

Abstract
Purpose of review
The well-recognized plasticity and diversity typical of monocytes and macrophages have recently been expanded by the knowledge that additional macrophage lineages originated directly from embryonic progenitors, populate and establish residency in all tissues examined so far. This review aims to summarize our current understanding on the diversity of monocyte/macrophage subtypes associated with the vasculature, their specific origins, and nature of their cross-talk with the endothelium.
Recent findings
Taking stock of the many interactions between the endothelium and monocytes / macrophages reveals a far more intricate and ever-growing depth. In addition to circulating and surveilling the endothelium, monocytes can specifically be differentiated into patrolling cells that crawl on the surface of the endothelium and promote homeostasis. The conversion of classical to patrolling is endothelium-dependent uncovering an important functional link. In addition to patrolling cells, the endothelium also recruits and harbor an intimal-resident myeloid population that resides in the tunica intima in the absence of pathological insults. Moreover, the adventitia is populated with resident macrophages that support blood vessel integrity and prevent fibrosis.
Summary
The last few years have witnessed a significant expansion in our knowledge of the many subtypes of monocytes and macrophages and their corresponding functional interactions with the vascular wall. In addition to surveying the endothelium for opportunities of diapedeses, monocyte and macrophages take residence in both the intima (as patrolling or resident) and in the adventitia. Their contributions to vascular function are broad and critical to homeostasis, regeneration, and expansion.
Keywords: Blood vessels, collaterals, perivascular, tunica adventitia, tunica intima, vascular inflammation
INTRODUCTION
As the selective gatekeeper between blood and tissues, the endothelium specifically interacts and activates immune cells to facilitate their trafficking. These heterotypic cell interactions are usually brief and are meant to gain access to specific tissue/organ sites. Nonetheless, in contrast to other immune cells, the interactions between endothelium and monocytes/macrophages appear to be far more diverse, interdependent, and specialized.
During developmental angiogenesis, macrophages establish bridges that bring tip cells from adjacent sprouts in proximity facilitating the formation of a new vascular branch [1]. In this manner, macrophages have been acknowledged to actively participate in vascular expansion. Monocyte / macrophages also contribute to angiogenesis through a variety of mechanisms including secretion of VEGF and other cytokines [2], promote intima regeneration [3*], and contribute to collateral growth under hypoxic conditions [2,4]. In small vessels, perivascular macrophages can also regulate permeability responses [5*,6**]. Clearly amongst all bone-marrow derived cells, heterotypic associations between endothelial and monocytes / macrophages are versatile and broad. This wide functional diversity has uncovered the realization of multiple subtypes of monocytes and macrophages that interact with the vascular wall and that are derived from distinct precursors. In fact, blood vessels, particularly in the adventitia are initially populated by macrophages directly derived from embryonic progenitors and supplemented by infiltrating monocytes that contribute to the resident pool of macrophages post-birth [7**]. How this distinct origin and long-term residency impacts their function in comparison to monocyte-derived macrophages is yet to be understood.
Here we examine a fast-growing body of new information on monocyte / macrophage interactions with the vascular wall. Our objective is to offer a timely and informed summary of the current knowledge on the subject, bringing to light differences in origin, molecular cross-talk with the endothelium and functional relationships.
PATROLLING MONOCYTES
Monocytes circulate and constantly survey the endothelium, in some cases this surveillance results in extravasation. Once in the extravascular space, these highly plastic cells can display wide functional diversity in response to their environment [8]. However, in other cases, interaction with the endothelium does not result in vascular extravasation instead, monocytes constantly crawl on the endothelial surface. These two major monocyte subpopulations can be molecularly identified by their cell surface profile which is shared by both mouse and human [9]. Specifically: Ly6chiCcr2hiCx3cr1lo (CD14+CD16− in humans) defines “inflammatory” or “classical” monocytes that are abundant and highly responsive to inflammatory signals, these cells can infiltrate tissues and differentiate into macrophages [8]. In contrast, Ly6cloCcr2loCx3cr11hi (CD14+CD16+ in humans), also referred as “non-classical” or “patrolling” monocytes patrol the endothelium by crawling along the luminal aspect of blood vessels in both the micro- and macro-vasculature (Fig. 1a). This second monocyte subtype is thought to support the endothelium and maintain its homeostasis [10,11]. Patrolling monocytes are generally long-lived and constantly attaching to the tunica intima of the circulatory system and interacting with the endothelium [9].
Figure 1. Recruitment and dynamics of vascular and perivascular monocyte/macrophages.
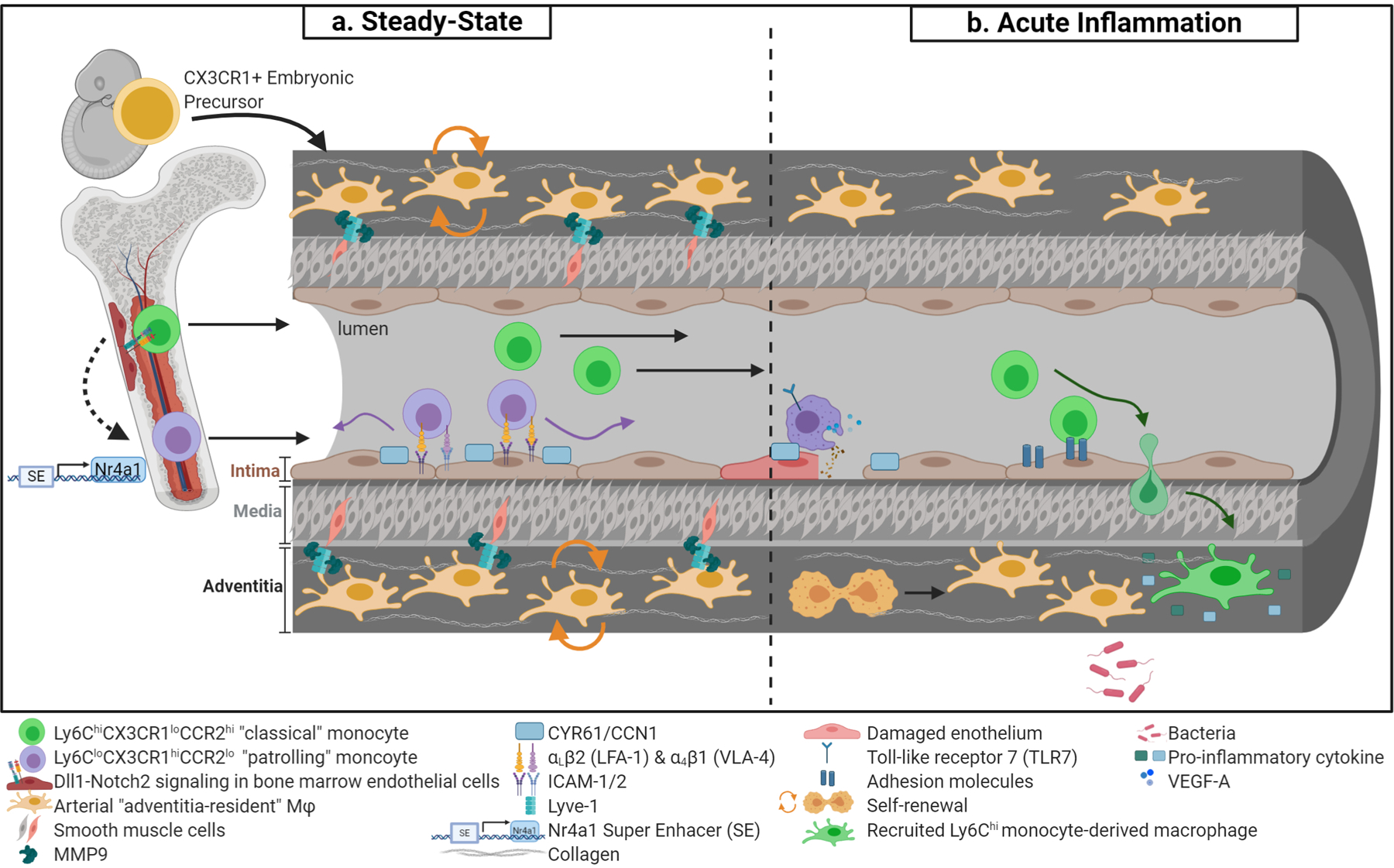
(a) Arterial “adventitia-resident” macrophages are derived from CX3CR1+ embryonic precursors and seed the perivasculature prior to birth (orange cells). Throughout adulthood, adventitia-resident macrophages are maintained by self-renewal. Adventitia-resident macrophages regulate collagen content via matrix metalloproteinase-p (Mmp9) proteolysis, which is dependent on Lyve-1 and smooth muscle cell binding. At stead-state, Ly6Chi classical monocytes (in green) are the precursors to Ly6Clo patrolling monocytes (in purple). Monocyte conversion is dependent on Dll1/Notch2 signaling in distinct vascular beds. Additionally, Nr4a1 super enhancer regulates patrolling monocyte development and is essential for monocyte survival. In the circulation, patrolling monocytes crawling requires αLβ2 integrin (or LFA-1) and other adhesion molecules highlighted. Furthermore, endothelial cystein-rich angiogenic inducer 61 (Cyr61 or Ccn1) promotes efficient surveillance. (b) Patrolling monocytes remove cellular debris from the endothelial surface at sites of necrosis in a TLR-7-dependent manner. At sites of injured endothelium, patrolling monocytes may facilitate endothelial proliferation through secretion of Vegfa. Moreover, activated endothelium upregulates adhesion molecules that enable monocyte capture, rolling, and extravasation into tissue and migrate to inflammatory stimuli. Recruited monocytes differentiate into macrophages as part of a pro-inflammatory response. Adventitia-resident macrophages are severely depleted during bacterial infection but are able to regain steady-state levels through self-renewal.
During steady-state, Ly6chi classical monocytes are the precursors to Ly6clow patrolling monocytes [12]. The process of conversion from classical to patrolling requires contact with the endothelium and activation of the Notch pathway [13*]. Specifically, activation of Notch2 in monocytes by Dll1 in endothelial cells effectively mediates the transition from classical to patrolling in several vascular beds [13*]. In addition to Notch, the transcription factor Nr4a1 (Nur77) is a critical regulator of patrolling, while not affecting classical monocytes [14]. Several studies have demonstrated that depletion in Nr4a1 impairs differentiation and survival of patrolling monocytes [14–16]. A caveat of these studies is the broad expression of Nr4a1 which could impact related cell subtypes. Recently, two studies further refined the link between Nr4a1 and monocytes. One study performed targeted inactivation of a sub-domain within the Nr4a1 enhancer that is exclusively utilized by patrolling but not other monocytes (Fig. 1a) [15**]. The other showed a critical role for C/EBPβ in the regulation of Nr4a1 expression during monocyte conversion using a single cell transcriptomics approach [17*]. Both studies confirmed the absolute requirement of Nr4a1 in the conversion of classical to patrolling monocytes. Furthermore, myeloid depletion of Klf2, an upstream regulator of Nr4a1, explicitly reduced patrolling monocytes [15**].
In the microvasculature of the dermis, kidney, and mesentery, patrolling monocytes crawl along the endothelium regardless of the direction of blood flow [16]. However, in large arteries like the carotid, patrolling monocytes preferentially migrate in the direction of blood flow [18**]. The crawling process requires αLβ2 integrin (or LFA-1), a highly expressed receptor in patrolling monocytes in all healthy tissues [10]. Intracellular adhesion molecule-1 (Icam-1) is the major endothelial ligand for αLβ2, while Icam-2 enhances, but it is dispensable for adhesion [16]. Besides αLβ2, the patrolling function of monocytes is thought to require α4β1 (or very-late antigen 4, VLA-4), but alone, α4β1 is unable to fully support this function (Fig. 1a) [18**]. Recently, α4β1 has been implicated in patrolling of renal glomeruli [19].
Patrolling monocytes express high levels of Cx3cr1. Consequently, monoallelic Cx3cr1-GFP knock-in mice (Cx3cr1GFP/+) are the most commonly used labeling strategy to distinguish patrolling versus other monocytes in vivo, although this reporter mouse also labels other immune cell populations (i.e. tissue-resident macrophages, dendritic cells, and subsets of NK and T cells) [20]. Deficiency of Cx3cr1 results in an overall reduction of patrolling monocytes; however, motility of these monocytes in the microvasculature and in large arteries is independent of Cx3cr1 [16,18**].
On the endothelial side, in addition to Icam-1/2, patrolling monocyte-endothelial interactions also require the cystein-rich angiogenic inducer 61 (Cyr61 or Ccn1), a matricellular protein with chemoattractant properties for monocytes that is produced and secreted by endothelial cells, fibroblasts, and other cell types [21]. Endothelium bound-Ccn1 promotes efficient surveillance of patrolling monocytes at steady-state, but this molecule is not required for adhesion to the endothelium [21].
Functionally, patrolling monocytes, which express high levels of toll-like receptors (TLRs), behave as the “housekeepers” of the vasculature [16,22]. They have phagocytosis capabilities that allows them to scavenge microparticles and remove cellular debris from the endothelial surface at sites of necrosis in a TLR-7-dependent manner [10,16] (Fig. 1b). In addition, patrolling monocytes have proangiogenic properties and aide in organ repair [3*,23,24*–26].
Recently, Gitzin et al. showed that following carotid injury, patrolling monocytes are recruited to the endothelium at wound sites to promote endothelial cell proliferation and tissue repair [3*]. In this study, the authors showed that carotid injury induces endothelial cell expression of Cx3cr1 at the wound site which results in the recruitment of patrolling monocytes. Depletion of patrolling monocytes using Cx3cr1 loss-of-function (Cx3cr1GFP/GFP) mice impaired endothelial regeneration. Lastly, in-vitro experiments suggest patrolling monocytes mediate endothelial cell proliferation through secretion of Vefg-a [3*].
In disease, patrolling monocytes largely play protective roles and contribute to the resolution of inflammation. During atherosclerosis, patrolling monocytes are functionally atheroprotective [18**,27,28]. Monocyte crawling is increased at early stages of the disease, through CD36-mediated uptake of oxidized lipids (OxLDL) [29*]. Depletion of patrolling monocytes in atherosclerotic models results in aggravated atherosclerosis [18**,28]. Following myocardium infarction, patrolling monocytes are part of a second-wave of inflammatory cell recruitment that aids to repair, promote angiogenesis and improve myofibroblast accumulation, as well as deposition of collagen necessary for granulation tissue formation [23,25]. In renal ischemia reperfusion injury CD169+ monocytes, which include both classical and patrolling, play an anti-inflammatory role by regulating Icam-1 expression on endothelial cells [26]. Mice depleted of Cd169 had exacerbated renal reperfusion injury [26]. To clarify the subset of CD169+ monocytes contributing to the resolution of the pathology, the authors performed adoptive transfer experiments. Transfer of Ly6clo patrolling monocytes into CD169-depleted mice rescued the mice from lethal renal injury and normalized renal endothelial cell Icam-1 expression levels. These findings indicate that CD169+ Ly6clo patrolling monocytes play a major role in regulating inflammation in renal reperfusion injury [26]. Additionally, patrolling monocytes contribute to cancer immune-surveillance by orchestrating an antitumor response through recruitment of NK cells, and thus, controlling lung metastasis in the mouse [30]. In other diseases, like systemic lupus erythematosus and arthritis, patrolling monocytes have been implicated in the pathogenesis of disease [31,32].
MONOCYTES/MACROPHAGE IN COLLATERAL ARTERIAL REMODELING
In adults, arteriogenesis refers to the growth or remodeling of arteries (collaterals) triggered by the occlusion of an upstream vascular branch. Although the mechanisms of collateral growth are still unclear [4], monocytes and macrophages have been identified as important players in the process.
Studies suggesting the importance of monocyte/macrophages in collateral arteriogenesis can be traced back to the 1970s, when Schaper et al. [33] showed a large number of monocytes adhering to the surface of endothelial cells following canine chronic coronary artery occlusion. Subsequently, monocyte/macrophage accumulation was also noted around collateral arteries in rabbit and murine hindlimb ischemia models [34,35]. Monocyte recruitment requires the upregulation of chemoattractant or activating cytokines and adhesion molecules [36]. During collateral arteriogenesis, the usually quiescent endothelium becomes activated, resulting in the upregulation of Icam-1, vascular cell adhesion molecule-1 (Vcam-1) and chemokines such as monocyte chemoattractant protein 1 (Mcp-1 or Ccl2), amongst others [2,4]. Mcp-1 is the ligand for C-C chemokine receptor (Ccr2), which is required for classical monocyte recruitment [37]. Mcp-1 plays a significant and beneficial role during collateral arteriogenesis. Mice depleted of Mcp-1 (Ccl2−/−) had lower monocyte recruitment, reduced collateral artery regeneration, and low perfusion recovery [37,38]. Furthermore, administration of Mcp-1 during hindlimb ischemia increased collateral artery regeneration [34,39]. Recently, it was shown that nuclear factor of activated T cells 5 (Nfat5) controls Mcp-1 release in endothelial cells during hindlimb ischemia [40*]. Specifically, reduction of Nfat5 by shRNA adenovirus resulted in attenuated arteriogenesis in rat hindlimb ischemia [40*].
Experiments using Ccr2-depleted (Ccr2−/−) mice, where classical monocyte infiltration into damaged tissues is abrogated has also supported a critical role for monocytes in collateral formation [37,41]. Bone-marrow transplantations of Ccr2−/− mice into wild-type recipients (Ccr2-WT) has revealed that both arteriolar remodeling and monocyte/macrophage recruitment are significantly impaired in a dorsal skinfold model of injury [42]. Importantly, not all macrophages are derived from Ccr2 classical monocytes. As previously discussed, a major subset of adventitia-resident macrophages originate from embryonic precursors and proliferate to maintain the lineage through self-renewal [7**]. This subtype of macrophage (tissue-resident) is not affected by Ccr2 depletion and abundantly populate the perivascular wall. It is also possible that these cells, in addition to monocyte-derived macrophages, contribute to collateral formation.
In myocardial infarction and hindlimb ischemia, early invading Ly6chi monocytes give rise to ischemic macrophages that undergo phenotypic changes overtime [25,43*]. What exactly promotes macrophage differentiation and maturation in ischemic tissue? Previously it has been shown that endothelial cells can promote macrophage polarization [5*]. Further supporting this concept, Krishnasamy et al. [43*] recently showed that macrophage maturation from Ly6chi monocytes and inflammatory polarization after hindlimb ischemia is regulated by endothelial Dll1. Mice with either endothelial-specific deletion of Dll1 (Dll1 i∆EC) or myeloid cell-specific Notch-deficiency not only exhibited a low perfusion recovery and a reduction in collateral branch formation, but also in low macrophage numbers and macrophage activation state [43*]. Therefore, monocyte-derived macrophage maturation is dependent on canonical Notch signaling, which is essential for collateral arteriogenesis following ischemia. Additionally, monocytes/macrophages are believed to be the major source of matrix metalloproteinases (Mmps) and other growth factors like vascular endothelial growth factor (Vegf), which leads to the rearrangement of extracellular matrix and remodeling of arterioles [37,44,45]. Whether monocyte-derived macrophages or adventitia-resident macrophages are the most critical source of these secreted proteins for collateral formation is not yet fully understood. Defined lineage-tracing experiments could help clarify the individual and overlapping contributions of these two subpopulations associated with the vascular wall.
ARTERIAL “ADVENTITIA-RESIDENT” MACROPHAGES
Genetic tracing studies and others have shown that arterial “adventitia-resident” macrophages densely populate the tunica adventitia: the outermost layer of connective tissue common to all blood vessels [6**,7**,46**]. These cells can be recognized by expression of Lyve-1, as well as Csf1r (CD115), CD11b, F4/80, and CD64 (FcγR1) [6**,7**,46**]. As other macrophages, adventitia-resident macrophages are dependent on Csfr1-Csf1 signaling for survival and their maintenance requires Cx3cr1 activation, where endothelial cells and Pdgfrα+ mesenchymal cells constitute the primary source of Cx3cl1 [7**,46**].
In recent years, our knowledge of the ontogeny of tissue-resident macrophages has changed drastically. It is now understood that most tissue-resident macrophages arise from embryonic precursors, that seed tissues during development. These cells are capable of self-renewal and fully independent of bone marrow-derived cells [12,47,48] (Fig. 1a). Two waves of precursors have been reported for adventitial macrophages. Tracing studies identified Cx3cr1+ embryonic progenitors as a definitive source of adventitial macrophages. These cells are observed by embryonic day 16.5 (E16.5) in the mouse aorta [7**]. Subsequently, the adventitia receives a second wave of precursors, this time originated from circulating monocytes. In adulthood, adventitia-resident macrophages are maintained via self-renewal, independent of circulating monocytes [7**]. In addition, following bacterial (LPS) infection, adventitia-resident macrophages are severely depleted, but eventually reach steady-state levels through self-renewal [7**].
In terms of cell-surface profile, a recent study has identified two independent populations of monocyte-derived tissue-resident macrophages characterized by distinct gene expression profiles: LyveloMHCIIhiCx3cr1hi (LyveloMHCIIhi) and Lyve1hiMHCIIloCx3cr1lo (Lyve1hiMHCIIlo), the latter speaks to adventitia-resident macrophages [6**]. These parallel populations were found in the heart, fat, dermis, and lung, as well as in human lung and fat. This study elegantly showed specific niches to these two macrophage populations, concluding that LyveloMHCIIhi macrophages preferentially reside adjacent to nerve bundles and fibers whereas Lyve1hiMHCIIlo macrophages are prevalent to blood vessels across tissues. Specific depletion of Lyve1hiMHCIIlo macrophages in a mouse model resulted in exacerbated experimental lung and heart fibrosis. The authors conclude that Lyve1hiMHCIIlo macrophages support blood vessel integrity at steady-state since depletion of these macrophages leads to better infiltration of monocytes and other inflammatory cells upon fibrosis.
In general, tissue-resident macrophages have a diverse array of functions; in addition to their conventional immune regulation, they also participate in homeostasis, repair and angiogenesis [5,47,48]. For example, embryonic tissue macrophages in the brain act as fusion cells that physically bridge tip cells of sprouting vessels to promote vascular anastomosis [1]. Specifically in relation to adventitial-resident macrophages, depletion studies using Csf1r inhibitors or Lyve1wt/cre;Csf1rflox/flox mice resulted in an increase in collagen deposition within the arterial wall and arterial stiffness [46**]. In vitro co-culture experiments further suggested that adventitia-resident macrophages regulate collagen content via Mmp9-dependent proteolysis [46**]. Since Lyve-1 is the receptor for the extracellular matrix glycosaminoglycan hyaluronan (HA), blocking antibodies were used against the Lyve-1 binding site, which resulted in the reduction of collagen degradation in vitro [46**]. Therefore, the authors speculate that adventitia-resident macrophages regulate collagen levels through HA-dependent smooth muscle cell adhesion and thus, modulate smooth muscle cell function.
INTIMAL CD11C+ MYELOID CELLS
Endothelial cells provide an antithrombotic surface by forming tight barriers through homotypic cell interactions that effectively separate the blood from the surrounding tissues. In general, only under inflammatory conditions are endothelial cells capable of recruiting immune cells and form transient heterotypic interactions that promote their extravasation at sites of inflammation. However, in large arteries there appears to be an exception to the transient nature of these interactions. Early morphological observations have suggested that a resident myeloid population might occupy the intimal layer of healthy (uninflamed) vessels in regions of turbulent flow like the lesser curvature of aortic arch, bifurcation, and branch openings [49,50].
In healthy C57BL/6 mice, an intimal CD11c+ myeloid cell population was also observed in the aorta at regions that experience disturbed blood flow (Fig. 2a) [51,52]. Very rarely were these CD11c+ myeloid cells described in the intima of the descending aorta, which experiences uniform laminar flow, suggesting that their recruitment and/or residency might be flow-type dependent [52]. Intimal CD11c+ myeloid cells increase with age in the ascending aorta and were found to populate the thoracic aorta of 60wk old mice [53]. These myeloid cells reside in the subintimal space with long cellular processes projecting into the lumen [51,52]. Collectively, these intimal cells are CD11c+, MHCII+, CD68+, Cx3cr1+ as validated by immunofluorescence and reporter mice [51–53]. Additionally, they express the dendritic markers αEβ7 (CD103) [54] and 33d1, although the latter remains controversial [54–56]. A previous study showed that FACS sorted CD11c+MHCII+ aortic cells have low phagocytic activity and are strong stimulators of T-cell proliferation, functionally supporting their classification as dendritic cells [54]. However, their identity as dendritic or macrophage is still in under debate. Using single-cell RNAseq Chakarov et al. [6**] recently characterized a tissue-resident macrophage population with a conserved phenotype across tissues. The profile identified by the authors matches the aortic intimal CD11c+ myeloid population. This finding prompts the question: Are the aortic intimal immune cells actually macrophages? Detailed transcriptomic profiling using single cell RNA-sequencing combined with precise immunofluorescence could help elucidate the cellular identity of the CD11c+ myeloid cells.
Figure 2. Intimal resident CD11c+ myeloid cells in arteries during health and disease.
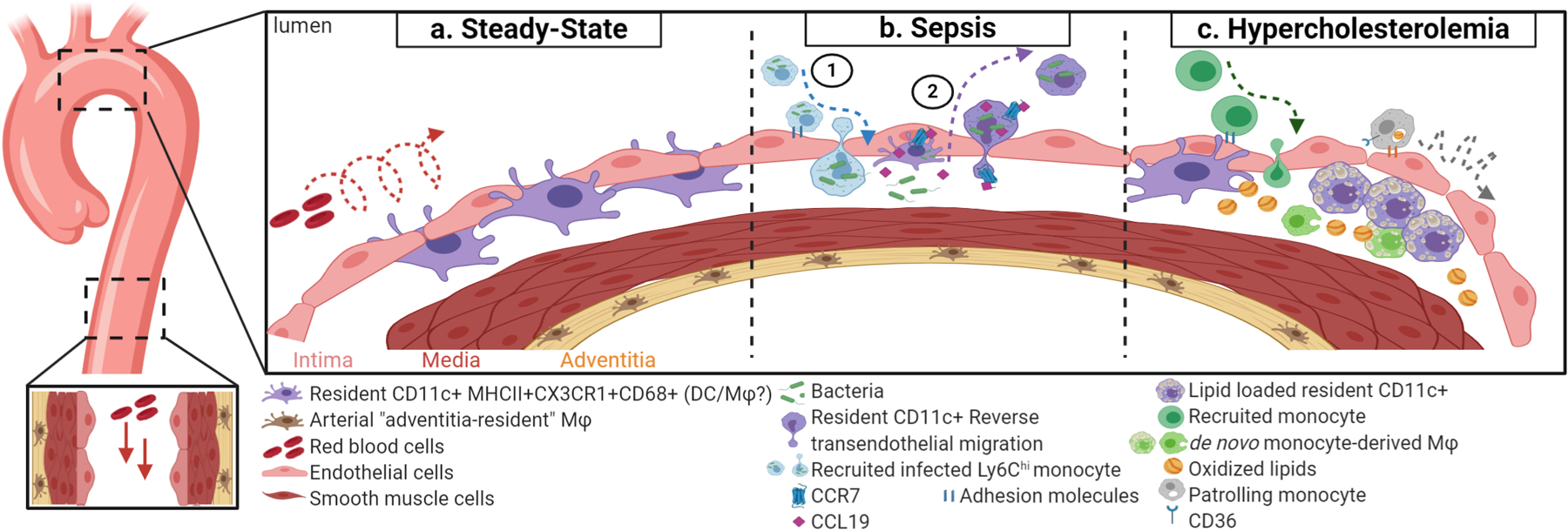
(a) In regions that experience disturb blood flow, like the lesser curvature of the aortic arch, resident CD11c+ myeloid cells (in purple) accumulate in the subintimal space of healthy aortae. Resident CD11c+ myeloid cells rarely accumulate in descending aortae, which experiences uniform laminar blood flow. (b) 1. Circulating monocytes (in blue) become infected with bacteria, gain access to the subintima space, and disseminate the infection. 2. Upon infection, resident CD11c+ myeloid cells upregulate Ccl19 and Ccr7, which is necessary for these cells clear the infection via reverse transendothelial migration, and enter the circulation. (c) At early stages of hypercholesterolemia, resident CD11c+ myeloid cells uptake oxidized lipids, contributing to the initial lesion bed. Additionally, CD36-mediate uptake of oxidized lipid by patrolling monocytes increases crawling.
The accumulation of intimal CD11c+ myeloid cells seems to be partially dependent on Cx3cr1. In Cx3cr1-defiecient mice, there is 40% less CD11c+ myeloid cells in the ascending aorta [53]. Furthermore, intimal CD11c+ myeloid cells are believed to be Flt3L-dependent [54]. Treatment of Flt3L increases intimal CD11c+ cells by 2.5 fold and the number of these intimal myeloid cells were remarkably reduced in Flt3−/− mice [54]. Using BrdU labeling experiments, it is believed that there is a low-grade contribution of recruited blood monocytes to the normal intima that serve as the predominant source of intimal CD11c+ myeloid cells [52]. However, additional studies involving parabiosis, transplants, and lineage tracing are needed to further advance our understanding on how intimal CD11c+ myeloid cells are maintained throughout adulthood. Moreover, at what age do CD11c+ myeloid cells seed the intima of the aorta? Understanding their origin could shed light into their physiological roles.
The function of intimal CD11c+ myeloid cells remains unknown. What is the relationship between these myeloid cells and endothelial cells? Are these myeloid cells necessary to maintain vascular homeostasis? Paulson et al. [55] showed that a single injection of tamoxifen to mice bearing the CD11c promoter-DTR transgene (CD11c-DTR mice) depletes the intimal myeloid cells by approximately 98% after 24hrs post-injection. Interestingly, approximately 75% of intimal CD11c+ myeloid cells are recovered by 21-days post-depletion and are re-localized to the lesser curvature of the aortic arch [55]. This implies the necessity for these cells by the endothelium in this region. Further studies to define the consequences of depletion of intimal CD11c+ cells in vascular homeostasis are still needed.
Recently, Roufaiel et al., [57*] showed that intimal CD11c+ myeloid cells have the potential to clear chlamydia muridarum in the arterial wall through reverse transendothelial migration (Fig. 2b). Upon infection of chlamydia muridarum (or with injection of LPS and PolyIC), there is a decrease in the number of intimal CD11c+ myeloid cells in the ascending aortic arch. Consequently, clearance of chlamydia muridarum 16S rRNA in the intima correlated with the reduction of DD11c+ myeloid cells present in the intima [57*]. These findings indicate that intimal CD11c+ myeloid cells have important innate-like functions in the normal artery.
The location of intimal CD11c+ myeloid cell makes them likely suspects in the initiation of atherosclerosis. Intimal CD11c+ myeloid cells are capable of uptaking neutral lipids just after 5 days of high-fat diet in Ldlr-depleted (Ldlr−/−) mice, a popular atherosclerotic model (Fig. 2c) [55]. Depleting these cells using CD11c-DTR mice crossed to Ldlr−/− mice prior to the induction of hypercholesterolemia resulted in the overall reduction of early lipid accumulation in the aortic wall [55]. In addition, lipid accumulation was found only in extracellular spaces, suggesting intimal CD11c+ myeloid cells play a role in lipid endocytosis during the earliest stages of plaque formation [55]. It has been suggested that the “resident” intimal CD11c+ cells significantly increase in number in advanced atherosclerosis based on the number of CD11c+MCII+ cells present [54]. Although expansion of cells matching this phenotype is evident, it is difficult to discriminate between the resident versus the recruited immune cell populations based on these markers alone since recruited cells can adopt these phenotypes [54,56]. Additionally, there is remarkable immune cell heterogeneity in atherosclerotic aorta. Recent scRNAseq studies identified up to 11 distinct leukocyte populations in the atherosclerotic aorta when compared to control aortas, with diverse macrophage and dendritic cell populations [58*,59**]. Therefore, lineage tracing experiments during the different stages of atherosclerosis are needed to enable the distinction between resident and de novo recruited cells.
CONCLUSIONS
In the last five years, our understanding of the spectrum of endothelial – monocyte / macrophage has significantly expanded along with the realization that macrophages do take long-term residence in organs, including the vascular wall. Their roles are broad and, not surprisingly, dependent on environmental conditions.
Research focus would benefit from combinatorial studies that bring lineage tracing and functional analysis in the context of both physiological and pathological conditions. Major obstacles in clarifying specific functions relate to the potential for functional compensation. Thus, studies that aim at understanding cellular redundancy by selectively eliminating lineages at specific times and sites would aid in clarifying to what degree can monocytes substitute for developmentally-derived long-term lineages. As the functional interdependencies between endothelium and mononuclear lineages become untangled, the information will expand our ability to systematically evaluate how cell-cell interactions impacts tissue adaptability and resilience to stressors.
KEY POINTS.
The vascular wall is home to heterogeneous populations of monocytes and macrophages, all with distinct phenotypes, functions, and origins.
The endothelium helps regulates monocyte conversion and macrophage maturation.
Resident, perivascular macrophages support blood vessel integrity.
Arterial regeneration requires monocytes and macrophages.
ACKNOWLEDGEMENT
We would like to acknowledge Dr. Jesse Williams for his review and edits. We would like to apologize to all the authors whose work could not be cited due to space limitations. Figures were created with BioRender.
FINANCIAL SUPPORT AND SPONSORSHIP
This work was supported from a grant from the National Institutes of Health (R35HL140014). GEH is supported by Howard Hughes Medical Institute Gilliam Fellowship (GT11560).
Funding: This work was supported by a grant from the National Institutes of Health R35HL140014 and Howard Hughes Medical Institute Gilliam Fellowship (GT11560).
Footnotes
CONFLICTS OF INTEREST
There are no conflicts of interests.
REFERENCES
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Fantin A, Vieira JM, Gestri G, et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010;116:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corliss BA, Azimi MS, Munson JM, et al. Macrophages: an inflammatory link between angiogenesis and lymphangiogenesis. Microcirculation 2016;23:95–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3*.Getzin T, Krishnasamy K, Gamrekelashvili J, et al. The chemokine receptor CX3CR1 coordinates monocyte recruitment and endothelial regeneration after arterial injury. EMBO Mol. Med 2018;10:151–159.Investigates the role of patrolling monocytes in endothelial regeneration following carotid injury using mice depleted of patrolling monocytes.
- 4.Cai W, Schaper W. Mechanisms of arteriogenesis. Acta Biochim. Biophys. Sin. (Shanghai) 2008;40:681–692. [PubMed] [Google Scholar]
- 5*.He H, Mack JJ, Güç E, et al. Perivascular macrophages limit permeability. Arterioscler. Thromb. Vasc. Biol 2016;36:2203–2212.Identifies that macrophages, in addition to other mural cells like pericytes, control vascular permeability.
- 6**.Chakarov S, Lim HY, Tan L, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 2019;363.Uncovers two distinct monocyte-derived tissue-resident macrophages populations across tissues. One population is localized adjacent to nerve bundles whereas the other population is a perivascular macrophage associated with blood vessels that contributes to vascular integrity.
- 7**.Ensan S, Li A, Besla R, et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat. Immunol 2016;17:159–168.Identifies the transcriptional signature of arterial “adventitia-resident” macrophages using RNA sequencing, determines their developmental origin using lineage tracing, and explores modes of replenishment of this population throughout adulthood.
- 8.Geissmann F, Manz MG, Jung S, et al. Development of monocytes, macrophages, and dendritic cells. Science 2010;327:656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003;19:71–82. [DOI] [PubMed] [Google Scholar]
- 10.Auffray C, Fogg D, Garfa M, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007;317:666–670. [DOI] [PubMed] [Google Scholar]
- 11.Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol 2009;27:669–692. [DOI] [PubMed] [Google Scholar]
- 12.Yona S, Kim K-W, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013;38:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.Gamrekelashvili J, Giagnorio R, Jussofie et al. Regulation of monocyte cell fate by blood vessels mediated by Notch signalling. Nat. Commun 2016;7:12597.Recognizes that monocyte conversion is regulated by endothelial cells in specific vascular beds and dependent on Dll1-Notch2 signaling. Conditional deletion of Notch2 in monocytes impairs Ly6Clo patrolling monocyte development.
- 14.Hanna RN, Carlin LM, Hubbeling HG, et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C-monocytes. Nat. Immunol 2011;12:778–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15**.Thomas GD, Hanna RN, Vasudevan NT, et al. Deleting an Nr4a1 Super-Enhancer Subdomain Ablates Ly6Clow Monocytes while Preserving Macrophage Gene Function. Immunity 2016;45:975–987.Identifies a specific enhancer at the Nr4a1locus exclusively utilized by patrolling monocytes. Taking advantage of this information, the work was able to dissect the biological impact of Nr4a1 function to patrolling monocytes. The study also shows that monocyte conversion is regulated by KLF2.
- 16.Carlin LM, Stamatiades EG, Auffray C, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 2013;153:362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Mildner A, Schönheit J, Giladi A, et al. Genomic Characterization of Murine Monocytes Reveals C/EBPβ Transcription Factor Dependence of Ly6C-Cells. Immunity 2017;46:849–862.e7.Highlights that transcription factor C/EBPβ regulates Nr4a1 expression, which is essential for monocyte conversion.
- 18**.Quintar A, McArdle S, Wolf D, et al. Endothelial protective monocyte patrolling in large arteries intensified by western diet and atherosclerosis. Circ. Res 2017;120:1789–1799.First to reveal that Ly6clo monocytes have unique patrolling characteristics in large arteries compared to the microvasculature. Additionally, depletion of patrolling monocytes exacerbates atherosclerosis.
- 19.Finsterbusch M, Hall P, Li A, et al. Patrolling monocytes promote intravascular neutrophil activation and glomerular injury in the acutely inflamed glomerulus. Proc. Natl. Acad. Sci. USA 2016;113:E5172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung S, Aliberti J, Graemmel P, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol 2000;20:4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imhof BA, Jemelin S, Ballet R, et al. CCN1/CYR61-mediated meticulous patrolling by Ly6Clow monocytes fuels vascular inflammation. Proc. Natl. Acad. Sci. USA 2016;113:E4847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cros J, Cagnard N, Woollard K, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 2010;33:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med 2007;204:3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24*.Olingy CE, San Emeterio CL, Ogle ME, et al. Non-classical monocytes are biased progenitors of wound healing macrophages during soft tissue injury. Sci. Rep 2017;7:447.Reveals that Ly6Clo patrolling monocytes preferentially give rise to CD206+ macrophages and examine their contributions to wound healing using a dorsal skinfold window chamber injury model.
- 25.Hilgendorf I, Gerhardt LMS, Tan TC, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res 2014;114:1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karasawa K, Asano K, Moriyama S, et al. Vascular-resident CD169-positive monocytes and macrophages control neutrophil accumulation in the kidney with ischemia-reperfusion injury. J. Am. Soc. Nephrol 2015;26:896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamers AAJ, Vos M, Rassam F, et al. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ. Res 2012;110:428–438. [DOI] [PubMed] [Google Scholar]
- 28.Hanna RN, Shaked I, Hubbeling HG, et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ. Res 2012;110:416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Marcovecchio PM, Thomas GD, Mikulski Z, et al. Scavenger receptor CD36 directs nonclassical monocyte patrolling along the endothelium during early atherogenesis. Arterioscler. Thromb. Vasc. Biol 2017;37:2043–2052.Uses intravital imaging to show that patrolling is increased in non-classical monocytes upon hypercholesterolemia. Additionally, oxidized lipid uptake increases patrolling in a CD36 dependent manner.
- 30.Hanna RN, Cekic C, Sag D, et al. Patrolling monocytes control tumor metastasis to the lung. Science 2015;350:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukherjee R, Kanti Barman P, Kumar Thatoi P, et al. Non-Classical monocytes display inflammatory features: Validation in Sepsis and Systemic Lupus Erythematous. Sci. Rep 2015;5:13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Misharin AV, Cuda CM, Saber R, et al. Nonclassical Ly6C(−) monocytes drive the development of inflammatory arthritis in mice. Cell Rep 2014;9:591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaper J, König R, Franz D, Schaper W. The endothelial surface of growing coronary collateral arteries. Intimal margination and diapedesis of monocytes. A combined SEM and TEM study. Virchows Arch A Pathol Anat Histol 1976;370:193–205. [DOI] [PubMed] [Google Scholar]
- 34.Heil M, Ziegelhoeffer T, Pipp F, et al. Blood monocyte concentration is critical for enhancement of collateral artery growth. Am. J. Physiol. Heart Circ. Physiol 2002;283:H2411–9. [DOI] [PubMed] [Google Scholar]
- 35.Arras M, Ito WD, Scholz D, et al. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J. Clin. Invest 1998;101:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao L-S, Bai Y-P. The dynamics of monocytes in the process of collateralization. Aging Med (Milton) 2019;2:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shireman PK. The chemokine system in arteriogenesis and hind limb ischemia. J. Vasc. Surg 2007;45 Suppl A:A48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voskuil M, Hoefer IE, van Royen N, et al. Abnormal monocyte recruitment and collateral artery formation in monocyte chemoattractant protein-1 deficient mice. Vasc Med 2004;9:287–292. [DOI] [PubMed] [Google Scholar]
- 39.Voskuil M, van Royen N, Hoefer IE, et al. Modulation of collateral artery growth in a porcine hindlimb ligation model using MCP-1. Am. J. Physiol. Heart Circ. Physiol 2003;284:H1422–8. [DOI] [PubMed] [Google Scholar]
- 40*.Lin X-C, Pan M, Zhu L-P, et al. NFAT5 promotes arteriogenesis via MCP-1-dependent monocyte recruitment. J. Cell Mol. Med 2019;Highlights an important role of NFAT5 in collateral arteriogenesis in a rat hindlimb ischemia model. Knockdown of NFAT5 inhibits monocyte recruitment by lowering MCP-1 expression in endothelial cells, which attenuates perfusion recovery.
- 41.Heil M, Ziegelhoeffer T, Wagner S, et al. Collateral artery growth (arteriogenesis) after experimental arterial occlusion is impaired in mice lacking CC-chemokine receptor-2. Circ. Res 2004;94:671–677. [DOI] [PubMed] [Google Scholar]
- 42.Nickerson MM, Song J, Meisner JK, et al. Bone marrow-derived cell-specific chemokine (C-C motif) receptor-2 expression is required for arteriolar remodeling. Arterioscler. Thromb. Vasc. Biol 2009;29:1794–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Krishnasamy K, Limbourg A, Kapanadze T, et al. Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat. Commun 2017;8:952.Shows that monocyte-derived macrophage maturation is dependent on endothelial Dll1 expression and canonical Notch signaling during hindlimb ischemia.
- 44.Dodd T, Jadhav R, Wiggins L, et al. MMPs 2 and 9 are essential for coronary collateral growth and are prominently regulated by p38 MAPK. J. Mol. Cell Cardiol 2011;51:1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morrison AR, Yarovinsky TO, Young BD, et al. Chemokine-coupled β2 integrin-induced macrophage Rac2-Myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J. Exp. Med 2014;211:1957–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46*.Lim HY, Lim SY, Tan CK, et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity 2018;49:326–341.e7.Elucidates the physiological role of Lyve-1+ arterial “adventitia-resident” macrophages. Lyve-1+ macrophages interact directly with smooth muscle cells via Lyve1 and release MMP9 to regulate collagen production in order to regulate arterial vessel homeostasis. Depletion of Lyve-1+ macrophages results in arterial stiffness/fibrosis.
- 47.Hoeffel G, Ginhoux F. Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol 2018;330:5–15. [DOI] [PubMed] [Google Scholar]
- 48.Williams JW, Giannarelli C, Rahman A, et al. Macrophage biology, classification, and phenotype in cardiovascular disease: JACC macrophage in CVD series (part 1). J. Am. Coll. Cardiol 2018;72:2166–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malinauskas RA, Herrmann RA, Truskey GA. The distribution of intimal white blood cells in the normal rabbit aorta. Atherosclerosis 1995;115:147–163. [DOI] [PubMed] [Google Scholar]
- 50.Millonig G, Niederegger H, Rabl W, et al. Network of vascular-associated dendritic cells in intima of healthy young individuals. Arterioscler. Thromb. Vasc. Biol 2001;21:503–508. [DOI] [PubMed] [Google Scholar]
- 51.Choi J-H, Do Y, Cheong C, Koh H, et al. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J. Exp. Med 2009;206:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jongstra-Bilen J, Haidari M, Zhu S-N, et al. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J. Exp. Med 2006;203:2073–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu P, Yu Y-RA, Spencer JA, et al. CX3CR1 deficiency impairs dendritic cell accumulation in arterial intima and reduces atherosclerotic burden. Arterioscler. Thromb. Vasc. Biol 2008;28:243–250. [DOI] [PubMed] [Google Scholar]
- 54.Choi J-H, Cheong C, Dandamudi DB, et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 2011;35:819–831. [DOI] [PubMed] [Google Scholar]
- 55.Paulson KE, Zhu S-N, Chen M, et al. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ. Res 2010;106:383–390. [DOI] [PubMed] [Google Scholar]
- 56.Zhu S-N, Chen M, Jongstra-Bilen J, Cybulsky MI. GM-CSF regulates intimal cell proliferation in nascent atherosclerotic lesions. J. Exp. Med 2009;206:2141–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57*.Roufaiel M, Gracey E, Siu A, et al. CCL19-CCR7-dependent reverse transendothelial migration of myeloid cells clears Chlamydia muridarum from the arterial intima. Nat. Immunol 2016;17:1263–1272.Uncovers the role of intimal dendritic cells during pathological processes. The authors demonstrated that intimal dendritic cells exhibit immunological functions and can undergo reverse transendothelial migration.
- 58*.Cochain C, Vafadarnejad E, Arampatzi P, et al. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res 2018;122:1661–1674.Reports the transcriptional heterogeneity of immune cells that accumulate in atherosclerotic aorta using single-cell RNA sequencing of FACS sorted CD45+ cells.
- 59**.Winkels H, Ehinger E, et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res 2018;122:1675–1688.Clarifies the transcriptional and phenotypic heterogeneity of immune cells in atherosclerotic plaques using single-cell RNA sequencing and Mass Cytometry.