

Abstract
Background:
Cerebral amyloid angiopathy (CAA) is a vascular neuropathology commonly reported in non-cognitively impaired (NCI), mild cognitive impairment, and Alzheimer’s disease (AD) brains. However, it is unknown whether similar findings are present in non-demented elderly subjects.
Objective:
This study determined the association between CAA and cognition among elderly NCI subjects with varying levels of AD pathology.
Methods:
Data from 182 cases that received a diagnosis of NCI at their first clinical assessment were obtained from the Rush Religious Orders study (RROS). A cognitive composite score was used to measure cognitive decline. CAA was dichotomized as present or absent. Cases were also dichotomized according to CERAD neuropathological diagnosis and Braak staging. A mixed model-repeated measures analysis assessed decline on the cognitive composite score.
Results:
CAA, alone, was not associated with cognitive decline [−0.87 (95% CI: −3.33, 1.58), p = 0.49]. However, among those with CAA, the High CERAD group had significantly greater decline relative to the Low CERAD group [−4.08 (95% CI: −7.10, −1.06), p = 0.008]. The High and Low CERAD groups were not significantly different [−1.77 (95% CI: −6.14, 2.60), p = 0.43] in those without CAA. Composite score decline in the High and Low Braak groups with [−1.32 (95% CI: −4.40, 1.75), p = 0.40] or without [0.27 (95% CI: −4.01, 4.56), p = 0.90] CAA was not significantly different.
Conclusion:
The current data shows that an interaction between CAA and plaque load is associated with greater decline on a cognitive composite score used to test non-cognitively impaired elderly participants in AD prevention trials.
Keywords: Amyloid, dementia, episodic memory, executive function, neuropathology, preclinical, prevention, vascular
INTRODUCTION
Cerebral amyloid angiopathy (CAA) is a common neuropathological finding within the cortex and leptomeninges resulting from amyloid deposition in cerebral blood vessels [1, 2]. Although CAA is associated with cortical amyloid-β (Aβ) deposition in Alzheimer’s disease (AD) [3, 4], it can occur independent of parenchymal Aβ plaque pathology [4, 5]. In a large community-based sample, CAA was present in over 75% of individuals at autopsy and was associated with an increased risk of incident dementia, independent of AD pathology [6]. Similar to parenchymal amyloid deposition, APOE ε4 allele status is a risk factor for both the presence and severity of CAA [1, 7]. The presence of CAA with amyloid plaque pathology is associated with an earlier age of progression to mild/moderate AD [8] and greater medial temporal lobe atrophy [9].
Although parenchymal amyloid deposition and CAA co-occur at a high rate [3–5, 9, 10], others report that individuals with severe CAA had significantly lower diffuse plaque loads than those without CAA suggesting that parenchymal and vascular amyloid clearance mechanisms may be independent processes [11]. In addition, a significant association between CAA and tau pathology has also been reported in AD [12, 13]. For example, significantly higher tau deposition was found around arteries relative to those without amyloid deposits [14]. Other findings suggest that tau-dependent degradation of brain vasculature results in an increased susceptibility to amyloid deposition, leading to the development of CAA [15]. Several studies have reported that increased CAA prevalence and severity is associated with those carrying the APOE ε4 allele [7, 14–16], suggesting an interaction between APOE allele status, CAA, and possibly tau and amyloid pathology.
Studies indicate that CAA, in the presence of AD pathology, exacerbates cognitive decline and is associated with clinical AD [17]. However, others report that vascular and amyloid pathologies have additive, not synergistic, effects on cognition in non-cognitively impaired (NCI) older individuals [18]. In terms of domain-specific cognitive performance, CAA is associated with decreased executive function in individuals with mild cognitive impairment (MCI), AD, and stroke [19]. CAA also affects episodic memory, semantic memory, and perceptual speed domains even after adjusting for the presence of dementia [6]. However, others have reported that CAA has only a minor effect on cognitive trajectories [20]. Interestingly, neuroimaging findings have shown that white matter vascular changes are associated with decreased functional connectivity in regions of the frontal cortex that support executive function in NCI cases [21].
Although findings indicate that CAA has a significant effect on cognition in the presence of AD pathology [6, 18–24], its interaction with measures of cognition employed in AD prevention trials has not been thoroughly investigated in individuals that died with a premortem clinical diagnosis of NCI but upon postmortem evaluation displayed AD pathology. Therefore, understanding the interaction between CAA and AD lesions upon cognitive outcomes used in AD prevention trials in NCI cases may aid in discerning treatment and placebo group differences. Therefore the aim of the present study is to determine the effect of CAA on longitudinal changes in a cognitive composite score similar to that used in AD prevention trials among cognitively intact elderly individuals with varying levels of AD pathology.
METHODS
Data examined was derived from 182 older deceased persons who were classified as NCI at their initial clinical evaluation upon entering the Rush Religious Order Study (RROS). Ninety eight individuals remained NCI at last testing within 12 months prior to death, while the remaining 84 progressed to MCI (n = 40) or AD (n = 44) (see Table 1). Among those who progressed to MCI, 13 were classified as amnestic and 27 were classified as non-amnestic. Previous work by our group has shown that plaque and tangle pathology does not differ significantly between amnestic and non-amnestic MCI subjects in this cohort [25]. The RROS participants [26, 27] had no coexisting clinical, cerebrovascular, or neurological conditions judged to contribute to cognitive impairment at their last clinical evaluation [26, 27], agreed to an annual clinical evaluation, signed an informed consent and an Anatomic Gift Act donating their brains at time of death. Data from these subjects have been used in numerous clinical pathological studies supported by our ongoing NIA program project grant entitled the “Neurobiology of Mild Cognitive Impairment in the Elderly” (PO1AG14449). At the time of these studies, individuals were chosen from all available RROS participants (n = 663) that came to autopsy during a rolling admission [26]. In addition, those taking anticholinesterases or medication for depression were also excluded from this study. The Human Investigation Committee of Rush University Medical Center approved this study.
Table 1.
Demographic, cognitive, and postmortem data by cerebral amyloid angiopathy status
| CAA – Present | CAA – Absent | p | |
|---|---|---|---|
| N | 137 | 45 | na |
| Gender (M/F) | 55/82 | 22/23 | 0.30 |
| APOE ε4 (Carrier/Non-Carrier) | 36/101 | 6/39 | 0.07 |
| Final Clinical Diagnosis (NCI/MCI/AD) | 70/31/36 | 28/9/8 | 0.24 |
| Age at Baseline (y) | 78.03 ± 6.13 | 75.37 ± 6.11 | 0.01 |
| Age at Death (y) | 86.67 ± 5.86 | 84.19 ± 6.03 | 0.02 |
| Education (y) | 18.34 ± 3.23 | 18.09 ± 3.89 | 0.66 |
| MMSE at Baseline | 28.40 ± 1.54 | 28.89 ± 1.07 | 0.05 |
| MMSE Proximate to Autopsy | 25.10 ± 5.71 | 26.02 ± 5.43 | 0.34 |
| Baseline Composite Score | 67.91 ± 6.84 | 69.55 ± 6.85 | 0.16 |
| Length of Follow-Up (y) | 7.67 ± 4.22 | 7.53 ± 4.58 | 0.84 |
| Interval Between Last Clinic Visit and Autopsy (y) | 0.68 ± 0.60 | 0.72 ± 0.50 | 0.69 |
| Postmortem Interval (h) | 6.93 ± 4.68 | 5.88 ± 3.08 | 0.09 |
| Brain Weight at Autopsy (g) | 1,197.79 ± 138.40 | 1,197.40 ± 147.78 | 0.99 |
Mean ± standard deviation.
Clinical evaluation
Each of the participants underwent a uniform, structured and clinical evaluation performed by a neurologist and a trained neuropsychological test technician [26, 28]. Medications used by the subjects within the previous fourteen days of the examination were reviewed and classified. A neurologist reviewed the medical history, medication use, neurologic examination, results of cognitive performance testing, and the neuropsychologist’s opinion of cognitive impairment and dementia. Each participant was evaluated in their home, emphasizing findings deemed clinically relevant. Clinical diagnostic classification was performed as described previously [22, 28]. At time of death individuals with a clinical diagnosis of MCI or AD were classified as progressors and NCI subjects were categorized as non-progressors. Petersen criteria [29] was used to diagnose MCI while NINCDS-ADRDA criteria were used to diagnose AD [30]. Individuals classified as NCI had cognitive test scores within normal limits for age and education and had no significant functional deficits.
Tissue preparation and neuropathological diagnosis
Brain accruement and processing was described previously [28, 31, 32]. Briefly, each brain was cut into 1 cm thick coronal slabs using a brain slice apparatus and hemisected. One hemisphere was immersion fixed in 4% paraformaldehyde (24–72 h) and cryoprotected (10% glycerol and 2% dimethyl sulfoxide in phosphate buffer solution) until processing for immunohistochemistry.
Diagnostic blocks (mid-frontal, superior temporal, entorhinal cortex, hippocampus, inferior parietal cortex, basal ganglia, thalamus, and substantia nigra) from the opposite hemisphere were paraffin embedded and sectionedat 6 μm. Examination for cerebral infarctions was conducted as described previously [33]. Bielschowsky silver stain was used to visualize neuritic plaques, diffuse plaques, and neurofibrillary tangles (NFTs). Sections were also immunostained for Aβ using antibody M0872 (1:100; Dako, CA) raised against Aβ1–40 and Aβ1–42. Paired helical filament tau (AT8; 1:800, Covance) immunohistochemistry was also used to label NFTs. Neuropathological diagnoses were determined according to CERAD [34] and Braak staging [35] as recommended by the NIA-Reagan criteria [36]. Exclusion criteria included mixed dementias, Parkinson’s disease, frontotemporal dementia, argyrophilic grain disease, vascular dementia, hippocampal sclerosis, stroke, and Lewy body disease. Cortical and subcortical Lewy bodies pathology was detected using α-synuclein immunohistochemistry as previously described [37] and scored semi-quantitatively according to the severity and anatomical distribution, separating brainstem predominant, limbic/transitional and diffuse neocortical types, depending on the anatomical distribution of α-synuclein-positivity [38, 39]. A board-certified neuropathologist or trained technician, blinded to clinical diagnosis, counted number of neuritic plaques and diffuse plaques revealed by Bielschowsky silver stain and tau immunohistochemistry using the phosphorylated paired helical filament tau AT8 marker for NFTs, respectively, in one square mm area (100x magnification) per cortical region as reported previously [28, 40]. CAA was assessed using a semiquantitative summary from the midfrontal, midtemporal, parietal, and calcarine cortices. Paraffin-embedded sections were immunostained using the beta-amyloid monoclonal antibody 6F/3D antibody (ThermoFisher). For each region, meningeal and parenchymal vessels were assessed for amyloid deposition and scored from 0 to 4, where:0 = no deposition, 1 = scattered segmental but no circumferential deposition, 2 = circumferential deposition up to 10 vessels, 3 = circumferential deposition up to 75% of the region, 4 = circumferential deposition over 75% of the total region. CAA score for each region was the maximum of the meningeal and parenchymal CAA scores. Scores were averaged across regions and summarized as a continuous measure of CAA pathology. CAA severity was then converted to a semi-quantitative summary and graded on a 0 to 3 scale based on the neuropathologist’s examination (0 = None, 1 = Mild, 2 = Moderate, 3 = Severe) [6].
Cognitive composite score
The composite score was comprised of eight cognitive tests that included: CERAD Word List Delayed Recall, WMS-R Logical Memory (delayed recall), Category Fluency (Fruits and Animals), Symbol Digit Modalities Test, Ravens Progressive Matrices (9-item), Judgment of Line Orientation (15-item), MMSE Orientation to Time, and MMSE Orientation to Place. The composite score used in this study is based on that of Langbaum et al. [41], but was refined in order to reflect the selection of tests being used in an ongoing AD prevention trial [42]. The tests that comprise this composite score are the same, or are analogous to, those used in other composite scores [42–44]. Individual raw scores for each test were standardized to a 0 to 1 scale by subtracting the minimum possible score for a test from the raw score and then dividing by the difference of the maximum and minimum possible scores. Since the Category Fluency test does not have an established maximum score, two standard deviations above the mean was used as the maximum. This method has been applied previously for a similar cognitive composite score [40]. No adjustments for directionality were needed since lower scores are indicative of decreased performance for all tests. The standardized scores for each test were then summed and divided by eight (number of tests) to obtain an unweighted average. Finally, for scaling purposes standardized scores were multiplied by 100.
Statistical analysis
Between-group frequency differences for categorical variables were analyzed using the Chi-square test while between-group differences for continuous variables were compared with a two-sample t-test. CAA status was converted to a dichotomous (Absent/Present) variable (CAA-Absent = None; CAA-Present = Mild, Moderate, Severe). The sample was also grouped by pathology severity based on CERAD neuropathological diagnosis and Braak stage. CERAD diagnosis was dichotomized into Low and High groups where the Low group included those with the No AD diagnosis and the High group included the Possible, Probable, and Definite AD diagnoses. Braak stage was also dichotomized into Low (0 – II) and High (III – V) groups. There were no Braak stage VI cases in our sample cohort.
Mixed model repeated measures (MMRM) analysis was used to examine change from baseline differences on the composite score when the sample was stratified by CAA and pathology status (CERAD and Braak groups). In these analyses time was treated as a categorical variable and data were restricted to the first six visits for each subject (baseline plus five years of follow-up). This follow-up length was selected in order to approximate the duration of current AD prevention trials [40]. Unstructured covariance structure was attempted for all models. In the event that the models did not converge, autoregressive order 1[AR(1)] followed by variance components structures were used. Kenward-Roger approximation for degrees of freedom was used for all models. All models adjusted forage at baseline, gender, education, APOE ε4 carrier status, and baseline composite score. The primary outcome for each analysis was the least-squares group difference in composite score change from baseline.
T-tests and ANOVAs were carried out using SYSTAT 13.1 (SYSTAT Software Inc., San Jose, CA). MedCalc 17.5 was used for the ROC analyses (MedCalc Software, Belgium). SAS Enterprise Guide 6.1 (SAS Institute, Cary, NC) was used for the MMRM analyses. Statistical significance was set at p ≤ 0.05.
RESULTS
Demographic, cognitive, and neuropathological characteristics
Demographic characteristics stratified by CAA status are shown in Table 1. Gender (p = 0.30), APOE ε4 carrier status, (p = 0.07), and clinical diagnosis (p = 0.24) frequencies were not significantly different between groups. The CAA-Present (CAA-P) group had significantly higher age at baseline (p = 0.01) and age at death (p = 0.02). Baseline MMSE (p = 0.05) was significantly greater for the CAA-Absent (CAA-A) group, however, this difference was less than one point, which is not meaningful from a clinical standpoint. MMSE proximate to autopsy (p = 0.34) and baseline composite score (p = 0.16) were not significantly different. Interval between last clinic visit and autopsy (p = 0.69), postmortem interval (p = 0.09), and brain weight at autopsy (p = 0.99) also showed no significant group differences. Between-group differences for CERAD diagnosis (p < 0.001) and Braak stage (p < 0.001) were also noted with the CAA-P group having a significantly higher frequency of more severe pathological classifications (Table 2).
Table 2.
CERAD diagnosis and Braak stage frequencies by cerebral amyloid angiopathy status
| CAA – Present | CAA – Absent | p | |
|---|---|---|---|
| CERAD Neuropathological Diagnosis | <0.001 | ||
| No AD | 29 | 26 | |
| Possible AD | 12 | 7 | |
| Probable AD | 68 | 7 | |
| Definite AD | 28 | 5 | |
| Braak Stage | <0.001 | ||
| 0 | 2 | 0 | |
| I | 12 | 14 | |
| II | 15 | 8 | |
| III | 39 | 13 | |
| IV | 41 | 10 | |
| V | 28 | 0 |
MMRM analyses
The overall composite score change from baseline difference between the CAA-A and CAA-P groups was not statistically significant (−0.87, 95% CI: −3.33, 1.58, p = 0.49; Fig. 1), even after adjusting for CERAD diagnosis and Braak stage (−1.32, 95% CI: −3.84, 1.19, p = 0.30). A secondary MMRM analysis using a slightly different CAA dichotomization (CAA-A = None and Mild, CAA-P = Moderate and Severe) also yielded no significant group difference (0.66, 95% CI: −1.63, 2.94, p = 0.57). Within the CAA-A group, no significant difference between APOE ε4 carriers and non-carriers was noted (2.10, 95% CI: −3.63, 7.84; p = 0.47); however the CAA-P ε4 carriers had significantly greater decline relative to non-carriers (−3.94, 95% CI: −6.81, −1.07; p = 0.007).
Fig. 1.
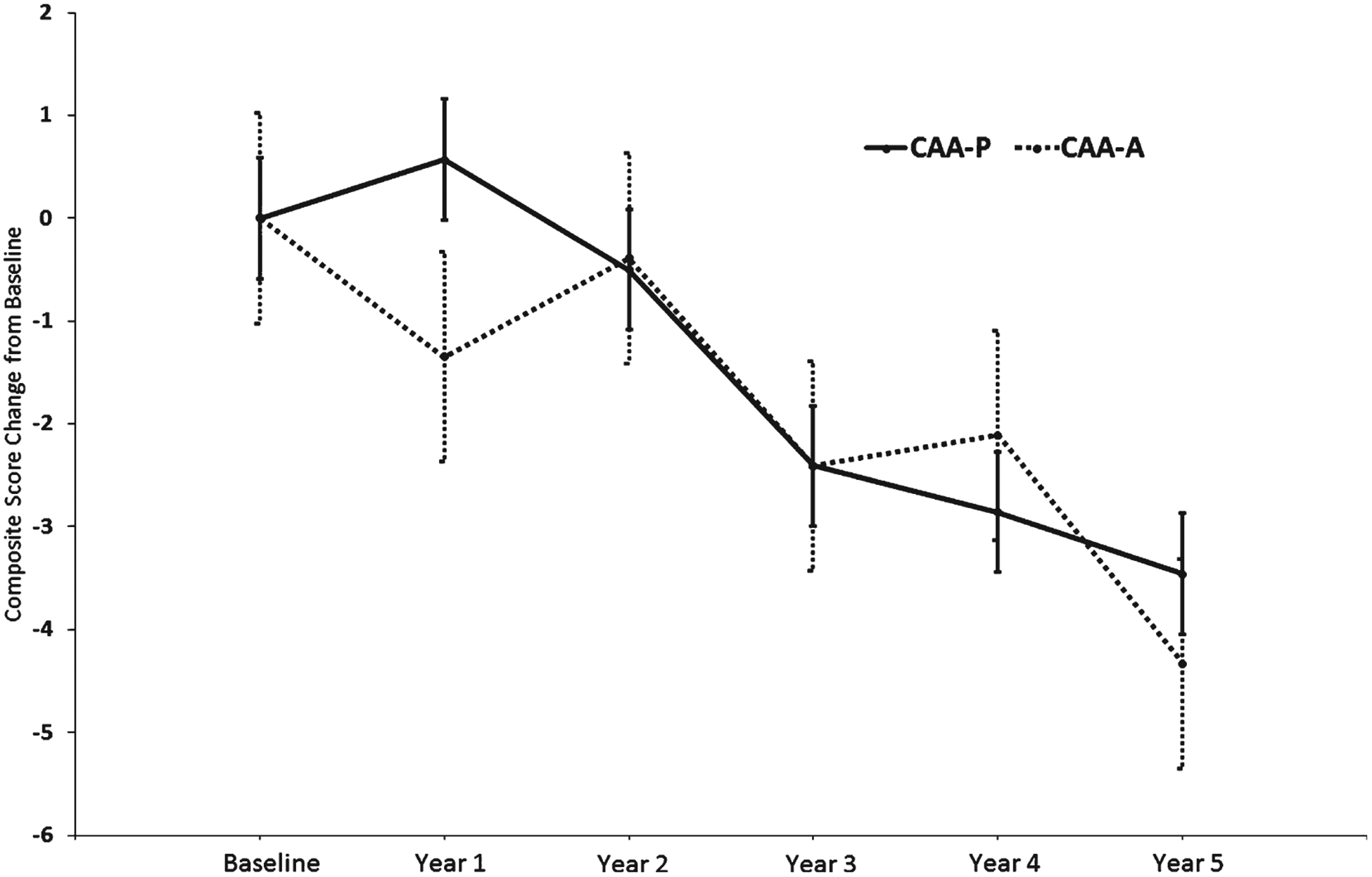
Composite Score Change from Baseline Estimates for CAA Status. Change from baseline group difference: −0.87, 95% CI (−3.33, 1.58), p = 0.49. Error bars represent the standard error.
Table 3 shows High and Low pathology group differences when stratified by CAA-P and CAA-A. Within the CAA-A group, the composite score change from baseline difference between High and Low CERAD groups was not significantly different (−1.61, 95% CI: −5.31, 2.09; p = 0.39; Fig. 2A). However, within the CAA-P group, the High CERAD group had significantly greater cognitive composite score change from baseline relative to the Low CERAD group (−3.92, 95% CI: −7.12, −0.71; p = 0.02; Fig. 2B). For the CAA-P group, the change from baseline difference between High and Low Braak groups was not significantly different (−1.43, 95% CI: −4.69, 1.83; p = 0.39). A similar result was also noted for the High/Low Braak group difference in the absence of CAA (0.90, 95% CI: −2.66, 4.45; p = 0.62).
Table 3.
MMRM-estimated cognitive composite score change from baseline results for CERAD and Braak stage by CAA status
| Δ from Baseline (within groups) | p | Δ from Low CERAD/Braak | p | |
|---|---|---|---|---|
| High versus Low CERAD | ||||
| CAA-A/High CERAD | −4.17 (−7.18, −1.17) | 0.007 | −1.61 (−5.31, 2.09) | 0.39 |
| CAA-A/ Low CERAD | −2.56 (−5.18, 0.06) | 0.06 | na | na |
| CAA-P/High CERAD | −4.30 (−5.75, −2.85) | <0.001 | −3.91 (−7.12, −0.71) | 0.02 |
| CAA-P/Low CERAD | −0.39 (−3.33, 2.56) | 0.80 | na | na |
| High versus Low Braak Stage | ||||
| CAA-A/High Braak | −2.82 (−5.71, 0.05) | 0.06 | 0.90 (−2.66, 4.45) | 0.50 |
| CAA-A / Low Braak | −3.72 (−6.32, −1.12) | 0.006 | na | na |
| CAA-P / High Braak | −3.89 (−5.33, −2.44) | <0.001 | −1.43 (−4.69, 1.83) | 0.39 |
| CAA-P / Low Braak | −2.46 (−5.46, 0.54) | 0.11 | na | na |
CAA-A, cerebral amyloid angiopathy absent; CAA-P, cerebral amyloid angiopathy present; Least-squares mean (95% Confidence Interval).
Fig. 2.
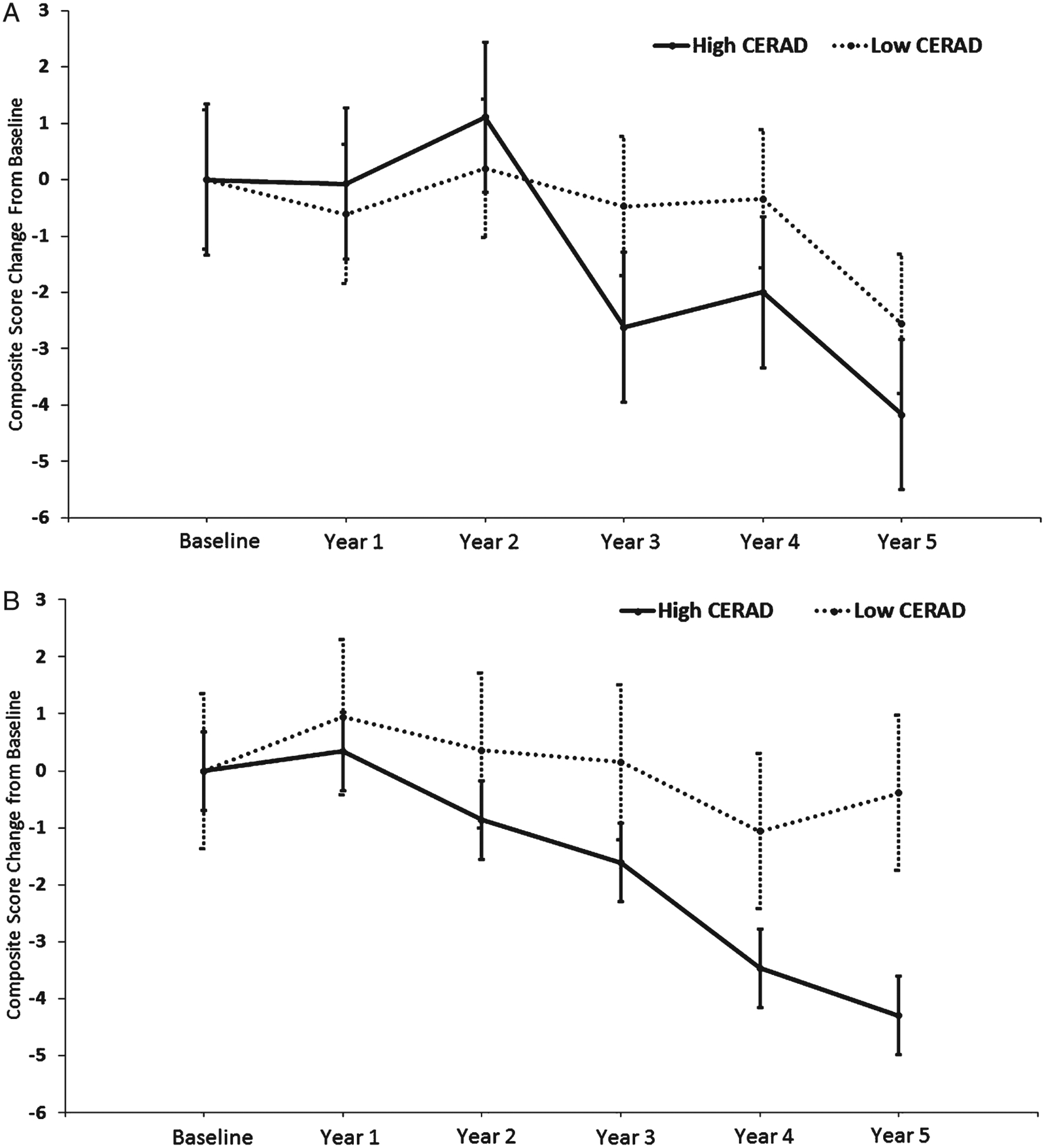
A Composite Score Change from Baseline Estimates for CERAD Status without CAA. Change from baseline group difference: −1.61, 95% CI (−5.31, 2.09), p = 0.39. Error bars represent the standard error. B Composite Score Change from Baseline Estimates for CERAD Status with CAA. Change from baseline group difference: −3.92, 95% CI (−7.12, −0.71), p = 0.02. Error bars represent the standard error.
Table 4 shows cognitive composite score change based on differences in CAA status stratified by Low and High pathology groups. For both the High and Low CERAD groups, the difference in composite score change from baseline between CAA-P and CAA-A was not statistically significant (−1.13, 95% CI: −4.83, 2.57; p = 0.55; Fig. 3A and −3.19, 95% CI: −6.95, 0.57; p = 0.10; Fig. 3B, respectively). For the High Braak group, the difference in change from baseline between CAA-P and CAA-A was not statistically significant (−0.82, 95% CI: −4.39, 2.75; p = 0.65). A similar result was found for the Low Braak group (−1.73, 95% CI: −5.24, 1.77; p = 0.33).
Table 4.
MMRM-estimated cognitive composite score change from baseline results for CAA status by CERAD and Braak stage
| Δ from Baseline (within groups) | p | Δ from CAA-P | p | |
|---|---|---|---|---|
| CERAD - CAA Absent versus Present | ||||
| High CERAD / CAA-A | −5.63 (−9.13, −2.13) | 0.002 | −1.13 (−5.31, 2.09) | 0.39 |
| High CERAD / CAA-P | −4.50 (−5.90, −3.09) | <0.001 | na | na |
| Low CERAD / CAA-A | −2.09 (−5.23, 1.04) | 0.19 | −3.19 (−6.95, 0.57) | 0.10 |
| Low CERAD / CAA-P | 1.10 (−1.97, 4.17) | 0.48 | na | na |
| Braak Stage - CAA Absent versus Present | ||||
| High Braak / CAA-A | −4.92 (−8.29, −1.55) | 0.004 | −0.82 (−4.39, 2.75) | 0.65 |
| High Braak / CAA-P | −4.09 (−5.53, −2.66) | <0.001 | na | na |
| Low Braak / CAA-A | −2.89 (−5.58, −0.02) | 0.04 | −1.73 (−5.24, 1.77) | 0.33 |
| Low Braak / CAA-P | −1.15 (−3.72, 1.42) | 0.38 | na | na |
CAA-A, cerebral amyloid angiopathy absent; CAA-P, cerebral amyloid angiopathy present; Least-squares mean (95% Confidence Interval).
Fig. 3.
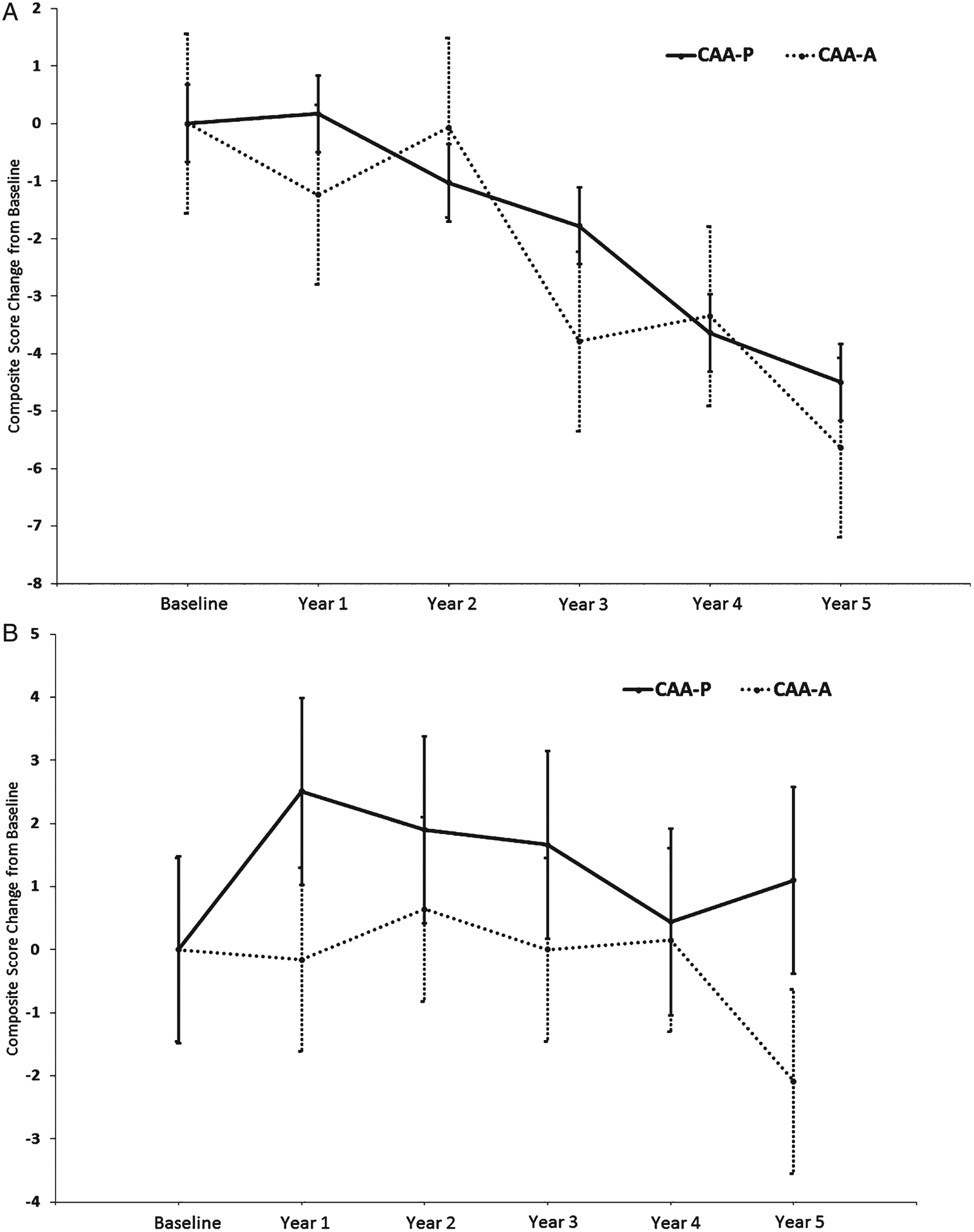
A Composite Score Change from Baseline Estimates for High CERAD by CAA Status. Change from baseline group difference: −1.13, 95% CI (−4.83, 2.57), p = 0.55. Error bars represent the standard error. B Composite Score Change from Baseline Estimates for Low CERAD by CAA Status. Change from baseline group difference: −3.19, 95% CI (−6.95, 0.57), p = 0.10. Error bars represent the standard error.
Episodic memory and executive function subanalysis
Given the significant composite score change from baseline difference between the High and Low CERAD groups for CAA-P (present study) combined with previous observations indicating that executive function decline is associated with the presence of CAA [12, 13], we further analyzed the episodic memory and executive function components, separately. Results of the episodic memory and executive function MMRM subanalyses are shown in Table 5. In the CAA-P group, the difference in change from baseline for episodic memory was statistically significant (−10.40, 95% CI: −17.10, −3.69; p = 0.002), but not for executive function (−1.52, 95% CI: −5.37, 2.33; p = 0.44).
Table 5.
MMRM-estimated episodic memory and executive function change from baseline results for CERAD status with CAA
| Δ from Baseline (within groups) | p | Δ from Low CERAD | p | |
|---|---|---|---|---|
| Episodic Memory Component | ||||
| CAA-P / High CERAD | −3.15 (−6.17, −0.13) | 0.04 | −10.40 (−17.10, −3.69) | 0.002 |
| CAA-P / Low CERAD | 7.25 (1.11, 13.39) | 0.02 | na | na |
| Executive Function Component | ||||
| CAA-P / High CERAD | −5.97 (−7.69 −4.26) | <0.001 | −1.52 (−5.37, 2.33) | 0.44 |
| CAA-P / Low CERAD | −4.45 (−7.98, −0.93) | 0.01 | na | na |
CAA-P, cerebral amyloid angiopathy present; Least-squares mean (95% Confidence Interval).
DISCUSSION
In the present study, we found that the presence of CAA and high parenchymal neuritic plaque pathology is associated with greater cognitive decline relative to those with low parenchymal neuritic plaque pathology and CAA in putative preclinical AD cases. The current finding shows an interactive effect of CAA and parenchymal plaque pathology on cognition. By contrast, others report either no interaction [18] or that this association is additive and not synergistic [17]. Here, we found that episodic memory decline drives the significant composite score change from baseline difference in the overall composite score within the CAA-P group with either high or low plaque pathology, whereas no significant differences were found for executive function. Early findings suggest that CAA differentially affects executive function [19, 21], while others indicate that CAA affects episodic memory [6, 22].
The present findings support and expand upon a previous investigation that examined the relationship of CAA to dementia using data also obtained from the RROS cohort [6]. In this regard, here we found a significant interaction between CAA and a cognition based upon the use of a cognitive composite score similar to one being used in an ongoing AD prevention trial [42], which provides greater ecological validity relative to the empirically-derived RROS cognitive domains [6, 26] as outcome measures. In addition, observation length approximated the treatment length of a prevention trial, and the statistical analysis treated time as a categorical and not a continuous variable, which is typical of clinical trial efficacy analyses. The inclusion of APOE ε4 carrier status as a covariate and the use of CERAD diagnosis and Braak stage in our analyses also differentiate our results from previous findings [6]. Since APOE ε4 status is associated with increased CAA [7, 14–16], the current results provide an enhanced estimate of the association between CAA and cognition beyond that previously reported [6]. Also, the separate analyses of CERAD diagnosis and Braak stage provide a more detailed investigation of the interaction between CAA, plaque and tangle pathology and cognitive performance.
The degree to which CAA influences different cognitive domains may depend on the severity of comorbid AD pathology, but different CAA phenotypes associated with AD [45] might underlie the observation that some cognitive domains are affected more than others. For example, sporadic CAA that occurs in the absence of parenchymal amyloid pathology shows a relatively high prevalence (20%) in autopsy studies [4, 6] and it is possible that these individuals may have a different clinical presentation relative to those with CAA and parenchymal amyloid pathology. In the current study, 29 individuals (16%) with CAA also had a ‘No AD’ CERAD diagnosis and did not display significant cognitive decline suggesting that CAA in the absence of parenchymal amyloid deposition results in a unique cognitive profile that differs from cognitive profiles related to parenchymal AD pathology. However, the cognitive decline observed in the CAA-P/High CERAD group may also reflect an interaction between CAA and neuritic dystrophy rather than CAA and parenchymal amyloid deposits. Since neuritic plaque load is used to determine CERAD diagnosis [34] and is correlated with cognitive decline [46] it is possible that the presence of dystrophic neurites associated with neuritic plaques contributed to the effects observed in this study.
The effect of CAA on cognitive outcomes must be considered in the context of data derived from clinical trials, particularly those involving anti-amyloid agents. In this regard, safety issues surrounding amyloid-related imaging abnormalities have been raised regarding the effect that amyloid-clearing therapies have on pre-existing CAA lesions and the subsequent risk of incident cerebrovascular hemorrhage [47]. A recent meta-analysis that pooled safety and efficacy data from several completed anti-amyloid clinical AD trials found no significant risk of cerebral microbleeds associated with treatment [48]. Despite this finding, it has been suggested that Aβ clearance via anti-amyloid therapies may lead to the development of new CAA lesions through the deposition of solubilized parenchymal Aβ on the walls of blood vessels while traversing the perivascular clearance pathway [49]. However, because Aβ is also cleared via microglial and transcytotic pathways mutli-drug therapeutic approaches have been proposed as a way to simultaneously target these amyloid clearance mechanisms [50]. By targeting these clearance pathways the deposition of solubilized Aβ upon the vasculature may be reduced, thereby minimizing the development of CAA lesions.
The impact of CAA on composite score change is most pronounced in subjects with high plaque pathology, suggesting that CAA and parenchymal plaque lesions act synergistically to affect cognitive decline. This relationship is supported by evidence from imaging studies [51–53] showing that both cognition and cortical network function are adversely affected by CAA. This finding suggests that individuals with imaging findings of amyloid positivity along with vascular abnormalities are likely to show the greatest decline on composite score outcomes and should be considered when designing and analyzing AD prevention trials. It is possible that measures from vascular-focused MRI sequences, such as fluid attenuation inversion recovery or gradient echo, could be used as covariates in efficacy analyses in order to account for their effect on cognitive outcomes. The effect, if any, this may have on statistical power and sample size estimates for AD prevention trials remains to be seen. Since amyloid-positivity is often used as part of inclusion criteria for AD prevention trials, the influence of parenchymal amyloid and CAA upon cognitive trajectories is an important factor to consider in the design and analysis of AD prevention trials.
A limitation of this study is its observational design and the ability to translate these findings to an interventional study. Although we designed the study and framed the results in the context of AD prevention trials, a direct inference from these results to those of intervention studies cannot be made. Another limitation of this study is the relatively small number of APOE ε4 carriers, particularly homozygous individuals, which may affect the associations reported here. Future studies with a greater balance of APOE ε4 carriers and non-carriers will extend these results. The subjects in this study were from a community-based group of highly educated retired clergy who had excellent health care and nutrition and were used in multiple clinical pathological [54, 55] and epidemiological investigations [26, 28, 29]. Individuals who volunteer may introduce bias by decreasing pathology but this is partially mitigated by high follow-up and autopsy rates of the RROS [32]. Strengths include uniform premortem clinical and postmortem pathological evaluation and that the final pathologic classification was performed without knowledge of the clinical evaluation. Additional strengths are the use of MMRM models, which are similar to those used in efficacy analyses of AD clinical trials [56–62]. Furthermore, the cognitive composite score used in the present investigation is similar to composites currently being used in ongoing AD prevention trials [42–44], which adds to the generalizability of our findings.
In summary, the present findings provide evidence of an interaction between CAA and neuritic plaque load, but not tangle pathology, on a cognitive outcome in preclinical AD. Since amyloid imaging is often used as an inclusion criterion for participation in AD prevention trials, understanding the extent that parenchymal amyloid and CAA contribute to observed treatment effects is critical to our understanding the results of these trials.
ACKNOWLEDGMENTS
We are indebted to the nuns, priests, and lay brothers who participated in the Rush Religious Orders Study and to the members of the Rush ADC.
This study was supported by grants PO1AG014449, RO1AG043375, P30AG010161, and P30AG042146 from the National Institute on Aging, National Institutes of Health and Barrow Neurological Institute Barrow and Beyond.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0765r1).
REFERENCES
- [1].Kumar-Sing S (2008) Cerebral amyloid angiopathy: Pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav 7(Suppl 1), 67–82. [DOI] [PubMed] [Google Scholar]
- [2].McLauchlan D, Malik GA, Robertson NP (2017) Cerebral amyloid angiopathy: Subtypes, treatment and role in cognitive impairment. J Neurol 264, 2184–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Attems J, Jellinger K (2014) The overlap between vascular disease and Alzheimer’s disease – lessons from pathology. BMC Med 12, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brenowitz WD, Nelson PT, Besser LM, Heller KB, Kukull WA (2015) Cerebral amyloid angiopathy and its co-occurrence with Alzheimer’s disease and other cerebrovascular neuropathologic changes. Neurobiol Aging 36, 2702–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kövari E, Herrmann FR, Hof PR, Bouras C (2013) The relationship between cerebral amyloid angiopathy and cortical microinfarcts in brain ageing and Alzheimer’s disease. Neuropathol Appl Neurobiol 39, 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Boyle PA, Yu L, Nag S, Leurgans S, Wilson RS, Bennett DA, Schneider JA (2015) Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 85, 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Caselli RJ, Walker D, Sue L, Sabbagh M, Beach T (2010) Amyloid load in nondemented brains correlates with APOE e4. Neurosci Lett 473, 168–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vidoni ED, Yeh H-W, Morris JK, Newell KL, Alqahtani A, Burns NC, Burns JM, Billinger SA (2016) Cerebral amyloid angiopathy is associated with earlier dementia onset in Alzheimer’s disease. Neurodegener Dis 16, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bos I, Verhey FR, Ramakers IHGB, Jacobs HIL, Soininen H, Freund-Levi Y, Hampel H, Tsolaki M, Wallin ÅK, van Buchem MA, Oleksik A, Verbeek MM, OldeRikkert M, van der Flier WM, Scheltens P, Aalten P, Visser PJ, Vos SJB (2017) Cerebrovascular and amyloid pathology in pre-dementia stages: The relationship with neurodegeneration and cognitive decline. Alzheimers Res Ther 9, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, part XV. Neurology 46, 1592–1596. [DOI] [PubMed] [Google Scholar]
- [11].Ringman JM, Sachs MC, Zhou Y, Monsell SE, Saver JL, Vinters HV (2014) Clinical predictors of severe cerebral amyloid angiopathy and influence of APOE genotype in persons with pathologically verified Alzheimer’s disease. JAMA Neurol 71, 878–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Williams S, Chalmers K, Wilcock GK, Love S (2005) Relationship of neurofibrillary pathology to cerebral amyloid angiopathy in Alzheimer’s disease. Neuropathol Appl Neurobiol 31, 414–421. [DOI] [PubMed] [Google Scholar]
- [13].Merlini M, Wanner D, Nitsch RM (2016) Tau pathology-dependent remodelling of cerebral arteriesprecedes Alzheimer’s disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol 131, 737–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chalmers K, Wilcock GK, Love S (2003) APOE ε4 influences the pathological phenotype of Alzheimer’s disease byfavouring cerebrovascular over parenchymal accumulation of Aβ protein. Neuropathol Appl Neurobiol 29, 231–238. [DOI] [PubMed] [Google Scholar]
- [15].Tanskanen M, Lindsberg PJ, Tienari PJ, Polvikoski T, Sulkava R, Verkkoniemi A, Rastas S, Paetau A, Kiuru-Enari S (2005) Cerebral amyloid angiopathy in a 95+cohort: Complement activation and apolipoprotein E (ApoE) genotype. Neuropathol Appl Neurobiol 31, 589–599. [DOI] [PubMed] [Google Scholar]
- [16].Yu L, Boyle PA, Nag S, Leurgans S, Buchman AS, Wilson RS, Arvanitakis Z, Farfel JM, De Jager PL, Bennett DA, Schneider JA (2015) APOE and cerebral amyloid angiopathy in community-dwelling older persons. Neurobiol Aging 36, 2946–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ (2002) Cerebral amyloid angiopathy and cognitive function: The HAAS autopsy study. Neurology 58, 1629–1634. [DOI] [PubMed] [Google Scholar]
- [18].Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Preboske GM, Kantarci K, Raman MR, Machulda MM, Mielke MM, Lowe VJ, Senjem ML, Gunter JL, Rocca WA, Roberts RO, Petersen RC, Jack CR Jr (2015) Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain 138(Pt 3), 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Case NF, Charlton A, Zwiers A, Batool S, McCreary CR, Hogan DB, Ismail Z, Zerna C, Coutts SB, Frayne R, Goodyear B, Haffenden A, Smith EE (2016) Cerebral amyloid angiopathy is associated with executive dysfunction and mild cognitive impairment. Stroke 47, 2010–2016. [DOI] [PubMed] [Google Scholar]
- [20].Kryscio RJ, Abner EL, Nelson PT, Bennett D, Schneider J, Yu L, Hemmy LS, Lim KO, Masaki K, Cairns N, Xiong C, Woltjer R, Dodge HH, Tyas S, Fardo DW, Lou W, Wan L, Schmitt FA (2016) The effect of vascular neuropathology on late-life cognition: Results from the SMART project. J Prev Alzheimers Dis 3, 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gold BT, Brown CA, Hakun JG, Shaw LM, Trojanowski JQ, Smith CD (2017) Clinically silent Alzheimer’s and vascular pathologies influence brain networks supporting executive function in healthy older adults. Neurobiol Aging 58, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA (2011) Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 69, 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Planton M, Raposo N, Albucher J-F, Pariente J (2017) Cerebral amyloid angiopathy-related cognitive impairment: The search for a specific neuropsychological pattern. Rev Neurol 173, 562–565. [DOI] [PubMed] [Google Scholar]
- [24].Schrag M, Kirshner H (2016) Neuropsychological effects of cerebral amyloid angiopathy. Curr Neurol Neurosci Rep 16, 76. [DOI] [PubMed] [Google Scholar]
- [25].Malek-Ahmadi M, Lu S, Chan Y, Perez SE, Chen K, Mufson EJ (2017) Cognitive domain dispersion association with Alzheimer’s pathology. J Alzheimers Dis 58, 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS (2012) Overview and findings from the Religious Order Study. Curr Alzheimer Res 9, 628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mufson EJ, Chen EY, Cochran EJ, Beckett LA, Bennett DA, Kordower JH (1999) Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp Neurol 158, 469–490. [DOI] [PubMed] [Google Scholar]
- [28].Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J (2002) Natural history of mild cognitive impairment in older persons. Neurology 59, 198–205. [DOI] [PubMed] [Google Scholar]
- [29].Petersen RC, Negash S (2008) Mild cognitive impairment: An overview. CNS Spect 13, 45–53. [DOI] [PubMed] [Google Scholar]
- [30].McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944. [DOI] [PubMed] [Google Scholar]
- [31].Bennett DA, Schneider JA, Buchman AS, Mendes de Leon C, Bienias JL, Wilson RS (2005) The Rush Memory and Aging Project: Study design and baseline characteristics of the study cohort. Neuroepidemiology 25, 163–175. [DOI] [PubMed] [Google Scholar]
- [32].Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE (2004) Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 61, 378–184. [DOI] [PubMed] [Google Scholar]
- [33].Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA (2009) The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis 18, 691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mirra SS (1997) The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: A commentary. Neurobiol Aging 18, S91–S94. [DOI] [PubMed] [Google Scholar]
- [35].Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- [36].The National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging 18, S1–S2. [PubMed] [Google Scholar]
- [37].Schneider JA, Arvanitakis Z, Bang W, Bennett DA (2007) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. [DOI] [PubMed] [Google Scholar]
- [38].McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology 47, 1113–1124. [DOI] [PubMed] [Google Scholar]
- [39].Jellinger KA (2009) A critical evaluation of current staging of [H9251]-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 1792, 730–740. [DOI] [PubMed] [Google Scholar]
- [40].Mitchell TW, Nissanov J, Han LY, Mufson EJ, Schneider JA, Cochran EJ, Bennett DA, Lee VM, Trojanowski JQ, Arnold SE (2000) Novel method to quantify neuropil threads in brains from elders with or without cognitive impairment. J Histochem Cytochem 48, 1627–1638. [DOI] [PubMed] [Google Scholar]
- [41].Langbaum JBS, Hendrix SB, Ayutyetnot N, Chen K, Fleisher AS, Shah RC, Bennett DA, Tariot PN, Reiman EM (2014) An empirically derived composite cognitive test score with improved power to track and evaluate treatments for preclinical Alzheimer’s disease. Alzheimers Dement 10, 666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].A study of CAD106 and CNP520 versus placebo in participants at risk for the onset of clinical symptoms of Alzheimer’s disease (Generation S1). Accessed electronically on 2/14/18 at https://clinicaltrials.gov/ct2/show/NCT02565511
- [43].Donohue MC, Sperling RA, Salmon DP, Rentz DM, Raman R, Thomas RG, Weiner M, Aisen PS, Australian Imaging, Biomarkers, and Lifestyle Flagship Study of Ageing; Alzheimer’s Disease Neuroimaging Initiative; Alzheimer’s Disease Cooperative Study (2014) The Preclinical Alzheimer Cognitive Composite: Measuring amyloid-related decline. JAMA Neurol 71, 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Coley N, Gallini A, Ousset PJ, Vellas B, Andrieu S; GuidAge study group (2016) Evaluating the clinical relevance of a cognitive composite outcome measure: An analysis of 1414 participants from the 5-year GuidAge Alzheimer’s prevention trial. Alzheimers Dement 12, 1216–1225. [DOI] [PubMed] [Google Scholar]
- [45].Allen N, Robinson AC, Snowden J, Davidson YS, Mann DMA (2014) Patterns of cerebral amyloid angiopathy define histopathological phenotypes in Alzheimer’s disease. Neuropathol Appl Neurobiol 40, 136–148. [DOI] [PubMed] [Google Scholar]
- [46].Malek-Ahmadi M, Perez SE, Chen K, Mufson EJ (2016) Neuritic and diffuse plaque associations with memory in non-cognitively impaired elderly. J Alzheimers Dis 53, 1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sperling RA, Jack CR Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ (2011) Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement 7, 367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Penninkilampi R, Brothers HM, Eslick GD (2017) Safety and efficacy of anti-amyloid-β immunotherapy in Alzheimer’s disease: A systematic review and meta-analysis. J Neuroimmune Pharmacol 12, 194–203. [DOI] [PubMed] [Google Scholar]
- [49].Yamada M (2012) Predicting cerebral amyloid angiopathy related intracerebral hemorrhages and other cerebrovascular disorders in Alzheimer’sdisease. Front Neurol 3, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Saito S, Ihara M (2014) New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front Aging Neurosci 6, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Poliakova T, Levin O, Arablinskiy A, Vasenina E, Zerr I (2016) Cerebral microbleeds in early Alzheimer’s disease. J Neurol 263, 1961–1968. [DOI] [PubMed] [Google Scholar]
- [52].Reijmer YD, Fotiadis P, Riley GA, Xiong L, Charidimou A, Boulouis G, Ayres AM, Schwab K, Rosand J, Gurol ME, Viswanathan A, Greenberg SM (2016) Progression of brain network alterations in cerebral amyloid angiopathy. Stroke 47, 2470–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Peréz JM, Evans AD, The Alzheimer’s Disease Neuroimaging Initiative (2016) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7, 11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mufson EJ, Binder L, Counts SE, DeKosky ST, de Toledo-Morrell L, Ginsberg SD, Ikonomovic MD, Perez SE, Scheff SW (2012) Mild cognitive impairment: Pathology and mechanisms. Acta Neuropathol 123, 13–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bennett DA, Wilson RS, Boyle PA, Buchman AS, Schneider JA (2012) Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol 72, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Turner RS, Thomas RG, Craft S, van Dyck CH, Mintzer J, Reynolds BA, Brewer JB, Rissman RA, Raman R, Aisen PS; Alzheimer’s Disease Cooperative Study (2015) A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 85, 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Abushakra S, Porsteinsson A, Scheltens P, Sadowsky C, Vellas B, Cummings J, Gauthier S, Hey JA, Power A, Wang P, Shen L, Tolar M (2017) Clinical effects of tramiprosate in APOE4/4 homozygous patients with mild Alzheimer’s disease suggest disease modification potential. J Prev Alzheimers Dis 4, 149–156. [DOI] [PubMed] [Google Scholar]
- [58].Wilkinson D, Windfeld K, Colding-Jørgensen E (2014) Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer’s disease (LADDER): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 13, 1092–1099. [DOI] [PubMed] [Google Scholar]
- [59].Gold M, Alderton C, Zvartau-Hind M, Egginton S, Saunders AM, Irizarry M, Craft S, Landreth G, Linnamägi U, Sawchak S (2010) Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: Results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 30, 131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Doraiswamy PM, Sperling RA, Johnson K, Reiman EM, Wong TZ, Sabbagh MN, Sadowsky CH, Fleisher AS, Carpenter A, Joshi AD, Lu M, Grundman M, Mintun MA, Skovronsky DA, Pontecorvo MJ, For the AV-45 A11 Study Group (2014) Florbetapir F 18 amyloid PET and 36-month cognitive decline: A prospective multicenter study. Mol Psychiatry 19, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H, Dowsett SA, Pontecorvo MJ, Dean RA, Demattos R (2016) Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement 12, 110–120. [DOI] [PubMed] [Google Scholar]
- [62].Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, Ashford E, Retout S, Hofmann C, Delmar P, Klein G, Andjelkovic M, Dubois B, Boada M, Blennow K, Santarelli L, Fontoura P; Scarlet RoAD Investigators (2017) A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 9, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]