

Abstract
β-D-2’-C-Methyl-2,6-diaminopurine ribonucleoside (2’-C-Me-DAPN) phosphoramidate prodrug (DAPN-PD) is a selective hepatitis C virus inhibitor that is metabolized intracellularly into two active metabolites: 2’-C-Methyl-DAPN triphosphate (2’-C-Me-DAPN-TP) and 2’-C-methyl-guanosine 5’-triphosphate (2’-C-Me-GTP). BMS-986094 and IDX-184 are also bioconverted to 2’-C-Me-GTP. A phase IIb clinical trial with BMS-986094 was abruptly halted due to adverse cardiac and renal effects. Herein, we developed an efficient large scale synthesis of DAPN-PD and determined intracellular pharmacology of DAPN-PD in comparison with BMS-986094 and IDX-184, versus Huh-7, HepG2 and interspecies primary hepatocytes and human cardiomyocytes. Imaging data of drug treated human cardiomyocytes was found to be most useful in determining toxicity potential as no obvious beating rate change was observed for IDX-184 up to 50 μM up at 48 h. However, with BMS-986094 and DAPN-PD at 10 μM changes to both beat rate and rhythm were noted.
Keywords: Nucleoside prodrug, hepatitis C virus, intracellular metabolism, hepatocytes, human cardiomyocytes, cardiotoxicity
Introduction
The nucleoside analog antiviral inhibitor (NI), sofosbuvir,[1] revolutionized the treatment of hepatitis C virus (HCV) infection and established NIs as an important class of anti-HCV agents due to their pan-genotypic potential and high barrier to the selection of resistant virus.[2] Despite this resounding success there have been many clinical failures with this class of anti-HCV agents,[3,4] most notably the failure of BMS-986094 (formerly known as INX-189 from Inhibitex, Inc., purchased by Bristol-Myers Squibb, Inc.)[5,6] which was halted in phase IIb clinical trials due to cardiac and renal adverse effects (Figure 1).[7] In this study, a patient receiving BMS-986094 for 40 days presented with rapidly progressive heart failure characterized by pronounced electrocardiographic changes and decreased ventricular ejection fraction accompanied by acute renal failure. Of the 34 patients treated with BMS-986094 in this phase IIb study, 14 had some evidence of cardiac dysfunction and there was one death.
Figure 1.
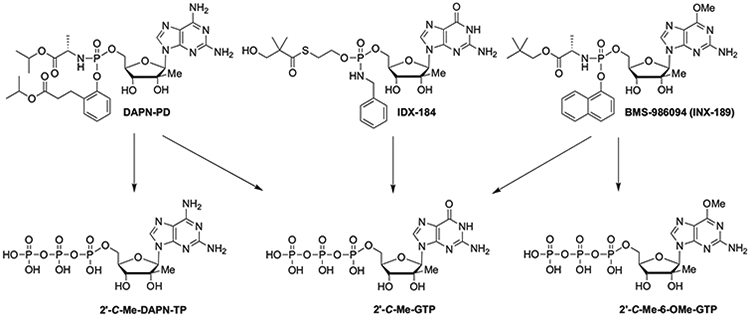
Structures of DAPN-PD, IDX-184 and BMS-986094. DAPN-PD was metabolized intracellularly into two nucleoside 5’-triphosphates (NI-TP): 2’-C-Me-DAPN-TP and 2’-C-Me-GTP. IDX-184 generated one NI-TP: 2’-C-Me-GTP. BMS-986094 was metabolized into two NI-TPs: 2’-C-Me-GTP and 2’-C-Me-6-OMe-GTP.
BMS-986094 is metabolized intracellularly to 2’-C-Me-GTP which is the actual HCV NS5B inhibitor (Figure 1) Mitochondrial toxicity has been associated with long-term use of some 2’-deoxynucleoside analogs,[8,9] especially in treatment of HIV and HBV, where the active 5’-triphosphorylated nucleoside metabolites can also serve as substrates for the mitochondrial DNA polymerase gamma. In the case of BMS-986094 the observed toxicity has been attributed to efficient incorporation of 2’-C-Me-GTP by mitochondrial RNA polymerase (POLRMT) resulting in mitochondrial toxicity[10,11] although some studies have come to a different conclusion.[12,13] However, incorporation by POLRMT, mitochondrial toxicity and increased lactic acid levels in HepG2 cells have been reported by other groups[14,15] including our own work.[16,17] Additionally, BMS-986094’s (but not its metabolites) blocking of the human ether-á-go-go-related (hERG) K+ ion channel was predicted computationally and was subsequently confirmed in two different biological assays.[18] A study done by BMS looking at the metabolism of BMS-986094 in human hepatocytes, human cardiomyocytes and ultimately studying various monkey tissues three weeks post a three-week dosing (week 6) found high levels of 2’-C-Me-GTP in the heart and kidney (2,610–4,280 ng/mL). They concluded “Persistent high concentrations of the active triphosphorylated metabolite, INX-09114 (2’-C-Me-GTP), in monkey heart and kidney appeared to correlate with toxicities in these tissues.”[19]
The FDA placed a clinical hold on IDX-184 (Idenix, Inc).[20] which was also undergoing phase IIb clinical studies at the time, as the same active metabolite, 2’-C-Me-GTP, was produced (Figure 1)[21,22] At the same time our group was in late stage preclinical studies of a novel anti-HCV NI, DAPN-PD,[23] that metabolized intracellularly into two active metabolites: 2’-C-Me-DAPN-TP and 2’-C-Me-GTP, (Figure 1)[15,16] Unlike the broad toxicity reported with BMS-986094,[24] we had observed no in vitro cytotoxicity in various cell lines up to 100 μM and more importantly no mitochondrial toxicity or increase in lactic acid levels up to 50 μM in HepG2 cells.[15,16] As we initiated animal studies we thought it worthy to compare the metabolism profile of our drug, DAPN-PD, with BMS-986094 and IDX-184 to determine intercellular delivery of prodrug, triphosphate(s) and various other metabolites versus liver cell lines, various species of primary hepatocytes and human cardiomyocytes. We also visually recorded cardiomyocytes up to 48 h after exposure to each drug to determine the contraction and rhythm and overall morphology. Our goal was to predict if DAPN-PD and IDX-184 would have a similar in vivo toxicity profile to that found with BMS-986094.
Materials and methods
Nucleoside analog compounds
The medicinal chemistry synthesis and characterization of DAPN-PD was described elsewhere,[15] and the process route is described below. 2’-C-Me-DAPN, 2’-C-Me-DAPN-monophosphate (−MP) 2’-C-Me-DAPN-TP, 2’-C-Me-G, 2’-C-Me-GMP, 2’-C-Me-GTP, BMS-986094 and IDX-184 were synthesized in house with a purity higher than 98%, as determined by HPLC-UV analysis (as an approximate 1:1 mixture of phosphorous diastereomers for DAPN-PD, BMS-9860945 and IDX-18425[25]). ddATP was purchased from Sigma-Aldrich (St. Louis, MO). All other reagents were the highest quality available from Thermo Fisher Scientific (Waltham, MA).
Cellular pharmacology studies in Huh-7 and HepG2 cells
The human hepatocellular carcinoma cell lines (Huh-7 and HepG2) were seeded at 1 × 106 per well in 12-well plates and incubated in a cell culture incubator at 37 °C with a humidified atmosphere of 5% CO2. Adherent cells were subsequently exposed to 50 μM of DAPN-PD, IDX-184 or BMS-986094. At 4 h, drug-containing medium was removed and cells were washed twice with ice-cold phosphate buffered saline (PBS). Cells were resuspended in 70% ice-cold methanol containing 20 nM ddATP overnight at −20 °C. The supernatants were then dried under a flow of air and dried samples stored at −20 °C until analyzed by LC-MS/MS.
Cellular pharmacology studies in primary mouse, rat, dog, monkey and human hepatocytes
Cryopreserved primary mouse, rat, dog, monkey and human hepatocytes and medium were purchased from Life Technologies (Grand Island, NY). After thawing in a 37 °C water bath for approximately 2 min, the cell suspension was immediately transferred to 48 mL pre-warmed cryopreserved hepatocyte recovery medium (for human) or Williams’ medium E and hepatocyte plating supplement pack (for animal) and then centrifuged at room temperature (human hepatocytes: 100 × g for 10 min, dog and monkey hepatocytes: 65 × g for 4 min, mouse and rat hepatocytes: 55 × g for 3 min, respectively) according to the supplier’s protocol. The resulting pellet was resuspended in pre-warmed incubation medium and cell viability and yield determined by Trypan blue exclusion dye. Cells were suspended at a target density of approximately 0.5 × 106 (mouse) or 1.0 × 106 cells/mL (rat, dog, monkey and human), respectively, for cellular pharmacology studies. Cell suspension (1 mL/well of 12-well plates) was incubated at 37 °C with a humidified atmosphere of 5% CO2 with DAPN-PD, IDX-184 or BMS-986094 at a final concentration of 50 μM. At 4 h, the extracellular medium was removed by centrifugation and cell pellets were washed with ice-cold PBS twice in order to remove any residual medium. Cells were resuspended in 70% ice-cold methanol containing 20 nM ddATP overnight at −20 °C. Supernatants were dried under a flow of air and dried samples stored at −20 °C until LC-MS/MS analysis.
Cellular accumulation of DAPN-PD, BMS-986094 and IDX-184 in human cardiomyocytes
Freshly thawed cryopreserved human cardiomyocytes (Cellular Dynamics International, Madison, WI) were seeded in 12-well collagen coated plates with iCell plating medium (Cellular Dynamics International, Madison, WI) at a density of 480,000 (plating efficiency 53%) and incubated in a cell culture incubator at 37 °C, 7% CO2. After 48 h, non-adhered cardiomyocytes and debris were removed by rinsing twice with iCell maintenance medium and the attached cells were incubated for an additional of 8 days in maintenance medium, with fresh medium replacement every other day. At day 10, when the cells beat spontaneously and had reached a stable beating rate, 10 μM of DAPN-PD, IDX-184, BMS-986094 and sofosbuvir (negative control) were added to each well respectively. At 4 h and 8 h after incubation, extracellular medium was removed and the cell layer washed with 1 mL of ice-cold PBS three times to remove any residual drug-containing medium. Cells were resuspended in 70% methanol containing 20 nM ddATP overnight at −20 °C. The supernatants were dried under a flow of air and dried samples stored at −20 °C until LC-MS/MS analysis.
Quantification of intracellular metabolites by LC-MS/MS
A rapid ion-pairing high performance liquid chromatography coupled with electrospray tandem mass spectrometry (LC-MS/MS) method[26] was adapted to quantify the intracellular metabolites. Prior to analysis, each sample was reconstituted in 200 μL mobile phase. An Ultimate 3000 HPLC system (Thermo Fisher Scientific, Waltham, MA) was used for separation. The processed samples were injected on a Hypersil GOLD column (100 × 1 mm) with a 3 μm particle size (Thermo Fisher Scientific, Waltham, MA). An ion-pair method (Mobile phase A, consisted of 2 mM ammonium phosphate and 3 mM hexylamine, and mobile phase B, acetonitrile) was used for a gradient elution. Mobile phase B was increased from 5 to 80% over 13 min, kept at 80% for 4 min, and returned to initial conditions without ramp. The flow rate was maintained at 50 μL/min. Mass spectrometric detection was performed on a TSQ Quantum Ultra triple quadrupole mass spectrometer equipped with electrospray ionization source (Thermo Fisher Scientific, Waltham, MA). Positive SRM detection mode was used with a spray voltage of 3.2 KV, sheath gas at 55 (arbitrary units), ion sweep gas at 0.3 (arbitrary units), auxiliary gas at 5 (arbitrary units), and a capillary temperature of 380 °C. The collision cell pressure was maintained at 1.5 mTorr. Xcalibur 2.0 was used to perform data analyses. The calibration curves were generated from standards of the three prodrugs, parent nucleosides, 2’-C-Me-DAPN-MP, 2’-C-Me-DAPN-TP, 2’-C-Me-GMP and 2’-C-Me-GTP using extracts from untreated cells. The lower limit of quantification for all analytes ranged from 0.5 to 1 nM.
Recording human cardiomyocyte beating patterns
Human cardiomyocytes were thawed and cultured as described above. Ten days post-plating when all cells demonstrated regular synchronous beating, cardiomyocytes were then exposed to 2, 10 and 50 μM of DAPN-PD, BMS-986094, IDX-184 or sofosbuvir (control) in duplicate. Cultures were imaged using a PerkinElmer spinning disc confocal microscope (PerkinElmer, Waltham, MA) and 60 second videos were recorded at 6 fps using a Hamamatsu Flash 4.0 sCMOS camera and Velocity Software at 2, 4, 8, 12, 24, and 48 h after drug exposure. Contractility of human cardiomyocytes was analyzed by manual counting to determine contraction rate and rhythm.
Results and discussion
Chemistry
With DAPN-PD progressing into animal studies we required kg quantities which necessitated reworking the synthesis versus what was developed for gram scale preparation.[16] Hydrolysis of 3 kg of dihydrocoumarin, 1, in anhydrous isopropanol was found to proceed in comparable yield and purity at room temperature in 18 h versus the small scale reaction which was done at reflux (Scheme 1) The intermediate phenyl dichlorophosphate, 2, was formed at −10 °C then 2 h toward room temperature whereas the small-scale reaction was done −70 °C then 18 h at room temperature. The solvent was also switched from ethyl ether to methyl t-butyl ether. The same adjustments to the conditions for the L-alanine isopropyl ester addition and reaction time provided intermediate 3, which was deemed too reactive to handle efficiently on large scale, hence, reaction with pentafluorophenol in the presence of triethylamine gave 4 as a stable solid with a 54/46 diastereomeric ratio at the phosphorous center (Scheme 1).
Scheme 1.
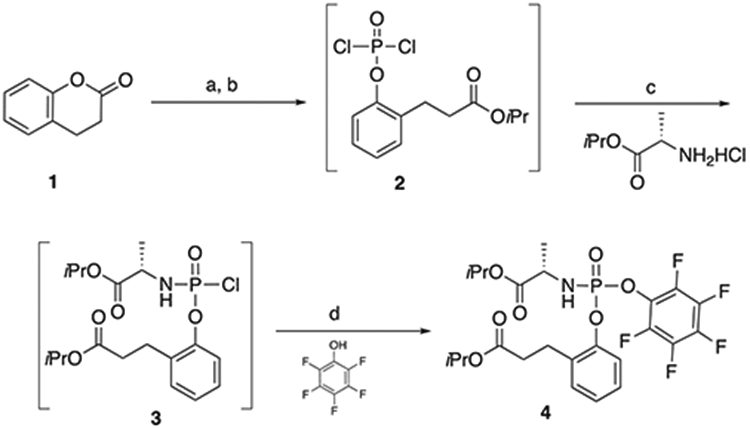
Synthesis of pentafluoro phosphoryl ester 4. Reagents and conditions: (a) (i) iPrOH, cat. H2SO4, rt, 18 h; (ii) NaHCO3 90%; (b) POCl3, t-BuOMe, rt 2 h; (c) Et3N, t-BuOMe, DCM, 2 h, rt; (d) Et3N, DCM, 5 oC, to rt, 2 h, 25% (2 steps).
For the large-scale synthesis of DAPN-PD the medicinal chemistry developed Boc protection/deprotection route.[16] was deemed to have too many steps that required purification for efficient scaleup. Ultimately a route was developed that relied on a transient intermediate 6-diazo purine nucleoside (Scheme 2)[27,28]
Scheme 2.
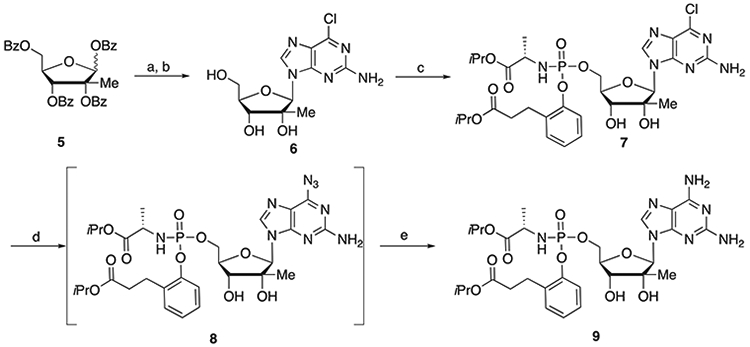
Synthesis of DAPN-PD. Reagents and conditions: (a) DBU, TMSOTf, CH3CN, 65 °C, 3 h, 79.5%; (b) NH3/MeOH, rt, 12 h, 69.9%; (c) 4, THF, t-BuMgCl, 0 °C to rt, 6 h, 80%; (d) NaN3, (Bu)4NI, DMF, 80 °C, 18 h; (e) H2, 20% Pd(OH)2, i-PrOH, rt, 52% (two steps).
The 6-chloro-2-aminopurine tribenzoylated nucleoside 6 was synthesized from known sugar 5[29] and 6-chloro-2-aminopurine in the presence of DBU with addition of TMSOTf at 0 °C as opposed to −40 °C used with the small-scale route. The unprotected 6-chloro-2-aminopurine nucleoside 6 was obtained by debenzylation with ammonia in methanol. Introduction of the prodrug moiety was accomplished with 950 grams of 6 and 2.56 kilograms of 4 by the addition of t-BuMgCl at 0 °C. Stirring was continued for 6 h during which time the temperature was maintained below 30 °C. The 6-azido intermediate 8 was prepared from 1.68 kilograms of 7 by reaction with sodium azide in the presence of tetrabutylammonium iodide. After the reaction was complete the excess sodium azide was removed by water extraction and the dried organic layer was stirred with charcoal and filtered through celite. Reduction of the 6-azido intermediate 8 was accomplished with 20% palladium hydroxide on carbon in the presence of hydrogen providing 847 grams of DAPN-PD.
Anti-HCV activity and cytotoxicity profile of DAPN-PD, IDX-184 and BMS-986094
DAPN-PD, IDX-184 and BMS-986094 were evaluated for inhibition of HCV genotype 1 b RNA replication in Huh-7 cells using a subgenomic HCV replicon system.[30] Cytotoxicity in Huh-7 cells was determined simultaneously by extraction and amplification of both HCV RNA and cellular ribosomal RNA (rRNA).[31] In addition, cytotoxicity was determined in primary human peripheral blood mononuclear (PBM) cells, human lymphoblastoid cells (CEM), and African Green monkey Vero cells.[32,33] The results are summarized in Table 1. While DAPN-PD and IDX-184 both display sub-micromolar EC50’s in our HCV replicon system, BMS-986094 has an impressive EC50 of 20 nM. However, as reported early on by Idenix,[21] BMS-986094 displays concerning toxicity versus multiple cell lines.
Table 1.
HCV replicon and cytotoxicity data for DAPN-PD, IDX-184 and BMS-986094.
| Compounds | Replicon Huh-7 (μM) | Cytotoxicity (CC50, μM) | ||||
|---|---|---|---|---|---|---|
| EC50 | EC90 | Huh-7 | PBM | Vero | CEM | |
| DAPN-PD | 0.7 | 2.5 | >10 | >100 | >100 | >100 |
| BMS-986094 | 0.02 | 0.04 | 0.8 | 4.7 | 14 | 8.7 |
| IDX-184 | 0.3 | 0.9 | >10 | >100 | >100 | >100 |
Intracellular metabolism of DAPN-PD, IDX-184 and BMS-986094 in Huh-7 and HepG2 cells.
Cellular uptake of DAPN-PD, IDX-184 and BMS-986094 were evaluated side-by-side in both Huh-7 and HepG2 cells to compare their intracellular metabolism. In both Huh-7 and HepG2 cell lines, DAPN-PD generated two distinct 5’-triphosphorylated nucleoside metabolites, 2’-C-Me-DAPN-TP and 2’-C-Me-GTP, with comparable total NI-TP levels to IDX-184. The major NI-TP metabolized from BMS-986094 was 2’-C-Me-GTP (240 and 350 pmol/million cells in Huh-7 and HepG2 cells, respectively) which was 94- and 88-fold higher than the total NI-TP generated from DAPN-PD, and 77- and 170-fold higher than that generated from IDX-184. Interestingly, for all three prodrugs, their respective metabolite profiles were similar in both cell lines (Figure 2 and Table 2).
Figure 2.
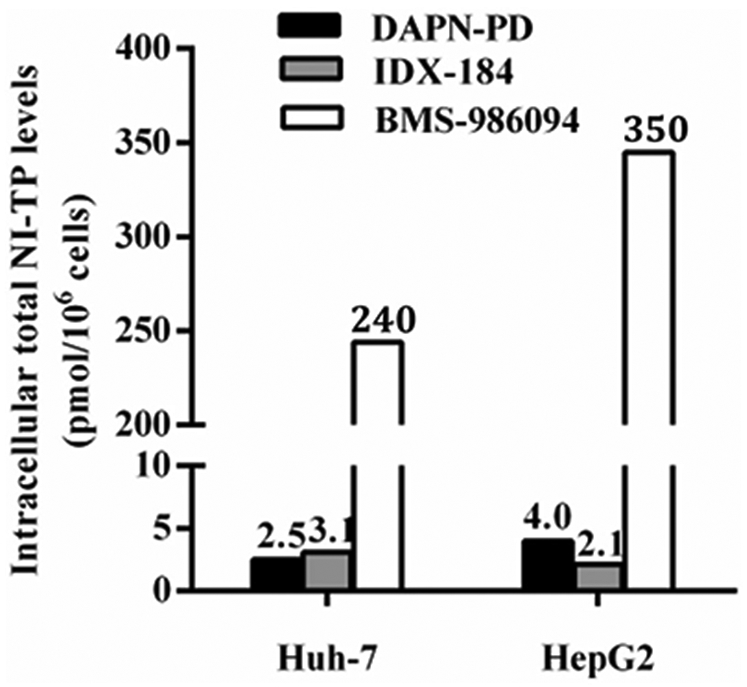
Total levels of NI-TP metabolites of DAPN-PD, IDX-184 and BMS-986094 in Huh-7 and HepG2 cells. 50 μM of each compound was incubated with Huh-7 or HepG2 cells for 4 h at 37 °C. Values represent the mean of three replicates. For DAPN-PD, total NI-TP is 2’-C-Me-DAPN-TP and 2’-C-Me-GTP. For IDX-184, NI-TP is 2’-C-Me-GTP. For BMS-986094, total NI-TP is 2’-C-Me-GTP and 2’-C-Me-6-OMe-GTP.
Table 2.
Comparison of DAPN-PD, IDX-184 and BMS-986094 intracellular metabolism in Huh-7 and HepG2 cells.
| Compounda | Cell type | Intracellular concentration of prodrugs and metabolites (pmol/106 cells ± SD)b | ||||
|---|---|---|---|---|---|---|
| Prodrugs | Nc | 2’-C-Me-G | NTPc | 2’-C-Me-GTP | ||
| DAPN-PD | Huh-7 | 170 ± 4 | 47 ± 3 | 7.5 ± 1.5 | 0.55 ± 0.11 | 2.0 ± 0.1 |
| HepG2 | 390 ± 33 | 27 ± 2 | 90 ± 12 | 1.2 ± 0.01 | 2.8 ± 0.4 | |
| IDX-184 | Huh-7 | 11 ± 1 | NA | 5.6 ± 1.6 | NA | 3.1 ± 0.3 |
| HepG2 | 21 ± 1 | NA | 20 ± 1 | NA | 2.1 ± 0.4 | |
| BMS-986094 | Huh-7 | 2,900 ± 358 | 220 ± 18 | 640 ± 36 | 0.32 ± 0.04 | 240 ± 13 |
| HepG2 | 2,400 ± 9 | 220 ± 1 | 4,900 ± 108 | 0.57 ± 0.10 | 350 ± 7 | |
Cells were incubated with each compound for 4 h at 50 μM.
Values represent the mean ± SD of three replicates.
For DAPN-PD, N is 2’-C-Me-DAPN; for BMS-986094, N is 2’-C-Me-6-OMe-G.
NA is not applicable.
As shown in Table 2, BMS-986094 also delivered very high level of the parent prodrug relative to DAPN-PD and IDX-184. In fact, the level detected for BMS-986094 were 260- and 110-times the levels observed for IDX-184 and 17- and 6-times the levels observed for DAPN-PD in Huh-7 and HepG2 cells, respectively. Also, the amount of 2’-C-Me-6-OMe-GTP generated from BMS-986094 is not significant relative to the levels of 2’-C-Me-GTP that were produced but in the case of DAPN-PD the levels of DAPN-TP are significant relative to the levels of 2’-C-Me-GTP produced. Based on the studies outlined above, the high levels of intracellular BMS-986094 would lead to blocking of the hERG potassium ion channel while high levels of 2’-C-Me-GTP would result in incorporation by POLRMT leading to mitochondrial toxicity and increased lactic acid levels.
NA is not applicable.
Intracellular metabolism of DAPN-PD, IDX-184 and BMS-986094 in mouse, rat, dog, monkey and human hepatocytes
The intracellular metabolism studies were conducted in primary hepatocytes from mouse, rat, dog, monkey and human. Accordingly, each primary cell culture was incubated with DAPN-PD, IDX-184 or BMS-986094 (50 μM, 4 h) and intracellular metabolites and intact prodrugs were readily detected form cell extracts by LC-MS/MS (Table 3). Surprisingly, BMS-986094 give the highest total TP concentration only in dog hepatocytes where IDX-184 and DAPN-PD produced their lowest levels among the five species tested. In all five species evaluated, DAPN-PD was metabolized into two nucleoside triphosphates: 2’-C-Me-DAPN-TP and 2’-C-Me-GTP but the ratio varied greatly depending on the species while the level of 2’-C-Me-6-OMe-GTP produced from BMS-986094 was quite low across all species and undetectable in mouse hepatocytes. The variation in NI-TP levels in different species and variation in the ratios of 2’-C-Me-DAPN-TP versus 2’-C-Me-GTP observed with DAPN-PD suggest that the interspecies differences may be attributed to the amount or efficiency of enzyme(s) responsible for the deamination of DAPN-PD or one of its metabolites.[34] In primary human hepatocytes, 2’-C-Me-DAPN-TP was the major metabolite produced by DAPN-PD, whereas 2’-C-Me-GTP was the primary metabolite observed with BMS-986094 and IDX-184. Additionally, the amount of 2’-C-Me-DAPN-TP produced by DAPN-PD (550 pmol/million cells) was markedly higher than the level of 2’-C-Me-GTP from BMS-986094 (240 pmol/million cells) or IDX-184 (200 pmol/million cells). Human hepatocytes showed the highest phosphorylation efficiency for DAPN-PD, followed by monkey, mouse and rat. In contrast, BMS-986094 had similar metabolism profiles in mouse, rat, dog and human hepatocytes.
Table 3.
Comparison of DAPN-PD, IDX-184 and BMS-986094 intracellular metabolism in mouse, rat, dog, monkey and human primary hepatocytes.
| Compounda | Hepatocyte species | Intracellular concentration prodrugs and metabolites (pmol/106 cells ± SD)b | ||||
|---|---|---|---|---|---|---|
| Prodrugs | Nc | 2’-C-Me-G | NTPc | 2’-C-Me-GTP | ||
| DAPN-PD | Mouse | 13 ± 1 | 1,300 ± 70 | BLOQ | 130 ± 4 | 48 ± 4 |
| Rat | 0.90 ± 0.50 | 310 ± 67 | 72 ± 47 | 99 ± 45 | 45 ± 5 | |
| Dog | 6.3 ± 1.5 | 113 ± 4 | 11 ± 4 | 14 ± 1 | 2.5 ± 0.2 | |
| Monkey | 160 ± 4 | 440 ± 43 | 6.8 ± 4.5 | 180 ± 6 | 13 ± 1 | |
| Human | 70 ± 6 | 2,100 ± 744 | 200 ± 13 | 550 ± 16 | 23 ± 2 | |
| IDX-184 | Mouse | 220 ± 3 | NA | 1,300 ± 137 | NA | 1,300 ± 29 |
| Rat | 1,100 ± 471 | NA | 320 ± 155 | NA | 780 ± 308 | |
| Dog | 110 ± 2 | NA | 230 ± 8 | NA | 150 ± 5 | |
| Monkey | 75 ± 3 | NA | 810 ± 160 | NA | 660 ± 3 | |
| Human | 160 ± 18 | NA | 2,500 ± 181 | NA | 200 ± 18 | |
| BMS-986094 | Mouse | 880 ± 135 | 190 ± 19 | 440 ± 53 | BLOQ | 320 ± 37 |
| Rat | 52 | 14 ± 3 | 210 ± 48 | 0.60 ± 0.10 | 250 ± 45 | |
| Dog | 2,200 ± 68 | 130 ± 13 | 1,800 ± 163 | 0.60 ± 0.04 | 310 ± 17 | |
| Monkey | 150 ± 19 | 200 ± 10 | 510 ± 32 | 6.2 ± 2.4 | 87 ± 3 | |
| Human | 4,100 ± 569 | 960 ± 13 | 9,500 ± 431 | 2.7 ± 0.3 | 240 ± 13 | |
Cells were incubated with each compound for 4 h at 50 μM.
Values represent the mean ± SD of three replicates.
For DAPN-PD, N is 2’-C-Me-DAPN; for BMS-986094, N is 2’-C-Me-6-OMe-G.
BLOQ, below limit of quantification. NA, not applicable.
For IDX-184, the mouse, rat and monkey hepatocytes produced higher 2’-C-Me-GTP levels than detected in dog and human hepatocytes and also produced significantly higher amounts of TP relative to the two other drugs (Figure 3).
Figure 3.
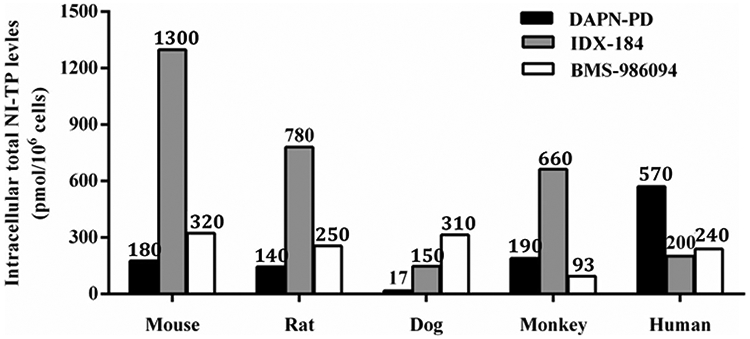
Total levels of active NI-TP metabolite of DAPN-PD, IDX-184 and BMS-986094 in interspecies primary hepatocytes. 50 μM of each compound was incubated with each species of hepatocytes for 4 h at 37 °C. Values represent the mean of three replicates. NI-TP: nucleoside 5’-triphosphate. For DAPN-PD, total NI-TP is 2’-C-Me-DAPN-TP and 2’-C-Me-GTP. For IDX-184, NI-TP is 2’-C-Me-GTP. For BMS-986094, total NI-TP is 2’-C-Me-GTP and 2’-C-Me-6-OMe-GTP.
Cellular accumulation of DAPN-PD, IDX-184 and BMS-986094 in human cardiomyocytes
As BMS-986094 was reported to have cardiotoxicity in humans,[6] we thought it was important to determine if NI-TP levels could be correlated with potential toxicity in human cardiomyocytes[18] with each of the three drugs. We initially dosed cardiomyocytes at 50 μM as we did for hepatocytes, Huh-7 and HepG2 cells, however, BMS-986094 treated cardiomyocytes stopped beating within 2 h after drug administration, indicating an acute cardiac effect. Therefore, we reduced the maximum concentration to 10 μM for this cellular pharmacology study. Using a similar approach, the intracellular metabolism of DAPN-PD, IDX-184 and BMS-986094 were assessed in human cardiomyocytes.[35] exposed to 10 μM concentrations of these prodrugs for 4 and 8 h. Intracellular metabolites and intact prodrug levels were detected in cell extracts by LC-MS/MS (Table 4).
Table 4.
Comparison of DAPN-PD, IDX-184, BMS-986094 and sofosbuvir intracellular metabolism in human cardiomyocytes.
| Compoundsa | Time (h) | Intracellular concentration of the metabolites and prodrugs in human cardiomyocytes (pmol/106 cells ± SD)b |
||||
|---|---|---|---|---|---|---|
| Prodrugs | Nc | 2’-C-Me-G | NTPc | 2’-C-Me-GTP | ||
| DAPN-PD | 4 | 70 ± 3 | 6.2 ± 0.1 | 8.0 ± 2.2 | 18 ± 3 | 28 ± 2 |
| 8 | 51 ± 1 | 4.6 ± 0.5 | 9.1 ± 0.7 | 16 ± 2 | 43 ± 1 | |
| IDX-184 | 4 | 21 ± 1 | NA | 3.6 ± 0.7 | NA | 2.9 ± 0.7 |
| 8 | 9.8 ± 1.4 | NA | 4.3 ± 0.3 | NA | 5.4 ± 0.3 | |
| BMS-986094 | 4 | 800 ± 118 | 16 ± 1 | 650 ± 74 | BLOQ | 410 ± 42 |
| 8 | 810 ± 80 | 11 ± 1 | 440 ± 41 | BLOQ | 540 ± 29 | |
| Sofosbuvir | 4 | 56 ± 3 | BLOQ | NA | 24 ± 1 | NA |
| (control) | 8 | 42 ± 4 | BLOQ | NA | 64 ± 1 | NA |
Cells were incubated with each compound for 4 and 8 h at 10 μM.
Values represent the mean ± SD of three replicates.
For DAPN-PD, N is 2’-C-Me-DAPN; for BMS-986094, N is 2’-C-Me-6-OMe-G; for sofosbuvir, N is 2’-F-2’-C-Me-U.
BLOQ, below limit of quantification. NA, not applicable.
In human cardiomyocytes, sofosbuvir produced 24 and 65 pmol/million cells of 2’-F-2’-C-Me-UTP at 4 h and 8 h respectively, while BMS-986094 produced a much higher level of 2’-C-Me-GTP (410 and 540 pmol/million cells at 4 h and 8 h). The total NI-TP levels for BMS-986094 were significantly higher than levels detected with DAPN-PD and IDX-184 at 4 h (9- and 138-fold, respectively) and at 8 h (9- and 100-fold, respectively) (Figure 4). Furthermore, the level of intact BMS-986094 was also significantly higher than levels of DAPN-PD and IDX-184 at 4 h (11- and 37-fold, respectively) and at 8 h (15- and 82-fold, respectively). BMS-986094 produced the highest level of 2’-C-Me-GTP in human cardiomyocytes of any cell line tested despite the fact that the incubations were done 5-fold less concentrated. Interestingly, the levels in human cardiomyocytes for 2’-C-Me-GTP from IDX-184 were among the lowest observed in any cell line even when the 5-fold reduction in incubation concentration is considered.
Figure 4.
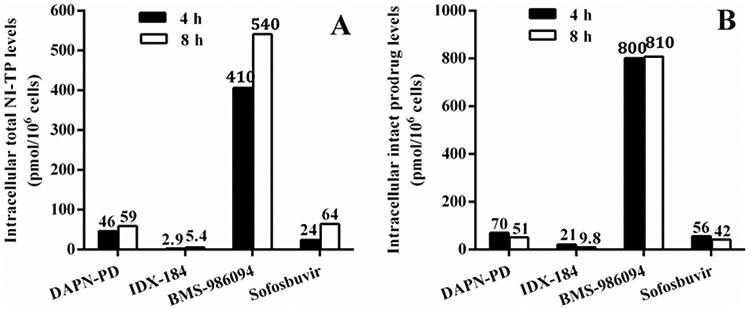
Intracellular accumulation of total NI-TP metabolite and intact prodrug of DAPN-PD, IDX-184, BMS-986094 and sofosbuvir at 4 and 8 h in human cardiomyocytes. (A) Intracellular level of total NI-TP metabolite(s) of DAPN-PD, IDX-184, BMS-986094 and sofosbuvir and (B) Intracellular level of intact prodrug of DAPN-PD, IDX-184, BMS-986094 and sofosbuvir at 4 h and 8 h in human cardiomyocytes at (10 μM, 37 °C). Values represent the mean of three replicates. NI-TP: nucleoside 5’-triphosphate. For DAPN-PD, total NI-TP is 2’-C-Me-DAPN-TP and 2’-C-Me-GTP. For IDX-184, NI-TP is 2’-C-Me-GTP. For BMS-986094, total NI-TP is 2’-C-Me-GTP and 2’-C-Me-6-OMe-GTP. For sofosbuvir, NI-TP is 2’-F-2’-C-Me-UTP.
Imaging of human cardiomyocytes after exposure to DAPN-PD, BMS-986094, and IDX-184
To assess potential acute cardiotoxicity of DAPN-PD, BMS-986094 and IDX-184 and determine if there is a correlate between 2’-C-Me-GTP levels and contraction rate and rhythm, we recorded the cardiomyocytes beating up to 48 h after treatment (2, 10 and 50 μM) each drug to observe the contraction and rhythm. For the control (sofosbuvir), no apparent change in beating rate or rhythm at all three concentrations up to 48 h was observed (Table 5). For BMS-986094, no clear change in beating rate or rhythm was noted at 2 μM up to 48 h; however, at 10 μM, higher beating rates were observed starting from 12 h compared with untreated cells. The higher beating rate was more obvious at 24 and 48 h where the blank control and SOF control beat rates have slowed notably. At 50 μM the cardiomyocytes stopped beating within 2 h, resumed briefly at 12 and 24 h, but with very unstable rhythm. For IDX-184, no apparent change in beating was observed up to 48 h at all three concentrations. And for DAPN-PD, we did not observe beating changes at 2 μM. However, at 10 and 50 μM, the beating rate dropped within 2 h then remained at a reduced rate out to 48 h. The rhythm was also altered and irregular at these two higher concentrations. Cardiomyocytes are non-dividing muscle cells with high-energy requirements for normal function which makes them more susceptible to mitochondrial toxicity.[36-38] The high level of the intact BMS-986094 prodrug at 4 h and equal amounts at 8 h demonstrate a rapid saturation of the cardiomyocytes and would likely maintain high levels of 2’-C-Me-GTP much longer than either DAPN-PD (16 times lower at 8 h) or IDX-184 (83 times lower at 8 h).[39] It is also worth highlighting that at 10 μM BMS-986094 caused a slight increase in beat rate while DAPN-PD at 10 μM caused the beat rate to decrease indicating that the toxic effects of DAPN-PD may be wholly or partially derived from different mechanisms.
Table 5.
Beating rate (beats/minute) of human cardiomyocytes after exposure to DAPN-PD, IDX-184, BMS-986094 and sofosbuvir.
| Compound | Conc. (μM) | 2 h | 4 h | 8 h | 12 h | 24 h | 48 h |
|---|---|---|---|---|---|---|---|
| DAPN-PD | 2 | 33.5 ± 0.7 | 34.0 ± 0 | 37.0 ± 0 | 36.0 ± 0 | 34.0 ± 1.4 | 27.5 ± 0.7 |
| 10 | 24.5 ± 0.7 | 23.5 ± 0.7 | 26.0 ± 1.4 | 25.5 ± 0.7 | 25.5 ± 0.7 | 25.5 ± 0.7 | |
| 50 | 21.5 ± 0.7 | 21.0 ± 0 | 22.0 ± 0 | 21.0 ± 0 | 19.0 ± 0 | 17.0 ± 0 | |
| IDX-184 | 2 | 32.0 ± 0 | 31.5 ± 0.7 | 37.0 ± 1.4 | 36.0 ± 1.4 | 32.5 ± 0.7 | 24.0 ± 1.4 |
| 10 | 30.0 ± 1.4 | 30.0 ± 0 | 34.0 ± 0 | 34.0 ± 1.4 | 31.0 ± 1.4 | 25.0 ± 1.4 | |
| 50 | 33.0 ± 0 | 31.5 ± 0.7 | 34.5 ± 0.7 | 34.5 ± 0.7 | 34.5 ± 4.9 | 27.5 ± 0.7 | |
| BMS-986094 | 2 | 31.0 ± 0 | 30.5 ± 0.7 | 34.5 ± 0.7 | 36.0 ± 0 | 32.5 ± 0.7 | 26.5 ± 0.7 |
| 10 | 31.0 ± 0 | 31.0 ± 0 | 35.0 ± 0 | 36.0 ± 0 | 34.5 ± 0.7 | 35.5 ± 2.1 | |
| 50 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 32.0 | 34.0 ± 0 | 0 ± 0 | |
| Sofosbuvir (control) | 2 | 36.5 ± 2.1 | 35.0 ± 1.4 | 38.5 ± 0.7 | 36.5 ± 0.7 | 33.0 ± 0 | 25.5 ± 0.7 |
| 10 | 34.0 ± 1.4 | 33.0 ± 1.4 | 35.0 ± 1.4 | 33.5 ± 0.7 | 30.0 ± 0 | 23.0 ± 0 | |
| 50 | 32.5 ± 0.7 | 32.0 ± 0 | 34.5 ± 0.7 | 33.0 ± 1.4 | 29.5 ± 0.7 | 23.5 ± 0.7 | |
| Blank control | 0 | 32.5 ± 1.7 | 31.8 ± 0.8 | 34.2 ± 1.6 | 34.2 ± 3.3 | 28.6 ± 1.5 | 23.6 ± 1.9 |
Conclusions
As earlier studies[14,18] concluded, the extremely high levels of 2’-C-Me-GTP in cardiomyocytes generated by incubation with BMS-986094 coupled with its incorporation by human mitochondrial RNA polymerase (POLRMT)[9,10,15] appears to play a major role in the clinically observed adverse cardiac effects. Our earlier work[15] showed DAPN-TP to have a similar incorporation level with POLRMT as 2’-C-Me-GTP, but its total NI-TP level in cardiomyocytes was 9 times lower than that from BMS-986094 after 4 and 8 h incubation. Thus, their similar level of toxicity observed upon visual observation of beat rate and rhythm of dosed cardio-myocytes would appear to result from a mechanism beyond or in combination with inhibition of POLRMT. Interestingly, no apparent changes to beat rate or rhythm were observed for IDX-184 treated cardiomyocytes, indicating that there may be a therapeutic window for IDX-184 in further human studies. In this study, we found cardiomyocyte imaging as a tool to predict cardiotoxicity, to provide the most valuable technique to rapidly predict cardiotoxicity prior to animal and eventually human studies.
Several subsequent animal studies in both rats and dogs were performed with DAPN-PD to determine the validity of our in vitro results and establish a potential therapeutic window. Single dose escalation studies set the maximum tolerated dose at a promising 1,000 mg/kg for both rats and dogs. However, seven day repeat dosing in both dogs and rats set the noobserved-adverse-effect-level (NOAEL) at <40 mg/kg/day with higher doses having adverse effects on multiple organs and tissues including cardiac tissue and function. Having determined a therapeutic windows of close to ten, DAPN-PD preclinical development was abandoned.
Experimental protocols
Chemistry
General information
Reagents were purchased from commercial sources. Unless noted otherwise, the materials used in the examples were obtained from readily available commercial suppliers or synthesized by standard methods known to one skilled in the art of chemical synthesis. Signal multiplicities are represented by s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quadruplet), br (broad), bs (broad singlet), m (multiplet). All J-values are in Hz.
Isopropyl 3-(2-(((((S)-1-isopropoxy-1-oxopropan-2-yl)amino)(perfluorophenoxy)phosphoryl)oxy)phenyl)propanoate (4)
A dry, N2 flushed, 100 L reactor was charged with dihydrocoumarin (3 kg, 20.2 mol) isopropyl alcohol (30.0 L) and H2SO4 (0.09 kg, 0.92 mol). The mixture was stirred at room temperature for 12 hours and then the pH of the mixture was adjusted to 6.5 ~ 7.0 by addition of NaHCO3 (6.21 kg, 73.9 mol). The resulting mixture was filtered through a pad of celite and the solvent was removed under reduced pressure. To the residue was added dichloromethane (6 L) and the mixture was dried over anhydrous Na2SO4 (3 kg). The mixture was filtered through a pad of silica gel, washed with dichloromethane (18 L) and evaporated under reduced pressure to afford 3.8 kg (90.4%) of crude isopropyl ester which was used directly in the next step. 1H-NMR (400 MHz, CDCl3) δ 7.44 (s, 1 H), 7.09-7.15 (m, 2 H), 6.85-6.90 (m, 2 H), 5.03 (m, 1 H), 2.92 (t, J = 6.8 Hz, 2 H), 2.72 (t, J = 6.8 Hz, 2 H), 1.23 (d, J = 6.0 Hz, 6 H); LRMS Calcd for C12H17O3 (M + 1)+ 209.12, found 209.20.
To a stirred solution of the above isopropyl ester (3.8 kg) in t-butyl methyl ether (38 L) was added POCl3 (2.8 kg, 11.1 mol). The mixture was cooled to −10 °C and triethylamine (2.4 kg, 23.7 mol) was added dropwise. After complete addition, the resulting mixture was allowed to warm to a temperature between 15 and 30 °C and stirred for an additional hour. The mixture was cooled to −10 °C and a solution of isopropyl L-alaninate hydrochloride (3.06 kg, 18.3 mol) and triethylamine (3.7 kg, 36.6 mol) in dichloromethane (14.3 L) was added dropwise to the reaction mixture and the internal temperature was raised to ambient temperature. After the reaction was complete, the mixture was filtered to remove the remaining salts followed by evaporation of the filtrate. To the resulting residue was added dichloromethane (26.6 L) and the mixture was cooled down to between 0 and 5 °C. Pentafluorophenol (3.36 kg, 18.3 mol) and triethylamine (1.85 kg, 18.3 mol) in dichloromethane (3.8 L) were added dropwise to the reaction mixture over 1 to 2 hours while maintaining the temperature between 15 and 30 °C. After one additional hour, the mixture was cooled to a temperature between 0 and 5 °C and 1 N HCl (30.4 L) was added and the layers were separated. The organic layer was further washed with saturated NaHCO3 (38 L X 2), brine (38 L X 2) and dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure to afford 4 (2.6 kg, 25.1%) (97.2% purity, PR/PS ratio = 54/46). 1H-NMR (Acetone-d6, 400 MHz) δ 7.40-6.77 (m, 3 H), 5.04-4.93 (m, 2 H), 4.12-3.84 (m, 3 H), 3.05-2.80 (m, 2 H), 2.78-2.42 (m, 2 H), 1.45-1.28 (m, 3 H), 1.25-1.14 (m, 12 H).
(2R,3R,4R,5R)-2-(2-Amino-6-chloro-9H-purin-9-yl)-5-(hydroxymethyl)-3-methyltetrahydrofuran-3,4-diol (6)
To a dry, N2 flushed, 100 L glass-lined reactor precooled (−10 °C) containing a solution of 5 (4.0 kg, 6.89 mol), 2-amino-6-chloropurine (1.29 kg, 7.58 mol), and 1,8-diazabicycl[5.4.0]undec-7-ene (DBU) (3.15 kg, 20.67 mol) in anhydrous acetonitrile (40 L) was added trimethylsilyl triflate (6.13 kg, 6.88 mmol) dropwise. After stirring at 0 °C for 30 minutes, the reaction mixture was heated at 65 °C with stirring for an additional 3 h, allowed to come to room temperature, then diluted with dichloromethane (40.0 L). The resulting solution was washed with saturated aqueous sodium bicarbonate (20 L), filtered through a pad of celite and the layers were separated. The organic phase was dried over anhydrous sodium sulfate (4.0 kg), filtered and the volatiles were removed under reduced pressure. To the resulting residue was added isopropyl alcohol (20.0 L) and the resulting slurry was stirred at 50 °C until all solids had dissolved. The homogeneous solution was cooled to room temperature with stirring, after 2 hours the resulting solid was collected by filtration and dried under vacuum. The tribenzoylated nucleoside (3.44 kg, 79.5%) was obtained and determined to be 98.6% pure by HPLC analysis. 1H-NMR (CD3OD, 400 MHz) δ 8.56 (s, 1 H), 6.01 (s, 1 H), 4.20 (d, J = 8.8 Hz, 1 H), 4.07-3.98 (m, 2 H), 3.85 (dd, J = 12.6 Hz, J = 3.0 Hz, 1 H), 0.97 (s, 3 H). 13C NMR (CD3OD, 100 MHz) δ 20.2, 61.0, 73.4, 80.3, 84.3, 92.7, 124.9, 142.6, 151.7, 154.7, 161.6; LRMS Calcd for C11H15ClN5O4 (M + 1)+ 316.08, found 316.21.
To a dry, N2 flushed, 100 L glass-lined reactor at room temperature containing a solution of tribenzoylated nucleoside (1.1 g, 1.66 mmol) from above was treated with a saturated solution of ammonia in methanol (66.0 L). The reaction mixture was stirred at room temperature for 12 h to reach completion. After removal of the solvent under reduced pressure, to the residue was added methanol (8.25 L) and the resulting mixture was heated at 50 °C with stirring for 30 minutes. After cooling to room temperature, dichloromethane (33.0 L) was added and stirring was continued for 2 h. The resulting solid was collected by filtration and dried under vacuum for 12 h. Compound 6 was obtained (1.16 kg, 69.9%) and determined to be 97.4% pure by HPLC analysis. 1H-NMR (400 MHz, DMSO-d6): 8.01 (s, 1 H), 6.73 (s, 2 H), 5.81 (s, 2 H), 5.76 (d, J = 2.8 Hz, 1 H), 5.18-5.22 (t, J = 6.8 Hz), 5.05 (s, 1 H), 0.78 (s, 3 H), 3.98-4.02 (m, 1 H), 3.78-3.84 (m, 2 H), 3.63-3.67 (m, 1 H). LC/MS calcd. for C11H16N6O4 (M + 1)+ 296.12, observed: 297.32.
Isopropyl 3-(2-(((((2 R,3R,4R,5R)-5-(2-amino-6-chloro-9H-purin-9-yl)-3,4-dihydroxy-4-methyltetrahydrofuran-2-yl)methoxy)(((S)-1-isopropoxy-1-oxopropan-2-yl)amino)phosphoryl)oxy)phenyl)propanoate (7)
A dry, N2 flushed, 100 L reactor was charged with 6 (0.95 kg, 3.0 mol), 4 (2.56 kg, 4.5 mol) and tetrahydrofuran (14.3 L) then the mixture was cooled to 0 °C. Next, t-BuMgCl (4.51 L, 9.02 mol, 2M solution in tetrahydrofuran) was added while maintaining the internal temperature below 30 °C. After complete addition, stirring was continued for 6 hours at ambient temperature. The mixture was cooled to between 0 and 5 °C and NH4Cl (9.5 L, 5 wt% aq.) was added and the organic solvent was removed under reduced pressure. To the remaining aqueous layer was added 9.5 L of ethyl acetate and the layers were separated. The organic layer was washed with saturated NaHCO3 (9.5 L X 2), brine (9.5 L X 2) and dried over anhydrous Na2SO4 (3.8 kg), filtered and evaporated under reduced pressure. The residue was purified by silica-gel column chromatography (dichloromethane/methanol = 30/1 to 10/1, v/v) to afford 7 (1.68 kg, 80% yield) (91.7% purity, Rp/Sp ratio = 51/49). 1HNMR (400 MHz, CD3OD) δ 8.16 (s, 1 H), 8.17 (s, 1 H), 7.37–7.04 (m, 4 H), 5.99 (s, 1 H), 5.97 4.97–4.91 (m, 2 H), 4.60–4.49 (m, 2 H), 4.32–4.18 (m, 2 H), 3.95–3.90 (m, 1 H), 2.99–2.96 (m, 2 H), 2.61–2.57 (m, 2 H), 1.34–1.31 (m, 3 H), 1.21–1.15 (m, 12 H), 1.00–0.99 (m, 3 H); 31PNMR (162 MHz, CD3OD) δ 3.80, 3.72; LRMS calcd for C29H40ClN6O10P (M + H)+ 699.23, found 699.40.
Isopropyl 3-(2-(((((2 R,3R,4R,5R)-5-(2,6-diamino-9H-purin-9-yl)-3,4-dihydroxy-4-methyltetrahydrofuran-2-yl)methoxy)(((S)-1-isopropoxy-1-oxopropan-2-yl)amino)phosphoryl)oxy)phenyl)propanoate (9)
A dry, N2 flushed, 100 L reactor was charged with 7 (1.68 kg, 2.4 mol), NaN3 (0.23 kg, 3.5 mol), tetrabutylammonium iodide (0.44 kg, 1.2 mol), dimethylformamide (8.4 L) and the mixture was heated to between 65 and 80 °C for 18 hours. The mixture was allowed to cool to room temperature and BuBr (0.42 kg, 3.1 mol) was added. After stirring for 1 hour, the solvent was removed under reduced pressure.
To the resulting mixture was added ethyl acetate (58.8 L) and aqueous NaCl (42 L, 10 wt% aqueous) and the layers were separated. To the separated organic layer was added 0.6 kg of Na2SO4 and 0.4 kg of decolorizing charcoal. The resulting mixture was then stirred for an hour, filtered through a pad of celite and the filtrate was evaporated under reduced pressure. To the residue were added isopropyl alcohol (16.8 L), 20% Pd(OH)2 on carbon (0.34 kg) and hydrogen gas was charged (1.0 to 1.5 kgf/cm2) with stirring for 11 hours at room temperature. The mixture was filtered through a pad celite and the filtrate was concentrated under reduced pressure. Ethyl acetate (25 L) and NaCl (17 L, 10 wt% aqueous) were added to the above residue and the layers were separated. The separated organic layer was washed with brine and dried over anhydrous Na2SO4 (0.6 kg), filtered and evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate/methanol, 30/1 to 10/1, v/v) to afford 9 (847 g, 52% over two steps) (99.0% purity, Rp/Sp ratio = 51:49). The two diasteroisomers were separated on small scale using silica gel column chromatography with a gradient of 0 to 10% MeOH in CH2Cl2. 14c-Sp: 1HNMR (400 MHz, CD3OD) δ 7.86 (s, 1 H), 7.37 (d, J = 8.4 Hz, 1 H), 7.25 (d, J = 7.2 Hz, 1 H), 7.17 (dt, J = 8.0 Hz, J = 1.6 Hz, 1 H), 7.08 (t, J = 7.2 Hz, 1 H), 5.94 (s, 1 H), 5.05-4.84 (m, 2 H), 4.62-4.46 (m, 2 H), 4.22 (s, 2 H), 3.95-3.91 (m, 1 H), 2.99 (t, J = 8.0 Hz, 2 H), 2.60 (t, J = 8.0 Hz, 2 H), 1.33 (d, J = 7.2 Hz, 3 H), 1.20 (d, J = 6.4 Hz, 3 H), 1.19 (d, J = 6.0 Hz, 3 H), 1.15 (d, J = 6.0 Hz, 3 H), 1.14 (t, J = 6.4 Hz, 3 H), 0.96 (s, 3 H); 31PNMR (162 MHz, CD3OD) δ 5.08; LRMS calcd for C29H43N7O10P (M + H)+ 680.28, found 680.12. 14c-Rp: 1HNMR (400 MHz, CD3OD) δ 8.03 (s, 1 H), 7.38 (d, J = 8.0 Hz, 1 H), 7.26 (d, J = 7.6 Hz, 1 H), 7.18 (dt, J = 7.6 Hz, J = 1.6 Hz, 1 H), 7.10 (t, J = 7.2 Hz, 1 H), 5.91 (s, 1 H), 4.95-4.79 (m, 2 H), 4.53-4.46 (m, 2 H), 4.18 (s, 2 H), 3.95-3.91 (m, 1 H), 2.99 (t, J = 8.0 Hz, 2 H), 2.61 (t, J = 8.0 Hz, 2 H), 1.35 (d, J = 7.2 Hz, 3 H), 1.19-1.16 (m, 12 H), 0.99 (s, 3 H); 31PNMR (162 MHz, CD3OD) δ 5.02; LRMS calcd for C29H43N7O10P (M + H)+ 680.28, found 680.14.
Acknowledgments
This paper is dedicated to our friend and colleague Professor Akira Matsuda’s in celebration of his 70th birthday.
Funding
This work was supported in part by CFAR (Center for AIDS Research, Emory University) grant NIH5P30-AI-50409 (to RFS).
References
- [1].Schinazi RF; Shi J; Whitaker T Sofosbuvir (Sovaldi): The First-in-Class HCV NS5B Nucleotide Polymerase Inhibitor In Innovative Drug Synthesis, 1st ed.; Li JJ, Johnson DS, Eds. John Wiley and Sons, Inc: Hoboken, New Jersey, 2016; pp 61–79. [Google Scholar]
- [2].McCown MF; Rajyaguru S; Le Pogam S; Ali S; Jiang WR; Kang H; Symons J; Cammack N; Najera I The Hepatitis C Virus Replicon Presents a Higher Barrier to Resistance to Nucleoside Analogs than to Nonnucleoside Polymerase or Protease Inhibitors. Antimicrob. Agents Chemother 2008, 52, 1604–1612. DOI: 10.1128/AAC.01317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Xie Y; Ogah CA; Jiang X; Li J; Shen J Nucleoside Inhibitors of Hepatitis C Virus NS5B Polymerase: A Systematic Review. Curr. Drug Targets 2016, 17, 1560–1576. DOI: 10.2174/1389450117666151209123751. [DOI] [PubMed] [Google Scholar]
- [4].Coats SJ; Garnier-Amblard EC; Amblard F; Ehteshami M; Amiralaei S; Zhang HW; Zhou LH; Boucle SR; Lu X; Bondada L; et al. Chutes and Ladders in Hepatitis C Nucleoside Drug Development. Antiviral Res. 2014, 102, 119–147. DOI: 10.1016/j.antiviral.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].McGuigan C; Madela K; Aljarah M; Gilles A; Brancale A; Zonta N; Chamberlain S; Vernachio J; Hutchins J; Hall A; et al. Design, Synthesis and Evaluation of a Novel Double Pro-Drug: INX-08189. A New Clinical Candidate for Hepatitis C Virus. Bioorg. Med. Chem. Lett 2010, 20, 4850–4854. DOI: 10.10/j.bmcl.2010.06.094. [DOI] [PubMed] [Google Scholar]
- [6].Neopentyl ((((2R,3R,4R,5R)-5-(2-amino-6-methoxy-9H-purin-9-yl)-3,4-dihydroxy-4-methyltetrahydrofuran-2-yl)methoxy)(naphthalen-1-yloxy)phosphoryl)-L-alaninate.
- [7].Ahmad T; Yin P; Saffitz J; Pockros PJ; Lalezari J; Shiffman M; Freilich B; Zamparo J; Brown K; Dimitrova D; et al. Cardiac Dysfunction Associated with a Nucleotide Polymerase Inhibitor for Treatment of Hepatitis C. Hepatology 2015, 62, 409–416. DOI: 10.1002/hep.27488. [DOI] [PubMed] [Google Scholar]
- [8].Fleischer R; Boxwell D; Sherman KE Nucleoside Analogues and Mitochondrial Toxicity. Clin. Infect. Dis 2004, 38, E79–E80. DOI: 10.1086/383151. [DOI] [PubMed] [Google Scholar]
- [9].Lewis W; Day BJ; Copeland WC Mitochondrial Toxicity of NRTI Antiviral Drugs: An Integrated Cellular Perspective. Nat. Rev. Drug Discov 2003, 2, 812–822. DOI: 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- [10].Feng JY; Xu Y; Barauskas O; Perry JK; Ahmadyar S; Stepan G; Yu H; Babusis D; Park Y; McCutcheon K; et al. Role of Mitochondrial RNA Polymerase in the Toxicity of Nucleotide Inhibitors of Hepatitis C Virus. Antimicrob. Agents Chemother 2016, 60, 806–817. DOI: 10.1128/AAC.01922-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Arnold JJ; Sharma SD; Feng JY; Ray AS; Smidansky ED; Kireeva ML; Cho A; Perry J; Vela JE; Park Y; Y.; et al. Sensitivity of Mitochondrial Transcription and Resistance of RNA Polymerase II Dependent Nuclear Transcription to Antiviral Ribonucleosides. PLoS Pathog. 2012, 8, e1003030 DOI: 10.1371/journal.ppat.1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Feng JY; Tay CH; Ray AS Role of Mitochondrial Toxicity in BMS-986094-Induced Toxicity. Toxicol. Sci 2017, 155, 2 DOI: 10.1093/toxsci/kfw224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Baumgart BR; Wang F; Kwagh J; Storck C; Euler C; Fuller M; Simic D; Sharma S; Arnold JJ; Cameron CE; et al. Effects of BMS-986094, a Guanosine Nucleotide Analogue, on Mitochondrial DNA Synthesis and Function. Toxicol. Sci 2016, 153, 396–408. DOI: 10.1093/toxsci/kfw135. [DOI] [PubMed] [Google Scholar]
- [14].Furman PA; Murakami E; Niu C; Lam AM; Espiritu C; Bansal S; Bao H; Tolstykh T; Micolochick Steuer H; Keilman M; et al. Activity and the Metabolic Activation Pathway of the Potent and Selective Hepatitis C Virus Pronucleotide Inhibitor PSI-353661. Antiviral Res. 2011, 91, 120–132. DOI: 10.1016/j.antiviral.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jin Z; Kinkade A; Behera I; Chaudhuri S; Tucker K; Dyatkina N; Rajwanshi VK; Wang G; Jekle A; Smith DB; et al. Structure-Activity Relationship Analysis of Mitochondrial Toxicity Caused by Antiviral Ribonucleoside Analogs. Antiviral Res 2017, 143, 151–161. DOI: 10.1016/j.antiviral.2017.04.005. [DOI] [PubMed] [Google Scholar]
- [16].Ehteshami M; Tao S; Ozturk T; Zhou L; Cho JH; Zhang H; Amiralaei S; Shelton JR; Lu X; Khalil A; et al. Biochemical Characterization of the Active anti-Hepatitis C Virus Metabolites of 2,6-Diaminopurine Ribonucleoside Prodrug Compared to Sofosbuvir and BMS-986094. Antimicrob. Agents Chemother 2016, 60, 4659–4669. DOI: 10.1128/AAC.00318-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhou L; Zhang H; Tao S; Bassit L; Whitaker T; McBrayer TR; Ehteshami M; Amiralaei S; Pradere U; Cho JH; et al. β-D-2’-C-Methyl-2,6-Diaminopurine Ribonucleoside Phosphoramidates Are Potent and Selective Inhibitors of Hepatitis C Virus (HCV) and Are Bioconverted Intracellularly to Bioactive 2,6-Diaminopurine and Guanosine 5’-Triphosphate Forms. J. Med. Chem 2015, 58, 3445–3458. DOI: 10.1021/jm501874e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Anwar-Mohamed A; Barakat KH; Bhat R; Noskov SY; Tyrrell DL; Tuszynski JA; Houghton M A Human Ether-á-Go-Go-Related (hERG) Ion Channel Atomistic Model Generated by Long Supercomputer Molecular Dynamics Simulations and Its Use in Predicting Drug Cardiotoxicity. Toxicol. Lett 2014, 230, 382–392. DOI: 10.1016/j.toxlet.2014.08.007. [DOI] [PubMed] [Google Scholar]
- [19].Li W; Trouba K; Ma L; Kwagh J; Storck C; Zhu Y; Flint O; Humphreys WG; Wang J; Liu A; et al. In Vitro Metabolite Formation in Human Hepatocytes and Cardiomyocytes and Metabolism and Tissue Distribution in Monkeys of the 2’-C-Methylguanosine Prodrug BMS-986094. Int. J. Toxicol 2017, 36, 35–49. DOI: 10.1177/1091581816683642. [DOI] [PubMed] [Google Scholar]
- [20].S-(2-(((((2R,3R,4R,5R)-5-(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-dihydroxy-4-methyltetrahydrofuran-2-yl)methoxy)(benzylamino)phosphoryl)oxy)ethyl) 3-hydroxy-2,2-dimethylpropanethioate.
- [21].Cretton-Scott E; Perigaud C; Peyrottes S; Licklider L; Camire M; Larsson M; La Colla M; Hildebrand E; Lallos L; Bilello J; et al. In Vitro Antiviral Activity and Pharmacology of IDX184, a Novel and Potent Inhibitor of HCV Replication. J. Hepatol 2008, 48, S220 DOI: 10.1016/S0168-8278(08)60590-5. [DOI] [Google Scholar]
- [22].Zhou XJ; Pietropaolo K; Chen J; Khan S; Sullivan-Bolyai J; Mayers D Safety and Pharmacokinetics of IDX184, a Liver-Targeted Nucleotide Polymerase Inhibitor of Hepatitis C Virus, in Healthy Subjects. Antimicrob. Agents Chemother 2011, 55, 76–81. DOI: 10.1128/AAC.01101-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Isopropyl 3-(2-(((((2R,3R,4R,5R)-5-(2,6-diamino-9H-purin-9-yl)-3,4-dihydroxy-4-methyltetrahydrofuran-2-yl)methoxy)(((S)-1-isopropoxy-1-oxopropan-2-yl)amino)phosphoryl)oxy)phenyl)propanoate.
- [24].Hutchins J; Chanberlain S; Chang C; Ganguly B; Gorovits E; Hall A; Henson G; Kolykhalov A; Liu Y; Muhammad J; et al. In Vitro Activity and in Vivo Pharmacokinetics of Highly Potent Phosphoramidate Nucleoside Analogue Inhibitors of Hepatitis C NS5B. Antivir. Res 2009, 82, A28 DOI: 10.1016/j.antiviral.2009.02.047. [DOI] [Google Scholar]
- [25].Sommadossi J-P Gosselin G; Pierra C; Perigaud C; Peyrottes S Compounds and Pharmaceutical Compositions for the Treatment of Viral Infections. WO2008082601 July 10, 2008. [Google Scholar]
- [26].Fromentin E; Gavegnano C; Obikhod A; Schinazi RF Simultaneous Quantification of Intracellular Natural and Antiretroviral Nucleosides and Nucleotides by Liquid Chromatography-Tandem Mass Spectrometry. Anal. Chem 2010, 82, 1982–1989. DOI: 10.1021/ac902737j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang H-w.; Zhou L; Coats SJ; McBrayer TR; Tharnish PM; Bondada L; Detorio M; Amichai SA; Johns MD; Whitaker T; Schinazi RF Synthesis of Purine Modified 2’-C-Methyl Nucleosides as Potential anti-HCV Agents. Bioorg. Med. Chem. Lett 2011, 21, 6788–6792. DOI: 10.1016/j.bmcl.2011.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhou L; Zhang H; Tao S; Ehteshami M; Cho JH; McBrayer TR; Tharnish P; Whitaker T; Amblard F; Coats SJ; Schinazi RF Synthesis and Evaluation of 2,6-Modified Purine 2′-C-Methyl Ribonucleosides as Inhibitors of HCV Replication. ACS Med. Chem. Lett 2016, 7, 17–22. DOI: 10.1021/acsmedchemlett.5b00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].McGuigan C; Perrone P; Madela K; Neyts J The Phosphoramidate ProTide Approach Greatly Enhances the Activity of β-2’-C-Methylguanosine against Hepatitis C Virus. Bioorg. Med. Chem. Lett 2009, 19, 4316–4320. DOI: 10.1016/j.bmcl.2009.05.122. [DOI] [PubMed] [Google Scholar]
- [30].Rondla R; Coats SJ; McBrayer TR; Grier J; Johns M; Tharnish PM; Whitaker T; Zhou L; Schinazi RF Anti-Hepatitis C Virus Activity of Novel β-D-2’-C-Methyl-4′-Azido Pyrimidine Nucleoside Phosphoramidate Prodrugs. Antiviral Chem. Chemother 2009, 20, 99–106. DOI: 10.3851/IMP1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Stuyver LJ; Whitaker T; McBrayer TR; Hernandez-Santiago BI; Lostia S; Tharnish PM; Ramesh M; Chu CK; Jordan R; Shi J; et al. A Ribonucleoside Analogue That Blocks the Replication of Bovine Viral Diarrhea and Hepatitis C Viruses in Culture. Antimicrob. Agents Chemother 2003, 47, 244–254. DOI: 10.1128/AAC.47.1.244-254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schinazi RF; Sommadossi JP; Saalmann V; Cannon DL; Xie M; Hart GC; Smith GA; Hahn EF Activities of 3′-Azido-3′-Deoxythymidine Nucleotide Dimers in Primary Lymphocytes Infected with Human Immunodeficiency Virus Type 1. Antimicrob. Agents Chemother 1990, 34, 1061–1067. DOI: 10.1128/AAC.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stuyver LJ; Lostia S; Adams M; Mathew J; Pai BS; Grier J; Tharnish P; Choi Y; Chong Y; Choo H; et al. Antiviral Activities and Cellular Toxicities of Modified 2′,3′-Dideoxy-2′,3′-Didehydrocytidine Analogues. Antimicrob. Agents Chemother 2002, 46, 3854–3860. DOI: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sofia MJ; Bao D; Chang W; Du JF; Nagarathnam D; Rachakonda S; Reddy PG; Ross BS; Wang PY; Zhang HR; et al. Discovery of a beta-D-2’-Deoxy-2’-Alpha-Fluoro-2’-beta-C-Methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J. Med. Chem 2010, 53, 7202–7218. DOI: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- [35].Mandenius CF; Steel D; Noor F; Meyer T; Heinzle E; Asp J; Arain S; Kraushaar U; Bremer S; Class R; et al. Cardiotoxicity Testing Using Pluripotent Stem Cell-Derived Human Cardiomyocytes and State-of-the-Art Bioanalytics: A Review. J. Appl. Toxicol 2011, 31, 191–205. DOI: 10.1002/jat.1663. [DOI] [PubMed] [Google Scholar]
- [36].Liu YM; Shim E; Nguyen P; Gibbons AT; Mitchell JB; Poirier MC Tempol Protects Cardiomyocytes from Nucleoside Reverse Transcriptase Inhibitor-Induced Mitochondrial Toxicity. Toxicol. Sci 2014, 139, 133–141. DOI: 10.1093/toxsci/kfu034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lund KC; Wallace KB Direct Effects of Nucleoside Reverse Transcriptase Inhibitors on Rat Cardiac Mitochondrial Bioenergetics. Mitochondrion 2004, 4, 193–202. 4, DOI: 10.1016/j.mito.2004.06.009. [DOI] [PubMed] [Google Scholar]
- [38].Lewis W Mitochondrial DNA Replication, Nucleoside Reverse-Transcriptase Inhibitors, and AIDS Cardiomyopathy. Prog. Cardiovasc. Dis 2003, 45, 305–318. DOI: 10.1053/pcad.2003.3. [DOI] [PubMed] [Google Scholar]
- [39].Tao S; Zhou L; Zhang H; Zhou S; Amiralaei S; Shelton J; Coats SJ; Schinazi RF Comparison of Three 2’-C-Methyl Guanosine Prodrugs for Hepatitis C Including a Novel β-D-2’-C-Me-2,6-Diaminopurine Ribonucleoside Phosphoramidate (RS-1389): Interspecies Hepatocyte and Human Cardiomyocyte Metabolism Profiles (Poster). Hepatology 2014, 60, 1170a–1171a.24833036 [Google Scholar]