

Abstract
Thiol dioxygenases are mononuclear non-heme FeII-dependent metalloenzymes that initiate the oxidative catabolism of thiol-containing substrates to their respective sulfinates. Cysteine dioxygenase (CDO), the best characterized mammalian thiol dioxygenase, contains a 3-histidine (3-His) coordination environment rather than the 2-His-1-carboxylate facial triad seen in most mononuclear non-heme FeII enzymes. A similar 3-His active site is found in the bacterial thiol dioxygenase 3-mercaptopropionate dioxygenase (MDO), which converts 3-mercaptopropionate (3-MPA) into 3-sulfinopropionic acid as part of bacterial sulfur metabolism pathway. In the present study, we have investigated the active site geometric and electronic structures of a third non-heme FeII-dependent thiol dioxygenase, cysteamine dioxygenase (ADO), by using a spectroscopic approach. Although a 3-His facial triad had previously been implicated on the basis of sequence alignment and site-directed mutagenesis studies, little is currently known about the active site environment of ADO. Our magnetic circular dichroism and electron paramagnetic resonance data provide compelling evidence that ADO features a 3-histidine facial triad, like CDO and MDO. Despite this similar coordination environment, spectroscopic results obtained for ADO incubated with substrate and various substrate analogues are distinct from those obtained for the other FeII-dependent thiol dioxygenases. This finding suggests that the secondary coordination sphere of ADO is distinct from those of CDO and MDO, demonstrating the significant role that secondary sphere residues play in dictating substrate specificity.
Graphical Abstract
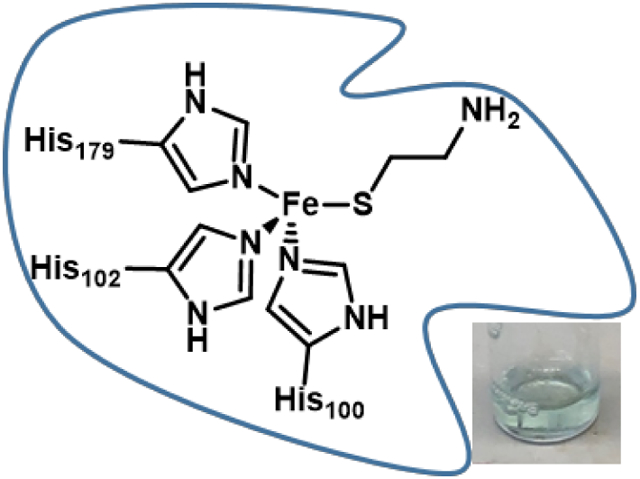
INTRODUCTION
Taurine plays many essential roles in mammalian metabolism.1-4 This sulfonic acid-containing metabolite helps to maintain homeostasis of important intracellular ions through osmotic regulation, preservation of cardiac and vascular functions, protection of neural cells from excitotoxicity, and stabilization of skeletal muscle membranes, among many functions.1-3 The best-studied pathway for taurine biosynthesis starts with the O2-dependent conversion of cysteine (Cys) to cysteine sulfinic acid by cysteine dioxygenase (CDO). Cysteine sulfinic acid is then further metabolized to hypotaurine and, finally, taurine.
Hypotaurine is also produced by the oxidation of cysteamine (2-aminoethanethiol, 2-AET), catalyzed by cysteamine dioxygenase (ADO, Scheme 1).4,5 Cys serves as a building block for the formation of coenzyme A, which is in turn degraded by pantetheinase into pantetheine and 2-AET. Although an enzyme responsible for the oxidation of 2-AET to hypotaurine has been postulated since the 1960’s,6-8 the investigation of ADO has been hindered by the reliance on isolating the enzyme from crude tissue. The gene responsible for ADO production was finally discovered in 2007 when Dominy et al. found a discrepancy between the levels of taurine biosynthesis and CDO expression in various tissues.9 This finding suggested that the generation of hypotaurine by ADO turnover may be responsible for the majority of taurine production in some tissues, particularly the brain. Thus, ADO appears to play a larger role in taurine biosynthesis and Cys catabolism than had originally been assumed. Interestingly, a recent report argued that the function of ADO has been misidentified and its primary function is instead to oxidize amino-terminal Cys residues of proteins in response to hypoxia.10
Scheme 1.

Simplified schematic of multiple pathways of Cys metabolism, where the number of steps required for the conversion of reactant to product is highlighted by the number of arrows. CDO catalyzes the first step in the conversion of Cys to cysteine sulfinic acid. ADO catalyzes the conversion of cysteamine (2-AET) to hypotaurine.4,5
Recently, four known non-heme FeII-dependent thiol dioxygenases [collectively TDOs: CDO, ADO, 3-mercaptopropionate dioxygenase (MDO), and mercaptosuccinate dioxygenase (MSDO)] have been identified as members of the cupin superfamily with roles in the metabolism of sulfur-containing compounds. Proteins in the cupin superfamily are typically metalloproteins that show low overall sequence identity but contain two short, highly conserved primary sequence motifs, Gx5HxHx3-6Ex6G (cupin motif 1) and Gx5-7PxGx2Hx3N (cupin motif 2), separated by an inter-motif region that varies both in length (~15-50 residues) and amino acid sequence.11 X-ray crystallographic studies have shown that the CDO and MDO active sites feature an FeII center that is coordinated by a 3-Histidine (3-His) triad (Figure 1).12,13 This binding motif is exceedingly rare in FeII-dependent oxygenases, which typically adopt a 2-His-1-carboxylate motif.14,15 Only a small number of other proteins have been definitively established by crystallography to feature this Fe binding motif, including diketone-cleaving dioxygenase,16 gentisate 1,2-dioxygenase,17 EgtB,18 and SznF.19 An unusual crosslink between Cys93 and Tyr157 present near the primary coordination sphere of CDO (Figure 1, left) plays a role in properly orienting substrate and suppressing water coordination to Cys-bound FeIICDO to preserve an open coordination site for O2.20,21 Cys-Tyr crosslinks have only been observed in two other enzymes; namely, galactose oxidase, where it serves as a reversible one-electron source,22 and NirA, where its function is unknown.23
Figure 1.

Left: Active site region of the crystal structure of Cys-bound Mus musculus CDO (PDB code: 4JTO).24 Right: Active site region of the crystal structure of resting MDO from Pseudomonas aeruginosa (PDB code: 4TLF).13
While the three-dimensional structure of ADO is not yet known, a sequence alignment with Mus musculus CDO revealed that strong similarities exist between these enzymes, including the two cupin motifs (despite the low sequence identity of 14%).11 Importantly, both CDO and ADO lack the highly conserved glutamate residue found in cupin motif 1 that is typical of other metal-binding cupin proteins. This sequence alignment of CDO and ADO also predicts the conservation of the 3-His facial triad that binds FeII in CDO. The substitution of one of these histidine residues of ADO by an alanine completely abolished catalytic activity, further supporting the hypothesis that this enzyme contains a 3-His facial triad.25
Interestingly, the iron incorporation during recombinant expression in as-isolated ADO is much higher than in CDO,26 suggesting important differences in the active site pocket. Indeed, residues deemed to play a role in the catalytic cycle of CDO or in maintaining the structural integrity, including a Ser153-His155-Tyr157 “catalytic triad” and the Cys93 involved in the Cys-Tyr crosslink of mammalian CDOs (Figure 1), are not conserved in ADO. The substitution of Cys93 in CDO by a Gly in ADO suggests that the latter might be more similar to MDOs or bacterial CDOs, which do not possess this crosslink (Figure 1).12,27 However, the existence of a Cys220-Tyr222 crosslink in ADO in a distinct location from the CDO crosslink was recently established via genetic incorporation of an unnatural amino acid, 3,5-difluoro-tyrosine, into ADO and characterization of this species with mass spectrometry and NMR spectroscopy.28 Interestingly, Arg60 (mouse CDO numbering, Figure 1, left), which has been postulated to play a role in regulating substrate specificity in CDO, is also not conserved in ADO. MDOs have been shown by X-ray crystallography to feature a Gln at the position corresponding to Arg60 of CDO (Figure 1, right)13,29 and are unable to efficiently produce cysteine sulfinic acid from Cys; instead, they have evolved to utilize 3-mercaptopropionic acid (3-MPA) as their native substrate.27 The absence of both of these residues in ADO (based on sequence alignment) reveals another important difference in the active sites between these paralogs and suggests that amino acid variations among the active sites may play an important role in modulating substrate specificity.
ADO displays high substrate specificity and is unable to convert Cys to cysteine sulfinic acid despite the structural similarities between 2-AET and Cys.25 To gain further understanding of the potential differences between the active sites of ADO and CDO, we have used magnetic circular dichroism (MCD) and electron paramagnetic resonance (EPR) spectroscopies to study complexes of ADO with its native substrate 2-AET, as well as the weakly competitive inhibitor Cys25 and another structurally similar substrate analogue, 3-MPA. Collectively, our data yield new insight into the geometric and electronic structures of the ADO active site and provide clues regarding the origin of the substrate specificity displayed by this enzyme.
MATERIALS AND METHODS
Recombinant Gene Expression and Protein Purification.
A codon-optimized Mus musculus ADO gBlock was ordered from Integrated DNA Technology and inserted into a pET small ubiquitin-related modifier (SUMO) expression vector via Gibson Assembly.30 The sequence of the cloned gene was verified at the University of Wisconsin-Madison Biotechnology Center. ADO was produced as previously described with minor changes.25 Escherichia coli Rosetta 2(DE3) cells were grown at 37 °C in 4 L of LB medium (containing 10 g tryptone, 10 g NaCl, 5 g of yeast extract per liter). All cultures were inoculated with 50 μg/mL kanamycin and 34 μg/mL chloramphenicol. The cells were grown to an OD600 of ~4, and then gene overexpression was induced by the addition of isopropyl-β-D-thiogalactopyranoside to a final concentration of 0.2 mM along with ferrous ammonium sulfate to a final concentration of 100 μM. Four hours post-induction, cells were harvested via centrifugation at 4,402×g for 20 min at 4 °C. The cell paste was flash frozen and stored at −80 °C overnight.
Frozen cell paste was thawed in 80 mL of 20 mM Tris buffer, pH 8.0, containing 250 mM NaCl, 5 mM imidazole, 1mM TCEP and four tablets of Roche Complete protease inhibitor (IMAC Buffer A). The cell suspension was lysed via pulsed sonication (15 s on, 30 s off for 10 min on ice). Soluble protein was separated from cellular debris by centrifugation for 30 min at 48,400×g at 4 °C. After filtering the supernatant with a 0.8 μm filter, the filtrate was applied to a 5-mL HisTrap-FF column. Protein was eluted in a linear gradient of IMAC Buffer A and 20 mM Tris, pH 8.0, containing 250 mM NaCl and 500 mM imidazole (IMAC Buffer B). As reported previously, two elution bands were observed,25 peaking at ~5 mM imidazole (0% IMAC Buffer B) and ~75 mM imidazole (15% IMAC Buffer B). Stain-free sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to verify the presence of the target protein heterodimer of ~42 kDa (28 kDa ADO protein and 14 kDa SUMO tag) in both peaks. Due to its higher purity, only protein associated with the second peak was kept for further purification. The fractions containing SUMO-tagged ADO were pooled, concentrated to 1 mL, and dialyzed overnight at 4 °C against a 20 mM Tris, pH 8.0, containing 250 mM NaCl to remove imidazole. The following morning the protein was concentrated to ~5 mg/mL, divided into two aliquots so as not to overload the column, and applied to a HiLoad 16/600 Superdex 200pg gel filtration column. The protein was separated into two elution peaks, with the second peak corresponding to 42 kDa expected for the folded, soluble fusion protein. Fractions containing the most pure and active ADO, as judged by SDS-PAGE and a qualitative activity assay, were pooled and concentrated (Figure S1, Supporting Information). Total protein yield was estimated using the absorbance at 280 nm and an extinction coefficient of 41.4 mM−1cm−1, calculated via ExPASy31 from the SUMO-tag and ADO protein sequences. The FeII and total Fe contents of the protein were determined through a colorimetric assay using the iron chelator 2,4,6-tris(2-pyridyl)-s-triazine (TPTZ) and an ε595 of 22.1 mM−1cm−1.32,33 The assay was performed in the presence and absence of a reductant (hydroxylamine) to determine the proportion of FeII versus FeIII initially present. As the fraction of Fe-bound ADO active sites was ~70-80%, the concentration of protein (0.6 mM for MCD spectroscopy and 0.5 mM for EPR spectroscopy) cited herein refers to the Fe-bound fraction, not that of the total protein. Protein aliquots to be used for spectroscopic characterization were flash frozen and stored at −80 °C.
Enzyme activity assays.
Enzymatic activity was determined qualitatively using thin layer chromatography (TLC), similar to methods established for CDO.34 Briefly, SUMO-tagged ADO was incubated aerobically with 20 mM 2-AET in Tris buffer, pH 8.0 at 37 °C for 30 min. Samples were heat denatured to stop the reaction and centrifuged for 5 min at 14,000 rpm in a table-top centrifuge to remove precipitated protein. The supernatant was spotted onto a silica TLC plate and placed in a beaker containing 20:20:60 (v/v) H2O/acetic acid/1-butanol running solution. After one hour, the plate was removed and developed with a 1.5% ninhydrin solution (w/v) in 3% (v/v) acetic acid in ethanol. Following heat activation of the stain, bands associated with 2-AET and hypotaurine were identified by comparison with control standard solutions (Figure S2, Supporting Information). Two bands arise from our 2-AET standard; while the origin of the lower feature is not conclusively known, we postulate that the second band could be due to cystamine formation.
Sample preparation.
Samples used for MCD spectroscopy were prepared anaerobically in a glovebox under a nitrogen atmosphere, while those used for EPR spectroscopy were prepared aerobically. Where applicable, 2-AET was added at 5-fold molar excess over Fe-bound ADO, while the substrate analogues Cys and 3-MPA were added at a 10-fold molar excess. For MCD spectroscopic studies of FeIIADO, a slightly sub-stoichiometric amount of dithionite relative to the total iron content was added to reduce the fraction of FeIII-bound sites to the FeII state. Samples for low-temperature (LT) MCD studies also contained 55% (v/v) glycerol as a glassing agent.
Spectroscopy.
LT MCD spectra were collected with a Jasco J-715 spectropolarimeter in conjunction with an Oxford Instruments SM4000-8T magnetocryostat. MCD spectra are presented as the difference between spectra obtained with the magnetic field aligned parallel and antiparallel to the light propagation axis to eliminate contributions from the CD background and glass strain. For Cys-bound ADO, variable temperature, variable field (VTVH) MCD data were collected by measuring the signal intensity as a function of magnetic field at a constant wavelength (477 nm = 20.964 cm−1) for several fixed temperatures. The data were fitted with the S=1/2 Brillouin function35
where , μj is the Bohr magneton, H is the field (in Tesla), kB is the Boltzmann constant, and T is the temperature.
X-band EPR data were collected using a Bruker ELEXSYS E500 spectrometer. The sample temperature was maintained at 20 K by an Oxford ESR 900 continuous flow liquid He cryostat regulated by an Oxford ITC-503S temperature controller. All spectra were obtained using the following experimental parameters: frequency = 9.386 GHz; microwave power = 12.62 mW; modulation amplitude = 3 G; modulation frequency = 100 kHz. The program EasySpin (version 5.2.25) was used to fit the experimental EPR spectra.36
RESULTS
As-isolated ADO is a translucent pale yellow in color, likely reflecting the small, but significant, fraction of active sites containing FeIII in addition to those featuring FeII.37 Upon addition of 2-AET, the protein solution turns faint blue in color, indicative of substrate coordinating to the FeIII-bound active sites. Consistent with this observation, a pair of positively signed features at 14,400 and ~17,800 cm−1 appear in the low-temperature (LT) MCD spectrum of the 2-AET adduct of as-isolated ADO (Figure 2A) that are absent in the MCD spectrum of substrate-free as-isolated ADO (Figure S3, Supporting Information). These results provide compelling evidence for the formation of an inner-sphere complex between FeIIIADO and 2-AET. The MCD features at 14,400 and ~17,800 cm−1 of 2-AET-bound ADO are characteristic of S→FeIII charge transfer (CT) transitions of high-spin (S=5/2) FeIII-thiolate complexes, which implies direct coordination of 2-AET to FeIIIADO via its thiolate group.38 The MCD spectrum of as-isolated ADO incubated with 2-AET also exhibits a large temperature dependent feature in the near UV-region, as well as the onset to a weaker temperature dependent feature in the near IR-region.
Figure 2.

MCD spectra at 4, 15, and 25 K of as-isolated ADO incubated with various substrate (analogues): A) ADO incubated with a 5-fold excess of 2-AET; B) ADO incubated with a 10-fold excess of 3-MPA; C) ADO incubated with a 10-fold excess of Cys. The feature indicated by * arises from a minor heme impurity.
To more definitively establish the origin of the features at 14,400 and 17,800 cm−1 in the LT MCD spectrum of as-isolated ADO in the presence of 2-AET, LT MCD spectra were also collected for dithionite-reduced enzyme (exclusively containing FeII-bound active sites) incubated with 2-AET (Figure 3). The loss of these features in the reduced sample corroborates their assignment as S→FeIII CT transitions arising from FeIIIADO. In contrast, the positively signed features in the near-UV and the near-IR region are retained upon FeIII reduction, indicating that they are associated with FeIIADO. The onset to the feature in the near-IR region at 10,000 cm−1 is likely due to an FeII d→d transition, based on a comparison with MCD data reported for other FeII complexes.39 However, further spectroscopic studies are needed to conclusively assign these features (note that the near UV feature is also present in the MCD spectrum of resting ADO).
Figure 3.

MCD spectra at 4, 15, and 25 K of FeIIADO incubated with a 5-fold excess of 2-AET.
Our research group and others gained important insights into the nature of substrate/active site interactions in the FeII-dependent thiol dioxygenases CDO and MDO by utilizing substrate analogues.33,34,37,40,41 Here, we have employed the same strategy by incubating as-isolated ADO with Cys and 3-MPA. Previous work has revealed that Cys is not a substrate for ADO; however, it does act as a weak competitive inhibitor, suggesting that it is capable of binding to the active site.25 Moreover, while both 2-aminoethanethiol and Cys contain amino groups that can coordinate to the iron center, 3-MPA does not. Thus, by comparing spectroscopic data obtained for ADO incubated with 2-AET, Cys, or 3-MPA, it is possible to infer whether the amino, thiolate, and/or carboxylate groups bind to FeII ion.
Incubation of as-isolated ADO with 3-MPA results in an MCD spectrum that is nearly identical to that of 2-AET-bound ADO (Figure 2B). Specifically, the onset to a positive feature displayed by ADO incubated with 2-AET in the near UV-region (>32,000 cm−1) is retained, as is the feature in the near-IR region. In addition, the two features in the visible region (at ~14,000 and 18,000 cm−1) display the same saturation behavior as their counterparts in the 2-AET-bound as-isolated ADO spectrum, indicating that 3-MPA binds in a similar manner to that of the native substrate. Thus, our results may indicate that 2-AET coordinates to the FeIIIADO active site in a monodentate fashion, via coordination of only the terminal thiolate. Notably, a similar monodentate, thiolate-only binding mode has previously been proposed by Pierce et al. for 3-MPA coordination to MDO on the basis of EPR experiments.27
Incubation of as-isolated ADO with a 10-fold excess of Cys leads to an MCD spectrum that is drastically different from those observed for ADO treated with 2-AET or 3-MPA (Figure 2C). While the onset to a positive feature in the near-UV region (>34,000 cm−1) is retained, numerous positively and negatively signed features appear in the visible region that have no counterparts in the other MCD spectra. Notably, these features display a distinctly different saturation behavior than those associated with 2-AET-bound FeIIIADO, implying that the Cys adduct of ADO possesses a different spin ground state. Indeed, the variable-temperature variable-field (VTVH) MCD data collected for the intense feature at 20,964 cm−1 can be fit with an S = 1/2 Brillouin function, consistent with the formation of a low-spin FeIII complex (Figure 4).
Figure 4.
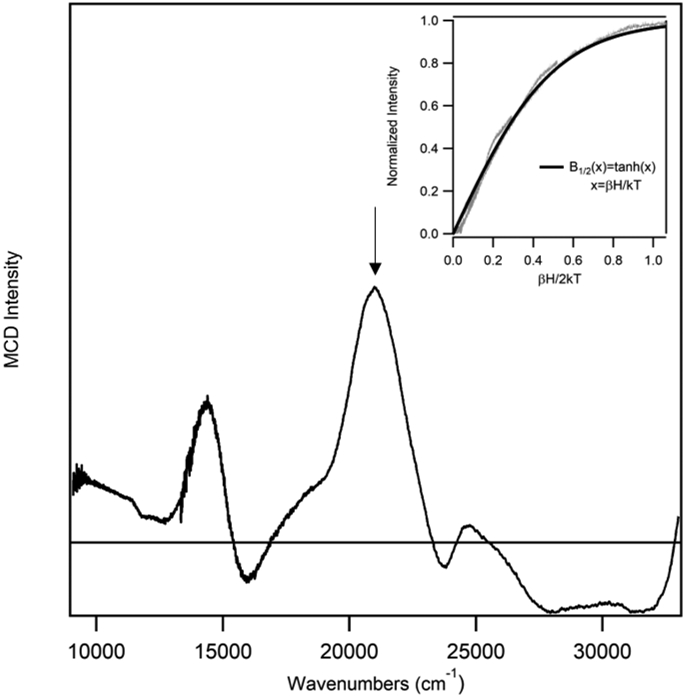
MCD spectrum at 4 K of as-isolated ADO incubated with a ~15-fold excess of Cys. Inset: VTVH MCD data (gray) collected for ADO + Cys at 20,964 cm−1 (indicated by vertical arrow) at 2.2, 4.5, 8, 15, and 25 K superimposed by a Brillouin function for S=1/2 (black).
EPR spectroscopy was employed as a complementary tool to probe the electronic structure of the FeIIIADO active site and to monitor changes in response to substrate (analogue) binding. As-isolated ADO exhibits a nearly rhombic, high-spin (S = 5/2) signal centered at geff ≈ 4.3 (Figure 5A), similar to that displayed by as-isolated CDO.34 The addition of 2-AET yields a new, more axial EPR signal attributable to an S=5/2 FeIII species (Figure 5B). Consistent with our MCD data, the appearance of this distinct high-spin EPR signal demonstrates the formation of an inner-sphere 2-AET-FeIIIADO complex (see Table S1 of the Supporting Information for EPR fit parameters). Incubation of as-isolated ADO with 3-MPA also results in a more axial high-spin (S = 5/2) EPR spectrum (Figure 5C). The nearly identical EPR spectra displayed by 3-MPA- and 2-AET-bound FeIII-ADO indicate that 3-MPA binds in a similar manner to the native substrate, supporting our hypothesis that 2-AET coordinates to the FeIIIADO active site solely via its terminal thiolate.
Figure 5.

X-band EPR spectra at 20 K of A) as-isolated ADO and ADO incubated with B) 2-AET, C) 3-MPA, and D) Cys. EPR spectral simulations (dotted lines) are overlaid on the experimental spectra (solid line) for comparison. Fit parameters are provided in the Supporting Information.
Incubation of as-isolated ADO with Cys results in the formation of a high-spin (S=5/2) species that exhibits a similar, albeit slightly more rhombic EPR signal than 2-AET-bound FeIII-ADO (Figure 5D), which seemingly conflicts with our MCD spectra of the Cys adduct. However, the high-field region of the EPR spectrum obtained for ADO incubated with the substrate analogue Cys also shows a new signal at g ≈ 2.4 (Figure 5D), consistent with the formation of a low-spin (S=1/2) FeIII species as suggested by our VTVH MCD data (Figure 4). The fact that both high- and low-spin Cys-bound FeIIIADO species contributed to the EPR spectrum seemed puzzling, as the MCD spectrum of Cys-bound ADO is dominated by a low-spin species.
This apparent discrepancy between the ratio of high-spin and low-spin species contributing to the MCD and EPR spectra of Cys-bound FeIIIADO prompted us to prepare EPR samples with glycerol, the glassing agent used for MCD studies. Upon incubation of ADO with Cys and glycerol, a drastic increase in the relative intensity of the S=1/2 signal is observed (Figure 6B). Notably, such a change in the relative intensities of the S = 5/2 and S = 1/2 signals was not seen with addition of glycerol to 2-AET-bound ADO (data not shown). To assess whether this shift in high-spin to low-spin ratio could be due to glycerol binding to the active site, polyethylene glycol 200 (PEG-200) was used as an alternative glassing agent. Cys was still able to coordinate to FeIIIADO and the low-spin species was found to be the major product (Figure 6C), though the formation of a distinct high-spin FeIII species was evident from the appearance of new signals in the 800-1700 Gauss range. Thus, coordination of glycerol to the Cys-bound FeIIIADO active site can be ruled out as being responsible for the formation of a low-spin (S = 1/2) FeIII complex.
Figure 6.

X-band EPR spectra at 20 K of as-isolated ADO incubated with A) a 10-fold excess of Cys, B) a 10-fold excess of Cys and 55% (v/v) glycerol, and C) a 10-fold excess of Cys and 55% (v/v) PEG-200. EPR spectral simulations (dotted lines) are overlaid on the experimental spectra (solid line) for comparison. Fit parameters are provided in the Supporting Information.
Finally, to further validate the 3-His triad implicated in ADO and demonstrate that our spectra are dominated by contributions from Fe residing in the ADO active site, the putative Fe ligand His100 was replaced by Ala and as-isolated H100A ADO was characterized by EPR spectroscopy. Importantly, the high-spin (S=5/2) EPR signals displayed by WT ADO incubated with 2-AET are noticeably absent in the spectrum of H100A ADO in the presence of substrate (Figure 7B). Thus, we conclude that in our samples of WT ADO the large majority of Fe ions are present in the active site and, thus, that non-specifically bound Fe ions contribute minimally to our MCD and EPR spectra of the WT enzyme.
Figure 7.

X-band EPR spectra at 20 K of A) as-isolated wild-type ADO and B) the H100A ADO variant incubated with a 5-fold excess of 2-AET.
DISCUSSION
Although ADO was discovered more than ten years ago, relatively little is known about this important sulfur-metabolism enzyme. Apart from one recent study demonstrating the existence of a Cys-Tyr crosslink in ADO that is formed in a very different region of the primary sequence than in CDO,28 little else has been published on ADO’s structure and function. To start filling this knowledge gap, we have used MCD and EPR spectroscopies to ascertain the coordination environment of the ADO active site.
The MCD spectrum of as-isolated ADO incubated with 2-AET is dominated by a pair of positively signed features at 14,400 and 17,800 cm−1 (Figure 2A) reminiscent of those arising from S→FeIII CT transitions in the MCD spectrum of Cys-bound CDO at 15,250 and 18,570 cm−1.38 As replacement of a neutral His ligand with an anionic carboxylate would likely result in significant blue-shifts of the S→FeIII CT features, our MCD data of 2-AET-bound ADO provide evidence that ADO also possesses a 3-His active site. In addition, the EPR spectrum of H100A incubated with 2-AET lacks the high-spin ferric signal observed in the EPR spectrum of 2-AET-bound ADO, further corroborating our hypothesis that ADO contains a neutral 3-His active site.
The striking similarities between the MCD spectra of ADO incubated with 2-AET and 3-MPA (Figure 2A and 2B), along with the fact that the S→FeIII CT transitions of 2-AET-bound ADO are slightly red-shifted from those of Cys-bound CDO (consistent with a lower coordination number in ADO), suggest that 2-AET and 3-MPA bind to ADO in the same manner, potentially in a monodentate fashion through the terminal thiolate. Thus, ADO appears to be the second TDO displaying monodentate substrate binding, as both an EPR study of NO and 3-MPA-bound MDO27 and a Mössbauer study of Cys-bound MDO13 afforded evidence for monodentate binding to FeIIMDO via its thiolate moiety. The EPR spectra of 2-AET- and 3-MPA-bound FeIIIADO (Figure 5A and 5B) reveal that these high-spin ferric species exhibit a more axial zero-field splitting (E/D = 0.248 and 0.233, respectively) relative to that reported for Cys-bound FeIIICDO (E/D = 0.171).34 This finding is consistent with a more dominant Fe─S bonding interaction in ADO, further corroborating our hypothesis that 2-AET and 3-MPA bind to FeIIIADO solely through the terminal thiolate.
Our MCD and EPR data indicate that Cys can bind directly to the ADO iron center, consistent with the notion that this substrate analogue is a weak competitive inhibitor.25 Interestingly, the VTVH MCD data of Cys-bound ADO (Figure 4) provide evidence for the formation of a low-spin (S=1/2) FeIII complex, previously only observed for substrate (analogue)-bound FeIIICDO incubated with the strong-field ligand cyanide.41,42 Our EPR data (Figure 6) reveal that the ratio of a low-spin to high-spin ferric species can be modulated by the addition of glycerol, with the high-spin Cys-FeIIIADO complex being the predominant form in the absence of a glassing agent (Figure 5). The addition of PEG-200, instead of glycerol, to Cys-bound ADO also yields a low-spin species, arguing against the possibility that a change in spin state is due to direct glycerol coordination to the active site. Rather, our data support the hypothesis that the glassing (i.e., molecular crowding) agents induce a global protein conformational change, which further suggests that the ADO active site environment differs from that of CDO. Such differences in active site architecture are likely responsible for the distinct substrate specificities displayed by the various TDOs.
Comparison of ADO to other TDOs.
Recently, the distinct substrate specificities displayed by CDO and MDO were attributed to key structural differences in the respective active site pockets; namely, the presence of an Arg or Gln at position 60 (Figure 1) and the presence of a cis-peptide bond between residues Ser158 and Pro159 in CDO (all Rattus norvegicus CDO numbering) that is absent in MDO.20,43,44 Cys binding to CDO occurs via bidentate coordination to the iron through the thiolate and amine groups, with the substrate carboxylate forming a salt bridge to Arg60. Because MDO contains a Gln at the position corresponding to Arg60 of RnCDO, it is unable to form an analogous salt bridge with the carboxylate moiety of its native substrate. Instead, MDO features a conserved Arg at position 164 (Rn CDO numbering) that is postulated to form a salt bridge with the 3-MPA carboxylate tail,20 potentially promoting bidentate substrate coordination to the FeII center. However, previous spectroscopic studies of MDO favor monodentate binding of 3-MPA via thiolate-only coordination.27 In contrast to Cys and 3-MPA, 2-AET does not contain a carboxylate. It is thus not surprising that based on sequence alignment (Figure S4 of the Supporting Information), the residue in ADO corresponding to Arg60 of CDO is a Val, which is unable to hydrogen bond with a carboxylate. Although 2-AET features an amine group that could potentially coordinate to the iron of ADO, our EPR and MCD data favor monodentate, thiolate-only coordination of 2-AET, similar to 3-MPA binding to MDO.
Mammalian CDO features a thioether crosslink between Cys93 and Tyr157 (Figure 1) that has been implicated in properly orienting the substrates and suppressing the binding of a water molecule to Cys-bound FeIICDO so as to preserve an open coordination site for O2. 20,21 Interestingly, the crosslinked and non-crosslinked forms of CDO can be distinguished via SDS-PAGE. Bacillus subtilis CDO does not form a thioether crosslink as it possesses a Gly at the position corresponding to Cys93, but does contain a cis-peptide bond that positions Tyr157 similarly as in RnCDO.9,20 Despite the lack of both the thioether crosslink and cis-peptide bond in MDO, X-ray crystal structures revealed that in this enzyme Tyr159 adopts essentially the same orientation relative to the iron center as Tyr157 does in CDO.20 Similar to MDO, ADO also appears to lack a thioether crosslink involving the Tyr residue corresponding to Tyr157 in CDO as well as the cis-peptide bond. However, ADO has been shown to form a different thioether crosslink between Cys220 and Tyr222 (ADO numbering).28 Although the exact role of this distal thioether crosslink in ADO remains to be elucidated, it is possible that it serves to impose an active site architecture similar to that of MDO, which also does not contain a cis-peptide bond.
CONCLUSIONS
The spectroscopic data obtained in the present study yield new insight into the active site geometry of ADO. The MCD spectra of the well-characterized Cys-bound FeII and FeIIICDO species are similar to those of 2-AET-bound ADO,38 strongly suggesting that these enzymes share the same 3-His active site coordination environment. As such, ADO is the fourth thiol dioxygenase that coordinates its metallocofactor via a 3-His triad instead of the more common 2-His-1carboxylate facial triad. Importantly, differences in substrate specificity between ADO and the other thiol dioxygenases likely stem from variations in key second-sphere residues present in CDO and MDO. ADO binds 2-AET and 3-MPA in a similar manner and does not appear to promote coordination of the amine functional group of 2-AET, supporting monodentate binding via the thiolate moiety. Finally, a change in spin state of the Cys-bound FeIII-ADO adduct in the presence of osmolytes may suggest that the active site of ADO is more sensitive to solution conditions than CDO.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for financial support from the National Institute of General Medical Sciences of the National Institutes of Health (Grant GM117120 to T.C.B.). This research was conducted in part while R.L.F. and S.L.D. were supported by the National Institute of General Medical Sciences of the National Institutes of Health (Grant T32GM008505).
Footnotes
Supporting Information
SDS-PAGE gel of SUMO-tagged ADO; TLC activity screen results; MCD spectra as-isolated wild-type ADO in the absence and presence of 2-AET; sequence alignment of select thiol dioxygenases; and EPR fit parameters. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Huxtable RJ (1992) Physiological Actions of Taurine. Physiol. Rev 72, 101–163. [DOI] [PubMed] [Google Scholar]
- (2).Schaffer SW, Ju Jong C, KC R, and Azuma J (2010) Physiological roles of taurine in heart and muscle. J. Biomed. Sci 17, 101–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Xu Y-J, Arneja AS, Tappia PS, and Dhalla NS (2008) The potential health benefits of taurine in cardiovascular disease. Exp. Clin. Cardiol 13, 57–65. [PMC free article] [PubMed] [Google Scholar]
- (4).Stipanuk MH (1986) Metabolism of sulfur-containing amino acids. Annu. Rev. Nutr 6, 179–209. [DOI] [PubMed] [Google Scholar]
- (5).Stipanuk MH, Dominy JE, Lee J-I, and Coloso RM (2006) Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J. Nutr 136, 1652S–1659S. [DOI] [PubMed] [Google Scholar]
- (6).Cavallini D, Scandurra R, and De Marco C (1963) The Enzymatic Oxidation of Cysteamine to Hypotaurine in the Presence of Sulfide. J. Biol. Chem 238, 2999–3005. [PubMed] [Google Scholar]
- (7).Rotilio G, Federici G, Calabrese L, Costa M, and Cavallini D (1970) An electron paramagnetic resonance study of the nonheme iron of cysteamine oxygenase. J. Biol. Chem 245, 6235–6236. [PubMed] [Google Scholar]
- (8).Jacobsen JG, and Smith LH (1968) Biochemistry and physiology of taurine and taurine derivatives. Physiol. Rev 48, 424–511. [DOI] [PubMed] [Google Scholar]
- (9).Dominy JE, Simmons CR, Karplus PA, Gehring AM, and Stipanuk MH (2006) Identification and characterization of bacterial cysteine dioxygenases: A new route of cysteine degradation for eubacteria. J. Bacteriol 188, 5561–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Masson N, Keeley TP, Giuntoli B, White MD, Lavilla Puerta M, Perata P, Hopkinson RJ, Flashman E, Licausi F, and Ratcliffe PJ (2019) Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science (80-. ). 364, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Stipanuk MH, Simmons CR, Karplus PA, and Dominy JE (2011) Thiol dioxygenases: Unique families of cupin proteins. Amino Acids 41, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, and Phillips GN (2006) Structure and mechanism of mouse cysteine dioxygenase. Proc. Natl. Acad. Sci 103, 3084–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tchesnokov EP, Fellner M, Siakkou E, Kleffmann T, Martin LW, Aloi S, Lamont IL, Wilbanks SM, and Jameson GNL (2015) The cysteine dioxygenase homologue from Pseudomonas aeruginosa is a 3-mercaptopropionate dioxygenase. J. Biol. Chem 290, 24424–24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Straganz GD, and Nidetzky B (2006) Variations of the 2-His-1-carboxylate theme in mononuclear non-heme Fe II oxygenases. ChemBioChem 7, 1536–1548. [DOI] [PubMed] [Google Scholar]
- (15).Koehntop KD, Emerson JP, and Que L (2005) The 2-His-1-carboxylate facial triad: A versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J. Biol. Inorg. Chem 10, 87–93. [DOI] [PubMed] [Google Scholar]
- (16).Diebold AR, Neidig ML, Moran GR, Straganz GD, and Solomon EI (2010) The three-his triad in dke1: Comparisons to the classical facial triad. Biochemistry 49, 6945–6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen J, Li W, Wang M, Zhu G, Liu D, Sun F, Hao N, Li X, Rao Z, and Zhang XC (2008) Crystal structure and mutagenic analysis of GDOsp, a gentisate 1,2-dioxygenase from Silicibacter Pomeroyi. Protein Sci. 17, 1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Goncharenko KV, Vit A, Blankenfeldt W, and Seebeck FP (2015) Structure of the sulfoxide synthase EgtB from the ergothioneine biosynthetic pathway. Angew. Chemie - Int. Ed 54, 2821–2824. [DOI] [PubMed] [Google Scholar]
- (19).Ng TL, Rohac R, Mitchell AJ, Boal AK, and Balskus EP (2019) An N-nitrosating metalloenzyme constructs the pharmacophore of streptozotocin. Nature 566, 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Aloi S, Davies CG, Karplus PA, Wilbanks SM, and Jameson GNL (2019) Substrate Specificity in Thiol Dioxygenases. Biochemistry 2398–2407. [DOI] [PubMed] [Google Scholar]
- (21).Driggers CM, Kean KM, Hirschberger LL, Cooley RB, Stipanuk MH, and Karplus PA (2016) Structure-Based Insights into the Role of the Cys–Tyr Crosslink and Inhibitor Recognition by Mammalian Cysteine Dioxygenase. J. Mol. Biol 428, 3999–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Whittaker MM, Ekberg CA, Peterson J, Sendova MS, Day EP, and Whittaker JW (2000) Spectroscopic and magnetochemical studies on the active site copper complex in galactose oxidase. J. Mol. Catal. - B Enzym 8, 3–15. [Google Scholar]
- (23).Schnell R, Sandalova T, Hellman U, Lindqvist Y, and Schneider G (2005) Siroheme- and [Fe4-S4]-dependent NirA from Mycobacterium tuberculosis is a sulfite reductase with a covalent Cys-Tyr bond in the active site. J. Biol. Chem 280, 27319–27328. [DOI] [PubMed] [Google Scholar]
- (24).Driggers CM, Cooley RB, Sankaran B, Hirschberger LL, Stipanuk MH, and Karplus PA (2013) Cysteine Dioxygenase Structures from pH4 to 9: Consistent Cys-Persulfenate Formation at Intermediate pH and a Cys-Bound Enzyme at Higher pH. J.Mol.Biol 425, 3121–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dominy JE, Simmons CR, Hirschberger LL, Hwang J, Coloso RM, and Stipanuk MH (2007) Discovery and characterization of a second mammalian thiol dioxygenase, cysteamine dioxygenase. J. Biol. Chem 282, 25189–25198. [DOI] [PubMed] [Google Scholar]
- (26).Tchesnokov EP, Wilbanks SM, and Jameson GNL (2012) A strongly bound high-spin iron(II) coordinates cysteine and homocysteine in cysteine dioxygenase. Biochemistry 51, 257–264. [DOI] [PubMed] [Google Scholar]
- (27).Pierce BS, Subedi BP, Sardar S, and Crowell JK (2015) The “Gln-Type” Thiol Dioxygenase from Azotobacter vinelandii Is a 3-Mercaptopropionic Acid Dioxygenase. Biochemistry 54, 7477–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang Y, Griffith WP, Li J, Koto T, Wherritt DJ, Fritz E, and Liu A (2018) Cofactor Biogenesis in Cysteamine Dioxygenase: C─F Bond Cleavage with Genetically Incorporated Unnatural Tyrosine. Angew. Chemie Int. Ed 57, 8149–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Driggers CM, Hartman SJ, and Karplus PA (2015) Structures of Arg- and Gln-type bacterial cysteine dioxygenase homologs. Protein Sci. 24, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, and Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- (31).Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, and Bairoch A (2005) Protein Identification and Analysis Tools on the ExPASy Server, in The Proteomics Protocols Handbook, pp 571–607. Humana Press, Totowa, NJ. [Google Scholar]
- (32).Fischer DS, and Price DC (1964) a Simple Serum Iron Method Using the New Sensitive Chromogen Tripyridyl-S-Triazine. Clin. Chem 10, 21–31. [PubMed] [Google Scholar]
- (33).Blaesi EJ, Fox BG, and Brunold TC (2015) Spectroscopic and computational investigation of the H155A variant of cysteine dioxygenase: Geometric and electronic consequences of a third-sphere amino acid substitution. Biochemistry 54, 2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Blaesi EJ, Fox BG, and Brunold TC (2014) Spectroscopic and Computational Investigation of Iron(III) Cysteine Dioxygenase: Implications for the Nature of the Putative Superoxo-Fe(III) Intermediate. Biochemistry 53, 5759–5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Arrott AS (2008) Approximations to Brillouin functions for analytic descriptions of ferromagnetism Calculating the spontaneous magnetization and defining the Curie temperature using a positive-feedback model Approximations to Brillouin functions for analytic descriptions . J. Appl. Phys. Rev. Sci. Instruments Infin. Spin Am. J. Phys. J. Appl. Phys 1031, 7–715. [Google Scholar]
- (36).Stoll S, and Schweiger A (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson 178, 42–55. [DOI] [PubMed] [Google Scholar]
- (37).Pierce BS, Gardner JD, Bailey LJ, Brunold TC, and Fox BG (2007) Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: Electronic structure of the active site and mechanistic implications. Biochemistry 46, 8569–8578. [DOI] [PubMed] [Google Scholar]
- (38).Gardner JD, Pierce BS, Fox BG, and Brunold TC (2010) Spectroscopic and computational characterization of substrate-bound mouse cysteine dioxygenase: Nature of the ferrous and ferric cysteine adducts and mechanistic implications. Biochemistry 49, 6033–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Solomon EI, Pavel EG, Loeb KE, and Campochiaro C (1995) Magnetic circular dichroism spectroscopy as a probe of the geometric and electronic structure of non-heme ferrous enzymes. Coord. Chem. Rev 144, 369–460. [Google Scholar]
- (40).Li W, and Pierce BS (2015) Steady-state substrate specificity and O2-coupling efficiency of mouse cysteine dioxygenase. Arch. Biochem. Biophys 565, 49–56. [DOI] [PubMed] [Google Scholar]
- (41).Li W, Blaesi EJ, Pecore MD, Crowell JK, and Pierce BS (2013) Second-sphere interactions between the C93-Y157 cross-link and the substrate-bound Fe site influence the O2 coupling efficiency in mouse cysteine dioxygenase. Biochemistry 52, 9104–9119. [DOI] [PubMed] [Google Scholar]
- (42).Fischer AA, Miller JR, Jodts RJ, Ekanayake DM, Lindeman SV, Brunold TC, and Fiedler AT (2019) Spectroscopic and Computational Comparisons of Thiolate-Ligated Ferric Nonheme Complexes to Cysteine Dioxygenase: Second-Sphere Effects on Substrate (Analogue) Positioning. Inorg. Chem 58, 16487–16499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Brandt U, Schürmann M, and Steinbüchel A (2014) Mercaptosuccinate dioxygenase, a cysteine dioxygenase homologue, from Variovorax paradoxus strain B4 is the key enzyme of mercaptosuccinate degradation. J. Biol. Chem 289, 30800–30809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Brandt U, Galant G, Meinert-Berning C, and Steinbüchel A (2019) Functional analysis of active amino acid residues of the mercaptosuccinate dioxygenase of Variovorax paradoxus B4. Enzyme Microb. Technol 120, 61–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.