

Abstract
Mitochondrial DNA (mtDNA) variant pathogenicity interpretation has special considerations given unique features of the mtDNA genome, including maternal inheritance, variant heteroplasmy, threshold effect, absence of splicing, and contextual effects of haplogroups. Currently there are insufficient standardized criteria for mtDNA variant assessment, which leads to inconsistencies in clinical variant pathogenicity reporting. An international working group of mtDNA experts was assembled within the Mitochondrial Disease Sequence Data Resource (MSeqDR) Consortium and obtained Expert Panel status from ClinGen. This group reviewed the 2015 American College of Medical Genetics (ACMG) and Association of Molecular Pathology (AMP) standards and guidelines that are widely used for clinical interpretation of DNA sequence variants and provided further specifications for additional and specific guidance related to mtDNA variant classification. These Expert Panel based consensus specifications allow for consistent consideration of the unique aspects of the mtDNA genome that directly influence variant assessment, including addressing mtDNA genome composition and structure, haplogroups and phylogeny, maternal inheritance, heteroplasmy, and functional analyses unique to mtDNA, as well specifications for utilization of mtDNA genomic databases and computational algorithms.
Keywords: mtDNA, mitochondria, heteroplasmy, pathogenicity, criteria, variant interpretation
INTRODUCTION
Primary mitochondrial disease is a heterogeneous collection of energy deficiency disorders presenting with highly variable phenotypes ranging from adult-onset isolated organ involvement to infantile-onset multi-system manifestations with high morbidity and mortality (Gorman et al., 2016; Landrum et al., 2020). Pathogenic variants in several hundred nuclear and all mitochondrial DNA (mtDNA) genes are currently recognized to cause primary mitochondrial disease (Marni J. Falk, 2020; E. M. McCormick, Zolkipli-Cunningham, & Falk, 2018). Determining the molecular etiology of primary mitochondrial disease in an affected individual can be particularly challenging given the extensive heterogeneity of clinical symptoms, poorly understood genotype-phenotype correlations, and non-specific nature of many symptoms with significant clinical overlap for other conditions (E.M. McCormick, Place, & Falk, 2013). Inconsistencies exist among clinical diagnostic laboratories in the pathogenicity interpretation and reporting of mtDNA variants, as a potentially disease-causing mtDNA variant may be reported as pathogenic by one laboratory but benign by another due to differing variant classification algorithms or practices. Challenges in variant pathogenicity classification are not exclusive to mtDNA, as this problem certainly exists for nuclear DNA (nDNA) variants. However, while it is rare to sequence a nuclear gene in multiple tissues with the exception of evaluation for mosaicism, tissue differences in heteroplasmy levels for a given variant are a common source of potential misinterpretation for mtDNA variant pathogenicity classification. As with many genetic conditions, confirming the correct molecular etiology in individuals with primary mitochondrial disease is important to end their diagnostic odyssey, facilitate proper medical management, enable participation in emerging clinical treatment trials, and determine accurate recurrence risk and prenatal options that may be available for affected individuals and their family members.
The American College of Medical Genetics (ACMG) and Association of Molecular Pathology (AMP) guidelines on variant interpretation were developed to provide a standardized framework with guidance to improve variant interpretation consistency among clinicians and laboratories (Richards et al., 2015). Considerations for mtDNA variants were briefly discussed in this initial framework, with general guidance provided for nomenclature and reporting of heteroplasmy level; and additional complicating factors for mtDNA variant interpretation were also reviewed.
To address the complexities of mtDNA variant assessment, an international working group of mtDNA experts was assembled within the Mitochondrial Disease Sequence Data Resource (MSeqDR) Consortium (Falk, Shen, & Gai, 2016; Falk et al., 2015; Shen et al., 2016) to critically review them in the context of the current ACMG/AMP guidelines. The mtDNA expert panel focused on evaluating the relevance of all existing guideline criteria for mtDNA and providing consensus specifications for mtDNA variant assessment. The mtDNA working group members were diverse in expertise and location, including clinical geneticists who care for patients in a mitochondrial medicine clinic; clinical, research, and clinical laboratory-based genetic counselors; laboratory directors from academic and commercial clinical diagnostic laboratories; mtDNA genome researchers; and experienced bioinformaticians who organize and develop mtDNA variant databases and interpretation tools.
Here, we have expanded upon the relevant considerations outlined in the 2015 initial variant interpretation guidelines and propose specifications for mtDNA variant interpretation. Each criterion for pathogenic or benign status was reviewed for its relevance to the mitochondrial genome and was unchanged, specified, or removed when appropriate. Furthermore, each criterion was evaluated for its applicability to protein-coding (messenger RNA, mRNA), transfer RNA (tRNA), and ribosomal RNA (rRNA) genes in the mitochondrial genome (see Supp Table S1 for list of mtDNA genes, genome coordinates, and gene category).
Our proposals were developed in close collaboration with the National Institutes of Health (NIH)-funded Clinical Genome Resource (ClinGen, http://www.clinicalgenome.org), who has encouraged disease-specific expert panels to carefully review current ACMG/AMP guidelines and propose necessary specifications based on the particular characteristics of a given disease (Rehm et al., 2015) while staying consistent with ACMG/AMP variant interpretation guidelines (Richards et al., 2015). ClinGen Sequence Variant Interpretation (SVI) committee oversees this process, and has approved these specifications (see Table 1), which are made publicly accessible on the ClinGen website (https://clinicalgenome.org/affiliation/50027/).
Table 1:
Specifications to the ACMG/AMG guidelines for mtDNA variant assessment.
| ACMG/AMP current description | mtDNA specifications | |||||||
|---|---|---|---|---|---|---|---|---|
| ACMG/AMP criteria codes | Original ACMG/AMP rule summary | Stand alone | Very strong | Strong | Moderate | Supporting | Comments | Relevant gene class |
| PVS1 | Null variant in a gene where LOF is a known mechanismof disease | - | Large heteroplasmic mtDNA deletions, where at least one gene is completely deleted | Assessment of small deletions, nonsense, and frameshift variants in protein-coding genes should follow established guidelines (Abou Tayoun et al., 2018) | Nonsense mediated decay is not known to occur for mtDNA, however ClinGen SVI PVS1 guidelines (Abou Tayoun et al., 2018) will be utilized when applicable (see Figure 2). | mRNA | ||
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change | - | - | Applied per original ACMG/AMP guidelines | - | - | mRNA | |
| PS2 | De novo (both maternity and paternity confirmed) in a patient with the disease and no family history | - | De novo (maternity confirmed or identical full mtDNA sequence) in a patient with the disease and no family history; with weighting per ClinGen SVI guidance | Older sequencing techniques such as Sanger sequencing cannot reliably detect heteroplasmy levels below 30–50%. Current NGS techniques can typically detect heteroplasmy levels as low as 1.5%. It is recommended to test several tissues in the mother to fully assess for the presence and level of the mtDNA variant in question. Utilize ClinGen SVI recommendation for applying these criteria (https://clinicalgenome.org/site/assets/files/3461/svi_proposal_for_de_novo_criteria_v1_0.pdf), the mitochondrial genome would best fit with Table 1 “phenotypic consistency” category of “phenotype consistent with gene but not highly specific.” | mRNA tRNA rRNA | |||
| PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect | - | - | - | - | Functional validation is present in cybrid studies or single fiber analysis | The following criteria should be met to apply: - a biochemical deficiency must be observed in patient cell line with mtDNA variant in question - whether the biochemical deficiency is transferred to mutant cybrids (in the case of enzymatic deficiency, <20% activity of control or a decrease in activity that is >2 standard deviations from control mean) - whether cybrid cells carry high mutant load (minimal 60%) - if studies have been reproduced and are consistent | mRNA tRNA rRNA |
| PS4 | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in control subjects | - | - | Variant present in ≥16 unrelated probands | Variant present in ≥4 unrelated probands | Variant present in 2 unrelated probands in different top-level haplogroups | Individuals are defined as affected if they: -meet diagnostic criteria for one of the classic mitochondrial disease clinical syndromes (MELAS, MERRF, MIDD, NARP, Pearson, KSS, CPEO, CPEO plus, Leigh, LHON, primary lactic acidosis) OR -have 1 “red flag” feature with 2 or more nonspecific features (Haas et al., 2007) OR - have 3 or more nonspecific features with lab abnormalities (Haas et al., 2008) | mRNA tRNA rRNA |
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain without benign variation | - | - | - | - | - | - | None |
| PM2 | Absent from control subjects | - | - | - | - | Frequency <0.00002 (0.002%, 1/50,000) | - | mRNA tRNA rRNA |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant | - | - | - | - | - | mtDNA variants are maternally inherited and not inherited in an autosomal recessive manner | None |
| PM4 | Protein length changes due to in-frame deletions / insertions in a nonrepeat region or stop-loss variants | - | - | - | Applied per original ACMG/AMP guidelines | - | - | mRNA |
| PM5 | Missense change at amino acid residue where a different missense change determined to be pathogenic has been seen before | - | - | - | Applied per original ACMG/AMP guidelines (mRNA) | Same nucleotide position as previously established pathogenic variant in a rRNA/tRNA | - | mRNA tRNA rRNA |
| PM6 | Assumed de novo (but without confirmation of maternity and paternity | - | Assumed de novo, but without confirmation of maternity (maternal testing done by targeted variant analysis and/or targeted gene sequencing) | See PS2. | mRNA tRNA rRNA | |||
| PP1 | Cosegregation in multiple affected family members in a gene definitively known to cause the disease | - | - | - | Cosegregation with disease in 5+ maternal family members and level of heteroplasmy segregating with disease manifestations | Cosegregation with disease in 2–4 maternal family members and level of heteroplasmy segregating with disease manifestations | Variant must not only segregate in maternal family members, but the level of heteroplasmy must also segregate with disease manifestations, where those individuals with more mild symptoms or appearing to be healthy have lower to undetectable levels of the variant and those more severely affected individuals and/or tissues have higher levels of the variant. This criterion cannot be applied when a variant is present at homoplasmy in multiple family members. | mRNA tRNA rRNA |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease | - | - | - | - | - | mtDNA exhibits lack of recombination and a relatively high mutation rate (due to lack of histones or other protective structures) that allows for mtDNA variants to accumulate over time. | None |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product | - | - | - | - | Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, etc) | See Figure 3. | mRNA tRNA |
| PP4 | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology | - | - | - | - | Decreased ETC enzyme activity (<20%) performed in a CLIA-approved (or equivalently-certified) laboratory in muscle, liver, and/or fibroblasts (for fibroblasts, must be seen in multiple unrelated probands and/or assayed in different individuals). | Other causes of ETC enzyme deficiency must be excluded, to the best of current ability, by comprehensive mtDNA and nDNA sequencing. Nuclear DNA genes including ETC complex subunits, assembly factors, and translation components should be thoroughly evaluated with no pathogenic or likely pathogenic variants (present in trans if autosomal recessive inheritance) that could be causative detected. See Supp Table S3 for list of genes. | mRNA tRNA rRNA |
| PP5 | Reputable source recently reports variant as pathogenic | - | - | - | - | - | Removed per ClinGen SVI recommendation (Biesecker et al., 2018). | None |
| BA1 | Allele frequency is > 0.05 (5%) | Top-level haplogroup defining variants in individuals that are members of that same top-level haplogroup OR frequency > 0.01 (1%) | - | - | - | - | - | mRNA tRNA rRNA |
| BS1 | Allele frequency is greater than expected for disorder | - | - | Frequency 0.005 – 0.0099 (0.5% – 0.99%) | - | - | - | mRNA tRNA rRNA |
| BS2 | Observed in a healthy adult individual for a recessive (homozygous), dominant heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age | - | - | Observed at a higher heteroplasmy in a healthy adult individual, especially in healthy maternal family members, than in same tissue tested in an affected individual | - | Observed at a higher heteroplasmy in a healthy adult individual, especially in healthy maternal family members, than in different tissue(s) tested in an affected individual | - | mRNA tRNA rRNA |
| BS3 | Well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing | - | - | - | - | No evidence of functional effect in cybrid studies or single fiber analysis is present (no statistically significant difference from control; mean values of <2 SD from control mean, or 50% enzyme activity compared to controls). | See PS3. | mRNA tRNA rRNA |
| BS4 | Lack of segregation in affected members of a family | - | - | Lack of segregation in affected members of a family and/or segregation of disease in paternal family members. | - | - | See PS4 for criteria to be met to be considered “affected.” | mRNA tRNA rRNA |
| BP1 | Missense variant in a gene for which primarily truncating variants are known to cause disease | - | - | - | - | - | Most variants in protein-coding mtDNA genes are not truncating, but rather missense variants. Even if truncating variants were more common, this would not preclude missense variants from also causing a loss of protein function. | None |
| BP2 | Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder or observed in cis with a pathogenic variant in any inheritance pattern | - | - | - | - | Other mtDNA variant is observed in individual’s mtDNA that has previously been confirmed to be pathogenic | - | mRNA tRNA rRNA |
| BP3 | In-frame deletions / insertions in a repetitive region without a known function | - | - | - | - | - | There are a few locations in the mtDNA genome where indels within a repetitive region are observed outside of two common locations: one is in the hypervariable region 1 (around position 16,189) and the other in hypervariable region 2 (around position 310). These indels are well-known benign findings. | None |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product | - | - | - | - | Multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, etc) | See Figure 3. | mRNA tRNA |
| BP5 | Variant found in a case with an alternate molecular basis for disease | - | - | - | - | Mitochondrial DNA variant found in a case with a nuclear DNA-related disease | - | mRNA tRNA rRNA |
| BP6 | Reputable source recently reports variant as benign | - | - | - | - | - | Removed per ClinGen SVI recommendation (Biesecker et al., 2018). | None |
| BP7 | A synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence nor the creation of a new splice site AND the nucleotide is not highly conserved | - | - | - | - | A synonymous (silent) variant. | Mitochondrial genes do not undergo splicing. Conservation is included in predictor algorithms used in PP3 and BP4, so conservation will be incorporated in this criterion. | mRNA |
Key: LOF - loss of function; SVI - Sequence Variant Interpretation Committee
CONSIDERATIONS FOR mtDNA GENOME VARIANT ASSESSMENT
The general criteria for variant assessment are the same for mtDNA variants as they are for nuclear gene variants. Indeed, segregation data, functional studies, and variant prevalence rates in patients versus controls are key determinants underlying the pathogenicity assertion of a mtDNA variant (whether pathogenic or benign).
The generalizable concepts of reduced penetrance, variable expressivity, and genetic heterogeneity are also relevant to diseases caused by mtDNA pathogenic variants. Leber hereditary optic neuropathy (LHON) is a classic example of reduced penetrance in mtDNA-related disorders (Yu-Wai-Man & Chinnery, 1993). Most instances of this condition, classically characterized as acute vision loss in one eye followed by vision loss in the other eye, are caused by one of three common pathogenic variants in mtDNA-encoded complex I subunits. Although most pathogenic mtDNA variants occur in a state of heteroplasmy, where the wild-type allele is also present, LHON-associated variants are frequently homoplasmic, where all copies of the mtDNA genome harbor the pathogenic variant. However, it is well-described that only 50% of males and 10% of females with a known pathogenic LHON variant will at any point in their life develop the characteristic vision loss. In addition, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome provides a classic mtDNA-associated example of variable expressivity, where individuals with the same variant can range from healthy, to having diabetes and sensorineural deafness, to suffering adult-onset occipital strokes or self-resolving stroke-like episodes, or even childhood-onset neurodevelopmental regression with basal ganglia lesions typical of Leigh syndrome (DiMauro & Hirano, 1993a). Genetic heterogeneity is also exemplified by Leigh syndrome, which is now recognized to be caused by pathogenic variants in at least 95 genes across both genomes (14 mtDNA genes and >81 nDNA genes) (Rahman, Noronha, Thiele, & Rahman, 2017).
However, multiple other ACMG/AMP nuclear variant interpretation criteria are not applicable to mtDNA variant analysis due to many unique features inherent to mtDNA: mtDNA genome composition (protein-coding genes, tRNA genes, rRNA genes, lack of introns), cytoplasmic maternal inheritance, haplogroups, and multiple genome copy number per cell leading to complex heteroplasmy and threshold effects (E. M. McCormick, Zolkipli-Cunningham, et al., 2018).
mtDNA genome composition and structure
mtDNA is a circular genome that contains 16,569 base pairs, encodes 37 genes, and is exclusively maternally inherited through the female germline (via oocytes) (Case & Wallace, 1981; Egger & Wilson, 1983; Giles, Blanc, Cann, & Wallace, 1980). While there have been occasional reports of biparental inheritance (Luo et al., 2018; Schwartz & Vissing, 2002), this remains a rare exception (Rius et al., 2019) and recently has been attributed to paternally-inherited Nuclear Mitochondrial DNA segments (NUMTs, or mtDNA pseudogenes) rather than paternal inheritance of the mitochondrial genome (Wei et al., 2020). The mitochondrial genome includes a non-coding D-loop region that regulates mtDNA transcription and replication, 13 protein-coding genes that encode for core subunits of complexes I, III, IV, and V (ATP synthase) of the electron transport chain (ETC), 2 rRNA genes, and 22 tRNA genes necessary for the translation of the 13 protein-coding gene products. mtDNA genes do not undergo splicing, mtDNA genomes do not undergo homologous recombination, and they replicate autonomously (Anderson et al., 1981). Many pathogenic mtDNA variants occur in the 22 tRNA genes crucial for translation of mitochondrial encoded genes. The impact of pathogenic variants in mt-tRNA and mt-rRNA genes is unique to mtDNA, because nuclear tRNA and rRNA genes are present with high copy number. For the tRNA and rRNA variants, the most important consideration is their impact on molecular structure. Relevance of the ACMG/AMP criteria were recently reviewed and updates and new criteria were proposed for mitochondrial tRNA variant interpretation (Wong et al., 2020). While there are unique aspects of mitochondrial tRNA variants that must be considered for interpretation, in this effort we specified existing criteria as outlined in the ACMG/AMP guidelines for these unique features rather than adding new criteria, to ensure consistency with current universal standards for variant interpretation (Richards et al., 2015).
mtDNA reference sequences and nomenclature
Correct mtDNA annotation and standardized naming conventions are critical to convey consistent, unambiguous information among clinicians, patients, and researchers. The recommended reference sequence for reporting mtDNA variation is the revised Cambridge Reference Sequence (rCRS), GenBank accession number NC_012920.1, which comprises 16,569 base pairs. The rCRS (Andrews et al., 1999) is the updated version of the first published human mitochondrial genome (Anderson et al., 1981), which has been used as a positional allele standard for reporting mtDNA variation by forensic and clinical researchers for the past three decades. Importantly, rCRS is not a “wild type” or “consensus” sequence, nor is it in a central or root position on the phylogenetic tree (Figure 1). Our Expert Panel consensus, reached after multiple rounds of extensive discussions, was that the rCRS should continue to be used for variant annotation to avoid accidental discordance in reporting. Use of other mtDNA reference sequences is not recommended for clinical purposes, although this sometimes is seen. Most notable was the use of a Yoruban mtDNA sequence, formerly listed in GenBank as NC_001807.4, as a standard by some commercial chip manufacturers beginning in 2007 and also included in the UCSC reference genome builds hg18 and hg19, causing much confusion with its discordant scoring. Another reference sequence, the Reconstructed Sapiens Reference Sequence (RSRS) was proposed as a standard in 2012 (Behar et al., 2012), with the intent to change the established mtDNA reference standard to an ancestrally-based inferred sequence. The MSeqDR Consortium has published detailed guidelines for how to preferentially convey the reference sequence genome used when generating mtDNA genome sequence variant calling files (VCF) (Falk et al., 2015). A full discussion of the pitfalls of using mtDNA genome sequences other than the rCRS as reference sequence was published in 2014 (Bandelt, Kloss-Brandstatter, Richards, Yao, & Logan, 2014). Of note, none of the positions corresponding to the RSRS-rCRS differences are associated with confirmed pathogenic variants (for a list of the RSRS-rCRS differences, see http://www.phylotree.org/resources/RSRS_vs_rCRS.htm).
Figure 1:
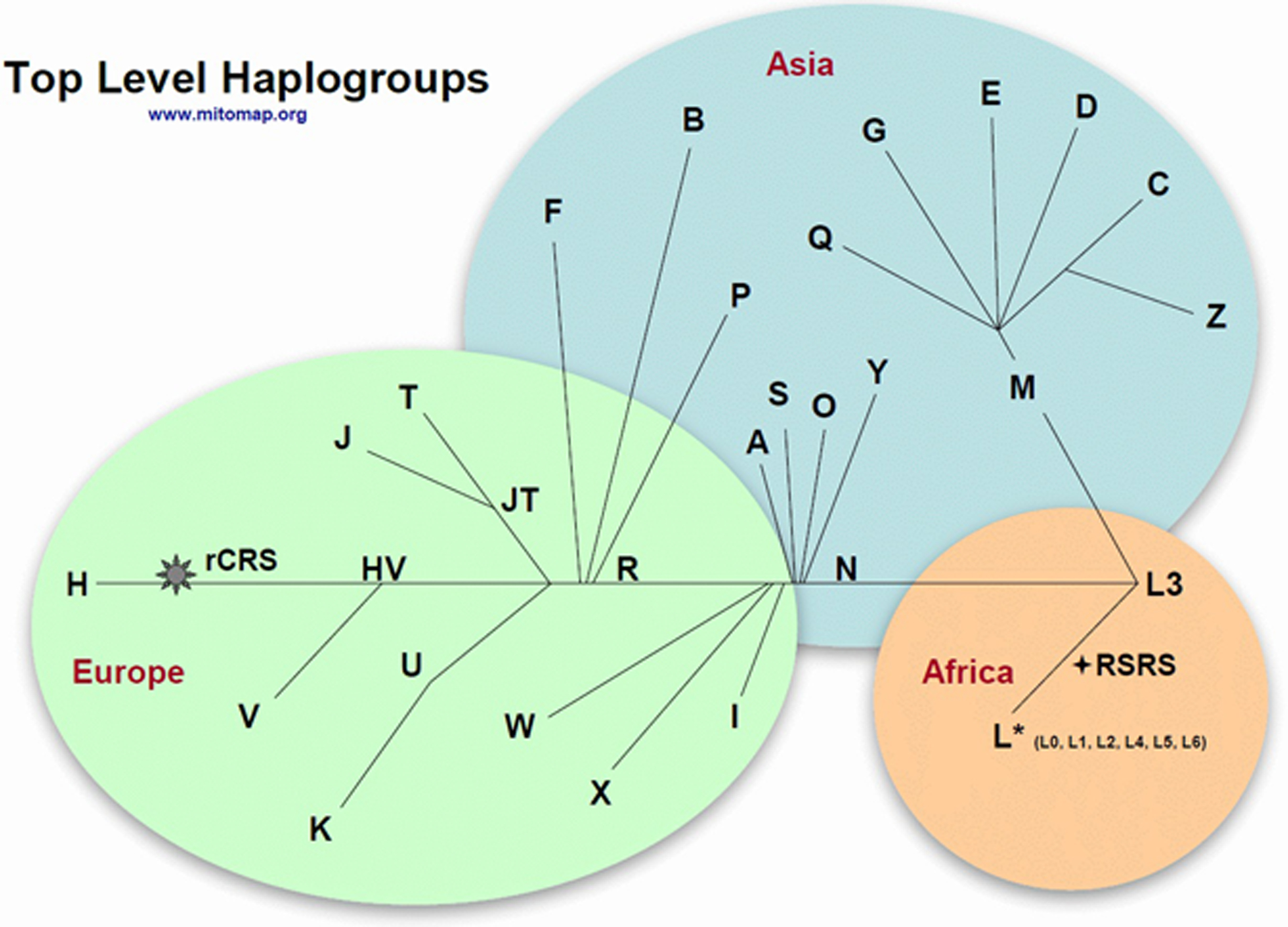
Overview of top-level haplogroups. Used with permission from www.mitomap.org. A full list of haplogroup-defining variants is available at https://www.mitomap.org/MITOMAP/HaplogroupMarkers.
As is the convention for clinical testing, all variant nomenclature should follow the recommendations of the Human Genome Variation Society (HGVS) (den Dunnen et al., 2016) and gene symbols should conform to the officially approved gene symbols curated by the Human Gene Nomenclature Committee (HGNC).
Haplogroups and phylogeny
There are sets of single nucleotide variants and/or small indels found within regions of mtDNA in any individual when compared to the rCRS. Owing to the lack of recombination among mitochondrial genomes, these variants cluster together in the global mtDNA phylogeny to form fixed mtDNA haplogroups. Because of their ancient origins, the mtDNA variants associated with haplogroups are homogeneous (or homoplasmic) and associated with particular lineages, having accumulated radiating maternal lineages as humans migrated around the globe.
In our proposed specifications, “top-level” haplogroups have a letter designation, or in the case of the African L lineage, a letter-number designation. Letter – number designations in the European/Asian lineages M and N are referred to as “branch-level haplogroups”. “Haplogroups” have a letter-number-letter designation, although phylogenetic tools will often subdivide these further using a nomenclature of alternating numbers and letters (for example, H2a vs. H2a2a1). In these specifications, we refer to “haplogroup” at the terse letter-number-letter level to include all sub-haplogroups contained within the larger haplogroup. However, it is important to note that haplogroups labeled as letter-number-letter do not universally indicate the same level of phylogeny (Blanc, Chen, D’Amore, & Wallace, 1983; Denaro et al., 1981; Merriwether et al., 1991; Navarro-Gomez et al., 2015; Schurr et al., 1990; Wallace, Garrison, & Knowler, 1985).
Variants defined here as “haplogroup markers” are present at an allele frequency of 80% or higher in individuals in that specific haplogroup branch at the letter-number-letter level. Variants referred to as “haplogroup-associated” are present at an allele frequency of 50–79.9% in individuals in that specific haplogroup branch.
Haplogroup markers and haplogroup-associated variants are by definition benign. However, an important nuance to recognize is that a particular variant that may be a haplogroup marker or haplogroup-associated variant for one haplogroup may be associated with disease manifestations in the setting of other haplogroups (Brown, Torroni, Reckord, & Wallace, 1995; Hudson et al., 2007; Torroni et al., 2003; Wei, Gomez-Duran, Hudson, & Chinnery, 2017). Unlike interpretation of nuclear gene variants, using mtDNA phylogeny in conjugation with minor allele frequencies is critical for proper mtDNA variant interpretation. While many haplotype blocks also exist in nDNA, there is a relatively limited number of mtDNA haplogroups given the small size of the mitochondrial genome.
mtDNA heteroplasmy and threshold effect
Mitochondria have many copies per cell, with multiple mtDNA genomes present in each mitochondrion, so that a mixture of mtDNA can exist in a given cell or tissue, of which a proportion are ‘healthy’ (wild-type) and a proportion harbor a given mtDNA variant. This mixture of mtDNA genomes creates a biologic phenomenon unique to mtDNA called heteroplasmy (Wallace & Chalkia, 2013). Due to random segregation of mitochondria during cellular division, heteroplasmy levels may differ between family members and between tissues or among the same tissue tested over time in a given individual. Most, including the more severe, pathogenic mtDNA variants tend to be heteroplasmic in nature (Gorman et al., 2016), where the presence and severity of clinical disease symptoms may directly relate to the pathogenic variant heteroplasmy levels in affected tissues (Shoffner et al., 1990). A phenotype associated with a specific pathogenic variant may only present when the variant reaches a particular level (or threshold) in a given tissue.
This phenomenon is exemplified by the MT-ATP6 m.8993T>C pathogenic variant which typically only causes disease manifestations in individuals with variant heteroplasmy levels in blood that exceed 90% (Thorburn, Rahman, & Rahman, 1993). The exact heteroplasmic threshold is not usually known for novel or uncommon pathogenic variants and may vary greatly based on the functional impact of a given variant. Typically, the heteroplasmic threshold is commonly quoted for many variants to be in the 60–80% range in a given tissue for it to cause severe clinical symptoms (Stewart & Chinnery, 2015). However, it is common for a mtDNA variant to cause a broad spectrum of clinical symptoms at variable heteroplasmy levels, such as the common MT-TL1 m.3243A>G pathogenic variant that leads to MELAS at 50–70% heteroplasmy levels, Leigh syndrome at levels above 90%, and maternally-inherited diabetes and deafness (MIDD) at levels below 40%; interpretation of this variant is further complicated by its being known to be selected against in blood over time even as symptom severity progresses (Kaufmann et al., 2009; Kaufmann et al., 2011). Only 10% of individuals that carry the m.3243A>G mtDNA pathogenic variant ever present with classical symptoms of MELAS (Nesbitt et al., 2013).
There is no one absolute threshold for all mtDNA pathogenic variants, as some variants may cause symptoms at lower levels and others must be present at much higher levels to cause disease. The heteroplasmic load of specific mtDNA variants varies among tissue types, such that the heteroplasmy observed in a readily sampled tissue source (such as blood or urine) may not always represent the heteroplasmy load in a symptomatic tissue (such as brain, gastrointestinal tract, or muscle). The type of tissue analyzed must be taken into account as analysis in some tissues, such as blood, has been shown to have limited sensitivity in the detection of mtDNA variants compared to other tissues, such as urinary epithelial cells (Fayssoil et al., 2017; Liu et al., 2013).
While there are overlapping concepts between heteroplasmy of the mitochondrial genome and either mosaicism or somatic variants associated with cancer in nuclear genes, these are distinctly different features of the genomes in which they occur.
The term “pathogenic” is typically defined as referring to a variant that is disease-causing, although it may be associated with either complete or variable penetrance depending on disease inheritance mode(s) for a given gene, specific variant, environmental or lifestyle factors, and genetic background. A specific mtDNA variant that may be definitely pathogenic in some individuals when identified in some tissues, is unlikely, when present at low heteroplasmy levels, to cause classical disease manifestations or may even cause an entirely different spectrum of symptoms. For example, high heteroplasmy levels of the MT-TK m.8344A>G variant can present with the classical myoclonic epilepsy and ragged red fibers (MERRF) syndrome, but at lower heteroplasmy levels can manifest as more mild symptoms such as myopathy and sensorineural hearing loss (Shoffner et al., 1990). Thus, the biological concept of heteroplasmy, with variable tissue levels that may change over time and be subject to variant-specific or tissue-specific thresholds, can underlie a broad spectrum of phenotypes that may differ between individuals. This uniquely complicates mtDNA variant interpretation.
Specificity of gene-disease pairs
Interpretation of mtDNA variants from published literature and in the laboratory can be hampered by the presumption that a variant will always be associated with a similar phenotype as that reported in individual cases. Rather, phenotypes associated with specific mtDNA pathogenic variants are notoriously variable and do not present in a specific, consistent manner, even among members of the same family. While some clinical syndrome presentations may strongly suggest an energetic functional problem with a likely mitochondrial cause, this may be due to disease causing variants influencing mitochondrial structure or function that are rooted in either genome. An exemplary case is Leigh syndrome, which is caused by pathogenic variants in more than 95 genes that influence mitochondrial function (Rahman et al., 2017). Moreover, different pathogenic variants within the same gene can produce a wide variety of phenotypes, such as mtDNA-encoded complex I subunit genes in which pathogenic variants can cause widely variable phenotypes ranging from LHON to Leigh syndrome (Blok et al., 2007; Chinnery, 1993; Debray et al., 2007). Therefore, distinct mtDNA gene-disease pair categorizations are often difficult to establish. Although certain pathogenic mtDNA variants may reproducibly cause specific phenotypes, this is rare in mtDNA disease, and variable expressivity needs to be considered when reviewing literature and family histories for evidence of variant-disease segregation.
SCOPE OF WORK
Here, we report the consensus findings of the MSeqDR-ClinGen mtDNA expert panel working group to modify existing ACMG/AMP variant interpretation guidelines for purposes of mtDNA variant curation (Table 1). These guidelines were formally reviewed and granted approval by the ClinGen Sequence Variant Interpretation (SVI) Committee and the ClinGen Clinical Domain Working Group (CDWG) Oversight Committee. Four important stipulations are noted:
These specifications are to be utilized to assess the pathogenicity of mtDNA variants in primary mitochondrial disease. Primary mitochondrial disease encompasses many classically-defined clinical syndromes (often labeled with a range of acronyms such as MELAS, MERRF, neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP), LHON, chronic progressive external ophthalmoplegia (CPEO), Kearns Sayre Syndrome (KSS), and many more) that display considerable phenotypic overlap and may also commonly include less well-defined groupings of multi-system clinical disorders (Barca et al., 2020; E. M. McCormick, Muraresku, & Falk, 2018). It is not reasonable to have distinct variant interpretation specifications for each clinical syndrome or constellation, as evidenced by each primary mitochondrial disease patient having on average 16 unique symptoms (Zolkipli-Cunningham et al., 2018).
Similarly, these specifications are not meant for interpretation of clinical significances of mtDNA variants in other diseases such as cancer. In many cases, the functional significance of a mtDNA variant is context dependent, as some cancers acquire the same variant(s) that are ancient haplogroup founding variants in human populations (Brandon, Baldi, & Wallace, 2006; Triska et al., 2019).
This working group recognizes certain mtDNA variants have been associated with several complex characteristics, including predisposition to common diseases and longevity. Such correlations are outside the scope of these mtDNA variant interpretation specifications.
This working group recognizes the goal of modifying the current ACMG/AMP variant interpretation guidelines is to provide universal standardization of mtDNA variant curation and interpretation. This task is distinct from answering the question of whether a given variant is causing disease manifestations in a specific individual. Given the unique phenomenon of heteroplasmy, a pathogenic variant may be present in a healthy individual who carries it at low heteroplasmy levels in tested tissue samples, but this does not confirm the variant itself is benign. Rather, the variant may not be present at a high enough level to impair mitochondrial function and cause disease in that individual. Ideally, additional information such as phenotype and heteroplasmy levels in different tissues of a given individual would be taken into account to accurately interpret variant pathogenicity. Using the analogy of a nuclear gene variant example, the deletion of a phenylalanine residue at position 508 in CFTR is the most common pathogenic variant in patients with cystic fibrosis, recognized as a pathogenic variant for autosomal recessive cystic fibrosis. In the context of someone who is a carrier of only this pathogenic variant, there is no clinical phenotype; however, the same person would have cystic fibrosis in the context of having another pathogenic variant on their other CFTR allele. Regardless of context, the variant is always pathogenic for autosomal recessive cystic fibrosis. By contrast, mtDNA heteroplasmy levels to be considered as definitive evidence for pathogenicity were not specified in our work as there are no absolute cut-offs that will be generalizable and accurate enough to account for every mtDNA variant in every family. Therefore, such personalized variant correlation is outside the scope of this work but needs to be considered. Future work is needed to develop guidelines that will facilitate the definitive diagnosis or exclusion of mtDNA disease in individuals with features of primary mitochondrial disease with differing levels of heteroplasmy in the context of their specific mtDNA variant, mtDNA haplogroup, and environmental/lifestyle exposures.
METHODS:
MSeqDR-ClinGen mtDNA expert panel working group meetings
Beginning in September 2016, regular Web meetings were held of the MSeqDR-ClinGen mtDNA Expert Panel working group. Close communication was regularly maintained throughout this process with ClinGen, including with the ClinGen SVI Committee, to ensure consistency of this process with the established variant curation rules and overall ClinGen framework.
mtDNA variant data review
As outlined below in the specific categories, mtDNA data from MITOMAP, HmtDB, and MSeqDR resources were extensively mined and analyzed for objective evidence of rule specification (Clima et al., 2017; Falk et al., 2016; Falk et al., 2015; Lott et al., 2013; Shen et al., 2016).
mtDNA variant analysis pilot
As detailed in Supp Table S2, variants that were prior considered as pathogenic, benign, or uncertain significance by MITOMAP (Lott et al., 2013), ClinVar (Landrum et al., 2020), and/or a clinical diagnostic laboratory were piloted against the specified mtDNA variant guidelines. The variants chosen for analysis were decided upon by this mitochondrial disease expert panel that includes clinical geneticists; clinical, research, and clinical laboratory-based genetic counselors; laboratory directors from academic and commercial clinical diagnostic laboratories; mtDNA genome researchers; and bioinformaticians from several sites across the world (https://clinicalgenome.org/affiliation/50027/).
Variants chosen for analysis were determined from consensus of this expert panel, were present in a variety of genes, and were varied in the amount of evidence available to review. Several universally-accepted pathogenic variants were selected to ensure these specifications were consistent with this classification. These variants included the most well-known mtDNA etiologies (m.3243A>G classically associated with MELAS and m.8344A>G classically associated with MERRF), variants known to cause a spectrum of primary mitochondrial disease manifestations with onset ranging from childhood to adulthood (m.10158T>C, m.13513G>A, and m.14459G>A), and a more rare variant that has been shown to be associated with primary mitochondrial disease (m.1644G>A). These variants are considered pathogenic both by this expert panel consensus and due to their classification as “confirmed” in MITOMAP. MITOMAP classifications have largely been performed by a single manual curator (author MTL), who reviewed all published literature involving mtDNA variants over the past three decades and made this information available in the public Web resource, MITOMAP. The head of MITOMAP’S clinical team (author VP) then reviewed this curated data to ensure agreement before publication in MITOMAP. To avoid bias, a third curator (author EMM), performed this variant pilot and presented outcome to the expert panel for review, discussion, and to reach classification consensus.
This process was also followed for variants that were prior considered to be variants of uncertain significance (VUS) or benign. However, given MITOMAP’s focus on curation of potentially pathogenic variants, classifications from ClinVar and/or clinical diagnostic laboratories were considered as the existing classification. The expert panel chose variants considered uncertain or benign based on their own clinical experiences, as well as variants that have been known to have been misclassified in the past.
All variants widely considered to be disease-causing by the mitochondrial community were classified as pathogenic, with the exception of m.1644G>A that was classified as likely pathogenic. This is acceptable as this variant is not one of the common mtDNA etiologies, has only been reported in approximately seven affected individuals to date, and had functional validation reported that showed the deleterious effect of the variant. Similarly, variants considered benign by these sources were classified as benign or likely benign by the Expert Panel mtDNA variant specifications, demonstrating their consistency with historic mtDNA variant assessment. Lastly, these specifications allowed for consensus determination on variants of uncertain significance, and resolved differences in reporting for some variants. The selected variants and pilot outcome were reviewed with ClinGen, as required to obtain ClinGen Variant Curation Expert Panel status and SVI approval of these specifications.
SPECIFIED GUIDELINES
Databases and predictive algorithms
The most comprehensive mtDNA disease sequence and variant resources are MITOMAP (Lott et al., 2013), HmtDB (Clima et al., 2017) and HmtVar (Preste, Vitale, Clima, Gasparre, & Attimonelli, 2019), MtSNPscore (Bhardwaj et al., 2009), and MSeqDR (Shen et al., 2018; Shen et al., 2016). Most mtDNA sequence resources utilize the GenBank full-length mtDNA dataset as, at the present time, GenBank has the most comprehensive set of mtDNA genome sequences (Shen et al., 2016). Caution must be used however given that not all individuals in these community-submitted datasets are healthy. Indeed, there are known cohorts of affected individuals in this data set, including 100 individuals with LHON.
MITOMAP
The MITOMAP database has been manually curating published mtDNA variants since 1996 (Kogelnik, Lott, Brown, Navathe, & Wallace, 1996). As of July 2020, MITOMAP includes 14,735 mtDNA nucleotide variants from 51,192 full-length human mtDNA genome sequences and 74,326 mtDNA control region-only sequences from the GenBank dataset. These sequences encompass the breadth of the mitochondrial phylogenetic tree and contain approximately 1,500 marker variants seen at ≥80% frequency in their respective haplogroups. These include 220 major markers for over 30 top-level haplogroups and an additional 1263 markers for over 400 haplogroups at the letter-number-letter level (https://mitomap.org/MITOMAP/HaplogroupMarkers). These markers represent 13% of the nucleotide variants found in the full-length sequence set, but in terms of frequency, make up 41% of the variant alleles present.
In MITOMAP there are currently 793 mtDNA variants reported with possible association with disease (391 in protein-coding regions, 23 in the control region, and 379 in tRNA and rRNA genes). Of these 793 variants, 93 are considered by MITOMAP as pathogenic (updated listing at https://mitomap.org/MITOMAP/ConfirmedMutations). MITOMAP’s pathogenicity assessment is based upon literature review, conservation, demonstrable biochemical and/or histochemical defects, mutant load/phenotype segregation, reports of multiple independent cases, and variant frequency (see https://mitomap.org/MITOMAP/ConfirmedCriteria). MITOMAP’s variant categories regarding disease involvement include “confirmed” where multiple independent laboratories and/or researchers have evaluated the variant and reported compelling functional evidence of its pathogenicity, and “reported” where a variant has been seen in affected individuals and is considered to be related to disease, although rigorous validation has not been performed. “Benign” status is not independently validated in MITOMAP at this time; rather variants without reported or confirmed status but present in the GenBank dataset are simply listed as sequence variations.
The MITOMASTER portion of MITOMAP provides a comprehensive set of analytical tools for the analysis of mtDNA variation, including variant frequency, conservation, and haplogroup determination (Lott et al., 2013). Queries can be readily initiated using single nucleotide variants (SNV), SNV sets, mtDNA genome sequences in fasta format, or by GenBank accession numbers.
MITOMAP also includes the MitoTIP tool for in-silico analysis of novel tRNA variants (Sonney et al., 2017). The MitoTIP score for a tRNA variant is intended as a starting point for assessment, and evaluates conservation, structural disruption, and variant location. Scores are given according to quartile rank, ranging from “likely benign” to “likely pathogenic”.
HmtDB
HmtDB (Clima et al., 2017), http://www.hmtdb.uniba.it, is an open resource created in 2005 to support population genetics and mitochondrial disease studies, hosting human mitochondrial genome sequences from both those with and without reported disease phenotypes. The primary source of HmtDB are the nucleotide databases within the International Nucleotide Sequence Database Collaboration (INSDC). Of note, the INSDC includes GenBank. As of July 2020, HmtDB includes 49,304 full length mtDNA genome sequences (comprises 44,058 reportedly healthy individuals and 5,246 affected individuals); 1,567 coding region only sequences (comprises 1,381 reportedly healthy individuals and 186 affected individuals), and more than 10,947 variant sites. Sequences are annotated with population information (allowing for ascertainment of continent-specific allele frequencies), and nucleotide and amino acid variability are estimated according to SiteVar and MitVar algorithms (Horner & Pesole, 2003; Pesole & Saccone, 2001). Moreover, HmtDB allows for the prediction of the haplogroup of any human mitochondrial genome, supports queries through various criteria, and offers an integrated API to programmatically access human mtDNA genome data.
HmtVar
HmtVar (Preste et al., 2019), https://www.hmtvar.uniba.it/, is a manually curated database offering variability and pathogenicity information for human mtDNA genome variants. Data are gathered from HmtVar’s twin database, HmtDB, described above, and integrated with pathogenicity predictions and information from several mtDNA-specific online resources. A pathogenicity prediction can then be generated for both nonsynonymous mRNA and tRNA variants (Diroma, Lubisco, & Attimonelli, 2016; Santorsola et al., 2016) and is enhanced by a literature mining pipeline (Vitale, Preste, Palmisano, & Attimonelli, 2020). As of July 2020, HmtVar includes 34,297 variants in protein-coding genes, 4,547 variants in mt-tRNAs, 803 variants in mt-rRNAs, and 1,329 variants in the regulatory region. The nonsynonymous and tRNA variants are further classified into five tiers (152 polymorphic, 9,666 likely polymorphic, 36 likely pathogenic, 15 VUS, and 18,899 pathogenic variants). These variants include both variants reported in humans and other potential nucleotide changes.
MtSNPscore
MtSNPscore (Bhardwaj et al., 2009) is a comprehensive weighted scoring system for identification of mtDNA variations that can impact pathogenicity and would likely be associated with disease. It identifies and scores disease-associated mtDNA variants by filtering out polymorphic sites and sites with no reported or predicted functional role. The method is available at http://ab-openlab.csir.res.in/snpscore/ and allows the end user to customize the weighted scores based on the disease understanding and the study design. The method has been tested on an Indian ataxia dataset (92 individuals), sequenced as part of the MtSNPscore study, as well as another publicly available mtSNP dataset comprising 576 mitochondrial genomes of Japanese individuals from six phenotypic groups. Although this tool provides useful information, it is labor-intensive, requiring a full patient data set and a priori knowledge of parameters of interest.
MSeqDR
MSeqDR, an online Web mitochondrial disease sequence data resource (Falk et al., 2015), compiles reference mitochondrial genome variant data from the general and mitochondrial disease populations through close collaboration with several resources including MITOMAP, HmtDB, researchers, and clinical diagnostic laboratories such as GeneDx (Shen et al., 2016). Different population-level allele frequency data sets (including from Asian populations and totaling over 200,000 mitochondrial genomes with additional data being actively collected and added when available for public access), comprehensive in silico variant effect predictions from MitoTIP and HmtDB, and variant pathogenicity assessment data such as from MITOMAP are made easily available through mvTool (Shen et al., 2018). MSeqDR also includes several variant-disease association bioinformatics resources such as the MSeqDR-LSDB (a mitochondrial disease locus specific database that efficiently organizes variant pathogenicity data), Quick-Mitome (to facilitate annotation and interpretation of individual patient and trio whole exome and mtDNA genome variant datasets through phenotype-guided tools), literature mining tools to facilitate analysis of genotype-phenotype associations, Phy-mer (to provide reference free haplogroup analysis) (Navarro-Gomez et al., 2015), and MToolbox (identifies variants, haplogroups, and variant effect annotations in Sanger and next-generation sequencing datasets, and can discriminate between possible NUMTs and mitochondrial DNA fragments) (Calabrese et al., 2014).
Other databases
ClinVar (http://www.ncbi.nlm.nih.gov/clinvar) is an NCBI-hosted database for the submission and curation of variants from both nuclear and mitochondrial DNA genomes in disease-related genes (Landrum et al., 2014). This freely accessible database hosts data from clinical laboratories and research projects that provide variant interpretations on observed genomic variants. ClinVar plays a crucial role in the sharing of variant interpretations among clinical laboratories, in particular, allowing for increased visibility of rare variant interpretation and providing a reliable means for efficient communication among labs that have observed rare variants in a clinical diagnostic setting. Cross-reference links between ClinVar and MSeqDR are provided for mtDNA variants reported in ClinVar.
The most useful databases and predictive algorithms for mtDNA variant curation are listed in Table 2. Utilization of smaller databases that are not actively updated is strongly discouraged, unless they provide unique and complementary data to that which is already available in the recommended comprehensive databases. One example is mtDB which is still occasionally cited as a reference for variant frequency but contains only 2,704 mtDNA genome sequences and has not been updated for 12 years (Ingman & Gyllensten, 2006).
Table 2:
Bioinformatics resources to support mtDNA variant assessment.
| Sequences | Variant pathogenicity | Sequence analysis | ||||
|---|---|---|---|---|---|---|
| Tool | mtDNA sequence repository | mtDNA sequence source | mtDNA variant patdogenicity assessment | mtDNA variant patdogenicity assessment source | mtDNA sequence analysis | mtDNA sequence analysis source |
| Mitomap https://www.mitomap.org/MITOMAP | + | GenBank | + | Manual curation MitoTIP | + | MITOMASTER |
| MSeqDR https://mseqdr.org/ | + | GenBank GeneDx HmtDB (sequences not in GenBank) User-entered sequences | + | MSeqDR-GBrowse MSeqDR-LSDB MitoMap (manual curation, MitoTip) | + | mvTool PhyMer HmtDB (MToolBox) |
| MtSNPScore http://ab-openlab.csir.res.in/snpscore/ | − | − | + | MtSNPScore | − | − |
| HmtDB https://www.hmtdb.uniba.it/ | + | International Nucleotide Sequence Database Collaboration (includes GenBank) | + | HmtVAR | + | MToolBox |
Many population databases that effectively describe nuclear gene variations are neither relevant nor appropriate for the interpretation of mtDNA variants. In particular, the 1000 Genomes Project (Auton et al., 2015), Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS/), Exome Aggregation Consortium (ExAC) (Karczewski et al., 2017), and genome Aggregation Database (gnomAD)(Lek et al., 2016) are important databases for cataloging genomic variations in several global populations and are commonly utilized for assessing the frequency of particular variants in these populations (Auton et al., 2015; Lek et al, 2016). However, the exome data in these databases are constructed from data produced using next generation sequencing (NGS) platforms that require hybridization-based targeted exome capture, and generally do not target mtDNA. Genomes in gnomAD are produced using the same short read sequences as exomes, which creates a problem for accurate mapping of sequences back to the nuclear and/or mitochondrial DNAs. The presence of NUMTs throughout the nuclear genome make proper mapping of short read NGS results difficult (Wei et al., 2020). While some NUMTs are fixed in humans, others are polymorphic among populations, and still others formed more recently. It is unclear what impact these newer (undocumented) NUMTs might have in their interference with proper short read mapping to the mtDNA genome. Based on these challenges, neither ExAC nor gnomAD currently provide population frequency data for mtDNA variants. Therefore, they cannot be used for the purpose of guiding mtDNA variant pathogenicity interpretation. This will need to be revisited in the future, as curators of these databases work to generate reliable mtDNA genome variant data (poster #1402, American Society of Human Genetics 2018 meeting).
Several pathogenicity annotation databases that are widely used and commonly known for collecting information on disease and normal states in nuclear genes are not relevant for interpreting mtDNA variants, including HGMD, DECIPHER, and LOVD. While the Human Gene Mutation Database (HGMD, http://www.hgmd.org) is a useful resource to identify specific variants in the published literature that may be implicated in causing disease (Stenson et al., 2017), its scope does not include mtDNA variants, but instead recommends consultation with the MITOMAP database. The DECIPHER database started as a collection of nuclear genome-wide copy number variants in patient cases and has since expanded to include SNVs and small indels (Firth et al., 2009). LOVD aggregates data from individual locus-specific databases that uses the Leiden open-source variation database system (Fokkema, den Dunnen, & Taschner, 2005). Although initially begun as a means to collect mtDNA pathogenic variants in a manner that includes genotype and phenotypic information (Elson et al., 2012; K, Jalali, Scaria, & Bhardwaj, 2013), the function of this database has largely been replaced by MSeqDR that remains actively curated with the support of the United Mitochondrial Disease Foundation (Falk et al., 2015; Shen et al., 2016; Shen, McCormick, Muraresku, Falk, & Gai, 2020).
Other in silico prediction algorithms
Although in silico prediction algorithms commonly utilized for nDNA variant assessment such as SIFT (P. Kumar, Henikoff, & Ng, 2009), Polyphen-2 (Adzhubei et al., 2010), FatHmm, PROVEAN, and MutationAssessor will assess mtDNA variants, comparative analyses have shown poor correlation among such prediction tools with MITOMAP and HmtVar manually-curated mtDNA variants (Bris et al., 2018; Castellana, Ronai, & Mazza, 2015). Therefore, these tools are not recommended for use in mtDNA variant pathogenicity assessment.
APOGEE, a meta-predictor (Castellana et al., 2017) for variants in protein-coding genes that incorporates predictions from HmtVAR and MitoTIP (described above) for tRNA variants are optimized for mtDNA and should be considered when assessing mtDNA variant pathogenicity (see PP3, BP4 below). There are currently no readily accessible informatics tools for prediction of rRNA variant effects.
Detailed descriptions of specified guidelines
All previous general recommendations of the ACMG/AMP variant interpretation guidelines are applied here for mtDNA variant guidelines, unless otherwise specified. We use the five-tier classification system used by ACMG/AMP for nuclear variant interpretation, where variants are classified into categories of “Pathogenic”, “Likely Pathogenic”, “Uncertain Significance”, “Likely Benign”, and “Benign”. We also continue to qualify the term “variant” with interpretative assertions, instead of using the terms “mutation” and “polymorphism”. Nomenclature should follow the recommendations of the Human Gene Variation Society (HGVS), where mtDNA variants start with the “NC_012920.1:m” or “m.” designation and use the revised Cambridge Reference Sequence (rCRS) as reference for positional information consistent with the existing ACMG/AMP recommendations.
The rules for combining criteria for pathogenicity assertions have not been modified from the current ACMG/AMP guidelines (Richards et al., 2015). All rules were specified for relevance to mtDNA protein-coding and rRNA/tRNA genes. 19 rules were further specified, and 7 that were deemed not applicable so removed (see Table 1).
PVS1: Null variant
Large heteroplasmic mtDNA deletions, where at least one gene is completely deleted, are a known cause of primary mitochondrial disease (Goldstein & Falk, 1993) and are always pathogenic. It is not currently possible to ascertain the prevalence of these deletions in the general population. There are no large deletions present in the GenBank set because “full length” sequences are limited to those that are greater than 15.4 kb (where the classically described common mtDNA deletion encompasses 5 kb of the 16 kb genome). Furthermore, even if there was no size limit, variants reported in population sequences are homoplasmic in nature whereas large deletions in affected individuals are heteroplasmic.
Assessment of small deletions, nonsense, and frameshift variants in protein-coding genes should follow established guidelines (Abou Tayoun et al., 2018) although some aspects of these guidelines are not applicable to mtDNA. Nonsense mediated decay is not known to occur for mtDNA genes therefore the length of the truncation and the missing residues requires assessment per established guidelines (Abou Tayoun et al., 2018) and there is no splicing of protein-coding genes (see Figure 2). There is a paucity of initiation coding variants leading to the inability to pilot a training set of these variant types on any potential specifications. With the exception of large deletions in which at least one gene is deleted, this criterion cannot be applied to tRNA or rRNA variants. Nonsense and frameshift variants are not relevant to non-protein coding genes. As tRNA and rRNA gene small deletions are rare, similar to initiation codon variants, it is not possible to test any potential specifications on a set of definitely pathogenic or benign variants. Similarly, duplications in any mtDNA gene are rare. Given these limitations, it is not possible for this expert panel to provide guidance on assessing these types of variants at the present time. Indeed, assessment of each variant requires review of the location and any possible structural effects.
Figure 2:
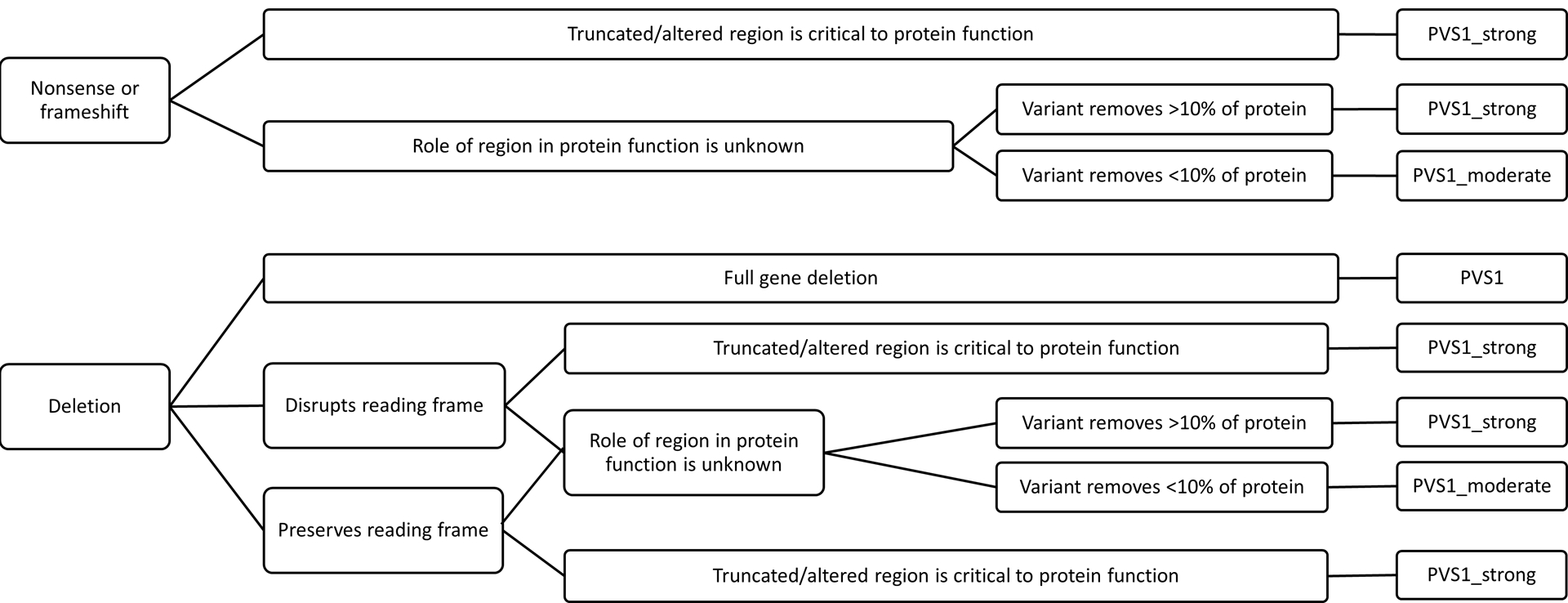
PVS1 decision tree, adapted from Abou Tayoun et al., 2018.
An analysis of stop-gain and frameshift variants in the coding regions of approximately 47,000 full length mtDNA sequences in GenBank has shown that these variant types are evidently poorly tolerated, as they are quite rare in the general population (32/874,880, or a frequency of 3.65764E−05). Of these 32 variants, half are found near the terminal end of the gene (with ≥90% of the coding region remaining). Several of the remaining few may in fact be haplogroup- or lineage-specific. Although stop-gain and frameshift are rare, PVS1_moderate might be the most frequently applied criterion given that half of them leave >90% of the coding region unperturbed (Abou Tayoun et al., 2018).
PS1, PM5, BP7: Nucleotide and/or amino acid position
Several of the existing criteria for amino acid changes are relevant for protein-coding mtDNA genes (PS1, PM5). These criteria are not relevant to non-protein coding genes. However, PM5_supporting was specified to capture a different nucleotide change at the same position in a tRNA. While no amino acid change would occur as these genes are not protein-coding, a nucleotide change at a position where a different nucleotide change was pathogenic would increase suspicion for its pathogenicity.
BP7 has been specified to only include synonymous variants. BP7, as written in the ACMG/AMP guidelines (Richards et al., 2015), is applied when a synonymous variant is not predicted to affect splicing and the nucleotide position is not highly conserved. This raises two separate issues for the mitochondrial genome. The first issue is that synonymous variants in nuclear genes are pathogenic most often due to generation of a cryptic splice site. Since this is not a concern in mtDNA genes, synonymous variants will be considered as supporting evidence for benign. Although no synonymous variants in mtDNA have been reported to be pathogenic, this finding cannot be up-weighted to stand-alone for benign given the chance that a synonymous variant may potentially effect translational efficiency or could inactivate a gene embedded within a larger gene (such as humanin within the 16S rRNA gene or MOTS-c within the 12S rRNA gene) (C. Lee, Kim, & Cohen, 2016; C. Lee, Yen, & Cohen, 2013; C. Lee et al., 2015). The second issue is that no consensus exists in the field of mitochondrial genomics as to which conservation groups to use and how to calculate conservation is not defined. Furthermore, conservation is included in predictor algorithms used in PP3 and BP4, so conservation will be incorporated in these criteria but not under BP7.
PS2, PM6: De novo variants
Minimal modifications were made to the ACMG/AMP rules regarding the de novo status of variants. Confirmation of paternity and testing paternal samples is not warranted as mtDNA is exclusively maternally inherited through the oocyte. PS2 has been specified to clarify confirmation of identical full mtDNA genome sequence with mother to confirm maternity. Routine nuclear markers are utilized to confirm maternity for nuclear variants however may introduce room for error if used to confirm maternity when assessing mtDNA variants. This expert panel agrees nuclear and mitochondrial DNA analysis should occur simultaneously, however this is not always the case and mtDNA sequencing can be performed in isolation. If not performed together, sample mix-up between mtDNA and nDNA could theoretically be possible (although this is also possible for any nuclear gene sequence data). Overall, since this is such a strongly-weighted line of evidence, this expert panel took the conservative approach and this criterion will only apply if mtDNA genome sequencing in mother is also performed. PM6 has been updated to capture circumstances when a mother’s mitochondrial genome is not sequenced but rather targeted variant analysis is undertaken which is in keeping with this criterion as described in the ACMG/AMP guidelines.
When assessing de novo status, particularly close attention must be paid to the testing method and tissue tested in the maternal sample, as older sequencing techniques such as Sanger sequencing cannot reliably detect heteroplasmy levels below 30–50% depending on the gene and laboratory (McCormick et al., 2013) while current NGS techniques can typically detect heteroplasmy levels as low as 1.5%. Therefore, it is recommended to test several tissues in the mother by NGS to fully assess for the presence and level of the mtDNA variant in question.
Furthermore, we recognize the ClinGen SVI recommendation for applying these criteria (https://clinicalgenome.org/working-groups/sequence-variant-interpretation/) and recommend utilization of this guidance for assessing de novo mtDNA variants. In particular, the mitochondrial genome would best fit with the phenotypic consistency category of “phenotype consistent with gene but not highly specific.”
PS3, BS3: Functional studies
Transmitochondrial cybrid studies involve generation of two cell lines with a common nuclear genome background that have the same mtDNA genome except for a specific mtDNA variant in question, one cell line is homoplasmic for the reference allele and the other is homoplasmic for the mtDNA variant in question. These reconstructed cell lines are generated by fusing standard cell lines depleted of mitochondria with cells of an individual that are heteroplasmic for a mtDNA variant, and then selecting homoplasmic mutant or reference clones for further functional analysis (Jun, Trounce, Brown, Shoffner, & Wallace, 1996; King & Attardi, 1989; King, Koga, Davidson, & Schon, 1992; Trounce, Neill, & Wallace, 1994). This approach allows for isolation and measurement of mtDNA variant specific effects, since the nuclear background is controlled. Biochemical studies to assess ETC and oxidative phosphorylation (OXPHOS) function, such as polarographic or spectrophotometric analyses, can then be performed in the two reconstructed cell lines to determine if there is a detectable functional effect of the mtDNA variant in question. Additional studies may be performed as appropriate, and may be considered for scoring as supporting (PS3_supporting) functional evidence for pathogenicity or supporting (BS3_supporting) evidence of benign impact. As no standard or universal parameters exist to objectively analyze cybrid studies, results of these studies cannot be considered strong or moderate evidence at this time.
Several parameters must be met to meet this line of evidence. First, a biochemical deficiency (for example, enzymatic deficiency, mitochondrial translation deficiency or respiratory chain complex assembly defect) must be observed in the primary patient cell line with the mtDNA variant in question. If there is not documented biochemical defect in the patient cell line, the transfer of the defect to cybrid cells cannot be shown. When this criterion is met, whether the biochemical deficiency is transferred to mutant cybrids needs to be taken into account. In the case of enzymatic deficiency, <20% of normal activity of controls or a decrease in activity that is >2 standard deviations below control mean should be shown, per mitochondrial disease consensus criteria (Parikh et al., 2015; Walker, Collins, & Byrne, 1996) and correlated to mutant load. Furthermore, whether cybrid cells carry a high mutant load (minimal heteroplasmy of 60% mutant (Stewart & Chinnery, 2015) and whether the studies have been reproduced and shown to be consistent should all be taken into consideration.
Single-fiber studies, where a muscle fiber is analyzed for OXPHOS activity and mtDNA variant heteroplasmy (Yarham et al., 2011) can also be utilized as functional evidence of pathogenicity. Similar to cybrids, as there are no standards to objectively analyze results, these studies will be used as supporting evidence for pathogenicity or benign status.
PM2, BA1, BS1: Variant frequency
See Table 2 for databases of particular importance to use when querying mtDNA variant allele frequencies. While these databases are currently the most useful to curate mtDNA variants, we recognize that databases change over time and new databases will be introduced, so we specify here several factors that need to be considered when utilizing data from a database for purposes of mtDNA variant curation. These factors include:
(1) Quality and nature of sequence databases is equally, if not more, important than their number of sequences as it cannot be assumed that all sequences in a given database are of good quality. In particular, data derived from ancient DNA sequences, sequences from tumor cell lines, and/or sequences from tumor tissue should not be utilized as a background reference. Utilizing allele frequencies from these ancient individuals or cell lines could skew data interpretation and allele frequency.
(2) Databases must be currently maintained and updated. When compared to older technology (such as autoradiographs), newer sequences have less noise if direct sequencing was performed. Furthermore, older or poorly maintained databases typically have small numbers of genomes that can skew allele frequency data and the sequences in these databases are typically not described in sufficient detail regarding data source and sequence data quality.
(3) Caution must be used when considering mtDNA sequences generated from exome or genome sequencing data, as depending on the methodology used, these may contain nuclear DNA mitochondrial pseudogenes (NUMTs as detailed above, which can be problematic in interpreting variant data from genome sequencing) or may contain regions of mtDNA without coverage (as is particularly problematic when using low-level, off-target mtDNA data obtained with targeted exome sequencing). Rigorous variant calling quality control and filtering criteria are essential.
(4) We are also mindful of certain ethnic groups and several mitochondrial lineages being underrepresented in current databases, which may skew variant frequency data. Tools such as MSeqDR are working to increase the compilation of complementary allele frequency data from Asian and other non-Caucasian populations to address this current limitation.
Variant frequency below 0.00002 (or 0.002%, 1/50,000) from controls in reliable mitochondrial genome databases is considered supporting evidence for pathogenicity (PM2_supporting). This conclusion was reached based on the following factors:
(1) mtDNA variants universally accepted to be pathogenic were assessed for their allele frequency in GenBank sequences, as supported by MITOMAP, and there were no such variants present at a frequency greater than 0.005 (0.5%, equivalent to 250/50,000). While a cut-off below 0.005 (0.5%) was acceptable to capture pathogenic variants, there are currently 14,045 variants in MITOMAP that would meet this criterion (95% of all variants in MITOMAP), making it ineffective to be considered as evidence for pathogenicity. Upon piloting a variant frequency cut-off of below 0.002%, 19% of the total variants in MITOMAP met this criterion, which led to this criterion being down-weighted to only supporting evidence. It is presently difficult to determine a lower cut-off, as reliable mitochondrial genome databases such as MITOMAP currently have only 51,192 full length mtDNA genome sequences from GenBank.
(2) The pathogenic variant with highest allele frequency was m.11778G>A, which was present at 0.27% (137/51,192 mtDNA sequences in MITOMAP). Of note, 117/137 sequences for m.11778G>A were identified in the sequence record as being from LHON studies. Counting only the remaining twenty sequences (all lacking specific mention of disease status) reduces the allele frequency of m.11778G>A to 0.04%.
Variant frequency was also assessed in GenBank sequences, as supported by MITOMAP, to look for trends that would infer benign status. The following factors were considered to support benign status:
(1) Reported disease variants present at above 1% were catalogued, and all were found to be either haplogroup-defining (at >80% frequency) or haplogroup-associated (at >50% frequency) for haplogroups or subgroups with a minimum of 10 sequences. As no additional pathogenic variants were captured with this cut-off, variant frequency exceeding 0.01 (1%) is considered stand-alone evidence for benign classification (BA1). However, there must be no additional conflicting evidence to support pathogenicity, such as a novel occurrence in a certain haplogroup.
(2) Out of 793 reported disease variants, only two (m.15942T>C and m.13637A>G) have been reported as possibly pathogenic that are above a 0.5% frequency cutoff and also not haplogroup-associated. These two variants are each seen at ~0.8% in 51,192 GenBank sequences. Therefore, an allele frequency of 0.005 – 0.0099 (0.5% – 0.99%) is strong evidence for benign classification (BS1).
PM4: Protein length changes
This rule can only be applied for variants in the 13 protein-coding mtDNA genes. Otherwise, this rule was not changed from the current ACMG/AMP variant interpretation guidelines.
PP1, PS4, BS2, and BS4: Segregation and presence in unrelated probands
Presence or absence of a mtDNA variant in maternal family members does not alone provide enough information to apply a supporting line of evidence for pathogenicity, as the clinical presentations and heteroplasmy level in different tissues of each family member also needs to be considered. While the presence of a mtDNA variant in several affected family members may seem to infer pathogenicity, the opposite would be true if the variant was present at higher heteroplasmy levels in those more mildly affected and at lower levels in those more severely affected or in more symptomatic tissues. Therefore, in order to apply segregation criteria as evidence of pathogenicity, the mtDNA variant in question must not only segregate in maternal family members, but the level of heteroplasmy must also correlate with disease manifestations, where those individuals with more mild symptoms or appearing to be healthy have lower to undetectable levels of the variant and those more severely affected individuals and/or tissues have higher levels of the variant.
Due to the quantitative nature of mtDNA variant inheritance, LOD scores cannot be calculated for this genome. Therefore, we evaluated the literature for reports of extended kindreds with universally-accepted pathogenic mtDNA variants. There are several extended families reported with m.8344A>G that is classically associated with MERRF. These family members had varying symptoms ranging from healthy to severely affected individuals with early death that correlated to variant load (Shoffner et al., 1990; Howell et al., 1996). Healthy or mildly affected individuals had low to undetectable heteroplasmy levels in various tissues and more severely affected individuals had higher heteroplasmy levels. When adding the number of segregations where this held true across the reported kindreds, there were greater than 10 segregations. This is further exemplified by kindreds with the m.3243A>G pathogenic variant that is classically associated with MELAS (Martinuzzi, et al., 1992; Van den Oueweland, et al., 1994; de Vries, et al., 1994), where adding the segregations across multiple kindreds equaled greater than 20 segregations. Furthermore, while these two examples occur in tRNA genes, there is also a report of an extended kindred with a pathogenic variant in the protein coding gene MT-ATP6 with several affected and healthy family members whose symptoms correlate with heteroplasmy level, with a total of more than five segregations (Castagna et al., 2007). However, given the inability to calculate LOD scoring, we will not include such high levels of segregation as strong evidence. Rather, we propose meeting the above criterion in five or more maternal family members is moderate evidence for pathogenicity (PP1_moderate), and in two to four maternal family members is supporting evidence for pathogenicity (PP1).
While the above specifications generally hold true, careful review of the family, testing methodology, and tissue tested must be performed. Furthermore, this rule cannot be applied when a variant is present at homoplasmy in all family members.
Alternatively, detecting higher heteroplasmy levels of a mtDNA variant in a healthy individual who is past the age of expected disease for typically severe diseases, or in a healthy matrilineal family member of an affected individual, would refute pathogenicity. Therefore, a variant seen at consistently higher heteroplasmy levels in a healthy unrelated adult or adult matrilineal family member (as some mtDNA conditions are adult-onset) compared to an affected individual would be evidence for benign classification (BS2). Due to varying threshold effect of different variants and the lack of complete understanding of this phenomenon, it is not possible to define how much fold higher a heteroplasmy level must be to apply this criterion. If the same tissue is tested in the proband and the healthy individual, a higher heteroplasmy level in an unaffected individual or healthy maternal family member in the same tissue tested in the affected individual or proband would meet this criterion (BS2). However, if a different tissue is assessed and the heteroplasmy level in an unaffected individual or healthy maternal family member is higher than in any other tissue tested in the affected individual or proband would meet this criterion as supporting (BS2_supporting). Furthermore, lack of mtDNA variant segregation in affected matrilineal family members or segregation of clinical disease manifestations in paternal family members would also be evidence for benign classification (BS4).
PS4 applies to case control studies or, in the absence or paucity of such studies, can be applied when a variant is absent from large population databases and present in multiple unrelated probands with a similar phenotype. As there are not numerous case control studies with mtDNA variants, this necessitates the latter application of this rule. This was also found to hold true for other ClinGen expert panels, who have adapted this criterion to count unrelated individuals with similar phenotypes and require the same variant to be absent in large population databases (Gelb et al., 2018; K. Lee et al., 2018; Mester et al., 2018). These calculations are based on likelihood and LOD scoring which is not calculable for mtDNA variants. Therefore, we will follow the Bayesian approach that has been utilized by the ClinGen SVI, recognizing this can be optimized for mtDNA variants in the future.
Individuals with primary mitochondrial disease have, on average, 16 major symptoms, with the most common reported symptoms being muscle weakness, chronic fatigue, exercise intolerance, imbalance, gastrointestinal problems, and developmental delay (Zolkipli-Cunningham et al., 2018). Other features commonly seen in those with primary mitochondrial disease include cerebral stroke-like lesions in a non-vascular pattern, Leigh syndrome, seizures (in particular, epilepsia partialis continua), myoclonus, ataxia, cardiomyopathy, heart block, Wolff-Parkinson-White arrhythmia, retinal dystrophy, optic atrophy, ophthalmoplegia, and anesthetic hypersensitivity. Additionally, diabetes mellitus, growth problems, sensorineural hearing loss, mood disorders, liver problems, renal problems, dysautonomia, immune dysfunction, anemia, and dementia can also be seen (Haas et al., 2007). While the presence of one or a combination of these features in an individual would be concerning for primary mitochondrial disease, these features are non-specific. Therefore, to apply PS4, individuals will be considered affected if diagnostic criteria is met for one of the classic mitochondrial disease clinical syndromes (MELAS, MERRF, MIDD, NARP, Pearson, KSS, CPEO, CPEO plus, Leigh syndrome spectrum, LHON, primary lactic acidosis). However, given the strict clinical criteria of several of these syndromes that many individuals with pathogenic mtDNA variants do not meet (Barca et al., 2020), an individual can also be defined as affected if one “red flag” feature is present along with two or more non-specific features (Haas et al., 2007), or they have three or more non-specific features with suggestive lab abnormalities (Haas et al., 2008).
Lastly, while identification of a mtDNA variant in unrelated probands would seem to be evidence for pathogenicity, the top level haplogroups of the probands must also be taken into consideration. If a mtDNA variant is present in two unrelated probands and both are members of the same top level mtDNA haplogroup, then it is possible the variant in question is related to that haplogroup. Therefore, in order to meet PS4_supporting criteria, the unrelated probands must be members of different top level mtDNA haplogroups (Figure 1).
PP3, BP4: Computational evidence
The previous section on databases and predictive algorithms discusses computational evidence to consider, and Table 2 details computation tools of particular importance when curating mtDNA variants. As in Richards et al., 2015, these rules can only be applied once when evaluating a variant, even if multiple predictors agree on pathogenic or benign impact. Furthermore, consistent with Richards et al., 2015, all predictors must agree to meet this criterion (Figure 3).
Figure 3:
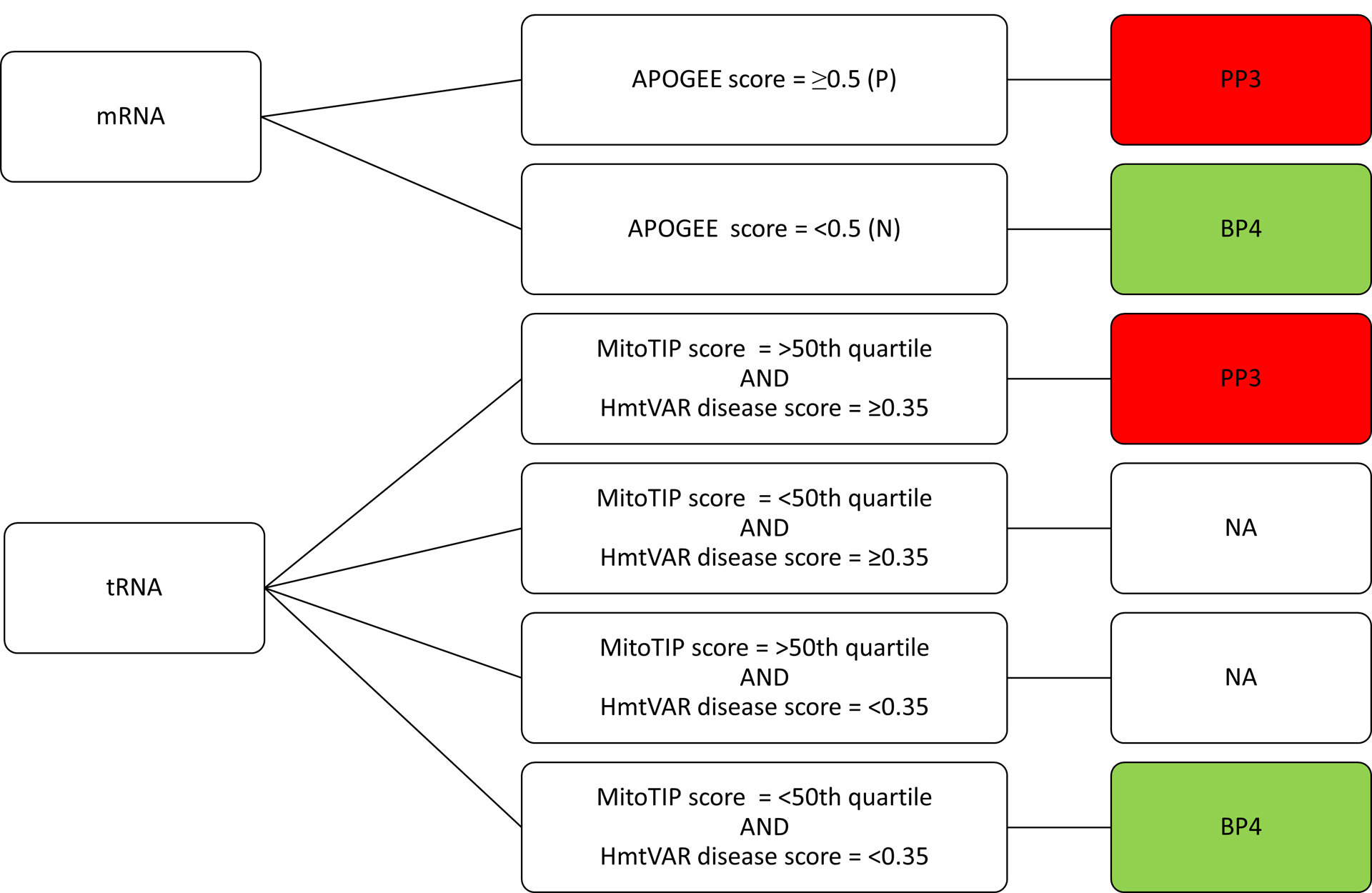
Decision tree to apply criteria for in silico prediction tools, PP3 and BP4. PP3/BP4 will be applied for tRNA single nucleotide deletions based on MitoTIP only. P = pathogenic and N = neutral, per APOGEE algorithm.
PP4: Phenotype
PP4 was specified because the manner in which primary mitochondrial disease presents is rarely, if ever, so specific that one can a priori identify a single, particular gene cause that would explain the patient’s phenotype. The reality is that there is extensive locus and allelic heterogeneity, with many variants occurring in a variety of different mtDNA and/or nuclear genes that could produce similar and overlapping disease presentations. However, ETC enzyme activity, a commonly utilized biochemical assay for ETC function, can relay some important information.
Evaluating enzymatic function of the mitochondrial ETC complexes can provide evidence of pathogenicity, should deficiencies in correlating an enzyme complex or complexes relevant to the variant in question be noted. For example, a pathogenic variant in a gene encoding a subunit of complex I may cause demonstrable complex I deficiency, or a pathogenic variant in a mt-tRNA needed for the translation of protein-coding mtDNA genes may cause deficiencies of complexes I, III, IV, and/or V that harbor mtDNA-encoded protein subunits (DiMauro & Hirano, 1993a, 1993b). However, it is important to note several caveats when applying this rule:
(1) ETC enzyme activity deficiencies should be ideally evaluated in muscle and liver, tissues with high energy demand and in which control ranges have been established, which are most likely to exhibit dysfunction in the setting with impaired ETC function and manifest clinical signs or symptoms. ETC enzyme deficiencies noted in skin-derived fibroblast cell lines can also be considered supporting evidence for pathogenicity. However, fibroblast cell line deficiencies must be seen in multiple unrelated probands and/or assayed in different individuals. Of note, ETC enzyme activities in buccal samples will not be considered as evidence for or against mtDNA variant pathogenicity given a lack of replicated and validated data demonstrating close correlation with ETC enzyme activities in other symptomatic tissues (Goldenthal et al., 2012).
(2) Benign criteria cannot be applied for normal ETC enzyme activities as it has been shown that individuals with pathogenic mtDNA variants can have normal ETC enzyme activities, and these studies may be influenced by the age of the individual when analysis is performed, as well as by the presence of mitochondrial content alterations (such as proliferation) that may compensate for and mask an underlying enzymatic deficiency.
(3) A decrease will be defined by a clinically-recognized laboratory performing the validated biochemical testing, and age- and tissue-matched controls must be used. While there are no international standards for what constitutes decreased ETC activity, consensus criteria define an ETC complex deficiency as falling below 20% of the control mean (Parikh et al., 2015; Walker et al., 1996).
(4) Other causes of ETC enzyme deficiency must be excluded, to the best of current ability, by comprehensive mtDNA and nDNA sequencing. Nuclear DNA genes encoding ETC complex subunits, assembly factors, and translation components should be thoroughly evaluated without detection of any pathogenic or likely pathogenic variants that could be causative of the individual’s clinical presentation (such as present in trans if autosomal recessive inheritance). Supp Table S3 provides a detailed list of genes to evaluate for this purpose (Marni J. Falk, 2020).
BA1: Haplogroup-defining variants
Haplogroup markers and haplogroup-associated variants are benign when present in an individual with that haplogroup. To facilitate variant curation and as outlined in Figure 1, MITOMAP has indexed ~300 haplogroup markers that are present in the top level haplogroups of the mitochondrial tree (top level haplogroups are defined in Figure 1 and above under haplogroups and phylogeny section). Haplogroup markers and haplogroup-associated variants are available at https://mitomap.org/MITOMAP/HaplogroupMarkers. Furthermore, a table of the most frequent variants in MITOMAP’s GenBank sequence set is available at https://mitomap.org//MITOMAP/TopVariants. Most of these extremely common variants are ancestral and scattered throughout the human mitochondrial tree. These ancestral variants are often not labeled on phylogenetic tree branches or by personal DNA testing services as haplogroup-defining markers, but are, in fact, present at extremely high population frequency.
Maintaining awareness of the common ancestral variants is essential as occasionally an extremely common variant is reported, without an understanding of its global frequency, as a possible cause for disease (Aikhionbare, Khan, Carey, Okoli, & Go, 2004; Houshmand et al., 2011; R. Kumar et al., 2007; Roshan et al., 2012). It is also important to be aware that while haplogroup markers or haplogroup-associated variants do not typically cause disease, occasionally such variants can have negative impact when found on the background of a different haplogroup. For example, m.3394T>C is a marker for haplogroup M9a (99.7% frequency) and is associated with high altitude adaptation in Tibet. However, when this same variant is found in haplogroups B4c and F1, it is associated with LHON (Ji et al., 2012; Kang et al., 2016). In such instances where a haplogroup-defining variant is identified in an individual of a different haplogroup, that variant should be curated as outlined in these guidelines.
BP2, BP5: Alternate disease cause
Identifying another disease-causing variant, either in nDNA or mtDNA, is considered supporting evidence for benign classification for a mtDNA variant being curated, especially when the mtDNA variant being curated is present at homoplasmy in all tissues of a proband and their healthy relatives tested. However, several additional factors must be taken into consideration before applying either criterion. Most pathogenic variants when present in an individual at low levels of heteroplasmy (generally below 20%) are unlikely to be causative of clinical disease manifestations in that individual. Indeed, more than 1 in 200 individuals harbor low levels (generally below 20%) of common mtDNA pathogenic variants in blood that are not causing known medical symptoms (Elliott, Samuels, Eden, Relton, & Chinnery, 2008), although it could be causal of some symptoms such as the common m.3243A>G variant when occurring below 10% heteroplasmy levels in blood or urine may cause MIDD (Laloi-Michelin et al., 2009). Overall, identification of another pathogenic mtDNA variant that is present only at low heteroplasmy levels (in multiple and/or symptomatic tissues) will not apply for either of these criterion. Furthermore, there have been several reports of individuals with two pathogenic mtDNA variants (Brown, Allen, Van Stavern, Newman, & Wallace, 2001; Howell et al., 2002), further substantiating these criteria must be carefully considered before applying them to mtDNA variant interpretation.
In addition, we recognize that there are more cases being identified with more than one genetic etiology for complex, multi-system disease manifestations (Craigen et al., 2013). Furthermore, it is quite possible to have a primary mitochondrial disease caused by a mtDNA variant and a separate, non-mitochondrial disease genetic condition. However, we will utilize these rules to keep consistent with the guidelines as written in Richards et al., 2015.
Detailed descriptions of original ACMG/AMP criteria removed from mtDNA-specified guidelines
Several criteria have been removed from the mtDNA guidelines on variant interpretation because they are not applicable to mtDNA variants.
While there are no well-defined pathogenic variant hotspots or clusters in protein-coding genes, there are positions in tRNAs where a variant would raise greater concern for pathogenicity. In particular, tRNAs harbor several structurally and/or functionally important areas that would raise concern if disrupted by a variant, such as in the stem loop structure, in the anticodon position where post-transcriptional modification occurs, or in positions involved in tertiary structure interactions. While this could be captured in PM1, this information is available in HmtVar and several tRNA domains, structures, and conserved sequences have been incorporated into the analysis algorithm of the MitoTIP tool (see Table 2). Therefore PM1 has been removed so as to not double count this line of evidence.
PM3 was removed because mtDNA variants are maternally inherited and not autosomal recessive. PP2 has been removed as high variability among mtDNA haplotypes is well-documented, owing to maternal inheritance with lack of recombination and a relatively high mutation rate (due to lack of histones or other protective structures) that allows for mtDNA variants to accumulate over time. It is this characteristic that makes mtDNA variation so amenable to evolutionary and forensic uses.
PP5 and BP6 are not useable for mtDNA variant interpretation, as any variant observed in a database (e.g., ClinVar) should be assessed using the information provided (such as segregation data, heteroplasmy load, etc.) and not rely on only a blind pathogenicity assertion. Even if an interpretation protocol is provided, the exact evidence used for the assertion should be examined, as it is possible that professional judgement pushed the classification of a variant from one category to another.
BP1 was not used in these guidelines because most variants in protein-coding mtDNA genes are not truncating, but rather are missense variants. Even if truncating variants were more common, this would not preclude missense variants from also causing a loss of protein function. BP3 was removed as there are a few locations in the mtDNA genome where indels within a repetitive region are observed outside of two common locations: one is in the hypervariable region 1 (around position 16,189) and the other in hypervariable region 2 (around position 310). These indels are well-known benign findings often associated with a T to C transition at position 16,189, for example, that leads to a run of cysteine nucleotides. Similarly the nine base pair insertion-deletion polymorphism between the MT-CO2 and MT-TK genes is a defining variant for Asian haplogroup B and of unknown functional significance. These are routinely excluded from clinical and/or phylogenetic considerations.
CONCLUSION
We report here MSeqDR-ClinGen Expert Panel reviewed, amended, and ClinGen approved ACMG/AMP variant interpretation consensus guideline specifications for purposes of mtDNA variant interpretation. Working within the original framework, the five-tier variant pathogenicity assertion categories were excluded or amended to allow for meaningful interpretation of the unique biological aspects of mtDNA. Key biological factors that uniquely complicate mtDNA variant interpretation include mtDNA haplogroups and heteroplasmy, with variable tissue levels that may change over time, be subject to variant-specific or tissue-specific thresholds, and underlie a broad spectrum of phenotypes that may differ between individuals. Detailed guidelines are provided for all variants throughout the mtDNA as they relate to all primary mitochondrial diseases, and we have included a detailed description of the appropriate reference genome, genomic databases, and functional analyses necessary to inform accurate mtDNA variant interpretation.
Supplementary Material
Acknowledgements;
We are grateful to the United Mitochondrial Disease Foundation for their organizational and administrative support, as well as ongoing partnership of MSeqDR Consortium activities since its inception. We thank Roberto Preste for his assistance incorporating HmtVar for purposes of this manuscript. We also are thankful to the ClinGen Sequence Variant Interpretation Committee and Clinical Domain Working Group Oversight Committee for their detailed review, suggestions, and approval of these mtDNA variant specifications.
Grant numbers;
This work was supported by the United Mitochondrial Disease Foundation and North American Mitochondrial Disease Consortium (NAMDC funded by NS078059 from NINDS, NICHD, and NCAT) funding for the Mitochondrial Disease Sequence Data Resource (MSeqDR), as well as NIH grants NS021328, MH108592, OD010944, U41-HG008634, and U24-HD093483 and U.S. Department of Defense grant W81XWH-16-1-0401. The content is solely the responsibility of the authors and does not necessarily represent the official views of funders including the National Institutes of Health.
Footnotes
Conflict of interest statement;
The authors have no relevant financial conflicts of interest to disclose. RB is employed by GeneDx, DEPA and SW are employed by Invitae, CMS is employed by Variantyx and QNA Diagnostics, and RM is employed by ARUP.
Data availability statement;
Data, including specifications and variant pilot outcome, are available at https://clinicalgenome.org/affiliation/50027 and https://erepo.clinicalgenome.org/evrepo/, respectively.
References;
- Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, … ClinGen Sequence Variant Interpretation Working, G. (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat, 39(11), 1517–1524. doi: 10.1002/humu.23626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, … Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat Methods, 7(4), 248–249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikhionbare FO, Khan M, Carey D, Okoli J, & Go R (2004). Is cumulative frequency of mitochondrial DNA variants a biomarker for colorectal tumor progression? Mol Cancer, 3, 30. doi: 10.1186/1476-4598-3-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, … Young IG (1981). Sequence and organization of the human mitochondrial genome. Nature, 290(5806), 457–465. [DOI] [PubMed] [Google Scholar]
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, & Howell N (1999). Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet, 23(2), 147. doi: 10.1038/13779 [DOI] [PubMed] [Google Scholar]
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, … Consortium GP (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Kloss-Brandstatter A, Richards MB, Yao YG, & Logan I (2014). The case for the continuing use of the revised Cambridge Reference Sequence (rCRS) and the standardization of notation in human mitochondrial DNA studies. J Hum Genet, 59(2), 66–77. doi: 10.1038/jhg.2013.120 [DOI] [PubMed] [Google Scholar]
- Barca E, Long Y, Cooley V, Schoenaker R, Emmanuele V, DiMauro S, … Hirano M (2020). Mitochondrial diseases in North America: An analysis of the NAMDC Registry. Neurol Genet, 6(2), e402. doi: 10.1212/NXG.0000000000000402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar DM, van Oven M, Rosset S, Metspalu M, Loogvali EL, Silva NM, … Villems R (2012). A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am J Hum Genet, 90(4), 675–684. doi: 10.1016/j.ajhg.2012.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj A, Mukerji M, Sharma S, Paul J, Gokhale CS, Srivastava AK, & Tiwari S (2009). MtSNPscore: a combined evidence approach for assessing cumulative impact of mitochondrial variations in disease. BMC Bioinformatics, 10 Suppl 8, S7. doi: 10.1186/1471-2105-10-S8-S7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG, Harrison SM, & ClinGen Sequence Variant Interpretation Working Group (2018). The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med, 20(12), 1687–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc H, Chen KH, D’Amore MA, & Wallace DC (1983). Amino acid change associated with the major polymorphic Hinc II site of Oriental and Caucasian mitochondrial DNAs. Am J Hum Genet, 35(2), 167–176. [PMC free article] [PubMed] [Google Scholar]
- Blok MJ, Spruijt L, de Coo IF, Schoonderwoerd K, Hendrickx A, & Smeets HJ (2007). Mutations in the ND5 subunit of complex I of the mitochondrial DNA are a frequent cause of oxidative phosphorylation disease. J Med Genet, 44(4), e74. doi: 10.1136/jmg.2006.045716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon M, Baldi P, & Wallace DC (2006). Mitochondrial mutations in cancer. Oncogene, 25(34), 4647–4662. doi: 10.1038/sj.onc.1209607 [DOI] [PubMed] [Google Scholar]
- Bris C, Goudenege D, Desquiret-Dumas V, Charif M, Colin E, Bonneau D, … Procaccio V (2018). Bioinformatics Tools and Databases to Assess the Pathogenicity of Mitochondrial DNA Variants in the Field of Next Generation Sequencing. Front Genet, 9, 632. doi: 10.3389/fgene.2018.00632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MD, Allen JC, Van Stavern GP, Newman NJ, & Wallace DC (2001). Clinical, genetic, and biochemical characterization of a Leber hereditary optic neuropathy family containing both the 11778 and 14484 primary mutations. Am J Med Genet, 104(4), 331–338. [PubMed] [Google Scholar]
- Brown MD, Torroni A, Reckord CL, & Wallace DC (1995). Phylogenetic analysis of Leber’s hereditary optic neuropathy mitochondrial DNA’s indicates multiple independent occurrences of the common mutations. Hum Mutat, 6(4), 311–325. doi: 10.1002/humu.1380060405 [DOI] [PubMed] [Google Scholar]
- Calabrese C, Simone D, Diroma MA, Santorsola M, Gutta C, Gasparre G, … Attimonelli M (2014). MToolBox: a highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics, 30(21), 3115–3117. doi: 10.1093/bioinformatics/btu483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case JT, & Wallace DC (1981). Maternal inheritance of mitochondrial DNA polymorphisms in cultured human fibroblasts. Somatic Cell Genet, 7(1), 103–108. doi: 10.1007/BF01544751 [DOI] [PubMed] [Google Scholar]
- Castellana S, Fusilli C, Mazzoccoli G, Biagini T, Capocefalo D, Carella M, … Mazza T (2017). High-confidence assessment of functional impact of human mitochondrial non-synonymous genome variations by APOGEE. PLoS Comput Biol, 13(6), e1005628. doi: 10.1371/journal.pcbi.1005628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellana S, Ronai J, & Mazza T (2015). MitImpact: an exhaustive collection of pre-computed pathogenicity predictions of human mitochondrial non-synonymous variants. Hum Mutat, 36(2), E2413–2422. doi: 10.1002/humu.22720 [DOI] [PubMed] [Google Scholar]
- Chinnery PF (1993). Mitochondrial Disorders Overview In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)). Seattle (WA). [Google Scholar]
- Clima R, Preste R, Calabrese C, Diroma MA, Santorsola M, Scioscia G, … Attimonelli M (2017). HmtDB 2016: data update, a better performing query system and human mitochondrial DNA haplogroup predictor. Nucleic Acids Res, 45(D1), D698–D706. doi: 10.1093/nar/gkw1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craigen WJ, Graham BH, Wong LJ, Scaglia F, Lewis RA, & Bonnen PE (2013). Exome sequencing of a patient with suspected mitochondrial disease reveals a likely multigenic etiology. BMC Med Genet, 14, 83. doi: 10.1186/1471-2350-14-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debray FG, Lambert M, Chevalier I, Robitaille Y, Decarie JC, Shoubridge EA, … Mitchell GA (2007). Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics, 119(4), 722–733. doi: 10.1542/peds.2006-1866 [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, … Taschner PE (2016). HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat, 37(6), 564–569. doi: 10.1002/humu.22981 [DOI] [PubMed] [Google Scholar]
- Denaro M, Blanc H, Johnson MJ, Chen KH, Wilmsen E, Cavalli-Sforza LL, & Wallace DC (1981). Ethnic variation in Hpa 1 endonuclease cleavage patterns of human mitochondrial DNA. Proc Natl Acad Sci U S A, 78(9), 5768–5772. doi: 10.1073/pnas.78.9.5768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, & Hirano M (1993a). Melas In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)). Seattle (WA). [Google Scholar]
- DiMauro S, & Hirano M (1993b). Merrf In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)). Seattle (WA). [Google Scholar]
- Diroma MA, Lubisco P, & Attimonelli M (2016). A comprehensive collection of annotations to interpret sequence variation in human mitochondrial transfer RNAs. BMC Bioinformatics, 17(Suppl 12), 338. doi: 10.1186/s12859-016-1193-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger J, & Wilson J (1983). Mitochondrial inheritance in a mitochondrially mediated disease. N Engl J Med, 309(3), 142–146. doi: 10.1056/NEJM198307213090304 [DOI] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, & Chinnery PF (2008). Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet, 83(2), 254–260. doi: 10.1016/j.ajhg.2008.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson JL, Sweeney MG, Procaccio V, Yarham JW, Salas A, Kong QP, … McFarland R (2012). Toward a mtDNA locus-specific mutation database using the LOVD platform. Hum Mutat, 33(9), 1352–1358. doi: 10.1002/humu.22118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ (Ed.) (2020). Mitochondrial Disease Genes Compendium: From Genes to Clinical Manifestations: Elsevier. [Google Scholar]
- Falk MJ, Shen L, & Gai X (2016). From case studies to community knowledge base: MSeqDR provides a platform for the curation and genomic analysis of mitochondrial diseases. Cold Spring Harb Mol Case Stud, 2(3), a001065. doi: 10.1101/mcs.a001065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ, Shen L, Gonzalez M, Leipzig J, Lott MT, Stassen AP, … Lee-Jun W (2015). Mitochondrial Disease Sequence Data Resource (MSeqDR): a global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities. Mol Genet Metab, 114(3), 388–396. doi: 10.1016/j.ymgme.2014.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayssoil A, Laforet P, Bougouin W, Jardel C, Lombes A, Becane HM, … Wahbi K (2017). Prediction of long-term prognosis by heteroplasmy levels of the m.3243A>G mutation in patients with the mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome. Eur J Neurol, 24(2), 255–261. doi: 10.1111/ene.13176 [DOI] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, … Carter NP (2009). DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet, 84(4), 524–533. doi: 10.1016/j.ajhg.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokkema IF, den Dunnen JT, & Taschner PE (2005). LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum Mutat, 26(2), 63–68. doi: 10.1002/humu.20201 [DOI] [PubMed] [Google Scholar]
- Gelb BD, Cave H, Dillon MW, Gripp KW, Lee JA, Mason-Suares H, … ClinGen RWG (2018). ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet Med, 20(11), 1334–1345. doi: 10.1038/gim.2018.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles RE, Blanc H, Cann HM, & Wallace DC (1980). Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A, 77(11), 6715–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenthal MJ, Kuruvilla T, Damle S, Salganicoff L, Sheth S, Shah N, … Legido A (2012). Non-invasive evaluation of buccal respiratory chain enzyme dysfunction in mitochondrial disease: comparison with studies in muscle biopsy. Mol Genet Metab, 105(3), 457–462. doi: 10.1016/j.ymgme.2011.11.193 [DOI] [PubMed] [Google Scholar]
- Goldstein A, & Falk MJ (1993). Mitochondrial DNA Deletion Syndromes In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)). Seattle (WA). [PubMed] [Google Scholar]
- Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, … Turnbull DM (2016). Mitochondrial diseases. Nat Rev Dis Primers, 2, 16080. doi: 10.1038/nrdp.2016.80 [DOI] [PubMed] [Google Scholar]
- Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, & Cohen BH (2007). Mitochondrial disease: a practical approach for primary care physicians. Pediatrics, 120(6), 1326–1333. doi: 10.1542/peds.2007-0391 [DOI] [PubMed] [Google Scholar]
- Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, … Diagnosis, M. M. S. s. C. o. (2008). The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab, 94(1), 16–37. doi: 10.1016/j.ymgme.2007.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner DS, & Pesole G (2003). The estimation of relative site variability among aligned homologous protein sequences. Bioinformatics, 19(5), 600–606. [DOI] [PubMed] [Google Scholar]
- Houshmand M, Montazeri M, Kuchekian N, Noohi F, Nozar G, & Zamani A (2011). Is 8860 variation a rare polymorphism or associated as a secondary effect in HCM disease? Arch Med Sci, 7(2), 242–246. doi: 10.5114/aoms.2011.22074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell N, Miller NR, Mackey DA, Arnold A, Herrnstadt C, Williams IM, & Kubacka I (2002). Lightning strikes twice: Leber hereditary optic neuropathy families with two pathogenic mtDNA mutations. J Neuroophthalmol, 22(4), 262–269. [DOI] [PubMed] [Google Scholar]
- Hudson G, Carelli V, Spruijt L, Gerards M, Mowbray C, Achilli A, … Chinnery PF, (2007). Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet, 81(2), 228–233. doi: 10.1086/519394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingman M, & Gyllensten U (2006). mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res, 34(Database issue), D749–751. doi: 10.1093/nar/gkj010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji F, Sharpley MS, Derbeneva O, Alves LS, Qian P, Wang Y, … Wallace DC (2012). Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc Natl Acad Sci U S A, 109(19), 7391–7396. doi: 10.1073/pnas.1202484109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun AS, Trounce IA, Brown MD, Shoffner JM, & Wallace DC (1996). Use of transmitochondrial cybrids to assign a complex I defect to the mitochondrial DNA-encoded NADH dehydrogenase subunit 6 gene mutation at nucleotide pair 14459 that causes Leber hereditary optic neuropathy and dystonia. Mol Cell Biol, 16(3), 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- K S, Jalali S, Scaria V, & Bhardwaj A (2013). MitoLSDB: a comprehensive resource to study genotype to phenotype correlations in human mitochondrial DNA variations. PLoS One, 8(4), e60066. doi: 10.1371/journal.pone.0060066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L, Zheng HX, Zhang M, Yan S, Li L, Liu L, … Jin L (2016). MtDNA analysis reveals enriched pathogenic mutations in Tibetan highlanders. Sci Rep, 6, 31083. doi: 10.1038/srep31083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, … The Exome Aggregation Consortium. (2017). The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res, 45(D1), D840–D845. doi: 10.1093/nar/gkw971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Battista V, … De Vivo DC (2009). Protean phenotypic features of the A3243G mitochondrial DNA mutation. Arch Neurol, 66(1), 85–91. doi: 10.1001/archneurol.2008.526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Sproule DM, … De Vivo DC (2011). Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology, 77(22), 1965–1971. doi: 10.1212/WNL.0b013e31823a0c7f [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MP, & Attardi G (1989). Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science, 246(4929), 500–503. [DOI] [PubMed] [Google Scholar]
- King MP, Koga Y, Davidson M, & Schon EA (1992). Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNA(Leu(UUR)) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol Cell Biol, 12(2), 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogelnik AM, Lott MT, Brown MD, Navathe SB, & Wallace DC (1996). MITOMAP: a human mitochondrial genome database. Nucleic Acids Res, 24(1), 177–179. doi: 10.1093/nar/24.1.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, & Ng PC (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc, 4(7), 1073–1081. doi: 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Kumar R, Bhat A, Bamezai RN, Shamsi MB, Kumar R, Gupta NP, … Dada R (2007). Necessity of nuclear and mitochondrial genome analysis prior to assisted reproductive techniques/intracytoplasmic sperm injection. Indian J Biochem Biophys, 44(6), 437–442. [PubMed] [Google Scholar]
- Laloi-Michelin M, Meas T, Ambonville C, Bellanné-Chantelot C, Beaufils S, Massin P, … Group, M. D. F. S. (2009). The clinical variability of maternally inherited diabetes and deafness is associated with the degree of heteroplasmy in blood leukocytes. J Clin Endocrinol Metab, 94(8), 3025–3030. doi: 10.1210/jc.2008-2680 [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, … Kattman BL (2020). ClinVar: improvements to accessing data. Nucleic Acids Res, 48(D1), D835–D844. doi: 10.1093/nar/gkz972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, & Maglott DR (2014). ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res, 42(Database issue), D980–985. doi: 10.1093/nar/gkt1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kim KH, & Cohen P (2016). MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free Radic Biol Med, 100, 182–187. doi: 10.1016/j.freeradbiomed.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Yen K, & Cohen P (2013). Humanin: a harbinger of mitochondrial-derived peptides? Trends Endocrinol Metab, 24(5), 222–228. doi: 10.1016/j.tem.2013.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, … Cohen P, (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab, 21(3), 443–454. doi: 10.1016/j.cmet.2015.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Krempely K, Roberts ME, Anderson MJ, Carneiro F, Chao E, … Karam R (2018). Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum Mutat, 39(11), 1553–1568. doi: 10.1002/humu.23650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … Consortium EA (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Ma Y, Fang F, Zhang Y, Zou L, Yang Y, … Qi Y (2013). Wild-Type Mitochondrial DNA Copy Number in Urinary Cells as a Useful Marker for Diagnosing Severity of the Mitochondrial Diseases. PLoS One, 8(6), e67146. doi: 10.1371/journal.pone.0067146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, Sarmady M, … Wallace DC (2013). mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr Protoc Bioinformatics, 44, 1.23.21–26. doi: 10.1002/0471250953.bi0123s44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Valencia CA, Zhang J, Lee NC, Slone J, Gui B, … Huang T (2018). Biparental Inheritance of Mitochondrial DNA in Humans. Proc Natl Acad Sci U S A, 115(51), 13039–13044. doi: 10.1073/pnas.1810946115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick EM, Muraresku CC, & Falk MJ (2018). Mitochondrial Genomics: A complex field now coming of age. Curr Genet Med Rep, 6(2), 52–61. doi: 10.1007/s40142-018-0137-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick EM, Place E, & Falk MJ (2013). Molecular genetic testing for mitochondrial disease: from one generation to the next. Neurotherapeutics, 10(2), 251–261. doi: 10.1007/s13311-012-0174-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick EM, Zolkipli-Cunningham Z, & Falk MJ (2018). Mitochondrial disease genetics update: recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr Opin Pediatr, 30(6), 714–724. doi: 10.1097/MOP.0000000000000686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriwether DA, Clark AG, Ballinger SW, Schurr TG, Soodyall H, Jenkins T, … Wallace DC (1991). The structure of human mitochondrial DNA variation. J Mol Evol, 33(6), 543–555. doi: 10.1007/BF02102807 [DOI] [PubMed] [Google Scholar]
- Mester JL, Ghosh R, Pesaran T, Huether R, Karam R, Hruska KS, … Eng C (2018). Gene-specific criteria for PTEN variant curation: Recommendations from the ClinGen PTEN Expert Panel. Hum Mutat, 39(11), 1581–1592. doi: 10.1002/humu.23636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Gomez D, Leipzig J, Shen L, Lott M, Stassen AP, Wallace DC, … Gai X (2015). Phy-Mer: a novel alignment-free and reference-independent mitochondrial haplogroup classifier. Bioinformatics, 31(8), 1310–1312. doi: 10.1093/bioinformatics/btu825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt V, Pitceathly RD, Turnbull DM, Taylor RW, Sweeney MG, Mudanohwo EE, … McFarland R (2013). The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m.3243A>G mutation--implications for diagnosis and management. J Neurol Neurosurg Psychiatry, 84(8), 936–938. doi: 10.1136/jnnp-2012-303528 [DOI] [PubMed] [Google Scholar]
- Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, … DiMauro S (2015). Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med, 17(9), 689–701. doi: 10.1038/gim.2014.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesole G, & Saccone C (2001). A novel method for estimating substitution rate variation among sites in a large dataset of homologous DNA sequences. Genetics, 157(2), 859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preste R, Vitale O, Clima R, Gasparre G, & Attimonelli M (2019). HmtVar: a new resource for human mitochondrial variations and pathogenicity data. Nucleic Acids Res, 47(D1), D1202–D1210. doi: 10.1093/nar/gky1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman J, Noronha A, Thiele I, & Rahman S (2017). Leigh map: A novel computational diagnostic resource for mitochondrial disease. Ann Neurol, 81(1), 9–16. doi: 10.1002/ana.24835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, … ClinGen. (2015). ClinGen--the Clinical Genome Resource. N Engl J Med, 372(23), 2235–2242. doi: 10.1056/NEJMsr1406261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 17(5), 405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius R, Cowley MJ, Riley L, Puttick C, Thorburn DR, & Christodoulou J (2019). Biparental inheritance of mitochondrial DNA in humans is not a common phenomenon. Genet Med. doi: 10.1038/s41436-019-0568-0 [DOI] [PubMed] [Google Scholar]
- Roshan M, Kabekkodu SP, Vijaya PH, Manjunath K, Graw J, Gopinath PM, & Satyamoorthy K (2012). Analysis of mitochondrial DNA variations in Indian patients with congenital cataract. Mol Vis, 18, 181–193. [PMC free article] [PubMed] [Google Scholar]
- Santorsola M, Calabrese C, Girolimetti G, Diroma MA, Gasparre G, & Attimonelli M (2016). A multi-parametric workflow for the prioritization of mitochondrial DNA variants of clinical interest. Hum Genet, 135(1), 121–136. doi: 10.1007/s00439-015-1615-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr TG, Ballinger SW, Gan YY, Hodge JA, Merriwether DA, Lawrence DN, … Wallace DC (1990). Amerindian mitochondrial DNAs have rare Asian mutations at high frequencies, suggesting they derived from four primary maternal lineages. Am J Hum Genet, 46(3), 613–623. [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, & Vissing J (2002). Paternal inheritance of mitochondrial DNA. N Engl J Med, 347(8), 576–580. doi: 10.1056/NEJMoa020350 [DOI] [PubMed] [Google Scholar]
- Shen L, Attimonelli M, Bai R, Lott MT, Wallace DC, Falk MJ, & Gai X (2018). MSeqDR mvTool: A mitochondrial DNA Web and API resource for comprehensive variant annotation, universal nomenclature collation, and reference genome conversion. Hum Mutat. doi: 10.1002/humu.23422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Diroma MA, Gonzalez M, Navarro-Gomez D, Leipzig J, Lott MT, … Gai X (2016). MSeqDR: A Centralized Knowledge Repository and Bioinformatics Web Resource to Facilitate Genomic Investigations in Mitochondrial Disease. Hum Mutat, 37(6), 540–548. doi: 10.1002/humu.22974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, McCormick EM, Muraresku CC, Falk MJ, & Gai X (2020). Clinical Bioinformatics in Precise Diagnosis of Mitochondrial Disease. Clin Lab Med, 40(2), 149–161. doi: 10.1016/j.cll.2020.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, & Wallace DC (1990). Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell, 61(6), 931–937. [DOI] [PubMed] [Google Scholar]
- Sonney S, Leipzig J, Lott MT, Zhang S, Procaccio V, Wallace DC, & Sondheimer N (2017). Predicting the pathogenicity of novel variants in mitochondrial tRNA with MitoTIP. PLoS Comput Biol, 13(12), e1005867. doi: 10.1371/journal.pcbi.1005867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, … Cooper DN (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet, 136(6), 665–677. doi: 10.1007/s00439-017-1779-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JB, & Chinnery PF (2015). The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet, 16(9), 530–542. doi: 10.1038/nrg3966 [DOI] [PubMed] [Google Scholar]
- Thorburn DR, Rahman J, & Rahman S (1993). Mitochondrial DNA-Associated Leigh Syndrome and NARP In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A(Eds.), GeneReviews((R)). Seattle (WA). [PubMed] [Google Scholar]
- Torroni A, Campos Y, Rengo C, Sellitto D, Achilli A, Magri C, … Scozzari R (2003). Mitochondrial DNA haplogroups do not play a role in the variable phenotypic presentation of the A3243G mutation. Am J Hum Genet, 72(4), 1005–1012. doi: 10.1086/373936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triska P, Kaneva K, Merkurjev D, Sohail N, Falk MJ, Triche TJ, … Gai X (2019). Landscape of Germline and Somatic Mitochondrial DNA Mutations in Pediatric Malignancies. Cancer Res, 79(7), 1318–1330. doi: 10.1158/0008-5472.CAN-18-2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trounce I, Neill S, & Wallace DC (1994). Cytoplasmic transfer of the mtDNA nt 8993 T-->G (ATP6) point mutation associated with Leigh syndrome into mtDNA-less cells demonstrates cosegregation with a decrease in state III respiration and ADP/O ratio. Proc Natl Acad Sci U S A, 91(18), 8334–8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale O, Preste R, Palmisano D, & Attimonelli M (2020). A data and text mining pipeline to annotate human mitochondrial variants with functional and clinical information. Mol Genet Genomic Med, 8(2), e1085. doi: 10.1002/mgg3.1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker UA, Collins S, & Byrne E (1996). Respiratory chain encephalomyopathies: a diagnostic classification. Eur Neurol, 36(5), 260–267. doi: 10.1159/000117269 [DOI] [PubMed] [Google Scholar]
- Wallace DC, & Chalkia D (2013). Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol, 5(11), a021220. doi: 10.1101/cshperspect.a021220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Garrison K, & Knowler WC (1985). Dramatic founder effects in Amerindian mitochondrial DNAs. Am J Phys Anthropol, 68(2), 149–155. doi: 10.1002/ajpa.1330680202 [DOI] [PubMed] [Google Scholar]
- Wei W, Gomez-Duran A, Hudson G, & Chinnery PF (2017). Background sequence characteristics influence the occurrence and severity of disease-causing mtDNA mutations. PLoS Genet, 13(12), e1007126. doi: 10.1371/journal.pgen.1007126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Pagnamenta AT, Gleadall N, Sanchis-Juan A, Stephens J, Broxholme J, … BioResource, N. (2020). Nuclear-mitochondrial DNA segments resemble paternally inherited mitochondrial DNA in humans. Nat Commun, 11(1), 1740. doi: 10.1038/s41467-020-15336-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LC, Chen T, Wang J, Tang S, Schmitt ES, Landsverk M, … Craigen WJ (2020). Interpretation of mitochondrial tRNA variants. Genet Med, 22(5), 917–926. doi: 10.1038/s41436-019-0746-0 [DOI] [PubMed] [Google Scholar]
- Yarham JW, Al-Dosary M, Blakely EL, Alston CL, Taylor RW, Elson JL, & McFarland R (2011). A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum Mutat, 32(11), 1319–1325. doi: 10.1002/humu.21575 [DOI] [PubMed] [Google Scholar]
- Yu-Wai-Man P, & Chinnery PF (1993). Leber Hereditary Optic Neuropathy In Adam MP, Ardinger HH Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)). Seattle (WA). [Google Scholar]
- Zolkipli-Cunningham Z, Xiao R, Stoddart A, McCormick EM, Holberts A, Burrill N, … Falk MJ (2018). Mitochondrial disease patient motivations and barriers to participate in clinical trials. PLoS One, 13(5), e0197513. doi: 10.1371/journal.pone.0197513 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.