

Abstract
Purpose of Review
The goal of this review is to obtain a better understanding of how chondrogenesis defines skeletal development via cell transdifferentiation from chondrocytes to bone cells.
Recent Findings
A breakthrough in cell lineage tracing allows bone biologists to trace the cell fate and demonstrate that hypertrophic chondrocytes can directly transdifferentiate into bone cells during endochondral bone formation. However, there is a knowledge gap for the biological significance of this lineage extension and the mechanisms controlling this process.
Summary
This review first introduces the history of the debate on the cell fate of chondrocytes in endochondral bone formation; then summarizes key findings obtained in recent years, which strongly support a new theory: the direct cell transdifferentiation from chondrocytes to bone cells precisely connects chondrogenesis (for providing a template of the future skeleton, classified as phase I) and osteogenesis (for finishing skeletal construction, or phase II) in a continuous lineage-linked process of endochondral bone formation and limb elongation; and finally outlines nutrition factors and molecules that regulate the cell transdifferentiation process during the relay from chondrogenesis to osteogenesis.
Keywords: Chondrocyte, Chondrogenesis, Osteogenesis, Cell transdifferentiation
Introduction
Most bones (80%) in mammals are formed through endochondrogenesis, which includes the appendicular and axial skeleton as well as some vertebrate cranial bones. In embryonic development, endochondrogenesis starts through the condensation of mesenchymal cells which then differentiate into chondrocytes. During chondrogenesis, the mesenchymal cells become round and then flattened chondrocytes, followed by pre-hypertrophic and hypertrophic chondrocytes. The purpose of this phase is to organize into a columnar structure within a growth plate responsible for longitudinal growth. These newly formed chondrocytes secrete unique matrix proteins and calcify cartilage matrices. Subsequently, hypertrophic chondrocytes, which have been considered terminally differentiated cells, undergo degeneration and apoptosis. Finally, the cartilage is thought to be resorbed and replaced with bone by the invasion of vascular associated osteoclasts (for the removal of dead chondrocytes and calcified cartilage) and osteoblast precursor cells (to deposit new bone) from underlying bone marrow and vasculature [1, 2]. However, during a period of over a century, a number of researchers have made a strong case for the capability of mature chondrocytes to transform into bone cells [3–6]. Furthermore, there is a knowledge gap in the link between chondrogenesis and osteogenesis, as it is unclear how the “dead chondrocytes” transmit the inherited skeletal template message to bone cells.
Recently, remarkable progress in imaging and cell lineage tracing technologies have shaded new light in revealing secrets of mouse skeletal biology, which may help to solve the above puzzles. In this paper, we will first review the debate on the history of chondrocyte cell fate in endochondral bone formation. Next, we will summarize the key findings obtained in recent years which strongly support a new theory: the direct cell transdifferentiation from chondrocytes to bone cells precisely connects chondrogenesis (for providing a template of the future skeleton, or phase I) and osteogenesis (for finishing skeletal construction, or phase II) during a continuous single lineage process of endochondral bone formation and limb elongation. Finally, we will outline nutrition factors and molecules that regulate this cell transdifferentiation process during development. We will especially present new findings on the diverse roles of BMP signaling in different chondrocyte populations (i.e., a stimulatory role in growth plate yet an inhibitory role in the perichondrium). These studies not only challenge conventional wisdom but also stimulate future research on why and how BMP signaling defines skeletal shape and size.
There Has Been a Debate on the Cell Fate of Chondrocytes in Endochondral Bone Formation for over a Century
Strelzoff first reported cartilage resorption and osteogenesis in chick embryos (1873) [7]. Twenty years later, Brachet (1893) provided a more detailed description of this phenomenon in both the embryonic and postnatal long bone of chicks [8]. However, these findings did not provide much detail on the cell cytology and histology of cartilage and bone as commented by Fell [9]. Thus, Fell then studied thirty chicks before and after hatching and documented a detailed change of long bone cell morphologies plus confirmation of chondroblast degeneration. Interestingly, all these authors observed a sign of cell transformation from chondroblasts into bone-like cells. Furthermore, a similar observation of the cell transformation from chondroblasts to bone-like cells was documented in pig long bone by Carey [10]. Due to these studies, there have already been three views proposed regarding the fate of chondroblasts: (1) giving rise to osteoblasts, (2) forming part of the marrow reticulum, and (3) degenerating and disappearing [9]. Furthermore, the in vitro embryonic long bone culture studies using the chick chorioallantoic membrane system agree with the above in vivo data with two key pieces of evidence—the cartilage is very unstable, and it directly transforms into osteoid tissue and bone [11]. Interestingly, in the discussion of the cell transformation of chondroblasts into bone-like cells, Fell stated: “The view that bone can develop by the direct transformation of cartilage has often been expressed, and it must be admitted that I have hitherto regarded such statements with scepticism … Nevertheless, a study of chondrogenic endosteal cultures both in life and by means of histological preparations has forced me to the conclusion that a direct transformation of cartilage into bone actually occurs in this material.” Many decades later, using the same chick chorioallantoic membrane culture technique, Jing et al. confirmed that the chondrocytes in newborn mouse mandibular condyle cartilage directly transform into bone cells with a mixture expression of type I- and type II-collagen proteins after 5-day cultures [12].
Although the view of chondrocyte cell death and cartilage replacement by bone has been dominant in the field for many decades, there always has been a dispute about the fate of hypertrophic chondrocytes. The main premise is that at least some of these cells directly transform into bone cells, rather than undergoing programmed cell death (please see an excellent review article by Tsang et al. [6]). Here, we included a few pieces of evidence obtained from studies of long bone and temporomandibular joint (referred to as TMJ and considered unique although similar to large joints in many aspects) to support this cell transformation during development. First, Yoshioka et al. used an electron microscope to observe that some of the hypertrophic chondrocytes in the deepest tissue layer of rat mandibular condylar cartilage appeared to be released from their lacunae into the primary spongiosa [13]. Second, a series of osteoblast-specific genes, such as those for alkaline phosphatase, osteocalcin, osteopontin, and bone sialoprotein, have been shown to be expressed in hypertrophic chondrocytes [5, 14–16]. Third, hypertrophic chondrocytes were able to proliferate and switch into bone-forming cells after vascular invasion was interposed in the normal growth plate [17]. Fourth, Jing et al. recently demonstrated that hypertrophic chondrocytes express high levels of the anti-apoptotic protein BCL-2 and are positive for BrdU (indicating a cell division in these mature chondrocytes) [12]. Fifth, the percentage of apoptotic rates for chondrocytes in response to fracture is rather small (approximately 5%) [18–20]. However, these data are largely a phenotypic description, which are not regarded as sufficiently conclusive to challenge current concepts surrounding the process of endochondral ossification. There are new methods used to more deeply analyze these varieties of cells, as expounded upon below.
Cell Lineage Tracing Studies Precisely Demonstrate that Hypertrophic Chondrocytes Directly Transdifferentiate into Bone Cells during Endochondral Bone Formation
The gold standard for investigating fates of particular cells in vivo within their native environment is lineage-tracing experiments using transgenic mice [21], in which the combination of a Cre-loxP system with fluorescent markers as reporters precisely trace cell fate in vivo [22–24]. For example, to prove the direct cell transdifferentiation from chondrocytes into bone cells, Jing et al. used Aggrecan-creERT2 (Acan-creERT2, which contains the recombinase and tamoxifen-responsive estrogen receptor driven by a cartilage-specific promoter and expressed in the cellular cytoplasm) and Rosa26-fluorescent protein (with a ubiquitous promoter driven-stop sequence, flanked by loxP sites upstream of a fluorescent protein sequence such as tomato) [25, 26]. Injections with tamoxifen (binding to the Cre-ERt2) translocated the Cre in the cytoplasm to the nucleus for initiation of recombination (i.e., chondrocytes and all descendants of those cells are permanently labeled via the fluorescent reporter). To further prove that these labelled descendants of chondrocytes are indeed bone cells, a mouse line expressing 2.3 Col1a1-GFP [27] is then added into the Cre-loxP compound mice (Fig.1, left panel). In this circumstance, the 2.3Col1a1-GFP represents a marker of mature osteoblasts and osteocytes. When all of the mouse lines are bred together, the red and green colors become superimposed, leading to yellow fluorescence (combination of red and green). A yellow bone cell indicates the presence of both the tomato reporter and GFP, which demonstrates that the transdifferentiation of bone cells from chondrocytes occurred (Fig.1, right panel, arrows). Utilizing this approach, several laboratories have confirmed the cellular transformation of chondrocytes into bone cells during long bone development and growth [28–31], in which hypertrophic chondrocytes constitute a source of osteoblasts distinct from the perichondrium [32] and periosteum [33]. In addition, implementing these compound lines, Park et al. demonstrated that hypertrophic chondrocytes in the lowest zone of limb cartilages give rise to cells that express osteogenic genes such as Col1a1, osteocalcin, and Runx2 [34]. Thus, these authors stated that these cells may correspond to the “dark” chondrocytes observed in ultrastructural studies that differ from classic apoptotic chondrocytes.
Fig. 1.
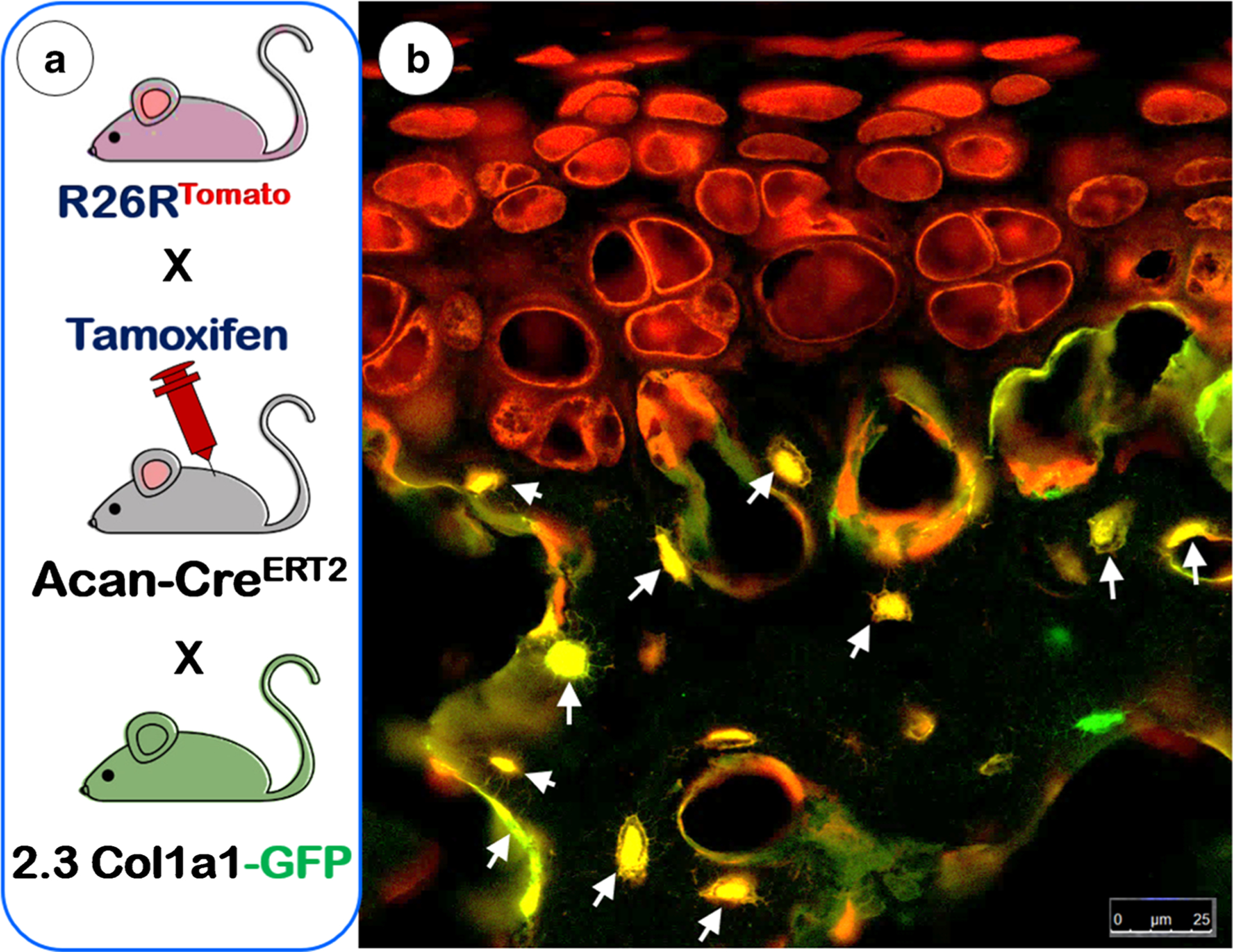
The principle and mechanism of the cell lineage tracing technique. a The schematic diagram illustrates the cross that generates triple mice containing Acan-CreERT2, R26RtdTomato, and 2.3Col1a1-GFP along with a tamoxifen induction to activate the Cre event. b The confocal image of articular cartilage from a 6-week-old mouse shows three types of bone cells in the subchondral bone. This examination revealed non-chondrocyte-derived bone cells (green), chondrocyte-derived bone cells with no GFP activation yet (red), chondrocyte-derived bone cells that produce tomato (reflecting cartilage origin), and type I collagen with GFP activation (reflecting a fully functioning bone cell in yellow as denoted with arrows)
To precisely address the time frame of the entire cell transdifferentiation process, Zhou et al. administered tamoxifen to pregnant females at age E15.5 with Acan-CreERT2; ROSA26R-lacZ embryos and traced the appearance of LacZ+ cells in the femur of these embryos at three timelines postinjection [31]. Nine hours after activation, weak LacZ signals were observed in both chondrocytes and hypertrophic chondrocytes. Next, at 18 h, many chondrocytes and hypertrophic chondrocytes plus very few LacZ+ bone cells in the primary spongiosa were detected. By 24 h postinjection, significantly more cells in the primary spongiosa were positive for LacZ. A separate study by Jing et al. using Acan-CreERT2; ROSA26R-tomato pups also revealed tomato+ dendritic bone cells in the primary spongiosa under TMJ cartilage columns 24 h after tamoxifen injection [12]. Together, these studies support the notion that the direct cell transdifferentiation event is a rapid process, in which there might be no stem or progenitor cell required.
In addition to the critical role of the cell transdifferentiation of chondrocytes in normal skeletal development, Zhou et al. demonstrated that chondrocytes in the repair callus are an essential source of osteoblasts responsible for bone formation during fracture healing [31]. Similar transformational events have been observed when using hypertrophic chondrocyte grafts to heal tibial defects [30, 35•] or mandibular fractures [36]. Furthermore, Hu et al. [37•] showed a spatially-dependent phenotypic overlap between hypertrophic chondrocytes and osteoblasts at the chondro-osseous border within the fracture callus, in a region defined as the transition zone. These authors found that hypertrophic chondrocytes in this zone activate expression of pluripotency factors such as Sox2, Oct4 (Pou5f1), and Nanog. Likewise, they discovered that during the fracture healing process, conditional knockout of Sox2 results in reduction of the fracture callus and a delay in the conversion of the cartilage to bone. Clearly, these results independently confirm that the transformation of hypertrophic chondrocytes directly into bone cells is a universal property of hypertrophic chondrocytes in both normal development and tissue repair.
Overall, this newly obtained tracing data is most likely to end over a century of debate on the cell fate of chondrocytes (cell death or survival for transformation into bone cells). However, a new theory is needed to fill the critical knowledge gap in connecting chondrogenesis and osteogenesis.
Chondrogenesis and Osteogenesis Are Two Sequentially Separate Phases in a Continuous Lineage Process During Endochondral Bone Formation
Literature appears to support a new theory on how bone cells build a skeleton identical to the cartilage template. First, the origin of the chondrocyte is evolutionarily essential for the formation of the vertebrate endoskeleton, as the cartilage appears prior to bone in both head and limb development [38, 39]. Second, chondrogenesis functions as the earliest step of skeletogenesis in the growth plate, which controls longitudinal growth of the skeleton [40]. Early mouse studies [41, 42] suggest that chondrogenesis and osteogenesis in the growth plate are closely coupled in time and space, in which hypertrophic chondrocytes are thought to provide a scaffold for the subsequent formation of trabecular bone by mineralizing their surrounding matrix. Nevertheless, these scenarios did not go on to precisely elaborate how bone cells build a bone structure identical to the cartilage template.
As of late, pulse-chase studies using a Rosa26R-LacZ reporter line crossed with either Osx-CreERt or 3.2 Col 1-CreERt mice revealed that Osx+ osteoblast precursors in the perichondrium directly contribute to trabecular osteoblasts, osteocytes, and stromal cells in the primary ossification center even before vascular invasion of the cartilage [32]. In contrast, the 3.2 Col 1+ osteoblasts remain in the cortical bone surface. Data seems to support no cell lineage connection between both chondrogenesis and osteogenesis except a coincidence of these two events in time and space during development. However, these authors mainly focused on the embryonic stage, with no extension to postnatal stages in endochondrogenesis studies [32].
Recent work has greatly improved this plight by providing strong evidence (that contradicts earlier claims) that chondrogenesis and osteogenesis are not lineage connected. Findings of direct cell transdifferentiation, which count for over 60–70% of bone cells from chondrocytes in the long bone metaphysis [29, 31] or in the mandibular condyle ramus [12] during endochondrogenesis, link both chondrogenesis and osteogenesis in the same lineage pathway for the first time. Importantly, these studies have covered both embryonic and postnatal stages. Furthermore, a genetic disruption or mutation of molecules essential for chondrogenesis leads to great changes of skeletal pattern and shape [2, 43••, 44••, 45•, 46–48]. Conversely, a gene knockout or a mutation in molecules vital for bone formation results in only osteopenia or osteomalacia with little impact on bone shape or size [49–51, 52••]. Based on these findings, a new theory is raised: chondrogenesis (phase I: for defining the skeletal template) and osteogenesis (phase II: for finalizing skeletal formation) are two sequential phases in a continuous lineage-defined process. This scenario takes place during endochondral bone formation, in which the cell transdifferentiation from chondrocytes into bone cells precisely secures the intrinsic transmission of the skeletal template message from the cartilage to bone [52••]. Indeed, there are a number of gene regulation and nutrition factor studies in both phase connections that support this new theory.
Vital Roles of Molecules and Nutrition Factors in Coupling Chondrogenesis and Osteogenesis
Diverse Roles of BMP Signaling in the Cartilage to Bone Transition
BMPs are vital for endochondral bone formation [49, 50, 53–55]. For example, an in vivo genetic model created using inducible Bmp2 conditional knockout (cKO) mice by Col2a1-CreERT2 showed severe defects in chondrocyte proliferation, differentiation, and apoptosis in the growth plates [56]. The deletion of two critical type I receptors Bmpr1a and Bmpr1b (expressed throughout the growth plate) also revealed not only severe chondrodysplasia but also the virtual absence of endochondral ossification during embryonic development [54, 57, 58]. Similarly, the removal of BMP downstream molecules such as Smad1/5 or Smad1/5/8 results in a nearly complete blockage of chondrocyte differentiation and an absence of the axial skeleton [54]. Postnatal studies support the critical role of BMP signaling in coupling chondrogenesis and osteogenesis as well. Removing Bmpr1a in chondrocytes using Acan-CreERT2 at postnatal day 3 via a one-time injection of tamoxifen leads to opposite changes in the metaphysis and epiphysis: (1) an arrest of long bone growth with no sign of a metaphysis (indicating a stimulatory role of BMP signaling in the growth plate) and (2) a massive increase in epiphysis mass (suggesting an inhibitory role of BMP signaling in the perichondrium) [48, 59] (Fig. 2a). This unexpected finding challenges the prevailing paradigm and raises a very interesting question: Why does the same gene play a totally different role in the growth plate and perichondrium during endochondrogenesis?
Fig. 2.

BMP signaling is a key regulator in chondrogenesis, which defines skeletal size and shape. a The images of radiology (left) and toluidine blue stain (right) of 2-month-old long bones with a one-time tamoxifen injection at postnatal day 3 showed a complete halt of long bone growth with a much expanded epiphysis, yet little sign of a metaphysis (*) in Aggrecan-CreERT2 induced Bmpr1a knockout (cKO) mice. b Confocal images of Bmpr1a cKO models from the cell lineage tracing background (containing Acan-CreERT2; R26RtdTomato; Bmpr1a loxP) with a co-localization of DMP1 immunostaining showed numerous tomato+ chondrocytes and chondrocyte-derived red bone cells in the expanded epiphysis (top panel). There was a complete arrest of cell transdifferentiation (*) in the cKO metaphysis (left lower panel) compared with the control, in which tomato+ bone cells accounted for a large portion of red bone cells (right lower panel)
Taking advantage of tracing and high-resolution imaging techniques, Jing et al. generated a compound mouse line consisting of Acan-CreERT2; R26RtdTomato; and Bmpr1afx/fx in which activation of the tomato reporter and deletion of Bmpr1a take place in chondrocyte only with one-time tamoxifen injection at postnatal day 3 [52••]. By 2 months, there were numerous tomato+ chondrocytes and chondrocyte-derived red bone cells in the cKO epiphysis responsible for a much expanded epiphysis (Fig. 2b, top panel). In contrast, the cKO metaphysis displayed a complete arrest in cell transdifferentiation with no sign of a metaphysis (Fig. 2b) compared with the control, inside which tomato+ bone cells accounted for a large portion of bone cells (Fig. 2b, right lower panel). These results reveal three key points: (1) Bmpr1a plays completely different roles in the formation of epiphysis (an inhibitory role) and metaphysis (a stimulatory role); (2) Bmpr1a directly controls cell transdifferentiation regardless of its inhibitory or stimulatory role in articular cartilage or a growth plate; and (3) removing Bmpr1a in chondrocytes results in remarkable changes in limb length and shape [52••] (Fig. 2a).
Meanwhile, Jing et al. conditionally removed Bmpr1a in bone cells only using a 3.6 Col 1-Cre line [52••]. These cKO mice displayed no apparent change in overall bone length or shape except an increase in bone volume (Fig. 3). Similarly, deletions of Bmpr1a by the 3.2 Col 1-Cre [49, 50] or the Dmp1-Cre line [60] lead to an increase in bone volume with little change in long bone structure or shape.
Fig. 3.

Conditionally removed Bmpr1a in bone cells achieved by using 3.6 Col1a1-Cre had no apparent effect on long bone size and shape. a X-ray images displayed an increase in bone volume in 2-month-old Bmpr1a cKO bone marrow (see arrows) with no apparent change in overall bone size and shape (modified from Jing et al. 2017 [52••]). b The backscattered SEM image confirmed this increase in bone formation (as shown by the arrow) in the cKO metaphysis
Together, removing Bmpr1a in chondrocytes results in severe deformities in skeletal shape and length, whereas deleting the same gene in bone cells has little impact on the skeleton. These results further support the notion that chondrocytes instead of osteoblasts or osteocytes determine skeletal shape and size. In addition, diverse roles of BMP signaling in different chondrocytes may partly explain why the skeleton mainly grows longitudinally. Perhaps this finding raises an interesting question for future exploration: chondrocytes in the growth plate are different from those in the perichondrium.
Collectively, the aforementioned studies of Bmpr1a cKO lines in chondrocytes and bone cells with the cell tracing background precisely confirm that chondrogenesis and osteogenesis are one continuous lineage-linked process. The former defines the skeletal pattern and shape whereas the latter subsequently builds the bone structure based on the inherited message from chondrocytes. Importantly, BMP signaling plays a key role in the coupling of these two phases via regulation of cell transdifferentiation in a totally different manner. It enhances transdifferentiation to bone cells in the metaphysis from chondrocytes in the growth plate and slows down transdifferentiation to bone cells in the epiphysis from chondrocytes in the perichondrium.
Ihh Pathway Controls Endochondral Bone Formation
Indian hedgehog (Ihh), a direct target of BMP pathways in chondrocytes [54], is a central regulator in endochondral bone formation [2, 61, 62]. Many studies also suggest that the Ihh pathway couples both chondrogenesis and osteogenesis, which is supported by the following evidence: (1) The deletion of Bmpr1a using Acan-CreERT2 leads to a dramatic decrease of Ihh expression in the growth plate [48]; (2) an inhibition of hedgehog signaling through deletion of Smoothened in chondrocytes blocks formation of the primary spongiosa [63]; (3) the hedgehog signal-responsive cells in the growth plate contribute to long bone formation [47]; (4) removing Ihh in chondrocytes with Col2a1-CreERT2 leads to a complete loss of the growth plate and trabecular bone in the metaphysis [43••]; and (5) thyroid hormones control the transit of proliferating immature chondrocytes into mature hypertrophic chondrocytes and then osteoblasts at the epiphysis via an increase of Ihh levels [45•]. There is also evidence to show that Ihh controls the cell transdifferentiation from chondrocytes to bone cells via an activation of the Wnt signaling pathway [43••].
Vital Roles of Wnt/β-catenin in Endochondral Bone Formation
β-catenin, the key downstream component of the canonical Wnt signaling pathway, is essential for regulating skeletal development and growth. Several independent studies showed a great impact on changes in the β-catenin levels of chondrocytes in both chondrogenesis and osteogenesis. For example, an earlier report demonstrated that ectopic expression of Wnt14 under the control of a Col2a1 promoter greatly enhances differentiation of hypertrophic chondrocytes and endochondral ossification [64]. By the same token, a late study showed that high Wnt signaling levels caused by stabilizing β-catenin in cartilage-specific Col11α2 expressing cells facilitate the replacement of hypertrophic chondrocytes by bone cells. In contrast, postnatal ablation of β-catenin using Col2a1-Cre resulted in a decrease in hypertrophic chondrocytes and a reduction in subsequent endochondral ossification [65]. Furthermore, Dao et al. demonstrated that β-catenin is a prominent inducer of chondrocyte hypertrophy and maturation, as well as osteoblast differentiation and perichondrial bone formation via BMP2 signaling during embryonic and early postnatal days [66]. Specific deletion of β-catenin in hypertrophic chondrocytes using Col10a1-Cre resulted in impaired terminal differentiation of chondrocytes, substantial deficiency of mature osteoblasts, and significant loss of subchondral bone [44••, 46]. These authors also revealed that the β-catenin in hypertrophic chondrocytes has little impact on activities of osteoprecursors in bone marrow [46].
A Low Phosphorus Level Accelerates Chondrogenesis and Cell Transdifferentiation of Chondrocytes to Bone Cells
Mutations of DMP1 in humans [67, 68] or deletions of Dmp1 in mice and rabbits [69, 70] lead to abnormalities in osteocyte maturation and an increase of fibroblast growth factor 23 (FGF23), a key factor for autosomal recessive hypophosphatemic rickets type I [71–73]. For a long time, hypophosphatemic rickets have been thought to be caused by an inhibition of chondrogenesis, leading to an accumulation of hypertrophic chondrocytes and a failure in the replacement of cartilage by bone [74]. To address this issue, Li et al. deleted Dmp1 in the background of Acan-CreERT2; R26RtdTomato and Col10a1-Cre; R26RtdTomato, respectively. Both KO mouse lines displayed an accelerated chondrogenesis (instead of inhibition) plus a sharp increase in Aggrecan+ or Col 10a1+ red bone cells (indicating an enhancement of cell transdifferentiation from chondrocytes to bone [75•]. As constitutive stabilized β-catenin in chondrocytes recapture a very similar defect in the TMJ condyle as shown in Dmp1 KO mice [76], this lab then used the tracing technique to further demonstrate that hypophosphatemia-induced acceleration of cell transdifferentiation is indeed caused by an increase in β-catenin [75•]. Based on the above results, these authors concluded that hypophosphatemic rickets starts from an acceleration of chondrogenesis (including cell transdifferentiation), leading to more chondrocyte-derived bone cells (instead of a reduction in bone cell numbers). Due to a decrease in Pi and the removal of Dmp1, the chondrocyte-derived bone cells are immature in cell differentiation, instigating a defect in mineralization [75•].
Conclusion and Future Research Direction
A debate of chondrocyte fate in endochondral bone formation (which accounts for most of the skeletal components in our body) has occurred over a period of a century, although the view of cell death and replacement of chondrocytes by bone cells is dominant in the field. A recent breakthrough in cell lineage tracing research using a more sensitive tracing reporter (tomato fluorescent protein) plus a high-resolution imaging technique revealed a first-time true picture of the cell transdifferentiation from chondrocytes into bone cells (within 24 h inside the growth plate or in the TMJ condyle). Furthermore, deletions of key molecules (such as BMP signaling) essential for endochondrogenesis or reducing phosphorous levels in the tracing background lead to great changes in labeled chondrocytes and chondrocyte-derived bone cells, which is responsible for defective skeletal shape and size (Figs. 2, 4). Conversely, removing the same key molecules in bone cells has little impact on the skeletal shape except for bone volume and mineralization (Fig. 3). These new findings support a new theory of “two phases and one lineage-defined process in endochondral bone formation”, in which chondrogenesis (phase I) defines skeletal template and osteogenesis (phase II) finalizes skeletal formation [52••]. Importantly, cell transdifferentiation couples these two sequential phases in a continuous lineage-defined process, in which the diverse roles of BMPR1A in chondrocytes of the growth plate (a stimulatory role) and the perichondrium (an inhibitory role) secure the longitudinal growth of long bone yet limit the periphery expansion of joints (Fig. 4).
Fig. 4.
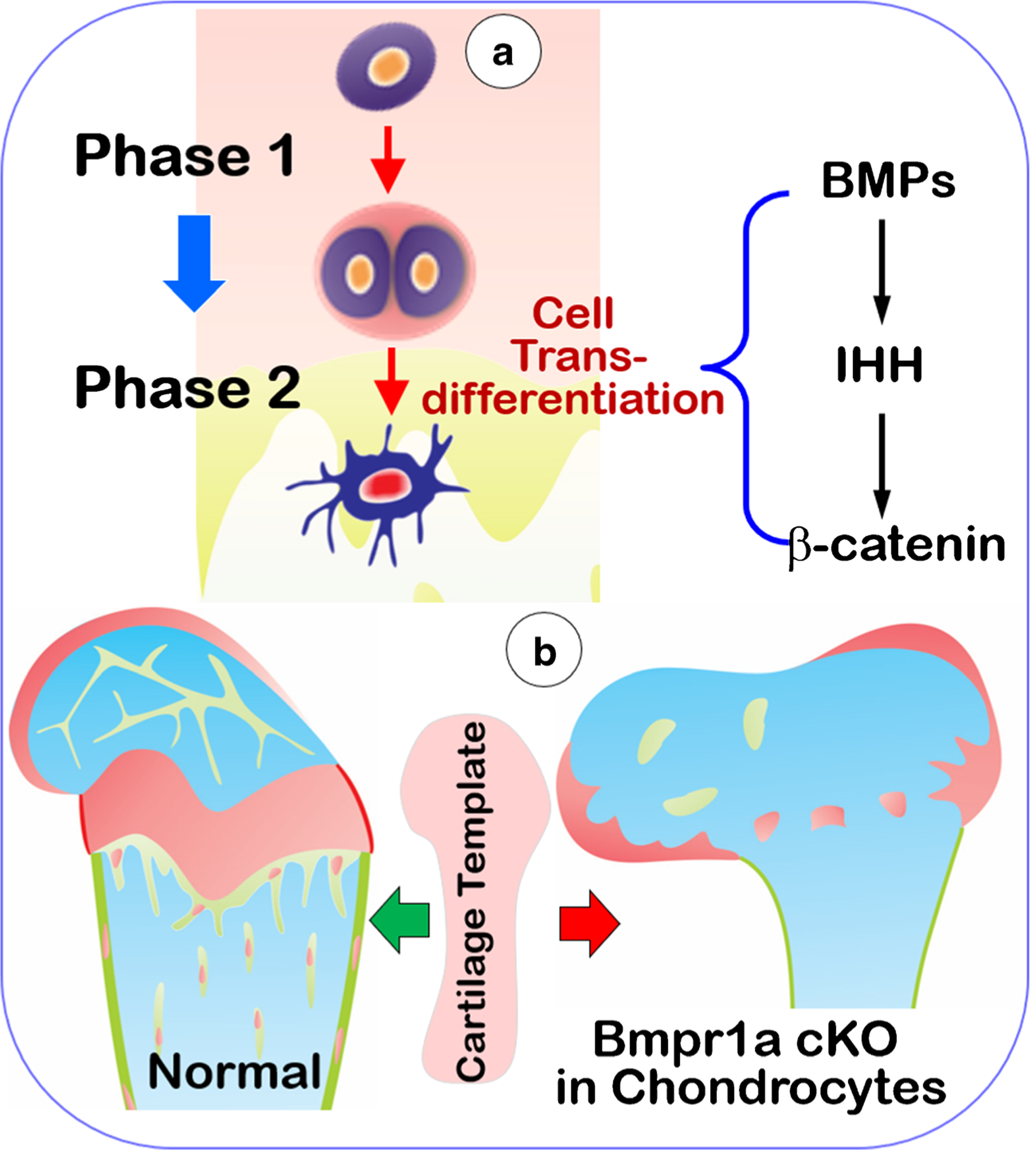
This schematic diagram outlines a new theory of two phases and one lineage process in endochondrogenesis. a Endochondral bone is formed by two phases in one lineage-linked process: chondrogenesis (phase I) and osteogenesis (phase II). In phase I, chondrocytes define the skeletal template via a series of morphological changes, including the formation of hypertrophic chondrocytes. In phase II, many mature chondrocytes directly transdifferentiate into bone cells in a day or even shorter periods of time, which finalize skeletal formation. These processes are made possible due to the vital role of BMPs, IHH, and Wnt/β-catenin pathways in control of the coupling of both phases. b Deletions of the above key molecules (such as BMP signaling) lead to a defective skeleton in shape and size
However, these new findings also raise two key questions. First, a so-called terminally differentiated cell such as the hypertrophic chondrocyte is not a terminal cell and is instead an intermediate cell prior to a true mature bone cell. If this is the case, what is the molecular mechanism behind the cell transdifferentiation? Second, rapid cell transformation (from a few hours to a day) gives little time for creation of a new stem cell or a progenitor cell. What are the key factors for the switch of gene expression profiles leading to this cell type switch?
Emerging single-cell RNA sequence technology [77] can unveil a full gene expression profile in different individual cells, which greatly revolutionizes transcriptomic studies. It is expected that this breakthrough in technology will hold the promise to answer the above questions in the future.
Footnotes
Conflict of Interest Yan Jing, Zheng Wang, Hui Li, Chi Ma, and Jian Feng declare no conflict of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Gibson G Active role of chondrocyte apoptosis in endochondral ossification. Microsc Res Tech. 1998;43:191–204. 10.1002/(SICI)1097-0029(19981015)43:2<191::AID-JEMT10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 2.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6. 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro IM, Adams CS, Freeman T, Srinivas V. Fate of the hypertrophic chondrocyte: microenvironmental perspectives on apoptosis and survival in the epiphyseal growth plate. Birth Defects Res C Embryo Today. 2005;75:330–9. 10.1002/bdrc.20057. [DOI] [PubMed] [Google Scholar]
- 4.Kahn AJ, Simmons DJ. Chondrocyte-to-osteocyte transformation in grafts of perichondrium-free epiphyseal cartilage. Clin Orthop Relat Res. 1977:299–304. [DOI] [PubMed] [Google Scholar]
- 5.Roach HI. Trans-differentiation of hypertrophic chondrocytes into cells capable of producing a mineralized bone matrix. Bone Miner. 1992;19:1–20. 10.1016/0169-6009(92)90840-a. [DOI] [PubMed] [Google Scholar]
- 6.Tsang KY, Chan D, Cheah KS. Fate of growth plate hypertrophic chondrocytes: death or lineage extension? Develop Growth Differ. 2015;57:179–92. 10.1111/dgd.12203. [DOI] [PubMed] [Google Scholar]
- 7.ZJ S Uber die Histogenese des Knochens. Cent f Med 1873:274. [Google Scholar]
- 8.A B. Etude sur la resorption du cartilage et le developpement des os longs chez les oiseaux. J Anat Physiol 1893:10:391–417. [Google Scholar]
- 9.Fell HB. The histogenesis of cartilage and bone in the long bones of the embryonic fowl. J Morphol Physiol. 1925;40:417–59. 10.1002/jmor.1050400302. [DOI] [Google Scholar]
- 10.Carey EJ. Direct observations on the transformation of the mesenchyme in the thigh of the pig embryo (Sus scrofa) were performed, especially in reference to the genesis of the thigh muscles, knee and hip-joints, and the primary bone of the femur. J Morphol. 1922;37: 1–77. [Google Scholar]
- 11.Fell HB. Chondrogenesis in cultures of endosteum. P R Soc Lond B Conta. 1933;112:417–27. 10.1098/rspb.1933.0019. [DOI] [Google Scholar]
- 12.Jing Y, Zhou X, Han X, Jing J, von der Mark K, Wang J, et al. Chondrocytes directly transform into bone cells in mandibular condyle growth. J Dent Res. 2015;94:1668–75. 10.1177/0022034515598135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshioka C, Yagi T. Electron microscopic observations on the fate of hypertrophic chondrocytes in condylar cartilage of rat mandible. J Craniofac Genet Dev Biol. 1988;8:253–64. [PubMed] [Google Scholar]
- 14.Descalzi Cancedda F, Gentili C, Manduca P, Cancedda R. Hypertrophic chondrocytes undergo further differentiation in culture. J Cell Biol. 1992;117:427–35. 10.1083/jcb.117.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerstenfeld LC, Shapiro FD. Expression of bone-specific genes by hypertrophic chondrocytes: implication of the complex functions of the hypertrophic chondrocyte during endochondral bone development. J Cell Biochem. 1996;62:1–9. 10.1002/(SICI)1097-4644(199607)62:1<1::AID-JCB1>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 16.Roach HI, Erenpreisa J, Aigner T. Osteogenic differentiation of hypertrophic chondrocytes involves asymmetric cell divisions and apoptosis. J Cell Biol. 1995;131:483–94. 10.1083/jcb.131.2.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Enishi T, Yukata K, Takahashi M, Sato R, Sairyo K, Yasui N. Hypertrophic chondrocytes in the rabbit growth plate can proliferate and differentiate into osteogenic cells when capillary invasion is interposed by a membrane filter. PLoS One. 2014;9:e104638 10.1371/journal.pone.0104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rundle CH, Wang X, Sheng MH, Wergedal JE, Lau KH, Mohan S. Bax deficiency in mice increases cartilage production during fracture repair through a mechanism involving increased chondrocyte proliferation without changes in apoptosis. Bone. 2008;43:880–8. 10.1016/j.bone.2008.07.239. [DOI] [PubMed] [Google Scholar]
- 19.Gaber S, Fischerauer EE, Frohlich E, Janezic G, Amerstorfer F, Weinberg AM. Chondrocyte apoptosis enhanced at the growth plate: a physeal response to a diaphyseal fracture. Cell Tissue Res. 2009;335:539–49. 10.1007/s00441-008-0735-0. [DOI] [PubMed] [Google Scholar]
- 20.Lee FY, Choi YW, Behrens FF, DeFouw DO, Einhorn TA. Programmed removal of chondrocytes during endochondral fracture healing. J Orthop Res. 1998;16:144–50. 10.1002/jor.1100160124. [DOI] [PubMed] [Google Scholar]
- 21.Kretzschmar K, Watt FM. Lineage tracing. Cell. 2012;148:33–45. 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Romagnani P, Rinkevich Y, Dekel B. The use of lineage tracing to study kidney injury and regeneration. Nat Rev Nephrol. 2015;11: 420–31. 10.1038/nrneph.2015.67. [DOI] [PubMed] [Google Scholar]
- 23.Humphreys BD, DiRocco DP. Lineage-tracing methods and the kidney. Kidney Int. 2014;86:481–8. 10.1038/ki.2013.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jing Y, Hinton RJ, Chan KS, Feng JQ. Co-localization of cell lineage markers and the tomato signal. J Vis Exp. 2016. 10.3791/54982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henry SP, Jang CW, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis. 2009;47:805–14. 10.1002/dvg.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM, et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci U S A. 2005;102:14665–70. 10.1073/pnas.0504750102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalajzic Z, Liu P, Kalajzic I, Du Z, Braut A, Mina M, et al. Directing the expression of a green fluorescent protein transgene in differentiated osteoblasts: comparison between rat type I collagen and rat osteocalcin promoters. Bone. 2002;31:654–60. 10.1016/s8756-3282(02)00912-2. [DOI] [PubMed] [Google Scholar]
- 28.Ono N, Ono W, Nagasawa T, Kronenberg HM. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat Cell Biol. 2014;16:1157–67. 10.1038/ncb3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang G, Zhu L, Hou N, Lan Y, Wu XM, Zhou B, et al. Osteogenic fate of hypertrophic chondrocytes. Cell Res. 2014;24:1266–9. 10.1038/cr.2014.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A. 2014;111:12097–102. 10.1073/pnas.1302703111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014;10:e1004820 10.1371/journal.pgen.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell. 2010;19:329–44. 10.1016/j.devcel.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debnath S, Yallowitz AR, McCormick J, Lalani S, Zhang T, Xu R, et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature. 2018;562:133–9. 10.1038/s41586-018-0554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park J, Gebhardt M, Golovchenko S, Perez-Branguli F, Hattori T, Hartmann C, et al. Dual pathways to endochondral osteoblasts: a novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol Open. 2015;4:608–21. 10.1242/bio.201411031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.•.Bahney CS, Hu DP, Taylor AJ, Ferro F, Britz HM, Hallgrimsson B, et al. Stem cell-derived endochondral cartilage stimulates bone healing by tissue transformation. J Bone Miner Res. 2014;29: 1269–82. 10.1002/jbmr.2148 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study represents cartilage can transform into bone by activating the pluripotent transcription factor Oct4A, which shifts the paradigm for the mechanism of endochondral bone repair.
- 36.Wong SA, Hu D, Miclau T, Bahney C, Marcucio R. Trans differentiation of chondrocytes to osteoblasts during endochondral ossification in the healing mandible. FASEB J. 2016;30(suppl 1): 1039.11(.Abstract). [Google Scholar]
- 37.•.Hu DP, Ferro F, Yang F, Taylor AJ, Chang W, Miclau T, et al. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development. 2017;144:221–34. 10.1242/dev.130807 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that chondrocytes can differentiate to become osteoblasts during fracture callus repair. This process involves expression of pluripotency genes such as Sox2.
- 38.Zhang G, Eames BF, Cohn MJ. Chapter 2. Evolution of vertebrate cartilage development. Curr Top Dev Biol. 2009;86:15–42. 10.1016/S0070-2153(09)01002-3. [DOI] [PubMed] [Google Scholar]
- 39.Zhang G An evo-devo view on the origin of the backbone: evolutionary development of the vertebrae. Integr Comp Biol. 2009;49: 178–86. 10.1093/icb/icp061. [DOI] [PubMed] [Google Scholar]
- 40.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406. 10.1016/s1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 41.Erlebacher A, Filvaroff EH, Gitelman SE, Derynck R. Toward a molecular understanding of skeletal development. Cell. 1995;80: 371–8. 10.1016/0092-8674(95)90487-5. [DOI] [PubMed] [Google Scholar]
- 42.Chung UI, Lanske B, Lee K, Li E, Kronenberg H. The parathyroid hormone/parathyroid hormone-related peptide receptor coordinates endochondral bone development by directly controlling chondrocyte differentiation. Proc Natl Acad Sci U S A. 1998;95:13030–5. 10.1073/pnas.95.22.13030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.••.Maeda Y, Nakamura E, Nguyen MT, Suva LJ, Swain FL, Razzaque MS, et al. Indian hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc Natl Acad Sci U S A. 2007;104:6382–7. 10.1073/pnas.0608449104 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that postnatal chondrocyte-derived Ihh is essential for maintaining the growth plate and articular surface and is required for sustaining trabecular bone and skeletal growth.
- 44.••.Houben A, Kostanova-Poliakova D, Weissenbock M, Graf J, Teufel S, von der Mark K, et al. Beta-catenin activity in late hypertrophic chondrocytes locally orchestrates osteoblastogenesis and osteoclastogenesis. Development. 2016;143:3826–38. 10.1242/dev.137489 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows β-catenin has dual functions in trabecular bone formation by regulating the osteoclastogenesis and involving in the transdifferentiation from chondrocytes to osteoblast precursors.
- 45.•.Aghajanian P, Xing W, Cheng S, Mohan S. Epiphyseal bone formation occurs via thyroid hormone regulation of chondrocyte to osteoblast transdifferentiation. Sci Rep. 2017, 7:10432 10.1038/s41598-017-11050-1 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that thyroid hormone regulates chondrocyte to osteoblast transformation in the secondary ossification centre through opposing SHH and IHH signaling.
- 46.Golovchenko S, Hattori T, Hartmann C, Gebhardt M, Gebhard S, Hess A, et al. Deletion of beta catenin in hypertrophic growth plate chondrocytes impairs trabecular bone formation. Bone. 2013;55: 102–12. 10.1016/j.bone.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 47.Haraguchi R, Kitazawa R, Imai Y, Kitazawa S. Growth plate-derived hedgehog-signal-responsive cells provide skeletal tissue components in growing bone. Histochem Cell Biol. 2018;149: 365–73. 10.1007/s00418-018-1641-5. [DOI] [PubMed] [Google Scholar]
- 48.Jing J, Ren Y, Zong Z, Liu C, Kamiya N, Mishina Y, et al. Feng JQ.BMP receptor 1A determines the cell fate of the postnatal growth plate. Int J Biol Sci. 2013;9:895–906. 10.7150/ijbs.7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamiya N, Ye L, Kobayashi T, Mochida Y, Yamauchi M, Kronenberg HM, et al. BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development. 2008;135:3801–11. 10.1242/dev.025825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamiya N, Ye L, Kobayashi T, Lucas DJ, Mochida Y, Yamauchi M, et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J Bone Miner Res. 2008;23:2007–17. 10.1359/jbmr.080809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu F, Woitge HW, Braut A, Kronenberg MS, Lichtler AC, Mina M, et al. Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. Int J Dev Biol. 2004;48:645–53. 10.1387/ijdb.041816fl. [DOI] [PubMed] [Google Scholar]
- 52.••.Jing Y, Jing J, Ye L, Liu X, Harris SE, Hinton RJ, et al. Chondrogenesis and osteogenesis are one continuous developmental and lineage defined biological process. Sci Rep. 2017;7:10020 10.1038/s41598-017-10048-z [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that chondrogenesis and osteogenesis are one continuous developmental and lineage-defined biological process, in which Bmpr1a signaling in chondrocytes is necessary for the formation of a pool of osteoprogenitors that then contributes in a major way to overall bone formation and growth.
- 53.Tsumaki N, Yoshikawa H. The role of bone morphogenetic proteins in endochondral bone formation. Cytokine Growth Factor Rev. 2005;16:279–85. [DOI] [PubMed] [Google Scholar]
- 54.Retting KN, Song B, Yoon BS, Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development. 2009;136:1093–104. 10.1242/dev.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shu B, Zhang M, Xie R, Wang M, Jin H, Hou W, et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J Cell Sci. 2011;124: 3428–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoon BS, Pogue R, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR. Lyons KM.BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development. 2006;133:4667–78. [DOI] [PubMed] [Google Scholar]
- 58.Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci. 2005;102:5062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jing J, Hinton RJ, Feng JQ. Bmpr1a signaling in cartilage development and endochondral bone formation. Vitam Horm. 2015;99: 273–91. 10.1016/bs.vh.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 60.Lim J, Shi Y, Karner CM, Lee SY, Lee WC, He G, et al. Dual function of Bmpr1a signaling in restricting preosteoblast proliferation and stimulating osteoblast activity in the mouse. Development. 2015. 10.1242/dev.126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8:727–38. 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 62.Lin GL, Hankenson KD. Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J Cell Biochem. 2011;112:3491–501. 10.1002/jcb.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Long F, Chung UI, Ohba S, McMahon J, Kronenberg HM, McMahon AP. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development. 2004;131: 1309–18. 10.1242/dev.01006. [DOI] [PubMed] [Google Scholar]
- 64.Abboud M, Koeck B, Stark H, Wahl G, Paillon R. Immediate loading of single-tooth implants in the posterior region. Int J Oral Maxillofac Implants. 2005;20:61–8. [PubMed] [Google Scholar]
- 65.Yuasa T, Kondo N, Yasuhara R, Shimono K, Mackem S, Pacifici M, et al. Transient activation of Wnt/{beta}-catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am J Pathol. 2009;175:1993–2003. 10.2353/ajpath.2009.081173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Li W, Wang Y, Huang Y. Growth inhibition of human lens epithelial cells by short hairpin RNA in transcription factor forkhead box E3 (FOXE3). Graefes Arch Clin Exp Ophthalmol. 2012;250:999–1007. 10.1007/s00417-012-1944-5. [DOI] [PubMed] [Google Scholar]
- 67.Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–5. 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38: 1248–50. 10.1038/ng1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ye L, Mishina Y, Chen D, Huang H, Dallas SL, Dallas MR, et al. Dmp1-deficient mice display severe defects in cartilage formation responsible for a chondrodysplasia-like phenotype. J Biol Chem. 2005;280:6197–203. 10.1074/jbc.M412911200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu T, Wang J, Xie X, Wang K, Sui T, Liu D, et al. DMP1 ablation in the rabbit results in mineralization defects and abnormalities in haversian canal/osteon microarchitecture. J Bone Miner Res. 2019;34:1115–28. 10.1002/jbmr.3683. [DOI] [PubMed] [Google Scholar]
- 71.Lu Y, Feng JQ. FGF23 in skeletal modeling and remodeling. Curr Osteoporos Rep. 2011;9:103–8. 10.1007/s11914-011-0053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carpenter TO, Shaw NJ, Portale AA, Ward LM, Abrams SA, Pettifor JM. Rickets. Nat Rev Dis Primers. 2017;3:17101 10.1038/nrdp.2017.101. [DOI] [PubMed] [Google Scholar]
- 73.Feng JQ, Clinkenbeard EL, Yuan B, White KE, Drezner MK. Osteocyte regulation of phosphate homeostasis and bone mineralization underlies the pathophysiology of the heritable disorders of rickets and osteomalacia. Bone. 2013;54:213–21. 10.1016/j.bone.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qin C, D’Souza R, Feng JQ. Dentin matrix protein 1 (DMP1): new and important roles for biomineralization and phosphate homeostasis. J Dent Res. 2007;86:1134–41. 10.1177/154405910708601202. [DOI] [PubMed] [Google Scholar]
- 75.•.Li H, Jing Y, Zhang R, Zhang Q, Wang J, Martin A, et al. Hypophosphatemic rickets accelerate chondrogenesis and cell trans-differentiation from TMJ chondrocytes into bone cells via a sharp increase in beta-catenin. Bone. 2019;131:115151 10.1016/j.bone.2019.115151 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that the Dmp1-null caused hypophosphatemia, leading to acceleration (instead of inhibition) of chondrogenesis and bone trans-differentiation from chondrocytes but inhibition of bone cell maturation due to a sharp increase in β-catenin.
- 76.Jing Y, Jing J, Wang K, Chan K, Harris SE, Hinton RJ, et al. Vital roles of beta-catenin in trans-differentiation of chondrocytes to bone cells. Int J Biol Sci. 2018;14:1–9. 10.7150/ijbs.23165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401. 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]