

Abstract
Global prevalence of coronavirus disease 2019 (COVID-19) calls for an urgent development of anti-viral regime. Compared with the development of new drugs, drug repurposing can significantly reduce the cost, time, and safety risks. Given the fact that coronavirus harnesses spike protein to invade host cells through angiotensin-converting enzyme 2 (ACE2), hence we see if any previous anti-virtual compounds can block spike-ACE2 interaction and inhibit the virus entry. The results of molecular docking and molecular dynamic simulations revealed that remdesivir exhibits better than expected anti-viral invasion potential against COVID-19 among the three types of compounds including remdesivir, tenofovir and lopinavir. In addition, a positive correlation between the surface area occupied by remdesivir and anti-viral invasion potential was also found. As such, the structure of remdesivir was modified by linking an N-benzyl substituted diamidine derivative to its hydroxyl group through an ester bond. It was found that this compound has a higher anti-viral invasion potential and greater specificity.
Keywords: COVID-19, remdesivir, diamidine, spike, angiotensin- converting enzyme 2 (ACE2)
Introduction
A novel coronavirus, which was officially named as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has caused the global spread of coronavirus disease 2019 (COVID-19) since 2019. As of May 30, there have been more than 6 000 000 diagnosed cases and 360 000 confirmed deaths worldwide. Many relevant institutions quickly responded to this challenge to prevent the spread of the epidemic and effectively treat those infected. Moreover, a series of COVID-19-related research work, such as the exploration of clinical treatment methods, the development and screening of vaccines and drugs, and the improvement in diagnostic tools and medical equipment, are all being carried out actively.
To identify potential drugs against SARS-CoV-2, drug repurposing has gained great attention because reusing the drugs that are already available has the advantages of less time and money costs and higher safety in comparison to the original development for a specific disease. For instance, remdesivir, an adenosine analogue for treating Ebola virus infection, and antiprotozoal drugs chloroquine, are being considered and investigated to control the COVID-19[1–2]. Other anti-viral or anti-inflammatory drugs, such as lopinavir, ritonavir, ribavirin, umifenovir, tenofovir, favipiravir, galidesivir, and disulfiram have been approved as drug repurposing options to test their anti-COVID-19 potential. Although the current treatment scheme witnessed some early success, none of these candidates exhibited reproducible effectiveness in a large cohort of patients, necessitating the screening of drugs in a high-through output fashion.
High-throughput screening and receptor structure-based drug design are inseparable from the selection of receptor proteins. Coronaviral proteases, such as main protease and papain-like protease, have been employed to some computational studies of drug repurposing against COVID-19[3]. Since these proteases are involved in the proteolytic processing of the polyproteins into individual non-structural proteins[4], employing these proteases as targets to perform drug design aims to control viral gene expression and replication. In addition to coronaviruses-encoded proteases, spike protein is also worthy of attention. It is the most important surface membrane protein of coronavirus, and its receptor binding domain can specifically recognize and bind to the angiotensin-converting enzyme 2 (ACE2), the receptor on host cells[5‒7]. Genomic sequence, phylogenetic and structural analysis have proved that SARS-CoV-2 can also rely on its spike protein to bind to the host cell surface receptor ACE2 similar to SARS/SARS-like coronaviruses[8‒10], yet there is much controversy about the binding strength. Some studies demonstrated that the binding capacity of SARS-CoV-2 spike protein to ACE2 is weaker than that between SARS spike and ACE2[9]. Nevertheless, the interaction strength of SARS-CoV-2 spike with ACE2 has reached the threshold for the virus to invade the host. But the kinetics investigation on spike-ACE2 interaction revealed that spike protein of SARS-CoV-2 exhibits 10 to 20-fold higher affinity toward ACE2 compared with SARS-CoV[11]. Blocking the spike-ACE2 binding is beneficial to inhibit virus entry or invasion, although there are still concerns to be settled such as the affinity issue, which necessitates an in-depth investigation.
In this study, we tried to screen and understand the mechanism of known drugs for COVID-19 for the purpose of designing new drugs with higher efficacy. The great progress in spike protein sequence of SARS-CoV-2[11] as well as its complex structure with ACE2[12‒14] provides a strong basis for the computational work[15‒18]. Our work follows three steps. Firstly, we conducted a virtual screening of the therapeutics approved by Food and Drug Administration (FDA) to test their efficacy against COVID-19. Considering that the spike-ACE2 interaction plays a vital role in SARS-CoV-2 entry, we focused on compounds that could function as inhibitors of such interaction. Those candidates being screened exhibit various structural characteristics and binding strength to the target (Supplementary Fig. 1 , available online), which raises two concerns: which protein, spike or ACE2, do they prefer to bind and why do they interact with their targets? To answer these questions, during the second step, we selected three proposed drugs including remdesivir, tenofovir and lopinavir (Fig. 1 ) to perform molecular dynamics (MD) simulations and binding energy calculations to understand their interaction details with targets. It was interesting to find remdesivir can block the interaction between spike protein and ACE2. Its preference to occupy ACE2 indicated that remdesivir has the potential to prevent the entry of SARS-CoV-2 spike. The results of protein-ligand interaction investigation indicated there is a positive correlation between the ACE2 surface area occupied by remdesivir and anti-viral invasion potential. Hence, during the third step, we attempted to improve the binding capacity of remdesivir to ACE2 by drug modification. Computational results showed that conjugating remdesivir with N-benzyl substituted diamidine derivative[19] through an ester bond dramatically increased the efficacy against SARS-CoV-2. We hope this study can provide a theoretical basis for future design and modification of anti-SARS-CoV-2 drugs.
1.
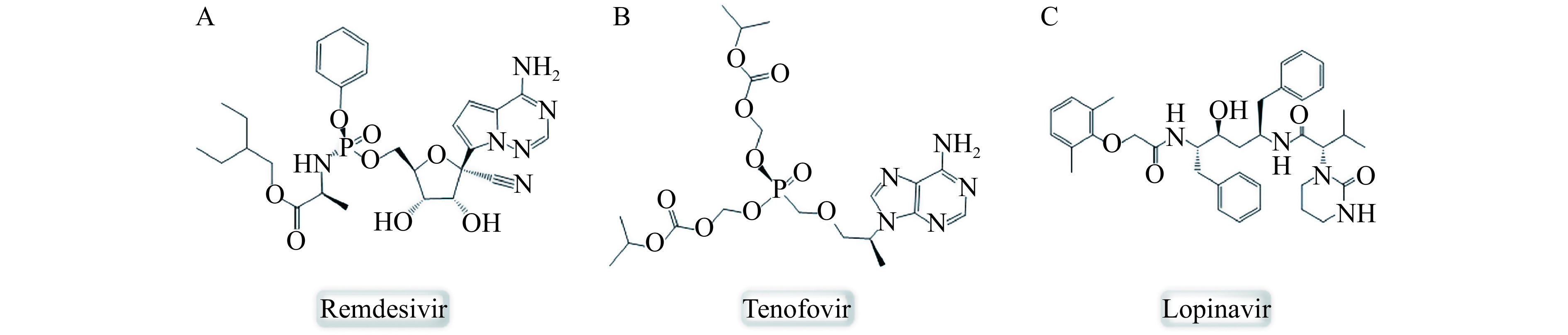
Structural formulas of three antiviral compounds.
A: Remdesivir; B: Tenofovir; C: Lopinavir.
Materials and methods
The overall methodology employed in this work was represented and summarized in Fig. 2 . It followed virtual screening, molecular docking, MD simulation, trajectory analysis, binding energy calculation, and drug design.
2.
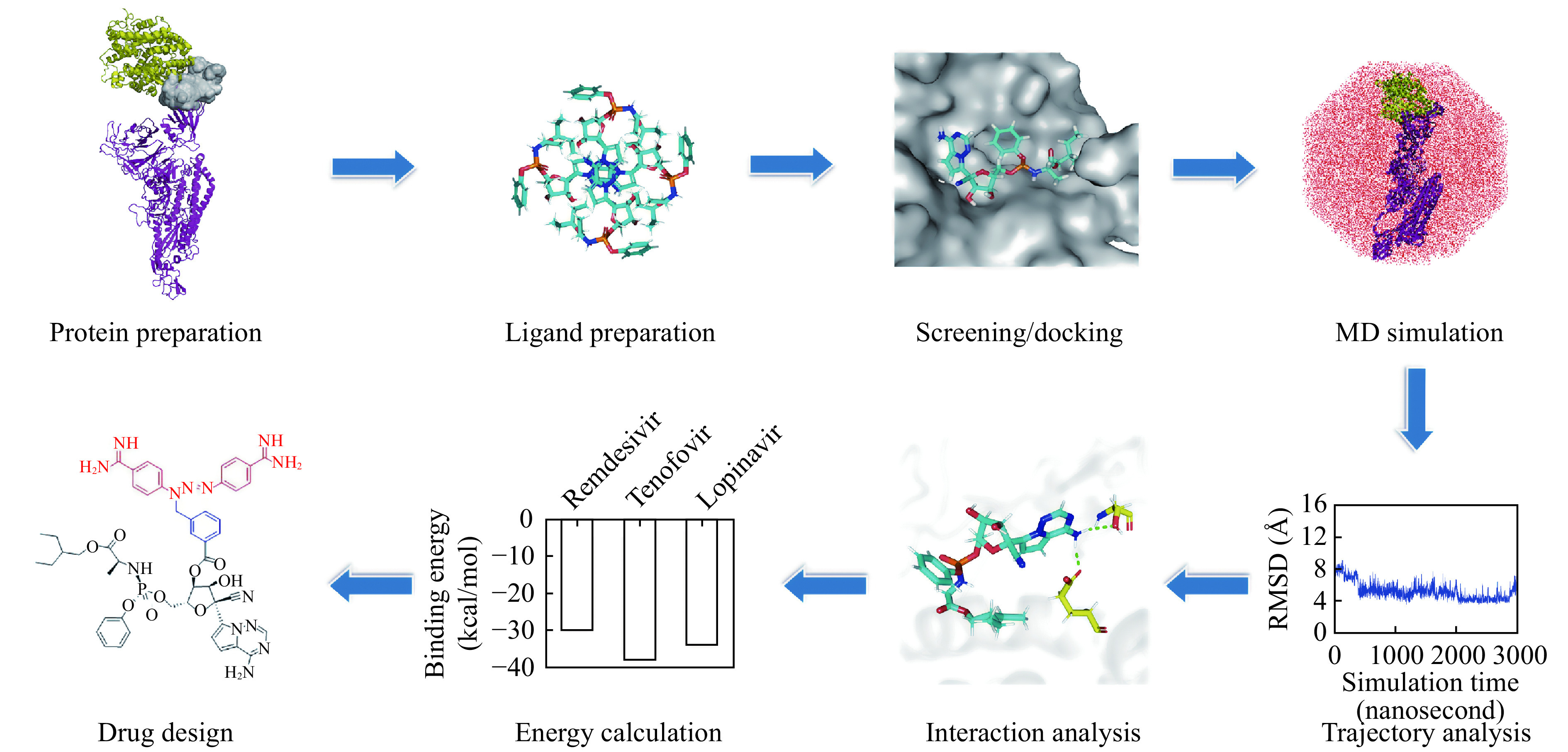
The flowchart diagram performed in the current study.
MD: molecular dynamics.
Virtual screening of drug library
To search inhibitors for COVID-19, a drug database containing 2080 FDA-approved molecules were applied to conduct the high-throughput virtual screening by using molecular docking. Due to the effectiveness against SARS coronavirus, remdesivir was also included in the screening database. These drug molecules were optimized through Powell conjugate gradient algorithm with Tripos force field and Gasteiger-Hückel charges. The convergence criterion was 0.005 kcal/(mol·Å) and the maximum iteration was set to 1000. As exhibited in the spike-ACE2 complex structure constructed by homologous modeling and MD simulation[20], spike protein of SARS-CoV-2 contains three subunits Ⅰ, Ⅱ, and Ⅲ (Supplementary Fig. 2A, available online). However, only subunit Ⅲ can interact with ACE2 and there is no interaction interface of subunits Ⅰ and Ⅱ with ACE2. As such, in order to strike a balance between computing resources and computational accuracy, both ACE2 and subunit Ⅲ of spike protein were included in our calculations (Supplementary Fig. 2B, available online).
Molecular docking was performed within Surflex-Dock Screen module[21] of SYBYL program[22]. The docking pocket was generated to the residues at the spike-ACE2 interaction interface with the threshold value and bloat value of 0.5 Å and 0 Å, respectively. Next, 2080 molecules were docked into the active pocket. The candidate molecules were selected on the basis of the following considerations: There were no clashes between the ligand and any residues on spike-ACE2; In a cluster of similar molecules, smaller ones were preferred because they allowed more room for optimizing structures; Molecules that could form distinguishable hydrogen bonds (with bond length and angle falling within 2.50 to 3.20 Å and 130° to 180°, respectively) and π-π stacking interactions (3.00 to 5.00 Å) with the residues in the binding pocket were preferred[23]. Subsequently, the affinity of small molecules bound to the spike-ACE2 complex was evaluated by consensus score (CScore), which integrates a number of popular scoring functions including G_Score, D_Score, PMF_Score, Chem_Score, Crash, Polar, and Global_Cscore.
Molecular dynamics simulations
MD simulations were performed with AMBER 16 program[24]. The starting conformation of protein-ligand complex came from the docking results. Subsequently, the complex was solvated in a periodic TIP3P octahedral box[25]. The minimum distance from the protein atom to the edge of the box was set to be about 10.0 Å. Twenty-four sodium counter-ions were added to neutralize the charge. A cut-off radius of 12.0 Å was applied for van der Waals and electrostatic interactions. The particle-mesh Ewald method[26] was used to evaluate the long-range electrostatic interactions. To relax the initial structures, a two-step energy minimization was performed: 2500 steps of the steeped descent and subsequent 2500 steps of conjugate gradient minimization, in which the protein was fixed by a 500 kcal/mol restraint force; 1000 steps of the steeped descent and 9000 steps of conjugate gradient minimization without restraints. Subsequently, the system was gradually heated to 300 K in 6 steps of 100-picoseconds MD simulations (from 0 to 50 K, 100 K, 150 K, 200 K, 250 K, and 300 K). Then the MD simulations were performed in the NVT ensemble (constant numbers of atoms [N], volume [V], and temperature [T]) with a time step of 2 femtoseconds. The temperature was kept constant using the Berendsen temperature algorithm[27]. The SHAKE algorithm[28] was employed to constrain all bonds involving hydrogen atoms.
For the protein receptor (spike-ACE2 complex), the FF03 force field[29] was applied. In the case of small molecule ligands, the force field parameters were obtained by quantum mechanism calculations with GAUSSIAN 09 program[30]. The general parameterization procedure included: Optimize the geometries of ligands in gas phase by Hartree−Fock (HF) method with 6−31G(d,p) basis set; Calculate electrostatic potential (ESP) at B3LYP/cc−pVTZ level. The polarizable continuum model (PCM) using the integral equation formalism (IEF) variant (IEFPCM) was applied to mimic an organic solvent environment (ε=4.0); Assign the force field parameters and restrained electrostatic potential (RESP) partial charges by using the ANTECHAMBER module[31] in the AMBER 16 package[24].
The evolutions of total, kinetic and potential energies with the simulation time for the studied complex systems including spike-remdesivir-ACE2, spike-tenofovir-ACE2, and spike-lopinavir-ACE2 were summarized in Supplementary Fig. 3 (available online). All the studied systems reach an equilibrium during the MD simulation. Besides, the evolution of root-mean-square deviations (RMSDs) of backbone atoms (C, N, and O atoms) compared with the initial complex structure along the simulation time were calculated and exhibited in Fig. 3 . The RMSDs of spike-remdesivir-ACE2 stabilize after 8 nanoseconds, and those of spike-tenofovir-ACE2 and spike-lopinavir-ACE2 become stable after 10 nanoseconds. In addition, the binding energies of the three ligands to receptor calculated from 11 to 15 nanoseconds are comparable to those calculated from 11 to 20 nanoseconds (Supplementary Fig. 4, available online). This also confirms the stability of MD simulations between 11 to 20 nanoseconds. As a result, 10-nanoseconds production MD runs from 11 to 20 nanoseconds were employed in the trajectory analysis and energetic calculations.
3.

Evolution of root-mean-square deviation (RMSD) of backbone C, N, and O atoms along the simulation time.
Binding free energy calculation
The binding free energies (ΔGbind) of the studied drugs with spike-ACE2 were calculated by using molecular mechanics/Poisson-Boltzman surface area (MM-PBSA) method[32–34] in AMBER16 program[24]. A total of 1000 snapshots in the MD trajectory from 11 to 20 nanoseconds were extracted for the calculation. In such a simulation of solvated states, the majority of the energy contributions would come from solvent-solvent interactions. The fluctuations in total energy would be an order of magnitude larger than binding energy. Therefore, according to the thermodynamic cycle, the binding free energy in solvent (ΔGbind, solv) can be calculated by equation (1):
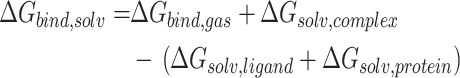 |
1 |
where solvation free energies were calculated by Poisson-Boltzmann (PB) equation.
The interaction energies between spike and ACE2 were calculated on the 10 lowest-energy conformations obtained through the MD trajectories. The water and counter-ions were excluded during the energetic computation. The interaction energies were calculated by equation (2):
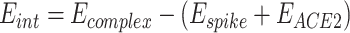 |
2 |
In this equation, Espike, EACE2, and Ecomplex are the energies of spike protein, ACE2, and their complex. They can be obtained through the following potential functional where U represents the potential energy of the system:
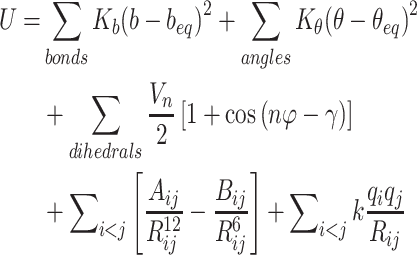 |
3 |
The five items in the potential function are bond stretching vibrational energy, angle bending vibrational energy, torsional rotating potential barrier, van der Waals and electrostatic interactions, respectively. The parameters of bond stretching (Kb and beq), angle bending (Kθ and θeq), torsional rotating (Vn and n) and Lennard-Jones (Aij and Bij) come from the Amber FF03 force field[29].
Results
Virtual screening results
After primary screening, 238 compounds dose-dependently bound to the spike-ACE2 interaction interface (Supplementary Table 1, available online), and subsequently they were subjected to the second-round confirmation. Ten of the 238 hit compounds have been reported to have antiviral activity (Supplementary Table 1), and thus other hits would not be further studied here. The screened 10 antiviral compounds were tenofovir, remdesivir, atazanavir, valacyclovir, valganciclovir, abacavir, doravirine, adefovir, lopinavir, and ritonavir (Supplementary Table 1 and Supplementary Fig. 1). As expected, several antiviral compounds have been found in other drug screening lists, such as remdesivir, lopinavir, and ritonavir[1,9,35].
To confirm the binding activity of those antiviral compounds, we further conducted an in-depth study regarding their docking scores and modes to the active site. The detailed views of docking were shown in Fig. 4 . It is obvious that these antiviral compounds bind to the spike-ACE2 interface through hydrogen bonding and hydrophobic interactions. It is interesting to find that these hit compounds exhibit different locations. For example, remdesivir and doravirine have stronger interaction with ACE2 than spike, indicating they are biased towards to ACE2. Adefovir, tenofovir, and valacyclovir tend to be close to spike, and are expected to interact strongly with spike than with ACE2.
4.
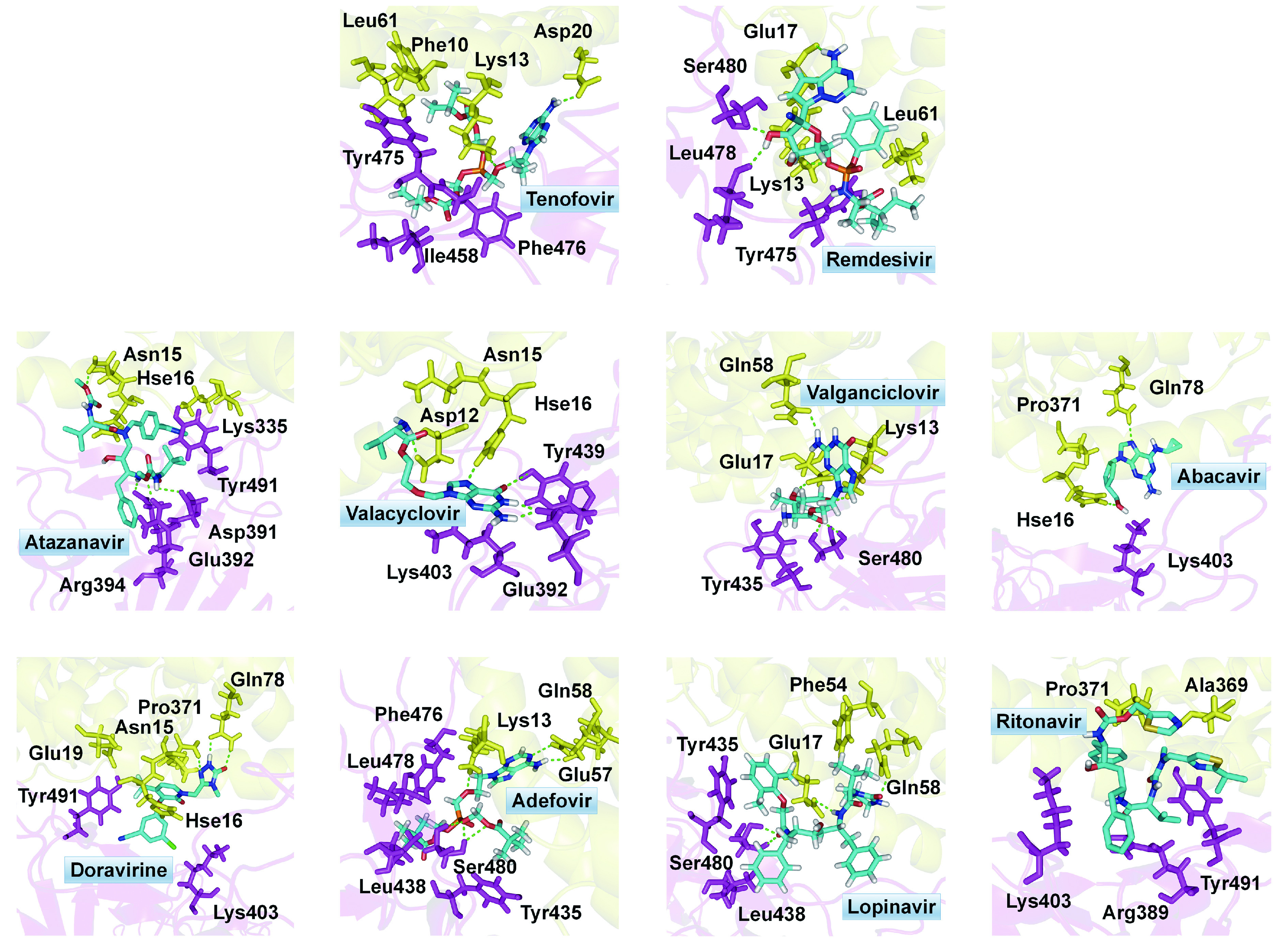
Docking conformations and interacting residues of the hit antiviral compounds in the binding pocket locating at spike-ACE2 interface.
These anti-viral compounds have some obvious structural features. For tenofovir and remdesivir, which possess the highest docking scores, they have a phosphate ester and a fused nitrogen-containing heterocyclic ring attached by an amide group (‒NH2). Furthermore, more than one cyclic side-chains are often observed. Especially for lopinavir, it has the most cyclic structures among these hit compounds. In order to evaluate the anti-viral invasion efficacy of these compounds and further perform drug modification, we selected remdesivir, tenofovir, and lopinavir for subsequent MD simulations to investigate their binding strength and binding conformations to the targeting protein.
Ligand-receptor interaction energies
The binding energies of the studied compounds to spike-ACE2 are displayed in Fig. 5 . All the binding energies are negative, indicating the binding possibilities of remdesivir, tenofovir, and lopinavir to the interface between spike and ACE2 (Fig. 5A ). Their binding energies increase in the sequence of tenofovir (−42.9 kcal/mol) < remdesivir (−36.4 kcal/mol) < lopinavir (−32.9 kcal/mol), demonstrating the binding strength follows the sequence of tenofovir > remdesivir > lopinavir. In order to investigate the influence of the studied compounds on the interaction between spike and ACE2, we analyzed the change in spike-ACE2 interaction energies resulted from the ligand binding. As shown in Fig. 5B , the spike-ACE2 interaction strength is decreased upon the ligand binding. This result demonstrates that remdesivir, tenofovir, and lopinavir may hinder the coupling between spike protein and ACE2.
5.
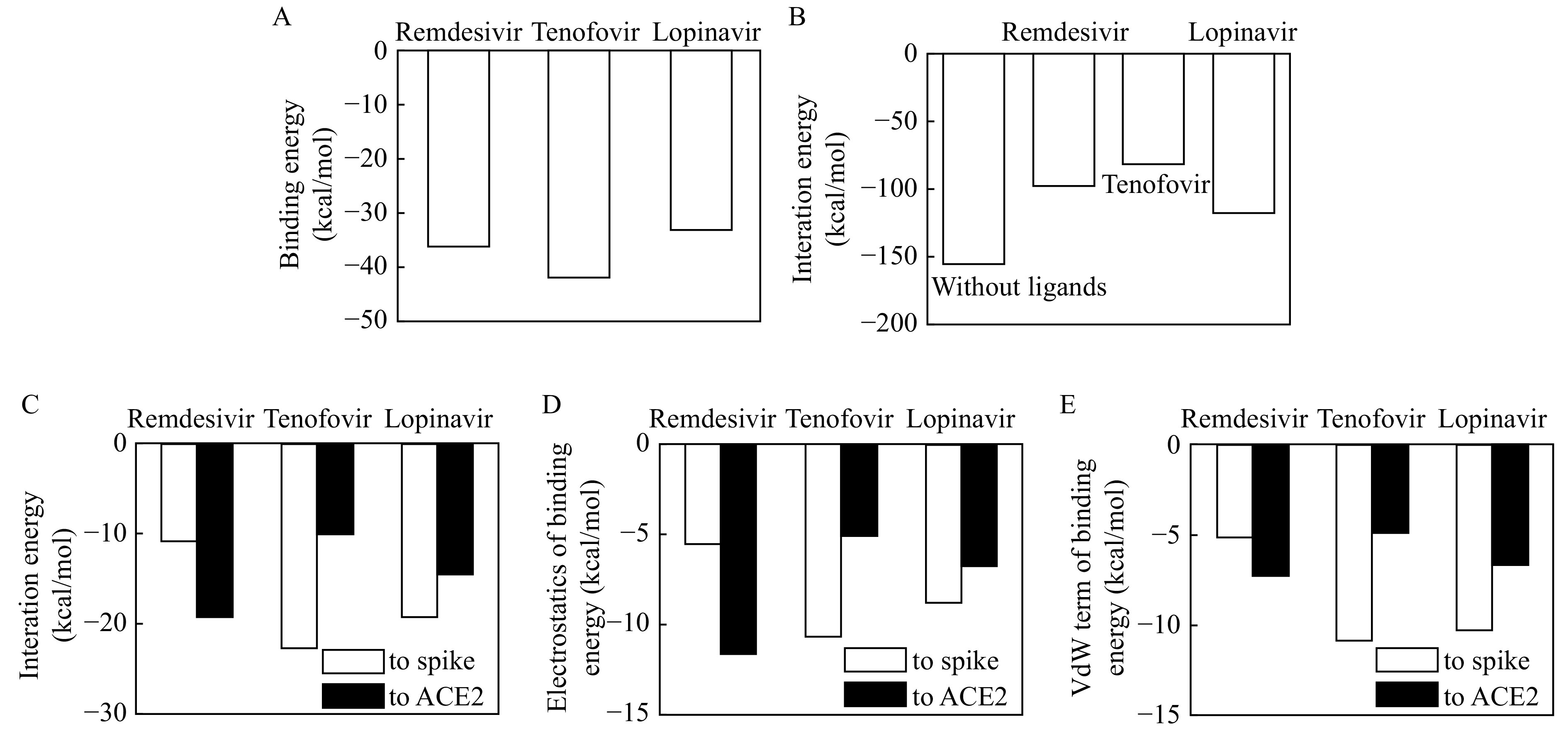
Binding energy diagrams.
A: Binding energies of studied compounds to spike-ACE2 coupling complex. B: Interaction energies between spike and ACE2 before and after compound binding. C: Bind energies of selected compounds to spike and ACE2. D and E: Electrostatic (D) and van der Waals (VdW) interaction energies (E) of selected compounds to spike and ACE2.
Meanwhile, these compounds exhibited distinct binding energies to spike and ACE2 (Fig. 5C ). remdesivir has higher binding strength to ACE2 (−19.2 kcal/mol) than spike protein (−10.8 kcal/mol), and tenofovir has higher binding strength to spike (‒21.8 kcal/mol) than ACE2 (‒10.2 kcal/mol, respectively). Different from them, lopinavir has comparable binding strength to spike (−18.7 kcal/mol) and ACE2 (−14.6 kcal/mol). Among these compounds, their binding capacities to ACE2 follows the sequences of remdesivir > lopinavir > tenofovir, while was reversed totally once interacted with spike.
To understand the contributions of different energetic terms to interaction of ligands with spike protein and ACE2, Fig. 5D and E give the electrostatic and van der Waals interaction energies, respectively. Electrostatic term plays an important role in the preference of remdesivir to ACE2, and the van der Waals interaction of remdesivir with spike and ACE2 are comparable to each other. In the case of tenofovir, both the electrostatic and van der Waals contribute more to the binding to spike than ACE2. Similarly, both van der Waals interaction and electrostatic term have contributed more to the binding of lopinavir to spike.
Ligand-receptor interaction modes
As demonstrated above, the studied potential drugs with various structures exhibited different binding strength with spike protein and ACE2. For further understanding of the influence of structures on the ligand-receptor interaction, the hydrogen bonding and hydrophobic interactions were investigated.
Hydrogen bonding occupancy
The hydrogen bond occupancy (the percent of snapshots containing hydrogen bonds in the MD trajectory) is an important parameter to characterize the difficulty of hydrogen bond formation, which can reflect the binding strength between drugs and proteins. Therefore, we calculated the hydrogen bond occupancies of the studied ligand-receptor systems (Table 1 and Fig. 6 ).
1. Hydrogen bonds between the studied antiviral compounds and spike-ACE2 with occupancy time more than 20%.
| Number | Acceptor | Donor H | Donor | Distance (Å) | Angle (°) | Occupancy (%) |
| a,b, and cAtom indexes were shown in Supplementary Fig. 5, 6, and 7 respectively, available online. | ||||||
| Between remdesivir and spike-ACE2a | ||||||
| 1 | OE@Glu17@ACE2 | H29@remdesivir | N6@remdesivir | 2.90 | 159.3 | 71.3 |
| 2 | OG@Ser480@spike | H30@ remdesivir | N6@remdesivir | 3.08 | 159.5 | 28.2 |
| Between tenofovir and spike-ACE2b | ||||||
| 1 | OE1@Gln479@spike | H29@tenoforvir | N3@tenoforvir | 2.97 | 161.6 | 49.1 |
| 2 | O@Ser480@spike | H29@tenoforvir | N3@tenoforvir | 2.92 | 154.8 | 34.2 |
| 3 | ND1@Hie16@ACE2 | H30@tenoforvir | N3@tenoforvir | 3.13 | 158.7 | 29.3 |
| 4 | N2@tenoforvir | H@Ser480@spike | N@Ser480@spike | 3.08 | 160.5 | 83.2 |
| 5 | O9@tenoforvir | H@Phe476@spike | N@Phe476@spike | 3.06 | 160.9 | 31.4 |
| Between lopinavir and spike-ACE2c | ||||||
| 1 | OE@Glu17@ACE2 | H33@lopinavir | N4@lopinavir | 2.98 | 156.9 | 87.9 |
| 2 | O1@lopinavir | H@Ser480@spike | N@Ser480@spike | 2.94 | 156.2 | 96.8 |
| 3 | O4@lopinavir | HZ@Lys13@ACE2 | NZ@Lys13@ACE2 | 2.90 | 155.9 | 71.3 |
6.
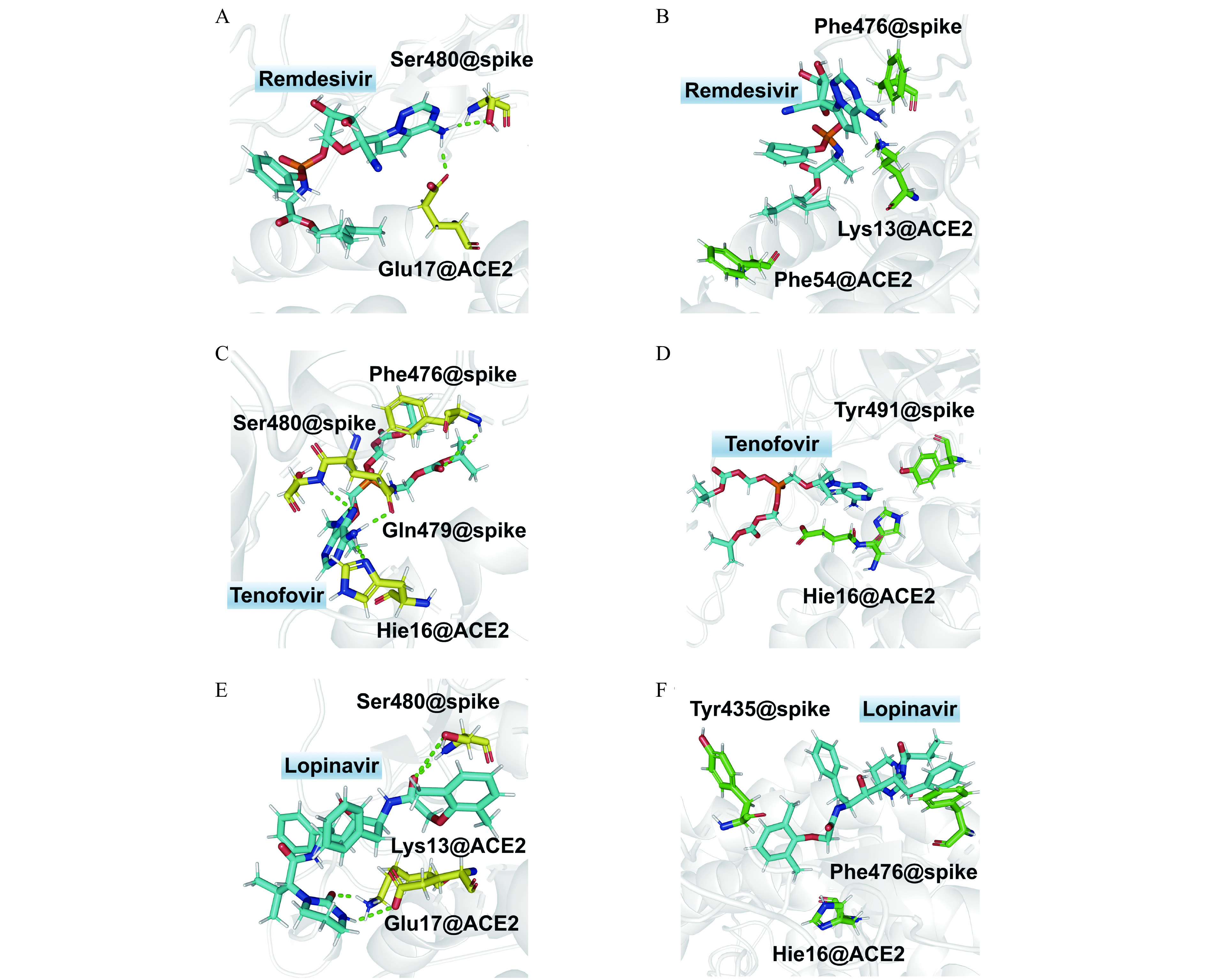
Hydrogen bonding and hydrophobic interaction diagrams of studied antiviral compounds with spike and ACE2.
A, C, and E: Hydrogen bonding in the complexes of remdesivir (A), tenofovir (C), and lopinavir (E) with receptors, respectively. B, D, and F: Hydrophobic interaction between remdesivir (B), tenofovir (D), and lopinavir (F) with receptors.
There are two hydrogen bonds between remdesivir and spike-ACE2, both of which are involved in the hydrogen atom of amide group bonding to the triazine ring in remdesivir (H29@remdesivir and H30@remdesivir, Table 1 , Fig. 6A , and Supplementary Fig. 5 , available online). Its hydrogen bond formed with side-chain carboxyl anion (‒COO−) in Glu17 of ACE2 (OE@Glu17@ACE2) has hydrogen bond length (donor-acceptor) of 2.90 Å and bond angle (donor‒H···acceptor) of 159.3°. The other hydrogen bond is formed between amide hydrogen in remdesivir with the hydroxyl oxygen atom in Ser480 of spike protein. The hydrogen bond length and angle are 3.08 Å and 159.5°, respectively. The higher occupancy of hydrogen bond between remdesivir with ACE2 (71.3%) than that with spike (28.2%) indicates that remdesivir has a strong tendency to form hydrogen bonding interaction with ACE2 than spike protein.
There are much more hydrogen bonding interactions of tenofovir with spike than ACE2. Five hydrogen bonds are formed with Gln479, Ser480, and Phe476, while only one hydrogen bond forms with Hie16 of ACE2 (Table 1 ). The amide hydrogen atoms (H29 and H30) in the purine ring of tenofovir can form hydrogen bonds not only with Gln479 and Ser480 of spike protein, but also with Hie16 of ACE2 (Fig. 6C and Supplementary Fig. 6 , available online). The long chains of phosphate ester increase the flexibility of tenofovir, resulting in the competition of the hydrogen bonding with spike and ACE2. This can be seen from the fluctuated and lower occupancies (smaller than 50%). In comparison, the hydrogen bonds lying at rigid purine ring (N2@tenofovir···H@Ser480 of spike) are much more stable with a high occupancy of 83.2%.
In the case of hydrogen bonds between lopinavir and spike-ACE2, the hydrogen bonding interaction between groups in the six-membered heterocyclic ring and some residues of ACE2 are found to be dominating (Table 1 , Fig. 6E and Supplementary Fig. 7 , available online). For instance, lopinavir uses amide hydrogen in six-membered heterocyclic ring to form hydrogen bond with side-chain carboxyl anion (‒COO‒) in Glu17 of ACE2. The occupancy is 87.9% with an average bonding length of 2.98 Å and an angle of 156.9°. It also forms a hydrogen bond between oxygen in the six-membered heterocyclic ring and Lys13 of ACE2 with an occupancy of 71.3%. The average length and angle of this hydrogen bond is 2.90 Å and 155.9°, respectively. In addition, the hydrogen bonding of ligand formed with hydroxyl hydrogen atom in Ser480 of spike is also found with the highest occupancy of 96.8% among these three hydrogen bonds.
Comparing the hydrogen bonding acceptors and donors in Table 1 , one can find that both Glu17 of ACE2 and Ser480 of spike protein are key residues to form hydrogen bonding with the studied ligands. For the ligands, the amide group attaching to the rigid fused nitrogen-containing heterocyclic ring plays an essential role in the hydrogen bonds with receptors. In addition, we also noticed that a rigid ligand skeleton is critical to support a stable hydrogen bonding.
Hydrophobic interaction
In addition to the hydrogen bonding, the ligand-receptor hydrophobic interactions were also investigated (Fig. 6 and Supplementary Fig. 8‒10 , available online). The results showed that there were hydrophobic interactions of methyl group of remdesivir with the alkyl side-chain of Lys13 of ACE2, as shown by the distance (4.81 Å) between hydrophobic centers in Supplementary Fig. 8. The interaction between isobutyl group at the end of the remdesivir and benzene ring of Phe54 is stronger with a smaller distance of 4.58 Å (Supplementary Fig. 8). In addition, the π-π stacking interaction between the triazine ring of remdesivir and benzene ring of Phe476 of spike protein was also found. The average value of distance between the mass centers of two rings along the simulation time is about 4.72 Å (Supplementary Fig. 8).
In the case of lopinavir, three kinds of hydrophobic interactions with spike protein and ACE2 were detected. T-shaped π-π interactions was formed between 2,6-dimethylphenyl group of lopinavir and 4-hydroxylphenyl side-chain of Tyr435 of spike protein. The phenyl group of lopinavir also formed a π-π stacking with phenmethyl group of Phe476 of spike protein. The distances between the stacked fragments were 4.45 and 4.67 Å, respectively (Fig. 6F and Supplementary Fig. 10). Furthermore, alkyl-π interaction involving methyl of 2,6-dimethylphenyl group of lopinavir and imidazole ring of Hie16 of ACE2 is weaker, with a longer distance of 5.22 Å.
In comparison with remdesivir and lopinavir, the hydrophobic interaction of tenofovir with its receptor is weaker. Only T-shaped π-π interactions were found between its purine ring and aromatic rings of Tyr491 of spike protein as well as Hie16 of ACE2. As exhibited in Supplementary Fig. 9, the distance between the stacked fragments were 4.21 and 4.44 Å, respectively.
Structural modification of remdesivir
In order to further improve the therapeutic efficacy, structural modification of remdesivir was conducted. Since the spike-ACE2 interaction involves a large interface (Supplementary Fig. 2B), it is expected that increasing the area occupied by remdesivir on ACE2 will help improve its ability to resist viral invasion. Hence the compounds with binding abilities to ACE2 were considered. Because of the side-effect of ACE2 antagonists such as lowering blood pressure, diminazene, which is an agonist of ACE2 and has myocardial protective effect, was employed to investigate its binding capacity to the spike-ACE2 interface. As shown in Fig. 7 , the binding capacity (with docking score of 8.0891) of diminazene is slightly lower than that of remdesivir (with docking score of 8.9948 in Supplementary Fig. 1). Compound JL-01 was obtained through structural modification by Nitrogen-benzyl substituted methyl benzoate. Its binding capacity was slightly increased to 8.3138 (Fig. 7 ). Subsequently, JL-01 was employed to modify remdesivir through an ester bond, which ended producing an N-benzyl substituted diamidine derivative (JL-02 in Fig. 7 ). This modification of remdesivir showed a significant improvement in the binding capacity to 10.5261. Considering that remdesivir is the phosphate prodrug of GS-441524, we also linked GS-441524 to JL-01 in the way of ester bond. The binding ability of the obtained compound (JL-03, Fig. 7 ) was not significantly improved.
7.
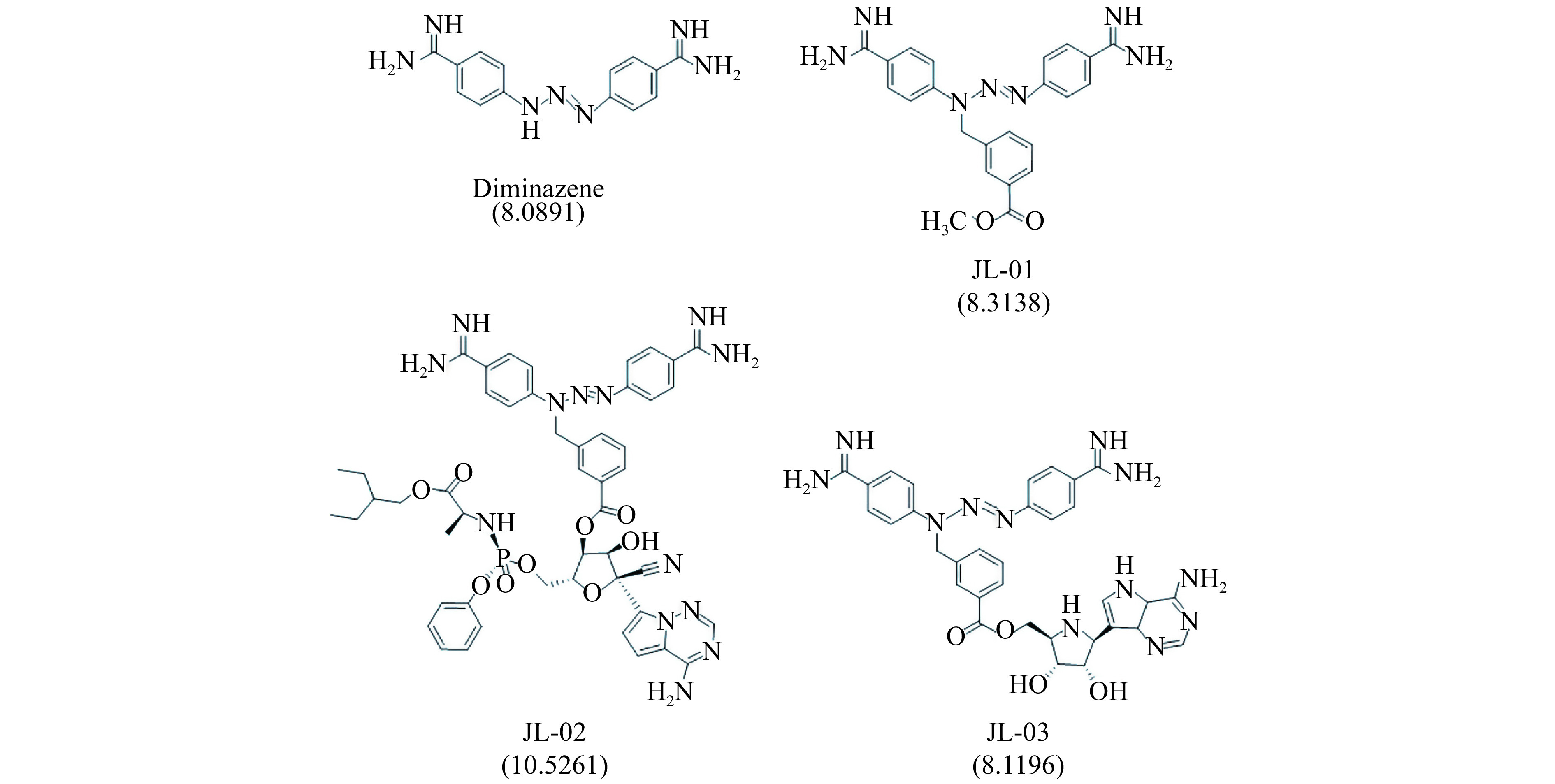
The modified compounds based on remdesivir as well as their docking score to the spike-ACE2 interface.
Discussion
It was reported that coronavirus uses its spike protein to bind to the host ACE2 and causes the infection. Therefore, we speculate that hindering the interaction between spike and ACE2 is a strategy to inhibit the viral invasion. A database of therapeutics including remdesivir and 2080 FDA-approved drugs were employed to screen candidates capable of blocking the spike-ACE2 interactions. The hit antiviral compounds could be divided into several classes through systematic analysis of their functions and targets. Remdesivir has been extensively studied and proved to be effective against SARS. Interestingly, other classes of drugs also attracted our attention since there were few reports about their antivirus activity against coronavirus. These drugs contained atazanavir, doravirine, lopinavir, and ritonavir used for the treatment of human immunodeficiency virus, valacyclovir employed for herpes simplex virus, valganciclovir applied for cytomegalovirus, adefovir for hepatitis B virus, tenofovir for chronic hepatitis B, and abacavir for retrovirus. These anti-viral compounds exhibit different molecular skeletons and side-chains. For example, remdesivir and tenofovir have the highest docking score, and both bear a phosphate ester and a fused N-containing heterocyclic ring attached by an amide group (‒NH2). It was also observed that more than one cyclic side-chains existed, especially for lopinavir, which possesses the most cyclic structures among these hit compounds (Supplementary Fig. 1). In the present work, remdesivir, tenofovir, and lopinavir were selected to investigate their effects on hindering the spike-ACE2 interactions, and the effects of other hit antiviral compounds will be studied and discussed in the future.
All the selected antiviral compounds can bind to the interface between spike and ACE2, and tenofovir exhibits the highest binding strength followed by remdesivir and lopinavir (Fig. 5A ). This binding weakens the interaction between spike and ACE2, gradually decreasing the spike-ACE2 interaction energies (as shown in Fig. 5B , the smaller the absolute values of interaction energies, the lower the interaction strength). This may indicate the competitive role of ligands in the spike-ACE2 interacting partners, which can be seen from the correlation between ligand binding energies and decreasing of spike-ACE2 interaction strength.
The selected compounds exhibited different preferences toward spike and ACE2. Remdesivir tended to bind ACE2, whereas tenofovir exhibited selectivity towards spike. This result indicates that remdesivir in those host cells can inhibit the combination of spike and ACE2 by occupying the surface of ACE2, and thus playing the role of blocking virus invasion. Lopinavir can bind to spike and ACE2 with comparable strength. It is like an adhesive that pulls spike and ACE2 together to form a ternary complex like a sandwich. This conforms to the fact that lopinavir decreases spike-ACE2 interaction strength to the lowest degree among the selected three compounds. This result might explain the limited power of lopinavir to uncouple the spike-ACE2 binding. Therefore, among those three compounds, remdesivir has the most potential to inhibit the SARS-CoV-2 invade the host cells.
Understanding the difference in action manners of these potential compounds can provide a clear direction for drug modification or design. Thereby, we further analyzed the interaction terms, including electrostatics and van der Waals, between the studied compounds and targeting. The results demonstrated that the N-containing heterocyclic rings played an important role in the ligand-receptor π-π stacking interactions, presumably, owing to the rigidity of these fused rings. This finding intrigues us to choose N-benzyl substituted diamidine derivative to modify remdesivir for the purpose of further increasing the efficacy. There are two reasons for choosing it as the modification unit. The first is that diamidine containing multi-rings, which play a very important role in binding of compounds with spike-ACE2 interface, as discussed above. The second reason is that diamidine is an agonist of ACE2 with myocardial protective effect, in contrast to the side-effect of ACE2 antagonists such as lowing blood pressure. The modification of remdesivir by N-benzyl substituted diamidine derivative (JL-02 in Fig. 7 ) showed a significant improvement in the binding capacity. However, for JL-03 which was obtained by linking N-benzyl substituted diamidine derivative to the metabolite of remdesivir (GS-441524), there was no obvious improvement in binding capacity (Fig. 7 ). This result may indicate three aspects. Firstly, this kind of structural modification is specific. Secondly, considering the larger size of remdesivir than its metabolite GS-441524, increasing size of anti-SARS-CoV-2 drugs may mitigate the likelihood of virus entry due to the larger occupancy of ACE2. Thirdly, due to the high binding capacity to ACE2 surface, JL-02 can enrich at the spike-ACE2 interface at high concentration. In a sense, JL-02 is targeted and has higher compatibility.
In summary, JL-02, which can be obtained by connecting remdesivir to N-benzyl substituted diamidine derivate (JL-01), shows excellent anti-viral invasion potential. It is a targeted compound with higher safety. Our study not only validated the possible therapeutic power of drugs against SARS-CoV-2 invasion from a computational viewpoint, but more importantly, rationalizing the combinational use of therapeutics that harness distinct anti-viral mechanism to benefit the outcome.
Acknowledgments
The supercomputing service from AM-HPC is gratefully acknowledged for high-performance computing.
References
- 1.Wang ML, Cao RY, Zhang LK, et al remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro . Cell Res. 2020;30(3):269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mwololo SW, Mutiso JM, Macharia JC, et al In vitro activity and in vivo efficacy of a combination therapy of diminazene and chloroquine against murine visceral leishmaniasis . J Biomed Res. 2015;29(3):214–223. doi: 10.7555/JBR.29.20140072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muralidharan N, Sakthivel R, Velmurugan D, et al Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. J Biomol Struct Dyn. 2020:doi: 10.1080/07391102.2020.1752802. [Epub ahead of print]. doi: 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- 4.Xue XY, Yu HW, Yang HT, et al Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82(5):2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu XT, Chen P, Wang JF, et al Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63(3):457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao D, Wang ZM, Wang LS Prevention of atrial fibrillation with renin-angiotensin system inhibitors on essential hypertensive patients: a meta-analysis of randomized controlled trials. J Biomed Res. 2015;29(6):475–485. doi: 10.7555/JBR.29.20140149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar Ramanathan AS, Karuppiah B, Vijayan M, et al Effect of angiotensin converting enzyme gene I/D polymorphism in South Indian children with nephrotic syndrome. J Biomed Res. 2019;33(3):201–207. doi: 10.7555/JBR.32.20150095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang YZ. Initial genome release of novel coronavirus[EB/OL]. [2020-01-10]. http://virological.org/t/initial-genome-release-of-novel-coronavirus/319?from=groupmessage.
- 9.Morse JS, Lalonde T, Xu SQ, et al Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. Chembiochem. 2020;21(5):730–738. doi: 10.1002/cbic.202000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong N, Yang XM, Ye LW, et al. Genomic and protein structure modelling analysis depicts the origin and infectivity of 2019-nCoV, a new coronavirus which caused a pneumonia outbreak in Wuhan, China[EB/OL]. [2020-01-21]. https://www.biorxiv.org/content/10.1101/2020.01.20.913368v1.
- 11.Wrapp D, Wang NS, Corbett KS, et al Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waterhouse A, Bertoni M, Bienert S, et al SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bienert S, Waterhouse A, de Beer TAP, et al The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017;45(D1):D313–D319. doi: 10.1093/nar/gkw1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertoni M, Kiefer F, Biasini M, et al Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep. 2017;7(1):10480. doi: 10.1038/s41598-017-09654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikram S, Ahmad J, Durdagi S Screening of FDA approved drugs for finding potential inhibitors against Granzyme B as a potent drug-repurposing target. J Mol Graph Model. 2020;95:107462. doi: 10.1016/j.jmgm.2019.107462. [DOI] [PubMed] [Google Scholar]
- 16.Kondagari B, Dulapalli R, Krishna Murthy D, et al Towards the virtual screening of BIK inhibitors with the homology-modeled protein structure. Med Chem Res. 2013;22(3):1184–1196. doi: 10.1007/s00044-012-0105-z. [DOI] [Google Scholar]
- 17.Kumar S, Jena L, Mohod K, et al Virtual screening for potential inhibitors of high-risk human papillomavirus 16 E6 protein. Interdiscip Sci Comput Life Sci. 2015;7(2):136–142. doi: 10.1007/s12539-015-0008-z. [DOI] [PubMed] [Google Scholar]
- 18.Umamaheswari A, Pradhan D, Hemanthkumar M Virtual screening for potential inhibitors of homology modeled Leptospira interrogans MurD ligase . J Chem Biol. 2010;3(4):175–187. doi: 10.1007/s12154-010-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan WSD, Liao WP, Zhou S, et al Targeting the renin-angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr Opin Pharmacol. 2018;40:9–17. doi: 10.1016/j.coph.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 20.MD simulated (37 °C, water, all-atom) model of Spike-ACE2(Spike is a homo-trimer in color, human ACE2 is in yellow)[EB/OL]. [2020-06-19]. https://ghddi-ailab.github.io/Targeting2019-nCoV/nCov_Structures/.
- 21.Spitzer R, Jain AN Surflex-dock: docking benchmarks and real-world application. J Comput Aided Mol Des. 2012;26(6):687–699. doi: 10.1007/s10822-011-9533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tripos Inc. SYBYL-X 1.3[EB/OL]. [2011-07-14]. https://sybyl-x.software.informer.com/1.3/.
- 23.Zhang G, Guo S, Cui HQ, et al Virtual screening of small molecular inhibitors against DprE1. Molecules. 2018;23(3):524. doi: 10.3390/molecules23030524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Case DA, Betz RM, Cerutti DS, et al. AMBER 2016[CP]. San Francisco, CA: University of California, 2016.
- 25.Jorgensen WL, Chandrasekhar J, Madura JD Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 26.de Souza ON, Ornstein RL Effect of periodic box size on aqueous molecular dynamics simulation of a DNA dodecamer with particle-mesh Ewald method. Biophys J. 1997;72(6):2395–2397. doi: 10.1016/S0006-3495(97)78884-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berendsen HJC, Postma JPM, van Gunsteren WF, et al Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 28.Ryckaert JP, Ciccotti G, Berendsen HJC Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23(3):327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 29.Duan Y, Wu C, Chowdhury S, et al A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24(16):1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 30.Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09 (revision D.01)[CP]. Wallingford, CT: Gaussian Inc., 2009.
- 31.Wang JM, Wolf RM, Caldwell JW, et al Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 32.Brice AR, Dominy BN Analyzing the robustness of the MM/PBSA free energy calculation method: application to DNA conformational transitions. J Comput Chem. 2011;32(7):1431–1440. doi: 10.1002/jcc.21727. [DOI] [PubMed] [Google Scholar]
- 33.Negri M, Recanatini M, Hartmann RW Computational investigation of the binding mode of bis(hydroxylphenyl)arenes in 17β-HSD1: molecular dynamics simulations, MM-PBSA free energy calculations, and molecular electrostatic potential maps. J Comput Aided Mol Des. 2011;25(9):795–811. doi: 10.1007/s10822-011-9464-7. [DOI] [PubMed] [Google Scholar]
- 34.Chéron N, Shakhnovich EI Effect of sampling on BACE-1 ligands binding free energy predictions via MM-PBSA calculations . J Comput Chem. 2017;38(22):1941–1951. doi: 10.1002/jcc.24839. [DOI] [PubMed] [Google Scholar]
- 35.Li GD, de Clercq E Therapeutic options for the 2019 novel coronavirus (2019-nCoV) Nat Rev Drug Discov. 2020;19(3):149–150. doi: 10.1038/d41573-020-00016-0. [DOI] [PubMed] [Google Scholar]