

Abstract
Although inflammation is necessary during the early phases of tissue repair, persistent inflammation contributes to fibrosis. Acute tendon injuries often heal through a fibrotic mechanism, which impedes regeneration and functional recovery. Because inflammation mediated by nuclear factor κB (NF-κB) signaling is implicated in this process, we examined the spatial, temporal, and cell type–specific activation profile of canonical NF-κB signaling during tendon healing. NF-κB signaling was maintained through all phases of tendon healing in mice, including the remodeling phase, and tenocytes and myofibroblasts from the Scleraxis (Scx)- lineage were the predominant populations that retained NF-κB activation into the late stages of repair. We confirmed persistent NF-κB activation in myofibroblasts in human tendon scar tissue. Deleting the canonical NF-κB kinase, IKKβ, in Scx-lineage cells in mice increased apoptosis and the deposition of the matrix protein periostin during the late stages of tendon repair, suggesting that persistent NF-κB signaling may facilitate myofibroblast survival and fibrotic progression. Consistent with this, myofibroblasts in human tendon scar samples displayed enhanced prosurvival signaling compared to control tissue. Together, these data suggest that NF-κB may contribute to fibrotic tendon healing through both inflammation-dependent and inflammation-independent functions, such as NF-κB–mediated cell survival.
Introduction
Tendons are dense connective tissues that facilitate skeleton locomotion by transmitting forces from muscle contraction to bone. The tendon extracellular matrix (ECM), which is predominantly type I collagen (1) is produced and maintained by a heterogeneous population of tendon-resident fibroblasts (2, 3) known as tenocytes. Many, but not all, adult tenocytes arise from Scleraxis-lineage (ScxLin) cells (3–5). Scx encodes a basic helix-loop-helix protein that is required for normal tendon development and elongation (6, 7). Acute tendon injuries commonly heal with excessive scar tissue deposition, which complicates repair and causes functional deficits both during active healing and after completion of the healing process. Scar-mediated tendon healing is especially problematic in flexor tendons of the hand because excessive scar tissue deposition can greatly reduce range of motion, negatively impacting patient quality of life (8). Both chronic inflammation and the persistence of matrix-producing cells have been implicated as predominant drivers of scar-mediated healing (9–13). Although the cellular and molecular contributions during tendon healing are not well defined (14), it is clear that healing occurs through the activity of both tendon-resident tenocytes and extrinsic cells, including immune cells and bone marrow–derived cells (3, 4, 15–19), and that ScxLin-tenocytes migrate into the repair site and align along an organized cellular bridge (Fig. 1A)(3).
Fig. 1. Canonical NF-κB signaling persists throughout tendon healing and is activated in tendon cells and αSMA+ myofibroblasts.
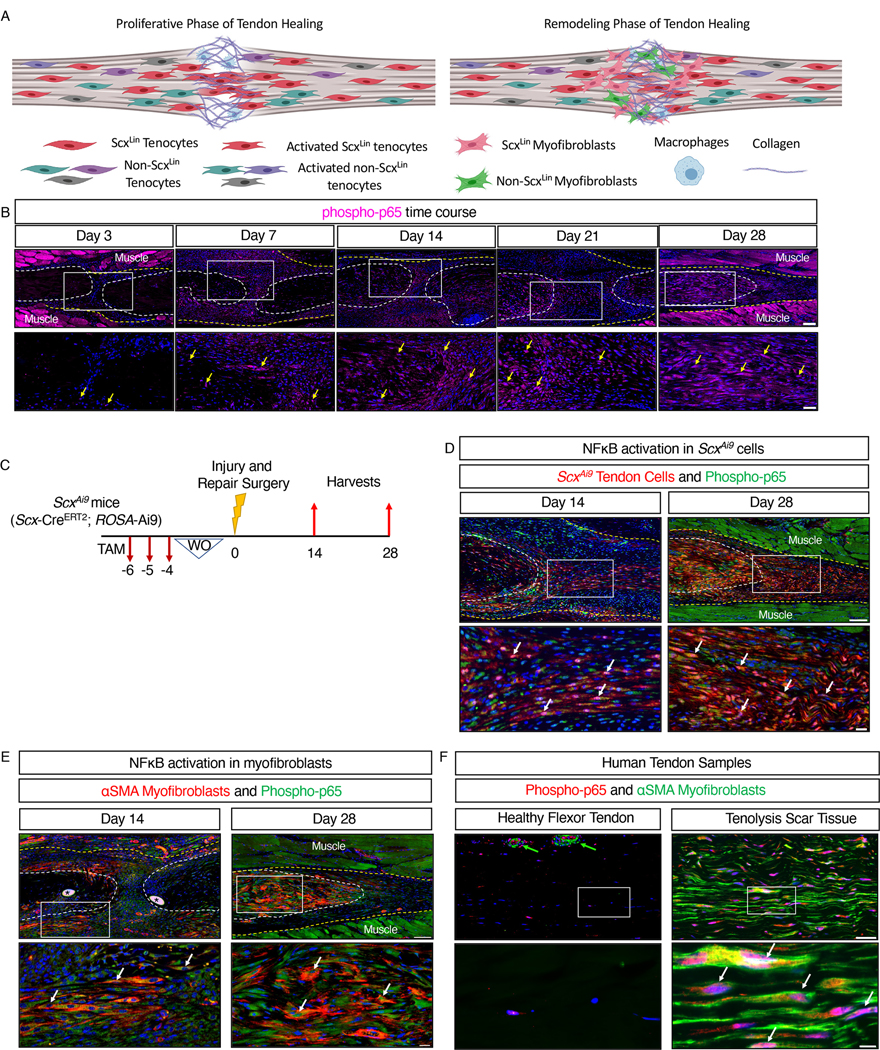
(A) Overview of the proliferative and remodeling phases of tendon healing. The heterogeneous population of tenocytes derived from both the Scx lineage (ScxLin) and other precursors becomes activated after injury, with ScxLin tenocytes migrating into the injury site to form an aligned cellular bridge across the damaged area. Some ScxLin and non- ScxLin cells then transdifferentiate into myofibroblasts, with both tenocytes and myofibroblasts interacting with macrophages throughout healing. Different color cells denote different subpopulations of cells. ScxLin cells are red, and ScxLin-derived myofibroblasts are pink. Image was created with Biorender.com (B) Immunofluorecence showing phosphorylated p65 (phospho-p65, pink) in sections of healing tendons from C57Bl/6J mice at 3, 7, 14, 21, and 28 days after tendon transection surgery. Yellow arrows indicate examples of positive stain. The tendon stubs are outlined by white dotted lines and the surrounding scar tissue by a yellow dotted line. n=3–4 mice per time point. Scale bars, 100 μM (upper) and 50 μM (lower). (C) Generation of ScxAi9 mice by crossing Scx-CreERT2 mice to the ROSA-Ai9 reporter line to enable tamoxifen (TAM)-inducible labeling of cells expressing Scx just prior to the time of tendon injury and repair with tdTomato (Ai9). (D and E) Co-immunofluorescence showing phospho-p65 (green) and tdTomato (red) in healing tendons of ScxAi9 mice (D) and phospho-p65 (green) and αSMA (red) in healing tendons of C57Bl/6J mice (E) at the indicated times after tendon transection. White arrows indicate examples of co-localization. Sutures are indicated by *. N=3–4 mice per timepoint. Scale bars, 100 μM (upper) and 20 μM (lower). (F) Co-immunofluorescence showing αSMA (green) and phospho-p65 (red) in human healthy flexor tendon and tenolysis scar tissue formed at and around flexor tendon repair. Co-localization is indicated by white arrows, and blood vessels by green arrows. N=2 healthy flexor tendons and N=2 tenolysis scar tissue samples. Scale bars, 50 μM (upper) and 20 μM (lower). For all images, nuclei were stained with DAPI (blue), and the white boxes in the upper images indicate the regions shown below in the higher magnification images. Auto-fluorescent muscle is labeled.
Modulating inflammation is a critical step in successful wound healing, which, like tendon healing, is characterized by overlapping phases of inflammation, cellular proliferation and matrix deposition, and matrix remodeling (20, 21). Appropriate induction of the inflammatory response is needed to initiate a proper healing response (22, 23), including the activation of tissue-resident cell populations (24), such as tenocytes. Cellular activation stimulates the conversion of fibroblasts and tenocytes to myofibroblasts (25, 26) (Fig. 1A), which facilitate successful tissue healing by producing, contracting, and remodeling the ECM (13, 27). However, if allowed to persist unabated, inflammation – and thereby myofibroblast activity – can result in tissue fibrosis. Myofibroblasts normally undergo apoptosis or revert to a more basal fibroblast state following the proliferative phase of tissue repair, but persistent inflammation maintains myofibroblasts in an activated state (4, 26–30)(Fig. 1A), which results in ECM production at a rate greater than that which tissue remodeling can accommodate, thereby leading to tissue fibrosis. Thus, understanding the mechanisms that regulate inflammation, myofibroblast activation, and myofibroblast persistence is critical to identifying therapeutic approaches to promote successful tissue healing.
The canonical nuclear factor kappa B (NF-κB) signaling cascade drives both inflammatory and pro-survival gene expression, greatly affecting the outcomes of healing tissues (31–34). In short, the NF-κB canonical heterodimer, most commonly a complex between proteins p50 and p65, is held in the cytoplasm by the inhibitor of κB (IκBα) protein, preventing translocation into the nucleus and subsequent transcriptional initiation of NF-κB-dependent genes. Through a wide variety of possible receptor-ligand interactions, the IκB kinase (IKK) complex becomes activated following the phosphorylation of its β subunit (IKKβ), which subsequently phosphorylates and marks IκBα for degradation, with IκBα degradation facilitating p50-p65 nuclear translocation and the initiation of NF-κB–dependent downstream gene expression.
Canonical NF-κB signaling has been implicated in human tendon disease (35, 36). Additionally, global overactivation of canonical NF-κB signaling in a mouse model of acute flexor tendon injury and repair drives deposition of collagen matrix at the repair site and increases the presence of myofibroblasts that produce α smooth muscle actin (αSMA+ myofibroblasts) and macrophages that produce the adhesion G protein–coupled receptor F4/80 (F4/80+ macrophages), two cell types that jointly drive scar tissue formation (9, 37). Global inhibition of canonical NF-κB signaling through either p65-directed small interfering RNAs (siRNAs) or a variety of pharmacological inhibitors result in decreased scar formation, diminished αSMA+ myofibroblast presence, and decreased collagen deposition (35). Additionally, cultured tendon fibroblasts treated with p65 inhibitors exhibit decreased proliferation and increased apoptosis, suggesting that canonical NF-κB signaling promotes tendon cell survival (35). Although informative, these studies do not address the specific roles of NF-κB signaling–positive cell populations during tendon healing in vivo. Abraham et al. demonstrated that IKKβ deletion in Scx-lineage (ScxLin) cells during healing of the enthesis, the transitional, connective region between tendon and bone, resulted in improved biomechanical property outcomes relative to wild-type mice (38); however, no study has assessed the cell type–specific effects of canonical NF-κB signaling on scar-mediated tendon healing in vivo.
Here, we tested the hypothesis that flexor tendon cells activate canonical NF-κB signaling following acute injury and repair, and that these NF-κB signaling–positive tendon cells contribute to excessive scar tissue formation during tendon healing. We characterized the spatial and temporal patterns of canonical NF-κB signaling in vivo and identified specific fibroblast populations that activate and maintain canonical NF-κB signaling, even after the inflammatory phase of healing has resolved, in both murine and human scar tissue, suggesting previously unappreciated, inflammation-independent roles for canonical NF-κB signaling in promoting the fibrotic response to tendon injury. Additionally, we deleted IKKβ specifically in ScxLin cells (IKKβKOScx) and examined the effects on the later phases of tendon healing using a combination of functional testing, analysis of signaling pathway activation, and histology.
Results
Canonical NF-κB signaling persists through all phases of fibrotic tendon healing
To assess the spatial localization and temporal pattern of canonical NF-κB signaling throughout flexor tendon healing, we stained healing tendons for phosphorylated p65 (phospho-p65), a marker of canonical NF-κB activation, at various time points from 3 to 28 days after surgical transection and suturing of the flexor digitorum longus (FDL) tendon (Fig. 1A). A small number of peripheral scar tissue cells were phospho-p65+ at day 3 post-surgery (Fig. 1B). The number of phospho-p65+ cells increased by day 7 post-surgery, primarily in cells residing within the developing scar tissue (Fig. 1B). The phospho-p65+ cells persisted at the injury site through days 14, 21, and 28 post-surgery (Fig. 1B), with a substantial portion of these cells residing within the native tendon stubs by D28. In addition, whereas most phospho-p65+ cells were observed in the tendon stub adjacent to the repair site, there was a progressive decrease in phospho-p65+ cells in the native tendon as the distance from the repair site increased (fig. S1. A to C). Collectively, these data suggest that sustained activation of canonical NF-κB signaling persists in tendon cell populations following the acute inflammatory phase of tendon healing. Although canonical NF-κB signaling is primarily associated with acute inflammation, this prolonged activation profile indicates a role for NF-κB in later phases of healing, and is consistent with other work demonstrating the involvement of other inflammatory pathways well into the later phases of tendon healing (39).
Canonical NF-κB signaling is maintained by adult tendon cells and myofibroblasts during tendon healing
NF-κB signaling during tissue repair is generally associated with the macrophages that drive the inflammatory phase of healing. The presence of phospho-p65+ cells within the tendon stubs following repair suggested that tendon fibroblast populations are an additional cell population that can activate canonical NF-κB signaling during healing. To assess canonical NF-κB activation specifically in adult tendon cells that express Scx just prior the time of tendon repair surgery, we used mice that express the fluorescent protein tdTomato (Ai9) in cells that express Scx at the time of tamoxifen (TAM) treatment (ScxAi9). ScxAi9 mice were injected with TAM for three days, with four days between the final injection and the day of surgery to ensure the washout of TAM (Fig. 1C). This ensured that all red-fluorescing cells were derived from adult tendon cells that expressed Scx prior to tendon injury and repair. Co-immunofluorescence between tdTomato (labels ScxAi9 cells) and phospho-p65 at days 14 and 28 post-surgery confirmed that ScxAi9 cells were a predominant population activating canonical NF-κB signaling during post-inflammatory phases of healing (Fig. 1D). We have previously shown that myofibroblasts, a contractile cell type that can contribute to both proper healing and pathologic tissue fibrosis (10, 40), are present at the healing tendon and originate in part from tendon cells (15, 41). Co-immunofluorescence between the myofibroblast marker αSMA and phospho-p65 demonstrated that myofibroblasts activated canonical NF-κB signaling at both days 14 and 28 post-surgery (Fig. 1E). To confirm these findings in human, we obtained both healthy human flexor tendon and scar tissue formed in response to flexor tendon repair surgery from patients undergoing a tenolysis procedure. Healthy human samples exhibited linear, organized collagen arrays with few cells, whereas tendon scar tissue samples exhibited high cellularity and an unorganized matrix (fig. S2). Healthy human flexor tendon did not contain αSMA+ myofibroblasts, and few phospho-p65+ tendon cells were observed within the body of the tendon (Fig. 1F). Blood vessels exhibited αSMA+ staining, as expected, with higher concentrations of phopho-p65+ cells existing around these blood vessels (Fig. 1F). Clinical tendon scar tissue obtained during tenolysis surgery contained phospho-p65-expressing αSMA+ myofibroblasts, demonstrating the clinical relevance of sustained NF-κB activation in tendon injury (Fig. 1F).
Adult ScxAi9 tendon cells contribute to cellular bridging and myofibroblast fate during tendon healing
We have previously shown that nearly 70% of adult flexor tendon cells express Scx (using the tamoxifen inducible Scx-CreERT2 mouse line; ScxAi9), and that these ScxAi9 cells account for a small percentage of αSMA+ myofibroblasts present at the healing tendon (41). However, the myofibroblast fate of ScxLin cells had not been assessed. Using the Scx-Cre; Rosa-Ai9 reporter mouse to label all Scx-lineage cells (ScxLinAi9), we found that the majority of tendon cells were ScxLinAi9+ in uninjured adult mouse flexor tendons (Fig. 2A). ScxLinAi9+ cells were found throughout the scar tissue at day 7 post-surgery (Fig. 2A). At days 14 and 21, ScxLinAi9+ cells had organized into a cellular bridge spanning the scar tissue between the tendon stubs (Fig. 2A), similar to what is seen with for ScxAi9+ cells during tendon repair (41). Virtually all αSMA+ myofibroblasts present at the healing tendon were ScxLinAi9+ and phospho-p65+ at days 14 and 28 post-surgery (Fig. 2B). Previous work has suggested that these are recruited αSMA+ cells that subsequently activate Scx expression after surgery (18). Therefore, Scx-Cre mice, which carry a transgene encoding Cre under the control of the Scx promoter, and therefore result in Cre-mediated recombination in all Scx-lineage cells regardless of active Scx expression, could be used to modulate canonical NF-κB signaling in both the tendon cell and myofibroblast populations during tendon healing to understand how canonical NF-κB signaling in fibroblast populations modulate healing.
Fig. 2. Scx-Cre mice can be used to target both tendon cells and myofibroblasts.
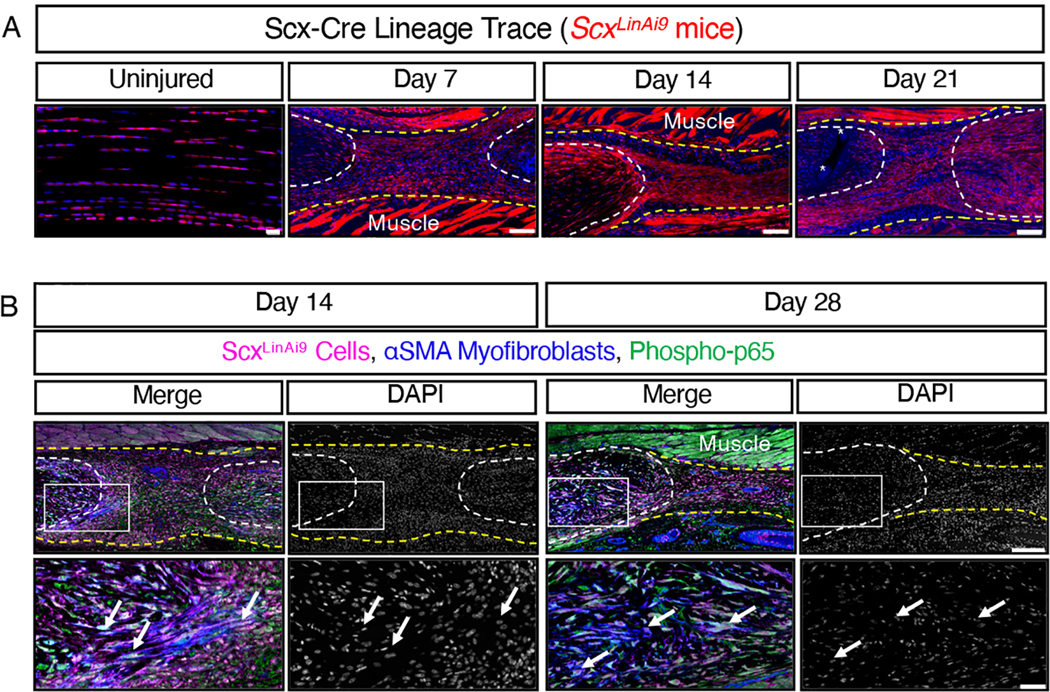
(A) Scx-Cre mice were crossed to ROSA-Ai9 reporter mice to label and trace Scx-lineage (ScxLinAi9) cells (red) during homeostasis (uninjured) and throughout healing at days 7, 14, and 21 post-surgery. N=3–4 mice per timepoint. Sutures labeled by *. Scale bars, 20 μM (uninjured) and 100 μM (post-surgery). (B) Co-immunofluorescence of tdTomato (labels ScxLinAi9 cells, pink), αSMA+ myofibroblasts (blue), and phospho-p65+ (green) cells in ScxLinAi9 mice at days 14 and 28 post-surgery. The white boxes in the upper images indicate the regions shown below in the higher magnification images. N=3–4 mice per timepoint. Scale bars, 200 μM (upper) and 50 μM (lower). For all images, nuclei were stained with DAPI (blue in (A), grey in (B)). Auto-fluorescent muscle is labeled. Examples of positive stain are indicated by white arrows. Tendon stubs are outlined by white dotted lines and scar tissue by yellow dotted line.
Reducing NF-κB signaling in tenocytes does not affect baseline tendon function or biomechanical properties
IKKβ is the predominant kinase driving downstream canonical NF-κB signaling (Fig. 3A). We generated mice that lacked IKKβ specifically in ScxLin cells (IKKβKOScx), that is, both copies of Ikbkb2, the gene encoding IKKβ, were deleted specifically in ScxLin cells. We also generated mice that had one copy of the Ikbkb gene deleted specifically in ScxLin cells (IKKβHetScx). Significantly decreased expression of Ikbkb, which encodes IKKβ, was detected in uninjured FDL tendons from IKKβKOScx mice relative to wild-type littermates (~64% decrease in Ikbkb gene expression) (Fig. 3B). Furthermore, uninjured FDL tendons from IKKβHetScx and IKKβKOScx mice exhibited decreased amounts of IKKβ protein compared to wild-type littermates (Fig. 3C). To confirm that IKKβKOScx did not significantly alter baseline tendon function or mechanical properties compared to wild type, uninjured tendons were assessed. We detected no significant differences between metatarsophalangeal (MTP) flexion angle (Fig. 3D), gliding resistance (Fig. 3E), stiffness (Fig. 3F), or maximum load at failure (Fig. 3G) between genotypes.
Fig. 3. IKKβKOScx does not negatively affect baseline tendon gliding function or biomechanical properties.

(A) IKKβ is a kinase that stimulates canonical NF-κB signaling and downstream gene expression by promoting the degradation of the inhibitor IκBα. (B) Quantitative PCR for Ikbkb in uninjured tendons from wild-type (WT) and IKKβKOScx mice normalized to β-actin expression. N=3–4 pooled samples per genotype, where each pooled sample consists of tendons from 3–4 mice. (C) Western blot for and quantification of IKKβ in protein extracted from uninjured tendons from wild-type, IKKβHetScx, and IKKβKOScx mice. N=2 independent experiments, with 3 mice per genotype pooled per independent experiment. Complete blots for one biological replicate are shown in fig. S3. (D to G) Measurement of metatarsophalangeal (MTP) joint flexion angle (D), gliding resistance (E), stiffness (F), and maximum load at failure (G) of uninjured tendons from wild-type and IKKβKOScx mice. N=10 mice per genotype. Students t-test was used to assess statistical significance between genotypes. *, p ≤ 0.05.
Reducing NF-κB signaling in tenocytes decreases JNK and Foxo3a signaling during tendon healing
IKKβ is involved in a variety of signaling cascades (42). We examined how IKKβKOScx affected the activation of several downstream signaling pathways relative to wild-type littermates during the later phases of tendon healing (days 14 and 28 post-surgery) by western blotting. IKKβKOScx tendon repairs exhibited less IKKβ protein relative to wild type, as expected (Fig. 4A). However, canonical NF-κB signaling (phospho-p65) was not substantially altered in IKKβKOScx healing tendons relative to wild type, which may suggest that diminution of phospho-p65 in ScxLin cells does not markedly decrease the overall amount of phospho-p65 in the healing tendon (Fig. 4B). Extracellular signal–regulated kinase (ERK) signaling was largely unaffected between genotypes (Fig. 4B). Jun N-terminal kinase (JNK) signaling appeared to be suppressed in IKKβKOScx healing tendons relative to wild-type, but phospho-p38 findings were inconclusive (Fig. 4C). IKKβKOScx healing tendons had less phospho-AKT at day 14 (Fig. 4D). β-Catenin signaling and phospho-S6K1 [an indicator of mechanistic target of rapamycin (mTOR signaling)] were inconclusive (Fig. 4D, E). To assess the effect of IKKβKOScx on apoptosis, we examined phospho-Foxo3a. Decreased amounts of phospho-Foxo3a were present in IKKβKOScx tendon repairs at 28 days post-surgery, indicative of increased apoptosis (Fig. 4E).
Fig. 4. IKKβKOScx affects several signaling cascades in addition to canonical NF-κB signaling.

Western blots and quantification for the indicated components of NF-κB, MAPK, mTOR, AKT, β-Catenin, and apoptosis signaling in tendons from wild-type and IKKβKOScx mice 14 and 28 days after tendon transection. (A) IKKβ. (B) Total p65, phosphorylated p65 (p-p65), total ERK1/2, and phosphorylated ERK1/2 (p-ERK1/2). (C) Total p38, phosphorylated p38 (p-p38), total JNK, and phosphorylated JNK (p-JNK). (D) Total AKT, phosphorylated AKT (p-AKT), total S6K1, and phosphorylated S6K1 (p-S6K1). (E) β-catenin and phosphorylated Foxo3a (p-Foxo3a). Complete blots for one biological replicate are shown in fig. S4. N=2 independent experiments, with 3 mice pooled per genotype per timepoint for each independent experiment.
Reducing NF-κB signaling in tenocytes promotes apoptosis during tendon healing
Given the decreased phospho-Foxo3a observed in IKKβKOScx tendon repairs, we further investigated the effects of IKKβ-dependent signaling on cell survival during healing, through assessment of the NF-κB–associated survival factors Bcl-2 and Bcl-xL in wild-type and IKKβKOScx healing tendons 28 days post-surgery. Although not significant, there was a trending decrease in Bcl-2+ cells in IKKβKOScx repairs relative to wild-type littermates (Fig. 5A). No difference in Bcl-xL was detected between genotypes (Fig. 5A). Cleaved caspase 3 was also assessed as a marker for apoptotic cells. There was a significant increase in cleaved caspase 3+ apoptotic cells in IKKβKOScx repairs relative to wild type (Fig. 5A). Together, these data suggest that IKKβ signaling in ScxLin mediates cell survival, possibly through NF-κB dependent Bcl-2 expression.
Fig. 5. IKKβKOScx increases apoptosis.

(A) Immunofluorescence and quantification of Bcl-2, Bcl-xL, and cleaved caspase 3 in tendons from wild-type and IKKβKOScx mice 28-days after tendon transection. N=4 mice per genotype. Scale bars, 100 μM (upper) and 50 μM (lower). Student’s t-test used to assess statistical significance between genotypes at a given time point. *, p ≤ 0.05. (B) Co-immunofluorescence of tdTomato (ScxLinAi9 cells, green), αSMA (red), and Bcl-2 (pink) in day 14 post-surgery mouse tendon. N=3–4 mice. Scale bars, 100 μM (upper) and 50 μM (lower). (C) Co-immunofluorescence of αSMA (red) and Bcl-2 (green) at day 28 post-surgery in C57Bl/6J mouse tendon. N=3–4 mice. Scale bars, 50 μM (upper) and 20 μM (lower). (D) Co-immunofluorescence of αSMA (green) and Bcl-2 (red) in healthy human flexor tendon and tenolysis scar tissue from flexor tendon. N=2 healthy human tendon samples and N=2 tenolysis scar tissue samples. Scale bars, 50 μM (upper) and 20 μM (lower). For all images, tendon is outlined by white dotted lines, scar tissue is outlined by a yellow dotted line, white arrows indicate examples of positive staining, and green arrows indicate auto-fluorescent blood cells. Boxes in the upper images indicate the regions shown below in higher magnification. Nuclei were stained with DAPI (blue).
We then confirmed that Bcl-2 was present in ScxLinAi9 αSMA+ myofibroblasts at 14 days post-surgery in the healing tendon (Fig. 5B). Additionally, Bcl-2 expression was maintained by αSMA+ myofibroblasts through day 28 post-surgery (Fig. 5C). Consistent with this, αSMA+ myofibroblasts in human tenolysis scar tissue also produced Bcl-2 (Fig. 5D), indicating both the clinical relevance of these findings and identifying pro-survival signaling in persistent myofibroblasts as a potential therapeutic target. A subset of tendon cells in healthy human flexor tendon contained Bcl-2+ cells, with higher concentrations of Bcl-2+ cells surrounding αSMA+ blood vessels (Fig. 5D).
Reducing NF-κB signaling in tenocytes negatively impacts gliding function in healed tendons
To determine the functional implications of IKKβ deletion in ScxLin cells during the later phases of tendon healing, we examined changes in tendon range of motion and mechanical properties. MTP range of motion was significantly decreased in IKKβKOScx healing tendons relative to wild-type littermates at 28 days post-surgery (wild type: 36.44 ± 4.04; KO: 25.61 ± 2.95) (Fig. 6A). Between days 14 and 28, wild-type tendon MTP range of motion improved 41.35%, whereas IKKβKOScx tendons only improved 3.31%. There was a trending increase in gliding resistance in IKKβKOScx tendons relative to wild-type littermates at day 28 post-surgery (wild type: 26.34 ± 4.89; KO: 47.36 ± 8.80) (Fig. 6B). Between days 14 and 28, wild-type tendon gliding resistance decreased 33.25%, and IKKβKOScx tendons increased by 7.20%. IKKβKOScx tendon repairs did not exhibit significantly different stiffness or maximum load at failure at either day 14 or 28 post-surgery relative to wild-type littermates (Fig. 6C,D).
Fig. 6. IKKβKOScx tendons heal with impaired gliding ability.

(A to D) Measurement of metatarsophalangeal (MTP) joint flexion angle (A), gliding resistance (B), stiffness (C), and maximum load at failure (D) of wild-type and IKKβKOScx tendons at 14 and 28 days post-surgery. N=10–14 mice per genotype per timepoint. Two-way ANOVA with Sidak’s multiple comparisons test was used to assess statistical significance between genotypes at a given time point. *, p ≤ 0.05.
Reducing NF-κB signaling in tenocytes increases periostin matrix deposition in healing tendons
Given that IKKβKOScx repairs exhibited trending deficits in gliding ability, we next assessed if tissue morphology or matrix deposition was affected. No differences in overall tissue morphology were detected using Alcian blue hematoxylin–orange G and Masson’s trichrome staining (Fig. 7A). Although there were no apparent differences in collagen content at day 14, IKKβKOScx repairs exhibited regions of disorganized collagen matrix at the tendon-muscle border that were not detected in wild-type littermates at day 28 (Fig. 7A), possibly suggesting less organized matrix. Staining for heat shock protein 47 (HSP47), a collagen chaperone protein indicative of collagen-producing cells, revealed no significant differences in the collagen-producing cells between genotypes at either time point (Fig. 7A, B). Although no differences in the pro-fibrotic matricellular protein periostin were detected at day 14, IKKβKOScx repairs had significantly increased periostin at day 28 post-surgery (Fig. 7A, B)(43).
Fig. 7. IKKβKOScx tendons heal with increased periostin deposition.
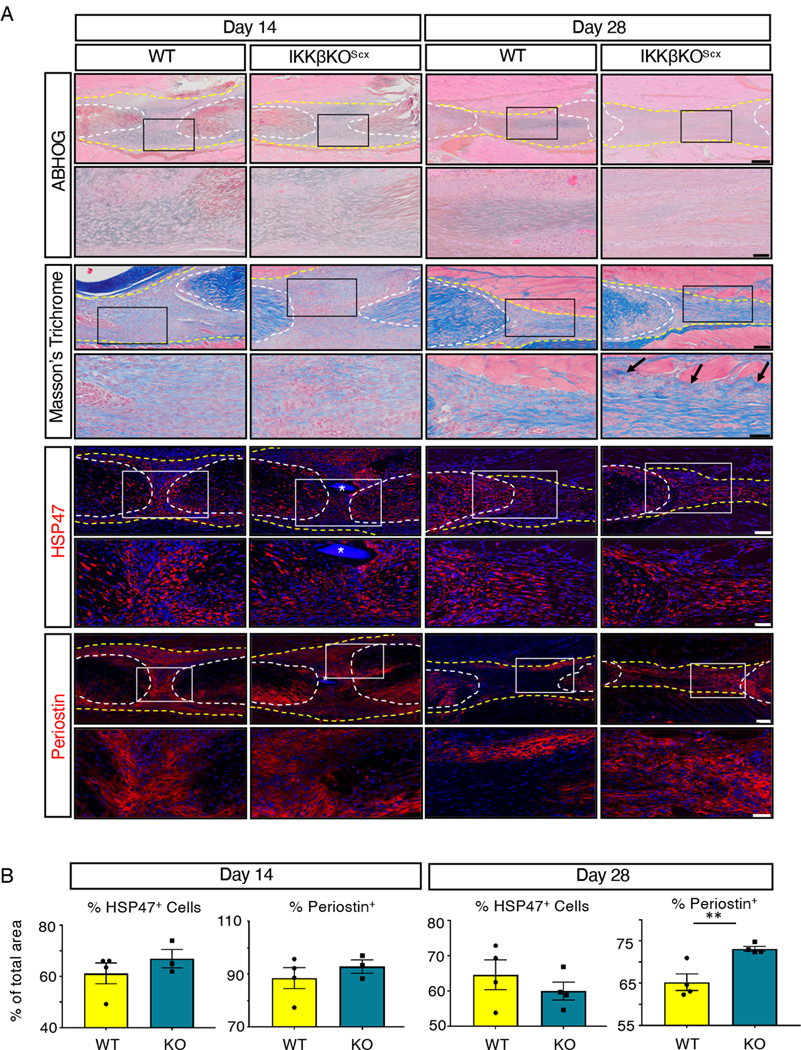
(A) Histology of wild -type and IKKβKOScx tendons 14 and 28 days post-surgery. Alcian blue/hematoxylin and Orange G stain (ABHOG) was used to assess overall morphology. Masson’s trichrome stain was used to visualize collagen content and organization. Example of collagen disorganization are indicated by black arrows. The collagen chaperone HSP47 and periostin matrix were assessed by immunofluorescence. For all images, tendon stubs are outlined by white dotted lines and scar tissue by yellow dotted lines, and boxes in the upper images indicate the regions shown below in the higher magnification images. Nuclei were stained with DAPI (blue). Sutures are labeled by *. N=3–4 mice per genotype per time point. Scale bars, 200 μM (upper) and 50 μM (lower) for ABHOG stains; 100 μM (upper) and 50 μM (lower) for Masson’s Trichrome stain, HSP47, and periostin. (B) Quantification of HSP47 and periostin at days 14 and 28 post-surgery. N=3–4 mice per genotype per timepoint. Student’s t-test used to assess statistical significance between genotypes at a given timepoint, except for day 14 HSP47 which required a Mann-Whitney test. ** indicates p ≤ 0.01.
Reducing NF-κB signaling in tenocytes does not affect macrophage recruitment during tendon healing
We previously described how global activation of canonical NF-κB signaling increases the presence of macrophages and myofibroblasts during the proliferative phase of tendon healing (37). Therefore, we assessed if IKKβKOScx resulted in altered presence of these cell types during the later phases of tendon healing. There was a trend towards increased F4/80+ macrophages in IKKβKOScx relative to wild type at 14 and 28 days post-surgery, but these differences were not significant (Fig. 8A,B). To examine whether IKKβKOScx affected macrophage polarization in vivo, we stained for the classically activated (M1) macrophage marker inducible nitric oxide synthase (iNOS) and alternatively activated (M2) marker interleukin-1 receptor antagonist (IL-1RA)(15). Although no differences in iNOS were detected between genotypes at either 14 or 28 days post-surgery, IL-1RA was significantly increased in IKKβKOScx day 28 repairs, suggesting that IKKβKOScx may drive macrophage polarization toward an M2-like, pro-resolving identity (Fig. 8A,B).
Fig. 8. IKKβKOScx enhances M2 macrophage polarization.
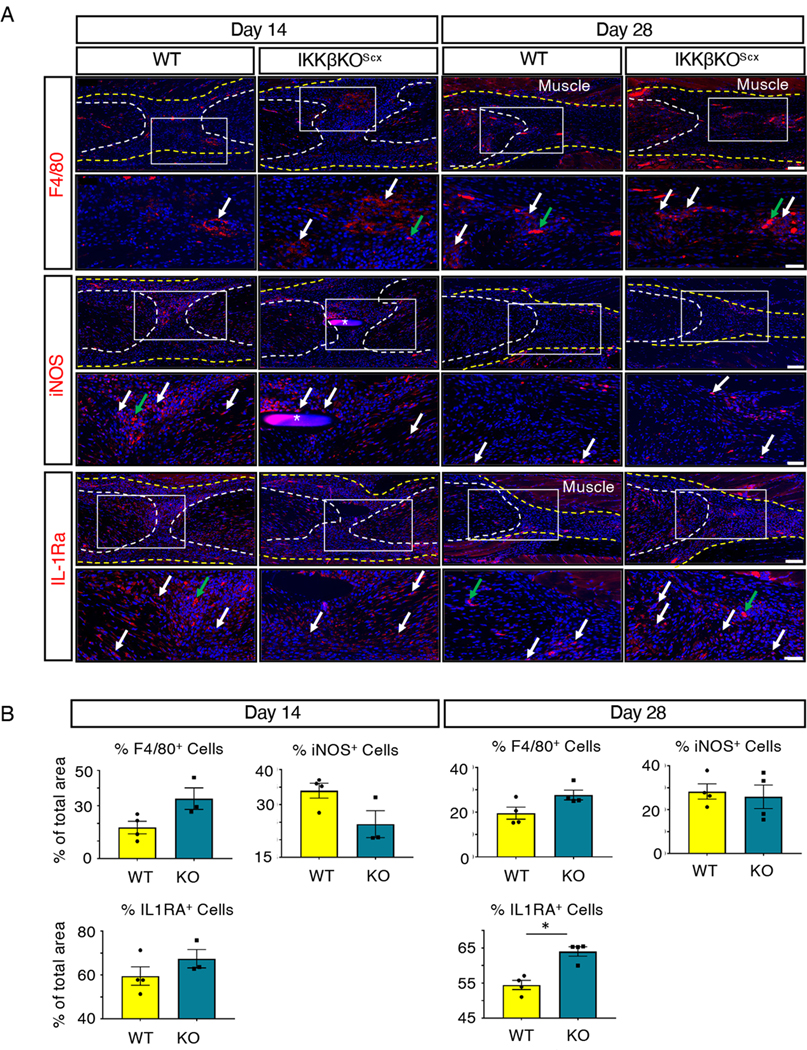
(A) Immunofluorescence of wild-type and IKKβKOScx tendons at 14 and 28 days post-surgery to assess F4/80+ macrophages, iNOS (M1 macrophages), and IL-1RA (M2 macrophages). Tendon stubs are outlined by white dotted lines and scar tissue by a yellow dotted line. White boxes in the upper images indicate the regions shown below in the higher magnification images. Examples of positive stain are indicated by white arrows, and examples of auto-fluorescent blood cells are indicated by green arrows. Auto-fluorescent muscle is labeled, and nuclei are stained with DAPI (blue). Sutures are labeled with *. N=3–4 mice per genotype per timepoint. Scale bars, 100 μM (upper) and 50 μM (lower). (B) Quantification of F4/80, iNOS, and IL-1RA fluorescence at days 14 and 28 post-surgery. N=3–4 mice per genotype per timepoint. Student’s t-test used to assess statistical significance between genotypes at a given time point, except for day 28 F4/80, day 14 iNOS, and day 28 IL-1RA, which required a Mann-Whitney test. *, p ≤ 0.05.
Reducing NF-κB signaling in tenocytes transiently increases fibroblast activation in healing tendons
IKKβKOScx healing tendons exhibited a significant, transient increase in αSMA+ myofibroblasts 14 days post-surgery relative to wild-type repairs that was no longer seen by day 28 (Fig. 9A,B). Notably, between days 14 and 28 the presence of αSMA+ cells decreased by 5.10% in wild-type repairs and 28.44% in IKKβKOScx repairs, highlighting a rapid decline in overall myofibroblast content in IKKβKOScx healing tendons. Because myofibroblasts are a type of activated fibroblast, we also examined other markers of fibroblast activation, including S100a4 (also known as fibroblast specific protein 1, FSP1), fibroblast activation protein (FAP), and vascular cell adhesion protein 1 (VCAM-1, also known as CD106). S100a4+ cell presence was also significantly increased at day 14 post-surgery in IKKβKOScx healing tendons relative to wild-type littermates, but this difference was no longer seen by day 28 (Fig. 9A,B). Both FAP+ and VCAM-1+ cells were increased at 14 days post-surgery in IKKβKOScx repairs relative to wild type (Fig. 9A,B). Although there were no differences in FAP+ cells between genotypes at day 28, there was a trend towards decreased VCAM-1+ cells by day 28 in IKKβKOScx repairs relative to wild type (Fig. 9A,B).
Fig. 9. IKKβKOScx drives increased fibroblast activation and myofibroblast presence at day 14 post-surgery relative to wild type.

(A) Immunofluorescence of wild-type and IKKβKOScx tendons at 14 and 28 days post-surgery to assess markers of myofibroblasts and activated fibroblasts: αSMA, S100a4, FAP, and VCAM-1. Tendon stubs are outlined by white dotted lines and scar tissue by a yellow dotted line. White boxes in the upper images indicate the regions shown below in the higher magnification images. Examples of positive stain are indicated by white arrows, and examples of auto-fluorescent blood cells and α-SMA+ blood vessels are indicated by green arrows. Auto-fluorescent muscle is labeled, and nuclei are stained with DAPI (blue). N=3–4 mice per genotype per timepoint. Scale bars, 100 μM (upper) and 50 μM (lower). (B) Quantification of αSMA, S100a4, FAP, and VCAM-1 fluorescence at days 14 and 28 post-surgery. N=3–4 mice per genotype per timepoint. Student’s t-test used to assess statistical significance between genotypes at a given time point, except for day 28 αSMA, which required a Mann-Whitney test. * indicates p ≤ 0.05, ** indicates p ≤ 0.01, **** indicates p ≤ 0.0001.
Discussion
Here, we characterized both the spatial localization and cell-type specific temporal activation profile of canonical NF-κB signaling following acute flexor tendon injury and repair. We determined that canonical NF-κB activation persisted beyond the acute inflammatory phase of healing in both mice and humans, remaining active in both ScxLin myofibroblasts during the later phases of healing in mice and in human myofibroblasts in clinical tendon scar samples long after the resolution of acute inflammation. IKKβ knockout in ScxLin cells decreased cell survival, and persistent NF-κB activation in wild-type myofibroblasts was associated with increased cell survival signaling in murine and human scar tissue. Contrary to our hypothesis, IKKβKOScx resulted in increased scar formation, increased periostin matrix deposition, and an increase in activated fibroblasts and αSMA+ myofibroblasts at day 14 post-surgery. However, a decline in myofibroblast content was observed by day 28, suggesting time-dependent functions of NF-κB during healing.
Inflammation has been implicated as the critical driver of excessive scar tissue formation, or fibrosis, in a variety of tissues (44). Inflammation is characterized by an influx of inflammatory cells such as neutrophils and macrophages. Macrophages have various functions during healing. Notably, they secrete cytokines and chemokines that recruit and activate fibroblast cell populations, the cells primarily responsible for laying down a provisional matrix that is necessary for repair (11). Macrophages are also involved in driving the differentiation of fibroblasts into myofibroblasts (9). Myofibroblasts have historically been viewed as a specialized contractile fibroblast cells that can lay down matrix and aid in wound closure but are deactivated or eliminated in later phases of healing when the provisional matrix is remodeled. Chronic persistence of either macrophages or myofibroblasts can result in continued, excessive deposition of matrix proteins, resulting in a fibrotic healing response (9, 10, 12). Although canonical NF-κB signaling is often viewed as an immune cell–activated inflammatory signaling cascade, it has previously been shown that fibroblast populations can activate NF-κB signaling to contribute to inflammation and promote the survival of myofibroblasts (33, 38), thereby promoting fibrotic healing. Here, we showed that tendon and ScxLin myofibroblast cell populations maintained the activation of canonical NF-κB signaling during post-inflammatory phases of healing, highlighting a previously unappreciated temporal contribution of NF-κB signaling to tendon healing. Additionally, we have provided evidence that IKKβ-mediated signaling influences cell survival during late phases of tendon healing, with IKKβKOScx resulting in increased apoptosis at day 28. It is also possible that the increased apoptosis is partly a result of increased proliferation in IKKβKOScx repairs, which results in an increased need for apoptosis, although we did not examine proliferation in this study. Our data also highlight the clinical importance of myofibroblast survival because we demonstrated sustained pro-survival signaling in myofibroblasts in both murine and human scar tissue. Future studies investigating the influence of canonical NF-κB signaling on tendon cell and myofibroblast survival as a mechanism of fibrotic healing is an important future direction to explore, especially considering our demonstration that phospho-p65+ and Bcl-2+ myofibroblasts were present in clinical samples of human tendon scar tissue (Figs. 1 and 5).
It has been demonstrated that IKKβ deletion in ScxLin cells results in improved biomechanical properties outcomes of the enthesis relative to wild type (38). The present study demonstrates that IKKβ deletion in the ScxLin population in vivo was not biomechanically beneficial to flexor tendon healing and drove a pro-fibrotic response. Collectively these data suggest that canonical NF-κB signaling in ScxLin tendon cells may have differential effects on the healing process that depend on the other cell populations with which ScxLin cells interact. That is, midsubstance tendon healing, as used in this study, involves a combination of tendon-derived and extrinsically derived cells such immune cells. Enthesis healing is further complicated by additional cellular contributions from the bone, such as osteoblasts and osteoclasts, and the fibrocartilaginous transition region, resulting in a much more diverse cellular environment. It has previously been shown that global overactivation of canonical NF-κB signaling drives increased matrix formation (37), whereas global inhibition of canonical NF-κB decreases matrix deposition and adhesion formation (35), suggesting that NF-κB signaling drives scar formation in vivo. However, our ScxLin-specific knockout of IKKβ resulted in an unexpected increase in scar formation. There are several possibilities that could explain this discrepancy. First, the increased presence of αSMA+ myofibroblasts at day 14 in IKKβKOScx repairs, relative to wild type, may drive excess scar formation during healing. IKKβKOScx cells in the healing tendon may also influence proliferation and recruitment patterns of both tendon cells and extrinsically recruited cells, resulting in a fundamentally altered cellular landscape in the repaired tendon compared to wild-type littermates. This in turn could lead to a more pro-fibrotic cellular environment during healing. Although changes in the acute inflammatory phase and proliferation were not assessed in this study, there were clear changes in cellular composition between wild-type and IKKβKOScx tendon repairs (Fig. 8). Additionally, the loss of IKKβ in ScxLin cells may affect intrinsic (cell autonomous) ScxLin cell function, resulting in altered proliferation or myofibroblast differentiation of ScxLin cells, which is likely to influence scar formation. Collectively, we believe the most likely explanation for the unexpected increase in scar tissue in the healing tendons of IKKβKOScx mice is that the non-inducible nature of the Scx-Cre used in this study prevented temporal control of IKKβ deletion. In future studies we plan to inducibly inhibit the sustained activation of canonical NF-κB signaling in ScxLin cells during late stages of healing, and we hypothesize that canonical NF-κB is primarily driving persistence and survival of matrix-producing cells such as myofibroblasts. We anticipate that temporally-controlled inhibition of NF-κB signaling at these later timepoints will allow healing to naturally occur through the inflammatory and proliferative phases with canonical NF-κB signaling only targeted during the remodeling phase.
Although no changes in baseline tendon function were observed between genotypes, some IKKβKOScx mice presented with developmental abnormalities. A small number of male and female IKKβKOScx mice exhibited skin with rough, scabby patches that became increasingly lesion-prone as they aged. Even though it has previously been reported that Scx is not detected in the skin (45), Scx is expressed in hair follicle cells (46) and keloid scars of the skin (47), which could account for the rare skin phenotypes that we observed. A couple of male IKKβKOScx mice presented with tumor-like growths around the groin area. This may be indicative of a testicular growth, because Scx has previously been implicated in Sertoli cell function and differentiation (48). These tendon-independent effects are an important reminder that although Scx is a highly useful tool for studying tendon development, biology, and healing, Scx is expressed by more than just tendon and ligament cells.
One limitation of this study is that the Scx-Cre targets all ScxLin cells, which contribute to other tissues besides tendons and ligaments, in a non-inducible manner. Initially this study crossed the IKKβF/F animals to the inducible Scx-CreERT2 mouse to generate conditional IKKβ knockout in Scx+ tendon cells. Unfortunately, insufficient DNA recombination was observed following TAM treatment, and so the Scx-Cre mouse was utilized instead. A second limitation is that IKKβ mediates signaling through more than just the canonical NF-κB cascade, complicating the interpretation of results. Moving forward, the p65F/F mouse (49), targeted by the inducible Scx-CreERT2, can be utilized to specifically examine canonical NF-κB signaling in flexor tendon healing at specific time points. Another limitation is that no time points beyond day 28 post-surgery were examined, making it impossible to know if IKKβKOScx tendon gliding function would continue to worsen over time or if the excess scar tissue at day 28 in IKKβKOScx healing tendons would eventually remodel successfully into stronger tendon with no negative impact on gliding function. Finally, although we demonstrate that nearly all phospho-p65 activation occurred in αSMA+ myofibroblasts from the ScxLin, there is also the possibility that non-ScxLin and non-tendon-derived myofibroblasts, such as those from the bone marrow (50, 51), can contribute to the healing process, and the role of those cells has not been assessed in this study.
Together, these data suggest that ScxLin cells activate canonical NF-κB signaling during late stages of tendon healing, whereas altered canonical NF-κB signaling by ScxLin cells influences scar-mediated tendon healing and cell survival. We also provide evidence that myofibroblasts within human tendon scar tissue have activated canonical NF-κB signaling and express the pro-survival protein Bcl-2, identifying the modulation of persistent myofibroblast survival as a potential target to inhibit fibrotic healing and promote tendon regeneration. Understanding the cellular contributors to scar-mediated tendon healing and the molecular mechanisms through which they drive scar formation is necessary to design effective therapies to combat excessive scar deposition and drive regenerative healing.
Materials and Methods
Study design
The goal of this study was to investigate the temporal and spatial activation pattern of canonical NF-κB signaling during tendon healing with the goal of delineating cell-type-specific effects of NF-κB knockdown on healing. Genetic mouse models were utilized to determine that tendon cells and myofibroblasts activate and maintain canonical NF-κB signaling throughout healing. We also confirmed that canonical NF-κB-expressing myofibroblasts are present in human tendon scar tissue. We hypothesized that ScxLin-specific knockdown of canonical NF-κB kinase IKKβ would result in decreased scar tissue formation during healing. We investigated these effects using gliding testing, tensile mechanical testing, histology, immunofluorescence, and western blot during the late proliferative, and remodeling phases of healing. Sample size and statistical methods were determined from previous studies and used to inform the experimental design of this work(15, 37, 41, 52) in order to determine significant differences in experimental outcomes. Samples for gliding and biomechanical testing, histology and immunofluorescence, and western blotting were randomized, and blinding occurred during analysis when possible. Specifics on statistical methods and sample size are included in the figure legends.
Animal ethics
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal procedures described were approved by the University Committee on Animal Research (UCAR) at the University of Rochester Medical Center.
Mice
Scx-CreERT2 and Scx-Cre mice were generously provided by Dr. Ronen Schweitzer. IKKβFlox/Flox mice (53) were generously provided by Dr. Michael Karin. C57Bl/6J (#000664) and ROSA-Ai9F/F (#007909) mice were obtained from the Jackson Laboratory. ROSA-Ai9F/F mice express TdTomato red fluorescence in the presence of Cre-mediated recombination, which excises a STOP cassette preventing TdTomato expression (54). Scx-CreERT2 and Scx-Cre mice were crossed to the ROSA-Ai9F/F reporter mouse to trace adult Scx+ tendon cells (ScxAi9) and Scx-lineage cells (ScxLinAi9), respectively. To label adult Scx+ tendon cells, ScxAi9 animals received three 100mg/kg i.p. tamoxifen (TAM) injections beginning six days prior to tendon injury and repair surgery with a four day washout period between the final injection and surgery, as previously described(41). ScxLinAi9 mice express Ai9 in all ScxLin cells, regardless of active Scx expression levels. In the IKKβF/F mouse strain, Cre-mediated recombination excises exons 6 and 7 from the Ikbkb locus, resulting in non-functional IKKβ protein(53). Scx-Cre mice were crossed to IKKβF/F animals to delete IKKβ specifically in ScxLin cells. IKKβKOScx, in which both Ikbkb alleles underwent excision were used for all studies. In Fig. 3C we also generated IKKβHetScx mice in which only one copy of Ikbkb was excised. All mouse studies were performed with 10–12 week-old male and female mice.
Human samples
Human scar tissue samples were collected from patients (N=2) undergoing tenolysis surgery (55). In short, many patients develop excess scar tissue formation in the months following a primary flexor tendon repair, which restricts digit movement. Many patients need to undergo a secondary surgical procedure, tenolysis, during which the surgeon removes excess scar tissue from around the repair site with the goal of improving digit range of motion. Collection of this human tenolysis scar tissue, as well as healthy human flexor tendon controls (N=2), was obtained following informed consent and was approved by the Research Subjects Review Board (RSRB) at the University of Rochester (Protocol #54231). Samples were collected from male patients undergoing tenolysis of digital flexor tendon scar tissue following an acute injury. Patient #1 was 37 years old and tendon scar tissue was collected during tenolysis surgery 12 months following initial flexor tendon repair surgery. Patient #2 was 69 years old and tendon scar tissue was collected during tenolysis surgery 21 months following initial flexor tendon repair surgery.
Flexor tendon repair
Mice underwent complete transection and repair of the flexor digitorum longus (FDL) tendon in the right hind paw as previously described (56). Prior to surgery, mice were injected with 15–20μg of sustained-release buprenorphine. Mice were anesthetized with Ketamine (60mg/kg) and Xylazine (4mg/kg). The FDL tendon was first transected at the myotendinous junction to reduce prevalence of rupture of the repair site and the skin was closed with a 5–0 suture. Immediately afterwards, a small incision was made on the posterior surface of the hind paw, the FDL tendon was located and completely transected. The tendon was repaired using 8–0 suture and the skin was closed with 5–0 suture. Following surgery, animals resumed prior cage activity, food intake, and water consumption.
RNA extraction and quantitative PCR
Total RNA was isolated from pools of intact FDL tendons (6–8 tendons per biological replicate) from groups of IKKβKOScx (n=3, biological replicates) and wild type mice (n=4, biological replicates) by column purification (TRIzol Reagent; Fisher Scientific 15596026, Direct-zol RNA Microprep kit; Zymo Research R2061) and converted to cDNA (qScript; Quantabio 84034). Primers were designed (Primer Express, Applied Biosystems) and validated to detect changes in the expression of exons 6–7 of the murine Ikbkb gene (deleted in the IKKβKOScx mice) (primer sequences provided in table S1) and used for qPCR (PerfeCTa SYBR Green; Quantabio 84069, CFX Connect Real-Time System; Bio-Rad). Relative expression was calculated using the ΔΔCt method and the housekeeping gene beta actin (Actb)(57). Data are shown relative to average Ikbkb expression in the wild type and IKKβKOScx groups.
Protein extraction and western blotting
Total protein was extracted from wild type, IKKβHetScx and IKKβKOScx littermate uninjured flexor tendons. Protein was also extracted from wild type and IKKβKOScx flexor tendons at days 14 and 28 post-surgery. Repaired samples consisted of the repair site and 1–2mm of native tendon on either side of the repair, with three tendons pooled per genotype per timepoint to ensure sufficient protein levels. Protein was extracted from two independent poolings of three tendons per genotype per time-point. As such, the protein contribution is reflective of both native tendon tissue and the surrounding scar. Tendons were homogenized using 0.5mm zirconium oxide beads and a Bullet Blender Gold Cell Disrupter (Next Advance Inc.), protein was extracted using radioimmunoprecipitation assay buffer (RIPA) buffer with added protease/phosphatase inhibitors, and 20μg were loaded into each well of a NuPAGE 4–12% Bis-Tris Gel (Invitrogen). Following transfer, membranes were probed with antibodies for IKKβ (Fig. 3C, 1:1000, Cat#: 2678, Cell Signaling), IKKβ (Fig. 4A, 1:500, Cat#: NB100–56509, Novus Biological), phospho-p65 (1:1000, Cat#: 3033, Cell Signaling), total p65 (1:1000, Cat#: 8242, Cell Signaling), phospho-ERK1/2 (1:1000, Cat#: 4377, Cell Signaling), total ERK1/2 (1:1000, Cat#: 9102, Cell Signaling), phospho-JNK (1:1000, Cat#: 4668, Cell Signaling), total JNK (1:1000, Cat#: 9252, Cell Signaling), phospho-p38 (1:1000, Cat#: 9216, Cell Signaling), total p38 (1:1000, Cat#: 9212, Cell Signaling), phospho-S6K1 (1:1000, Cat#: 9234, Cell Signaling), total S6K1 (1:1000, Cat#: 9202, Cell Signaling), phospho-AKT (1:1000, Cat#: 4060, Cell Signaling), total AKT (1:1000, Cat#: 9272, Cell Signaling), β-catenin (1:1000, Cat#: 610154, BD Biosciences), p-FOXO3A (1:500, Cat#: ab47285, Abcam) and GAPDH (1:1000, Cat#: 2118, Cell Signaling). Full length gels provided in figs. S3 and S4. Band intensity quantification performed using ImageJ. Protein was extracted from n=3 tendons per genotype per time-point.
Direct fluorescence in frozen sections
Hind paws from ScxLin mice were harvested from uninjured contralateral controls and at days 7, 14, and 21 post-surgery for frozen sectioning. Hind paws were fixed in 10% NBF for 24 hours at 4°C, decalcified in 14% EDTA for 4 days at 4°C, and processed in 30% sucrose for 24 hours at 4°C to cryo-protect the tissue. Samples were then embedded in Cryomatrix (ThermoFisher) and sectioned into 8μm sagittal sections using a cryotape-transfer method(58). Sections were mounted on glass slides using 1% chitosan in 0.25% acetic acid and counterstained with the nuclear stain DAPI. Endogenous fluorescence was imaged on a VS120 Virtual Slide Microscope (Olympus). Images are representative of 3–4 specimens per time-point and are presented in figure 2A.
Histology and immunofluorescence
Hind paws were harvested at days 14 and 28 post-surgery (n = 3–4 per genotype). Hind paws were fixed in 10% neutral buffered formalin (NBF) at room temperature for 72 hours, decalcified in Webb Jee EDTA (pH 7.2–7.4) for 1 week at room temperature, processed, and embedded in paraffin. All samples were cut into three-micron sagittal sections, followed by de-waxing and dehydrating for analysis. Sections were stained with Alcian blue/hematoxylin and Orange G (ABHOG) for tissue morphology and Masson’s Trichrome for collagen content. Chromogen immunohistochemistry was performed on paraffin sections for phospho-p65 (1:200, Cat#: ab86299, Abcam), with a rabbit polymer kit (#MP-7401, Vector Laboratories). For immunofluorescence, paraffin sections were probed with antibodies for phospho-p65 (1:200, Cat#: ab86299, Abcam), tdTomato (1:500, Cat#: AB8181, SICGEN), α-SMA-CY3 (1:200, Cat#: C6198, Sigma Life Sciences), α-SMA-FITC (1:500, Cat#: F3777, Sigma Life Sciences), periostin (1:250, Cat#: ab14041, Abcam), F4/80 (1:500, Cat#: sc-26643, Santa Cruz), S100a4 (1:2000, Cat#: ab197896, Abcam), Bcl2 (1:500, Cat#: ab182858, Abcam), Bcl-xL (1:400, Cat#: ab32370, Abcam), Cleaved Caspase 3 (1:100, Cat#: 9661, Cell Signaling), HSP47 (1:250, Cat#: ab109117, Abcam), iNOS (1:100, Cat#: ab15323, Abcam), IL-1RA (1:10000, Cat#: ab124962, Abcam), FAP (1:500, Cat#: ab53066, Abcam), and VCAM-1 (1:1000, Cat#: ab134047, Abcam) overnight at 4°C. The following secondary antibodies were used: donkey anti-rabbit 647 (1:200, Cat#: 711-606-152, Jackson ImmunoResearch), Donkey anti-rabbit FITC (1:200, Cat#: 711-;546-152, Jackson ImmunoResearch), Donkey anti-goat Rhodamine-Red-X (1:200, Cat#: 705-296-147, Jackson ImmunoResearch), Donkey anti-rabbit Rhodamine-Red-X (1:200, Cat#: 711-296-152, Jackson ImmunoResearch). Sections were counterstained with nuclear DAPI stain and imaged using a VS120 Virtual Slide Microscope (Olympus). These images are presented in Figures 1, 2B, 5, 7, 8, 9 and figures S1 and S2.
Quantification of fluorescence
Fluorescent images were processed using Visiopharm image analysis software v.6.7.0.2590 (Visiopharm, Hørsholm, Denmark). Automatic segmentation using a threshold classifier was used to define discrete cell populations based on fluorescent channel. Regions of interest were drawn to include the tendon stubs and the entire scar tissue region for processing. The area of each fluorescent signal was calculated, as was the area of each region of interest, and these values were used to determine overall percentage of each cell type. An n=3–4 was used for quantification.
Gliding function and biomechanical properties assessment
Tendon gliding function was assessed as previously described(59). Hindlimbs were harvested at the knee-joint and the proximal end of the FDL tendon was detached from the myotendinous junction. The FDL tendon was secured between two pieces of tape and was loaded incrementally with small weights ranging from 0 to 19g, with images captured after each load. Measurement of the flexion angle of the metatarsophalangeal (MTP) joint relative to the unloaded position were made using Image J. Gliding resistance was derived from the changes in MTP flexion angle over the range of applied loads. An increase in Gliding Resistance and decrease in MTP Flexion Angle is associated with restricted range of motion and increased scar tissue. Following gliding testing, the FDL tendon was released from the tarsal tunnel. The proximal end of the tendon and the toes of the hind paw was secured into an Instron 8841 uniaxial testing system (Instron Corporation, Norwood, MA). The tendon was loaded until failure at a rate of 30mm/minute. Eight-14 samples per genotype per time point were analyzed.
Statistical analyses
Quantitative data was analyzed in GraphPad Prism and is presented as mean ± standard error of the mean (SEM). Normality was assessed using the Shapiro-Wilk normality test for all data. Two-way ANOVA with Sidak’s multiple comparisons tests were used to determine significance between genotypes at a given time point for functional analyses (gliding and biomechanics). Students t-test were used to determine significance between normally distributed imaging and qPCR data. When data failed the normality test (HSP47 at D14, iNOS at D14, IL-1RA at D28, αSMA at D28, and F4/80 at D28), a Mann-Whitney (Wilcoxon) test was used was used as the primary statistical analysis. To account for the potential that the small sample sizes used for quantifying imaging data and qPCR could impact the Shapiro-Wilk normality test, p-values were derived from both unpaired student’s t-tests and Mann-Whitney (Wilcoxon) tests, and p-values are provided in table S2.
For all studies p values ≤ 0.05 were considered significant, with the following conventions: *=p ≤ 0.05, **=p ≤ 0.01, ***=p ≤ 0.001, ****=p ≤ 0.0001.
Supplementary Material
Fig. S1. Phosphorylated p65 staining is localized at injury site.
Fig. S2. Comparison of healthy and injured human flexor tendon.
Fig. S3. Full-length western blots for data from uninjured tendons.
Fig. S4. Full-length western blots for data from repaired tendons.
Table S1. qPCR primer sequences.
Table S2. P-values for student’s t-test and Mann-Whitney test when n ≤ 4.
Acknowledgements:
We would like to thank the Histology, Biochemistry and Molecular Imaging (HBMI) and the Biomechanics, Biomaterials and Multimodal Tissue Imaging (BBMTI) for technical assistance with the histology and biomechanical testing, respectively.
Funding: This work was supported in part by NIH/ NIAMS K01AR068386 and R01AR073169 (to AEL), F31 AR074815 (to KTB), T32 AR076950 (to AECN and HAA), and R01AR056696 (to HAA). The HBMI and BBMTI Cores were supported by NIH/ NIAMS P30AR069655. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and material availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Thorpe CT, Screen HR, Tendon Structure and Composition. Advances in experimental medicine and biology 920, 3–10 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Kendal AR, Layton T, Al-Mossawi H, Appleton L, Dakin S, Brown R, Loizou C, Rogers M, Sharp R, Carr A, Multi-omic single cell analysis resolves novel stromal cell populations in healthy and diseased human tendon. Sci Rep 10, 13939 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Best KT, Loiselle AE, Scleraxis lineage cells contribute to organized bridging tissue during tendon healing and identify a subpopulation of resident tendon cells. FASEB J 33, 8578–8587 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howell K, Chien C, Bell R, Laudier D, Tufa SF, Keene DR, Andarawis-Puri N, Huang AH, Novel Model of Tendon Regeneration Reveals Distinct Cell Mechanisms Underlying Regenerative and Fibrotic Tendon Healing. Sci Rep 7, 45238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshimoto Y, Takimoto A, Watanabe H, Hiraki Y, Kondoh G, Shukunami C, Scleraxis is required for maturation of tissue domains for proper integration of the musculoskeletal system. Sci Rep 7, 45010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murchison ND, Price BA, Conner DA, Keene DR, Olson EN, Tabin CJ, Schweitzer R, Regulation of tendon differentiation by scleraxis distinguishes force-transmitting tendons from muscle-anchoring tendons. Development, (2007). [DOI] [PubMed] [Google Scholar]
- 7.Huang AH, Watson SS, Wang L, Baker BM, Akiyama H, Brigande JV, Schweitzer R, Requirement for scleraxis in the recruitment of mesenchymal progenitors during embryonic tendon elongation. Development 146, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loiselle AE, Kelly M, Hammert WC, Biological Augmentation of Flexor Tendon Repair: A Challenging Cellular Landscape. The Journal of hand surgery 41, 144–149; quiz 149 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Pakshir P, Hinz B, The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix biology : journal of the International Society for Matrix Biology, (2018). [DOI] [PubMed] [Google Scholar]
- 10.Hinz B, Lagares D, Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nature reviews. Rheumatology 16, 11–31 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wynn TA, Barron L, Macrophages: master regulators of inflammation and fibrosis. Seminars in liver disease 30, 245–257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pradere J-P, Kluwe J, De Minicis S, Jiao J-J, Gwak G-Y, Dapito DH, Jang M-K, Guenther ND, Mederacke I, Friedman R, Dragomir A-C, Aloman C, Schwabe RF, Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wynn TA, Vannella KM, Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nichols AEC, Best KT, Loiselle AE, The cellular basis of fibrotic tendon healing: challenges and opportunities. Translational research : the journal of laboratory and clinical medicine 209, 156–168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ackerman JE, Nichols AE, Studentsova V, Best KT, Knapp E, Loiselle AE, Cell non-autonomous functions of S100a4 drive fibrotic tendon healing. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loiselle AE, Frisch BJ, Wolenski M, Jacobson JA, Calvi LM, Schwarz EM, Awad HA, O’Keefe RJ, Bone marrow-derived matrix metalloproteinase-9 is associated with fibrous adhesion formation after murine flexor tendon injury. PloS one 7, e40602 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kajikawa Y, Morihara T, Watanabe N, Sakamoto H, Matsuda K, Kobayashi M, Oshima Y, Yoshida A, Kawata M, Kubo T, GFP chimeric models exhibited a biphasic pattern of mesenchymal cell invasion in tendon healing. J Cell Physiol 210, 684–691 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Dyment NA, Hagiwara Y, Matthews BG, Li Y, Kalajzic I, Rowe DW, Lineage tracing of resident tendon progenitor cells during growth and natural healing. PloS one 9, e96113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehner C, Spitzer G, Gehwolf R, Wagner A, Weissenbacher N, Deininger C, Emmanuel K, Wichlas F, Tempfer H, Traweger A, Tenophages: a novel macrophage-like tendon cell population expressing CX3CL1 and CX3CR1. Dis Model Mech 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eming SA, Martin P, Tomic-Canic M, Wound repair and regeneration: mechanisms, signaling, and translation. Science translational medicine 6, 265sr266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurtner GC, Werner S, Barrandon Y, Longaker MT, Wound repair and regeneration. Nature 453, 314–321 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Goren I, Allmann N, Yogev N, Schurmann C, Linke A, Holdener M, Waisman A, Pfeilschifter J, Frank S, A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am J Pathol 175, 132–147 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mirza R, DiPietro LA, Koh TJ, Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol 175, 2454–2462 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanasaki K, Taduri G, Koya D, Diabetic nephropathy: the role of inflammation in fibroblast activation and kidney fibrosis. Front Endocrinol (Lausanne) 4, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baum J, Duffy HS, Fibroblasts and myofibroblasts: what are we talking about? J Cardiovasc Pharmacol 57, 376–379 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klingberg F, Hinz B, White ES, The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol 229, 298–309 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pakshir P, Hinz B, The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix biology : journal of the International Society for Matrix Biology 68–69, 81–93 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA, Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3, 349–363 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD, Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest 128, 2127–2143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desmouliere A, Redard M, Darby I, Gabbiani G, Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 146, 56–66 (1995). [PMC free article] [PubMed] [Google Scholar]
- 31.Lawrence T, The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor perspectives in biology 1, a001651 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu T, Zhang L, Joo D, Sun S-C, NF-κB signaling in inflammation. Signal Transduction and Targeted Therapy 2, 17023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watson MR, Wallace K, Gieling RG, Manas DM, Jaffray E, Hay RT, Mann DA, Oakley F, NF-kappaB is a critical regulator of the survival of rodent and human hepatic myofibroblasts. Journal of hepatology 48, 589–597 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Pan HW, Cui YH, Zeng JW, NF-kappaB mediates the survival of corneal myofibroblast induced by angiotensin II. Invest Ophthalmol Vis Sci 55, 4220–4228 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Chen S, Jiang S, Zheng W, Tu B, Liu S, Ruan H, Fan C, RelA/p65 inhibition prevents tendon adhesion by modulating inflammation, cell proliferation, and apoptosis. Cell Death Dis 8, e2710 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dakin SG, Martinez FO, Yapp C, Wells G, Oppermann U, Dean BJ, Smith RD, Wheway K, Watkins B, Roche L, Carr AJ, Inflammation activation and resolution in human tendon disease. Science translational medicine 7, 311ra173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Best KT, Lee FK, Knapp E, Awad HA, Loiselle AE, Deletion of NFKB1 enhances canonical NF-κB signaling and increases macrophage and myofibroblast content during tendon healing. Scientific reports 9, 10926 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abraham AC, Shah SA, Golman M, Song L, Li X, Kurtaliaj I, Akbar M, Millar NL, Abu-Amer Y, Galatz LM, Thomopoulos S, Targeting the NF-kappaB signaling pathway in chronic tendon disease. Science translational medicine 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freeberg MAT, Easa A, Lillis JA, Benoit DSW, van Wijnen AJ, Awad HA, Transcriptomic Analysis of Cellular Pathways in Healing Flexor Tendons of Plasminogen Activator Inhibitor 1 (PAI-1/Serpine1) Null Mice. J Orthop Res 38, 43–58 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darby IA, Laverdet B, Bonte F, Desmouliere A, Fibroblasts and myofibroblasts in wound healing. Clinical, cosmetic and investigational dermatology 7, 301–311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Best KT, Loiselle AE, Scleraxis lineage cells contribute to organized bridging tissue during tendon healing and identify a subpopulation of resident tendon cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, fj201900130RR (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hinz M, Scheidereit C, The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO reports 15, 46–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker JT, McLeod K, Kim S, Conway SJ, Hamilton DW, Periostin as a multifunctional modulator of the wound healing response. Cell and tissue research 365, 453–465 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borthwick LA, Wynn TA, Fisher AJ, Cytokine mediated tissue fibrosis. Biochimica et biophysica acta 1832, 1049–1060 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cserjesi P, Brown D, Ligon KL, Lyons GE, Copeland NG, Gilbert DJ, Jenkins NA, Olson EN, Scleraxis: a basic helix-loop-helix protein that prefigures skeletal formation during mouse embryogenesis. Development 121, 1099–1110 (1995). [DOI] [PubMed] [Google Scholar]
- 46.Pryce BA, Brent AE, Murchison ND, Tabin CJ, Schweitzer R, Generation of transgenic tendon reporters, ScxGFP and ScxAP, using regulatory elements of the scleraxis gene. Dev Dyn 236, 1677–1682 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Naitoh M, Kubota H, Ikeda M, Tanaka T, Shirane H, Suzuki S, Nagata K, Gene expression in human keloids is altered from dermal to chondrocytic and osteogenic lineage. Genes to cells : devoted to molecular & cellular mechanisms 10, 1081–1091 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Muir T, Sadler-Riggleman I, Skinner MK, Role of the basic helix-loop-helix transcription factor, scleraxis, in the regulation of Sertoli cell function and differentiation. Mol Endocrinol 19, 2164–2174 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, Klein U, Germinal center B cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. The Journal of experimental medicine 211, 2103–2118 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, Friedman R, Varro A, Tycko B, Wang TC, Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19, 257–272 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Broekema M, Harmsen MC, van Luyn MJ, Koerts JA, Petersen AH, van Kooten TG, van Goor H, Navis G, Popa ER, Bone marrow-derived myofibroblasts contribute to the renal interstitial myofibroblast population and produce procollagen I after ischemia/reperfusion in rats. J Am Soc Nephrol 18, 165–175 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Ackerman JE, Best KT, O’Keefe RJ, Loiselle AE, Deletion of EP4 in S100a4-lineage cells reduces scar tissue formation during early but not later stages of tendon healing. Scientific reports 7, 8658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, Krampert M, Goebeler M, Gillitzer R, Israel A, Krieg T, Rajewsky K, Haase I, TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature 417, 861–866 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H, A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao LL, Chang J, Wide Awake Secondary Tendon Reconstruction. Hand clinics 35, 35–41 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Ackerman JE, Loiselle AE, Murine Flexor Tendon Injury and Repair Surgery. Journal of visualized experiments : JoVE, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.) 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 58.Dyment NA, Jiang X, Chen L, Hong SH, Adams DJ, Ackert-Bicknell C, Shin DG, Rowe DW, High-Throughput, Multi-Image Cryohistology of Mineralized Tissues. Journal of visualized experiments : JoVE, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hasslund S, Jacobson JA, Dadali T, Basile P, Ulrich-Vinther M, Soballe K, Schwarz EM, O’Keefe RJ, Mitten DJ, Awad HA, Adhesions in a murine flexor tendon graft model: Autograft versus allograft reconstruction. J Orthop Res 26, 824–833 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Phosphorylated p65 staining is localized at injury site.
Fig. S2. Comparison of healthy and injured human flexor tendon.
Fig. S3. Full-length western blots for data from uninjured tendons.
Fig. S4. Full-length western blots for data from repaired tendons.
Table S1. qPCR primer sequences.
Table S2. P-values for student’s t-test and Mann-Whitney test when n ≤ 4.