

The Drp1-HK1-NLRP3 axis is a key mechanism of oligodendrocyte damage and white matter degeneration in AD.
Abstract
Myelin degeneration and white matter loss resulting from oligodendrocyte (OL) death are early events in Alzheimer’s disease (AD) that lead to cognitive deficits; however, the underlying mechanism remains unknown. Here, we find that mature OLs in both AD patients and an AD mouse model undergo NLR family pyrin domain containing 3 (NLRP3)–dependent Gasdermin D–associated inflammatory injury, concomitant with demyelination and axonal degeneration. The mature OL-specific knockdown of dynamin-related protein 1 (Drp1; a mitochondrial fission guanosine triphosphatase) abolishes NLRP3 inflammasome activation, corrects myelin loss, and improves cognitive ability in AD mice. Drp1 hyperactivation in mature OLs induces a glycolytic defect in AD models by inhibiting hexokinase 1 (HK1; a mitochondrial enzyme that initiates glycolysis), which triggers NLRP3-associated inflammation. These findings suggest that OL glycolytic deficiency plays a causal role in AD development. The Drp1-HK1-NLRP3 signaling axis may be a key mechanism and therapeutic target for white matter degeneration in AD.
INTRODUCTION
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder that is characterized by extracellular senile plaques that are composed of deposits of β-amyloid (Aβ) and neurofibrillary tangles (NFTs) of hyperphosphorylated tau protein (1). Despite decades of progress in understanding the pathogenesis of AD, the mechanisms that underlie AD-related cellular damage and cognitive deficits remain elusive. Before plaques and tangles become apparent in AD patients, white matter loss and demyelination are evident, often manifesting early in the course of the disease (2) and predicting disease status (3). Such brain injury is attributable to the damage of oligodendrocytes (OLs), myelin-forming glial cells in the central nervous system. The loss of OLs and a reduction of myelin density, axonal loss, and astrogliosis are major changes in white matter that occur in the brains of both AD patients and AD animals (4, 5). Therefore, OL impairment and white matter degeneration have been hypothesized to be primary events in AD pathology (2), given the failure of anti-amyloid therapy.
Similar to astrocytes, mature OLs rely heavily on glycolysis for energy production even in the presence of oxygen. OLs consume large amounts of glucose and lactate for energy-demanding myelination tasks. They also provide pyruvate and lactate to ensheathed neurons to maintain long-term axonal integrity through a lactate shuttle process (6). In human AD, OL alterations precede neuronal impairment, and the loss of OLs may lead to cognitive deficits (5, 7). Furthermore, a genome-wide association study found that Bin1 and Got2, genes enriched in OLs, are dysregulated in AD and associated with AD risk (8). Unbiased proteomics and biochemical analyses of postmortem brains from AD patients and AD animals also found decreases in proteins of mature OLs (8), and a recent study reported that OL precursor cells (OPCs) exhibited a senescence-like phenotype that was concomitant with the formation of Aβ plaques (9). However, the mechanisms by which OLs become dysfunctional and lead to AD pathology have remained unknown.
Mitochondria are the hubs of cellular metabolic activity, innate and adaptive immune responses, and programmed cell death. Mitochondria continually change their morphology and location to adapt to functional changes via a delicate balance between fission and fusion. Mitochondrial fission fragments the organelle, which, if excessive, can generate reactive oxygen species, impair bioenergetics, cause aberrant mitophagy, and ultimately cell death (10). This process is primarily controlled by dynamin-related protein 1 (Drp1), a cytosolic guanosine triphosphatase. Upon activation, Drp1 is recruited from the cytosol to the outer mitochondrial membrane (OMM), where it assembles by self-oligomerization to initiate mitochondrial division (11). Posttranslational modification (e.g., phosphorylation) is a driving force that mediates Drp1 recruitment to the mitochondria (12). Elevated Drp1 activation is observed in brain tissue from AD patients and AD mice, neurons exposed to Aβ peptide, and fibroblasts derived from AD patients (13–15). Drp1 also interacts with monomeric and oligomeric Aβ in brain tissue from early-onset and severe AD patients (16), proposing a mechanism of aberrant Drp1 activation in AD. Notably, genetic and pharmacological inhibition of global Drp1 hyperactivation reduces neuropathology and synaptic deficits and attenuates behavioral abnormalities in AD mouse models (13, 17, 18), consistent with a causal role for Drp1 hyperactivation in AD. In addition, Drp1 can regulate postsynaptic endocytosis, neuronal morphology, and brain function independent of mitochondrial fission (19). While Drp1 has been intensively studied in neuronal cells, recent studies highlight a driving role for Drp1 activation in cell metabolism and the immune response in nonneuronal cells as well, including astrocytes (20), microglia (21), macrophages (22), and T cells (23). Thus, Drp1 has been implicated in regulation of broad cellular functions. However, whether Drp1 is activated in OLs and its exact role in white matter degeneration in AD are unknown.
The NLR family pyrin domain containing 3 (NLRP3) inflammasome is crucial for long-term inflammatory response (24). The NLRP3 inflammasome consists of NLRP3, the adaptor protein apoptosis–associated speck-containing protein with a CARD (ASC), and pro–caspase-1. Upon stress exposure, it assembles inside cells and leads to increased cleavage and activity of caspase-1, as well as downstream release of interleukin-1β (IL-1β). Activated IL-1β then stimulates downstream cytokines and chemokine receptors to trigger inflammatory responses (24). Activation of proinflammatory caspase-1 drives Gasdermin D (GSDMD)–mediated pyroptosis (i.e., proinflammatory programmed cell death) (25). The NLRP3 inflammasome can be activated by components of glycolysis and reactive oxygen species and also senses cellular metabolites and damage-associated molecular patterns, positioning it as an intracellular sensor of metabolic stress (26). It has also been demonstrated that the NLRP3 inflammasome drives development and progression of amyloidogenesis (27) and tau hyperphosphorylation-related NFTs (28). Unknown, however, is whether NLRP3 inflammasome signaling can sense metabolic alterations of OLs in AD and thereby contribute to AD-associated neuropathology.
In the present study, we found that mature OLs in human AD patients and AD mice exhibited NLRP3-dependent GSDMD-associated inflammatory injury, concomitant with demyelination and axonal degeneration. OL-specific down-regulation of Drp1 in cell culture and AD transgenic mice abolished NLRP3 inflammasome activation and inflammatory response. Notably, heterozygous knockout of Drp1 in mature OLs of AD mice corrected myelin loss, reduced axonal degeneration, and improved cognitive performance. Mechanistically, we found that Drp1 hyperactivation in mature OLs suppressed glycolysis by inhibiting hexokinase 1 (HK1), the enzyme that initiates glycolysis by catalyzing phosphorylation of glucose to glucose-6-phosphate. This, in turn, activates the NLRP3 inflammasome. Overall, our findings have revealed a Drp1-HK1-NLRP3 signaling axis in mature OLs that elicits metabolic stress in OLs, thereby triggering inflammation and OL injury that culminates in demyelination, white matter degeneration, and cognitive impairment in models of AD.
RESULTS
NLRP3 inflammasome activation and GSDMD-marked cell injury in mature OLs of AD models
OL injury is a prerequisite to demyelination and white matter degeneration. To evaluate changes in OLs in AD, we first performed triple-label immunofluorescence staining of the postmortem cortex from five AD patients and five neurologically normal subjects (see the information of human subjects in Table 1). In the AD patient cortex, we found that Aβ plaques marked with the anti-6E10 antibody contained myelin basic protein (MBP)+ or 2′,3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase)+ mature OLs (Fig. 1, A and C). In the core of Aβ plaques, the area covered by anti-MBP or anti-CNPase immune-positive signal was reduced, which was not observed in plaque-free areas of AD patient brains (Fig. 1, A to D). These data are consistent with previous studies (29) and indicate that damaged mature OLs and focal demyelination are associated with Aβ plaques in AD. OLs in the core and neighboring areas of Aβ plaques were immunopositive for anti-GSDMD antibody, indicating the occurrence of NLRP3-dependent pyroptotic injury. GSDMD-labeled OLs were rarely observed in control subjects (Fig. 1, A to D).
Table 1. AD patients’ and normal subjects’ postmortem brain samples (paraffin-embedded).
| ID | Age | Gender | Brain region | Diagnosis |
| A98-78 | 75 | M | Cortex | Unaffected control |
| A97-197 | 77 | F | Cortex | Unaffected control |
| A02-29 | 77 | M | Cortex | Unaffected control |
| A97-83 | 74 | F | Cortex | Unaffected control |
| HCT15HAU_19_04_PC | 65 | M | Cortex | Unaffected control |
| BEB18005_19_04_PC | 75 | M | Cortex | AD |
| HBAX_19_04_PC | 86 | F | Cortex | AD |
| HBBD_19_04_PC | 70 | M | Cortex | AD |
| HBGB_19_04_PC | 85 | F | Cortex | AD |
| HBGS_19_04_PC | 78 | F | Cortex | AD |
Fig. 1. Mature OLs undergo GSDMD-marked pyroptotic injury and inflammation.
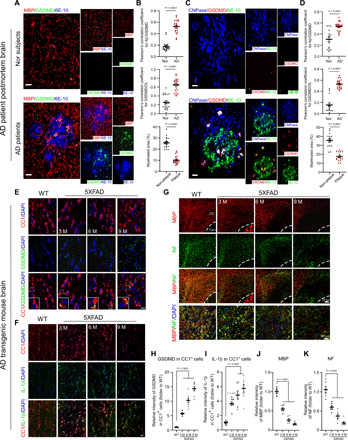
(A) Postmortem cortex brain sections from normal (Nor) subjects and AD patients (Table 1) were stained with anti-MBP, anti-GSDMD, and anti–6E-10 antibodies. (B) Quantification: (top) colocalization of GSDMD and Aβ plaques; (middle) localization of GSDMD in MBP+ OLs; (bottom) the area covered by MBP+ OLs in Aβ plaques and non-Aβ plaque area. Student’s t test. (C) Postmortem cortex brain sections from normal subjects and AD patients were stained with anti-CNPase, anti-GSDMD, and anti–6E-10 antibodies. (D) Quantification: (top) the colocalization of GSDMD in Aβ plaques; (middle) the localization of GSDMD in CNPase+ OLs; (bottom) the area covered by CNPase+ OLs in Aβ plaques and non-Aβ plaque area. Student’s t test. Note: To quantify the colocalization of proteins, Pearson’s correlation coefficient of two proteins per 45,000 μm2 was determined using ImageJ plug-in JaCoP. Fifteen to 21 images per group were analyzed. The brains of WT and 5XFAD mice were harvested at the ages of 3, 6, and 9 months (M). Brain sections were stained with (E) anti-GSDMD (green) and anti-CC1 (a marker of mature OL, red) antibodies, (F) anti–IL-1β (green) and anti-CC1 (red), and (G) anti-neurofilament (NF, green) and anti-MBP (red). n = 3 to 5 mice per group. The histogram shows the intensities of GSDMD (H) and IL-1β (I) in the CC1+ OLs, and the intensities of MBP (J) and NF (K) in the corpus callosum of mice. Scale bar, 20 μm. The data were compared by one-way analysis of variance (ANOVA) with Tukey’s post hoc test. All data are expressed as the means ± SEM.
These findings prompted us to examine NLRP3 inflammasome activation in OLs in AD transgenic mice. We used 5XFAD transgenic mice that expressed amyloid precursor protein (APP) Swedish, Florida, and London mutations, as well as presenilin (PS1) M146L and L286V mutations. These mice accumulate deposits of Aβ plaques throughout vulnerable brain regions and also develop cognitive deficits (30). We harvested mouse brains at 3, 6, and 9 months of age. Immunostaining revealed an increase in the intensity of GSDMD in the adenomatous polyposis coli clone CC1 (CC1)–positive mature OLs in the corpus callosum (i.e., a region that is enriched in OLs and myelin) of 5XFAD mice, starting at 3 months old, which continued to increase with age (Fig. 1, E and H). The immunodensities of NLRP3 and IL-1β concomitantly increased in CC1+ OLs in the same brain regions of 5XFAD mice (fig. S1, A and B, and Fig. 1, F and I). Increases in the immunodensities of Iba1 and glial fibrillary acidic protein (GFAP) confirmed the induction of inflammation (fig. S1, C and D). By contrast, GSDMD immunosignal was rarely observed in the GFAP+ astrocytes in the corpus callosum of the 5XFAD mice (fig. S1E). Consistent with a previous study (27), we observed increased NLRP3 and GSDMD immunodensities in the Iba1+ microglia of the corpus callosum of 5XFAD mice (fig. S1F). Western blot revealed that NLRP3 protein levels also age-dependently increased in the corpus callosum in 5XFAD mice. The cleavage of pro–caspase-1, GSDMD, and pro–IL-1β, reflecting activation of the NLRP3 inflammasome, was present in the corpus callosum in 5XFAD mice and accumulated with age (fig. S1G). We also observed age-dependent demyelination, reflected by lower immunodensity of anti-MBP (Fig. 1, G and J) and lower myelin-covered area that was assessed by anti-MBP immunohistochemistry and Black Gold II staining in 5XFAD mice (fig. S1, H and I). The immunodensity of neurofilaments (NFs) in MBP+ OLs also age-dependently decreased in the corpus callosum in 5XFAD mice (Fig. 1, G and K).
In OLs that differentiated from OPCs, the addition of oligomeric Aβ1-42 (i.e., a toxic form of the Aβ peptide) caused an increase in NLRP3 protein levels, the cleavage of GSDMD, and the release of IL-1β (fig. S2, A to C). These changes were not observed in mature OLs treated with the oligomeric control peptide Aβ42-1 (fig. S2, D and E). Treatment with oligomeric Aβ1-42 peptides did not cause rapid cell death (fig. S2F), suggesting that the toxic Aβ1-42 peptide induced a noncanonical pyroptotic injury, similar to that previously observed in macrophages (31) and neutrophils (32). These data indicate that in the AD models, mature OLs undergo GSDMD-mediated pyroptotic injury and inflammation via NLRP3 inflammasome activation, which is associated with demyelination and axonal degeneration.
Drp1 hyperactivation in mature OLs in AD models
We previously reported that Drp1 is hyperactivated in mature OLs exposed to tumor necrosis factor–α and oxidative stress and that systemic inhibition of Drp1 hyperactivation reduced mature OL death and demyelination in animal models of multiple sclerosis (33). In the present study, we examined whether Drp1 was activated in the OLs in AD models. Drp1 oligomerization and Drp1 phosphorylation at Ser616 residue (p-Drp1Ser616) are markers of Drp1 activation. In the postmortem cortex samples from AD patients (Table 2) and the corpus callosum of 5XFAD mice, Drp1 tetramer levels were much higher than in the corresponding control samples (Fig. 2, A and C). The number of p-Drp1Ser616+ CC1+ OLs of the AD patients was also significantly increased compared to the control subjects (Fig. 2B). In the corpus callosum of the 5XFAD mice, we observed an increase in the levels of mitochondrial fission factor (Mff), a Drp1 mitochondrial fission adaptor protein, but few changes in the levels of other fission adaptor proteins [e.g., mitochondrial fission 1 protein (Fis1) and mitochondrial elongation factor 1 (MIEF)] and fusion components [e.g., mitofusin 1 (Mfn1) and Mfn2] (fig. S2G). In addition, Drp1 tetramer and p-Drp1Ser616 levels increased in mature OLs exposed to oligomeric Aβ1-42 compared to control cells (Fig. 2, D and E). The increase in Drp1 activation positively correlated with GSDMD cleavage (Fig. 2F). Note that there was no Drp1 oligomerization induced in the mature OLs following treatment with the oligomeric control peptide Aβ42-1 (fig. S2H). While the protein level of Mff increased, no changes in other mitochondrial fusion or fission components in mature OLs exposed to oligomeric Aβ1-42 were observed (fig. S2I). Thus, Drp1-mediated mitochondrial fission was overactivated in human AD brains, the white matter of AD mice, and mature OLs exposed to the toxic Aβ peptide. Mff, rather than other mitochondrial fission adaptor proteins, may be a primary factor in mature OLs that mediates the hyperactivation of mitochondrial fission.
Table 2. AD patients’ and normal subjects’ postmortem brain samples (frozen).
| ID | Age | Gender | Brain region | Diagnosis |
| HCTYP 1801 | 75 | M | Cortex | Control |
| HCTYP 1803 | 76 | M | Cortex | Control |
| HCTYP 1805 | 74 | M | Cortex | Control |
| HCTYP 1807 | 76 | M | Cortex | Control |
| S17925-AN18943 | 90 | M | Cortex | Control |
| HBRC_19_02_PC | 86 | F | Cortex | AD |
| HBBD_19_02_PC | 70 | M | Cortex | AD |
| HBGT_19_02_PC | 68 | F | Cortex | AD |
| HBGZ_19_02_PC | 91 | F | Cortex | AD |
| S13006-AN07603 | 88 | M | Cortex | AD |
Fig. 2. Drp1 hyperactivation in mature OLs of AD models.
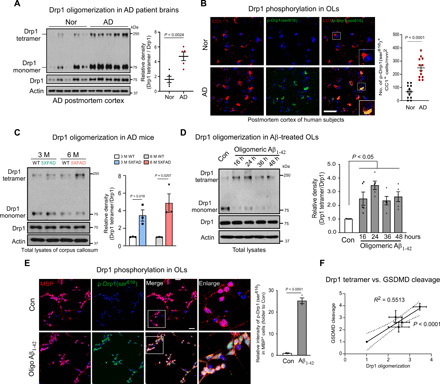
(A) Total protein lysates were harvested from the frozen postmortem cortex of AD patients and control subjects (Table 2). Western blot analysis was performed in the absence of 2-mercaptoethanol (β-ME) to examine Drp1 oligomers. Histogram: the relative density of Drp1 tetramer versus Drp1 monomer is shown. Student’s t test. (B) Postmortem cortex sections from normal subjects and AD patients (Table 1) were stained with anti–p-Drp1 Ser616 and anti-CC1 antibodies. Scale bar, 50 μm. The number of p-Drp1 Ser616+ CC1+ cells per square millimeter was quantified and is presented in the histogram. Student’s t test. (C) Total protein lysates from the corpus callosum of WT and 5XFAD mouse brains were harvested at the ages of 3 and 6 months. Drp1 tetramer was analyzed by Western blot analysis in the absence of β-ME. The relative density of Drp1 tetramer versus the Drp1 monomer is shown in the histogram. n = 3 mice per group. Student’s t test. (D) Mature OLs were differentiated from OPCs and exposed to oligomeric Aβ1-42 for the indicated periods. Drp1 tetramer was analyzed by Western blot analysis of total protein lysates in the absence of β-ME. The relative density of Drp1 tetramer versus the Drp1 monomer is shown in the histogram. The data from five independent experiments were analyzed using one-way ANOVA with Tukey’s post hoc test. (E) Mature OLs were treated with oligomeric Aβ1-42 for 24 hours and stained with anti–p-Drp1 Ser616 (green) and anti-MBP (red) antibodies. The intensity of p-Drp1Ser616 in the MBP+OLs was quantitated and is presented in the histogram. The data from three independent experiments were compared using the Student’s t test. Scale bar, 20 μm. (F) A linear correlation between the relative Drp1 tetramer density and the cleavage of GSDMD is shown. All data are expressed as the mean ± SEM.
Suppression of Drp1 hyperactivation in mature OLs abolishes AD-associated NLRP3 inflammasome activation and pyroptotic injury
In mature OLs exposed to oligomeric Aβ1-42, both Drp1 down-regulation by lentiviral Drp1 short-hairpin RNA (shRNA; fig. S3A) and Drp1 inhibition by the small-molecule inhibitor mitochondrial division inhibitor 1 (Mdivi-1) (34) inhibited Drp1 hyperactivation (fig. S3, B to D), prevented the increase in NLRP3 protein level, and blocked cleavage of pro–caspase-1 and GSDMD (Fig. 3A and fig. S3E). The presence of lentiviral Drp1 shRNA and treatment with Mdivi-1 significantly reduced the release of IL-1β in mature OLs exposed to oligomeric Aβ1-42 (Fig. 3A and fig. S3F), suggesting the inhibition of Aβ-induced inflammation. In parallel, we evaluated the effects of adding lentivirus encoding green fluorescent protein (GFP) and Drp1–wild-type (WT) or Drp1-K38A mutant, a mutant that eliminates Drp1-mediated fission activity (35), to mature OLs. Here, expression of Drp1-WT induced GSDMD cleavage, whereas the Drp1-K38A mutant had no effect (Fig. 3B). These data suggest that Drp1 is upstream of NLRP3 inflammasome activation.
Fig. 3. Suppression of Drp1 hyperactivation attenuates NLRP3-dependent inflammasome activation and GSDMD-mediated pyroptotic injury in cell culture and AD mice.
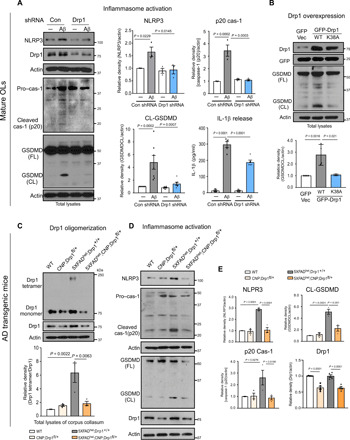
(A) Mature OLs were infected with lentivirus encoding Drp1 or control shRNA for 48 hours and maintained in the cell culture medium with puromycin (1 μg/ml), followed by treatment with oligomeric Aβ1-42 (5 μM) for 24 hours. Western blot analysis of total protein lysates was carried out with the indicated antibodies. The densities of NLRP3, cleaved caspase-1, and GSDMD relative to actin are shown in the histograms. The released IL-1β into the medium was measured by ELISA. The data from at least three independent experiments were analyzed. (B) Total lysates of mature OLs expressing GFP control vector (GFP Vec), GFP-Drp1 WT (WT), or GFP-Drp1 K38A (K38A) were subjected to Western blot analysis with anti-GSDMD antibody. The density of GSDMD cleavage relative to actin is shown in the histogram. The data are from four independent experiments. Total brain lysates were harvested from 6-month-old WT, CNP;Drp1fl/+, 5XFADhet;Drp1+/+, and 5XFADhet;CNP;Drp1fl/+ mice. n = 3 to 5 mice per group. (C) Drp1 tetramer was analyzed by Western blot analysis in the absence of β-ME. The relative density of Drp1 tetramer versus the Drp1 monomer is shown in the histogram. (D) Western blot analysis of mouse corpus callosum lysates was carried out with the indicated antibodies. (E) The densities of Drp1, NLRP3, cleaved caspase-1, and GSDMD (CL) relative to actin are shown in the histograms. All the data are expressed as the mean ± SEM and compared by one-way ANOVA with Tukey’s post hoc test.
Pharmacologic inhibition of Drp1 hyperactivation by P110 peptide or Mdivi-1 had protective effects in AD mouse models (13, 17). However, none of the Drp1 inhibitors could reveal a selective role for Drp1 in mature OLs of AD mice. Thus, we used a mature OL-specific genetic knockdown approach. Drp1 homozygous knockout is embryonically lethal (36, 37), but heterozygous knockout mice are healthy and fertile (38). We heterozygously knocked out Drp1 from mature OLs in Drp1fl/fl mice by expressing Cre recombinase at the endogenous CNP locus (hereafter referred to as CNP; Drp1fl/+). We then generated double-mutant 5XFADhet;CNP;Drp1fl/+ mice by crossing 5XFAD heterozygous mice with CNP;Drp1fl/+ mice (fig. S4A). 5XFADhet;CNP;Drp1fl/+ double-mutant mice were born at the expected Mendelian ratio and were indistinguishable from WT and 5XFAD littermates, suggesting lack of overt developmental deficits. No differences in body weight were observed between double-mutant and CNP;Drp1fl/+ mice compared to the WT and 5XFADhet/Drp1+/+ mice (fig. S4B).
Immunostaining confirmed the down-regulation of Drp1 in the CC1+ OLs of both CNP;Drp1fl/+ and double-mutant 5XFADhet;CNP;Drp1fl/+ mice (fig. S4C). There was no decrease in Drp1 immunoreactivity observed in either the GFAP+ astrocytes of the corpus callosum or the NeuN+ neurons in the cortex of both CNP;Drp1fl/+ mice and WT littermates (fig. S4D), supporting the specific heterozygous knockout of Drp1 in the mature OLs. Western blot analysis also revealed that total protein levels of Drp1 were lower in the corpus callosum in CNP;Drp1fl/+ and 5XFADhet;CNP;Drp1fl/+ mice than in the WT littermates and 5XFADhet/Drp1+/+ mice (Fig. 3, C to E). We focused on the corpus callosum because it is an OL- and myelin-rich region, meaning that its protein expression more accurately reflects the effect of Drp1 knockdown on OL protein. The oligomers of Drp1 in the corpus callosum in 5XFADhet/Drp1+/+ mice were higher than in the age-matched WT littermates. The level of Drp1 oligomers in 5XFADhet;CNP;Drp1fl/+ mice of the same age returned to the levels of their WT littermates (Fig. 3C). Thus, removing one copy of the Drp1 gene in mature OLs reduced Drp1 oligomerization in 5XFAD mice. The protein level of NLRP3 and the cleavage of pro–caspase-1 and GSDMD consistently increased in the corpus callosum in 5XFADhet/Drp1+/+ mice at 6 months of age, all of which were abolished in 5XFADhet;CNP;Drp1fl/+ mice (Fig. 3, D and E). Thus, the mature OL-specific heterozygous knockout of Drp1 suppressed NLRP3 inflammasome activation in AD mice.
Mature OL-specific heterozygous knockout of Drp1 reduces AD pathology and cognitive deficits
Next, we examined the AD pathology in double-mutant 5XFADhet;CNP;Drp1fl/+ mice. First, we performed triple-label immunostaining of brain sections from the 5XFADhet;Drp1+/+ mice. Consistent with the observations made in human AD patient postmortem brains (Fig. 1), GSDMD+ and MBP+ OLs increased in the core and neighboring areas of anti–6E10-positive Aβ plaques. This aberrant event was diminished in 5XFADhet;CNP;Drp1fl/+ mice with limited MBP+ and GSDMD+ OLs (Fig. 4, A and B). Iba1+ microglia appeared to surround the GSDMD that accumulated in the Aβ plaques (fig. S1J), which is likely due to the recruitment of the microglia to the plaques (39). The immunodensity of IL-1β in CC1+ OLs (Fig. 4, C and D) and immunodensities of GFAP and Iba1 in the corpus callosum (fig. S4, H and I) were significantly lower in 5XFADhet;CNP;Drp1fl/+ mice than in 5XFADhet/Drp1+/+ mice. No changes in the immunodensity of IL-1β, GFAP, or Iba1 were observed between the WT and CNP;Drp1fl/+ mice (Fig. 4, C and D, and fig. S4, H and I). The myelin-covered area, assessed by anti-MBP immunohistochemistry and Black Gold II myelin staining, in 5XFADhet;CNP;Drp1fl/+ mice was significantly larger than that in 5XFADhet/Drp1+/+ mice (Fig. 4, E and F), indicating an improvement in myelination. The lower intensity of NFs in MBP+ OLs was evident in 5XFADhet/Drp1+/+ mice, which was corrected in 5XFADhet;CNP;Drp1fl/+ mice (fig. S4, E to G).
Fig. 4. Mature OL-specific Drp1 heterozygous knockout is protective in 5XFAD mice.
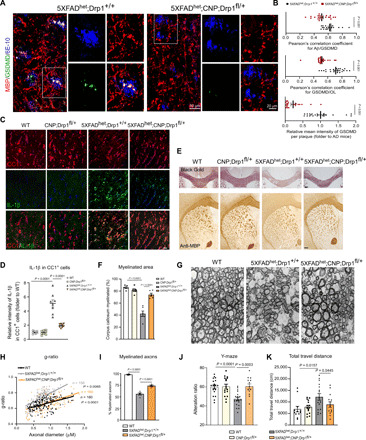
(A) Brain sections containing the corpus callosum from 6-month-old 5XFADhet;Drp1+/+ and 5XFADhet;CNP;Drp1fl/+ mice were stained with anti-GSDMD, anti-MBP, and anti-6E10 antibodies. (B) The colocalization of GSDMD and Aβ plaques (top) and the localization of GSDMD in MBP+OLs (middle) were quantified. The intensity of GSDMD in the Aβ plaque area was analyzed (lower). n = 3 to 4 mice per group. (C) Brain sections from 6-month-old WT, CNP;Drp1fl/+, 5XFADhet;Drp1+/+, and 5XFADhet;CNP;Drp1fl/+ mice were stained with anti–IL-1β (green) and anti-CC1 (red) antibody for immunofluorescence analysis. n = 4 mice per group. Scale bar, 20 μm. (D) The intensity of IL-1β in CC1+OLs was quantified. (E) Mouse brain sections were stained with the Black Gold II myelin staining kit (top) and anti-MBP for immunohistochemistry analysis (bottom). Scale bar, 200 μm. (F) The area of positive anti-MBP staining was quantified and is presented in the histogram. (G) The corpus callosum of each mouse of the indicated genotypes was harvested for TEM analysis. (H) The g-ratio of the myelin of the mice was measured. (I) The percentage of myelinated axons was quantified. (J) Cognitive activity was assessed in 6-month-old mice using the Y-maze. Nine female and 10 male WT, 8 female and 8 male CNP;Drp1fl/+, 9 female and 10 male 5XFADhet;Drp1+/+, and 7 female and 6 male 5XFADhet;CNP;Drp1fl/+ mice were used. (K) Movement activity was assessed in 6-month-old mice using the open-field chamber. The total travel distance of mice is shown. Six female and 6 male WT, 8 female and 7 male CNP;Drp1fl/+, 8 female and 8 male 5XFADhet;Drp1+/+, and 7 female and 7 male 5XFADhet;CNP;Drp1fl/+ mice were used. All the data are expressed as the mean ± SEM and compared by one-way ANOVA with Tukey’s post hoc test.
To further examine the changes in myelin and axon integrity in AD mice, we next performed transmission electron microscopy (TEM) analysis of the corpus callosum in mice. Compared to the WT mice, 5XFADhet/Drp1+/+ mice exhibited abnormal myelin, including ballooned myelin, degenerated sheaths, degenerated axons that were surrounded by degenerated sheaths, and the loss of myelinated axons. These abnormalities were diminished in 5XFADhet;CNP;Drp1fl/+ mice (Fig. 4G). Quantitative TEM analysis revealed that 5XFADhet/Drp1+/+ mice had significantly thinner myelin, which was reflected in the increase in the g-ratio, compared to the WT mice, and the lower percentage of axons that were surrounded by myelin (Fig. 4, H and I). By contrast, 5XFADhet;CNP;Drp1fl/+ mice exhibited a decreased g-ratio and a higher percentage of axons surrounded by myelin (Fig. 4, H and I). These data further support our observation that heterozygous knockout of Drp1 in mature OLs prevented myelin degeneration and axonal loss in AD mice.
In parallel, we examined the behavior of animals representing all of the genotypes. 5XFADhet/Drp1+/+ mice had decreased short-term cognitive ability as assessed by the Y-maze at 6 months of age. Consistent with a previous report (40), the 5XFAD mice were hyperactive during the open-field test. By contrast, 5XFADhet;CNP;Drp1fl/+ mice showed improvement in the alteration ratio of the Y-maze test and normalization of the total distance traveled in the open-field test to levels similar to those observed with the WT mice (Fig. 4, J and K). CNP;Drp1fl/+ mice did not exhibit alterations of behavior compared to the WT mice (Fig. 4, J and K).
Synaptic loss more strongly correlates with cognitive decline in AD than the numbers of plaques and tangles or neuronal loss (41). To determine the impact of the heterozygous knockout of Drp1 in mature OLs of AD mice on synapse loss, we immunostained mouse brain sections with antibodies against synaptophysin and postsynaptic density–95 kDa (PSD95) and assessed the colocalization of pre- and postsynaptic puncta, which reflect the synaptic density (41). Confocal imaging revealed a significant decrease in the synaptic density in the cortex of 5XFADhet;Drp1+/+ mice at 6 months of age. By contrast, the loss of synapses was attenuated in age-matched 5XFADhet;CNP;Drp1fl/+ mice (fig. S4, J to L). These findings are consistent with the improvement in cognitive activity observed with 5XFADhet;CNP;Drp1fl/+ mice (Fig. 4J). In summary, our results demonstrate that the mature OL-specific heterozygous knockout of Drp1 reduced AD-associated pathology and behavioral phenotypes in 5XFAD mice.
Oligodendroglial Drp1 hyperactivation induces a glycolytic defect in AD models
To determine the molecular basis by which mature OL Drp1 hyperactivation induces NLRP3 inflammasome activation, inflammation, and pyroptotic injury, we harvested the corpus callosum from 6-month-old mice of all genotypes and performed unbiased label-free proteomic analysis. Among the 1098 proteins that were identified (fig. S5A), we focused on those that were altered in 5XFADhet/Drp1+/+ mice (i.e., >2-fold down- or up-regulated relative to the WT mice) and simultaneously modified in 5XFADhet;CNP;Drp1fl/+ mice (i.e., >2-fold up- or down-regulated relative to 5XFADhet/Drp1+/+ mice; Fig. 5A). A total of 300 proteins were used for the protein pathway analysis (Fig. 5A, marked with *). A graphical comparison of the top 10 enriched KEGG pathways and the top 10 enriched biological pathways showed significant alterations in metabolic pathways, in which proteins enriched in glycolysis were mostly affected (Fig. 5B). Heatmap analysis further showed that the proteins enriched in glycolysis and the TCA cycle were all down-regulated in the 5XFADhet/Drp1+/+ mice and up-regulated in 5XFADhet;CNP;Drp1fl/+ mice (Fig. 5C and fig. S5B). We thus hypothesized that an impairment in mature OL glycolysis might be a primary molecular event resulting from Drp1 hyperactivation in AD models.
Fig. 5. Mature OL-specific Drp1 hyperactivation induces glycolytic stress in AD mice.
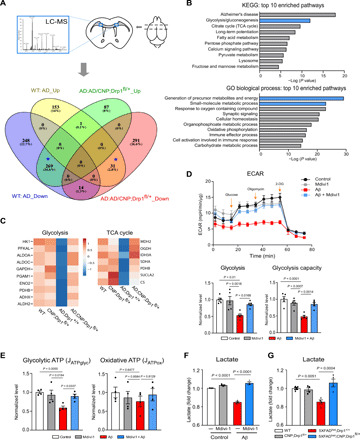
(A) The corpus callosum was harvested from 6-month-old WT, CNP;Drp1fl/+, 5XFADhet;Drp1+/+, and 5XFADhet;CNP;Drp1fl/+ mice (shown in scheme). Label-free tandem mass spectrometry analysis was conducted to identify the proteins changed by mature OL-specific Drp1 heterozygous knockout in 5XFAD mice. The number of proteins changed in mice of the four genotypes is shown. In particular, 269 proteins accounting for 24.6% of total identified proteins were decreased in 5XFAD mice but elevated in 5XFADhet;CNP;Drp1fl/+ mice. Thirty-one proteins were increased in 5XFAD mice and decreased in 5XFADhet;CNP;Drp1fl/+ mice. These 300 proteins were used for bioinformatics analysis. (B) The molecular and cellular functions of the 300 proteins from the pool marked as * in (A) were analyzed. The proteins were enriched for proteins involved in glycolysis and the TCA cycles and significantly regulated by mature OL-specific Drp1 heterozygous knockout. (C) Heatmap analysis for these enriched proteins is shown. (D) Mature OLs were treated with oligomeric Aβ1-42 for 24 hours in the presence or absence of Mdivi-1 (5 μM). The ECAR was analyzed with the seahorse analyzer (n = 2 replicates per group). The data are from four to five independent experiments. (E) Glycolytic and oxidative ATP production rates were calculated as described in Materials and Methods. (F) The lactate content of mature OLs was measured. The data are from at least three independent experiments. (G) Total protein lysates were prepared from the corpus callosum of mice with the indicated genotypes, and the lactate content was measured. n = 4 mice per group. The data are from at least three independent experiments. All the data are expressed as the mean ± SEM and compared by one-way ANOVA with Tukey’s post hoc test.
We next monitored metabolic dynamics of mature OLs in vitro by treating them with oligomeric Aβ1-42 in the presence or absence of Mdivi-1, a small-molecule inhibitor of mitochondrial fission. The real-time extracellular acidification rate (ECAR; i.e., a measure of glycolysis) decreased in mature OLs after exposure to Aβ1-42 for 24 hours, a cell culture condition that mimics chronic damage in AD before severe cell damage. By contrast, treatment with Mdivi-1 significantly restored the ECAR and improved the glycolytic capacity of mature OLs exposed to oligomeric Aβ1-42 (Fig. 5D). Mitochondrial function was also examined by evaluating changes in the oxygen consumption rate (OCR) under the same treatment conditions. Oligomeric Aβ1-42 treatment lowered the maximal respiratory capacity of mitochondria in mature OLs, whereas treatment with Mdivi-1 improved this, albeit not significantly (fig. S5C). To determine whether the glycolytic rates were truly different between conditions, we used the basal ECAR and OCR obtained above to calculate the glycolytic and oxidative adenosine triphosphate (ATP) production rates, respectively, using the method described in (42). Mature OLs exposed to oligomeric Aβ1-42 for 24 hours resulted in a decrease in the glycolytic ATP production rate, which was abrogated by treatment with Mdivi-1 (Fig. 5E). By contrast, the oxidative ATP production rate under the same treatment condition was comparable among the experimental groups (Fig. 5E). The decrease in the oligodendroglial glycolytic rate induced by Aβ was further confirmed by measuring the level of lactate, a major by-product of glycolysis. Mdivi-1 treatment resulted in lactate levels similar to control cells (Fig. 5F). Consistent with these data, lactate levels in the corpus callosum of 5XFADhet/Drp1+/+ mice were lower than those in their WT littermates (Fig. 5G). The lactate levels were restored in the 5XFADhet;CNP;Drp1fl/+ mice. Together, our data demonstrated that Drp1 hyperactivation caused low glycolytic rate and decreased lactate content in mature OLs from AD models. However, since oligomeric Aβ1-42 decreased both ECAR and OCR, we cannot exclude the possibility that the improvement in ECAR by Mdivi-1 treatment is an indirect consequence of reduced cell damage. Future studies on the specificity of Drp1 for glycolysis in mature OLs may help us understand the role of Drp1 in the regulation of energy metabolism.
Oligodendroglial Drp1 inhibits HK1, eliciting glycolytic stress and inflammation in AD models
Glycolysis mediates glucose metabolism to provide metabolites for energy generation. HK1 is the rate-limiting enzyme that initiates the first step of glycolysis by the phosphorylation of glucose (43). As shown in Fig. 5, the proteins that were enriched in the glycolysis pathway were all down-regulated in AD mice but up-regulated by heterozygous knockout of Drp1 in mature OLs. We speculated that Drp1 hyperactivation might influence oligodendroglial glycolysis by inhibiting HK1, resulting in a sequential decrease in proteins downstream of this enzyme. Western blot analysis confirmed that the HK1 protein levels were lower in the corpus callosum of 5XFAD mice compared to the WT mice, and heterozygous knockout of Drp1 corrected this defect (fig. S5D). To determine the molecular involvement of HK1 in AD, we first computationally analyzed the priority of HK1 in AD phenotypes, genes, and pathways by performing a virtual screening of a total of 10,072 prioritized disease phenotypes and 23,499 prioritized genes. We prioritized biomedical entities using the context-sensitive network–based ranking algorithm (see Supplementary Methods). Data mining suggested that HK1 is associated with AD-specific phenotypes and AD-associated genes, ranking in the top 27.75 and 22.60%, respectively, which were significantly higher than random ranking (P < 0.01; fig. S5, E to G). The top-ranked pathways for HK1 are related to the immune response and metabolism (fig. S5H), consistent with the molecular action of mature OL Drp1 hyperactivation in AD (Fig. 5B).
We found that HK1 was enriched in the CC1+ OLs of the control subjects who were neurologically normal, whereas the immunodensity of HK1 significantly decreased in the CC1+ OLs in the postmortem cortex samples from the AD patients (Fig. 6A), indicating the loss of HK1 in AD. HK1 immunodensity also decreased in the CC1+ OLs of the corpus callosum and the adjacent gray matter of 5XFADhet/Drp1+/+ mice, which was corrected in the 5XFADhet;CNP;Drp1fl/+ mice (Fig. 6B and fig. S6A). In the brain, HK1 is the major isozyme present, whereas HK2 is typically induced by stress. We failed to detect HK2 in the CC1+ OLs in either WT or 5XFADhet/Drp1+/+ mice (fig. S6B), consistent with the cell-type specificity of HK isoforms (44). To determine whether the decrease of HK1 also occurred in other cells of the corpus callosum, we focused on astrocytes that also rely on glycolysis. Different from that in mature OLs, the HK1 immunodensity in GFAP+ astrocytes in the corpus callosum was low, consistent with a previous report (45). The HK1 levels in the GFAP+ astrocytes of WT and 5XFAD mice were comparable (fig. S6C). Given that heterozygous knockout of Drp1 was restricted to mature OLs (fig. S4, C and D), we inferred that the improved HK1 levels in 5XFADhet;CNP;Drp1fl/+ mice were likely from the mature OLs. However, we cannot exclude a possible cell nonautonomous effect of the heterozygous mature OLs-specific Drp1 loss on HK1 levels, such as in the neurons where HK1 is highly expressed.
Fig. 6. Drp1-HK1 axis induces NLRP3 inflammasome activation in OLs of AD.
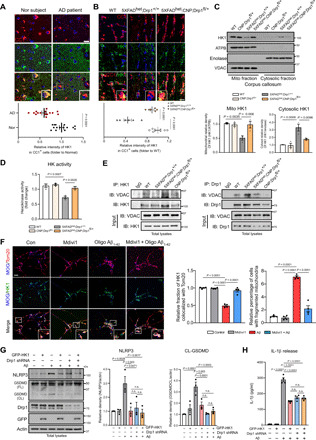
(A) Postmortem cortex sections from normal subjects and AD patients (Table 1) were stained with anti-HK1 and anti-CC1 antibodies. Scale bar, 20 μm. The HK1 intensity in CC1+ OLs was quantified. (B) Brain sections from mice of the indicated genotypes were stained with anti-HK1 and anti-CC1 antibodies. Scale bar, 20 μm. The HK1 intensity in CC1+ OLs of mice was quantified. n = 3 mice per group. (C) The corpus callosum was harvested from 6-month-old mice. Western blot analysis of mitochondrial and cytosolic fractions was performed. The HK1 density on mitochondrial and cytosolic fractions was quantified. n = 3 mice per group. ATPB, a mitochondrial inner membrane protein. Enolase, a cytosolic protein. VDAC, a mitochondrial outer membrane protein. (D) HK1 activity was measured using protein lysates harvested from mouse corpus callosum. n = 3 mice per group. (E) The corpus callosum was harvested from 6-month-old mice. Immunoprecipitation with anti-HK1 or anti-Drp1 was carried out. n = 3 mice per group. (F) Mature OLs were treated with Mdivi-1 (5 μM) 4 hours followed by the addition of oligomeric Aβ1-42 (5 μM). After 24 hours, cells were stained with anti-Tom20, anti-HK1, and anti-MOG antibodies. Left: Representative images from three independent experiments. Right: Percentage of mitochondrial HK1 density out of total HK1 density in MOG+ cells and percentage of MOG+ cells with fragmented mitochondria. Scale bar, 20 μm. (G) Mature OLs were infected with Drp1 shRNA followed by overexpression of GFP-HK1 for 48 hours, and cells were treated with oligomeric Aβ1-42 (5 μM) for 24 hours. Western blot analysis was performed. n.s., not significant. The densities of NLRP3 and cleaved GSDMD relative to actin are shown in histograms. (H) The release of IL-1β was analyzed by ELISA. All the data are expressed as the mean ± SEM and compared by one-way ANOVA with Tukey’s post hoc test.
HK1 localizes to the OMM, where it interacts with voltage-dependent anion channels (VDACs) to regulate glycolysis (46). The dissociation of HK1 from the mitochondrial VDACs causes a significant decrease in its enzyme activity, which is sufficient to inhibit glycolysis (46) and induce NLRP3 inflammasome activation (47). We isolated the mitochondrial and cytosolic fractions from the corpus callosum of mice. The purity of the mitochondrial fractions was determined by the enrichment of mitochondrial protein [Mitochondrial ATP Synthase Beta Subunit (ATPB] and the relative absence of myelin (MBP), presynapse (synaptophysin), axon (Tau), and cytosolic (enolase) proteins (fig. S6D). Western blot analysis revealed that HK1 protein levels decreased in the mitochondrial fractions and increased in the cytosolic fractions in the corpus callosum of 5XFADhet/Drp1+/+ mice beginning at 3 months of age (fig. S6E), suggesting that HK1 dissociated from mitochondria at an early stage of AD. By contrast, Drp1 began to accumulate on the mitochondria of the corpus callosum in the 5XFAD mice at 3 months old and continued to accumulate with age. Its levels were inversely correlated with the mitochondrial HK1 levels (fig. S6E). The dissociation of HK1 from the mitochondria in the 5XFAD mice was abolished by the mature OL-specific heterozygous knockout of Drp1 (Fig. 6C). The overall HK enzyme activity in the corpus callosum was decreased in 5XFADhet/Drp1+/+ mice but restored in the 5XFADhet;CNP;Drp1fl/+ mice (Fig. 6D).
Immunoprecipitation of total corpus callosum proteins revealed a decrease in the interaction between HK1 and VDAC in 5XFAD mice (Fig. 6E and fig. S6F) but not in 5XFADhet;CNP;Drp1fl/+ mice (Fig. 6E). By contrast, increased binding between Drp1 and VDAC in the corpus callosum occurred in the 5XFAD mice. The mature OL-specific heterozygous knockout of Drp1 in 5XFAD mice corrected this aberrant interaction between Drp1 and VDAC and restored the formation of the HK1 and VDAC complex (Fig. 6E). These results indicate that oligodendroglial Drp1 hyperactivation inhibited glycolytic activity of HK1, likely by competitively binding to the VDACs to disrupt the HK1/VDAC complex on mitochondria. Furthermore, in myelin OL glycoprotein (MOG)+ mature OLs, the addition of oligomeric Aβ1-42 peptide induced extensive mitochondrial fragmentation and reduced HK1 localization on mitochondria compared to control cells (Fig. 6F). Under the same stress conditions, treatment with Mdivi-1 greatly reduced the percentage of cells with fragmented mitochondria and enhanced HK1 association with mitochondria (Fig. 6F). In addition, overexpression of GFP-HK1 abrogated the degree of mitochondrial fragmentation in MOG+ mature OLs exposed to oligomeric Aβ1-42 (fig. S6I). These findings suggest that competitive binding of Drp1 and HK1 to mitochondria simultaneously affects mitochondrial morphology. Compensation for HK1 loss on mitochondria of mature OLs corrected mitochondrial fragmentation, which may result from blocking Drp1 mitochondrial accumulation.
2-Deoxy-d-glucose (2-DG) is a potent inhibitor of glycolysis via its action on HKs. Treatment of mature OLs with 2-DG decreased cellular lactate levels and increased NLRP3 protein levels (fig. S6, G and H), confirming that glycolysis inhibition was sufficient to activate the NLRP3 inflammasome (47). Aβ triggers HK1 detachment from mitochondria, decreasing HK1 activity (48). Thus, we transfected GFP-HK1 plasmid into mature OLs followed by addition of oligomeric Aβ1-42. HK1 overexpression alone abolished the increases in NLRP3 protein levels and the cleavage of GSDMD (Fig. 6G) and the release of IL-1β (Fig. 6H) induced by oligomeric Aβ1-42. These results were similar to those observed with mature OLs infected with Drp1 shRNA or treated with Mdivi-1 (Fig. 3 and fig. S3). Down-regulation of Drp1 by either Drp1 shRNA expression or Mdivi-1 treatment followed by HK1 overexpression had no additive effects on NLRP3 levels or the release of IL-1β (Fig. 6, G and H, and fig. S6, J and K), suggesting that Drp1 and HK1 are part of the same signaling pathway. Thus, Drp1 hyperactivation-induced HK1-dependent glycolytic stress elicited NLRP3 activation in mature OLs, leading to inflammation.
DISCUSSION
In the present study, aberrant Drp1 activation was a primary signal that disturbed the glycolytic homeostasis of mature OLs in AD, triggering sequential inflammatory damage and subsequent white matter loss and axonal degeneration (Fig. 7). These findings were further corroborated by our results that showed that the mature OL-specific heterozygous knockout of Drp1 corrected the HK1-mediated glycolytic defect in mature OLs, reduced NLRP3 inflammasome activation, attenuated the loss of myelin and axons, and improved cognitive performance in AD mice. Thus, our work here provides evidence of a crucial role for the Drp1-HK1-NLRP3 signaling axis in maintaining the metabolic homeostasis of mature OLs. Overall, our findings provide previously unidentified insights into the mechanism of white matter degeneration in AD and thus identify a new area for potential therapeutic intervention.
Fig. 7. The scheme of the study.
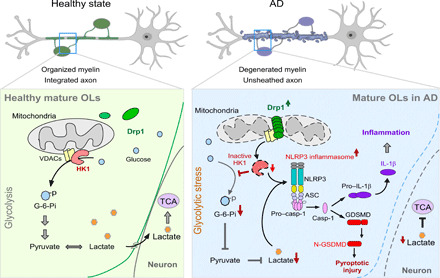
Under healthy conditions, HK1 localizes on the mitochondria outer membrane where it binds to VDAC. Mitochondrial HK1 converts glucose to glucose-6-phosphate (G-6-Pi) via phosphorylation, which initiates glycolysis, generating pyruvate and lactate as energy resources. Lactate can also enter neurons to support the TCA cycle and neuronal survival. In AD, Drp1 in mature OLs is hyperactivated by stress (e.g., soluble Aβ). Hyperactivated Drp1 translocates to the mitochondria, where it preferentially binds to VDAC, dissociating HK1 from VDAC. This dissociation inactivates HK1 and suppresses glycolysis. HK1 inactivation and glycolytic inhibition results in glycolytic stress, which induces NLRP3 inflammasome activation, cleavage of GSDMD and pro–IL-1β, and inflammatory injury of the mature OLs. The results are demyelination and axonal degeneration. Down-regulation of Drp1 in the mature OLs blocks these abnormal events from their start and, therefore, reduces the OL inflammation, myelin loss, and white matter degeneration observed in AD.
White matter tracts are composed of myelinated white fibers that provide the connectivity central to cognition, emotion, and consciousness. The preservation of axonal integrity is maintained by the myelin sheath and critical for facilitating synaptic function and neuronal transmission (49). Thus, functional disturbance of OLs may result in synaptic deficits and cognitive impairment apparent in the early stages of AD. OLs are highly susceptible to Aβ peptide (50). Studies of 3XTg-AD and APP/PS1 mice, for example, showed that OL damage and myelin disruption correlate with the earliest appearance of Aβ accumulation before disease symptom onset (4, 51). Cellular accumulation of Aβ was also shown to age-dependently drive degeneration of myelin and axons, as well as overall shrinkage of the corpus callosum in 5XFAD mice (52, 53). A recent study showed thread-like Aβ in the periaxonal space underneath the myelin sheath in the white matter of 5XFAD mice before plaque deposition (54). Thread-like Aβ may disturb the activity of OLs and initiate early white matter pathology. In our study, soluble Aβ–induced Drp1 hyperactivation in mature OLs was a driving force in the downstream cellular damage, as the genetic and pharmacological inhibition of Drp1 in mature OLs abolished Aβ-induced inflammatory and metabolic stress. A decline of trophic support in dysregulated OLs might lead to axonal energy deprivation, manifested by impairments in axonal transport and disturbances in lipid homeostasis and neuronal metabolism, exacerbating Aβ accumulation and AD pathology. The soluble Aβ–induced metabolic damage of OLs via the Drp1-HK1-NLRP3 signaling axis may contribute to neuroinflammation and development of mature Aβ plaques that observed later in the gray matter in both animals and patients. Our observations propose a signaling pathway that supports a vicious cycle between Aβ accumulation and OL metabolic deficiency.
Mature OLs arise from a large population of precursor cells that are defined by the expression of OL transcriptional factor 2 and the chondroitin sulfate proteoglycan Neuron glia antigen-2 (NG2) (55). In the present study, we marked mature OLs with specific markers (i.e., MBP and CC1) to ensure that our observations were made in defined cells. In contrast to mature OLs, previous studies found that NG2-glia expression and proliferation increased in AD mice (56), reflecting a compensatory response to OL damage. In human AD, NG2-glia exhibit senescence-like phenotypes (9), suggesting an impairment in the continual capacity of OL precursors to repair and proliferate under prolonged AD-related stress. OPCs mainly use oxidative phosphorylation to maintain an energy supply, whereas mature OLs rely heavily on glycolysis (6). The different metabolic programs of these cells may reflect different vulnerabilities to AD insult. In AD, mature OLs might be more vulnerable to changes in metabolic homeostasis because of the lack of self-renewal or a loss of compensation from the OPCs. We previously showed that OPCs are resistant to the damage induced by Drp1 hyperactivation in multiple sclerosis (33), suggesting that there might be an OL lineage-selective cell death mechanism driven by aberrant Drp1 activation. This hypothesis requires further investigation, especially in the context of AD.
We found that inhibition of HK1 by Drp1 activation in mature OLs could be one of the mechanisms to induce NLRP3 inflammasome activation in AD. HK inhibition and dissociation from the mitochondria is an essential step in slowing the rate of glycolysis. HK1 binds to and stabilizes VDACs on OMM (46). Dissociation of HK1 from VDACs causes instability of the mitochondrial permeability transition pore that releases large molecules such as mitochondrial DNA into the cytosol, induces mitochondrial reactive oxygen species production, and causes cardiolipin externalization to the OMM (44). All these events could be activators of the NLRP3 inflammasome (24). HK1 inactivation due to dissociation from the mitochondria is sufficient to induce NLRP3 inflammasome activation (47). We further found that either 2-DG or toxic Aβ-induced inhibition of glycolysis in mature OLs was sensed by NLRP3, triggering subsequent inflammation. Wolf et al. (47) reported that injection of HKVBD, a cell-permeable peptide that blocks the interaction between VDACs and HK1, could induce inflammation and the recruitment of inflammatory cells in mice. This response was reduced in mice deficient in caspase-1, providing an in vivo causative link between HK1 dissociation from the mitochondria and NLRP3 inflammasome activation. Thus, energy sensing and dysregulated glycolysis are prerequisites for NLRP3 inflammasome activation; however, the factors that connect HK1 and NLRP3 have not been identified. We showed that Drp1 competitively bound to VDAC, which resulted in the dissociation of HK1 from the complex, and that HK1 could cooperate with Drp1 to induce higher NLRP3 levels and the release of IL-1β. In OLs, HK1 could be a bridging molecule that links Drp1 hyperactivation and NLRP3 activation. Note that the cell culture model does not replicate the development of chronic pathology observed in the AD animal model. Future studies using mice overexpressing HK1 in mature OLs of AD mice will help address the impact of HK1 restoration on NLRP3 inflammasome activity and AD-associated neuropathology.
NLRP3 inflammasome activation and pyroptosis were previously observed in myelin-forming OLs in multiple sclerosis (57), suggesting that OLs conserve their ability to respond to NLRP3 signaling like other immune cells. Unlike many strong NLRP3 inflammasome activators, such as ATP and nigericin, oligomeric Aβ did not stimulate rapid pyroptosis but induced GSDMD cleavage via NLRP3 inflammasome activation. This cellular damage likely resulted from the low level of chronic inflammasome activation induced by Aβ in OLs compared to the strong acute NLRP3 activators. Because there was no observable loss of CC1+ OLs in 6-month-old 5XFAD mice, we exclude the possibility that the increased NLRP3/GSDMD levels in the mature OLs of the AD mice at this age resulted from other cells. Zhang et al. suggest the role of Aβ-induced oligodendroglial senescence in triggering local inflammatory damage (9). Although not causing death, glycolytic stress could exacerbate myelination withdrawal (58). Consistently, here, we demonstrated the functional damage of mature OLs as a result of glycolytic stress–induced inflammasome activation. Our speculation is also supported by a recent study that showed that chronic injection of Aβ peptide in rat eyes induces prolonged NLRP3 inflammasome activation, which causes pyroptotic death in the retinal pigment epithelium only in the very late stage of the injury (59).
Mature OL-specific Drp1 heterozygous knockout mice did not exhibit metabolic, gross physical, or behavioral abnormalities. This is consistent with previous findings of a lack of obvious phenotypes in global Drp1 heterozygous knockout mice (37) or synaptic and mitochondrial deficiencies (38). By contrast, Drp1 heterozygous knockout in 5XFAD mice attenuated mature OL injury, demyelination, and axonal degeneration, echoing a recent finding that partial global reduction in Drp1 reduces Aβ production and corrects both mitochondrial dysfunction and synaptic activity in APP transgenic mice (18). Thus, our results provide additional evidence that targeting Drp1 could be a therapeutic strategy against AD. Notably, we demonstrated that OL Drp1 is a key factor that mediates white matter degeneration in AD. Drp1 interaction with Aβ is likely to induce Drp1 activation in AD (16). We speculate that soluble Aβ–induced Drp1 hyperactivation may result in a decrease in mitochondrial energy deficits in OLs that is caused by excessive mitochondrial fission. To correct this redox imbalance, OLs require more pyruvate and lactate as energy fuel sources that can enter mitochondria to restore redox balance and increase flux through glycolysis to restore ATP levels. Simultaneously, Drp1 translocation to mitochondria disrupts the binding of HK1/VDACs, leading to HK1 dissociation from mitochondria and subsequently suppressing glycolysis. The competitive binding of Drp1 and HK2 (an isozyme of HK in the heart) to the mitochondria was observed in cardiomyocytes exposed to ischemic stress (60). Yang et al. reported that inhibition of Drp1 activation with siRNA or Mdivi-1 increased mitochondrial HK2 in cardiomyocytes under ischemic stress. By contrast, inhibition of HK2 by 2-DG, a modified glucose molecule that induces HK2 detachment from the mitochondria, recruited Drp1 to the mitochondria. Notably, the competitive binding of Drp1 and HK2 to the mitochondria concomitantly occurred with a change of mitochondrial morphology; 2-DG treatment enhanced mitochondrial fragmentation, whereas both Mdivi-1 and Drp1 siRNA reduced the abnormal mitochondrial morphology (60). Thus, Drp1 activation under stress conditions could be a central node that regulates both mitochondrial morphology and glycolysis. In summary, in our proposed model, soluble Aβ induced Drp1 activation in mature OLs, which induced Drp1 translocation to the mitochondria where Drp1 formed oligomers. The oligomeric Drp1 on the mitochondria competed with HK1 to bind to VDACs, which caused HK1 dissociation from the mitochondria and subsequent glycolysis stress and NLRP3 inflammasome activation. A decrease in the production of metabolites worsens energy deficiency and exacerbates glycolytic impairment, leading to the inflammatory injury of mature OLs, myelin loss, and axonal degeneration. The suppression of Drp1 hyperactivation in AD attenuated these abnormal events.
Mitochondrial adaptors including Fis1, Mff, and MiD49/51 recruit Drp1 to the mitochondria via transient protein-protein interactions. These fission adaptors may play different roles in basal mitochondrial fission and fission under pathological conditions. We found that Mff was more sensitive to AD-associated stress than other fission adaptors in mature OLs, suggesting a possible mechanism by which Drp1 is recruited to the mitochondria in mature OLs in the context of AD. Furthermore, we showed that Drp1 bound to the VDACs in the corpus callosum of AD mice, which suppressed HK1-mediated glycolysis. Future work will investigate whether Mff cooperates with VDACs to recruit Drp1 on the mitochondria, and the role of Mff in OL metabolic homeostasis in AD.
In summary, we demonstrate here the importance of OL glycolytic deficiency in both initiation and development of AD and highlight activation of the Drp1-HK1-NLRP3 signaling axis as a key mechanism of white matter degeneration in AD. Glycolysis is an important step in maintaining glucose metabolism, and glucose metabolism deficiency is a notable characteristic of the pathogenesis of AD. HK can be regarded as the gatekeeper of glycolysis in the brain, as its expression correlates directly with the rate of glucose metabolism across multiple brain regions. Given the low expression of HK1 in astrocytes (fig. S6) (45), our findings may also provide insights into where and how this shift in glucose metabolism occurs in AD. Future studies on the lactate shuttle between neurons and OLs via the Drp1-HK1 axis may warrant a better understanding of the mechanism that underlies the blockade of neuronal glucose uptake in AD.
MATERIALS AND METHODS
Antibodies and reagents
Protein phosphatase inhibitor and protease inhibitor cocktails were purchased from Sigma-Aldrich. Antibodies against Drp1 (catalog no. 611113, 1:3000) and OPA1 (catalog no. 612607, 1:2000) were purchased from BD Biosciences. Antibodies against IL-1β (catalog no. ab9722, 1:500), synaptophysin (catalog no. ab32127, 1:1000), and VDAC (catalog no. ab34726, 1:3000) were purchased from Abcam. Antibodies against GSDMDC1 (catalog no. NBP2-33422, 1:500), AIF1/Iba1 (catalog no. NB100-1028, 1:1000), and MBP (catalog no. NB600717, 1:500) were purchased from Novus. Antibodies against phospho-Drp1 (Ser616, catalog no. 3455, 1:500), HK1 (catalog no. 2024, 1:2000), cleaved GSDMD (Asp275, catalog no. 36425, 1:500), and caspase-1 (D7F10, catalog no. 3866, 1:500) were purchased from Cell Signaling Technology. Antibodies against DLP1/Drp1 (catalog no. bs-4100R, 1:500) and IL-1β (catalog no. bs-6319R, 1:500) were purchased from Bioss. Antibodies against Fis1 (catalog no. 10956-1-AP, 1:2000), Mff (catalog no. 17090-AP, 1:2000), MIEF1 (catalog no. 20164-1-AP, 1:1000), HK1 (catalog no. 19662-1-AP, 1:500), HK2 (catalog no. 22029-1-AP, 1:500), Tau (catalog no. 10274-1-AP, 1:500), NLRP3 (catalog no. 19771-1-AP, 1:500), and Tom20 (catalog no. 11802-1-AP, 1:1000) were purchased from ProteinTech. Antibodies against Mfn1 (catalog no. H00055669-M04, 1:2000) and Mfn2 (catalog no. H00009927-M01, 1:2000) were purchased from Abnova. Antibodies against caspase-1 (catalog no. sc-56036, 1:500), Enolase (catalog no. sc-56036, 1:3000), and Tom20 (catalog no. sc-17764, 1:500) were purchased from Santa Cruz Biotechnology. Antibodies against GFAP (catalog no. MAB360, 1:1000) and CC1 (catalog no. OP80, 1:500) were purchased from Millipore. The antibody against Iba1 (catalog no. 019-19741, 1:1000) was purchased from Wako. Antibodies against β-tubulin TUBB3 (catalog no. 801201, 1:1000) and amyloid1-16 (catalog no. 803015, 1:1000) were purchased from BioLegend. The antibody against MBP (catalog no. SMI-99P, 1:500) was purchased from Covance. Antibodies against β-actin (catalog no. A1978, 1:10,000), NF (catalog no. N4142, 1:1000), NeuN (catalog no. MAB377, 1:500), and 4′,6-diamidino-2-phenylindole (DAPI) (catalog no. D9542, 1:10,000) were purchased from Sigma-Aldrich. The antibody against CNPase (catalog no. GTX85451, 1:100) was purchased from Genetex. The antibodies against PSD95 (catalog no. MA1-045, 1:500) and MOG (catalog no. PIPA547319, 1:300) were purchased from Invitrogen. NLRP3 (catalog no. AG-20B-0014-C100, 1:500) was purchased from AdipoGen. Horseradish peroxidase (HRP)–conjugated anti-mouse and anti-rabbit secondary antibodies; Alexa 405/488/568 goat anti-mouse, anti-rabbit, and anti-rat secondary antibodies; and Alexa 488/568 donkey anti-mouse and anti-rabbit secondary antibodies were purchased from Thermo Fisher Scientific. Donkey anti-goat Alexa 405 secondary antibody was purchased from Abcam. Dylight 405 donkey anti-chicken immunoglobulin G (IgG) (H + L) was purchased from Jackson ImmunoResearch. Mdivi-1 (catalog no. M0199) and 2-DG were purchased from Sigma-Aldrich.
Preparation of oligomeric Aβ1-42 and control peptide Aβ42-1
The Aβ1-42 (GenicBio Limited) and Aβ42-1 peptide (AnaSpec) peptides were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; Sigma-Aldrich) to a final concentration of 5 mM and placed under a chemical hood overnight. The next day, HFIP was further evaporated using a SpeedVac Concentrator for 1 hour. Monomer Aβ (5 mM) was prepared by dissolving Aβ peptide in anhydrous dimethyl sulfoxide (Sigma-Aldrich). The oligomeric Aβ and control peptides were prepared by diluting the monomer Aβ solution in Dulbecco’s modified Eagle’s medium (DMEM)/F12 and then incubating at 4°C for 24 hours.
Cell culture
Human OPCs (catalog no. SCR600) were purchased from Millipore. The differentiation of progenitor cells into mature OLs followed the protocols of the Human Oligodendrocyte Differentiation Kit (catalog no. SCR600, Millipore). The immunostaining of anti-MBP antibody was used to confirm the presence and yield rate of mature OLs. The differentiation protocol yielded 67.72 ± 4.8% MBP+ OLs and 37.01 ± 2.73% β-tubulin III+ neuronal cells. Neither Iba1+ nor GFAP+ immunosignals were observed in the differentiated cells.
Constructs and transfection
To construct the lentiviral Drp1 plasmid encoding enhanced GFP, human WT or mutant Drp1 cDNA was amplified using a standard polymerase chain reaction method and then inserted into pHRIG-AktDN (catalog no. 53597, Addgene) between the Sal I and Bam HI restriction sites upstream of the IRES sequence. Cells were transfected with plasmids using TransIT-2020 (Mirus Bio) according to the manufacturer’s protocol. Lentiviruses were generated by transfecting human embryonic kidney–293T cells with plasmids that encoded the envelope (pCMV-VSV-G; catalog no. 8454, Addgene), packing (psPAX2; catalog no. 12260, Addgene), and targeted open reading frame. After 12 hours of transfection, the medium was changed, and the lentiviruses were harvested after 36 hours. The lentiviruses were diluted with the corresponding medium at a 1:1 ratio, and the cells of interest were infected in the presence of Polybrene (8 μg/ml, Sigma-Aldrich) for 48 hours.
To knock down Drp1 in OLs, the OLs were transduced with Drp1 shRNA (catalog no. TRCN0000318425, Sigma-Aldrich) or control shRNA (catalog no. 8453, Addgene), followed by selection with puromycin (1 μg/ml) for 48 hours. For HK1 overexpression, OLs were transfected with FLHK1-pGFPN3 (catalog no. 21917, Addgene) using TransIT-2020 (Mirus Bio, LLC) for 48 hours, following the manufacturer’s protocol.
Generation of mature OL-specific Drp1 heterozygous knockout AD mice
All of the animal experiments were conducted in accordance with protocols that were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University and were performed according to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Sufficient procedures were used to reduce pain and discomfort of the mice during the experiments. The mice were mated, bred, and genotyped in the animal facility of Case Western Reserve University. All of the mice were maintained under a 12-hour light/12-hour dark cycle (lights on at 6 a.m. and lights off at 6 p.m.). All of the mice that were used in this study were maintained on a C57BL/6J background.
C57BL/6N-Atm1Brd Dnm1ltm1a(KOMP)Wtsi/Ics mice (EM: 08240) were generated by the Mouse Clinical Institute in France and purchased from the KOMP Repository. The mouse strain is Dnm1L (Drp1) heterozygous knockout-first mice. A pair of loxP sites were inserted, flanking Drp1 exon 2, and a LacZ-neomycin cassette that flanked FRT was inserted in intron 1, which terminated Drp1 transcription. The knockout-first mice were then bred with Flp recombinase transgenic mice (ACTB:FLPe B6J, catalog no. 005703, the Jackson Laboratory) to remove the LacZ-neomycin cassette, yielding Drp1fl/+ heterozygous Drp1 conditional knockout mice that contained the Drp1 allele with exon 2 flanked by LoxP sites. Homozygous Drp1fl/fl mice were generated by crossing Drp1fl/+ mice. Drp1fl/fl mice were then bred with the CNP-Cre+ mouse line to generate CNP-Cre;Drp1fl/+ mice. Double-mutant 5XFADhet;CNP;Drp1fl/+ mice were generated by crossing heterozygous 5XFAD mice (B6.Cg-Tg[APPSwFlLon, PSEN1*M146L*L286V] 6799Vas/Mmjax, catalog no. 008730, the Jackson Laboratory) with CNP;Drp1fl/+ mice. Inbred age-matched and sex-balanced CNP;Drp1fl/+, 5XFADhet;Drp1+/+, and 5XFADhet;CNP;Drp1fl/+ mice and WT littermates were used for further experiments.
Behavioral analysis
All of the behavioral analyses were conducted by an experimenter who was blinded to genotypes. Locomotor activity was assessed in an open field in all mouse genotypes at 6 months of age. Briefly, the mice were placed in the center of an activity chamber (Omnitech Electronics) and allowed to explore the chamber while being tracked by an automated infrared tracking system (Vertax, Omnitech Electronics). A 12-hour locomotor activity analysis was performed. For the Y-maze assessment, each mouse was placed in the middle of three arms of the Y-maze and allowed to freely explore the apparatus. Entries into limbs that passed through each half arm were recorded for 6 min to obtain the total number of arm entries and the percentage of alternations. Body weight and survival rate were recorded throughout the study period.
Enzyme-linked immunosorbent assay
Mature OLs were treated with Aβ (5 μM), and the conditioned medium was collected 16, 24, 36, and 48 hours after Aβ treatment. The OLs were incubated with Mdivi-1 (5 μM) 4 hours before Aβ treatment, and the conditioned medium was collected 24 hours after treatment. Drp1 knockdown OLs were treated with Aβ (5 μM), and the conditioned medium was collected 24 hours after treatment. IL-1β that was released from OLs was measured using enzyme-linked immunosorbent assay (ELISA) kits (DY201-05, R&D Systems) according to the manufacturer’s protocol. Absorbance at 450 nm was read using a microplate reader.
ECAR and OCR measurements
The ECAR and OCR were measured using the Seahorse Bioscience XFp Extracellular Flux Analyzer, according to the manufacturer’s instructions. Briefly, the cells (5000 per well) were seeded in XFp six-well microplates in 100 μl of growth medium and treated with Aβ (5 μM) with or without Mdivi-1 (5 μM) for 24 hours. For the OCR measurement, 1 hour before measuring oxygen consumption, the cell culture medium was replaced with XF assay medium (1 mM pyruvate, 2 mM glutamine, and 10 mM glucose in basal DMEM) and maintained in a non-CO2 incubator for 1 hour, and then, the sensor cartridges were placed in the XFp Analyzer for the Mito Stress Test. Mitochondrial function was determined by sequentially injecting 5 μM oligomycin A, 5 μM Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), and 2.5 μM rotenone and antimycin A. For the ECAR measurement, the cell culture medium was replaced with XF assay medium (2 mM glutamine in basal DMEM). Glycolysis was measured by sequentially injecting 10 mM glucose, 5 μM oligomycin, and 50 mM 2-DG. Results were normalized to the amount of protein in each group. Glycolytic and oxidative ATP production rates were calculated as previously described (42).
Lactate dehydrogenase assays
Cells were treated with Aβ (1, 5, or 10 μM) for 24 hours, and cell death was determined using the Cytotoxicity Detection Kit (lactate dehydrogenase) according to the manufacturer’s protocols (Roche, REF 11 644 793 001).
Measurement of lactate
Mouse tissues and the cells were homogenized in the lactate assay buffer, and lactate levels were measured using a Lactate Colorimetric Assay kit (catalog no. K627-100, BioVision) according to the manufacturer’s instructions.
Measurement of HK activity
Mouse tissues were homogenized in the HK assay buffer, and HK activity was measured using the Hexokinase Colorimetric Assay Kit (catalog no. K789-100, BioVision) according to the manufacturer’s instructions.
Immunohistochemistry
The mice were deeply anesthetized and transcardially perfused with 4% paraformaldehyde in phosphate-buffered saline (PBS). Dissected brains were postfixed in 4% paraformaldehyde overnight at 4°C and equilibrated in 30% sucrose. Frozen coronal brain sections (12 μm) were hydrated and treated with 3% H2O2 in methanol and then treated with 5% normal goat serum for 1 hour at room temperature. The sections were incubated with mouse anti-MBP antibody (1:500; Covance) in a humidified chamber overnight at 4°C. After being washed with PBS, the IHC Select HRP/DAB Kit (catalog nos. 20774 and 20775, Millipore) was used to examine the immunohistochemical localization of MBP. Images were captured using a digital microscope (VHX-7000, Keyence). MBP immunostaining was quantified using ImageJ software (NIH). The same image exposure times and threshold settings were used for sections from all groups. An experimenter who was blinded to the experimental groups conducted the quantification.
Immunofluorescence analysis
Cells were grown on coverslips, fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized with 0.1% Triton X-100 in PBS, and blocked with 2% normal goat serum or donkey serum. The cells were incubated with the following primary antibodies overnight at 4°C: (i) mouse anti-MBP (1:500; Covance) with rabbit anti–phospho-Drp1 (Ser616, 1:500; Cell Signaling Technology) and mouse anti–β-tubulin TUBB3 (1:1000; BioLegend); (ii) goat anti-MOG (1:300; Invitrogen), mouse anti-Tom20 (1:500, Santa Cruz Biotechnology), and rabbit anti-HK1 (1:1000, ProteinTech); and (iii) goat anti-MOG (1:300, Invitrogen) with rabbit anti-Tom20 (1:1000, Proteintech). After being washed with PBS, the cells were incubated with Alexa Fluor 488/568, 405/568 secondary antibody (1:500; Thermo Fisher Scientific) for 2 hours at room temperature, followed by Nuclei staining with DAPI (1:10,000; Sigma-Aldrich) for double staining, and incubated with Alexa Fluor 405/488/568 secondary antibody for triple staining. After mounting the cells on slides, the images were captured under a microscope.
Frozen mouse coronal brain sections (12 μm) were prepared and triple- or double-stained with the following antibodies: (i) mouse anti-amyloid1-16 (catalog no. 6E10, 1:1000; BioLegend) and rabbit anti-GSDMDC1 (1:500; Novus) with rat anti-MBP (1:500; Novus); (ii) mouse anti-CC1 (1:500; Millipore) with rabbit anti-GSDMDC1 (1:500; Novus), rabbit anti–IL-1β (1:500; Bioss), rabbit anti-NLRP3 (1:500; ProteinTech), rabbit anti-Drp1 (1:500; Bioss), rabbit anti-HK1 (1:500; ProteinTech), or rabbit anti-HK2 (1:500; ProteinTech); (iii) mouse anti-MBP (1:500; Covance) with rabbit anti-NF (1:1000; Sigma-Aldrich); (iv) mouse anti-GFAP (1:1000; Millipore) with rabbit anti-Iba1 (1:1000; Wako), rabbit anti-HK1 (1:500; ProteinTech), rabbit anti-Drp1 (1:500; Bioss), or rabbit anti-GSDMDC1 (1:500; Novus); (v) rabbit anti-Iba1 (1:1000; Wako) with mouse anti-NLRP3 (1:500; Adipogen); (vi) mouse anti-amyloid1-16 (catalog no. 6E10, 1:1000; BioLegend) with rabbit anti-GSDMDC1 (1:500; Novus) or goat anti-Iba1 (1:1000; Novus); (vii) goat anti-Iba1 (1:1000; Novus) with rabbit anti-GSDMDC1 (1:500; Novus); (viii) mouse anti-PSD95 (1:500; Invitrogen) with rabbit anti-synaptophysin (1:1000; Abcam); and (ix) mouse anti-NeuN (1:500; Sigma-Aldrich) with rabbit anti-Drp1 (1:500; Bioss). For the immunofluorescent analysis of postmortem brains from normal subjects and AD patients, paraffin-embedded sections (5 μm) were triple- or double-stained with the following antibodies: (i) mouse anti-amyloid1-16 (catalog no. 6E10, 1:1000; BioLegend) and rabbit anti-GSDMDC1 (1:500; Novus) with rat anti-MBP (1:500; Novus) or chicken anti-CNPase (1:100; Genetex) and (ii) mouse anti-CC1 (1:500; Millipore) with rabbit anti–phospho-Drp1 (Ser616, 1:500; Cell Signaling Technology) or rabbit anti-HK1 (1:500; Cell Signaling Technology). The sections were incubated with these antibodies in a humidified chamber overnight at 4°C. After being washed with PBS, the sections were incubated with a mixture of secondary antibodies that were conjugated with goat anti-mouse or rabbit Alexa 405/488/568 (1:500; Thermo Fisher Scientific), donkey anti-mouse or rabbit Alexa 488/568 (1:500; Thermo Fisher Scientific), donkey anti-goat Alexa 405 (1:500; Abcam), or donkey anti-chicken Dylight 405 (1:500; Jackson ImmunoResearch) for 2 hours at room temperature.
To minimize autofluorescence, the human and mouse tissue sections were incubated with 0.1% Sudan Black B for 20 min (diluted in 70% ethanol). After three washes with sterile water, the tissue sections were subject to immunostaining.
All of the sections were counterstained with DAPI (1:10,000; Sigma-Aldrich) and mounted with mounting medium (Dako). Images were acquired using a Fluoview FV 1000 confocal microscope (Olympus). Immunostaining quantification was performed using ImageJ software. The same image exposure times and threshold settings were used for sections from all of the groups. An experimenter who was blinded to the experimental groups conducted the quantification.
Black Gold II myelin staining
Black Gold II myelin staining was performed according to the manufacturer’s instructions (catalog no. AG105, Millipore). Briefly, 20-μm sections were incubated in prewarmed 0.3% Black Gold II solution at 60°C for 12 min. The sections were rinsed and incubated with 1% sodium thiosulfate solution for 3 min at 60°C. After rinsing with water, the sections were dehydrated in a graded series of alcohol, air-dried, and mounted in mounting solution. Images were acquired from matched areas of stained sections using a digital microscope (VHX-7000, Keyence). The data represent pooled results from at least three brains per experimental group. Black Gold II myelin staining was quantified using ImageJ software. An experimenter who was blinded to genotypes of the samples conducted the quantification.
Transmission electron microscopy
The mice were transcardially perfused with quarter-strength Karnovsky’s fixative solution at a flow rate of 10 ml/min for 3 min. Small pieces of the mouse corpus callosum were fixed by immersion in quarter-strength Karnovsky’s fixative solution for 2 hours at room temperature. The specimens were thoroughly rinsed in 0.1 M phosphate buffer (pH 7.4) and then postfixed for 2 hours in an unbuffered 1:1 mixture of 2% osmium tetroxide and 3% potassium ferricyanide. After rinsing with distilled water, the specimens were soaked overnight in an acidified solution of 0.25% uranyl acetate. After another rinse in distilled water, the sections were dehydrated in an ascending series of alcohol, passed through propylene oxide, and embedded in EMbed 812 resin mixture (Electron Microscopy Sciences). Thin sections were sequentially stained with acidified uranyl acetate, followed by a modification of Sato’s triple lead stain, and examined under an FEI Tecnai Spirit (T12) TEM with a Gatan US4000 4K × 4K charge-coupled device camera. The g-ratio was calculated from 155 to 160 randomly selected myelinated axons and measured as the axon diameter/total diameter of the axon plus the myelin sheath using ImageJ software.
Label-free proteomics
Each frozen mouse corpus callosum (n = 5 mice per group) was collected in a 1.5-ml tube containing 300 μl of 2% SDS and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). The samples were incubated on ice for 30 min and then sonicated with a probe sonicator at 50% amplitude followed by vortexing; this cycle was repeated four times with samples sitting on ice between each round. Following lysis, the samples were processed using a filter-aided sample preparation cleanup protocol with Amicon Ultra MWCO 3K filters (Millipore, Billerica, MA). Samples were reduced and alkylated on the filters with 10 mM dithiothreitol (Acros, Fair Lawn, NJ) and 25 mM iodoacetamide (Acros, Fair Lawn, NJ), respectively, and then concentrated to a final volume of 40 μl in 8 M urea. Protein concentration was measured using the Bradford method according to the manufacturer’s instructions (Bio-Rad, Hercules, CA).
Following reduction and alkylation, total protein (10 mg) was subjected to enzymatic digestion. The urea concentration was adjusted to 4 M using 50 mM tris (pH 8) and then proteins were digested with mass spectrometry–grade lysyl endopeptidase (Wako Chemicals, Richmond, VA) in an enzyme/substrate ratio of 1:40 for 2 hours at 37°C. The urea concentration was further adjusted to 2 M using 50 mM tris (pH 8), and the lysyl peptides were then digested with sequencing-grade trypsin (Promega, Madison, WI) in an enzyme/substrate ratio of 1:40 at 37°C overnight. Last, the samples were diluted in 0.1% formic acid (Thermo Fisher Scientific, Rockford, IL) before liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis.
The peptide digests (320 mg) were loaded onto a column in an 8-μl injection volume with blanks in between for a total of four LC/MS/MS runs. The resulting data were acquired with an Orbitrap Velos Elite mass spectrometer (Thermo Electron, San Jose, CA) equipped with a Waters nanoACQUITY LC system (Waters, Taunton, MA). The peptides were desalted on a trap column (180 μm by 20 mm, packed with C18 Symmetry, 5 μm, 100 Å; Waters, Taunton, MA) and subsequently resolved on a reversed-phase column (75 μm by 250 mm nano column, packed with C18 BEH130, 1.7 μm, 130 Å; Waters, Taunton, MA). LC was carried out at ambient temperature at a flow rate of 300 nl/min using a gradient mixture of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). The gradient ranged from 4 to 44% solvent B over 210 min. The peptides eluting from the capillary tip were introduced into the nanospray mode with a capillary voltage of 2.4 kV. A full scan was obtained for the eluted peptides in the range of 380 to 1800 atomic mass units followed by 25 data-dependent MS/MS scans. The MS/MS spectra were generated by collision-induced dissociation of the peptide ions at a normalized collision energy of 35% to generate a series of b and y ions as major fragments. In addition, a 1-hour wash was included between each sample. The proteins were identified and quantified using PEAKS 8.5 (Bioinformatics Solutions Inc., Waterloo, ON, Canada).
Isolation of subcellular fractions
Mouse brains were minced and homogenized in lysis buffer [250 mM sucrose, 20 mM Hepes-NaOH (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM ethylenediaminetetraacetic acid, protease inhibitor cocktail, and phosphatase inhibitor cocktail] and then placed on ice for 30 min. The collected tissues were disrupted 20 times by repeated aspiration through a 25-gauge needle, followed by a 30-gauge needle. The homogenates were centrifuged at 800g for 10 min at 4°C. The resulting supernatants were centrifuged at 10,000g for 20 min at 4°C. The pellets were washed with lysis buffer and centrifuged at 10,000g again for 20 min at 4°C. The final pellets were suspended in lysis buffer that contained 1% Triton X-100, yielding mitochondrion-rich lysate fractions. The supernatant contained cytosolic fractions. Mitochondrial ATPB protein, VDACs, and cytosolic enolase protein were used as loading controls for the mitochondrial and cytosolic fractions, respectively.
Coimmunoprecipitation
Tissues were lysed in total cell lysate buffer [50 mM tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail]. Total lysates were incubated with the indicated antibodies overnight at 4°C, followed by the addition of protein A/G beads for 2 hours at room temperature. Immunoprecipitates were washed four times with cell lysate buffer and analyzed by Western blot analysis.
Western blot
Protein concentrations were determined by protein assay dye reagents (Bio-Rad). Proteins were resuspended in Laemmli buffer, loaded on sodium dodecyl sulfate-polyacrylamide gels, and transferred to nitrocellulose membranes. Membranes were probed with the indicated antibodies, followed by visualization by electrochemiluminescence.
Human brain tissues
The postmortem normal subject brain tissues (A98-78, A97-197, A02-29, and A97-83) were provided by the Neuropathology Core facility of Northwestern University, and the postmortem human brain tissue was acquired in accordance with Northwestern University institutional review board approved protocols. The fixed postmortem normal subject (HCT15HAU_19_04_PC), all frozen normal subject brain tissues, and all fixed and frozen AD patient brain tissues were obtained from the NIH NeuroBioBank under a material transfer agreement between the NIH and Case Western Reserve University.
Quantification and statistical analysis
Sample sizes were determined by a power analysis on the basis of pilot data that were collected in our laboratory or published studies. In the animal studies, we used n = 12 to 19 mice per group for the behavioral tests, n = 3 to 6 mice per group for the biochemical analyses, and n = 3 to 6 mice per group for the pathology studies. In the cell culture studies, we performed each experiment with at least three independent replications. For all of the animal studies, we ensured randomization and blinded evaluations. For all of the imaging analyses, an observer who was blinded to the experimental groups conducted the quantification. No samples or animals were excluded from the analysis.
The data were analyzed using GraphPad Prism 8.0 software. Student’s t test was used for comparisons between two groups. Comparisons between three or more independent groups were performed using one-way analysis of variance, followed by the Tukey’s post hoc test. The data are expressed as the mean ± SEM. Statistical parameters are presented in each figure legend. Values of P < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
Funding: This study was supported by a pilot grant from the Department of Physiology and Biophysics, Case Western Reserve University, and a research grant from Dr. Ralph and Marian Falk Medical Research Foundation Transformative Award to X.Q. The postmortem brain tissues were provided by the Neuropathology Core of Northwestern University (NIH P30 AG013854 24) and the NIH NeuroBioBank. The study with the mouse genotypes was supported by a P30 core grant for vision research (NIH 5P30EY011373). We thank Translational Therapeutics Core of the Cleveland Alzheimer’s Disease Research Center (NIH/NIA: 1 P30 AGO62428-01) for assisting in the study with human postmortem brain tissues. R.X. and Q.W. acknowledge funding from NIH National Institute of Aging (AG057557, AG061388, and AG062272). Author contributions: X.Z. performed experiments in cell cultures and biochemical analyses of animal models and patient samples. R.W. maintained mice with the four genotypes, conducted blinded animal behavioral analysis, and collected mouse tissues. D.H. assisted in the metabolic analysis of cell culture and mouse tissue, and provided technical support on in vitro and in vivo experiments. X.S. bred heterozygous knockout Drp1 mice, 5XFAD mice, and the triple-crossed mice. H.F. carried out EM analysis. K.L. performed proteomics analysis. E.R.C. analyzed proteomic database. Q.W. and R.X. performed computational analysis of HK1 with AD human database. M.E.F. assisted in the study of human postmortem brains. A.A.P. assisted in the study of human postmortem brains and edited manuscript. X.Q. conceived, designed, and supervised all the studies and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The datasets generated under the current study are available from the corresponding author upon reasonable request. The proteomic database was submitted to figshare (https://figshare.com/) (https://doi.org/10.6084/m9.figshare.12720824.v1).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabb8680/DC1
REFERENCES AND NOTES
- 1.Jagust W., Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 19, 687–700 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nasrabady S. E., Rizvi B., Goldman J. E., Brickman A. M., White matter changes in Alzheimer’s disease: A focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 6, 22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salat D. H., Greve D. N., Pacheco J. L., Quinn B. T., Helmer K. G., Buckner R. L., Fischl B., Regional white matter volume differences in nondemented aging and Alzheimer’s disease. Neuroimage 44, 1247–1258 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desai M. K., Sudol K. L., Janelsins M. C., Mastrangelo M. A., Frazer M. E., Bowers W. J., Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia 57, 54–65 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhan X., Jickling G. C., Ander B. P., Liu D., Stamova B., Cox C., Jin L.-W., DeCarli C., Sharp F. R., Myelin injury and degraded myelin vesicles in Alzheimer’s disease. Curr. Alzheimer Res. 11, 232–238 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Philips T., Rothstein J. D., Oligodendroglia: Metabolic supporters of neurons. J. Clin. Invest. 127, 3271–3280 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behrendt G., Baer K., Buffo A., Curtis M. A., Faull R. L., Rees M. I., Gotz M., Dimou L., Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia 61, 273–286 (2013). [DOI] [PubMed] [Google Scholar]
- 8.McKenzie A. T., Moyon S., Wang M., Katsyv I., Song W. M., Zhou X., Dammer E. B., Duong D. M., Aaker J., Zhao Y., Beckmann N., Wang P., Zhu J., Lah J. J., Seyfried N. T., Levey A. I., Katsel P., Haroutunian V., Schadt E. E., Popko B., Casaccia P., Zhang B., Multiscale network modeling of oligodendrocytes reveals molecular components of myelin dysregulation in Alzheimer’s disease. Mol. Neurodegener. 12, 82 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang P., Kishimoto Y., Grammatikakis I., Gottimukkala K., Cutler R. G., Zhang S., Abdelmohsen K., Bohr V. A., Misra Sen J., Gorospe M., Mattson M. P., Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 22, 719–728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knott A. B., Perkins G., Schwarzenbacher R., Bossy-Wetzel E., Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 9, 505–518 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kageyama Y., Zhang Z., Sesaki H., Mitochondrial division: Molecular machinery and physiological functions. Curr. Opin. Cell Biol. 23, 427–434 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jahani-Asl A., Slack R. S., The phosphorylation state of Drp1 determines cell fate. EMBO Rep. 8, 912–913 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi A. U., Saw N. L., Shamloo M., Mochly-Rosen D., Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 9, 6128–6143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X., Su B., Lee H.-g., Li X., Perry G., Smith M. A., Zhu X., Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manczak M., Reddy P. H., Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 21, 2538–2547 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manczak M., Calkins M. J., Reddy P. H., Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 20, 2495–2509 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baek S. H., Park S. J., Jeong J. I., Kim S. H., Han J., Kyung J. W., Baik S. H., Choi Y., Choi B. Y., Park J. S., Bahn G., Shin J. H., Jo D. S., Lee J. Y., Jang C.-G., Arumugam T. V., Kim J., Han J.-W., Koh J.-Y., Cho D.-H., Jo D.-G., Inhibition of Drp1 ameliorates synaptic depression, Aβ deposition, and cognitive impairment in an Alzheimer’s disease model. J. Neurosci. 37, 5099–5110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manczak M., Kandimalla R., Fry D., Sesaki H., Reddy P. H., Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 25, 5148–5166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itoh K., Murata D., Kato T., Yamada T., Araki Y., Saito A., Adachi Y., Igarashi A., Li S., Pletnikov M., Huganir R. L., Watanabe S., Kamiya A., Iijima M., Sesaki H., Brain-specific Drp1 regulates postsynaptic endocytosis and dendrite formation independently of mitochondrial division. eLife 8, e44739 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motori E., Puyal J., Toni N., Ghanem A., Angeloni C., Malaguti M., Cantelli-Forti G., Berninger B., Conzelmann K. K., Götz M., Winklhofer K. F., Hrelia S., Bergami M., Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 18, 844–859 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Park J., Choi H., Min J.-S., Park S.-J., Kim J. H., Park H. J., Kim B., Chae J.-I., Yim M., Lee D.-S., Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 127, 221–232 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Sanchez-Lopez E., Zhong Z., Stubelius A., Sweeney S. R., Booshehri L. M., Antonucci L., Liu-Bryan R., Lodi A., Terkeltaub R., Lacal J. C., Murphy A. N., Hoffman H. M., Tiziani S., Guma M., Karin M., Choline uptake and metabolism modulate macrophage IL-1β and IL-18 production. Cell Metab. 29, 1350–1362.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buck M. D., O’Sullivan D., Klein Geltink R. I., Curtis J. D., Chang C.-H., Sanin D. E., Qiu J., Kretz O., Braas D., van der Windt G. J., Chen Q., Huang S.-C., O’Neill C. M., Edelson B. T., Pearce E. J., Sesaki H., Huber T. B., Rambold A. S., Pearce E. L., Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swanson K. V., Deng M., Ting J. P.-Y., The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broz P., Pelegrin P., Shao F., The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20, 143–157 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Hughes M. M., O’Neill L. A. J., Metabolic regulation of NLRP3. Immunol. Rev. 281, 88–98 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Heneka M. T., Kummer M. P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., Griep A., Axt D., Remus A., Tzeng T.-C., Gelpi E., Halle A., Korte M., Latz E., Golenbock D. T., NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ising C., Venegas C., Zhang S., Scheiblich H., Schmidt S. V., Vieira-Saecker A., Schwartz S., Albasset S., McManus R. M., Tejera D., Griep A., Santarelli F., Brosseron F., Opitz S., Stunden J., Merten M., Kayed R., Golenbock D. T., Blum D., Latz E., Buée L., Heneka M. T., NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitew S., Kirkcaldie M. T., Halliday G. M., Shepherd C. E., Vickers J. C., Dickson T. C., Focal demyelination in Alzheimer’s disease and transgenic mouse models. Acta Neuropathol. 119, 567–577 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., Berry R., Vassar R., Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kayagaki N., Stowe I. B., Lee B. L., O’Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q. T., Liu P. S., Lill J. R., Li H., Wu J., Kummerfeld S., Zhang J., Lee W. P., Snipas S. J., Salvesen G. S., Morris L. X., Fitzgerald L., Zhang Y., Bertram E. M., Goodnow C. C., Dixit V. M., Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Karmakar M., Minns M., Greenberg E. N., Diaz-Aponte J., Pestonjamasp K., Johnson J. L., Rathkey J. K., Abbott D. W., Wang K., Shao F., Catz S. D., Dubyak G. R., Pearlman E., N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat. Commun. 11, 2212 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo F., Herrup K., Qi X., Yang Y., Inhibition of Drp1 hyper-activation is protective in animal models of experimental multiple sclerosis. Exp. Neurol. 292, 21–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cassidy-Stone A., Chipuk J. E., Ingerman E., Song C., Yoo C., Kuwana T., Kurth M. J., Shaw J. T., Hinshaw J. E., Green D. R., Nunnari J., Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song W., Chen J., Petrilli A., Liot G., Klinglmayr E., Zhou Y., Poquiz P., Tjong J., Pouladi M. A., Hayden M. R., Masliah E., Ellisman M., Rouiller I., Schwarzenbacher R., Bossy B., Perkins G., Bossy-Wetzel E., Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 17, 377–382 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S. O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y.-I., Taguchi N., Morinaga H., Maeda M., Takayanagi R., Yokota S., Mihara K., Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Wakabayashi J., Zhang Z., Wakabayashi N., Tamura Y., Fukaya M., Kensler T. W., Iijima M., Sesaki H., The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 186, 805–816 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manczak M., Sesaki H., Kageyama Y., Reddy P. H., Dynamin-related protein 1 heterozygote knockout mice do not have synaptic and mitochondrial deficiencies. Biochim. Biophys. Acta 1822, 862–874 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.ElAli A., Rivest S., Microglia in Alzheimer’s disease: A multifaceted relationship. Brain Behav. Immun. 55, 138–150 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Son Y., Kim J. S., Jeong Y. J., Jeong Y. K., Kwon J. H., Choi H.-D., Pack J. K., Kim N., Lee Y.-S., Lee H. J., Long-term RF exposure on behavior and cerebral glucose metabolism in 5xFAD mice. Neurosci. Lett. 666, 64–69 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Hong S., Beja-Glasser V. F., Nfonoyim B. M., Frouin A., Li S., Ramakrishnan S., Merry K. M., Shi Q., Rosenthal A., Barres B. A., Lemere C. A., Selkoe D. J., Stevens B., Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mookerjee S. A., Gerencser A. A., Nicholls D. G., Brand M. D., Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 293, 12649–12652 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson J. E., Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 206, 2049–2057 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Robey R. B., Hay N., Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 25, 4683–4696 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Lundgaard I., Li B., Xie L., Kang H., Sanggaard S., Haswell J. D., Sun W., Goldman S., Blekot S., Nielsen M., Takano T., Deane R., Nedergaard M., Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun. 6, 6807 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pastorino J. G., Hoek J. B., Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 40, 171–182 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolf A. J., Reyes C. N., Liang W., Becker C., Shimada K., Wheeler M. L., Cho H. C., Popescu N. I., Coggeshall K. M., Arditi M., Underhill D. M., Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166, 624–636 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saraiva L. M., Seixas da Silva G. S., Galina A., da-Silva W. S., Klein W. L., Ferreira S. T., De Felice F. G., Amyloid-β triggers the release of neuronal hexokinase 1 from mitochondria. PLOS ONE 5, e15230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stassart R. M., Möbius W., Nave K.-A., Edgar J. M., The axon-myelin unit in development and degenerative disease. Front. Neurosci. 12, 467 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu J., Chen S., Ahmed S. H., Chen H., Ku G., Goldberg M. P., Hsu C. Y., Amyloid-β peptides are cytotoxic to oligodendrocytes. J. Neurosci. 21, RC118 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong Y.-X., Zhang H.-Y., Li H.-Y., Liu P.-H., Sui Y., Sun X.-H., Association between Alzheimer’s disease pathogenesis and early demyelination and oligodendrocyte dysfunction. Neural Regen. Res. 13, 908–914 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spencer N. G., Bridges L. R., Elderfield K., Amir K., Austen B., Howe F. A., Quantitative evaluation of MRI and histological characteristics of the 5xFAD Alzheimer mouse brain. Neuroimage 76, 108–115 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Gu L., Wu D., Tang X., Qi X., Li X., Bai F., Chen X., Ren Q., Zhang Z., Myelin changes at the early stage of 5XFAD mice. Brain Res. Bull. 137, 285–293 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Chu T.-H., Cummins K., Sparling J. S., Tsutsui S., Brideau C., Nilsson K. P. R., Joseph J. T., Stys P. K., Axonal and myelinic pathology in 5xFAD Alzheimer’s mouse spinal cord. PLOS ONE 12, e0188218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang S. H., Fukaya M., Yang J. K., Rothstein J. D., Bergles D. E., NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nielsen H. M., Ek D., Avdic U., Orbjörn C., Hansson O., Netherlands Brain B., Veerhuis R., Rozemuller A. J., Brun A., Minthon L., Wennström M., NG2 cells, a new trail for Alzheimer’s disease mechanisms? Acta Neuropathol. Commun. 1, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McKenzie B. A., Mamik M. K., Saito L. B., Boghozian R., Monaco M. C., Major E. O., Lu J.-Q., Branton W. G., Power C., Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 115, E6065–E6074 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rone M. B., Cui Q.-L., Fang J., Wang L.-C., Zhang J., Khan D., Bedard M., Almazan G., Ludwin S. K., Jones R., Kennedy T. E., Antel J. P., Oligodendrogliopathy in multiple sclerosis: Low glycolytic metabolic rate promotes oligodendrocyte survival. J. Neurosci. 36, 4698–4707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao J., Cui J. Z., To E., Cao S., Matsubara J. A., Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J. Neuroinflammation 15, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang Y.-L., Li J., Liu K., Zhang L., Liu Q., Liu B., Qi L.-W., Ginsenoside Rg5 increases cardiomyocyte resistance to ischemic injury through regulation of mitochondrial hexokinase-II and dynamin-related protein 1. Cell Death Dis. 8, e2625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabb8680/DC1