japanese individuals. Drug Metab. Pharmacokinet 2009, 24 (6), 557–564. [DOI] [PubMed] [Google Scholar]
- (78).Petit E; Langouet S; Akhdar H; Nicolas-Nicolaz C; Guillouzo A; Morel F Differential toxic effects of azathioprine, 6-mercaptopurine and 6-thioguanine on human hepatocytes. Toxicol. In Vitro 2008, 22 (3), 632–642. [DOI] [PubMed] [Google Scholar]
- (79).Aarbakke J; Janka-Schaub G; Elion GB Thiopurine biology and pharmacology. Trends Pharmacol. Sci 1997, 18 (1), 3–7. [DOI] [PubMed] [Google Scholar]
- (80).Nelson JA; Carpenter JW; Rose LM; Adamson DJ Mechanisms of action of 6-thioguanine, 6-mercaptopurine, and 8-azaguanine. Cancer Res 1975, 35 (10), 2872–2878. [PubMed] [Google Scholar]
- (81).Karran P Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br. Med. Bull 2006, 79–80, 153–170. [DOI] [PubMed] [Google Scholar]
- (82).Chrzanowska M; Kuehn M; Januszkiewicz-Lewandowska D; Kurzawski M; Drozdzik M Thiopurine S-methyltransferase pheno-type-genotype correlation in children with acute lymphoblastic leukemia. Acta. Polym. Pharm 2012, 69 (3), 405–410. [PubMed] [Google Scholar]
- (83).de Boer NK; van Bodegraven AA; Jharap B; de Graaf P; Mulder CJ Drug insight: Pharmacology and toxicity of thiopurine therapy in patients with IBD. Nat. Clin. Pract. Gastroenterol. Hepatol 2007, 4 (12), 686–694. [DOI] [PubMed] [Google Scholar]
- (84).Karim H; Ghalali A; Lafolie P; Vitols S; Fotoohi AK Differential role of thiopurine methyltransferase in the cytotoxic effects of 6-mercaptopurine and 6-thioguanine on human leukemia cells. Biochem. Biophys. Res. Commun 2013, 437 (2), 280–286. [DOI] [PubMed] [Google Scholar]
- (85).Chen ZS; Lee K; Kruh GD Transport of cyclic nucleotides and estradiol 17-β-D-glucuronide by multidrug resistance protein 4. Resistance to 6-mercaptopurine and 6-thioguanine. J. Biol. Chem 2001, 276 (36), 33747–33754. [DOI] [PubMed] [Google Scholar]
- (86).Wielinga PR; Reid G; Challa EE; van der Heijden I; van Deemter L; de Haas M; Mol C; Kuil AJ; Groeneveld E; Schuetz JD; et al. Thiopurine metabolism and identification of the thiopurine metabolites transported by MRP4 and MRP5 overexpressed in human embryonic kidney cells. Mol. Pharm 2002, 62 (6), 1321–1331. [DOI] [PubMed] [Google Scholar]
- (87).Peng XX; Chen ZS; Tiwari AK; Damaraju VL; Fu L; Cass CE; Ashby CR Jr.; Kruh GD; Shi Z Up-regulation of P-glycoprotein confers acquired resistance to 6-mercaptopurine in human chronic myeloid leukemia cells. Oncol. Lett 2011, 2 (3), 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).J Pharmacol Exp Ther; Baskin F; Rosenberg RN Decreased 6-mercaptopurine retention by two resistant variants of mouse neuroblastoma with normal hypoxanthine-guanine-phospho-ribosyltransferase activities. J. Pharmacol. Exp. Ther 1975, 193 (1), 293–300. [PubMed] [Google Scholar]
- (89).Chouchana L; Fernandez-Ramos AA; Dumont F; Marchetti C; Ceballos-Picot I; Beaune P; Gurwitz D; Loriot MA Molecular insight into thiopurine resistance: transcriptomic signature in lymphoblastoid cell lines. Genome Med 2015, 7 (1), 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Tzoneva G; Perez-Garcia A; Carpenter Z; Khiabanian H; Tosello V; Allegretta M; Paietta E; Racevskis J; Rowe JM; Tallman MS; et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat. Med 2013, 19 (3), 368–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Meyer JA; Wang J; Hogan LE; Yang JJ; Dandekar S; Patel JP; Tang Z; Zumbo P; Li S; Zavadil J; et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat. Genet 2013, 45 (3), 290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Elion GB; Burgi E; Hitchings GH Studies on condensed pyrimidine systems. IX. The synthesis of some 6-substituted purines. J. Am. Chem. Soc 1952, 74 (2), 411–414. [Google Scholar]
- (93).Albert A; Brown DJ; Cheeseman G 103. Pteridine studies. Part I. Pteridine, and 2- and 4-amino- and 2- and 4-hydroxy-pteridines. J. Chem. Soc 1951, 474–485. [Google Scholar]
- (94).Lin J; Zhang C; Deng B CN Patent 101817822, 2010.
- (95).Bergman J; Pettersson B; Hasimbegovic V; Svensson PH Thionations using a P4S10-pyridine complex in solvents such as acetonitrile and dimethyl sulfone. J. Org. Chem 2011, 76 (6), 1546–1553. [DOI] [PubMed] [Google Scholar]
- (96).Elion GB; Hitchings GH The synthesis of 6-thioguanine. J. Am. Chem. Soc 1955, 77 (6), 1676. [Google Scholar]
- (97).Ma Q CN Patent 1690058, 2005.
- (98).Kulikowski T; Grynkiewicz G PL Patent 162708, 1990.
- (99).Kramata P; Downey KM; Paborsky LR Incorporation and excision of 9-(2-phosphonylmethoxyethyl)guanine (PMEG) by DNA polymerase delta and epsilon in vitro. J. Biol. Chem 1998, 273 (34), 21966–21971. [DOI] [PubMed] [Google Scholar]
- (100).Romeo R; Carnovale C; Rescifina A; Chiacchio MA In Chemical synthesis of nucleoside analogues; Wiley: Hoboken, NJ, 2013; pp 163–208. [Google Scholar]
- (101).Hatse S; De Clercq E; Balzarini J Role of antimetabolites of purine and pyrimidine nucleotide metabolism in tumor cell differentiation. Biochem. Pharmacol 1999, 58 (4), 539–555. [DOI] [PubMed] [Google Scholar]
- (102).Kramata P; Downey KM 9-(2-phosphonylmethoxyethyl) derivatives of purine nucleotide analogs: A comparison of their metabolism and interaction with cellular DNA synthesis. Mol. Pharm 1999, 56 (6), 1262–1270. [DOI] [PubMed] [Google Scholar]
- (103).Reiser H; Wang J; Chong L; Watkins WJ; Ray AS; Shibata R; Birkus G; Cihlar T; Wu S; Li B; et al. GS-9219–a novel acyclic nucleotide analogue with potent antineoplastic activity in dogs with spontaneous non-Hodgkin’s lymphoma. Clin. Cancer Res 2008, 14 (9), 2824–2832. [DOI] [PubMed] [Google Scholar]
- (104).Pradere U; Garnier-Amblard EC; Coats SJ; Amblard F; Schinazi RF Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev 2014, 114 (18), 9154–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Schinkmanova M; Votruba I; Holy A N6-methyl-AMP aminohydrolase activates N6-substituted purine acyclic nucleoside phosphonates. Biochem. Pharmacol 2006, 71 (9), 1370–1376. [DOI] [PubMed] [Google Scholar]
- (106).Frey CR; Andrei G; Votruba I; Cannizzaro C; Han B; Fung W; Hung M; Liu X; Geleziunas R; Fiten P; et al. Mutations in adenosine deaminase-like (ADAL) protein confer resistance to the antiproliferative agents N6-cyclopropyl-PMEDAP and GS-9219. Anticancer Res 2013, 33 (5), 1899–1912. [PubMed] [Google Scholar]
- (107).Mertlikova-Kaiserova H; Rumlova M; Tloustova E; Prochazkova E; Holy A; Votruba I Point mutations in human guanylate kinase account for acquired resistance to anticancer nucleotide analogue PMEG. Biochem. Pharmacol 2011, 82 (2), 131–138. [DOI] [PubMed] [Google Scholar]
- (108).Thamm DH; Vail DM; Kurzman ID; Babusis D; Ray AS; Sousa-Powers N; Tumas DB GS-9219/VDC-1101 - a prodrug of the acyclic nucleotide PMEG has antitumor activity inspontaneous canine multiple myeloma. BMC Vet. Res 2014, 10 (30), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Lawrence JA; Huelsmeyer MK; Thamm DH; Tumas DB; Birkus G; Kurzman I; Vail DM Novel acyclic nucleotide analogues GS-343074 and GS-424044 demonstrate antiproliferative and pro-apoptotic activity in canine neoplastic cell lines. Vet. Comp. Oncol 2015, 13 (3), 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Morges MA; Burton JH; Saba CF; Vail DM; Burgess KE; Thamm DH Phase II evaluation of VDC-1101 in canine cutaneous T-cell lymphoma. J. Vet. Intern. Med 2014, 28 (5), 1569–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Cheng X; Cook GP; Desai MC; Doerffler E; He G-X; Kim CU; Lee WA; Rohloff JC; Wang J; Yang Z-Y WO Patent 2005066189, 2005.
- (112).Jansa P; Baszczynski O; Dracinsky M; Votruba I; Zidek Z; Bahador G; Stepan G; Cihlar T; Mackman R; Holy A; Janeba Z A novel and efficient one-pot synthesis of symmetrical diamide (bisamidate) prodrugs of acyclic nucleoside phosphonates and evaluation of their biological activities. Eur. J. Med. Chem 2011, 46 (9), 3748–3754. [DOI] [PubMed] [Google Scholar]
- (113).Horejsi K; Andrei G; De Clercq E; Snoeck R; Pohl R; Holy A Tricyclic etheno analogs of PMEG and PMEDAP: Synthesis and biological activity. Bioorg. Med. Chem 2006, 14 (23), 8057–8065. [DOI] [PubMed] [Google Scholar]
- (114).Rutman RJ; Cantarow A; Paschkis KE Studies in 2-acetylaminofluorene carcinogenesis. III. The utilization of uracil-2-C14 by preneoplastic rat liver and rat hepatoma. Cancer Res 1954, 14 (2), 119–123. [PubMed] [Google Scholar]
- (115).Callery P; Gannett P Cancer and Cancer Chemotherapy; Lippincott Williams & Wilkins: Philadelphia, PA, 2002; pp 924–951. [Google Scholar]
- (116).Heidelberger C; Griesbach L; Cruz O; Schnitzer RJ; Grunberg E Fluorinated pyrimidines. VI. Effects of 5-fluorouridine and 5-fluoro-2′-deoxyuridine on transplanted tumors. Exp. Biol. Med 1958, 97 (2), 470–475. [DOI] [PubMed] [Google Scholar]
- (117).Bosch L; Harbers E; Heidelberger C Studies on fluorinated pyrimidines. V. Effects on nucleic acid metabolism in vitro. Cancer Res 1958, 18 (3), 335–343. [PubMed] [Google Scholar]
- (118).Chaudhuri NK; Montag BJ; Heidelberger C Studies on fluorinated pyrimidines III. The metabolism of 5-fluorouracil-2-C14 and 5-fluoroorotic-2-C14 acid in vivo. Cancer Res 1958, 18 (3), 318–328. [PubMed] [Google Scholar]
- (119).Longley DB; Harkin DP; Johnston PG 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3 (5), 330–338. [DOI] [PubMed] [Google Scholar]
- (120).Guo Y; Shi M; Shen X; Yang C; Yang L; Zhang J Capecitabine plus irinotecan versus 5-FU/leucovorin plus irinotecan in the treatment of colorectal cancer: A meta-analysis. Clin. Colorectal Cancer 2014, 13 (2), 110–118. [DOI] [PubMed] [Google Scholar]
- (121).Du Z; Wang Y; Zhou Y; Wen F; Li Q First-line irinotecan combined with 5-fluorouracil and leucovorin for high-grade metastatic gastrointestinal neuroendocrine carcinoma. Tumori 2013, 99 (1), 57–60. [DOI] [PubMed] [Google Scholar]
- (122).Diasio RB; Harris BE Clinical pharmacology of 5-fluorouracil. Clin. Pharmacokinet 1989, 16 (4), 215–237. [DOI] [PubMed] [Google Scholar]
- (123).Wohlhueter RM; McIvor RS; Plagemann PG Facilitated transport of uracil and 5-fluorouracil, and permeation of orotic acid into cultured mammalian cells. J. Cell. Physiol 1980, 104 (3), 309–319. [DOI] [PubMed] [Google Scholar]
- (124).Grem JL 5-Fluorouracil: forty-plus and still ticking. A review of its preclinical and clinical development. Invest. New Drugs 2000, 18 (4), 299–313. [DOI] [PubMed] [Google Scholar]
- (125).Ghoshal K; Jacob ST An alternative molecular mechanism of action of 5-fluorouracil, a potent anticancer drug. Biochem. Pharmacol 1997, 53 (11), 1569–1575. [DOI] [PubMed] [Google Scholar]
- (126).Santi DV; Hardy LW Catalytic mechanism and inhibition of tRNA (uracil-5-)methyltransferase: evidence for covalent catalysis. Biochemistry 1987, 26 (26), 8599–8606. [DOI] [PubMed] [Google Scholar]
- (127).Huehls AM; Huntoon CJ; Joshi PM; Baehr CA; Wagner JM; Wang X; Lee MY; Karnitz LM Genomically incorporated 5-fluorouracil that escapes UNG-initiated base excision repair blocks DNA replication and activates homologous recombination. Mol. Pharmacol 2016, 89 (1), 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Berger SH; Pittman DL; Wyatt MD Uracil in DNA: consequences for carcinogenesis and chemotherapy. Biochem. Pharmacol 2008, 76 (6), 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Guillet M; Van Der Kemp PA; Boiteux S dUTPase activity is critical to maintain genetic stability in Saccharomyces cerevisiae. Nucleic Acids Res 2006, 34 (7), 2056–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Nilsen H; Rosewell I; Robins P; Skjelbred CF; Andersen S; Slupphaug G; Daly G; Krokan HE; Lindahl T; Barnes DE Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol. Cell 2000, 5 (6), 1059–1065. [DOI] [PubMed] [Google Scholar]
- (131).Pratt S; Shepard RL; Kandasamy RA; Johnston PA; Perry W 3rd; Dantzig AH The multidrug resistance protein 5 (ABCC5) confers resistance to 5-fluorouracil and transports its monophosphorylated metabolites. Mol. Cancer Ther 2005, 4 (5), 855–863. [DOI] [PubMed] [Google Scholar]
- (132).Guo Y; Kotova E; Chen ZS; Lee K; Hopper-Borge E; Belinsky MG; Kruh GD MRP8, ATP-binding cassette C11 (ABCC11), is a cyclic nucleotide efflux pump and a resistance factor for fluoropyrimidines 2′,3′-dideoxycytidine and 9′-(2′-phosphonylmethoxyethyl)adenine. J. Biol. Chem 2003, 278 (32), 29509–29514. [DOI] [PubMed] [Google Scholar]
- (133).Wang W; Zhao L; Wei X; Wang L; Liu S; Yang Y; Wang F; Sun G; Zhang J; Ma Y; et al. MicroRNA-320a promotes 5-FU resistance in human pancreatic cancer cells. Sci. Rep 2016, 6 (27641), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Wei X; Wang W; Wang L; Zhang Y; Zhang X; Chen M; Wang F; Yu J; Ma Y; Sun G MicroRNA-21 induces 5-fluorouracil resistance in human pancreatic cancer cells by regulating PTEN and PDCD4. Cancer Med 2016, 5 (4), 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Xie X; Liu H; Wang Y; Zhou Y; Yu H; Li G; Ruan Z; Li F; Wang X; Zhang J Nicotinamide N-methyltransferase enhances resistance to 5-fluorouracil in colorectal cancer cells through inhibition of the ASK1-p38 MAPK pathway. Oncotarget 2016, 45837–45848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Zhang X; Chen Y; Hao L; Hou A; Chen X; Li Y; Wang R; Luo P; Ruan Z; Ou J; et al. Macrophages induce resistance to 5-fluorouracil chemotherapy in colorectal cancer through the release of putrescine. Cancer Lett 2016, 381 (2), 305–313. [DOI] [PubMed] [Google Scholar]
- (137).Zhao T; Xu Z; Gu D; Wu P; Huo X; Wei X; Tang Y; Gong W; He ML; Chen J The effects of genomic polymorphisms in one-carbon metabolism pathways on survival of gastric cancer patients received fluorouracil-based adjuvant therapy. Sci. Rep 2016, 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Mader RM; Muller M; Steger GG Resistance to 5-fluorouracil. Gen. Pharmacol 1998, 31 (5), 661–666. [DOI] [PubMed] [Google Scholar]
- (139).Schoffski P The modulated oral fluoropyrimidine prodrug S-1, and its use in gastrointestinal cancer and other solid tumors. Anti-Cancer Drugs 2004, 15 (2), 85–106. [DOI] [PubMed] [Google Scholar]
- (140).Kono A; Hara Y; Sugata S; Karube Y; Matsushima Y; Ishitsuka H Activation of 5′-deoxy-5-fluorouridine by thymidine phosphorylase in human tumors. Chem. Pharm. Bull 1983, 31 (1), 175–178. [DOI] [PubMed] [Google Scholar]
- (141).Nishioka M; Miyamoto H; Kurita N; Higashijima J; Yoshikawa K; Miyatani T; Shimada M Pyrimidine nucleoside phosphorylase and dihydropyrimidine dihydrogenase activities as predictive factors for the efficacy of doxifluridine together with mitomycin C as adjuvant chemotherapy in primary colorectal cancer. Hepatogastroenterology 2007, 54 (76), 1089–1093. [PubMed] [Google Scholar]
- (142).Osanai T; Ichikawa W; Takagi Y; Uetake H; Nihei Z; Sugihara K Expression of pyrimidine nucleoside phosphorylase (PyNPase) in colorectal cancer. Jnp. J. Clin. Onco 2001, 31 (10), 500–505. [DOI] [PubMed] [Google Scholar]
- (143).Noguchi T; Moriyama H; Wada S; Takeno S; Kimura Y; Uchida Y; Gabbert HE High level concentration of pyrimidine nucleoside phosphorylase in esophageal squamous cell carcinoma but no correlation with clinicopathological parameters. Dis. Esophagus 2003, 16 (4), 307–311. [DOI] [PubMed] [Google Scholar]
- (144).Mori K; Hasegawa M; Nishida M; Toma H; Fukuda M; Kubota T; Nagasue N; Yamana H; Hirakawa YSCK; Ikeda T; et al. Expression levels of thymidine phosphorylase and dihydropyrimidine dehydrogenase in various human tumor tissues. Int. J. Oncol 2000, 17 (1), 33–38. [DOI] [PubMed] [Google Scholar]
- (145).Ogata Y; Sasatomi T; Mori S; Matono K; Ishibashi N; Akagi Y; Fukushima T; Murakami H; Ushijima M; Shirouzu K Significance of thymidine phosphorylase in metronomic chemotherapy using CPT-11 and doxifluridine for advanced colorectal carcinoma. Anticancer Res 2007, 27 (4C), 2605–2611. [PubMed] [Google Scholar]
- (146).Lamont EB; Schilsky RL The oral fluoropyrimidines in cancer chemotherapy. Clin. Cancer Res 1999, 5 (9), 2289–2296. [PubMed] [Google Scholar]
- (147).Alberto P; Mermillod B; Germano G; Kaplan S; Weber W; Joss R; Spati B; Martz G; Cavalli F A randomized comparison of doxifluridine and fluorouracil in colorectal carcinoma. Eur. J. Cancer Clin. Oncol 1988, 24 (3), 559–563. [DOI] [PubMed] [Google Scholar]
- (148).Kang YK; Yook JH; Chang HM; Ryu MH; Yoo C; Zang DY; Lee JL; Kim TW; Yang DH; Jang SJ; et al. Enhanced efficacy of postoperative adjuvant chemotherapy in advanced gastric cancer: results from a phase 3 randomized trial (AMC0101). Cancer Chemother. Pharmacol 2014, 73 (1), 139–149. [DOI] [PubMed] [Google Scholar]
- (149).Herr RJ In Modern Drug Synthesis; Wiley: Hoboken, NJ, 2010; pp 57–71. [Google Scholar]
- (150).Shimma N; Umeda I; Arasaki M; Murasaki C; Masubuchi K; Kohchi Y; Miwa M; Ura M; Sawada N; Tahara H; Kuruma I; Horii I; Ishitsuka H The design and synthesis of a new tumor-selective fluoropyrimidine carbamate, capecitabine. Bioorg. Med. Chem 2000, 8 (7), 1697–1706. [DOI] [PubMed] [Google Scholar]
- (151).Walko CM; Lindley C Capecitabine: A review. Clin. Ther 2005, 27 (1), 23–44. [DOI] [PubMed] [Google Scholar]
- (152).Ishitsuka H In Discovery and preclinical pharmacology of capecitabine; Humana Press: Totowa, NJ, 2003; pp 249–259. [Google Scholar]
- (153).Ishikawa T; Utoh M; Sawada N; Nishida M; Fukase Y; Sekiguchi F; Ishitsuka H Tumor selective delivery of 5-fluorouracil by capecitabine, a new oral fluoropyrimidine carbamate, in human cancer xenografts. Biochem. Pharmacol 1998, 55 (7), 1091–1097. [DOI] [PubMed] [Google Scholar]
- (154).Richardson MA Complementary and alternative therapy use in gynecologic oncology: implications for clinical practice. Gynecol. Oncol 2002, 84 (3), 360–362. [DOI] [PubMed] [Google Scholar]
- (155).Kosuri KV; Wu X; Wang L; Villalona-Calero MA; Otterson GA An epigenetic mechanism for capecitabine resistance in mesothelioma. Biochem. Biophys. Res. Commun 2010, 391 (3), 1465–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).McGuigan C; Murziani P; Slusarczyk M; Gonczy B; Vande Voorde J; Liekens S; Balzarini J Phosphoramidate ProTides of the anticancer agent FUDR successfully deliver the preformed bioactive monophosphate in cells and confer advantage over the parent nucleoside. J. Med. Chem 2011, 54 (20), 7247–7258. [DOI] [PubMed] [Google Scholar]
- (157).Vande Voorde J; Liekens S; McGuigan C; Murziani PG; Slusarczyk M; Balzarini J The cytostatic activity of NUC-3073, a phosphoramidate prodrug of 5-fluoro-2′-deoxyuridine, is independent of activation by thymidine kinase and insensitive to degradation by phosphorolytic enzymes. Biochem. Pharmacol 2011, 82 (5), 441–452. [DOI] [PubMed] [Google Scholar]
- (158).Tsume Y; Hilfinger JM; Amidon GL Enhanced cancer cell growth inhibition by dipeptide prodrugs of floxuridine: increased transporter affinity and metabolic stability. Mol. Pharmaceutics 2008, 5 (5), 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).DiMasi JA; Paquette C The economics of follow-on drug research and development: trends in entry rates and the timing of development. PharmacoEconomics 2004, 22 (Suppl 2), 1–14. [DOI] [PubMed] [Google Scholar]
- (160).Homsi J; Garrett CR Hepatic arterial infusion of chemotherapy for hepatic metastases from colorectal cancer. Cancer Control 2006, 13 (1), 42–47. [DOI] [PubMed] [Google Scholar]
- (161).Landowski CP; Song X; Lorenzi PL; Hilfinger JM; Amidon GL Floxuridine amino acid ester prodrugs: enhancing Caco-2 permeability and resistance to glycosidic bond metabolism. Pharm. Res 2005, 22 (9), 1510–1518. [DOI] [PubMed] [Google Scholar]
- (162).Power DG; Kemeny NE The role of floxuridine in metastatic liver disease. Mol. Cancer Ther 2009, 8 (5), 1015–1025. [DOI] [PubMed] [Google Scholar]
- (163).Konstantinidis IT; Groot Koerkamp B; Do RK; Gonen M; Fong Y; Allen PJ; D’Angelica MI; Kingham TP; DeMatteo RP; Klimstra DS; et al. Unresectable intrahepatic cholangiocarcinoma: Systemic plus hepatic arterial infusion chemotherapy is associated with longer survival in comparison with systemic chemotherapy alone. Cancer 2016, 122 (5), 758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Li C; Gu Y; Zhao M; Yuan Y; Wang F; Wang Z; Li W; Luo H; Chen C; Chen G; et al. Phase I trial of hepatic arterial infusion (HAI) of floxuridine with modified oxaliplatin, 5-fluorouracil and leucovorin (m-FOLFOX6) in Chinese patients with unresectable liver metastases from colorectal cancer. Cancer Chemother. Pharmacol 2014, 74 (5), 1079–1087. [DOI] [PubMed] [Google Scholar]
- (165).Ko YJ; Karanicolas PJ Hepatic arterial infusion pump chemotherapy for colorectal liver metastases: An old technology in a new era. Curr. Oncol 2014, 21 (1), e116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Murakami Y; Kazuno H; Emura T; Tsujimoto H; Suzuki N; Fukushima M Different mechanisms of acquired resistance to fluorinated pyrimidines in human colorectal cancer cells. Int. J. Oncol 2000, 17 (2), 277–283. [DOI] [PubMed] [Google Scholar]
- (167).Libra M; Navolanic PM; Talamini R; Cecchin E; Sartor F; Tumolo S; Masier S; Travali S; Boiocchi M; Toffoli G Thymidylate synthetase mRNA levels are increased in liver metastases of colorectal cancer patients resistant to fluoropyrimidine-based chemotherapy. BMC Cancer 2004, 4 (11), 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Beumer JH; Eiseman JL; Gilbert JA; Holleran JL; Yellow-Duke AE; Clausen DM; D’Argenio DZ; Ames MM; Hershberger PA; Parise RA; et al. Plasma pharmacokinetics and oral bioavailability of the 3,4,5,6-tetrahydrouridine (THU) prodrug, triacetyl-THU (taTHU), in mice. Cancer Chemother. Pharmacol 2011, 67 (2), 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Ebrahem Q; Mahfouz RZ; Ng KP; Saunthararajah Y High cytidine deaminase expression in the liver provides sanctuary for cancer cells from decitabine treatment effects. Oncotarget 2012, 3 (10), 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Funamizu N; Lacy CR; Fujita K; Furukawa K; Misawa T; Yanaga K; Manome Y Tetrahydrouridine inhibits cell proliferation through cell cycle regulation regardless of cytidine deaminase expression levels. PLoS One 2012, 7 (5), e37424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Neil GL; Moxley TE; Manak RC Enhancement by tetrahydrouridine of 1-β-D-arabinofuranosylcytosine (cytarabine) oral activity in L1210 leukemic mice. Cancer Res 1970, 30 (8), 2166–2172. [PubMed] [Google Scholar]
- (172).Kelly CJ; Gaudio L; Yesair DW; Schoenemann PT; Wodinsky I Pharmacokinetic considerations in evaluating the effects of tetrahydrouridine on 5-azacytidine chemotherapy in L1210 leukemic mice. Cancer Treat. Rep 1978, 62 (7), 1025–1032. [PubMed] [Google Scholar]
- (173).Laliberte J; Marquez VE; Momparler RL Potent inhibitors for the deamination of cytosine arabinoside and 5-aza-2′-deoxycytidine by human cytidine deaminase. Cancer Chemother. Pharmacol 1992, 30 (1), 7–11. [DOI] [PubMed] [Google Scholar]
- (174).Beumer JH; Eiseman JL; Parise RA; Florian JA Jr.; Joseph E; D’Argenio DZ; Parker RS; Kay B; Covey JM; Egorin MJ Plasma pharmacokinetics and oral bioavailability of 3,4,5,6-tetrahydrouridine, a cytidine deaminase inhibitor, in mice. Cancer Chemother. Pharmacol 2008, 62 (3), 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Sugiyama E; Kaniwa N; Kim SR; Kikura-Hanajiri R; Hasegawa R; Maekawa K; Saito Y; Ozawa S; Sawada J; Kamatani N; et al. Pharmacokinetics of gemcitabine in Japanese cancer patients: the impact of a cytidine deaminase polymorphism. J. Clin. Oncol 2006, 25 (1), 32–42. [DOI] [PubMed] [Google Scholar]
- (176).Yonemori K; Ueno H; Okusaka T; Yamamoto N; Ikeda M; Saijo N; Yoshida T; Ishii H; Furuse J; Sugiyama E; et al. Severe drug toxicity associated with a single-nucleotide polymorphism of the cytidine deaminase gene in a Japanese cancer patient treated with gemcitabine plus cisplatin. Clin. Cancer Res 2005, 11 (7), 2620–2624. [DOI] [PubMed] [Google Scholar]
- (177).Micozzi D; Carpi FM; Pucciarelli S; Polzonetti V; Polidori P; Vilar S; Williams B; Costanzi S; Vincenzetti S Human cytidine deaminase: a biochemical characterization of its naturally occurring variants. Int. J. Biol. Macromol 2014, 63, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Beumer JH; Eiseman JL; Parise RA; Joseph E; Holleran JL; Covey JM; Egorin MJ Pharmacokinetics, metabolism, and oral bioavailability of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine in mice. Clin. Cancer Res 2006, 12 (24), 7483–7491. [DOI] [PubMed] [Google Scholar]
- (179).Kaysen J; Spriggs D; Kufe D Incorporation of 5-fluorodeoxycytidine and metabolites into nucleic acids of human MCF-7 breast carcinoma cells. Cancer Res 1986, 46 (9), 4534–4538. [PubMed] [Google Scholar]
- (180).Beumer JH; Joseph E; Egorin MJ; Covey JM; Eiseman JL Quantitative determination of zebularine (NSC 309132), a DNA methyltransferase inhibitor, and three metabolites in murine plasma by high-performance liquid chromatography coupled with on-line radioactivity detection. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci 2006, 831 (1–2), 147–155. [DOI] [PubMed] [Google Scholar]
- (181).Morfouace M; Nimmervoll B; Boulos N; Patel YT; Shelat A; Freeman BB 3rd; Robinson GW; Wright K; Gajjar A; Stewart CF; et al. Preclinical studies of 5-fluoro-2′-deoxycytidine and tetrahydrouridine in pediatric brain tumors. J. Neuro-Oncol 2016, 126 (2), 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Holleran JL; Eiseman JL; Parise RA; Kummar S; Beumer JH LC-MS/MS assay for the quantitation of FdCyd and its metabolites FdUrd and FU in human plasma. J. Pharm. Biomed. Anal 2016, 129, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Newman EM; Morgan RJ; Kummar S; Beumer JH; Blanchard MS; Ruel C; El-Khoueiry AB; Carroll MI; Hou JM; Li C; et al. A phase I, pharmacokinetic, and pharmacodynamic evaluation of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine, administered with tetrahydrouridine. Cancer Chemother. Pharmacol 2015, 75 (3), 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Holleran JL; Beumer JH; McCormick DL; Johnson WD; Newman EM; Doroshow JH; Kummar S; Covey JM; Davis M; Eiseman JL Oral and intravenous pharmacokinetics of 5-fluoro-2′-deoxycytidine and THU in cynomolgus monkeys and humans. Cancer Chemother. Pharmacol 2015, 76 (4), 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Ferraris D; Duvall B; Delahanty G; Mistry B; Alt J; Rojas C; Rowbottom C; Sanders K; Schuck E; Huang KC; et al. Design, synthesis, and pharmacological evaluation of fluorinated tetrahydrouridine derivatives as inhibitors of cytidine deaminase. J. Med. Chem 2014, 57 (6), 2582–2588. [DOI] [PubMed] [Google Scholar]
- (186).Sakamoto J; Oba K; Matsui T; Kobayashi M Efficacy of oral anticancer agents for colorectal cancer. Dis. Colon Rectum 2006, 49 (10 Suppl), S82–91. [DOI] [PubMed] [Google Scholar]
- (187).Hsu CH; Shen YC; Lin ZZ; Chen PJ; Shao YY; Ding YH; Hsu C; Cheng AL Phase II study of combining sorafenib with metronomic tegafur/uracil for advanced hepatocellular carcinoma. J. Hepatol 2010, 53 (1), 126–131. [DOI] [PubMed] [Google Scholar]
- (188).Osugi H; Takada N; Takemura M; Kaseno S; Lee S; Ueno M; Tanaka Y; Fukuhara K; Fujiwara Y; Kinoshita H Oral fluoropyrimidine anticancer drug TS-1 for gastric cancer patients with peritoneal dissemination. Oncol. Rep 2002, 9 (4), 811–815. [PubMed] [Google Scholar]
- (189).Rich TA; Shepard RC; Mosley ST Four decades of continuing innovation with fluorouracil: current and future approaches to fluorouracil chemoradiation therapy. J. Clin. Oncol 2004, 22 (11), 2214–2232. [DOI] [PubMed] [Google Scholar]
- (190).Utsugi T New challenges and inspired answers for anticancer drug discovery and development. Jpn. J. Clin. Oncol 2013, 43 (10), 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Yamada T; Okamura S; Okazaki T; Ushiroyama T; Yanagawa Y; Ueki M; Sugimoto O; Yamazaki H; Sugino M; Masui Y Leukoencephalopathy following treatment with carmofur: a case report and review of the Japanese literature. Asia-Oceania J. Obstet. Gynaecol 1989, 15 (2), 161–168. [DOI] [PubMed] [Google Scholar]
- (192).Mizutani T [Leukoencephalopathy caused by antineoplastic drugs]. Brain Nerve 2008, 60 (2), 137–141. [PubMed] [Google Scholar]
- (193).Baehring JM; Fulbright RK Delayed leukoencephalopathy with stroke-like presentation in chemotherapy recipients. J. Neurol., Neurosurg. Psychiatry 2008, 79 (5), 535–539. [DOI] [PubMed] [Google Scholar]
- (194).Yamamoto M; Arii S; Sugahara K; Tobe T Adjuvant oral chemotherapy to prevent recurrence after curative resection for hepatocellular carcinoma. Br. J. Surg 1996, 83 (3), 336–340. [DOI] [PubMed] [Google Scholar]
- (195).Lee CH; Wu CL; Shiau AL Hypoxia-induced cytosine deaminase gene expression for cancer therapy. Hum. Gene Ther 2007, 18 (1), 27–38. [DOI] [PubMed] [Google Scholar]
- (196).Matuskova M; Baranovicova L; Kozovska Z; Durinikova E; Pastorakova A; Hunakova L; Waczulikova I; Nencka R; Kucerova L Intrinsic properties of tumour cells have a key impact on the bystander effect mediated by genetically engineered mesenchymal stromal cells. J. Gene Med 2012, 14 (12), 776–787. [DOI] [PubMed] [Google Scholar]
- (197).Wheeler HL; Merriam HF Über einige kondensationsprodukte des pseudothioharnstoffes: Synthese von uracil, thymin und ähnlichen verbindungen. Am. Chem. J 1903, 29, 378. [Google Scholar]
- (198).Dornow A; Boberg F; Schurer L Various condensations with chloroformylacetic acid ethyl ester. VIII. Synthesis of nitrogenous heterocycles. Arch. Pharm 1953, 286 (10), 494–501. [DOI] [PubMed] [Google Scholar]
- (199).Duschinsky R; Edward Pleven E; Heidelberger C The synthesis of 5-fluoropyrimides. J. Am. Chem. Soc 1957, 79 (16), 4559–4560. [Google Scholar]
- (200).Robins MJ; MacCoss M; Naik SR; Ramani G Nucleic acid related compounds. 21. Direct fluorination of uracil and cytosine bases and nucleosides using trifluoromethyl hypofluorite. Mechanism, stereochemistry, and synthetic applications. J. Am. Chem. Soc 1976, 98 (23), 7381–7389. [DOI] [PubMed] [Google Scholar]
- (201).Yamazaki A; Morisawa H; Oda Y; Uchida K US Patent 4186266, 1980.
- (202).Baasner B; Klauke E A new route to the synthesis of 5-fluorouracil. J. Fluorine Chem 1989, 45 (3), 417–430. [Google Scholar]
- (203).Kamiya T; Ishiduka I; Nakajima H US Patent 5453497, 1995.
- (204).Robins MJ; Naik SR A direct synthesis of 5-fluorocytosine and its nucleosides using trifluoromethyl hypofluorite. J. Chem. Soc., Chem. Commun 1972, No. 1, 18–19. [Google Scholar]
- (205).Brinkman HR; Kalaritis P; Morrissey JF US Patent 5476932, 1995.
- (206).Gore VG; Patkar LN; Bhalerao R; Hublikar MG; Pokharkar KS WO Patent 2011104540, 2011.
- (207).Cook AF Fluorinated Pyrimidine nucleosides. 1. Synthesis of a nitrogen analogue of the antitumor agent 2,2′-anhydro-1-beta-D-arabinofuranosyl-5-fluorocytosine hydrochloride. J. Med. Chem 1977, 20 (3), 344–348. [DOI] [PubMed] [Google Scholar]
- (208).Cook AF; Holman MJ; Kramer MJ; Trown PW Fluorinated pyrimidine nucleosides. 3. Synthesis and antitumor activity of a series of 5′-deoxy-5-fluoropyrimidine nucleosides. J. Med. Chem 1979, 22 (11), 1330–1335. [DOI] [PubMed] [Google Scholar]
- (209).Shen B; Jamison TF Rapid continuous synthesis of 5′-deoxyribonucleosides in flow via Bronsted acid catalyzed glycosylation. Org. Lett 2012, 14 (13), 3348–3351. [DOI] [PubMed] [Google Scholar]
- (210).Duschinsky R; Pleven E; Malbica J; Heidelberger C Synthesis of 5-fluorouracil nucleosides. 132nd Meeting Am. Chem. Soc., New York, 1957; p 19c. [Google Scholar]
- (211).Fox JJ; Wempen I; Duschinsky R Nucleosides of 5-fluorocytosine. 4th Intl. Cong. of Biochem., Vienna, 1958; p 6. [Google Scholar]
- (212).Hoffer M; Duschinsky R; Fox JJ; Yung N Simple syntheses of pyrimidine-2′-deoxy-ribonucleosides. J. Am. Chem. Soc 1959, 81 (15), 4112–4113. [Google Scholar]
- (213).Hanze AR Nucleic acids. IV. The catalytic reduction of pyrimidine nucleosides (human liver deaminase inhibitors). J. Am. Chem. Soc 1967, 89 (25), 6720–6725. [DOI] [PubMed] [Google Scholar]
- (214).Aoyama H Stereoselective synthesis of anomers of 5-substituted 2′-deoxyuridines. Bull. Chem. Soc. Jpn 1987, 60 (6), 2073–2077. [Google Scholar]
- (215).Wang ZX; Duan W; Wiebe LI; Balzarini J; De Clercq E; Knaus EE Synthesis of 1-(2-deoxy-β-D-ribofuranosyl)-2,4-difluoro-5-substituted-benzene thymidine mimics, some related alpha-anomers, and their evaluation as antiviral and anticancer agents. Nucleosides, Nucleotides Nucleic Acids 2001, 20 (1–2), 11–40. [DOI] [PubMed] [Google Scholar]
- (216).Kotick MP; Szantay C; Bardos TJ Synthesis of 5-S-substituted 2′-deoxyuridines. Study of the factors influencing stereoselectivity of the silyl modification of the Hilbert-Johnson reaction. J. Org. Chem 1969, 34 (12), 3806–3813. [DOI] [PubMed] [Google Scholar]
- (217).Yasumoto M; Marunaka T; Hashimoto S; Harima K; Suzue T JP Patent 76146482, 1976.
- (218).Miyashita O; Masumura K; Shimadzu H; Hashimoto N Studies on fluorinated pyrimidines. I. A new method of synthesizing 5-fluorouracil and its derivatives. Chem. Pharm. Bull 1981, 29 (11), 3181–3190. [Google Scholar]
- (219).Ozaki S; Ike Y; Mizuno H; Ishikawa K; Mori H The synthesis of 1-carbamoyl-5-fluorouracils. Bull. Chem. Soc. Jpn 1977, 50 (9), 2406–2412. [Google Scholar]
- (220).Ozaki S Synthesis and antitumor activity of 5-fluorouracil derivatives. Med. Res. Rev 1996, 16 (1), 51–86. [DOI] [PubMed] [Google Scholar]
- (221).Xiong J; Zhou S; Huang Z; Chao G; Wang L; Xu J CN Patent 102229571, 2011.
- (222).Kolder CR; den Hertog HJ Synthesis and reactivity of 5-chloro-2,4-dihydroxypyridine. Rec. Trav. Chim 1953, 72, 285–295. [Google Scholar]
- (223).Yano S; Ohno T; Ogawa K Convenient and practical synthesis of 5-chloro-4-hydroxy-2(1H)-pyridinone. Heterocycles 1993, 36, 145–148. [Google Scholar]
- (224).Mittelbach M; Kastner G; Junek H Synthesen mit Nitrilen, 71. Mitt. Zur Synthese von 4-Hydroxynicotinsaure aus Butadiendicarbonitrilen. Arch. Pharm 1985, 318 (6), 481–486. [Google Scholar]
- (225).Xu Y; Mao D; Zhang F CN Patent 1915976, 2007.
- (226).Poje M; Sokolić-Maravić L The mechanism for the conversion of uric acid into allantoin and dehydro-allantoin: A new look at an old problem. Tetrahedron 1986, 42 (2), 747–751. [Google Scholar]
- (227).Sugi M; Igi M EP Patent 0957096, 1999.
- (228).Temmink OH; Emura T; de Bruin M; Fukushima M; Peters GJ Therapeutic potential of the dual-targeted TAS-102 formulation in the treatment of gastrointestinal malignancies. Cancer Sci 2007, 98 (6), 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Heidelberger C; Anderson SW Fluorinated pyrimidines. XXI. The tumor-inhibitory activity of 5-trifluoromethyl-2′-deoxyuridine. Cancer Res 1964, 24, 1979–1985. [PubMed] [Google Scholar]
- (230).Heidelberger C; Boohar J; Kampschroer B Fluorinated pyrimidines. XXIV. In vivo metabolism of 5-trifluoromethyluracil-2-C-14 and 5-trifluoromethyl-2′-deoxyuridine-2-C-14. Cancer Res 1965, 25, 377–381. [PubMed] [Google Scholar]
- (231).Heidelberger C; Parsons DG; Remy DC Syntheses of 5-trifluoromethyluracil and 5-trifluoromethyl-2′-deoxyuridine. J. Med. Chem 1964, 7, 1–5. [DOI] [PubMed] [Google Scholar]
- (232).Dexter DL; Wolberg WH; Ansfield FJ; Helson L; Heidelberger C The clinical pharmacology of 5-trifluoromethyl-2′-deoxyuridine. Cancer Res 1972, 32 (2), 247–253. [PubMed] [Google Scholar]
- (233).Doi T; Ohtsu A; Yoshino T; Boku N; Onozawa Y; Fukutomi A; Hironaka S; Koizumi W; Sasaki T Phase I study of TAS-102 treatment in Japanese patients with advanced solid tumours. Br. J. Cancer 2012, 107 (3), 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Emura T; Nakagawa F; Fujioka A; Ohshimo H; Yokogawa T; Okabe H; Kitazato K An optimal dosing schedule for a novel combination antimetabolite, TAS-102, based on its intracellular metabolism and its incorporation into DNA. Int. J. Mol. Med 2004, 13 (2), 249–255. [PubMed] [Google Scholar]
- (235).Reyes P; Heidelberger C Fluorinated pyrimidines. XXVI. Mammalian thymidylate synthetase: its mechanism of action and inhibition by fluorinated nucleotides. Mol. Pharm 1965, 1 (1), 14–30. [PubMed] [Google Scholar]
- (236).Eckstein JW; Foster PG; Finer-Moore J; Wataya Y; Santi DV Mechanism-based inhibition of thymidylate synthase by 5-(trifluoromethyl)-2′-deoxyuridine 5′-monophosphate. Biochemistry 1994, 33 (50), 15086–15094. [DOI] [PubMed] [Google Scholar]
- (237).Peters GJ; Backus HH; Freemantle S; van Triest B; Codacci-Pisanelli G; van der Wilt CL; Smid K; Lunec J; Calvert AH; Marsh S; et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim. Biophys. Acta, Mol. Basis Dis 2002, 1587 (2–3), 194–205. [DOI] [PubMed] [Google Scholar]
- (238).Emura T; Suzuki N; Yamaguchi M; Ohshimo H; Fukushima M A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FU-resistant human cancer cells through a mechanism involving FTD incorporation in DNA. Int. J. Oncol 2004, 25 (3), 571–578. [PubMed] [Google Scholar]
- (239).Suzuki N; Emura T; Fukushima M Mode of action of trifluorothymidine (TFT) against DNA replication and repair enzymes. Int. J. Oncol 2011, 39 (1), 263–270. [DOI] [PubMed] [Google Scholar]
- (240).Suzuki N; Nakagawa F; Nukatsuka M; Fukushima M Trifluorothymidine exhibits potent antitumor activity via the induction of DNA double-strand breaks. Exp. Ther. Med 2011, 2 (3), 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Markley JC; Chirakul P; Sologub D; Sigurdsson ST Incorporation of 2′-deoxy-5-(trifluoromethyl)uridine and 5-cyano-2′-deoxyuridine into DNA. Bioorg. Med. Chem. Lett 2001, 11 (18), 2453–2455. [DOI] [PubMed] [Google Scholar]
- (242).Emura T; Suzuki N; Fujioka A; Ohshimo H; Fukushima M Potentiation of the antitumor activity of alpha, alpha, alpha-trifluorothymidine by the co-administration of an inhibitor of thymidine phosphorylase at a suitable molar ratio in vivo. Int. J. Oncol 2005, 27 (2), 449–455. [PubMed] [Google Scholar]
- (243).Fukushima M; Suzuki N; Emura T; Yano S; Kazuno H; Tada Y; Yamada Y; Asao T Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2′-deoxyribonucleosides. Biochem. Pharmacol 2000, 59 (10), 1227–1236. [DOI] [PubMed] [Google Scholar]
- (244).Ursem C; Van Loon K; Venook A Adjuvant Therapy Trials. Cancer J 2016, 22 (3), 196–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).De Clercq E Antiviral drugs in current clinical use. J. Clin. Virol 2004, 30 (2), 115–133. [DOI] [PubMed] [Google Scholar]
- (246).De Clercq E Selective anti-herpesvirus agents. Antiviral Chem. Chemo 2013, 23 (3), 93–101. [DOI] [PubMed] [Google Scholar]
- (247).Komatsu H; Umetani H Synthesis of trifluorothymidine: Green tlycosylation condition using neither chloroform nor transition metals. Org. Process Res. Dev 2002, 6, 847–850. [Google Scholar]
- (248).Hubbard AJ; Jones AS; Walker RT An investigation by 1H NMR spectroscopy into the factors determining the β:α ratio of the product in 2′-deoxynucleoside synthesis. Nucleic Acids Res 1984, 12 (17), 6827–6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Yano S; Kazuno H; Sato T; Suzuki N; Emura T; Wierzba K; Yamashita J; Tada Y; Yamada Y; Fukushima M; Asao T Synthesis and evaluation of 6-methylene-bridged uracil derivatives. Part 2: Optimization of inhibitors of human thymidine phosphorylase and their selectivity with uridine phosphorylase. Bioorg. Med. Chem 2004, 12, 3443–3450. [DOI] [PubMed] [Google Scholar]
- (250).Moriconi EJ; Cevasco AA Synthesis and reactions of cyclic amidines. J. Org. Chem 1968, 33 (5), 2109–2111. [Google Scholar]
- (251).Sun L; Li J; Bera H; Dolzhenko AV; Chiu GN; Chui WK Fragment-based approach to the design of 5-chlorouracil-linked-pyrazolo[1,5-a][1,3,5]triazines as thymidine phosphorylase inhibitors. Eur. J. Med. Chem 2013, 70, 400–410. [DOI] [PubMed] [Google Scholar]
- (252).Yano S; Kazuno H; Suzuki N; Emura T; Wierzba K; Yamashita J; Tada Y; Yamada Y; Fukushima M; Asao T Synthesis and evaluation of 6-methylene-bridged uracil derivatives. Part 1: Discovery of novel orally active inhibitors of human thymidine phosphorylase. Bioorg. Med. Chem 2004, 12, 3431–3441. [DOI] [PubMed] [Google Scholar]
- (253).Sorm F; Piskala A; Cihak A; Vesely J 5-azacytidine, a new, highly effective cancerostatic. Experientia 1964, 20 (4), 202–203. [DOI] [PubMed] [Google Scholar]
- (254).Sorm F; Vesely J Effect of 5-aza-2′-deoxycytidine against leukemic and hemopoietic tissues in AKR mice. Neoplasma 1968, 15 (4), 339–343. [PubMed] [Google Scholar]
- (255).Hubeek I; Stam RW; Peters GJ; Broekhuizen R; Meijerink JP; van Wering ER; Gibson BE; Creutzig U; Zwaan CM; Cloos J; et al. The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br. J. Cancer 2005, 93 (12), 1388–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Bouchard J; Momparler RL Incorporation of 5-Aza-2′-deoxycytidine-5′-triphosphate into DNA. Interactions with mammalian DNA polymerase alpha and DNA methylase. Mol. Pharm 1983, 24 (1), 109–114. [PubMed] [Google Scholar]
- (257).Singh V; Sharma P; Capalash N DNA methyltransferase-1 inhibitors as epigenetic therapy for cancer. Curr. Cancer Drug Targets 2013, 13 (4), 379–399. [DOI] [PubMed] [Google Scholar]
- (258).Hamm CA; Costa FF Epigenomes as therapeutic targets. Pharmacol. Ther 2015, 151, 72–86. [DOI] [PubMed] [Google Scholar]
- (259).Yoo J; Choi S; Medina-Franco JL Molecular modeling studies of the novel inhibitors of DNA methyltransferases SGI-1027 and CBC12: implications for the mechanism of inhibition of DNMTs. PLoS One 2013, 8 (4), e62152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Galm O; Herman JG; Baylin SB The fundamental role of epigenetics in hematopoietic malignancies. Blood Rev 2006, 20 (1), 1–13. [DOI] [PubMed] [Google Scholar]
- (261).Baylin SB DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol 2005, 2 (Suppl 1), S4–11. [DOI] [PubMed] [Google Scholar]
- (262).Legendre CR; Demeure MJ; Whitsett TG; Gooden GC; Bussey KJ; Jung S; Waibhav T; Kim S; Salhia B Pathway implications of aberrant global methylation in adrenocortical cancer. PLoS One 2016, 11 (3), e0150629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Wong KY; Chim CS DNA methylation of tumor suppressor protein-coding and non-coding genes in multiple myeloma. Epigenomics 2015, 7 (6), 985–1001. [DOI] [PubMed] [Google Scholar]
- (264).Qin T; Si J; Raynal NJ; Wang X; Gharibyan V; Ahmed S; Hu X; Jin C; Lu Y; Shu J; et al. Epigenetic synergy between decitabine and platinum derivatives. Clin. Epigenet 2015, 7 (97), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Al-Salihi M; Yu M; Burnett DM; Alexander A; Samlowski WE; Fitzpatrick FA The depletion of DNA methyltransferase-1 and the epigenetic effects of 5-aza-2′deoxycytidine (decitabine) are differentially regulated by cell cycle progression. Epigenetics 2011, 6 (8), 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Stresemann C; Lyko F Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123 (1), 8–13. [DOI] [PubMed] [Google Scholar]
- (267).Takahashi A; Imai Y; Yamakoshi K; Kuninaka S; Ohtani N; Yoshimoto S; Hori S; Tachibana M; Anderton E; Takeuchi T; et al. DNA damage signaling triggers degradation of histone methyltransferases through APC/C(Cdh1) in senescent cells. Mol. Cell 2012, 45 (1), 123–131. [DOI] [PubMed] [Google Scholar]
- (268).Ghoshal K; Datta J; Majumder S; Bai S; Kutay H; Motiwala T; Jacob ST 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell Biol 2005, 25 (11), 4727–4741. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (269).Rogstad DK; Herring JL; Theruvathu JA; Burdzy A; Perry CC; Neidigh JW; Sowers LC Chemical decomposition of 5-aza-2′-deoxycytidine (Decitabine): kinetic analyses and identification of products by NMR, HPLC, and mass spectrometry. Chem. Res. Toxicol 2009, 22 (6), 1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Gnyszka A; Jastrzebski Z; Flis S DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res 2013, 33 (8), 2989–2996. [PubMed] [Google Scholar]
- (271).Qin T; Castoro R; El Ahdab S; Jelinek J; Wang X; Si J; Shu J; He R; Zhang N; Chung W; et al. Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PLoS One 2011, 6 (8), e23372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Saunthararajah Y; Hillery CA; Lavelle D; Molokie R; Dorn L; Bressler L; Gavazova S; Chen YH; Hoffman R; DeSimone J Effects of 5-aza-2′-deoxycytidine on fetal hemoglobin levels, red cell adhesion, and hematopoietic differentiation in patients with sickle cell disease. Blood 2003, 102 (12), 3865–3870. [DOI] [PubMed] [Google Scholar]
- (273).Jones PA; Taylor SM Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20 (1), 85–93. [DOI] [PubMed] [Google Scholar]
- (274).Jansen RS; Rosing H; Wijermans PW; Keizer RJ; Schellens JH; Beijnen JH Decitabine triphosphate levels in peripheral blood mononuclear cells from patients receiving prolonged low-dose decitabine administration: a pilot study. Cancer Chemother. Pharmacol 2012, 69 (6), 1457–1466. [DOI] [PubMed] [Google Scholar]
- (275).Lavelle D; Chin J; Vaitkus K; Redkar S; Phiasivongsa P; Tang C; Will R; Hankewych M; Roxas B; Singh M; et al. Oral decitabine reactivates expression of the methylated gamma-globin gene in Papio anubis. Am. J. Hematol 2007, 82 (11), 981–985. [DOI] [PubMed] [Google Scholar]
- (276).Lavelle D; Vaitkus K; Ling Y; Ruiz MA; Mahfouz R; Ng KP; Negrotto S; Smith N; Terse P; Engelke KJ; et al. Effects of tetrahydrouridine on pharmacokinetics and pharmacodynamics of oral decitabine. Blood 2012, 119 (5), 1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Plagemann PG; Behrens M; Abraham D Metabolism and cytotoxicity of 5-azacytidine in cultured Novikoff rat hepatoma and P388 mouse leukemia cells and their enhancement by preincubation with pyrazofurin. Cancer Res 1978, 38 (8), 2458–2466. [PubMed] [Google Scholar]
- (278).Meininger V Clinical trials in ALS: what did we learn from recent trials in humans? Neurodegener. Dis 2005, 2 (3–4), 208–214. [DOI] [PubMed] [Google Scholar]
- (279).Lee TT; Momparier RL Inhibition of uridine-cytidine kinase by 5-azacytidine 5′-triphosphate. Med. Pediatr. Oncol 1976, 2 (3), 265–270. [DOI] [PubMed] [Google Scholar]
- (280).Garcia-Manero G Myelodysplastic syndromes: 2014 update on diagnosis, risk-stratification, and management. Am. J. Hematol 2014, 89 (1), 97–108. [DOI] [PubMed] [Google Scholar]
- (281).Raj K; Mufti GJ Azacytidine (Vidaza(R)) in the treatment of myelodysplastic syndromes. Ther. Clin. Risk. Manag 2006, 2 (4), 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Van Camp JA Decitabine (Dacogen): A DNA methyltransferace inhibitor for cancer; Wiley: Hoboken, NJ, 2010; pp 47–55. [Google Scholar]
- (283).Fanciullino R; Mercier C; Serdjebi C; Venton G; Colle J; Fina F; Ouafik L; Lacarelle B; Ciccolini J; Costello R Yin and yang of cytidine deaminase roles in clinical response to azacitidine in the elderly: a pharmacogenetics tale. Pharmacogenomics 2015, 16 (17), 1907–1912. [DOI] [PubMed] [Google Scholar]
- (284).Fanciullino R; Mercier C; Serdjebi C; Berda Y; Fina F; Ouafik L; Lacarelle B; Ciccolini J; Costello R Lethal toxicity after administration of azacytidine: implication of the cytidine deaminase-deficiency syndrome. Pharmacogenet. Genomics 2015, 25 (6), 317–321. [DOI] [PubMed] [Google Scholar]
- (285).Serdjebi C; Milano G; Ciccolini J Role of cytidine deaminase in toxicity and efficacy of nucleosidic analogs. Expert Opin. Drug Metab. Toxicol 2015, 11 (5), 665–672. [DOI] [PubMed] [Google Scholar]
- (286).Brueckner B; Rius M; Markelova MR; Fichtner I; Hals PA; Sandvold ML; Lyko F Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol. Cancer Ther 2010, 9 (5), 1256–1264. [DOI] [PubMed] [Google Scholar]
- (287).Hummel-Eisenbeiss J; Hascher A; Hals PA; Sandvold ML; Muller-Tidow C; Lyko F; Rius M The role of human equilibrative nucleoside transporter 1 on the cellular transport of the DNA methyltransferase inhibitors 5-azacytidine and CP-4200 in human leukemia cells. Mol. Pharmacol 2013, 84 (3), 438–450. [DOI] [PubMed] [Google Scholar]
- (288).Chabner BA; Drake JC; Johns DG Deamination of 5-azacytidine by a human leukemia cell cytidine deaminase. Biochem. Pharmacol 1973, 22 (21), 2763–2765. [DOI] [PubMed] [Google Scholar]
- (289).Scholl J; Joshi-Hangal R; Inloes R; Shi C; Tavema P; Choy GS; Redkar S; Azab M SGI-110, a novel second generation potent DNA methylation inhibitor, in development for the treatment of MDS and AML. Preclinical safety, pharmacokinetics, and DNA methylation results of a low volume subcutaneous (SC) formulation. Blood 2010, 116, 1872. [Google Scholar]
- (290).Tellez CS; Grimes MJ; Picchi MA; Liu Y; March TH; Reed MD; Oganesian A; Taverna P; Belinsky SA SGI-110 and entinostat therapy reduces lung tumor burden and reprograms the epigenome. Int. J. Cancer 2014, 135 (9), 2223–2231. [DOI] [PubMed] [Google Scholar]
- (291).Coral S; Parisi G; Nicolay HJ; Colizzi F; Danielli R; Fratta E; Covre A; Taverna P; Sigalotti L; Maio M Immunomodulatory activity of SGI-110, a 5-aza-2′-deoxycytidine-containing demethylating dinucleotide. Cancer Immunol. Immunother 2013, 62 (3), 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Srivastava P; Paluch BE; Matsuzaki J; James SR; Collamat-Lai G; Karbach J; Nemeth MJ; Taverna P; Karpf AR; Griffiths EA Immunomodulatory action of SGI-110, a hypomethylating agent, in acute myeloid leukemia cells and xenografts. Leuk. Res 2014, 38 (11), 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Fang F; Munck J; Tang J; Taverna P; Wang Y; Miller DF; Pilrose J; Choy G; Azab M; Pawelczak KS; et al. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clin. Cancer Res 2014, 20 (24), 6504–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Covre A; Parisi G; Nicolay HJ; Fonsatti E; Fratta E; Sigalotti L; Taverna P; Coral S; Maio M In vivo immunomodulatory activity of SGI-110, a second generation hypomethylating agent, in hematologic malignancies. AACR 104th Annual Meeting, Washington, DC, 2013. [Google Scholar]
- (295).Pískala A; Šorm F Nucleic acids components and their analogues. LI. Synthesis of 1-glycosyl derivatives of 5-azauracil and 5-azacytosine. Collect. Czech. Chem. Commun 1964, 29 (9), 2060–2076. [Google Scholar]
- (296).Ionescu D; Blumbergs P US Patent 7038038, 2006.
- (297).Vujjini SK; Varanasi G; Arevelli S; Kandala SC; Tirumalaraju SR; Bandichhor R; Kagga M; Cherukupally P An improved and scalable process for the synthesis of 5-azacytidine: An antineoplastic drug. Org. Process Res. Dev 2013, 17, 303–306. [Google Scholar]
- (298).Pliml J;Šorm F Synthesis of a 2-deoxy-D-ribofuranosyl-5-azacytosine. Collect. Czech. Chem. Commun 1964, 29 (10), 2576–2578. [Google Scholar]
- (299).Winkley MW; Robins RK Direct glycosylation of 1,3,5-triazinones. A new approach to the synthesis of the nucleoside antibiotic 5-azacytidine (4-amino-1-beta-D-ribofuranosyl-1,3,5-triazin-2-one) and related derivatives. J. Org. Chem 1970, 35 (2), 491–495. [DOI] [PubMed] [Google Scholar]
- (300).Lin KT; Momparler RL; Rivard GE High-performance liquid chromatographic analysis of chemical stability of 5-aza-2′-deoxycytidine. J. Pharm. Sci 1981, 70 (11), 1228–1232. [DOI] [PubMed] [Google Scholar]
- (301).Henschke JP; Zhang X; Yu J; Hu K; Mei L US Patent 8586729, 2013.
- (302).Kolla NK; Neelam UK; Baddam SR; Gangula S EP Patent 2424845, 2012.
- (303).Redkar S Scale up and development of a process for a low volume subcutaneous formulation of SGI-110, a potent hypomethylating agent. AACR Annual Meeting 2012, Chicago, IL, 2012. [Google Scholar]
- (304).Hertel LW; Boder GB; Kroin JS; Rinzel SM; Poore GA; Todd GC; Grindey GB Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine). Cancer Res 1990, 50 (14), 4417–4422. [PubMed] [Google Scholar]
- (305).Abratt RP; Bezwoda WR; Falkson G; Goedhals L; Hacking D; Rugg TA Efficacy and safety profile of gemcitabine in non-small-cell lung cancer: a phase II study. J. Clin. Oncol 1994, 12 (8), 1535–1540. [DOI] [PubMed] [Google Scholar]
- (306).Heinemann V Gemcitabine: progress in the treatment of pancreatic cancer. Oncology 2001, 60 (1), 8–18. [DOI] [PubMed] [Google Scholar]
- (307).Ansari D; Tingstedt B; Andersson R Pancreatic cancer - cost for overtreatment with gemcitabine. Acta Oncol 2013, 52 (6), 1146–1151. [DOI] [PubMed] [Google Scholar]
- (308).Ritzel MW; Ng AM; Yao SY; Graham K; Loewen SK; Smith KM; Hyde RJ; Karpinski E; Cass CE; Baldwin SA; et al. Recent molecular advances in studies of the concentrative Na +-dependent nucleoside transporter (CNT) family: identification and characterization of novel human and mouse proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides (system cib). Mol. Membr. Biol 2001, 18 (1), 65–72. [DOI] [PubMed] [Google Scholar]
- (309).Garcia-Manteiga J; Molina-Arcas M; Casado FJ; Mazo A; Pastor-Anglada M Nucleoside transporter profiles in human pancreatic cancer cells: role of hCNT1 in 2′,2′-difluorodeoxycytidine- induced cytotoxicity. Clin. Cancer Res 2003, 9 (13), 5000–5008. [PubMed] [Google Scholar]
- (310).Perez-Torras S; Garcia-Manteiga J; Mercade E; Casado FJ; Carbo N; Pastor-Anglada M; Mazo A Adenoviral-mediated overexpression of human equilibrative nucleoside transporter 1 (hENT1) enhances gemcitabine response in human pancreatic cancer. Biochem. Pharmacol 2008, 76 (3), 322–329. [DOI] [PubMed] [Google Scholar]
- (311).Bergman AM; Peters GJ Gemcitabine; Humana Press: Totowa, NJ, 2006; pp 225–251. [Google Scholar]
- (312).Huang P; Chubb S; Hertel LW; Grindey GB; Plunkett W Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res 1991, 51 (22), 6110–6117. [PubMed] [Google Scholar]
- (313).Mini E; Nobili S; Caciagli B; Landini I; Mazzei T Cellular pharmacology of gemcitabine. Ann. Oncol 2006, 17 (Suppl 5), v7–12. [DOI] [PubMed] [Google Scholar]
- (314).Wickremsinhe ER; Lutzke BS; Jones BR; Schultz GA; Freeman AB; Pratt SE; Bones AM; Ackermann BL Quantification of gemcitabine incorporation into human DNA by LC/MS/MS as a surrogate measure for target engagement. Anal. Chem 2010, 82 (15), 6576–6583. [DOI] [PubMed] [Google Scholar]
- (315).Ruiz van Haperen VW; Veerman G; Vermorken JB; Peters GJ 2′,2′-Difluoro-deoxycytidine (gemcitabine) incorporation into RNA and DNA of tumour cell lines. Biochem. Pharmacol 1993, 46 (4), 762–766. [DOI] [PubMed] [Google Scholar]
- (316).Modrak DE; Cardillo TM; Newsome GA; Goldenberg DM; Gold DV Synergistic interaction between sphingomyelin and gemcitabine potentiates ceramide-mediated apoptosis in pancreatic cancer. Cancer Res 2004, 64 (22), 8405–8410. [DOI] [PubMed] [Google Scholar]
- (317).Heinemann V; Xu YZ; Chubb S; Sen A; Hertel LW; Grindey GB; Plunkett W Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol. Pharm 1990, 38 (4), 567–572. [PubMed] [Google Scholar]
- (318).Muggia F; Diaz I; Peters GJ Nucleoside and nucleobase analogs in cancer treatment: not only sapacitabine, but also gemcitabine. Expert Opin. Invest. Drugs 2012, 21 (4), 403–408. [DOI] [PubMed] [Google Scholar]
- (319).Plunkett W; Huang P; Searcy CE; Gandhi V Gemcitabine: preclinical pharmacology and mechanisms of action. Semin. Oncol 1996, 23 (5 Suppl 10), 3–15. [PubMed] [Google Scholar]
- (320).Cerqueira NM; Fernandes PA; Ramos MJ Understanding ribonucleotide reductase inactivation by gemcitabine. Chem. - Eur. J 2007, 13 (30), 8507–8515. [DOI] [PubMed] [Google Scholar]
- (321).Morgan MA; Parsels LA; Maybaum J; Lawrence TS Improving gemcitabine-mediated radiosensitization using molecularly targeted therapy: a review. Clin. Cancer Res 2008, 14 (21), 6744–6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Gilbert JA; Salavaggione OE; Ji Y; Pelleymounter LL; Eckloff BW; Wieben ED; Ames MM; Weinshilboum RM Gemcitabine pharmacogenomics: cytidine deaminase and deoxycytidylate deaminase gene resequencing and functional genomics. Clin. Cancer Res 2006, 12 (6), 1794–1803. [DOI] [PubMed] [Google Scholar]
- (323).Hodge LS; Taub ME; Tracy TS The deaminated metabolite of gemcitabine, 2′,2′-difluorodeoxyuridine, modulates the rate of gemcitabine transport and intracellular phosphorylation via 2′-deoxycytidine kinase. Drug Metab. Dispos 2011, 39 (11), 2013–2016. [DOI] [PubMed] [Google Scholar]
- (324).Adema AD; Losekoot N; Smid K; Kathmann I; Myhren F; Sandvold ML; Peters GJ Induction of resistance to the lipophilic cytarabine prodrug elacytarabine (CP-4055) in CEM leukemic cells. Nucleosides, Nucleotides Nucleic Acids 2010, 29 (4–6), 394–399. [DOI] [PubMed] [Google Scholar]
- (325).Aksoy P; Zhu MJ; Kalari KR; Moon I; Pelleymounter LL; Eckloff BW; Wieben ED; Yee VC; Weinshilboum RM; Wang L Cytosolic 5′-nucleotidase III (NT5C3): Gene sequence variation and functional genomics. Pharmacogenet. Genomics 2009, 19 (8), 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Hagmann W; Jesnowski R; Lohr JM Interdependence of gemcitabine treatment, transporter expression, and resistance in human pancreatic carcinoma cells. Neoplasia 2010, 12 (9), 740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Pan MR; Hsu MC; Chen LT; Hung WC G9a orchestrates PCL3 and KDM7A to promote histone H3K27 methylation. Sci. Rep 2015, 5, 18709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Garcia-Cano J; Roche O; Cimas FJ; Pascual-Serra R; Ortega-Muelas M; Fernandez-Aroca DM; Sanchez-Prieto R p38MAPK and chemotherapy: We always need to hear both sides of the story. Front. Cell Dev. Biol 2016, 4 (69), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Wu Q; Wang X; Liu J; Zheng J; Liu Y; Li Y; Su F; Ou W; Wang R Nutlin-3 reverses the epithelial-mesenchymal transition in gemcitabine-resistant hepatocellular carcinoma cells. Oncol. Rep 2016, 36 (3), 1325–1332. [DOI] [PubMed] [Google Scholar]
- (330).Hayes GM; Carrigan PE; Beck AM; Miller LJ Targeting the RNA splicing machinery as a novel treatment strategy for pancreatic carcinoma. Cancer Res 2006, 66 (7), 3819–3827. [DOI] [PubMed] [Google Scholar]
- (331).Serdjebi C; Gagniere J; Desrame J; Fein F; Guimbaud R; Francois E; Andre T; Seitz JF; Monterymard C; Arsene D; et al. FFCD-1004 clinical trial: Impact of cytidine deaminase activity on clinical outcome in gemcitabine-monotherapy treated patients. PLoS One 2015, 10 (8), e0135907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Serdjebi C; Seitz JF; Ciccolini J; Duluc M; Norguet E; Fina F; Lacarelle B; Ouafik L; Dahan L Rapid deaminator status is associated with poor clinical outcome in pancreatic cancer patients treated with a gemcitabine-based regimen. Pharmacogenomics 2013, 14 (9), 1047–1051. [DOI] [PubMed] [Google Scholar]
- (333).Bepler G; Kusmartseva I; Sharma S; Gautam A; Cantor A; Sharma A; Simon G RRM1 modulated in vitro and in vivo efficacy of gemcitabine and platinum in non-small-cell lung cancer. J. Clin. Oncol 2006, 24 (29), 4731–4737. [DOI] [PubMed] [Google Scholar]
- (334).Rosell R; Danenberg KD; Alberola V; Bepler G; Sanchez JJ; Camps C; Provencio M; Isla D; Taron M; Diz P; et al. Ribonucleotide reductase messenger RNA expression and survival in gemcitabine/cisplatin-treated advanced non-small cell lung cancer patients. Clin. Cancer Res 2004, 10 (4), 1318–1325. [DOI] [PubMed] [Google Scholar]
- (335).Duxbury MS; Ito H; Benoit E; Waseem T; Ashley SW; Whang EE RNA interference demonstrates a novel role for integrin-linked kinase as a determinant of pancreatic adenocarcinoma cell gemcitabine chemoresistance. Clin. Cancer Res 2005, 11 (9), 3433–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Noble S; Goa KL Gemcitabine. A review of its pharmacology and clinical potential in non-small cell lung cancer and pancreatic cancer. Drugs 1997, 54 (3), 447–472. [DOI] [PubMed] [Google Scholar]
- (337).Barton-Burke M Gemcitabine: a pharmacologic and clinical overview. Cancer Nursing 1999, 22 (2), 176–183. [DOI] [PubMed] [Google Scholar]
- (338).Toschi L; Finocchiaro G; Bartolini S; Gioia V; Cappuzzo F Role of gemcitabine in cancer therapy. Future Oncol 2005, 1 (1), 7–17. [DOI] [PubMed] [Google Scholar]
- (339).Sun C; Ansari D; Andersson R; Wu DQ Does gemcitabine-based combination therapy improve the prognosis of unresectable pancreatic cancer? World J. Gastroenterol 2012, 18 (35), 4944–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (340).Ciliberto D; Botta C; Correale P; Rossi M; Caraglia M; Tassone P; Tagliaferri P Role of gemcitabine-based combination therapy in the management of advanced pancreatic cancer: a meta-analysis of randomised trials. Eur. J. Cancer 2013, 49 (3), 593–603. [DOI] [PubMed] [Google Scholar]
- (341).Qi WX; Tang LN; He AN; Shen Z; Lin F; Yao Y Doublet versus single cytotoxic agent as first-line treatment for elderly patients with advanced non-small-cell lung cancer: a systematic review and meta-analysis. Lung 2012, 190 (5), 477–485. [DOI] [PubMed] [Google Scholar]
- (342).Bai B; Huang HQ; Cai QQ; Wang XX; Cai QC; Lin ZX; Gao Y; Xia Y; Bu Q; Guo Y Promising long-term outcome of gemcitabine, vinorelbine, liposomal doxorubicin (GVD) in 14-day schedule as salvage regimen for patients with previously heavily treated Hodgkin’s lymphoma and aggressive non-Hodgkin’s lymphoma. Med. Oncol 2013, 30 (1), 350. [DOI] [PubMed] [Google Scholar]
- (343).Chan SL; Chan ST; Chan EH; He ZX Systemic treatment for inoperable pancreatic adenocarcinoma: review and update. Aizheng 2014, 33 (6), 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (344).Wagner AD; Buechner-Steudel P; Moehler M; Schmalenberg H; Behrens R; Fahlke J; Wein A; Behl S; Kuss O; Kleber G; et al. Gemcitabine, oxaliplatin and 5-FU in advanced bile duct and gallbladder carcinoma: two parallel, multicentre phase-II trials. Br. J. Cancer 2009, 101 (11), 1846–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Aurilio G; Macarulla T; Ramos JF; Fazio N; Nole F; Iglesias C Successful treatment with GEMOX in patient with metastatic pancreatic adenosquamous carcinoma. Tumori 2011, 97 (2), 239–242. [DOI] [PubMed] [Google Scholar]
- (346).Fang P; Hu JH; Cheng ZG; Liu ZF; Wang JL; Jiao SC Efficacy and safety of bevacizumab for the treatment of advanced hepatocellular carcinoma: a systematic review of phase II trials. PLoS One 2012, 7 (12), e49717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Prince SK; Forgeson G Bleeding from gastrointestinal tract recurrence of non-seminomatous germ cell tumour testis, showing temporary response to gemcitabine and oxaliplatin chemotherapy. N. Z. Med. J 2013, 126 (1385), 76–80. [PubMed] [Google Scholar]
- (348).Vici P; Sergi D; Pizzuti L; Mariani L; Arena MG; Barba M; Maugeri-Sacca M; Vincenzoni C; Vizza E; Corrado G; et al. Gemcitabine-oxaliplatin (GEMOX) as salvage treatment in pretreated epithelial ovarian cancer patients. J. Exp. Clin. Cancer Res 2013, 32 (1), 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Jordheim LP; Durantel D; Zoulim F; Dumontet C Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discovery 2013, 12 (6), 447–464. [DOI] [PubMed] [Google Scholar]
- (350).Bender DM; Bao J; Dantzig AH; Diseroad WD; Law KL; Magnus NA; Peterson JA; Perkins EJ; Pu YJ; Reutzel-Edens SM; Remick DM; Starling JJ; Stephenson GA; Vaid RK; Zhang D; McCarthy JR Synthesis, crystallization, and biological evaluation of an orally active prodrug of gemcitabine. J. Med. Chem 2009, 52 (22), 6958–6961. [DOI] [PubMed] [Google Scholar]
- (351).Pratt SE; Durland-Busbice S; Shepard RL; Heinz-Taheny K; Iversen PW; Dantzig AH Human carboxylesterase-2 hydrolyzes the prodrug of gemcitabine (LY2334737) and confers prodrug sensitivity to cancer cells. Clin. Cancer Res 2013, 19 (5), 1159–1168. [DOI] [PubMed] [Google Scholar]
- (352).Pratt SE; Durland-Busbice S; Shepard RL; Donoho GP; Starling JJ; Wickremsinhe ER; Perkins EJ; Dantzig AH Efficacy of low-dose oral metronomic dosing of the prodrug of gemcitabine, LY2334737, in human tumor xenografts. Mol. Cancer Ther 2013, 12 (4), 481–490. [DOI] [PubMed] [Google Scholar]
- (353).Koolen SL; Witteveen PO; Jansen RS; Langenberg MH; Kronemeijer RH; Nol A; Garcia-Ribas I; Callies S; Benhadji KA; Slapak CA; et al. Phase I study of oral gemcitabine prodrug (LY2334737) alone and in combination with erlotinib in patients with advanced solid tumors. Clin. Cancer Res 2011, 17 (18), 6071–6082. [DOI] [PubMed] [Google Scholar]
- (354).Williams R Discontinued in 2013: Oncology drugs. Expert Opin. Invest. Drugs 2015, 24, 95–110. [DOI] [PubMed] [Google Scholar]
- (355).Infante JR; Benhadji KA; Dy GK; Fetterly G; Ma WW; Bendell J; Callies S; Adjei AA Phase 1b study of the oral gemcitabine ‘pro-drug’ LY2334737 in combination with capecitabine in patients with advanced solid tumors. Invest. New Drugs 2015, 33 (2), 432–439. [DOI] [PubMed] [Google Scholar]
- (356).Bergman AM; Adema AD; Balzarini J; Bruheim S; Fichtner I; Noordhuis P; Fodstad O; Myhren F; Sandvold ML; Hendriks HR; et al. Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Invest. New Drugs 2011, 29 (3), 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Stuurman FE; Voest EE; Awada A; Witteveen PO; Bergeland T; Hals PA; Rasch W; Schellens JH; Hendlisz A Phase I study of oral CP-4126, a gemcitabine derivative, in patients with advanced solid tumors. Invest. New Drugs 2013, 31 (4), 959–966. [DOI] [PubMed] [Google Scholar]
- (358).Poplin E; Wasan H; Rolfe L; Raponi M; Ikdahl T; Bondarenko I; Davidenko I; Bondar V; Garin A; Boeck S; et al. Randomized, multicenter, phase II study of CO-101 versus gemcitabine in patients with metastatic pancreatic ductal adenocarcinoma: including a prospective evaluation of the role of hENT1 in gemcitabine or CO-101 sensitivity. J. Clin. Oncol 2013, 31 (35), 4453–4461. [DOI] [PubMed] [Google Scholar]
- (359).Stuurman FE; Lolkema MP; Huitema AD; Soetekouw PM; Rosing H; Rolfe L; Kaur P; Beijnen JH; van Tinteren H; Voest EE; et al. A phase 1 comparative pharmacokinetic and cardiac safety study of two intravenous formulations of CO-101 in patients with advanced solid tumors. J. Clin. Pharmacol 2013, 53 (8), 878–883. [DOI] [PubMed] [Google Scholar]
- (360).Venugopal B; Awada A; Evans TR; Dueland S; Hendlisz A; Rasch W; Hernes K; Hagen S; Aamdal S A first-in-human phase I and pharmacokinetic study of CP-4126 (CO-101), a nucleoside analogue, in patients with advanced solid tumours. Cancer Chemother. Pharmacol 2015, 76 (4), 785–792. [DOI] [PubMed] [Google Scholar]
- (361).Malfanti A; Miletto I; Bottinelli E; Zonari D; Blandino G; Berlier G; Arpicco S Delivery of gemcitabine prodrugs employing mesoporous silica nanoparticles. Molecules 2016, 21 (4), 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (362).Tsume Y; Incecayir T; Song X; Hilfinger JM; Amidon GL The development of orally administrable gemcitabine prodrugs with D-enantiomer amino acids: enhanced membrane permeability and enzymatic stability. Eur. J. Pharm. Biopharm 2014, 86 (3), 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Dasari M; Acharya AP; Kim D; Lee S; Rhea J; Molinaro R; Murthy N H-gemcitabine: a new gemcitabine prodrug for treating cancer. Bioconjugate Chem 2013, 24 (1), 4–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (364).Wi LI US Patent 20140134160, 2014.
- (365).Mehellou Y; Balzarini J; McGuigan C Aryloxy phosphoramidate triesters: a technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009, 4 (11), 1779–1791. [DOI] [PubMed] [Google Scholar]
- (366).Saif MW; Lee Y; Kim R Harnessing gemcitabine metabolism: a step towards personalized medicine for pancreatic cancer. Ther. Adv. Med. Oncol 2012, 4 (6), 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).McGuigan C; Habib N; Wasan H; Gabra H; Jiao L; Slusarczyk M; Chabot J; Saif MW A phosphoramidate ProTide (NUC-1031) and acquired and intrinsic resistance to gemcitabine. 2011 ASCO Annual Meeting, Chicago, IL, 2011. [Google Scholar]
- (368).Slusarczyk M; Lopez MH; Balzarini J; Mason M; Jiang WG; Blagden S; Thompson E; Ghazaly E; McGuigan C Application of ProTide technology to gemcitabine: a successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem 2014, 57 (4), 1531–1542. [DOI] [PubMed] [Google Scholar]
- (369).Ghazaly EA; Simon J; Gribben JG; Mohammad T; Oluwadunni E; Stavraka C; Hopkins T; Gabra H; harpreet W; Habib NA et al. proGem1: Phase 1 first-in-human study of the novel nucleotide analogue NUC-1031 in adult patients with advanced solid tumors. ASCO Annual Meeting, Chicago, IL, 2013. [Google Scholar]
- (370).Hertel LW US Patent 4808614, 1989.
- (371).Chang Y-K; Lee J; Park G-S; Lee M; Park CH; Kim HK; Lee G; Lee B-Y; Baek JY; Kim KS An efficient large-scale synthesis of gemcitabine employing a crystalline 2,2-difluoro-α-ribofuranosyl bromide. Tetrahedron 2010, 66, 5687–5691. [Google Scholar]
- (372).Jiang X; Li J; Zhang R; Zhu Y; Shen J An improved preparation process for gemcitabine. Org. Process Res. Dev 2008, 12, 888–891. [Google Scholar]
- (373).Myhren F; Borretzen B; Dalen A; Sandvold M EP Patent 0986570, 2004.
- (374).McGuigan C US Patent 20060142238, 2005.
- (375).Dammalapati VLNR; Kotala MB; Madhabaram S WO Patent 2016012781, 2016.
- (376).Matsuda A; Nakajima Y; Azuma A; Tanaka M; Sasaki T Nucleosides and nucleotides. 100. 2′-C-cyano-2′-deoxy-1-β-D-arabinofuranosyl-cytosine (CNDAC): design of a potential mechanism-based DNA-strand-breaking antineoplastic nucleoside. J. Med. Chem 1991, 34 (9), 2917–2919. [DOI] [PubMed] [Google Scholar]
- (377).Tener GM 2-Cyanoethyl phosphate and its use in the synthesis of phosphate esters. J. Am. Chem. Soc 1961, 83, 159–168. [Google Scholar]
- (378).Liu XJ; Nowak B; Wang YQ; Plunkett W Sapacitabine, the prodrug of CNDAC, is a nucleoside analog with a unique action mechanism of inducing DNA strand breaks. Aizheng 2012, 31 (8), 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Hanaoka K; Suzuki M; Kobayashi T; Tanzawa F; Tanaka K; Shibayama T; Miura S; Ikeda T; Iwabuchi H; Nakagawa A; et al. Antitumor activity and novel DNA-self-strand-breaking mechanism of CNDAC (1-(2-C-cyano-2-deoxy-β-D-arabino-pentofuranosyl) cyto-sine) and its N4-palmitoyl derivative (CS-682). Int. J. Cancer 1999, 82 (2), 226–236. [DOI] [PubMed] [Google Scholar]
- (380).Yoshida T; Endo Y; Obata T; Kosugi Y; Sakamoto K; Sasaki T Influence of cytidine deaminase on antitumor activity of 2′-deoxycytidine analogs in vitro and in vivo. Drug Metab. Dispos 2010, 38 (10), 1814–1819. [DOI] [PubMed] [Google Scholar]
- (381).Serova M; Galmarini CM; Ghoul A; Benhadji K; Green SR; Chiao J; Faivre S; Cvitkovic E; Le Tourneau C; Calvo F; et al. Antiproliferative effects of sapacitabine (CYC682), a novel 2′-deoxycytidine-derivative, in human cancer cells. Br. J. Cancer 2007, 97 (5), 628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Azuma A; Huang P; Matsuda A; Plunkett W 2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine: a novel anticancer nucleoside analog that causes both DNA strand breaks and G(2) arrest. Mol. Pharm 2001, 59 (4), 725–731. [DOI] [PubMed] [Google Scholar]
- (383).Liu X; Guo Y; Li Y; Jiang Y; Chubb S; Azuma A; Huang P; Matsuda A; Hittelman W; Plunkett W Molecular basis for G2 arrest induced by 2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine and consequences of checkpoint abrogation. Cancer Res 2005, 65 (15), 6874–6881. [DOI] [PubMed] [Google Scholar]
- (384).Wang Y; Liu X; Matsuda A; Plunkett W Repair of 2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine-induced DNA single-strand breaks by transcription-coupled nucleotide excision repair. Cancer Res 2008, 68 (10), 3881–3889. [DOI] [PubMed] [Google Scholar]
- (385).Liu X; Wang Y; Benaissa S; Matsuda A; Kantarjian H; Estrov Z; Plunkett W Homologous recombination as a resistance mechanism to replication-induced double-strand breaks caused by the antileukemia agent CNDAC. Blood 2010, 116 (10), 1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Arnaudeau C; Lundin C; Helleday T DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J. Mol. Biol 2001, 307 (5), 1235–1245. [DOI] [PubMed] [Google Scholar]
- (387).Liu X; Kantarjian H; Plunkett W Sapacitabine for cancer. Expert Opin. Invest. Drugs 2012, 21 (4), 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Kantarjian H; Faderl S; Garcia-Manero G; Luger S; Venugopal P; Maness L; Wetzler M; Coutre S; Stock W; Claxton D; et al. Oral sapacitabine for the treatment of acute myeloid leukaemia in elderly patients: a randomised phase 2 study. Lancet Oncol 2012, 13 (11), 1096–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Hansske F; Madej D; Robins MJ 2′ and 3′-ketonucleosides and their arabino and xylo reduction products: Convenient access via selective protection and oxidation of ribonucleosides. Tetrahedron 1984, 40 (1), 125–135. [Google Scholar]
- (390).Kaneko M; Hotoda H; Shibata T; Kobayashi T; Mitsuhashi Y; Matsuda A; Sasaki T EP Patent 0536936, 1996.
- (391).Westwood R; Wood G; Meades CK GB Patent 2459779, 2009.
- (392).Wood GJ; Westwood R WO Patent 2009136162, 2009.
- (393).Hattori H; Nozawa E; Iino T; Yoshimura Y; Shuto S; Shimamoto Y; Nomura M; Fukushima M; Tanaka M; Sasaki T; et al. Nucleosides and nucleotides. 175. Structural requirements of the sugar moiety for the antitumor activities of new nucleoside antimetabolites, 1-(3-C-ethynyl-β-D-ribo-pentofuranosyl)cytosine and -uracil. J. Med. Chem 1998, 41 (15), 2892–2902. [DOI] [PubMed] [Google Scholar]
- (394).Hammond-Thelin LA; Thomas MB; Iwasaki M; Abbruzzese JL; Lassere Y; Meyers CA; Hoff P; de Bono J; Norris J; Matsushita H; et al. Phase I and pharmacokinetic study of 3′-C-ethynylcytidine (TAS-106), an inhibitor of RNA polymerase I, II and III,in patients with advanced solid malignancies. Invest. New Drugs 2012, 30 (1), 316–326. [DOI] [PubMed] [Google Scholar]
- (395).Meike S; Yamamori T; Yasui H; Eitaki M; Matsuda A; Morimatsu M; Fukushima M; Yamasaki Y; Inanami O A nucleoside anticancer drug, 1-(3-C-ethynyl-β-D-ribo-pentofuranosyl)cytosine (TAS106), sensitizes cells to radiation by suppressing BRCA2 expression. Mol. Cancer 2011, 10, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Ahmed NK Multiple forms and inhibitors of uridine-cytidine kinase in neoplastic cells. Int. J. Biochem 1982, 14 (4), 259–262. [DOI] [PubMed] [Google Scholar]
- (397).Shimamoto Y; Koizumi K; Okabe H; Kazuno H; Murakami Y; Nakagawa F; Matsuda A; Sasaki T; Fukushima M Sensitivity of human cancer cells to the new anticancer ribo-nucleoside TAS-106 is correlated with expression of uridine-cytidine kinase 2. Jpn. J. Cancer Res 2002, 93 (7), 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (398).Kanda H; Takatori S; Matsuda A; Sasaki T; Tanaka M; Fukushima M; Wataya Y Cytotoxic mechanisms of new antitumor nucleoside analogues, 3′-ethynylcytidine (ECyd) and 3′-ethynyluridine (EUrd). Nucleic Acids Symp. Ser 1997, 37, 137–138. [PubMed] [Google Scholar]
- (399).Hattori H; Tanaka M; Fukushima M; Sasaki T; Matsuda A Nucleosides and nucleotides. 158. 1-(3-C-ethynyl-β-D-ribo-pentofuranosyl)-cytosine, 1-(3-C-ethynyl-β-D-ribo-pentofuranosyl)uracil, and their nucleobase analogues as new potential multifunctional antitumor nucleosides with a broad spectrum of activity. J. Med. Chem 1996, 39 (25), 5005–5011. [DOI] [PubMed] [Google Scholar]
- (400).Takatori S; Kanda H; Takenaka K; Wataya Y; Matsuda A; Fukushima M; Shimamoto Y; Tanaka M; Sasaki T Antitumor mechanisms and metabolism of the novel antitumor nucleoside analogues, 1-(3-C-ethynyl-beta-D-ribo-pentofuranosyl)cytosine and 1-(3-C-ethynyl-beta-D-ribo-pentofuranosyl)uracil. Cancer Chemother. Pharmacol 1999, 44 (2), 97–104. [DOI] [PubMed] [Google Scholar]
- (401).Weinreich J; Schott S; Konigsrainer I; Zieker D; Konigsrainer A; Schott H Cytostatic activity of the duplex drug linking 2′-deoxy-5-fluorouridine (5FdU) with 3′-C-ethynylcytidine (ECyd) against gastric adenocarcinoma cell lines. Invest. New Drugs 2011, 29 (6), 1294–1302. [DOI] [PubMed] [Google Scholar]
- (402).Eicher C; Dewerth A; Ellerkamp V; Fuchs J; Schott S; Armeanu-Ebinger S Effect of duplex drugs linking 2′-deoxy-5-fluorouridine (5-FdU) with 3′-C-ethynylcytidine (ECyd) on hepatoblastoma cell lines. Pediatr. Surg. Int 2013, 29 (2), 121–127. [DOI] [PubMed] [Google Scholar]
- (403).Tsao A; Hui EP; Juergens R; Marur S; Huat TE; Cher GB; Hong R-L; Hong WK; Chan AT-C Phase II study of TAS-106 in patients with platinum-failure recurrent or metastatic head and neck cancer and nasopharyngeal cancer. Cancer Med 2013, 2 (3), 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (404).Naing A; Fu S; Zinner RG; Wheler JJ; Hong DS; Arakawa K; Falchook GS; Kurzrock R Phase I dose-escalating study of TAS-106 in combination with carboplatin in patients with solid tumors. Invest. New Drugs 2014, 32 (1), 154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (405).Yoshimura Y; Sano T; Matsuda A; Ueda T Nucleosides and nucleotides. LXXVII: Synthesis of 6,3′-methanocytidine, 6,3′-methanouridine, and Their 2′-deoxyribonucleosides. Chem. Pharm. Bull. (Tokyo) 1988, 36, 162–167. [Google Scholar]
- (406).Nomura M; Sato T; Washinosu M; Tanaka M; Asao T; Shuto S; Matsuda A Nucleosides and nucleotides. Part 212: Practical large-scale synthesis of 1-(3-C-ethynyl-β-D-ribo-pentofuranosyl)-cytosine (ECyd), a potent antitumor nucleoside. Isobutyryloxy group as an efficient anomeric leaving group in the Vorbrüggen glycosylation reaction. Tetrahedron 2002, 58, 1279–1288. [Google Scholar]
- (407).Bitonti AJ; Bush TL; Lewis MT; Sunkara PS Response of human colon and prostate tumor xenografts to (E)-2′-deoxy-2′-(fluoromethylene) cytidine, an inhibitor of ribonucleotide reductase. Anticancer Res 1995, 15 (4), 1179–1182. [PubMed] [Google Scholar]
- (408).Avendano C; Menendez JA Antimetabolites; Elsevier: Oxford, UK, 2008; pp 9–52. [Google Scholar]
- (409).Stubbe J; van der Donk WA Ribonucleotide reductases: radical enzymes with suicidal tendencies. Chem. Biol 1995, 2 (12), 793–801. [DOI] [PubMed] [Google Scholar]
- (410).Seley KL Tezacitabine Hoechst Marion Roussel. Curr. Opin. Investig. Drugs 2000, 1 (1), 135–140. [PubMed] [Google Scholar]
- (411).Takahashi T; Nakashima A; Kanazawa J; Yamaguchi K; Akinaga S; Tamaoki T; Okabe M Metabolism and ribonucleotide reductase inhibition of (E)-2′-deoxy-2′-(fluoromethylene)cytidine, MDL 101,731, in human cervical carcinoma HeLa S3 cells. Cancer Chemother. Pharmacol 1998, 41 (4), 268–274. [DOI] [PubMed] [Google Scholar]
- (412).Rodriguez GI; Jones RE; Orenberg EK; Stoltz ML; Brooks DJ Phase I clinical trials of tezacitabine [(E)-2′-deoxy-2′-(fluoromethylene)cytidine] in patients with refractory solid tumors. Clin. Cancer Res 2002, 8 (9), 2828–2834. [PubMed] [Google Scholar]
- (413).Piepmeier JM; Rabidou N; Schold SC Jr.; Bitonti AJ; Prakash NJ; Bush TL In vitro and in vivo inhibition of glioblastoma and neuroblastoma with MDL101731, a novel ribonucleoside diphosphate reductase inhibitor. Cancer Res 1996, 56 (2), 359–361. [PubMed] [Google Scholar]
- (414).Bitonti AJ; Dumont JA; Bush TL; Cashman EA; Cross-Doersen DE; Wright PS; Matthews DP; McCarthy JR; Kaplan DA Regression of human breast tumor xenografts in response to (E)-2′-deoxy-2′-(fluoromethylene)cytidine, an inhibitor of ribonucleoside diphosphate reductase. Cancer Res 1994, 54 (6), 1485–1490. [PubMed] [Google Scholar]
- (415).Coucke PA; Cottin E; Decosterd LA Simultaneous alteration of de novo and salvage pathway to the deoxynucleoside triphosphate pool by (E)-2′-deoxy-(fluoromethylene)cytidine (FMdC) and zidovudine (AZT) results in increased radiosensitivity in vitro. Acta Oncol 2007, 46 (5), 612–620. [DOI] [PubMed] [Google Scholar]
- (416).Woessner RD; Loudy DE; Wallace CD; Montgomery LR; Cross-Doersen DE; Bush TL; Lewis MT; Prakash N; Bitonti AJ; Wright PS Decreased vascular endothelial growth factor expression associated with tumor regression induced by (E)-2′-deoxy-2′-(fluoromethylene)cytidine (MDL 101,731). Oncol. Res 1997, 9 (10), 543–552. [PubMed] [Google Scholar]
- (417).Bendell JC; Eder JP; Clark JW; Fidias P; Lynch TJ; Seiden MV; Ryan DP Phase I dose-escalation study of tezacitabine in combination with 5-fluorouracil in patients with advanced solid tumors. Cancer 2005, 103 (9), 1925–1931. [DOI] [PubMed] [Google Scholar]
- (418).McCarthy JR; Matthews DP; Stemerick DM; Huber EW; Bey P; Lippert BJ; Snyder RD; Sunkara PS Stereospecific method to (E) and (Z) terminal fluoroolefins and its application to the synthesis of 2′-deoxy-2′-fluoromethylenenucleosides as potential inhibitors of ribonucleoside diphosphate reductase. J. Am. Chem. Soc 1991, 113, 7439–7440. [Google Scholar]
- (419).Matthews DP; Persichetti RA; Sabol JS; Stewart KT; McCarthy JR Improved synthesis of (E)-2′-deoxy-2′-(fluoromethylene)cytidine - a potent inhibitor of ribonucleotide diphosphate reductase. Nucleosides Nucleotides 1993, 12, 115–123. [Google Scholar]
- (420).Chou KM; Kukhanova M; Cheng YC A novel action of human apurinic/apyrimidinic endonuclease: excision of L-configuration deoxyribonucleoside analogs from the 3′ termini of DNA. J. Biol. Chem 2000, 275 (40), 31009–31015. [DOI] [PubMed] [Google Scholar]
- (421).Quintas-Cardama A; Cortes J Evaluation of the L-stereo-isomeric nucleoside analog troxacitabine for the treatment of acute myeloid leukemia. Expert Opin. Invest. Drugs 2007, 16 (4), 547–557. [DOI] [PubMed] [Google Scholar]
- (422).Griffith DA; Jarvis SM Nucleoside and nucleobase transport systems of mammalian cells. Biochim. Biophys. Acta, Rev. Biomembr 1996, 1286 (3), 153–181. [DOI] [PubMed] [Google Scholar]
- (423).Lin CC; Beeram M; Rowinsky EK; Takimoto CH; Ng CM; Geyer CE Jr.; Denis LJ; De Bono JS; Hao D; Tolcher AW; et al. Phase I and pharmacokinetic study of cisplatin and troxacitabine administered intravenously every 28 days in patients with advanced solid malignancies. Cancer Chemother. Pharmacol 2009, 65 (1), 167–175. [DOI] [PubMed] [Google Scholar]
- (424).Gourdeau H; Jolivet J [Troxacitabine]. Bull. Cancer 2004, 91 (3), 213–218. [PubMed] [Google Scholar]
- (425).Moore EC; Hurlbert RB; Boss GR; Massia SP Inhibition of two enzymes in de novo purine nucleotide synthesis by triciribine phosphate (TCN-P). Biochem. Pharmacol 1989, 38 (22), 4045–4051. [DOI] [PubMed] [Google Scholar]
- (426).Krishnan P; Fu Q; Lam W; Liou JY; Dutschman G; Cheng YC Phosphorylation of pyrimidine deoxynucleoside analog diphosphates: selective phosphorylation of L-nucleoside analog diphosphates by 3-phosphoglycerate kinase. J. Biol. Chem 2002, 277 (7), 5453–5459. [DOI] [PubMed] [Google Scholar]
- (427).Dent SF; Arnold A; Stewart DJ; Gertler S; Ayoub J; Batist G; Goss G; Nevile A; Soulieres D; Jolivet J; et al. Phase II study of troxacitabine (BCH-4556) in patients with advanced non-small-cell lung cancer. Lung 2005, 183 (4), 265–272. [DOI] [PubMed] [Google Scholar]
- (428).Kukhanova M; Liu SH; Mozzherin D; Lin TS; Chu CK; Cheng YC L- and D-enantiomers of 2′,3′-dideoxycytidine 5′-triphosphate analogs as substrates for human DNA polymerases. Implications for the mechanism of toxicity. J. Biol. Chem 1995, 270 (39), 23055–23059. [DOI] [PubMed] [Google Scholar]
- (429).Adema AD; Zuurbier L; Floor K; Hubeek I; Kaspers GJ; Albertoni F; Peters GJ Cellular resistance against troxacitabine in human cell lines and pediatric patient acute myeloid leukemia blast cells. Nucleosides, Nucleotides Nucleic Acids 2006, 25 (9–11), 981–986. [DOI] [PubMed] [Google Scholar]
- (430).Giles FJ; Garcia-Manero G; Cortes JE; Baker SD; Miller CB; O’Brien SM; Thomas DA; Andreeff M; Bivins C; Jolivet J; et al. Phase II study of troxacitabine, a novel dioxolane nucleoside analog, in patients with refractory leukemia. J. Clin. Oncol 2002, 20 (3), 656–664. [DOI] [PubMed] [Google Scholar]
- (431).Lapointe R; Letourneau R; Steward W; Hawkins RE; Batist G; Vincent M; Whittom R; Eatock M; Jolivet J; Moore M Phase II study of troxacitabine in chemotherapy-naive patients with advanced cancer of the pancreas: gastrointestinal tumors. Ann. Oncol 2005, 16 (2), 289–293. [DOI] [PubMed] [Google Scholar]
- (432).Townsley CA; Chi K; Ernst DS; Belanger K; Tannock I; Bjarnason GA; Stewart D; Goel R; Ruether JD; Siu LL; et al. Phase II study of troxacitabine (BCH-4556) in patients with advanced and/or metastatic renal cell carcinoma: a trial of the National Cancer Institute of Canada-Clinical Trials Group. J. Clin. Oncol 2003, 21 (8), 1524–1529. [DOI] [PubMed] [Google Scholar]
- (433).Adema AD; Radi M; Daft J; Narayanasamy J; Hoebe EK; Alexander LE; Chu CK; Peters GJ Troxacitabine prodrugs for pancreatic cancer. Nucleosides, Nucleotides Nucleic Acids 2007, 26 (8–9), 1073–1077. [DOI] [PubMed] [Google Scholar]
- (434).Jimeno A; Messersmith WA; Lee CK; Ma WW; Laheru D; Donehower RC; Baker SD; Hidalgo M Phase I study of troxacitabine administered by continuous infusion in subjects with advanced solid malignancies. Ann. Oncol 2008, 19 (2), 374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Roboz GJ; Giles FJ; Ritchie EK; Allen-Bard S; Curcio TJ; Wilkes MA; Park SL; Kantarjian HM; Faderl S; Ravandi F; et al. Phase I/II study of continuous-infusion troxacitabine in refractory acute myeloid leukemia. J. Clin. Oncol 2006, 25 (1), 10–15. [DOI] [PubMed] [Google Scholar]
- (436).Vose JM; Panwalkar A; Belanger R; Coiffier B; Baccarani M; Gregory SA; Facon T; Fanin R; Caballero D; Ben-Yehuda D; et al. A phase II multicenter study of troxacitabine in relapsed or refractory lymphoproliferative neoplasms or multiple myeloma. Leuk. Lymphoma 2007, 48 (1), 39–45. [DOI] [PubMed] [Google Scholar]
- (437).Belleau BR; Evans CA; Tse HLA; Jin H; Dixit DM; Mansour TS Oxidative degradation of L-ascorbic acid acetals to 2′,3′-dideoxy-3′-oxaribofuranosides. Synthesis of enantiomerically pure 2′,3′-dideoxy-3′-oxacytidine stereoisomers as potential antiviral agents. Tetrahedron Lett 1992, 33 (46), 6949–6952. [Google Scholar]
- (438).Kim HO; Schinazi RF; Shanmuganathan K; Jeong LS; Beach JW; Nampalli S; Cannon DL; Chu CK L-β-(2S,4S)- and L-α-(2S,4R)-dioxolanyl nucleosides as potential anti-HIV agents: asymmetric synthesis and structure-activity relationships. J. Med. Chem 1993, 36 (5), 519–528. [DOI] [PubMed] [Google Scholar]
- (439).Whistler RL; Doner LW; Nayak UG 4-thio-D-arabinofuranosylpyrimidine nucleosides. J. Org. Chem 1971, 36 (1), 108–110. [DOI] [PubMed] [Google Scholar]
- (440).Ototani N; Whistler RL Preparation and antitumor activity of 4′-thio analogs of 2,2′-anhydro-1-β-D-arabinofuranosylcytosine. J. Med. Chem 1974, 17, 535–537. [DOI] [PubMed] [Google Scholar]
- (441).Secrist JA 3rd; Tiwari KN; Shortnacy-Fowler AT; Messini L; Riordan JM; Montgomery JA; Meyers SC; Ealick SE Synthesis and biological activity of certain 4′-thio-D-arabinofuranosylpurine nucleosides. J. Med. Chem 1998, 41 (20), 3865–3871. [DOI] [PubMed] [Google Scholar]
- (442).Roy AM; Tiwari KN; Parker WB; Secrist JA 3rd; Li R; Qu Z Antiangiogenic activity of 4′-thio-β-D-arabinofuranosylcytosine. Mol. Cancer Ther 2006, 5 (9), 2218–2224. [DOI] [PubMed] [Google Scholar]
- (443).Tiwari KN; Shortnacy-Fowler AT; Cappellacci L; Parker WB; Waud WR; Montgomery JA; Secrist JA 3rd Synthesis of 4′-thio-β-D-arabinofuranosylcytosine (4′-thio-ara-C) and comparison of its anticancer activity with that of ara-C. Nucleosides, Nucleotides Nucleic Acids 2000, 19 (1–2), 329–340. [DOI] [PubMed] [Google Scholar]
- (444).Parker WB; Shaddix SC; Rose LM; Waud WR; Shewach DS; Tiwari KN; Secrist JA 3rd Metabolism of 4′-thio-β-D-arabinofuranosylcytosine in CEM cells. Biochem. Pharmacol 2000, 60 (12), 1925–1932. [DOI] [PubMed] [Google Scholar]
- (445).Someya H; Shaddix SC; Tiwari KN; Secrist JA 3rd; Parker WB Phosphorylation of 4′-thio-β-D-arabinofuranosylcytosine and its analogs by human 2′-deoxycytidine kinase. J. Pharmacol. Exp. Ther 2003, 304 (3), 1314–1322. [DOI] [PubMed] [Google Scholar]
- (446).Someya H; Waud WR; Parker WB Long intracellular retention of 4′-thio-arabinofuranosylcytosine 5′-triphosphate as a critical factor for the anti-solid tumor activity of 4′-thio-arabinofuranosylcytosine. Cancer Chemother. Pharmacol 2006, 57 (6), 772–780. [DOI] [PubMed] [Google Scholar]
- (447).Robak T New nucleoside analogs for patients with hematological malignancies. Expert Opin. Invest. Drugs 2011, 20 (3), 343–359. [DOI] [PubMed] [Google Scholar]
- (448).Chen YW; Chou KM DNA lesion bypass polymerases and 4′-thio-β-D-arabinofuranosylcytosine (T-araC). Int. J. Biochem. Mol. Biol 2011, 2 (4), 340–346. [PMC free article] [PubMed] [Google Scholar]
- (449).Parker WB; Waud WR; Secrist JA 3rd Thiarabine, 1-(4-thio-β-D-arabinofuranosyl)cytosine. A deoxycytidine analog with excellent anticancer activity. Curr. Med. Chem 2015, 22 (34), 3881–3896. [DOI] [PubMed] [Google Scholar]
- (450).Thottassery JV; Westbrook L; Someya H; Parker WB c-Abl-independent p73 stabilization during gemcitabine- or 4′-thio-β-D-arabinofuranosylcytosine-induced apoptosis in wild-type and p53-null colorectal cancer cells. Mol. Cancer Ther 2006, 5 (2), 400–410. [DOI] [PubMed] [Google Scholar]
- (451).Waud WR; Gilbert KS; Secrist JA 3rd Lack of in vivo cross-resistance with 4′-thio-ara-C against drug-resistant murine P388 and L1210 leukemias. Cancer Chemother. Pharmacol 2011, 68 (2), 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (452).Waud WR; Gilbert KS; Secrist JA 3rd Preclinical combination therapy of thiarabine plus various clinical anticancer agents. Nucleosides, Nucleotides Nucleic Acids 2012, 31 (8), 630–646. [DOI] [PubMed] [Google Scholar]
- (453).Faderl S; Gandhi V; O’Brien S; Bonate P; Cortes J; Estey E; Beran M; Wierda W; Garcia-Manero G; Ferrajoli A; et al. Results of a phase 1–2 study of clofarabine in combination with cytarabine (araC) in relapsed and refractory acute leukemias. Blood 2005, 105 (3), 940–947. [DOI] [PubMed] [Google Scholar]
- (454).Lee CP; de Jonge MJ; O’Donnell AE; Schothorst KL; Hanwell J; Chick JB; Brooimans RA; Adams LM; Drolet DW; de Bono JS; et al. A phase I study of a new nucleoside analogue, OSI-7836, using two administration schedules in patients with advanced solid malignancies. Clin. Cancer Res 2006, 12 (9), 2841–2848. [DOI] [PubMed] [Google Scholar]
- (455).Goss G; Siu LL; Gauthier I; Chen EX; Oza AM; Goel R; Maroun J; Powers J; Walsh W; Maclean M; et al. A phase I, first in man study of OSI-7836 in patients with advanced refractory solid tumors: IND.147, a study of the Investigational New Drug Program of the National Cancer Institute of Canada Clinical Trials Group. Cancer Chemother. Pharmacol 2006, 58 (5), 703–710. [DOI] [PubMed] [Google Scholar]
- (456).Nayak UG; Whistler RL Synthesis of 5-thio-D-glucose. J. Org. Chem 1969, 34 (1), 97–100. [Google Scholar]
- (457).Meyer AS; Reichstein T l-Idomethylose. Desoxyzucker. 8. Mitteilung. Helv. Chim. Acta 1946, 29 (1), 139–152. [Google Scholar]
- (458).Whistler RL; Nayak UG; Perkins AW Anomeric methyl 4-thio-D-arabinofuranosides. J. Org. Chem 1970, 35 (2), 519–521. [Google Scholar]
- (459).Ness RK; Fletcher HG Jr. The anomeric 2,3,5-tri-O-benzoyl-D-arabinosyl bromides and other D-arabinofuranose derivatives. J. Am. Chem. Soc 1958, 80 (8), 2007–2010. [Google Scholar]
- (460).Jeong LS; Zhao LX; Choi WJ; Pal S; Park YH; Lee SK; Chun MW; Lee YB; Ahn CH; Moon HR Synthesis and antitumor activity of fluorocyclopentenyl-pyrimidines. Nucleosides, Nucleotides Nucleic Acids 2007, 26 (6–7), 713–716. [DOI] [PubMed] [Google Scholar]
- (461).Zhao LX; Yun M; Kim HO; Lee JA; Choi WJ; Lee KM; Lee SK; Lee YB; Ahn CH; Jeong LS Design, synthesis, and anticancer activity of fluorocyclopentenyl-pyrimidines. Nucleic Acids Symp. Ser. (Oxf) 2005, 49, 107–108. [DOI] [PubMed] [Google Scholar]
- (462).Peters GJ; Smid K; Vecchi L; Kathmann I; Sarkisjan D; Honeywell RJ; Losekoot N; Ohne O; Orbach A; Blaugrund E; et al. Metabolism, mechanism of action and sensitivity profile of fluorocyclopentenylcytosine (RX-3117; TV-1360). Invest. New Drugs 2013, 31 (6), 1444–1457. [DOI] [PubMed] [Google Scholar]
- (463).Choi WJ; Chung HJ; Chandra G; Alexander V; Zhao LX; Lee HW; Nayak A; Majik MS; Kim HO; Kim JH; Lee YB; Ahn CH; Lee SK; Jeong LS Fluorocyclopentenyl-cytosine with broad spectrum and potent antitumor activity. J. Med. Chem 2012, 55 (9), 4521–4525. [DOI] [PubMed] [Google Scholar]
- (464).Yang MY; Lee YB; Ahn CH; Kaye J; Fine T; Kashi R; Ohne O; Smid K; Peters GJ; Kim DJ A novel cytidine analog, RX-3117, shows potent efficacy in xenograft models, even in tumors that are resistant to gemcitabine. Anticancer Res 2014, 34 (12), 6951–6959. [PubMed] [Google Scholar]
- (465).Ahn CH; Choi WJ; Lee YB; Jeong LS; Lee SK US Patent 20050222185, 2005.
- (466).Cohen SS Sponges, cancer chemotherapy, and cellular aging. Perspect. Biol. Med 1963, 6 (2), 215–227. [DOI] [PubMed] [Google Scholar]
- (467).Sundaram M; Yao SY; Ingram JC; Berry ZA; Abidi F; Cass CE; Baldwin SA; Young JD Topology of a human equilibrative, nitrobenzylthioinosine (NBMPR)-sensitive nucleoside transporter (hENT1) implicated in the cellular uptake of adenosine and anti-cancer drugs. J. Biol. Chem 2001, 276 (48), 45270–45275. [DOI] [PubMed] [Google Scholar]
- (468).Clarke ML; Mackey JR; Baldwin SA; Young JD; Cass CE The role of membrane transporters in cellular resistance to anticancer nucleoside drugs. Cancer Treat. Res 2002, 112, 27–47. [DOI] [PubMed] [Google Scholar]
- (469).Liliemark JO; Plunkett W Regulation of 1-β-D-arabinofuranosylcytosine 5′-triphosphate accumulation in human leukemia cells by deoxycytidine 5′-triphosphate. Cancer Res 1986, 46 (3), 1079–1083. [PubMed] [Google Scholar]
- (470).Cohen SS The mechanisms of lethal action of arabinosyl cytosine (araC) and arabinosyl adenine (araA). Cancer 1977, 40 (1 Suppl), 509–518. [DOI] [PubMed] [Google Scholar]
- (471).Guchelaar HJ; Vermes I; Koopmans RP; Reutelingsperger CP; Haanen C Apoptosis- and necrosis-inducing potential of cladribine, cytarabine, cisplatin, and 5-fluorouracil in vitro: a quantitative pharmacodynamic model. Cancer Chemother. Pharmacol 1998, 42 (1), 77–83. [DOI] [PubMed] [Google Scholar]
- (472).Ross DD; Cuddy DP; Cohen N; Hensley DR Mechanistic implications of alterations in HL-60 cell nascent DNA after exposure to 1-β-D-arabinofuranosylcytosine. Cancer Chemother. Pharmacol 1992, 31 (1), 61–70. [DOI] [PubMed] [Google Scholar]
- (473).Drake JC; Hande KR; Fuller RW; Chabner BA Cytidine and deoxycytidylate deaminase inhibition by uridine analogs. Biochem. Pharmacol 1980, 29 (5), 807–811. [DOI] [PubMed] [Google Scholar]
- (474).Gandhi V; Plunkett W Modulation of arabinosylnucleoside metabolism by arabinosylnucleotides in human leukemia cells. Cancer Res 1988, 48 (2), 329–334. [PubMed] [Google Scholar]
- (475).Cai J; Damaraju VL; Groulx N; Mowles D; Peng Y; Robins MJ; Cass CE; Gros P Two distinct molecular mechanisms underlying cytarabine resistance in human leukemic cells. Cancer Res 2008, 68 (7), 2349–2357. [DOI] [PubMed] [Google Scholar]
- (476).Yamauchi T; Negoro E; Urasaki Y; Nishi R; Hori H; Ueda T Characterization of cytarabine-resistant leukemic cell lines established from five different blood cell lineages using gene expression and proteomic analyses. Int. J. Oncol 2011, 38 (4), 911–919. [DOI] [PubMed] [Google Scholar]
- (477).Lamba JK Genetic factors influencing cytarabine therapy. Pharmacogenomics 2009, 10 (10), 1657–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (478).Guo Y; Kock K; Ritter CA; Chen ZS; Grube M; Jedlitschky G; Illmer T; Ayres M; Beck JF; Siegmund W; et al. Expression of ABCC-type nucleotide exporters in blasts of adult acute myeloid leukemia: Relation to long-term survival. Clin. Cancer Res 2009, 15 (5), 1762–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (479).Wang JJ; Selawry OS; Vietti TJ; Bodey GP Sr. Prolonged infusion of arabinosyl cytosine in childhood leukemia. Cancer 1970, 25 (1), 1–6. [DOI] [PubMed] [Google Scholar]
- (480).Keane N; Freeman C; Swords R; Giles FJ Elacytarabine: lipid vector technology under investigation in acute myeloid leukemia. Expert Rev. Hematol 2013, 6 (1), 9–24. [DOI] [PubMed] [Google Scholar]
- (481).Walsby EJ; Coles SJ; Knapper S; Burnett AK The topoisomerase II inhibitor voreloxin causes cell cycle arrest and apoptosis in myeloid leukemia cells and acts in synergy with cytarabine. Haematologica 2011, 96 (3), 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (482).Fernandez HF New trends in the standard of care for initial therapy of acute myeloid leukemia. Hematology Am. Soc. Hematol. Educ. Program 2010, 2010, 56–61. [DOI] [PubMed] [Google Scholar]
- (483).Fathi AT; Karp JE New agents in acute myeloid leukemia: beyond cytarabine and anthracyclines. Curr. Oncol. Rep 2009, 11 (5), 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (484).Burke AC; Giles FJ Elacytarabine–lipid vector technology overcoming drug resistance in acute myeloid leukemia. Expert Opin. Invest. Drugs 2011, 20 (12), 1707–1715. [DOI] [PubMed] [Google Scholar]
- (485).O’Brien S; Rizzieri DA; Vey N; Ravandi F; Krug UO; Sekeres MA; Dennis M; Venditti A; Berry DA; Jacobsen TF; et al. Elacytarabine has single-agent activity in patients with advanced acute myeloid leukaemia. Br. J. Haematol 2012, 158 (5), 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (486).Adema AD; Smid K; Losekoot N; Honeywell RJ; Verheul HM; Myhren F; Sandvold ML; Peters GJ Metabolism and accumulation of the lipophilic deoxynucleoside analogs elacytarabine and CP-4126. Invest. New Drugs 2012, 30 (5), 1908–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (487).Bruheim S; Sandvold ML; Maelandsmo GM; Fodstad O Antitumor activity of elacytarabine combined with bevacizumab, cetuximab and trastuzumab in human NSCLC xenografts. Anticancer Res 2013, 33 (9), 3615–3621. [PubMed] [Google Scholar]
- (488).Giles F; Rizzieri D; Ravandi F; Swords R; Jacobsen TF; O’Brien S Elacytarabine, a novel 5′-elaidic acid derivative of cytarabine, and idarubicin combination is active in refractory acute myeloid leukemia. Leuk. Res 2012, 36 (4), e71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (489).Giles FJ; Vey N; Rizzieri D; Ravandi F; Prebet T; Borthakur G; Jacobsen TF; Hagen S; Nilsson B; O’Brien S Phase I and pharmacokinetic study of elacytarabine, a novel 5′-elaidic acid derivative of cytarabine, in adults with refractory hematological malignancies. Leukemia 2012, 26 (7), 1686–1689. [DOI] [PubMed] [Google Scholar]
- (490).Roboz GJ; Rosenblat T; Arellano M; Gobbi M; Altman JK; Montesinos P; O’Connell C; Solomon SR; Pigneux A; Vey N; et al. International randomized phase III study of elacytarabine versus investigator choice in patients with relapsed/refractory acute myeloid leukemia. J. Clin. Oncol 2014, 32 (18), 1919–1926. [DOI] [PubMed] [Google Scholar]
- (491).Walwick ER; Roberts WK; Dekker CA Cyclisation during the phosphorylation of uridine and cytidine by polyphosphoric acid: A new route to the O-2,2′-cyclonucleosides. Proc. Chem. Soc 1959, 84. [Google Scholar]
- (492).Mehellou Y; Valente R; Mottram H; Walsby E; Mills KI; Balzarini J; McGuigan C Phosphoramidates of 2′-β-D-arabinouridine (AraU) as phosphate prodrugs; design, synthesis, in vitro activity and metabolism. Bioorg. Med. Chem 2010, 18 (7), 2439–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (493).Qu G; Guo H; Yang X; Zhang X; Wang D; Niu H; Xia R; Li T; Wang F; Zhang Q, Zeng C CN Patent 101948492, 2011.
- (494).Jin C; Wu Z; Jia G; Li H; Wu X CN Patent 1583776, 2005.
- (495).Zong M CN Patent 1661030, 2005.
- (496).Erion MD; Reddy KR; Boyer SH; Matelich MC; Gomez-Galeno J; Lemus RH; Ugarkar BG; Colby TJ; Schanzer J; Van Poelje PD Design, synthesis, and characterization of a series of cytochrome P(450) 3A-activated prodrugs (HepDirect prodrugs) useful for targeting phosph(on)ate-based drugs to the liver. J. Am. Chem. Soc 2004, 126 (16), 5154–5163. [DOI] [PubMed] [Google Scholar]
- (497).Boyer SH; Sun Z; Jiang H; Esterbrook J; Gomez-Galeno JE; Craigo W; Reddy KR; Ugarkar BG; MacKenna DA; Erion MD Synthesis and characterization of a novel liver-targeted prodrug of cytosine-1-β-D-arabinofuranoside monophosphate for the treatment of hepatocellular carcinoma. J. Med. Chem 2006, 49 (26), 7711–7720. [DOI] [PubMed] [Google Scholar]
- (498).Bentley HR; Cunningham KG; Spring FS 509. Cordycepin, a metabolic product from cultures of cordyceps militaris (Linn.)Link. Part II. The structure of cordycepin. J. Chem. Soc 1951, 2301–2305. [Google Scholar]
- (499).Cheng Z; He W; Zhou X; Lv Q; Xu X; Yang S; Zhao C; Guo L Cordycepin protects against cerebral ischemia/reperfusion injury in vivo and in vitro. Eur. J. Pharmacol 2011, 664 (1–3), 20–28. [DOI] [PubMed] [Google Scholar]
- (500).Sugar AM; McCaffrey RP Antifungal activity of 3′-deoxyadenosine (cordycepin). Antimicrob. Agents Chemother 1998, 42 (6), 1424–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Wong YY; Moon A; Duffin R; Barthet-Barateig A; Meijer HA; Clemens MJ; de Moor CH Cordycepin inhibits protein synthesis and cell adhesion through effects on signal transduction. J. Biol. Chem 2010, 285 (4), 2610–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (502).Kraupp M; Marz R Nucleobase and nucleoside transport in mammalian cells. Wiener klinische Wochenschrift 1995, 107 (22), 677–680. [PubMed] [Google Scholar]
- (503).Holbein S; Wengi A; Decourty L; Freimoser FM; Jacquier A; Dichtl B Cordycepin interferes with 3′ end formation in yeast independently of its potential to terminate RNA chain elongation. RNA 2009, 15 (5), 837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Muller WE; Seibert G; Beyer R; Breter HJ; Maidhof A; Zahn RK Effect of cordycepin on nucleic acid metabolism in L5178Y cells and on nucleic acid-synthesizing enzyme systems. Cancer Res 1977, 37 (10), 3824–3833. [PubMed] [Google Scholar]
- (505).Overgaard-Hansen K The inhibition of 5-phosphoribosyl-1-pyrophosphate formation by cordycepin triphosphate in extracts of Ehrlich ascites tumor cells. Biochim. Biophys. Acta, Spec. Sect. Nucleic Acids Relat. Subj 1964, 80, 504–507. [DOI] [PubMed] [Google Scholar]
- (506).Rottman F; Guarino AJ The Inhibition of phosphoribosylpyrophosphate amidotransferase activity by cordycepin monophosphate. Biochim. Biophys. Acta, Spec. Sect. Enzymol. Subj 1964, 89, 465–472. [DOI] [PubMed] [Google Scholar]
- (507).Lee HJ; Burger P; Vogel M; Friese K; Bruning A The nucleoside antagonist cordycepin causes DNA double strand breaks in breast cancer cells. Invest. New Drugs 2012, 30 (5), 1917–1925. [DOI] [PubMed] [Google Scholar]
- (508).Lee SJ; Kim SK; Choi WS; Kim WJ; Moon SK Cordycepin causes p21WAF1-mediated G2/M cell-cycle arrest by regulating c-Jun N-terminal kinase activation in human bladder cancer cells. Arch. Biochem. Biophys 2009, 490 (2), 103–109. [DOI] [PubMed] [Google Scholar]
- (509).Nakamura K; Yoshikawa N; Yamaguchi Y; Kagota S; Shinozuka K; Kunitomo M Antitumor effect of cordycepin (3′-deoxyadenosine) on mouse melanoma and lung carcinoma cells involves adenosine A3 receptor stimulation. Anticancer Res 2006, 26 (1A), 43–47. [PubMed] [Google Scholar]
- (510).Tian X; Li Y; Shen Y; Li Q; Wang Q; Feng L Apoptosis and inhibition of proliferation of cancer cells induced by cordycepin. Oncol. Lett 2015, 10 (2), 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (511).Yao WL; Ko BS; Liu TA; Liang SM; Liu CC; Lu YJ; Tzean SS; Shen TL; Liou JY Cordycepin suppresses integrin/FAK signaling and epithelial-mesenchymal transition in hepatocellular carcinoma. Anti-Cancer Agents Med. Chem 2014, 14 (1), 29–34. [DOI] [PubMed] [Google Scholar]
- (512).Lee WW; Benitez A; Anderson CD; Goodman L; Baker BR Potential anticancer agents. LV. Synthesis of 3′-amino-2′,3′-dideoxyadenosine and related analogs. J. Am. Chem. Soc 1961, 83 (8), 1906–1911. [Google Scholar]
- (513).Hansske F; Robins MJ Regiospecific and stereoselective conversion of ribonucleosides to 3′-deoxynucleosides. A high yield three-stage synthesis of cordycepin from adenosine. Tetrahedron Lett 1985, 26, 4295–4298. [Google Scholar]
- (514).Aman S; Anderson DJ; Connolly TJ; Crittall AJ; Ji GJ From adenosine to 3′-deoxyadenosine: Development and scale up. Org. Process Res. Dev 2000, 4 (6), 601–605. [Google Scholar]
- (515).Grunebaum E; Cohen A; Roifman CM Recent advances in understanding and managing adenosine deaminase and purine nucleoside phosphorylase deficiencies. Curr. Opin. Allergy Clin. Immu 2013, 13 (6), 630–638. [DOI] [PubMed] [Google Scholar]
- (516).Carson DA; Wasson DB; Taetle R; Yu A Specific toxicity of 2-chlorodeoxyadenosine toward resting and proliferating human lymphocytes. Blood 1983, 62 (4), 737–743. [PubMed] [Google Scholar]
- (517).Cory JG; Rey DA; Carter GL; Bacon PE Nucleoside 5′-diphosphates as effectors of mammalian ribonucleotide reductase. J. Biol. Chem 1985, 260 (22), 12001–12007. [PubMed] [Google Scholar]
- (518).Carson DA; Wasson DB; Kaye J; Ullman B; Martin DWJ; Robins RK; Montgomery JA 2′-deoxycytidine kinase-mediated toxicity of deoxyadenosine analogs toward malignant human lympho-blasts in vitro and toward murine L1210 leukemia in vivo. Proc. Natl. Acad. Sci. U. S. A 1980, 77, 6865–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (519).Bryson HM; Sorkin EM Cladribine. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in haematological malignancies. Drugs 1993, 46 (5), 872–894. [DOI] [PubMed] [Google Scholar]
- (520).Ghanem H; Jabbour E; Faderl S; Ghandhi V; Plunkett W; Kantarjian H Clofarabine in leukemia. Expert Rev. Hematol 2010, 3 (1), 15–22. [DOI] [PubMed] [Google Scholar]
- (521).Lindemalm S; Liliemark J; Juliusson G; Larsson R; Albertioni F Cytotoxicity and pharmacokinetics of cladribine metabolite, 2-chloroadenine in patients with leukemia. Cancer Lett 2004, 210 (2), 171–177. [DOI] [PubMed] [Google Scholar]
- (522).King KM; Damaraju VL; Vickers MF; Yao SY; Lang T; Tackaberry TE; Mowles DA; Ng AM; Young JD; Cass CE A comparison of the transportability, and its role in cytotoxicity, of clofarabine, cladribine, and fludarabine by recombinant human nucleoside transporters produced in three model expression systems. Mol. Pharm 2006, 69 (1), 346–353. [DOI] [PubMed] [Google Scholar]
- (523).Wang L; Karlsson A; Arner ES; Eriksson S Substrate specificity of mitochondrial 2′-deoxyguanosine kinase. Efficient phosphorylation of 2-chlorodeoxyadenosine. J. Biol. Chem 1993, 268 (30), 22847–22852. [PubMed] [Google Scholar]
- (524).Huang MC; Ashmun RA; Avery TL; Kuehl M; Blakley RL Effects of cytotoxicity of 2-chloro-2′-deoxyadenosine and 2-bromo-2′-deoxyadenosine on cell growth, clonogenicity, DNA synthesis, and cell cycle kinetics. Cancer Res 1986, 46 (5), 2362–2368. [PubMed] [Google Scholar]
- (525).Parker WB; Bapat AR; Shen JX; Townsend AJ; Cheng YC Interaction of 2-halogenated dATP analogs (F, Cl, and Br) with human DNA polymerases, DNA primase, and ribonucleotide reductase. Mol. Pharmacol 1988, 34 (4), 485–491. [PubMed] [Google Scholar]
- (526).Tanabe K; Hiraoka W; Kuwabara M; Sato F; Matsuda A; Ueda T Induction of DNA double-strand breaks in Chinese hamster V79 cells by 2-chlorodeoxyadenosine. Chem.-Biol. Interact 1989, 71 (2–3), 167–175. [DOI] [PubMed] [Google Scholar]
- (527).Wataya Y; Watanabe K; Yoshida S; Hiramoto-Yoshioka A dNTP imbalance and DNA double strand breaks in mouse FM3A cells and the mechanism of cell death. Nucleic Acids Symp. Ser 1989, 21, 11–12. [PubMed] [Google Scholar]
- (528).Ghai V; Sharma K; Abbi KK; Shimko S; Epner EM Current approaches to epigenetic therapy for the treatment of mantle cell lymphoma. Adv. Exp. Med. Biol 2013, 779, 257–266. [DOI] [PubMed] [Google Scholar]
- (529).Lubecka-Pietruszewska K; Kaufman-Szymczyk A; Stefanska B; Cebula-Obrzut B; Smolewski P; Fabianowska-Majewska K Clofarabine, a novel adenosine analogue, reactivates DNA methylation-silenced tumour suppressor genes and inhibits cell growth in breast cancer cells. Eur. J. Pharmacol 2014, 723, 276–287. [DOI] [PubMed] [Google Scholar]
- (530).Stefanska B; Rudnicka K; Bednarek A; Fabianowska-Majewska K Hypomethylation and induction of retinoic acid receptor β2 by concurrent action of adenosine analogues and natural compounds in breast cancer cells. Eur. J. Pharmacol 2010, 638 (1–3), 47–53. [DOI] [PubMed] [Google Scholar]
- (531).Conrad DM; Robichaud MR; Mader JS; Boudreau RT; Richardson AM; Giacomantonio CA; Hoskin DW 2-Chloro-2′-deoxyadenosine-induced apoptosis in T leukemia cells is mediated via a caspase-3-dependent mitochondrial feedback amplification loop. Int. J. Oncol 2008, 32 (6), 1325–1333. [DOI] [PubMed] [Google Scholar]
- (532).Kopadze T; Dobert M; Leussink VI; Dehmel T; Kieseier BC Cladribine impedes in vitro migration of mononuclear cells: a possible implication for treating multiple sclerosis. Eur. J. Neurol 2009, 16 (3), 409–412. [DOI] [PubMed] [Google Scholar]
- (533).Mansson E; Spasokoukotskaja T; Sallstrom J; Eriksson S; Albertioni F Molecular and biochemical mechanisms of fludarabine and cladribine resistance in a human promyelocytic cell line. Cancer Res 1999, 59 (23), 5956–5963. [PubMed] [Google Scholar]
- (534).Scheible H; Laisney M; Wimmer E; Javornik A; Dolgos H Comparison of the in vitro and in vivo metabolism of cladribine (Leustatin, Movectro) in animals and human. Xenobiotica 2013, 43 (12), 1084–1094. [DOI] [PubMed] [Google Scholar]
- (535).Takenaka K; Morgan JA; Scheffer GL; Adachi M; Stewart CF; Sun D; Leggas M; Ejendal KF; Hrycyna CA; Schuetz JD Substrate overlap between Mrp4 and Abcg2/Bcrp affects purine analogue drug cytotoxicity and tissue distribution. Cancer Res 2007, 67 (14), 6965–6972. [DOI] [PubMed] [Google Scholar]
- (536).Spurgeon S; Yu M; Phillips JD; Epner EM Cladribine: not just another purine analogue? Expert Opin. Invest. Drugs 2009, 18 (8), 1169–1181. [DOI] [PubMed] [Google Scholar]
- (537).Singh V; Prajeeth CK; Gudi V; Benardais K; Voss EV; Stangel M 2-Chlorodeoxyadenosine (cladribine) induces apoptosis in human monocyte-derived dendritic cells. Clin. Exp. Immunol 2013, 173 (2), 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (538).Gerrie AS; Zypchen LN; Connors JM Fludarabine and rituximab for relapsed or refractory hairy cell leukemia. Blood 2012, 119 (9), 1988–1991. [DOI] [PubMed] [Google Scholar]
- (539).Beutler E; Sipe JC; Romine JS; Koziol JA; McMillan R; Zyroff J The treatment of chronic progressive multiple sclerosis with cladribine. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 1716–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (540).Pardanani A Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am. J. Hematol 2013, 88 (7), 612–624. [DOI] [PubMed] [Google Scholar]
- (541).Lajolo C; Campisi G; Deli G; Littarru C; Guiglia R; Giuliani M Langerhans’s cell histiocytosis in old subjects: two rare case reports and review of the literature. Gerodontology 2012, 29 (2), e1207–1214. [DOI] [PubMed] [Google Scholar]
- (542).Oh J; O’Connor PW Safety, Tolerability, and Efficacy of Oral Therapies for Relapsing-Remitting Multiple Sclerosis. CNS Drugs 2013, 27 (8), 591–609. [DOI] [PubMed] [Google Scholar]
- (543).Venner H Synthese der den natürlichen entsprechenden 2-desoxy-nucleoside des adenins, guanins und hypoxanthins. Chem. Ber 1960, 93 (1), 140–149. [Google Scholar]
- (544).Ikehara M; Tada H Studies of nucleosides and nucleotides. XXIV. Purine cyclonucleosides. I. 8,2′-cyclonucleoside derived from 2-chloro-8-mercapto-9-β-D-xylofuranosyladenine. J. Am. Chem. Soc 1965, 87 (3), 606–610. [DOI] [PubMed] [Google Scholar]
- (545).Christensen LF; Broom AD; Robins MJ; Bloch A Synthesis and biological activity of selected 2, 6-disubstituted (2-deoxy-α-and-β-D-erythro-pentofuranosyl) purines. J. Med. Chem 1972, 15 (7), 735–739. [DOI] [PubMed] [Google Scholar]
- (546).Zhong M; Nowak I; Robins MJ Regiospecific and highly stereoselective coupling of 6-(substituted-imidazol-1-yl)purines with 2-deoxy-3,5-di-O-(p-toluoyl)-α-D-erythro-pentofuranosyl chloride. Sodium-salt glycosylation in binary solvent mixtures: improved synthesis of cladribine. J. Org. Chem 2006, 71 (20), 7773–7779. [DOI] [PubMed] [Google Scholar]
- (547).Hoffer M α-Thymidin. Chem. Ber 1960, 93 (12), 2777–2781. [Google Scholar]
- (548).Jaworski JS Looking for a contribution of the non-equilibrium solvent polarization to the activation barrier of the SN2 reaction. J. Phys. Org. Chem 2002, 15 (6), 319–323. [Google Scholar]
- (549).Henschke JP; Zhang X; Huang X; Mei L; Chu G; Hu K; Wang Q; Zhu G; Wu M; Kuo C; Chen Y A stereoselective process for the manufacture of a 2′-deoxy-β-D-ribonucleoside using the Vorbruggen glycosylation. Org. Process Res. Dev 2013, 17 (11), 1419–1429. [Google Scholar]
- (550).Woo PWK; Dion HW; Lange SM; Dahl LF; Durham LJ A novel adenosine and ara-A deaminase inhibitor, (R)-3-(2-deoxy-β-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5 -d] [1,3]-diazepin-8-ol. J. Heterocycl. Chem 1974, 11, 641–643. [Google Scholar]
- (551).Chen SF; Stoeckler JD; Parks RE Jr. Transport of deoxycoformycin in human erythrocytes. Measurement by adenosine deaminase titration and radioisotope assays. Biochem. Pharmacol 1984, 33 (24), 4069–4079. [DOI] [PubMed] [Google Scholar]
- (552).Hunt SW 3rd; Hoffee PA Adenosine deaminase from deoxycoformycin-sensitive and -resistant rat hepatoma cells. Purification and characterization. J. Biol. Chem 1982, 257 (23), 14239–14244. [PubMed] [Google Scholar]
- (553).Merkler DJ; Brenowitz M; Schramm VL The rate constant describing slow-onset inhibition of yeast AMP deaminase by coformycin analogues is independent of inhibitor structure. Biochemistry 1990, 29 (36), 8358–8364. [DOI] [PubMed] [Google Scholar]
- (554).Kajander EO; Kubota M; Willis EH; Carson DA S-adenosylmethionine metabolism as a target for adenosine toxicity. Adv. Exp. Med. Biol 1986, 131 (Pt B), 221–226. [DOI] [PubMed] [Google Scholar]
- (555).Renshaw J; Harrap KR In vivo inhibition of mouse liver methyltransferase enzymes following treatment with 2′-deoxycoformycin and 2′-deoxyadenosine. Adv. Exp. Med. Biol 1986, 131 (Pt B), 673–675. [DOI] [PubMed] [Google Scholar]
- (556).Hermes M; Osswald H; Kloor D Role of S-adenosylhomocysteine hydrolase in adenosine-induced apoptosis in HepG2 cells. Exp. Cell Res 2007, 313 (2), 264–283. [DOI] [PubMed] [Google Scholar]
- (557).Grever MR; Doan CA; Kraut EH Pentostatin in the treatment of hairy-cell leukemia. Best Pract. Res. Clin. Haematol 2003, 16 (1), 91–99. [DOI] [PubMed] [Google Scholar]
- (558).Kurzrock R Therapy of T cell lymphomas with pentostatin. Ann. N. Y. Acad. Sci 2001, 941, 200–205. [DOI] [PubMed] [Google Scholar]
- (559).Anglin JL; Song Y A medicinal chemistry perspective for targeting histone H3 lysine-79 methyltransferase DOT1L. J. Med. Chem 2013, 56 (22), 8972–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (560).Showalter HD; Bunge RH; French JC; Hurley TR; Leeds RL; Leja B; McDonnell PD; Edmunds CR Improved production of pentostatin and identification of fermentation comet-abolites. J. Antibiot 1992, 45 (12), 1914–1918. [DOI] [PubMed] [Google Scholar]
- (561).Baker DC; Putt SR A total synthesis of pentostatin, the potent inhibitor of adenosine deaminase. J. Am. Chem. Soc 1979, 101 (20), 6127–6128. [Google Scholar]
- (562).Chan E; Putt SR; Showalter HDH; Baker DC Total synthesis of (8R)-3-(2-deoxy-β-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol (pentostatin), the potent inhibitor of adenosine deaminase. J. Org. Chem 1982, 47, 3457–3464. [Google Scholar]
- (563).Zhang H-R; Wang W US Patent 20090012288, 2009.
- (564).Phiasivongsa P; Redkar S WO Patent 2005027838, 2005.
- (565).Majda K; Lubecka K; Kaufman-Szymczyk A; Fabianowska-Majewska K Clofarabine (2-chloro-2′-fluoro-2′-deoxyarabinosylade-nine)-biochemical aspects of anticancer activity. Acta. Polym. Pharm 2011, 68 (4), 459–466. [PubMed] [Google Scholar]
- (566).Montgomery JA; Shortnacy-Fowler AT; Clayton SD; Riordan JM; Secrist JA 3rd Synthesis and biologic activity of 2′-fluoro-2-halo derivatives of 9-β-D-arabinofuranosyladenine. J. Med. Chem 1992, 35 (2), 397–401. [DOI] [PubMed] [Google Scholar]
- (567).Parker WB; Allan PW; Hassan AE; Secrist JA 3rd; Sorscher EJ; Waud WR Antitumor activity of 2-fluoro-2′-deoxyadenosine against tumors that express Escherichia coli purine nucleoside phosphorylase. Cancer Gene Ther 2003, 10 (1), 23–29. [DOI] [PubMed] [Google Scholar]
- (568).Bonate PL; Arthaud L; Cantrell WR Jr.; Stephenson K; Secrist JA 3rd; Weitman S Discovery and development of clofarabine: a nucleoside analogue for treating cancer. Nat. Rev. Drug Discovery 2006, 5 (10), 855–863. [DOI] [PubMed] [Google Scholar]
- (569).Takahashi T; Kanazawa J; Akinaga S; Tamaoki T; Okabe M Antitumor activity of 2-chloro-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl) adenine, a novel deoxyadenosine analog, against human colon tumor xenografts by oral administration. Cancer Chemother. Pharmacol 1999, 43 (3), 233–240. [DOI] [PubMed] [Google Scholar]
- (570).Carson DA; Wasson DB; Esparza LM; Carrera CJ; Kipps TJ; Cottam HB Oral antilymphocyte activity and induction of apoptosis by 2-chloro-2′-arabino-fluoro-2′-deoxyadenosine. Proc. Natl. Acad. Sci. U. S. A 1992, 89 (7), 2970–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (571).Parker WB; Shaddix SC; Chang CH; White EL; Rose LM; Brockman RW; Shortnacy AT; Montgomery JA; Secrist JA 3rd; Bennett LL Jr. Effects of 2-chloro-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)adenine on K562 cellular metabolism and the inhibition of human ribonucleotide reductase and DNA polymerases by its 5′-triphosphate. Cancer Res 1991, 51 (9), 2386–2394. [PubMed] [Google Scholar]
- (572).Lotfi K; Mansson E; Spasokoukotskaja T; Pettersson B; Liliemark J; Peterson C; Eriksson S; Albertioni F Biochemical pharmacology and resistance to 2-chloro-2′-arabino-fluoro-2′-deoxyadenosine, a novel analogue of cladribine in human leukemic cells. Clin. Cancer Res 1999, 5 (9), 2438–2444. [PubMed] [Google Scholar]
- (573).Xie C; Plunkett W Metabolism and actions of 2-chloro-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-adenine in human lymphoblastoid cells. Cancer Res 1995, 55 (13), 2847–2852. [PubMed] [Google Scholar]
- (574).Xie KC; Plunkett W Deoxynucleotide pool depletion and sustained inhibition of ribonucleotide reductase and DNA synthesis after treatment of human lymphoblastoid cells with 2-chloro-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl) adenine. Cancer Res 1996, 56 (13), 3030–3037. [PubMed] [Google Scholar]
- (575).Genini D; Adachi S; Chao Q; Rose DW; Carrera CJ; Cottam HB; Carson DA; Leoni LM Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood 2000, 96 (10), 3537–3543. [PubMed] [Google Scholar]
- (576).Locke F; Agarwal R; Kunnavakkam R; van Besien K; Larson RA; Odenike O; Godley LA; Liu H; Le Beau MM; Gurbuxani S; et al. A novel clofarabine bridge strategy facilitates allogeneic transplantation in patients with relapsed/refractory leukemia and high-risk myelodysplastic syndromes. Bone Marrow Transplant 2013, 48 (11), 1437–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (577).Tiley S; Claxton D Clofarabine in the treatment of acute myeloid leukemia in older adults. Ther. Adv. Hematol 2013, 4 (1), 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (578).Abramson JS; Takvorian RW; Fisher DC; Feng Y; Jacobsen ED; Brown JR; Barnes JA; Neuberg DS; Hochberg EP Oral clofarabine for relapsed/refractory non-Hodgkin lymphomas: results of a phase 1 study. Leuk. Lymphoma 2013, 54 (9), 1915–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (579).Faderl S; Garcia-Manero G; Estrov Z; Ravandi F; Borthakur G; Cortes JE; O’Brien S; Gandhi V; Plunkett W; Byrd A; et al. Oral clofarabine in the treatment of patients with higher-risk myelodysplastic syndrome. J. Clin. Oncol 2010, 28 (16), 2755–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (580).Montgomery JA; Shortnacy AT; Carson DA; Secrist JA 3rd 9-(2-Deoxy-2-fluoro-β-D-arabinofuranosyl)guanine: a metabolically stable cytotoxic analogue of 2′-deoxyguanosine. J. Med. Chem 1986, 29 (11), 2389–2392. [DOI] [PubMed] [Google Scholar]
- (581).Reichman U; Watanabe KA; Fox JJ A practical synthesis of 2-deoxy-2-fluoro-D-arabinofuranose derivatives. Carbohydr. Res 1975, 42 (2), 233–240. [DOI] [PubMed] [Google Scholar]
- (582).Bauta WE; Schulmeier BE; Burke B; Puente JF; Cantrell WR; Lovett D; Goebel J; Anderson B; Ionescu D; Guo R A new process for antineoplastic agent clofarabine. Org. Process Res. Dev 2004, 8, 889–896. [Google Scholar]
- (583).Tann CH; Brodfuehrer PR; Brundidge SP; Sapino C; Howell HG Fluorocarbohydrates in synthesis. An efficient synthesis of 1-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-5-iodouracil (β-FIAU) and 1-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)thymine (β-FMAU). J. Org. Chem 1985, 50 (19), 3644–3647. [Google Scholar]
- (584).Brodfuehrer PR; Sapino CJ; Howell HG A stereo-controlled synthesis of 1,3,5-tri-O-benzoyl-α-D-ribofuranose. J. Org. Chem 1985, 50 (14), 2597–2598. [Google Scholar]
- (585).Gandhi V; Keating MJ; Bate G; Kirkpatrick P Nelarabine. Nat. Rev. Drug Discovery 2006, 5 (1), 17–18. [DOI] [PubMed] [Google Scholar]
- (586).Osborne WR; Chen SH; Giblett ER; Biggar WD; Ammann AA; Scott CR Purine nucleoside phosphorylase deficiency. Evidence for molecular heterogeneity in two families with enzyme-deficient members. J. Clin. Invest 1977, 60 (3), 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (587).Curbo S; Kalsson A In Deoxynucleoside Analogs in Cancer Therapy; Humana Press: Totowa, NJ, 2006; pp 215–224. [Google Scholar]
- (588).Gokbuget N; Basara N; Baurmann H; Beck J; Bruggemann M; Diedrich H; Guldenzoph B; Hartung G; Horst HA; Huttmann A; et al. High single-drug activity of nelarabine in relapsed T-lymphoblastic leukemia/lymphoma offers curative option with subsequent stem cell transplantation. Blood 2011, 118 (13), 3504–3511. [DOI] [PubMed] [Google Scholar]
- (589).Ravandi F; Gandhi V Novel purine nucleoside analogues for T-cell-lineage acute lymphoblastic leukaemia and lymphoma. Expert Opin. Invest. Drugs 2006, 15 (12), 1601–1613. [DOI] [PubMed] [Google Scholar]
- (590).Mahmoudian M; Eaddy J; Dawson M Enzymic acylation of 506U78 (2-amino-9-β-D-arabinofuranosyl-6- methoxy-9H-purine), a powerful new anti-leukaemic agent. Biotechnol. Appl. Biochem 1999, 29 (Pt 3), 229–233. [PubMed] [Google Scholar]
- (591).Balakrishnan K; Nimmanapalli R; Ravandi F; Keating MJ; Gandhi V Forodesine, an inhibitor of purine nucleoside phosphorylase, induces apoptosis in chronic lymphocytic leukemia cells. Blood 2006, 108 (7), 2392–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (592).Robak T; Lech-Maranda E; Korycka A; Robak E Purine nucleoside analogs as immunosuppressive and antineoplastic agents: mechanism of action and clinical activity. Curr. Med. Chem 2006, 13 (26), 3165–3189. [DOI] [PubMed] [Google Scholar]
- (593).Rodriguez CO Jr.; Mitchell BS; Ayres M; Eriksson S; Gandhi V Arabinosylguanine is phosphorylated by both cytoplasmic 2′-deoxycytidine kinase and mitochondrial deoxyguanosine kinase. Cancer Res 2002, 62 (11), 3100–3105. [PubMed] [Google Scholar]
- (594).Rodriguez CO Jr.; Gandhi V Arabinosylguanine-induced apoptosis of T-lymphoblastic cells: incorporation into DNA is a necessary step. Cancer Res 1999, 59 (19), 4937–4943. [PubMed] [Google Scholar]
- (595).Rodriguez CO Jr.; Stellrecht CM; Gandhi V Mechanisms for T-cell selective cytotoxicity of arabinosylguanine. Blood 2003, 102 (5), 1842–1848. [DOI] [PubMed] [Google Scholar]
- (596).Gravatt LC; Chaffee S; Hebert ME; Halperin EC; Friedman HS; Kurtzberg J Efficacy and toxicity of 9-β-D-arabinofuranosylguanine (araG) as an agent to purge malignant T cells from murine bone marrow: application to an in vivo T-leukemia model. Leukemia 1993, 7 (8), 1261–1267. [PubMed] [Google Scholar]
- (597).Cohen MH; Johnson JR; Massie T; Sridhara R; McGuinn WD Jr.; Abraham S; Booth BP; Goheer MA; Morse D; Chen XH; et al. Approval summary: Nelarabine for the treatment of T-cell lymphoblastic leukemia/lymphoma. Clin. Cancer Res 2006, 12 (18), 5329–5335. [DOI] [PubMed] [Google Scholar]
- (598).Krenitsky TA; Koszalka GW; Jones LA; Averett DR; Moorman AR EP Patent 0294114, 1988.
- (599).Zong Z; Wang X; Zhao J; Chen L; Wei J CN Patent 101402662, 2009.
- (600).Hanna NB; Ramasamy K; Robins RK; Revankar GR A convenient synthesis of 2′-deoxy-6-thioguanosine, ara-guanine, ara-6-thioguanine and certain related purine nucleosides by the stereospecific sodium salt glycosylation procedure. J. Heterocycl. Chem 1988, 25 (6), 1899–1903. [Google Scholar]
- (601).Blumbergs P; Kalamas RL; Khan MS WO Patent 1991008215, 1991.
- (602).Sagar S; Kaur M; Minneman KP Antiviral lead compounds from marine sponges. Mar. Drugs 2010, 8 (10), 2619–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (603).Gandhi V; Chen W; Ayres M; Rhie JK; Madden TL; Newman RA Plasma and cellular pharmacology of 8-chloro-adenosine in mice and rats. Cancer Chemother. Pharmacol 2002, 50 (2), 85–94. [DOI] [PubMed] [Google Scholar]
- (604).Niitsu N; Umeda M; Honma Y Myeloid and monocytoid leukemia cells have different sensitivity to differentiation-inducing activity of deoxyadenosine analogs. Leuk. Res 2000, 24 (1), 1–9. [DOI] [PubMed] [Google Scholar]
- (605).Avramis VI; Plunkett W 2-fluoro-ATP: A toxic metabolite of 9-β-D-arabinosyl-2-fluoroadenine. Biochem. Biophys. Res. Commun 1983, 113 (1), 35–43. [DOI] [PubMed] [Google Scholar]
- (606).Rivero A; Rapado I; Tomas JF; Montalban C; de Ona R; Paz-Carreira J; Canales M; Martinez R; Sanchez-Godoy P; de Sevilla AF; et al. Relationship between 2′-deoxycytidine kinase (DCK) genotypic variants and fludarabine toxicity in patients with follicular lymphoma. Leuk. Res 2011, 35 (4), 431–437. [DOI] [PubMed] [Google Scholar]
- (607).Brockman RW; Cheng YC; Schabel FM Jr.; Montgomery JA Metabolism and chemotherapeutic activity of 9-β-D-arabinofuranosyl-2-fluoroadenine against murine leukemia L1210 and evidence for its phosphorylation by 2′-deoxycytidine kinase. Cancer Res 1980, 40 (10), 3610–3615. [PubMed] [Google Scholar]
- (608).Lotfi K; Karlsson K; Fyrberg A; Juliusson G; Jonsson V; Peterson C; Eriksson S; Albertioni F The pattern of deoxycytidineand deoxyguanosine kinase activity in relation to messenger RNA expression in blood cells from untreated patients with B-cell chronic lymphocytic leukemia. Biochem. Pharmacol 2006, 71 (6), 882–890. [DOI] [PubMed] [Google Scholar]
- (609).Lukenbill J; Kalaycio M Fludarabine: A review of the clear benefits and potential harms. Leuk. Res 2013, 37 (9), 986–994. [DOI] [PubMed] [Google Scholar]
- (610).Huang P; Sandoval A; Van Den Neste E; Keating MJ; Plunkett W Inhibition of RNA transcription: a biochemical mechanism of action against chronic lymphocytic leukemia cells by fludarabine. Leukemia 2000, 14 (8), 1405–1413. [DOI] [PubMed] [Google Scholar]
- (611).Pepper C; Lowe H; Fegan C; Thurieau C; Thurston DE; Hartley JA; Delavault P Fludarabine-mediated suppression of the excision repair enzyme ERCC1 contributes to the cytotoxic synergy with the DNA minor groove crosslinking agent SJG-136 (NSC 694501) in chronic lymphocytic leukaemia cells. Br. J. Cancer 2007, 97 (2), 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (612).Ferracin M; Zagatti B; Rizzotto L; Cavazzini F; Veronese A; Ciccone M; Saccenti E; Lupini L; Grilli A; De Angeli C; et al. MicroRNAs involvement in fludarabine refractory chronic lymphocytic leukemia. Mol. Cancer 2010, 9, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (613).Moussay E; Palissot V; Vallar L; Poirel HA; Wenner T; El Khoury V; Aouali N; Van Moer K; Leners B; Bernardin F; et al. Determination of genes and microRNAs involved in the resistance to fludarabine in vivo in chronic lymphocytic leukemia. Mol. Cancer 2010, 9, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (614).Molica S Progress in the treatment of elderly/unfit chronic lymphocytic leukemia patients: results of the German CLL-11 trial. Expert Rev. Anticancer Ther 2015, 15, 9–15. [DOI] [PubMed] [Google Scholar]
- (615).Montgomery JA; Hewson K Nucleosides of 2-fluoroadenine. J. Med. Chem 1969, 12 (3), 498–504. [DOI] [PubMed] [Google Scholar]
- (616).Barker R; Fletcher HG Jr. 2,3,5-tri-O-benzyl-D-ribosyl and -L-arabinosyl bromides. J. Org. Chem 1961, 26 (11), 4605–4609. [Google Scholar]
- (617).Glaudemans CPJ; Fletcher HG Jr. Syntheses with partially benzylated sugars. III. A simple pathway to a “cis- nucleoside,” 9-β-D-arabinofuranosyladenine (Spongoadenosine). J. Org. Chem 1963, 28 (11), 3004–3006. [Google Scholar]
- (618).Montgomery JA US Patent 4188378, 1980.
- (619).Montgomery JA; Shortnacy AT US Patent 4357324, 1982.
- (620).Blumbergs P; Khan MS; Kalamas RL US Patent 5110919, 1992.
- (621).Blumbergs P US Patent 5296589, 1994.
- (622).Kshirsagar SW; Deshpande MS; Sonawane SP; Maikap GC; Gurjar MK Simple modification to obtain high quality fludarabine. Org. Process Res. Dev 2012, 16 (5), 840–842. [Google Scholar]
- (623).Farina P; Petrucciani L; Colombo P; Caprioli G EP Patent 1464708, 2004.
- (624).Stellrecht CM; Ayres M; Arya R; Gandhi V A unique RNA-directed nucleoside analog is cytotoxic to breast cancer cells and depletes cyclin E levels. Breast Cancer Res. Treat 2010, 121 (2), 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (625).Katsaros D; Tortora G; Tagliaferri P; Clair T; Ally S; Neckers L; Robins RK; Cho-Chung YS Site-selective cyclic AMP analogs provide a new approach in the control of cancer cell growth. FEBS Lett 1987, 223 (1), 97–103. [DOI] [PubMed] [Google Scholar]
- (626).Tortora G; Ciardiello F; Ally S; Clair T; Salomon DS; Cho-Chung YS Site-selective 8-chloroadenosine 3′,5′-cyclic monophosphate inhibits transformation and transforming growth factor alpha production in Ki-ras-transformed rat fibroblasts. FEBS Lett 1989, 242 (2), 363–367. [DOI] [PubMed] [Google Scholar]
- (627).Ally S; Tortora G; Clair T; Grieco D; Merlo G; Katsaros D; Ogreid D; Doskeland SO; Jahnsen T; Cho-Chung YS Selective modulation of protein kinase isozymes by the site-selective analog 8-chloroadenosine 3′,5′-cyclic monophosphate provides a biological means for control of human colon cancer cell growth. Proc. Natl. Acad. Sci. U. S. A 1988, 85 (17), 6319–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (628).Halgren RG; Traynor AE; Pillay S; Zell JL; Heller KF; Krett NL; Rosen ST 8Cl-cAMP cytotoxicity in both steroid sensitive and insensitive multiple myeloma cell lines is mediated by 8Cladenosine. Blood 1998, 92 (8), 2893–2898. [PubMed] [Google Scholar]
- (629).Lang TT; Young JD; Cass CE Interactions of nucleoside analogs, caffeine, and nicotine with human concentrative nucleoside transporters 1 and 2 stably produced in a transport-defective human cell line. Mol. Pharm 2004, 65 (4), 925–933. [DOI] [PubMed] [Google Scholar]
- (630).Balakrishnan K; Stellrecht CM; Genini D; Ayres M; Wierda WG; Keating MJ; Leoni LM; Gandhi V Cell death of bioenergetically compromised and transcriptionally challenged CLL lymphocytes by chlorinated ATP. Blood 2005, 105 (11), 4455–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (631).Chen LS; Sheppard TL Chain termination and inhibition of Saccharomyces cerevisiae poly(A) polymerase by C-8-modified ATP analogs. J. Biol. Chem 2004, 279 (39), 40405–40411. [DOI] [PubMed] [Google Scholar]
- (632).Stellrecht CM; Rodriguez CO Jr.; Ayres M; Gandhi V RNA-directed actions of 8-chloro-adenosine in multiple myeloma cells. Cancer Res 2003, 63 (22), 7968–7974. [PubMed] [Google Scholar]
- (633).Stellrecht CM; Phillip CJ; Cervantes-Gomez F; Gandhi V Multiple myeloma cell killing by depletion of the MET receptor tyrosine kinase. Cancer Res 2007, 67 (20), 9913–9920. [DOI] [PubMed] [Google Scholar]
- (634).Yang SY; Jia XZ; Feng LY; Li SY; An GS; Ni JH; Jia HT Inhibition of topoisomerase II by 8-chloro-adenosine triphosphate induces DNA double-stranded breaks in 8-chloro-adenosine-exposed human myelocytic leukemia K562 cells. Biochem. Pharmacol 2009, 77 (3), 433–443. [DOI] [PubMed] [Google Scholar]
- (635).Dennison JB; Balakrishnan K; Gandhi V Preclinical activity of 8-chloroadenosine with mantle cell lymphoma: Roles of energy depletion and inhibition of DNA and RNA synthesis. Br. J. Haematol 2009, 147 (3), 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (636).Ahn YH; Jung JM; Hong SH 8-Chloro-cyclic AMP-induced growth inhibition and apoptosis is mediated by p38 mitogen-activated protein kinase activation in HL60 cells. Cancer Res 2005, 65 (11), 4896–4901. [DOI] [PubMed] [Google Scholar]
- (637).Stellrecht CM; Vangapandu HV; Le XF; Mao W; Shentu S ATP directed agent, 8-chloro-adenosine, induces AMP activated protein kinase activity, leading to autophagic cell death in breast cancer cells. J. Hematol. Oncol 2014, 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (638).McBrayer SK; Yarrington M; Qian J; Feng G; Shanmugam M; Gandhi V; Krett NL; Rosen ST Integrative gene expression profiling reveals G6PD-mediated resistance to RNA-directed nucleoside analogues in B-cell neoplasms. PLoS One 2012, 7 (7), e41455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (639).Shanmugam M; McBrayer SK; Qian J; Raikoff K; Avram MJ; Singhal S; Gandhi V; Schumacker PT; Krett NL; Rosen ST Targeting glucose consumption and autophagy in myeloma with the novel nucleoside analogue 8-aminoadenosine. J. Biol. Chem 2009, 284 (39), 26816–26830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (640).Brentnall HJ; Hutchinson DW Preparation of 8-chloroadenosine and its phosphate esters. Tetrahedron Lett 1972, 13 (25), 2595–2596. [Google Scholar]
- (641).Ryu EK; MacCoss M New procedure for the chlorination of pyrimidine and purine nucleosides. J. Org. Chem 1981, 46 (13), 2819–2823. [Google Scholar]
- (642).Woo NT; Jin SY; Cho DJ; Kim NS; Bae EH; Jung JH; Ham WH; Jung YH The facile and efficient synthesis of 8-chloroadenosine 3′,5′-cyclic monophosphate by phosphorylative cyclization of 8-chloroadenosine and its characterization by(1)H and (13)C NMR spectroscopy. Arch. Pharmacal Res 1997, 20 (2), 176–179. [DOI] [PubMed] [Google Scholar]
- (643).Schmidt CL; Townsend LB Purine nucleosides. XXXI. The directive effect which certain exocyclic substituents at C-8 of adenine have on the site of ribosylation. J. Org. Chem 1972, 37 (14), 2300–2302. [DOI] [PubMed] [Google Scholar]
- (644).Morris PE; Kamath VP The design of forodesin HCl and other purine nucleoside phosphorylase inhibitors; Wiley-VCH: Weinheim, 2008; pp 451–472. [Google Scholar]
- (645).Al-Kali A; Gandhi V; Ayoubi M; Keating M; Ravandi F Forodesine: review of preclinical and clinical data. Future Oncol 2010, 6 (8), 1211–1217. [DOI] [PubMed] [Google Scholar]
- (646).Homminga I; Zwaan CM; Manz CY; Parker C; Bantia S; Smits WK; Higginbotham F; Pieters R; Meijerink JP In vitro efficacy of forodesine and nelarabine (ara-G) in pediatric leukemia. Blood 2011, 118 (8), 2184–2190. [DOI] [PubMed] [Google Scholar]
- (647).Ogura M; Tsukasaki K; Nagai H; Uchida T; Oyama T; Suzuki T; Taguchi J; Maruyama D; Hotta T; Tobinai K Phase I study of BCX1777 (forodesine) in patients with relapsed or refractory peripheral T/natural killer-cell malignancies. Cancer Sci 2012, 103 (7), 1290–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (648).Balakrishnan K; Ravandi F; Bantia S; Franklin A; Gandhi V Preclinical and clinical evaluation of forodesine in pediatric and adult B-cell acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk 2013, 13 (4), 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (649).Evans GB; Furneaux RH; Gainsford GJ; Schramm VL; Tyler PC Synthesis of transition state analogue inhibitors for purine nucleoside phosphorylase and N-riboside hydrolases. Tetrahedron 2000, 56 (19), 3053–3062. [Google Scholar]
- (650).Batra H; Moriarty RM; Penmasta R; Sharma V; Stanciuc G; Staszewski JP; Tuladhar SM; Walsh DA; Datla S; Krishnaswamy S A concise, efficient and production-scale synthesis of a protected l-lyxonolactone derivative: An important aldonolactone core. Org. Process Res. Dev 2006, 10, 484–486. [Google Scholar]
- (651).Clinch K; Evans GB; Fleet GWJ; Furneaux RH; Johnson SW; Lenz DH; Mee SPH; Rands PR; Schramm VL; Taylor Ringia EA; Tyler PC Syntheses and bio-activities of the L-enantiomers of two potent transition state analogue inhibitors of purine nucleoside phosphorylases. Org. Biomol. Chem 2006, 4 (6), 1131–1139. [DOI] [PubMed] [Google Scholar]
- (652).Winslow CD; Morris PE Jr.. US Patent 6972331, 2005.
- (653).Evans GB; Furneaux RH; Hutchison TL; Kezar HS; Morris PE Jr.; Schramm VL; Tyler PC Addition of lithiated 9-deazapurine derivatives to a carbohydrate cyclic imine: convergent synthesis of the aza-C-nucleoside immucillins. J. Org. Chem 2001, 66 (17), 5723–5730. [DOI] [PubMed] [Google Scholar]
- (654).Kamath VP; Xue J; Juarez-Brambila JJ; Morris PE Jr. Alternative route towards the convergent synthesis of a human purine nucleoside phosphorylase inhibitor—forodesine HCl. Tetrahedron Lett 2009, 50 (37), 5198–5200. [Google Scholar]
- (655).Resek JE; Meyers AI Unsaturation of ketones, nitriles and lactams with methyl phenylsulfinate. Tetrahedron Lett 1995, 36 (39), 7051–7054. [Google Scholar]
- (656).Biocryst Pharmaceuticals. WO Patent 2016110527, 2016.
- (657).Schram KH; Townsend LB The synthesis of 6-amino-4-methyl-8-(β-D-ribofuranosyl) (4-H,8-H)pyrrolo-[4,3,2-de]pyrimido-[4,5-c]pyridazine, a new tricyclic nucleoside. Tetrahedron Lett 1971, 12 (49), 4757–4760. [Google Scholar]
- (658).Kucera LS; Iyer NP; Puckett SH; Buckheit RW Jr.; Westbrook L; Toyer BR; White EL; Germany-Decker JM; Shannon WM; Chen RC; et al. Activity of triciribine and triciribine-5′-monophosphate against human immunodeficiency virus types 1 and 2. AIDS Res. Hum. Retroviruses 1993, 9 (4), 307–314. [DOI] [PubMed] [Google Scholar]
- (659).Shen W; Kim J-S; Hilfinger J Expedient total synthesis of triciribine and its prodrugs. Synth. Commun 2012, 42 (3), 358–374. [Google Scholar]
- (660).Welker ME; Kulik G Recent syntheses of PI3K/Akt/mTOR signaling pathway inhibitors. Bioorg. Med. Chem 2013, 21 (14), 4063–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (661).Owonikoko TK; Khuri FR Targeting the PI3K/AKT/mTOR pathway. Am. Soc. Clin. Oncol. Educ. Book 2013, 2013, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (662).Garrett CR; Coppola D; Wenham RM; Cubitt CL; Neuger AM; Frost TJ; Lush RM; Sullivan DM; Cheng JQ; Sebti SM Phase I pharmacokinetic and pharmacodynamic study of triciribine phosphate monohydrate, a small-molecule inhibitor of AKT phosphorylation, in adult subjects with solid tumors containing activated AKT. Invest. New Drugs 2011, 29 (6), 1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (663).Wotring LL; Townsend LB; Jones LM; Borysko KZ; Gildersleeve DL; Parker WB Dual mechanisms of inhibition of DNA synthesis by triciribine. Cancer Res 1990, 50 (16), 4891–4899. [PubMed] [Google Scholar]
- (664).Sagatys E; Garrett CR; Boulware D; Kelley S; Malafa M; Cheng JQ; Sebti S; Coppola D Activation of the serine/threonine protein kinase Akt during the progression of Barrett neoplasia. Hum. Pathol 2007, 38 (10), 1526–1531. [DOI] [PubMed] [Google Scholar]
- (665).Costa C; Pereira S; Lima L; Peixoto A; Fernandes E; Neves D; Neves M; Gaiteiro C; Tavares A; Gil da Costa RM; et al. Abnormal protein glycosylation and activated PI3K/Akt/mTOR pathway: Role in bladder cancer prognosis and targeted therapeutics. PLoS One 2015, 10 (11), e0141253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (666).Houede N; Pourquier P Targeting the genetic alterations of the PI3K-AKT-mTOR pathway: Its potential use in the treatment of bladder cancers. Pharmacol. Ther 2015, 145, 1–18. [DOI] [PubMed] [Google Scholar]
- (667).Hoffman K; Holmes FA; Fraschini G; Esparza L; Frye D; Raber MN; Newman RA; Hortobagyi GN Phase I-II study: Triciribine (tricyclic nucleoside phosphate) for metastatic breast cancer. Cancer Chemother. Pharmacol 1995, 37 (3), 254–258. [DOI] [PubMed] [Google Scholar]
- (668).Wang Q; Li SH; Wang H; Xiao Y; Sahin O; Brady SW; Li P; Ge H; Jaffee EM; Muller WJ; et al. Concomitant targeting of tumor cells and induction of T-cell response synergizes to effectively inhibit trastuzumab-resistant breast cancer. Cancer Res 2012, 72 (17), 4417–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (669).Townsend LB; Lewis AF; Roti-Roti LW US Patent 4123524, 1978.
- (670).Soucy TA; Dick LR; Smith PG; Milhollen MA; Brownell JE The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer 2010, 1 (7), 708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (671).Yang X; Brownell JE; Xu Q; Zhu F; Ma J; Loke HK; Rollins N; Soucy TA; Minissale JJ; Thomas MP; et al. Absolute quantification of E1, ubiquitin-like proteins and Nedd8-MLN4924 adduct by mass spectrometry. Cell Biochem. Biophys 2013, 67 (1), 139–147. [DOI] [PubMed] [Google Scholar]
- (672).Luo Z; Pan Y; Jeong LS; Liu J; Jia L Inactivation of the Cullin (CUL)-RING E3 ligase by the NEDD8-activating enzyme inhibitor MLN4924 triggers protective autophagy in cancer cells. Autophagy 2012, 8 (11), 1677–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (673).Jin HS; Liao L; Park Y; Liu YC Neddylation pathway regulates T-cell function by targeting an adaptor protein Shc and a protein kinase Erk signaling. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (2), 624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (674).Chang FM; Reyna SM; Granados JC; Wei SJ; Innis-Whitehouse W; Maffi SK; Rodriguez E; Slaga TJ; Short JD Inhibition of neddylation represses lipopolysaccharide-induced proinflammatory cytokine production in macrophage cells. J. Biol. Chem 2012, 287 (42), 35756–35767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (675).Eriksen BC; Hagen B; Eik-Nes SH; Molne K; Mjolnerod OK; Romslo I Long-term effectiveness of the Burch colposuspension in female urinary stress incontinence. Acta Obstet. Gynecol. Scand 1990, 69 (1), 45–50. [DOI] [PubMed] [Google Scholar]
- (676).Blank JL; Liu XJ; Cosmopoulos K; Bouck DC; Garcia K; Bernard H; Tayber O; Hather G; Liu R; Narayanan U; et al. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res 2013, 73 (1), 225–234. [DOI] [PubMed] [Google Scholar]
- (677).Milhollen MA; Traore T; Adams-Duffy J; Thomas MP; Berger AJ; Dang L; Dick LR; Garnsey JJ; Koenig E; Langston SP; et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-κB-dependent lymphoma. Blood 2010, 116 (9), 1515–1523. [DOI] [PubMed] [Google Scholar]
- (678).Toth JI; Yang L; Dahl R; Petroski MD A gatekeeper residue for NEDD8-activating enzyme inhibition by MLN4924. Cell Rep 2012, 1 (4), 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (679).Zhao Y; Xiong X; Jia L; Sun Y Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis 2012, 3, e386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (680).Nawrocki ST; Kelly KR; Smith PG; Espitia CM; Possemato A; Beausoleil SA; Milhollen M; Blakemore S; Thomas M; Berger A; et al. Disrupting protein NEDDylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin. Cancer Res 2013, 19 (13), 3577–3590. [DOI] [PubMed] [Google Scholar]
- (681).Nawrocki ST; Griffin P; Kelly KR; Carew JS MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin. Invest. Drugs 2012, 21 (10), 1563–1573. [DOI] [PubMed] [Google Scholar]
- (682).Choi WJ; Moon HR; Kim HO; Yoo BN; Lee JA; Shin DH; Jeong LS Preparative and stereoselective synthesis of the versatile intermediate for carbocyclic nucleosides: effects of the bulky protecting groups to enforce facial selectivity. J. Org. Chem 2004, 69 (7), 2634–2636. [DOI] [PubMed] [Google Scholar]
- (683).Lee HW; Nam SK; Choi WJ; Kim HO; Jeong LS Stereoselective synthesis of MLN4924, an inhibitor of NEDD8-activating enzyme. J. Org. Chem 2011, 76 (9), 3557–3561. [DOI] [PubMed] [Google Scholar]
- (684).Langston SP; Olhava EJ; Vyskocil S WO Patent 2007092213, 2007.
- (685).An GI; Rhee H A facile synthesis of cis-4-amino-2-cyclopentene-1-methanol, a key intermediate for the synthesis of carbocyclic nucleosides. Nucleosides, Nucleotides Nucleic Acids 2002, 21 (1), 65–72. [DOI] [PubMed] [Google Scholar]
- (686).Armitage I; Elliott EL; Hicks F; Langston M; McCarron A; McCubbin QJ; O’Brien E; Stirling M; Zhu L Process development and GMP production of a potent NAE inhibitor Pevonedistat. Org. Process Res. Dev 2015, 19 (9), 1299–1307. [Google Scholar]
- (687).Nguyen AT; Zhang Y The diverse functions of Dot1 and H3K79 methylation. Genes Dev 2011, 25 (13), 1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (688).Anglin JL; Deng L; Yao Y; Cai G; Liu Z; Jiang H; Cheng G; Chen P; Dong S; Song Y Synthesis and structure-activity relationship investigation of adenosine-containing inhibitors of histone methyltransferase DOT1L. J. Med. Chem 2012, 55 (18), 8066–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (689).Krivtsov AV; Armstrong SA MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7 (11), 823–833. [DOI] [PubMed] [Google Scholar]
- (690).Krivtsov AV; Feng Z; Lemieux ME; Faber J; Vempati S; Sinha AU; Xia X; Jesneck J; Bracken AP; Silverman LB; et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008, 14 (5), 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (691).Muntean AG; Hess JL The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol.: Mech. Dis 2012, 7, 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (692).Tamai H; Inokuchi K 11q23/MLL acute leukemia: Update of clinical aspects. J. Clin. Exp. Hematop 2010, 50 (2), 91–98. [DOI] [PubMed] [Google Scholar]
- (693).Hilden JM; Dinndorf PA; Meerbaum SO; Sather H; Villaluna D; Heerema NA; McGlennen R; Smith FO; Woods WG; Salzer WL; et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: Report on CCG 1953 from the Children’s Oncology Group. Blood 2006, 108 (2), 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (694).Yu W; Chory EJ; Wernimont AK; Tempel W; Scopton A; Federation A; Marineau JJ; Qi J; Barsyte-Lovejoy D; Yi J; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun 2012, 3, 1288. [DOI] [PubMed] [Google Scholar]
- (695).Daigle SR; Olhava EJ; Therkelsen CA; Majer CR; Sneeringer CJ; Song J; Johnston LD; Scott MP; Smith JJ; Xiao Y; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20 (1), 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (696).Daigle SR; Olhava EJ; Therkelsen CA; Basavapathruni A; Jin L; Boriack-Sjodin PA; Allain CJ; Klaus CR; Raimondi A; Scott MP; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122 (6), 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (697).Waters NJ; Daigle SR; Rehlaender BN; Basavapathruni A; Campbell CT; Jensen TB; Truitt BF; Olhava EJ; Pollock RM; Stickland KA; et al. Exploring drug delivery for the DOT1L inhibitor pinometostat (EPZ-5676): Subcutaneous administration as an alternative to continuous IV infusion, in the pursuit of an epigenetic target. J. Controlled Release 2015, 220 (Pt B), 758–765. [DOI] [PubMed] [Google Scholar]
- (698).Basavapathruni A; Olhava EJ; Daigle SR; Therkelsen CA; Jin L; Boriack-Sjodin PA; Allain CJ; Klaus CR; Raimondi A; Scott MP; et al. Nonclinical pharmacokinetics and metabolism of EPZ-5676, a novel DOT1L histone methyltransferase inhibitor. Biopharm. Drug Dispos 2014, 35 (4), 237–252. [DOI] [PubMed] [Google Scholar]
- (699).Waters NJ; Smith SA; Olhava EJ; Duncan KW; Burton RD; O’Neill J; Rodrigue ME; Pollock RM; Moyer MP; Chesworth R Metabolism and disposition of the DOT1L inhibitor, pinometostat (EPZ-5676), in rat, dog and human. Cancer Chemother. Pharmacol 2016, 77 (1), 43–62. [DOI] [PubMed] [Google Scholar]
- (700).Lipka DB; Kuck D; Kliem C; Gerhauser C Substituted purine and 7-deazapurine compounds as modulators of epigenetic enzymes: a patent evaluation (WO2012075381). Expert Opin. Ther. Pat 2013, 23 (4), 537–543. [DOI] [PubMed] [Google Scholar]
- (701).Olhava EJ; Chesworth R; Kuntz KW; Richon VM; Pollck RM; Daigle SR WO Patent 2012075381, 2012.
- (702).Olhava EJ WO Patent 2014152566, 2014.
- (703).Bar-Yehuda S; Stemmer SM; Madi L; Castel D; Ochaion A; Cohen S; Barer F; Zabutti A; Perez-Liz G; Del Valle L; et al. The A3 adenosine receptor agonist CF102 induces apoptosis of hepatocellular carcinoma via de-regulation of the Wnt and NF-kappaB signal transduction pathways. Int. J. Oncol 2008, 33 (2), 287–295. [PubMed] [Google Scholar]
- (704).Cohen S; Stemmer SM; Zozulya G; Ochaion A; Patoka R; Barer F; Bar-Yehuda S; Rath-Wolfson L; Jacobson KA; Fishman P CF102 an A3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J. Cell. Physiol 2011, 226 (9), 2438–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (705).Young HW; Molina JG; Dimina D; Zhong H; Jacobson M; Chan LN; Chan TS; Lee JJ; Blackburn MR A3 adenosine receptor signaling contributes to airway inflammation and mucus production in adenosine deaminase-deficient mice. J. Immunol 2004, 173 (2), 1380–1389. [DOI] [PubMed] [Google Scholar]
- (706).Muller CE; Jacobson KA Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta, Biomembr 2011, 1808 (5), 1290–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (707).Madi L; Cohen S; Ochayin A; Bar-Yehuda S; Barer F; Fishman P Overexpression of A3 adenosine receptor in peripheral blood mononuclear cells in rheumatoid arthritis: involvement of nuclear factor-kappaB in mediating receptor level. J. Rheumatol 2007, 34 (1), 20–26. [PubMed] [Google Scholar]
- (708).Fishman P; Bar-Yehuda S; Liang BT; Jacobson KA Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discovery Today 2012, 17 (7–8), 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (709).Stemmer SM; Benjaminov O; Medalia G; Ciuraru NB; Silverman MH; Bar-Yehuda S; Fishman S; Harpaz Z; Farbstein M; Cohen S; et al. CF102 for the treatment of hepatocellular carcinoma: a phase I/II, open-label, dose-escalation study. Oncologist 2013, 18 (1), 25–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (710).Kim HO; Ji XD; Siddiqi SM; Olah ME; Stiles GL; Jacobson KA 2-substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3 adenosine receptors. J. Med. Chem 1994, 37 (21), 3614–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (711).Hou X; Lee HW; Tosh DK; Zhao LX; Jeong LS Alternative and improved syntheses of highly potent and selective A3 adenosine receptor agonists, Cl-IB-MECA and thio-Cl-IB-MECA. Arch. Pharmacal Res 2007, 30 (10), 1205–1209. [DOI] [PubMed] [Google Scholar]
- (712).Beutner KR; Friedman DJ; Forszpaniak C; Andersen PL; Wood MJ Valaciclovir compared with acyclovir for improved therapy for herpes zoster in immunocompetent adults. Antimicrob. Agents Chemother 1995, 39 (7), 1546–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (713).Elion GB Mechanism of action and selectivity of acyclovir. Am. J. Med 1982, 73 (1A), 7–13. [DOI] [PubMed] [Google Scholar]
- (714).Kieback DG Adenovirus-mediated thymidine kinase gene therapy induces apoptosis in human epithelial ovarian cancer cells and damages PARP-1. In Vivo 2009, 23 (1), 77–80. [PubMed] [Google Scholar]
- (715).Mesnil M; Yamasaki H Bystander effect in herpes simplex virus-thymidine kinase/ganciclovir cancer gene therapy: role of gapjunctional intercellular communication. Cancer Res 2000, 60 (15), 3989–3999. [PubMed] [Google Scholar]
- (716).Hasenburg A; Tong XW; Rojas-Martinez A; Nyberg-Hoffman C; Kieback CC; Kaplan AL; Kaufman RH; Ramzy I; Aguilar-Cordova E; Kieback DG Thymidine kinase (TK) gene therapy of solid tumors: valacyclovir facilitates outpatient treatment. Anticancer Res 1999, 19 (3B), 2163–2165. [PubMed] [Google Scholar]
- (717).Soderberg-Naucler C; Rahbar A; Stragliotto G Survival in patients with glioblastoma receiving valganciclovir. N. Engl. J. Med 2013, 369 (10), 985–986. [DOI] [PubMed] [Google Scholar]
- (718).Lawler SE Cytomegalovirus and glioblastoma; controversies and opportunities. J. Neuro-Oncol 2015, 123 (3), 465–471. [DOI] [PubMed] [Google Scholar]
- (719).Cobbs CS Cytomegalovirus and brain tumor: epidemiology, biology and therapeutic aspects. Curr. Opin. Oncol 2013, 25 (6), 682–688. [DOI] [PubMed] [Google Scholar]
- (720).Martin JC; Dvorak CA; Smee DF; Matthews TR; Verheyden JP 9-[(1,3-Dihydroxy-2-propoxy)methyl]guanine: a new potent and selective antiherpes agent. J. Med. Chem 1983, 26 (5), 759–761. [DOI] [PubMed] [Google Scholar]
- (721).Babu JS; Bay PC; Kumar Y; Khanduri CH WO Patent 2004048380, 2004.
- (722).Arzeno HB; Humphreys ER US Patent 5756736, 1998.
- (723).Khanduri CH; Kumar Y; Panda AK; Raina S; Sharma MK WO Patent 2005092891, 2005.
- (724).Krenitsky TA; Beauchamp LM EP Patent 0308065, 1989.
- (725).Etinger MY; Yudovich LM; Yuzefovich M; Nisnevich GA; Dolitzky BZ; Pertsikov B; Tishin B; Blasberger D US Patent 6849737, 2005.
- (726).Vetukuri PRVNKV; Vedantham R; Mathad VT; Padi PR; Ramasamy VA A concise route to valacyclovir hydrochloride. Helv. Chim. Acta 2011, 94 (4), 592–596. [Google Scholar]
- (727).Sidwell RW; Huffman JH; Khare GP; Allen LB; Witkowski JT; Robins RK Broad-spectrum antiviral activity of Virazole: 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 1972, 177 (4050), 705–706. [DOI] [PubMed] [Google Scholar]
- (728).Sidwell RW; Khare GP; Allen LB; Huffman JG; Witkowski JT; Simon LN; Robins RK In vitro and in vivo effect of 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide (ribavirin) on types 1 and 3 parainfulenza virus infections. Chemotherapy 2004, 21 (3–4), 205–220. [DOI] [PubMed] [Google Scholar]
- (729).James JS Ribavirin approved for hepatitis C combination treatment. AIDS Treat. News 1998, No. No 297, 7. [PubMed] [Google Scholar]
- (730).Eriksson B; Helgstrand E; Johansson NG; Larsson A; Misiorny A; Noren JO; Philipson L; Stenberg K; Stening G; Stridh S; et al. Inhibition of influenza virus ribonucleic acid polymerase by ribavirin triphosphate. Antimicrob. Agents Chemother 1977, 11 (6), 946–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (731).Sintchak MD; Nimmesgern E The structure of inosine 5′-monophosphate dehydrogenase and the design of novel inhibitors. Immunopharmacology 2000, 47 (2–3), 163–184. [DOI] [PubMed] [Google Scholar]
- (732).Jayaram HN; Cooney DA; Grusch M; Krupitza G Consequences of IMP dehydrogenase inhibition, and its relationship to cancer and apoptosis. Curr. Med. Chem 1999, 6 (7), 561–574. [PubMed] [Google Scholar]
- (733).Borden KL Targeting the oncogene eIF4E in cancer: From the bench to clinical trials. Clin. Invest. Med 2011, 34 (6), E315. [DOI] [PubMed] [Google Scholar]
- (734).Borden KL; Culjkovic-Kraljacic B Ribavirin as an anti-cancer therapy: acute myeloid leukemia and beyond? Leuk. Lymphoma 2010, 51 (10), 1805–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (735).Volpon L; Osborne MJ; Zahreddine H; Romeo AA; Borden KL Conformational changes induced in the eukaryotic translation initiation factor eIF4E by a clinically relevant inhibitor, ribavirin triphosphate. Biochem. Biophys. Res. Commun 2013, 434 (3), 614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (736).Holm N; Byrnes K; Johnson L; Abreo F; Sehon K; Alley J; Meschonat C; Md QC; Li BD A prospective trial on initiation factor 4E (eIF4E) overexpression and cancer recurrence in node-negative breast cancer. Ann. Surg. Oncol 2008, 15 (11), 3207–3215. [DOI] [PubMed] [Google Scholar]
- (737).Coleman LJ; Peter MB; Teall TJ; Brannan RA; Hanby AM; Honarpisheh H; Shaaban AM; Smith L; Speirs V; Verghese ET; et al. Combined analysis of eIF4E and 4E-binding protein expression predicts breast cancer survival and estimates eIF4E activity. Br. J. Cancer 2009, 100 (9), 1393–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (738).Pettersson F; Yau C; Dobocan MC; Culjkovic-Kraljacic B; Retrouvey H; Puckett R; Flores LM; Krop IE; Rousseau C; Cocolakis E; et al. Ribavirin treatment effects on breast cancers overexpressing eIF4E, a biomarker with prognostic specificity for luminal B-type breast cancer. Clin. Cancer Res 2011, 17 (9), 2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (739).Witkowski JT; Robins RK; Sidwell RW; Simon LN Design, synthesis, and broad spectrum antiviral activity of 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide and related nucleosides. J. Med. Chem 1972, 15 (11), 1150–1154. [DOI] [PubMed] [Google Scholar]
- (740).Banfi A; Dall’Oro B; Frigerio M; Mancini A US Patent 7285659, 2007.
- (741).Li YS; Zhang JJ; Mei LQ; Tan CX An improved procedure for the preparation of ribavirin. Org. Prep. Proced. Int 2012, 44 (4), 387–391. [Google Scholar]
- (742).Hennen WJ; Wong CH A new method for the enzymic synthesis of nucleosides using purine nucleoside phosphorylase. J. Org. Chem 1989, 54 (19), 4692–4695. [Google Scholar]
- (743).Taverna-Porro M; Bouvier LA; Pereira CA; Montserrat JM; Irivarren AM Chemoenzymatic preparation of nucleosides from furanoses. Tetrahedron Lett 2008, 49 (16), 2642–2645. [Google Scholar]
- (744).Galinanes M; Bullough D; Mullane KM; Hearse DJ Sustained protection by acadesine against ischemia- and reperfusion-induced injury. Studies in the transplanted rat heart. Circulation 1992, 86 (2), 589–597. [DOI] [PubMed] [Google Scholar]
- (745).Favero CB; Mandell JW A pharmacological activator of AMP-activated protein kinase (AMPK) induces astrocyte stellation. Brain Res 2007, 1168, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (746).Sengupta TK; Leclerc GM; Hsieh-Kinser TT; Leclerc GJ; Singh I; Barredo JC Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-β−4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: implication for targeted therapy. Mol. Cancer 2007, 6, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (747).Cronstein BN; Kamen BA 5-aminoimidazole-4-carbox-amide-1-β−4-ribofuranoside (AICA-riboside) as a targeting agent for therapy of patients with acute lymphoblastic leukemia: are we there and are there pitfalls? J. Pediatr. Hematol./Oncol 2007, 29 (12), 805–807. [DOI] [PubMed] [Google Scholar]
- (748).Montraveta A; Xargay-Torrent S; Lopez-Guerra M; Rosich L; Perez-Galan P; Salaverria I; Bea S; Kalko SG; de Frias M; Campas C; et al. Synergistic anti-tumor activity of acadesine (AICAR) in combination with the anti-CD20 monoclonal antibody rituximab in in vivo and in vitro models of mantle cell lymphoma. Oncotarget 2014, 5 (3), 726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (749).Van Den Neste E; Cazin B; Janssens A; Gonzalez-Barca E; Terol MJ; Levy V; Perez de Oteyza J; Zachee P; Saunders A; de Frias M; et al. Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (CLL): a multicenter phase I/II study. Cancer Chemother. Pharmacol 2013, 71 (3), 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (750).Nara T; Misawa M JP Patent 46033196, 1971.
- (751).Sakai S; Niwa K; Sasaki H; Hirose Y; Kinoshita K JP Patent 46028821, 1971.
- (752).Kanemitsu O; Akimoto T; Ito M; Mimura S JP Patent 43020717, 1968.
- (753).Baddiley J; Buchanan JG; Hardy FE; Stewart J Chemical studies in the biosynthesis of purine nucleotides. Part III. The synthesis of 5-amino-1-(β-D-ribofuranosyl)glyoxaline-4-carboxyamide and 4-amino-1-(β-D-ribofuranosyl)glyoxaline-5-carboxyamide. J. Chem. Soc 1959, 0, 2893–2901. [Google Scholar]
- (754).Kissman HM; Pidacks C; Baker BR Puromycin. Synthetic studies. XI. D-ribofuranosyl derivatives of 6-dimethylaminopurine. J. Am. Chem. Soc 1955, 77 (1), 18–24. [Google Scholar]
- (755).Windaus A; Langenbeck W Über die 4(5)-nitro-imidazol-5(4)-carbonsäure. Ber. Dtsch. Chem. Ges. B 1923, 56 (3), 683–686. [Google Scholar]
- (756).Kohyama N; Yamamoto Y A facile synthesis of AICAR from inosine. Synthesis 2003, 17, 2639–2642. [Google Scholar]
- (757).Coats SJ; Garnier-Amblard EC; Amblard F; Ehteshami M; Amiralaei S; Zhang H; Zhou L; Boucle SR; Lu X; Bondada L; et al. Chutes and ladders in hepatitis C nucleoside drug development. Antiviral Res 2014, 102, 119–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (758).Ji X; Wu Y; Yan J; Mehrens J; Yang H; DeLucia M; Hao C; Gronenborn AM; Skowronski J; Ahn J; et al. Mechanism of allosteric activation of SAMHD1 by dGTP. Nat. Struct. Mol. Biol 2013, 20 (11), 1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (759).Behrendt R; Schumann T; Gerbaulet A; Nguyen LA; Schubert N; Alexopoulou D; Berka U; Lienenklaus S; Peschke K; Gibbert K; et al. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Rep 2013, 4 (4), 689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (760).Hollenbaugh JA; Tao S; Lenzi GM; Ryu S; Kim DH; Diaz-Griffero F; Schinazi RF; Kim B dNTP pool modulation dynamics by SAMHD1 protein in monocyte-derived macrophages. Retrovirology 2014, 11, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (761).de Silva S; Wang F; Hake TS; Porcu P; Wong HK; Wu L Downregulation of SAMHD1 expression correlates with promoter DNA methylation in Sezary syndrome patients. J. Invest. Dermatol 2014, 134 (2), 562–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (762).Sjoblom T; Jones S; Wood LD; Parsons DW; Lin J; Barber TD; Mandelker D; Leary RJ; Ptak J; Silliman N; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314 (5797), 268–274. [DOI] [PubMed] [Google Scholar]
- (763).Rentoft M; Lindell K; Tran P; Chabes AL; Buckland RJ; Watt DL; Marjavaara L; Nilsson AK; Melin B; Trygg J; et al. Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (17), 4723–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (764).Kohnken R; Kodigepalli KM; Wu L Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol. Cancer 2015, 14, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (765).Rampazzo C; Tozzi MG; Dumontet C; Jordheim LP The druggability of intracellular nucleotide-degrading enzymes. Cancer Chemother. Pharmacol 2016, 77 (5), 883–893. [DOI] [PubMed] [Google Scholar]
- (766).Porta R; Benaglia M; Puglisi A Flow chemistry: Recent developments in the synthesis of pharmaceutical products. Org. Process Res. Dev 2016, 20 (1), 2–25. [Google Scholar]
- (767).Rodrigues T; Schneider P; Schneider G Accessing new chemical entities through microfluidic systems. Angew. Chem., Int. Ed 2014, 53 (23), 5750–5758. [DOI] [PubMed] [Google Scholar]
- (768).Hessel V; Kralisch D; Kockmann N; Noel T; Wang Q Novel process windows for enabling, accelerating, and uplifting flow chemistry. ChemSusChem 2013, 6 (5), 746–789. [DOI] [PubMed] [Google Scholar]