

Abstract
According to the 2020 Metrics of the HUPO Human Proteome Project (HPP), expression has now been detected at the protein level for >90% of the 19 773 predicted proteins coded in the human genome. The HPP annually reports on progress made throughout the world toward credibly identifying and characterizing the complete human protein parts list and promoting proteomics as an integral part of multiomics studies in medicine and the life sciences. NeXtProt release 2020–01 classified 17 874 proteins as PE1, having strong protein-level evidence, up 180 from 17 694 one year earlier. These represent 90.4% of the 19 773 predicted coding genes (all PE1,2,3,4 proteins in neXtProt). Conversely, the number of neXtProt PE2,3,4 proteins, termed the “missing proteins” (MPs), was reduced by 230 from 2129 to 1899 since the neXtProt 2019–01 release. PeptideAtlas is the primary source of uniform reanalysis of raw mass spectrometry data for neXtProt, supplemented this year with extensive data from MassIVE. PeptideAtlas 2020–01 added 362 canonical proteins between 2019 and 2020 and MassIVE contributed 84 more, many of which converted PE1 entries based on non-MS evidence to the MS-based subgroup. The 19 Biology and Disease-driven B/D-HPP teams continue to pursue the identification of driver proteins that underlie disease states, the characterization of regulatory mechanisms controlling the functions of these proteins, their proteoforms, and their interactions, and the progression of transitions from correlation to coexpression to causal networks after system perturbations. And the Human Protein Atlas published Blood, Brain, and Metabolic Atlases.
Keywords: Human Proteome Project (HPP), neXtProt protein existence (PE) metrics, missing proteins (MPs), non-MS PE1 proteins, uncharacterized protein existence 1 (uPE1), Chromosome-Centric HPP (C-HPP), Biology and Disease-HPP (B/D-HPP), PeptideAtlas, Mass Spectrometry Interactive Virtual Environment (MassIVE), Human Protein Atlas
Graphical Abstact

INTRODUCTION
The Human Proteome Project (HPP) of the Human Proteome Organization (HUPO) has provided a useful framework for international collaboration, data sharing and reanalysis, quality assurance, guidelines, communication, and acceleration of progress in building and utilizing proteomic knowledge since its launch in 2010.1 Remarkable progress has been documented in neXtProt, PeptideAtlas, and MassIVE and the annual Metrics paper in the HPP special issues of the Journal of Proteome Research (JPR).2–9 Our complementary decadal “A High-Stringency Blueprint of the Human Proteome” draws a striking parallel to the celebration of the completion of 90% of the Human Genome sequence in 2001.10
To celebrate the announcement of the completion of the first draft of the human proteome by HUPO and the HPP, JPR has published a Virtual Issue of the most significant papers in the HPP that have appeared in the past 10 years in the Journal.
PROGRESS ON THE HUMAN PROTEOME PARTS LIST: ACHIEVING THE MILESTONE OF DETECTING 90% OF PREDICTED PROTEIN-CODING GENES
Table 1 shows the increase from 13 975 PE1 proteins in neXtProt release 2012–02 to 17 874 in release 2020–01. This PE1 total now represents 90.4% of the 19 773 PE1,2,3,4 proteins predicted from the Human Genome version GRCh38. Conversely, the number of PE2,3,4 “Missing Proteins” has declined from 5511 in 2012 to 1899 in 2020, including a reduction of 230 from 2019.
Table 1.
Numbers of Proteins Classified in neXtProt Protein Existence Evidence Levels PE1,2,3,4 from 2012 to 2020, Showing Remarkable Progress in Confidently Identifying Predicted Human Proteins as PE1a
| PE level/neXtProt release | 2012–02 | 2013–09 | 2014–10 | 2016–01 | 2017–01 | 2018–01 | 2019–01 | 2020–01 |
|---|---|---|---|---|---|---|---|---|
| l: Evidence at Protein level | 13 975 | 15 646 | 16 491 | 16 518 | 17 008 | 17 470 | 17 694 | 17 874 |
| 2: Evidence at transcript level | 5205 | 3570 | 2647 | 2290 | 1939 | 1660 | 1548 | 1596 |
| 3: Inferred from homology | 218 | 187 | 214 | 565 | 563 | 452 | 510 | 253 |
| 4: Predicted | 88 | 87 | 87 | 94 | 77 | 74 | 71 | 50 |
| MP = PE2 + PE3 + PE4 | 5511 | 3844 | 2948 | 2949 | 2579 | 2186 | 2129 | 1899 |
| Human PeptideAtlas canonical proteins | 12 509 | 13 377 | 14 928 | 14 569 | 15 173 | 15 798 | 16 293 | 16 655 |
As of 2020–01, PE1/PE1 + 2 + 3 + 4 = 17 874/19 773 = 90.4% now PE1. The PE2,3,4 proteins are still “missing proteins” (MP). Dates represent the neXtProt releases used as baseline for the annual HPP metrics papers.2–9 More stringent Guidelines 2.1 were imposed in time for the 2016–01 release.11 Since 2016, the January release of neXtProt has served as the annual baseline. The final row shows the critical contribution of canonical proteins resulting from PeptideAtlas reanalyses of available MS datasets (see text and Figure 1).
The components of the HPP are the Chromosome-centric C-HPP, with 25 teams addressing the 24 chromosomes and mitochondria; the Biology and Disease-driven B/D-HPP, with 19 teams investigating organs, diseases, and biological functions; and the four Resource Pillars for Mass Spectrometry, Antibody-profiling, Knowledgebases, and Pathology. The C-HPP held its 21st Workshop on Future Strategies for the Human Proteome Project at Saint-Malo, France, in May 2019. The HPP Mass Spectrometry Data Interpretation Guidelines 3.0 were finalized, and integration of the MassIVE data resource was agreed upon for the neXtProt 2020–01 release. On the basis of the Guidelines 3.0,12 neXtProt has 16 836 entries that were validated by PeptideAtlas, 15 214 entries validated by MassIVE, 16 920 validated by one resource or the other, 15 130 by both resources, 84 only by MassIVE, and 1706 only by PeptideAtlas. There are four additional entries identified and curated by neXtProt from PTM proteomics papers. Supplementary Table S1 contains Excel files with all of these lists from neXtProt. In order to call a protein validated by MS, neXtProt requires that the minimum MS-evidence requirements described in the HPP Guidelines 3.0 are satisfied by either PeptideAtlas or MassIVE (or both) independently; i.e., insufficient evidence in both PeptideAtlas and MassIVE, such as one peptide in each, may not be combined to form sufficient evidence.
A major adjustment had occurred in 2015 as the HPP Guidelines 1.0 based on false-discovery rates ≤0.01 at the protein level were superseded by Guidelines 2.1, requiring two proteotypic (uniquely mapping) non-nested peptides of at least 9 amino acids in length.11 These requirements removed many proteins that were “one-hit wonders”, were based on peptides as short as 6 or 7 amino acids, or matched better to well-established proteins. That caused 485 proteins that would previously have been classified as PE1 to be put in the PE2,3,4 missing proteins set (438 PE2, 40 PE3, 7 PE4); as shown in Table 1, that substantially slowed the identification of PE1 proteins from 2014 to 2016. Meanwhile, in 2016 there was a large increase in PE3 (214 to 565) due to a decision of UniProt/SwissProt to remove upgrades to PE2 that relied on inclusion in the ArrayExpress or CleanEx transcriptome repositories, while greatly increasing the number of PE3 entries based only on homology (expression detected in nonhuman species).5 For the 2020–01 release neXtProt incorporated the merged RNA_seq data sets from Human Protein Atlas, GTEx, and FANTOM513 to upgrade PE3 and PE4 to PE2 if the expression values were ≥1 FPKM (fragments per kilobase of exon model per million reads mapped).
Figure 1 shows the major categories of experimental evidence upon which neXtProt has classified the human proteome. Of the 17 874 PE1, 16 924 are now based on validated mass spectrometry results (green bar). The yellow bar in Figure 1 represents 950 PE1 proteins identified by non-MS methods and curated by SwissProt/neXtProt based on the published literature: 73 from Edman sequencing, 122 from disease mutations, 35 from 3D structures, 342 from protein-protein interactions, 49 from antibody-based techniques, 127 from PTMs or proteolytic processing, and 202 from biochemical studies; because many proteins have multiple lines of evidence, these numbers represent the first evidence reported for each protein. Supplementary Table S2 provides a comprehensive list of the kinds of evidence used by SwissProt curators to classify 906 of these 950 as PE1, while Supplementary Table S3 further annotates 820 entries of this list with information from the Human Protein Atlas (see below). There are no HPP Guidelines for Data Interpretation for these different studies grouped in the non-MS category; SwissProt describes their PE process at https://www.uniprot.org/help/protein_existence. The yellow bar has declined quite dramatically from 1860 in 2016 to 950 in 2020, as more and more proteins have been confirmed with independent identification by increasingly sensitive and accurate mass spectrometry analyses. The missing proteins PE2,3,4 (Table 1) now number 1899 (red bar in Figure 1), hence 9.6% of the total predicted proteins.
Figure 1.
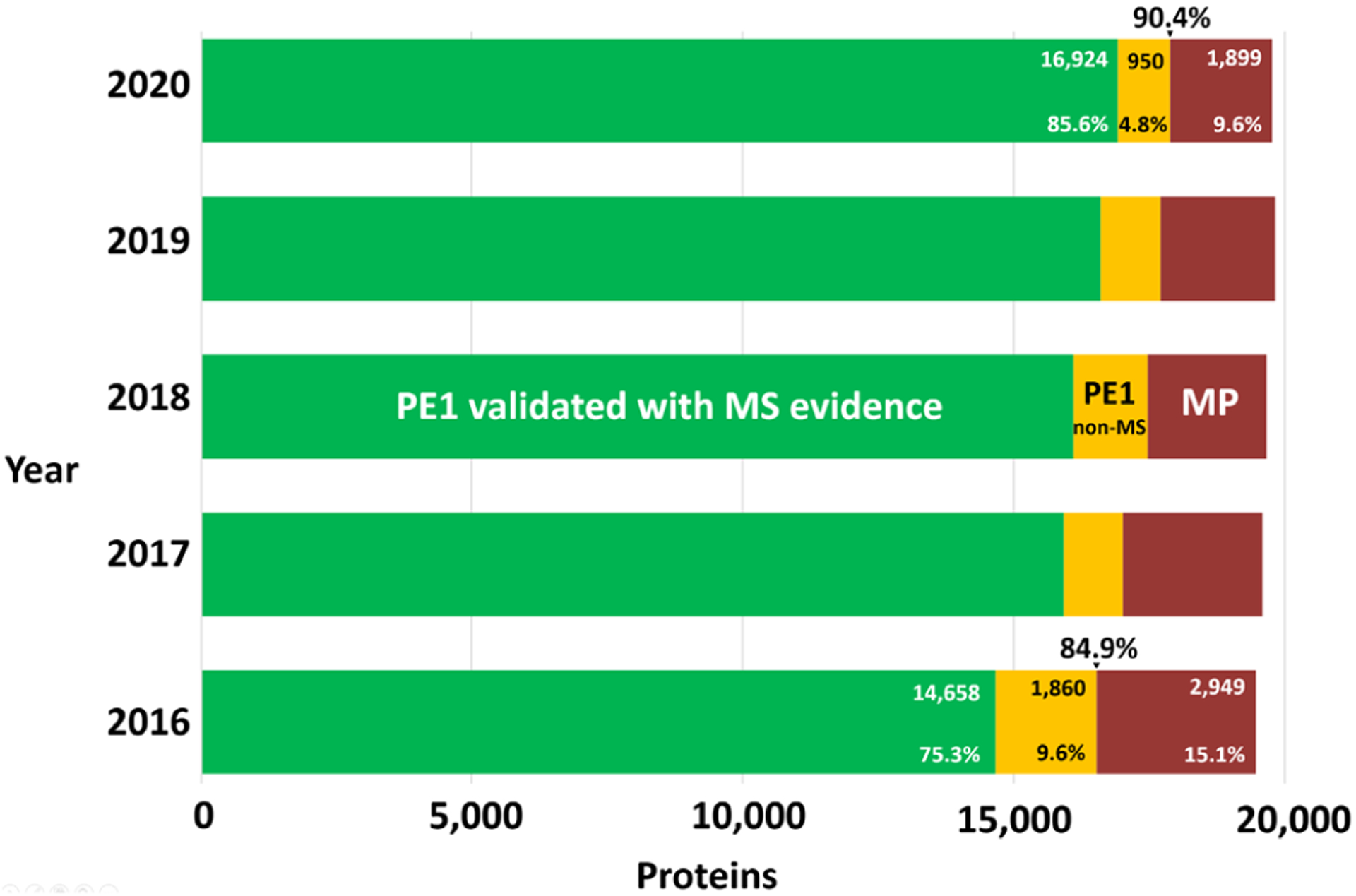
Bar chart showing the progression of the state of protein validation for the past five years, since the HPP Guidelines 2.1 have been in effect. The PE1 proteins validated with mass spectrometry (MS) evidence are depicted in green, PE1 proteins based on other types of protein evidence in yellow, and the PE2,3,4 missing proteins (MPs) that have no or insufficient protein-level evidence in human specimens and cell lines in red. The green bars for 2016–2019 are from PeptideAtlas, while for 2020 it is a combination with MassIVE. Data for the green + yellow (PE1) and red (PE2,3,4) bars are provided in Table 1.
From its beginning in 2012 neXtProt has relied on the standardized reanalysis of available data sets by PeptideAtlas. From these, there are now 16 655 canonical proteins in the 2020–01 Human PeptideAtlas build (Table 1). To the 2019–01 build, PeptideAtlas added 257 samples from 24 ProteomeXchange data sets comprising 101 million new spectrum identifications that passed stringent thresholds. This subset of data sets submitted to ProteomeXchange was selected to contribute new identifications reflecting new instrumentation, methods, or sample types. Note that the more data that are added, the more stringent the threshold must be for all data sets in order to maintain a constant FDR; thus, including data sets that do not contribute new identifications is counterproductive.
Figure 2 shows the top 10 newly analyzed data sets located through ProteomeXchange contributing to the increased number of canonical proteins in PeptideAtlas during 2019; they account for 369 of the 413 added canonical proteins. Note that, while 413 canonical proteins were added in PeptideAtlas, 51 previously canonical proteins were removed from the reference proteome due to Swiss-Prot entry merging as discussed below, yielding the net increase of 362 in Table 1. While the HPP and JPR encourage investigators to validate their findings of newly detected missing proteins with synthetic peptides, neither neXtProt nor PeptideAtlas nor MassIVE requires confirmatory synthetic peptide MS matching results for their annual updates of community MS data for the human proteome. Efforts to incorporate synthetic peptide MS matching are underway, but PeptideAtlas does not have an automated method to incorporate SRM or PRM results. Nevertheless, several PRM data sets are now included in PeptideAtlas, applying the HUPO/PSI Universal Spectrum Identifier in spectrum-based guidelines, as PRM acquires full MS2 spectra.
Figure 2.

Top 10 data sets that contributed to the increment in canonical proteins in between 2019 and 2020 builds of PeptideAtlas. These new MS-based canonical proteins represent both new PE1 proteins that previously were PE2,3,4 (red in Figure 1) and previous non-MS-based PE1 proteins (yellow in Figure 1) that are now validated with MS evidence in PeptideAtlas. Each data set is labeled with the ProteomeXchange identifier (PXD), first*last authors, publication year, number of new canonical proteins, and method highlighted.14–23
The largest gain came from PeptideAtlas reprocessing of PXD004452, a deep analysis of the extensively studied HeLa cell,24–26 using shotgun proteomics, three proteases, high peptide load, and 2D high-resolution reversed-phase peptide fractionation to saturate the sequencing speed with short gradients, as well as modern MS instruments.23 The original paper reported 584 000 unique peptide sequences and 14 200 protein isoforms from 12 200 protein-coding genes, 7000 N-acetylation sites, and 10 000 phosphorylation sites. This depth is comparable to the depth of next-generation RNA sequencing. Enhancing the sensitivity of the MS protocol reaches less-abundant proteins.
The second largest gain was from PeptideAtlas reprocessing of PXD010154, with combined RNA-seq and MS analyses of adjacent cryosections from 29 tissues.22 They reported detection of 13 640 proteins, including 37 PE2,3,4 MPs, based on “at least 1 unique peptide” confirmed with a synthetic peptide; only 8 met the HPP Guidelines 2.1 of two uniquely mapping peptides of at least 9 aa. Correlation of mRNA and protein abundance was generally low; 478 proteins were not detected in testis and 1408 across all tissues even when mRNA FPKM values were above the mean values. Moreover, 300 of the 478 not detected in testis by MS were not detected in HPA with antibodies. Among transcripts below the mean FPKM, GPCR, ion channels, and cytokine-related proteins were notable missing proteins. An extensive analysis showed challenges in finding protein-level evidence of sequence variants or splice isoforms; not a single lncRNA peptide was substantiated. The 93 new canonical proteins found by PeptideAtlas predominantly represent proteins previously classified as PE1 by non-MS methods.
The third largest gain with 53 new canonical proteins came from PeptideAtlas reanalysis of PXD005445, data from the BrainSpan Project, which examined 7 regions of the brain in individuals in 6 age intervals from infancy to age 42.21 After testis, brain is the most promising organ for deeper analysis to find MPs.22 Their analysis revealed that 8980 proteins were detected in at least one region and 6529 in all 7 regions. Differential analysis of protein expression by region showed prominence for nuclear functions, including RNA processing, especially in the cerebellum. Our Supplementary Table S3 provides information from the Human Protein Atlas of transcript expression and immunohistochemical localization of 820 of the 950 non-MS-based PE1 proteins, ordered alphabetically by tissue type.
DYNAMIC CHANGES IN neXtProt CLASSIFICATION OF PROTEINS
The year-to-year changes in neXtProt categories, including their inputs from UniProt/SwissProt, are quite dynamic and quite challenging to track, as shown in Figure 3. Note that 255 PE2,3,4 MPs were promoted by new evidence to PE1, while 10 MPs were deleted, 8 were demoted to PE5, 15 were gained from PE1, and 25 were newly added to neXtProt. PE1 had an unusual reason for a smaller increase than what came from PE2,3,4 MPs (a net of 180): 71 PE1 entries were merged into 7, mostly due to a major reclassification of HLA-A, HLA-B, and HLA-C genes and their protein products. In 2013, we noted that there were 21 genes for HLA-A, 35 for HLA-B, 14 for HLA-C, and 13 for HLA-DRB1 as neXtProt entries for Chromosome 6.2 Also, 15 previous PE1 proteins were demoted to PE2,3,4. Further details show 12 PE1 entries new to neXtProt, of which 6 involved the family of T Cell Receptor proteins, from which 13 more were added to the MPs according to predictive models (PE4). Previous annual metrics papers have reported corresponding dynamic year-to-year changes.6–8
Figure 3.
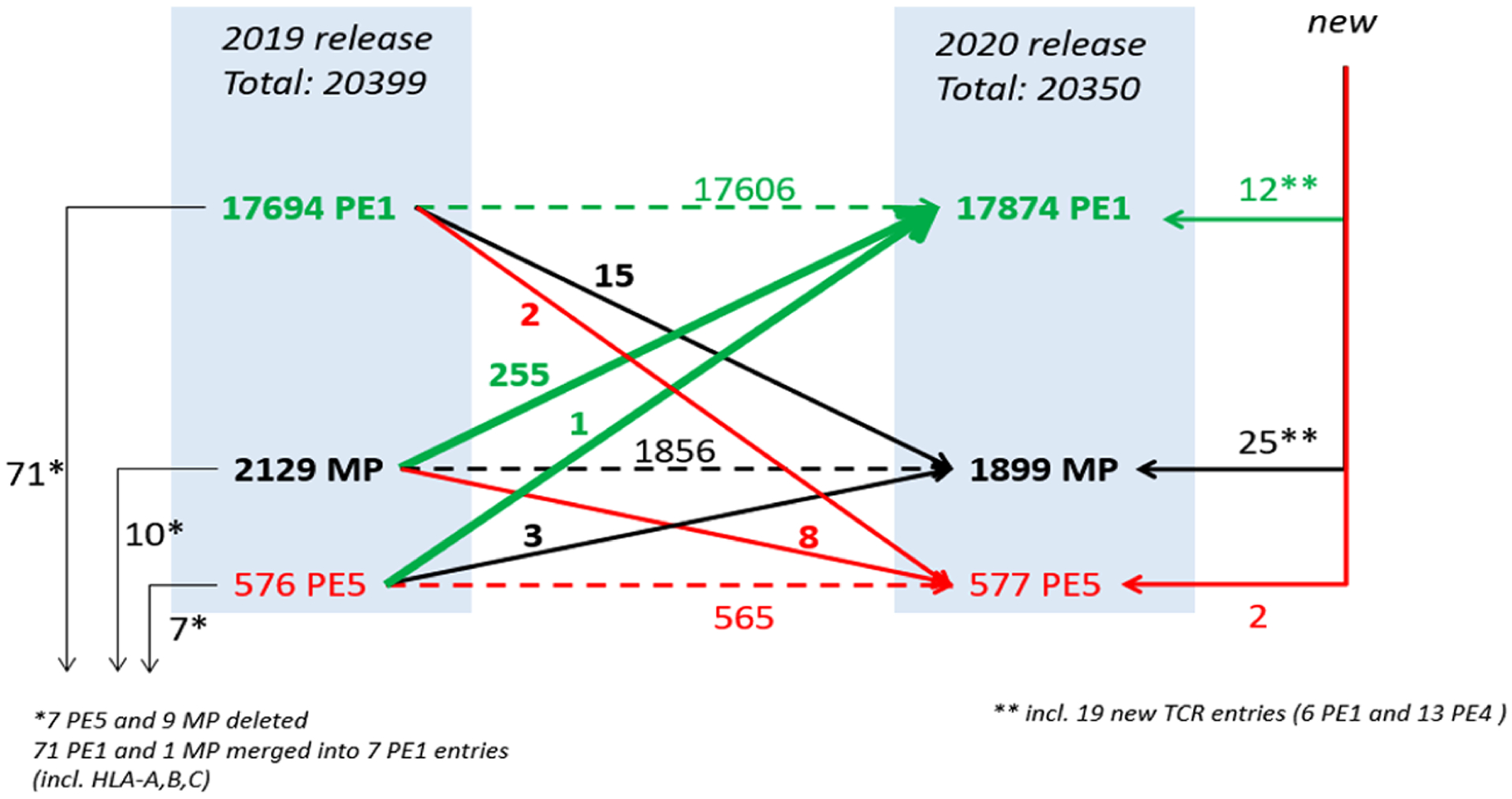
Flowchart depicts the detailed changes in neXtProt PE1–4 categories (plus PE5) from neXtProt release 2019–01 to neXtProt release 2020–01 (see text).
There were multiple minor changes in the category of PE5. In 2013, the HPP excluded category PE5 from the denominator of all predicted proteins, due to the status of these sequences as “dubious or uncertain” genes with a high proportion of pseudogenes.3 When C-HPP announced the MP50 Missing Proteins Challenge, PE5 entries were excluded.6
UPDATE FROM THE CHROMOSOME-CENTRIC HPP (C-HPP)
The 25 teams of the C-HPP have mounted two initiatives to characterize “the Dark Proteome”: the 2016 neXt-MP50 Challenge to find or document the finding of at least 50 PE2,3,4 missing proteins per chromosome5 and the 2018 neXt-CP50 Challenge to use experimental and informatics approaches to annotate functions for 1260 unannotated PE1 proteins (uPE1).7 Both challenges reflect the overall goal of the HPP to both detect and characterize the predicted human proteins. Among the advances on missing proteins that came from the entire community, there were 32 MPs identified in the 2019 HPP special issue of JPR and proposed to be reviewed for PE1: 1 from rarely studied bone (plus 5 non-MS PE1),27 3 from placenta,28 5 from testis,29 which has been a rich source of MPs, and 23 from matching peptides to their counterparts in SRM Atlas.30 Of the 9 PE1 candidates from the first three papers, Q6NT89/TMF-regulated nuclear protein (chr 1) from Lin et al., and Q8TAA1/RNase11 (chr 14) and Q8WW27/ABOBEC-4 (chr 1) from Sun et al. became PE1 in neXtProt 2020–01, as did Q5T5N4/C6orf118, which initially did not quite meet Guidelines criteria; the other 6 of the 9 remained PE2. None of these 3 data sets was released publicly in time to be included in the PeptideAtlas update release of 2020–01. Of the 23 from Elguoshy et al.,30 19 became PE1.
As of release 2019–01,8 chromosomes 19, 1, 5, and 17 had each noted a reduction in their number of PE2,3,4 MPs by 50 or more; as of release 2020–01, MPs had been reduced by at least 50 for Chromosomes 2, 3, 6, 10, 11, 12, 22, and X, as well (www.nextprot.org/about/protein-existence.), as shown in Table 2, Chromosome-by-Chromosome Status of Predicted Proteins in neXtProt 2020–01.
Table 2.
Chromosome-by-Chromosome Status of Predicted Proteins in neXtProt 2020–01a
| Chr | PE1 | PE2 | PE3 | PE4 | PE2 + 3 + 4 | PE1/PE1–4 | % PE1/PE1–4 | net decrease PE2,3,4(2016–2020) | uPE1 proteins |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1819 | 171 | 27 | 3 | 201 | 1819/2020 | 90.0% | 108 (309/201) | 145 |
| 2 | 1193 | 68 | 9 | 2 | 79 | 1193/1272 | 93.8% | 55 (134/79) | 87 |
| 3 | 981 | 72 | 9 | 2 | 83 | 981/1064 | 92.2% | 82 (141/59) | 55 |
| 4 | 688 | 44 | 15 | 0 | 59 | 688/747 | 92.1% | 36 (95/59) | 50 |
| 5 | 817 | 43 | 2 | 1 | 46 | 817/863 | 94.7% | 76 (122/46) | 50 |
| 6 | 930 | 72 | 7 | 4 | 83 | 930/1013 | 91.8% | 53 (136/83) | 59 |
| 7 | 828 | 81 | 37 | 16 | 134 | 828/962 | 86.1% | 3 (137/134) | 53 |
| 8 | 612 | 42 | 7 | 2 | 51 | 612/663 | 92.3% | 44 (95/51) | 35 |
| 9 | 683 | 80 | 8 | 3 | 91 | 683/774 | 88.2% | 38 (129/91) | 59 |
| 10 | 670 | 61 | 2 | 1 | 64 | 670/734 | 91.3% | 51 (115/64) | 52 |
| 11 | 1029 | 205 | 58 | 1 | 264 | 1029/1293 | 79.6% | 55 (319/264) | 71 |
| 12 | 942 | 66 | 3 | 0 | 69 | 942/1011 | 93.2% | 50 (119/69) | 52 |
| 13 | 304 | 17 | 1 | 0 | 18 | 304/322 | 94.4% | 25 (43/18) | 25 |
| 14 | 614 | 50 | 43 | 5 | 98 | 614/712 | 86.2% | −5 (93/98) | 32 |
| 15 | 524 | 47 | 2 | 0 | 49 | 524/573 | 91.4% | 24 (73/49) | 38 |
| 16 | 756 | 60 | 2 | 0 | 62 | 756/818 | 92.4% | 37 (99/62) | 47 |
| 17 | 1061 | 83 | 2 | 2 | 87 | 1061/1148 | 92.4% | 61 (148/87) | 66 |
| 18 | 252 | 12 | 1 | 0 | 13 | 252/265 | 95.1% | 11 (24/13) | 10 |
| 19 | 1281 | 112 | 10 | 1 | 123 | 1281/1404 | 91.2% | 138 (261/123) | 80 |
| 20 | 490 | 45 | 0 | 3 | 48 | 490/538 | 91.1% | 34 (82/48) | 42 |
| 21 | 191 | 36 | 5 | 1 | 42 | 191/233 | 82.0% | 7 (49/42) | 11 |
| 22 | 439 | 33 | 2 | 1 | 36 | 439/475 | 92.4% | 28 (64/36) | 33 |
| X | 731 | 82 | 1 | 2 | 85 | 731/816 | 89.6% | 60 (145/85) | 101 |
| Y | 27 | 13 | 0 | 0 | 13 | 27/40 | 67.5% | 3 (16/13) | 1 |
| Mito | 15 | 0 | 0 | 0 | 0 | 15/15 | 100.0% | 0 (0/0) | 0 |
| Unk | 2 | 2 | 0 | 0 | 2 | 2/4 | 50.0% | 0 (2/2) | 0 |
| ALL | 17 874 | 1596 | 253 | 50 | 1899 | 17 879/19 779 | 90.4% | 1050 (2949/1899) | 1254 |
| Sums | 17 879 | 1597 | 253 | 50 | 1900 | 17 699/19 829 | 90.2% | 1050 (2950/1900) | 1254 |
Note: There are discrepancies between the true total numbers of proteins in each PE category (ALL) and the Sums because six proteins are derived from two genes on two different chromosomes, and thus appear twice under the per-chromosome table values.
The C-HPP also monitors progress on demonstrating and predicting functions for the 1260 uPE1 proteins unannotated for function as of 2018, when C-HPP initiated the next-CP50 Challenge seeking to stimulate discovery and confirmation of function. Table 2 (column 10) gives the uPE1 numbers for each chromosome. The column sum of 1254 is identical with that for 2019. While many chromosome uPE1 numbers in 2020 are very close to 2019,8 chromosomes 1, 6, and 19 stand out with increases from 138 to 145, 54 to 59, and 77 to 80, respectively, as the PE1 total grew larger.
Another significant development was the application of the I-TASSER/COFACTOR algorithms to predict Gene Ontology terms for uPE1 proteins.31,32 neXtProt added a link for individual protein entries to facilitate submission of uPE proteins for a report of predicted functions. As of May 15, 2020, documented requests for C-I-TASSER function predictions totaled 561 proteins from 181 users from 35 countries, including 201 neXtProt proteins [https://zhanglab.ccmb.med.umich.edu/C-I-TASSER/bin/stat.cgi].
HIGHLIGHTS FROM PEPTIDE ATLAS AND MassIVE
The PeptideAtlas processing pipeline at a high level includes conversion of vendor files to mzML33 with the ProteoWizard34 msconvert tool. The mzML files are processed through Comet35,36 using the most recent THISP37 Level 3 database and SpectraST38 using the most relevant NIST spectral library. The search results are validated with PeptideProphet39 and iProphet40 tools within the Trans-Proteomic Pipeline41–43 environment. Data sets encompass all the most common instrument types, fragmentation modes, and labeling methods, including iTRAQ and TMT.44 Most data sets are available in ProteomeXchange,45 although there remain 114 older samples that predated ProteomeXchange and are not available there. All 1804 samples are thresholded at a constant iProphet-model estimated 0.0002 PSM-level FDR in order to achieve a 0.01 protein-level FDR. Canonical proteins require two non-nested uniquely mapping peptides of length 9 or more residues, as described in the HPP Guidelines 3.0.12 However, all peptides are reported to neXtProt and undergo remapping and filtering there. All spectra supporting the Human PeptideAtlas 2020–01 Build are available interactively via the web interface. All spectra that belong to a data set deposited in ProteomeXchange are referenced by their HUPO Protein Standards Initiative Universal Spectrum Identifier (USI).12
The MassIVE processing pipeline (Mass Spectrometry Interactive Virtual Environment)46 also starts with ProteoWizard conversion of raw files into mzML using msConvert, and follows it with MS-GF+ database search against the UniProt human reference proteome with isoforms as well as common contaminants. To ensure strict FDR controls at multiple levels, the MassIVE pipeline follows the MassIVE-KB process for construction of spectral libraries, with 0.1% spectrum-level FDR, <1% length-specific peptide level FDR, and 1% FDR for all proteins matched by at least one unique peptide, which corresponds to 0.013% protein-level FDR for proteins matched by 2+ peptides. In total, 227 unlabeled data sets from 27 404 LC-MS runs encompassing 658 million MS/MS HCD spectra were reprocessed. All spectra were mapped to exons in ENSEMBL; we noted whether the peptide maps uniquely to an exon, covers a splice junction, or is mapped to an exon at all. Canonical proteins require two non-nested uniquely mapping peptides of length 9 or more residues, per HPP Guidelines 3.0. However, all peptides are reported to neXtProt and undergo remapping and filtering there. All spectra are viewable online using MassIVE’s ProteinExplorer,47 including the complete provenance from each sequence to spectra, searches, and data sets that support it. Additionally, all matching synthetic spectra can be viewed for each sequence.
A major asset of the HPP is ProteomeXchange.45 All MS data sets submitted to the JPR annual issues are required to have a PXD identifier for manuscript reviewers and readers to access the primary data sets and metadata in one of the participating repositories: PRIDE,48,49 PeptideAtlas,50,51 MassIVE,47 jPOSTdb,52 iProX,53 or Panorama Public.54 As of October 3, 2020, ProteomeXchange listed 12 801 all-species data sets and 5296 human data sets [www.proteomexchange.org].
HIGHLIGHTS FROM THE BIOLOGY AND DISEASE-DRIVEN HPP (B/D-HPP)
The B/D-HPP comprises 19 teams (https://hupo.org/B/D-HPP) that focus on distinct biomedical research areas relevant to understanding basic biology and the development of human diseases. The B/D teams are collaborative communities that are open to the participation of scientists from around the globe and welcome productive interactions that disseminate the use of proteomics across the research community. The B/D-HPP has been active in developing the SRMAtlas, the use of DIA-SWATH-MS, and the search for organ-specific priority proteins.
Themes at the core of all B/D-HPP teams include (1) the identification of driver proteins that underlie health and disease states, and (2) the characterization of regulatory mechanisms that control the functions of these proteins, including posttranslational modifications, protein interactions, physicochemical properties, and their dynamic organization within a cell or tissue.
The identification and characterization of driver proteins provide critical insights into the molecular underpinning of health and disease, then guide the development of assays for early detection of disease and innovative therapeutic interventions. Two examples in 2019 of collaboration between B/D-HPP teams included the Plasma and Diabetes teams applying a robust workflow to monitor 1508 plasma proteins and cellular pathways associated with weight loss and weight maintenance,55 and the Rheumatic and Autoimmune Disorders and Cardiovascular teams defining a panel of priority proteins that reveal both relevant mechanistic insights and promising biomarker candidates.56
The ability to profile proteomes with high sensitivity and accuracy has uncovered disease- and age-associated signatures.52–54 Another study identified changes in alternative splicing and autophagy connected to a decline in skeletal muscle function.57 As members of the B/D-Cancer team, Jiang et al. identified novel therapeutic targets of early stage hepatocellular carcinoma,15 and the NCI Clinical Proteomics Tumor Analysis Consortium has provided vast publicly accessible data sets and major resource papers in 2019 on proteogenomic and immune characterization of colon cancers58 and clear cell renal cell carcinomas,59 with others in the pipeline. Phosphoproteome analyses of kinases and kinase substrates informed prediction of promising targeted therapies. The Infectious Diseases team identified secreted proteins as microbial biomarkers for early detection of infection with the pathogen causing Lyme disease, Borrelia burgdorferi.60
Several elegant studies demonstrate the fundamental role of proteomics in biological discoveries, such as characterizing phenotypes associated with distinct protein aggregation states: pathological TDP-43 conformers that have differential toxicities in neurons and correlate with frontotemporal lobar neurodegeneration;61 cullin-RING ubiquitin ligase and diacyl glycerol biosynthesis associated with protein aggregation events in neurodegenerative diseases;62,63 and the necessity of nuclear aggregation of a DNA sensing protein, IFI16, for human cells to elicit immune signaling and suppress viral gene expression during a herpesvirus infection.64
Protein posttranslational modifications (PTMs) have received special attention: glycosylated immunoglobulin light chain is a biomarker for early diagnosis of amyloidosis;65 tyrosine-nitration of cystathionine β-synthase blocks the trans-sulfuration pathway in acute pancreatitis, promoting homocysteine accumulation upon S-adenosylmethionine treatment;66 a top-down approach revealed deamidation, methylation, acetylation, trimethylation, phosphorylation, and S-glutathionylation of sarcomeric proteins of nonhuman primate skeletal muscle;67 and protein lysine hyperacetylation contributes to development of type 2 diabetes when mitochondrial activity of beta cells or insulin target tissues is perturbed.68
These accomplishments were aided by the methods development components of many B/D-HPP teams to promote enhanced proteomic and multiomic experimental workflows, as well as computational platforms for data analysis and integration. The cancer team reported a data integration pipeline for predicting kinase activities from phosphoproteome data sets,69 while the cardiovascular team published a method for automated contaminant removal from peptide samples.70 A broadly applicable strategy from the Aebersold laboratory explores the transitions from correlation to coexpression to causal networks,71 using an experimental design with at least two systems perturbations to identify causal relationships of proteins in complex processes. This approach could identify new proteins involved in biological processes and their functions, thereby breaking the trend of a high percentage of omics-era investigations focused on the already-most-studied proteins.
To promote the dissemination and application of proteomics methods within diverse biological and clinical studies, members of B/D-HPP teams have organized educational activities, conferences, and symposia, including during 2019 the Brain Proteome Project Workshop in Amsterdam, a FASEB conference on protein acetylation in Lisbon, the Human Immunopeptidome satellite meeting at HUPO-Adelaide, a virtual EMBL Workshop on Proteomics in Cell Biology and Disease Mechanisms, the Applied Public-Private Research enabling OsteoArthritis Clinical Headway (APPROACH) project, and a journal special issue on Understanding Proteome Organization in Space and Time.72
HIGHLIGHTS FROM HUMAN PROTEIN ATLAS
The prolific output from the Human Protein Atlas (HPA), which serves as the Antibody-profiling Resource Pillar of the HPP, continues. In 2019 the Tissue, Cell, and Pathology Atlas series was complemented by releases of the Blood, Brain, and Metabolic Atlases.73–75 The Blood Atlas focuses on expression of all protein-coding genes in different blood cell types, as well as a separate analysis on The Human Secretome,76 which provides a comprehensive annotation of 2600 proteins predicted to be secreted by human cells. This represents a large fraction of the targets for pharmaceutical drugs, current clinical chemistry, and future diagnostics, and categorizes the different secreted proteins as secreted locally from different tissues or secreted into blood. For the 729 proteins that were found to be secreted into blood, concentration levels based on mass spectrometry or antibody-based immune assays are provided.76 Besides classical plasma proteins, cytokines, interleukins, hormones and receptors, about 100 have no known function (presumably uPE1 or uPE2,3,4; candidates for I-TASSER analysis). Many of these circulating proteins are not currently detected with mass spectrometry-based proteomics or antibody-based immunoassays. The analysis revealed that assays are lacking for a large fraction of the secreted blood proteins.
HPA applied its vast array of antibody reagents to COVID-19 studies. A separate portal has been created with direct links to antibody-based protein data for >300 proteins suggested to interact with SARS-CoV-2. For a full understanding of the susceptibility for SARS-CoV-2 infection and aid in development of effective treatments, it is necessary to study the cell type-specific expression of COVID-19 related proteins on the tissue, cellular and subcellular levels. Surprisingly, an in-depth characterization in >150 different cell types of the angiotensin I converting enzyme 2 (ACE2), the widely presumed target of the SARS-CoV-2 spike protein, revealed very limited expression in human lung and respiratory epithelia.77 Instead, highest ACE2 expression was observed in enterocytes, renal tubules, gallbladder, cardiomyocytes, male reproductive cells, placental trophoblasts, ductal cells, eye and vasculature. These other organs show prominent damage in some COVID-19 patients. The study highlights the need for further studies on samples from COVID-19 patients to confirm the histological colocalization of the virus with ACE2 and other SARS-CoV-2 related proteins, including the host serum protease TMPRRS2, as these proteins constitute targets for ongoing drug development efforts. There is evidence that ACE2 expression in airway epithelial cells is stimulated by interferon.78
While neXtProt includes information from HPA for protein characterization, and the HPA utilizes neXtProt information, the antibody-based data generated by HPA are currently not sufficient for PE1 classification in the absence of other compelling evidence. From version 19 of the HPA, 56 proteins defined as missing proteins and 171 uPE1 proteins were targeted by antibodies of high reliability meeting the criteria for enhanced validation as defined by the International Working Group for Antibody Validation (IWGAV).79,80 Further special efforts are being developed to link antibody-based protein detection and localization with MS-based identification of proteins, especially missing proteins, led by Cecilia Lindskog and Charles Pineau. The initial focus will be on testis, known to harbor a large number of missing proteins.81 This represents a special version of the general strategy of enrichment of low abundance proteins the HPP has been encouraging for several years.
We have created Supplementary Tables S2 and S3 listing the 950 non-MS-based PE1 proteins (see above). For 906 it was feasible to retrieve results from up to 10 different types of studies contributing to the PE1 assignment. Of the 950, 820 proteins matched entries in HPA version 19 based on Ensembl version 92. Only 10 entries were solely based on antibody information, but 127 have available information on high confidence (reliability score = Enhanced or Supported) in the HPA. These could be priority targets for further studies. The table also includes information on tissue specificity based on mRNA expression data across 37 different organs based on HPA, GTEx and FANTOM5 data sets; 707 had elevated expression in 1–5 tissues, which could guide tissue-specific efforts to target these proteins, while 96 had low tissue specificity, presumably representing widely expressed housekeeping proteins.
DISCUSSION
The neXtProt and PeptideAtlas releases of 2020–01 mark a major milestone in identifying and characterizing the human proteome, with stringent protein-level PE1 detections exceeding 90% of the predicted proteins according to neXtProt. For historical perspective, the Human Genome Project had a major public celebration hosted by President Clinton on June 26, 2000 when the sequencing “approached 90%” of the goal. Of course, much more research is underway to capture and characterize splice variants, PTMs, proteolytic products, and protein-protein, protein-nucleic acid, and protein-lipid interactions and their roles in biological networks in health and disease. For nearly a decade we have called this phase “building the parts list”.3
Simultaneously, the proteomics community strives to make proteomics a regular and major part of multiomics research. The critical parameters of dynamics, localization, and function of proteins and especially their splice variants, sequence variants, and various PTMs can be clarified only by direct study of proteins individually and as part of the cellular proteome; they cannot be predicted from DNA and RNA studies. Thus, the emergence of proteogenomics as a strategic approach to molecular characterization of cancers, cardiovascular diseases, brain circuitry, reproductive processes, and infectious agent/host interactions is a harbinger of a productive future.
Proteomics is poised to play an increasing role in “precision medicine” and “precision health”, including use of proteogenomic results to predict clinically useful targets for small molecule and protein or antibody therapeutics in many disease categories. Proteins likewise are critical to development of companion individual cells, thereby revealing the heterogeneity of tissue and organ function. As always, advances in instrumentation and methods are critical for asking new questions about the biology and creating new diagnostics and therapeutics.diagnostic tests to guide use of chemo- and immunotherapies. At the cellular level, there is a major impetus to conduct RNA-seq, protein, and proteoform analyses in
A specific initiative of the HUPO Human Proteome Project since 2016 has been the neXt-MP50 Challenge to each HPP chromosome team to identify or document the identification by others of at least 50 neXtProt PE2,3,4 “missing proteins”, proteins not yet detected in any human specimen with credible high-quality protein evidence, either by mass spectrometry meeting HPP Guidelines or by expert curation of a variety of non-MS protein methods (see Figure 1 and Supplementary Table S2). This Challenge now has been met for 12 of the 24 chromosomes (Table 2). Over the past three years, as some investigators have reported finding only small numbers of missing proteins, we have wondered whether the ability to detect more MPs is becoming saturated. However, we see in Table 1 and Figures 1 and 3 that the number and proportion of PE2,3,4 missing proteins continue to decline and the number of PE1 proteins continues to grow. In the most recent year, PE1 proteins grew by a net of 180 proteins and 255 PE2,3,4 proteins were promoted to PE1 (Figure 3). It is noteworthy that detection by MS has also reduced the number of PE1 proteins identified only or primarily by non-MS methods from 1860 to 950 in the past four years.
There are several major reasons for Missing Proteins: transcription and translation may fail to generate sufficient protein to be detected with current MS or other methods; proteins’ sequences may not yield two non-nested, uniquely mapping tryptic peptides of at least 9 amino acids, especially among highly homologous families of proteins; and/or membrane proteins may not be solubilized. There are large differences in gene expression across tissue types and the life span. A review of HPA, GTEx, and FANTOM5 transcript databases led to an estimate of 800 to 1000 PE2,3,4 predicted proteins lacking detectable transcript expression across all tissues.13 There are 399 olfactory receptor genes for which no proteins have been found, even when long-sought supra-nasal olfactory epithelium specimens were analyzed,82 though that study did find 5 other MPs. Some distinctive tissue types may not yet have been studied, illustrated by the report of 15 MP candidates in fallopian tube.22 For proteins expressed at levels below the sensitivity of current methods, there is progress as enrichment strategies, fractionation methods, and MS instruments continue to increase sensitivity, illustrated in Figure 2 by the HeLa cell proteome.23 Data on transcript expression levels can guide researchers to the cell or organ types most likely to be expressing the protein, of which testis and brain have been most productive. Several clever methods have been applied by HPP investigators to enrich low-abundance proteins, including adsorption to ProteoMiner hexapeptide-covered beads,83 and the plans to utilize HPA antibodies together with MS to capture and thereby enrich still-missing proteins in testis, guided by transcript expression data (see HPA, above).
Meanwhile, some proteins whose sequences lack two predicted proteotypic tryptic peptides of at least 9 aa have been detected by using additional proteases with different cleavage specificity84 or finding N- and C-terminal peptides and peptides with missed cleavages.7 Low MW proteins also are under-represented in PE1; specific efforts to enrich for these proteins have not yet yielded many PE1 candidates, nor have top-down methods revealed a new PE1 protein. Detection based on protein-protein interactions must avoid claiming recombinant bait proteins as expressed. An analysis of homologous protein families was a special feature of a paper about how the first 43 newly detected PE1 proteins from Chromosome 17 were found as of 2018 in the neXt-MP50 Challenge.85
Major efforts have been directed at membrane proteins.86,87 Hydrophobic proteins account for about 40% of missing proteins; Lys and Arg residues tend to be few and either too close together in hydrophilic loops or too far apart for tryptic digestion to be useful. Other proteases have similar problems, generally generating higher sequence coverage but few additional protein identifications. Large families or groups include GPCR, zinc finger, homeobox, keratin-associated, and coiled-coil domain proteins.88,89
We expect all of these strategies for finding missing proteins, characterizing the functions of already-detected proteins, and utilizing proteogenomics in precision medicine to be fruitful in the coming years.
Supplementary Material
ACKNOWLEDGMENTS
We appreciate the guidance from the HPP Executive Committee and the participation of all HPP investigators. We thank the UniProt groups at SIB, EBI, and PIR for providing high-quality annotations for the human proteins in UniProtKB/Swiss-Prot. The neXtProt server is hosted by VitalIT in Switzerland, ProteomeXchange and PRIDE at the European Bioinformatics Institute in Cambridge, UK, PeptideAtlas at the Institute for Systems Biology in Seattle, and MassIVE at the University of California San Diego. G.S.O. acknowledges support from National Institutes of Health grants P30ES017885-01A1 and U24CA210967; E.W.D. from National Institutes of Health grants R01GM087221, R24GM127667, U19AG023122, and from National Science Foundation grant DBI-1933311; L.L. and neXtProt from the SIB Swiss Institute of Bioinformatics; C.M.O. by Canadian Institutes of Health Research Foundation Grant 148408 and a Canada Research Chair in Protease Proteomics and Systems Biology; M.S.B. by NHMRC Project Grant APP1010303; C.L. by the Knut and Alice Wallenberg Foundation for the Human Protein Atlas; and Y.-K.P. by grants from the Korean Ministry of Health and Welfare HI13C22098 and HI16C0257.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00485.
Supplementary Table S1: neXtProt Analysis of the 17 874 MS-based PE1 Proteins from PeptideAtlas and MassIVE (organized by PeptideAtlas only, MassIVE only, additional neXtProt entries, and All) (XLSX)
Supplementary Table S2: Evidence for the 950 non-MS-based PE1 neXtProt Proteins (ordered by neXtProt accession code, Column A, for 906 with retrievable data); Column headings nearly match columns A to O in Supplementary Table S3 (XLS)
Supplementary Table S3: Human Protein Atlas Immunohistochemistry and Transcript Expression Findings for 820 of the 950 non-MS-based PE1 neXtProt Proteins (ordered alphabetically by tissue with highest expression, Column S) (XLS)
Contributor Information
Gilbert S. Omenn, University of Michigan, Ann Arbor, Michigan 48109, United States; Institute for Systems Biology, Seattle, Washington 98109, United States;.
Lydie Lane, CALIPHO Group, SIB Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland;.
Christopher M. Overall, University of British Columbia, Vancouver, BC V6T 1Z4, Canada
Ileana M. Cristea, Princeton University, Princeton, New Jersey 08544, United States;.
Fernando J. Corrales, Centro Nacional de Biotecnologia, 28049 Madrid, Spain;.
Cecilia Lindskog, Uppsala University, 752 36 Uppsala, Sweden;.
Young-Ki Paik, Yonsei Proteome Research Center, Seoul 03722, Korea;.
Jennifer E. Van Eyk, Cedars Sinai, Los Angeles, California 90048, United States;.
Siqi Liu, BGI Group, Shenzhen 518083, China;.
Stephen R. Pennington, University College Dublin, Dublin 4, Ireland
Michael P. Snyder, Stanford University, Stanford, California 94305, United States
Mark S. Baker, Macquarie University, Macquarie Park, NSW 2109, Australia;.
Nuno Bandeira, University of California, San Diego, La Jolla, California 92093, United States;.
Ruedi Aebersold, ETH-Zurich and University of Zurich, 8092 Zurich, Switzerland.
REFERENCES
- (1).Legrain P; Aebersold R; Archakov A; Bairoch A; Bala K; Beretta L; Bergeron J; Borchers CH; Corthals GL; Costello CE; Deutsch EW; Domon B; Hancock W; He F; Hochstrasser D; Marko-Varga G; Salekdeh GH; Sechi S; Snyder M; Srivastava S; Uhlen M; Wu CH; Yamamoto T; Paik YK; Omenn GS The human proteome project: current state and future direction. Mol. Cell. Proteomics 2011, 10 (7), M111.009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Marko-Varga G; Omenn GS; Paik YK; Hancock WS A first step toward completion of a genome-wide characterization of the human proteome. J. Proteome Res 2013, 12 (1), 1–5. [DOI] [PubMed] [Google Scholar]
- (3).Lane L; Bairoch A; Beavis RC; Deutsch EW; Gaudet P; Lundberg E; Omenn GS Metrics for the human proteome project 2013–2014 and strategies for finding missing proteins. J. Proteome Res 2014, 13 (1), 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Omenn GS; Lane L; Lundberg EK; Beavis RC; Nesvizhskii AI; Deutsch EW Metrics for the human proteome project 2015: Progress on the human proteome and guidelines for high-confidence protein identification. J. Proteome Res 2015, 14 (9), 3452–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Omenn GS; Lane L; Lundberg EK; Beavis RC; Overall CM; Deutsch EW Metrics for the human proteome project 2016: Progress on identifying and characterizing the human proteome, including post-translational modifications. J. Proteome Res 2016, 15(11), 3951–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Omenn GS; Lane L; Lundberg EK; Overall CM; Deutsch EW Progress on the HUPO draft human proteome: 2017 metrics of the human proteome project. J. Proteome Res 2017, 16 (12), 4281–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Omenn GS; Lane L; Overall CM; Corrales FJ; Schwenk JM; Paik YK; Van Eyk JE; Liu S; Snyder M; Baker MS; Deutsch EW Progress on identifying and characterizing the human proteome: 2018 metrics from the HUPO human proteome project. J. Proteome Res 2018, 17 (12), 4031–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Omenn GS; Lane L; Overall CM; Corrales FJ; Schwenk JM; Paik YK; Van Eyk JE; Liu S; Pennington S; Snyder MP; Baker MS; Deutsch EW Progress on identifying and characterizing the human proteome: 2019 metrics from the HUPO human proteome project. J. Proteome Res 2019, 18 (12), 4098–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zahn-Zabal M; Michel PA; Gateau A; Nikitin F; Schaeffer M; Audot E; Gaudet P; Duek PD; Teixeira D; Rech de Laval V; Samarasinghe K; Bairoch A; Lane L The neXtProt knowledgebase in 2020: data, tools and usability improvements. Nucleic Acids Res 2019, 48 (D1), D328–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Adhikari S; Nice EC; Deutsch EW; Lane L; Omenn GS; Pennington S; Paik Y-K; Overall CM; Corrales F; Cristea IM; Van Eyk JE; Uhlén M; Lindskog C; Chan DW; Bairoch A; Waddington JC; Justice JL; Labaer J; Rodriguez H; He F; Kostrzewa M; Ping P; Gundry RL; Stewart P; Srivastava S; Srivastava S; Nogueira FCS; Domont GB; Vandenbrouck Y; Lam MPY; Wennersten S; Vizcaino JA; Wilkins M; Schwenk JM; Lundberg E; Bandeira N; Marko-Varga G; Weintraub ST; Pineau C; Kusebauch U; Moritz RL; Ahn SB; Palmblad M; Snyder MP; Aebersold R; HPP Consortium; Baker, M. S. A High-stringency blueprint of the human proteome. Nat. Commun 2020, DOI: 10.1038/s41467-020-19045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Deutsch EW; Overall CM; Van Eyk JE; Baker MS; Paik YK; Weintraub ST; Lane L; Martens L; Vandenbrouck Y; Kusebauch U; Hancock WS; Hermjakob H; Aebersold R; Moritz RL; Omenn GS Human proteome project mass spectrometry data interpretation guidelines 2.1. J. Proteome Res 2016, 15 (11), 3961–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Deutsch EW; Lane L; Overall CM; Bandeira N; Baker MS; Pineau C; Moritz RL; Corrales F; Orchard S; Van Eyk JE; Paik YK; Weintraub ST; Vandenbrouck Y; Omenn GS Human proteome project mass spectrometry data interpretation guidelines 3.0. J. Proteome Res 2019, 18 (12), 4108–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sjostedt E; Sivertsson A; Hikmet Noraddin F; Katona B; Nasstrom A; Vuu J; Kesti D; Oksvold P; Edqvist PH; Olsson I; Uhlen M; Lindskog C Integration of transcriptomics and antibody-based proteomics for exploration of proteins expressed in specialized tissues. J. Proteome Res 2018, 17 (12), 4127–4137. [DOI] [PubMed] [Google Scholar]
- (14).McKetney J; Runde RM; Hebert AS; Salamat S; Roy S; Coon JJ Proteomic atlas of the human brain in alzheimer’s disease. J. Proteome Res 2019, 18 (3), 1380–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jiang Y; Sun A; Zhao Y; Ying W; Sun H; Yang X; Xing B; Sun W; Ren L; Hu B; Li C; Zhang L; Qin G; Zhang M; Chen N; Zhang M; Huang Y; Zhou J; Zhao Y; Liu M; Zhu X; Qiu Y; Sun Y; Huang C; Yan M; Wang M; Liu W; Tian F; Xu H; Zhou J; Wu Z; Shi T; Zhu W; Qin J; Xie L; Fan J; Qian X; He F Chinese human proteome project, Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567 (7747), 257–261. [DOI] [PubMed] [Google Scholar]
- (16).Pernemalm M; Sandberg A; Zhu Y; Boekel J; Tamburro D; Schwenk JM; Bjork A; Wahren-Herlenius M; Amark H; Ostenson CG; Westgren M; Lehtio J In-depth human plasma proteome analysis captures tissue proteins and transfer of protein variants across the placenta. eLife 2019, DOI: 10.7554/eLife.41608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shraibman B; Kadosh DM; Barnea E; Admon A Human leukocyte antigen (HLA) peptides derived from tumor antigens induced by inhibition of DNA methylation for development of drug-facilitated immunotherapy. Mol. Cell. Proteomics 2016, 15 (9), 3058–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Leung KK; Nguyen A; Shi T; Tang L; Ni X; Escoubet L; MacBeth KJ; DiMartino J; Wells JA Multiomics of azacitidine-treated AML cells reveals variable and convergent targets that remodel the cell-surface proteome. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (2), 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mertins P; Qiao JW; Patel J; Udeshi ND; Clauser KR; Mani DR; Burgess MW; Gillette MA; Jaffe JD; Carr SA Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10 (7), 634–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Schiza C; Korbakis D; Panteleli E; Jarvi K; Drabovich AP; Diamandis EP Discovery of a human testis-specific protein complex TEX101-DPEP3 and selection of its disrupting antibodies. Mol. Cell. Proteomics 2018, 17 (12), 2480–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Carlyle BC; Kitchen RR; Kanyo JE; Voss EZ; Pletikos M; Sousa AMM; Lam TT; Gerstein MB; Sestan N; Nairn AC A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci 2017, 20 (12), 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang D; Eraslan B; Wieland T; Hallstrom B; Hopf T; Zolg DP; Zecha J; Asplund A; Li LH; Meng C; Frejno M; Schmidt T; Schnatbaum K; Wilhelm M; Ponten F; Uhlen M; Gagneur J; Hahne H; Kuster B A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol 2019, 15 (2), e8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bekker-Jensen DB; Kelstrup CD; Batth TS; Larsen SC; Haldrup C; Bramsen JB; Sorensen KD; Hoyer S; Orntoft TF; Andersen CL; Nielsen ML; Olsen JV An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst 2017, 4 (6), 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Meier F; Brunner AD; Koch S; Koch H; Lubeck M; Krause M; Goedecke N; Decker J; Kosinski T; Park MA; Bache N; Hoerning O; Cox J; Rather O; Mann M Online parallel accumulation-serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteomics 2018, 17 (12), 2534–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Robin T; Bairoch A; Muller M; Lisacek F; Lane L Large-scale reanalysis of publicly available HeLa cell proteomics data in the context of the human proteome project. J. Proteome Res 2018, 17 (12), 4160–4170. [DOI] [PubMed] [Google Scholar]
- (26).Liu Y; Mi Y; Mueller T; Kreibich S; Williams EG; Van Drogen A; Borel C; Frank M; Germain PL; Bludau I; Mehnert M; Seifert M; Emmenlauer M; Sorg I; Bezrukov F; Bena FS; Zhou H; Dehio C; Testa G; Saez-Rodriguez J; Antonarakis SE; Hardt WD; Aebersold R Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat. Biotechnol 2019, 37 (3), 314–322. [DOI] [PubMed] [Google Scholar]
- (27).Bell PA; Solis N; Kizhakkedathu JN; Matthew I; Overall CM Proteomic and N-Terminomic TAILS analyses of human alveolar bone proteins: Improved protein extraction methodology and LysargiNase digestion strategies increase proteome coverage and missing protein identification. J. Proteome Res 2019, 18 (12), 4167–4179. [DOI] [PubMed] [Google Scholar]
- (28).Lin Z; Zhang Y; Pan H; Hao P; Li S; He Y; Yang H; Liu S; Ren Y Alternative strategy to explore missing proteins with low molecular weight. J. Proteome Res 2019, 18 (12), 4180–4188. [DOI] [PubMed] [Google Scholar]
- (29).Sun J; Shi J; Wang Y; Wu S; Zhao L; Li Y; Wang H; Chang L; Lyu Z; Wu J; Liu F; Li W; He F; Zhang Y; Xu P Open-pFind enhances the identification of missing proteins from human testis tissue. J. Proteome Res 2019, 18 (12), 4189–4196. [DOI] [PubMed] [Google Scholar]
- (30).Elguoshy A; Hirao Y; Yamamoto K; Xu B; Kinoshita N; Mitsui T; Yamamoto T Utilization of the proteome data deposited in SRMAtlas for validating the existence of the human missing proteins in GPM. J. Proteome Res 2019, 18 (12), 4197–4205. [DOI] [PubMed] [Google Scholar]
- (31).Zhang C; Wei X; Omenn GS; Zhang Y Structure and protein interaction-based gene ontology annotations reveal likely functions of uncharacterized proteins on human chromosome 17. J. Proteome Res 2018, 17 (12), 4186–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhang C; Lane L; Omenn GS; Zhang Y Blinded testing of function annotation for uPE1 proteins by I-TASSER/COFACTOR pipeline using the 2018–2019 additions to neXtProt and the CAFA3 challenge. J. Proteome Res 2019, 18 (12), 4154–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Martens L; Chambers M; Sturm M; Kessner D; Levander F; Shofstahl J; Tang WH; Rompp A; Neumann S; Pizarro AD; Montecchi-Palazzi L; Tasman N; Coleman M; Reisinger F; Souda P; Hermjakob H; Binz PA; Deutsch EW mzML-a community standard for mass spectrometry data. Mol. Cell. Proteomics 2011, 10 (1), R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chambers MC; Maclean B; Burke R; Amodei D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J; Hoff K; Kessner D; Tasman N; Shulman N; Frewen B; Baker TA; Brusniak MY; Paulse C; Creasy D; Flashner L; Kani K; Moulding C; Seymour SL; Nuwaysir LM; Lefebvre B; Kuhlmann F; Roark J; Rainer P; Detlev S; Hemenway T; Huhmer A; Langridge J; Connolly B; Chadick T; Holly K; Eckels J; Deutsch EW; Moritz RL; Katz JE; Agus DB; MacCoss M; Tabb DL; Mallick P A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol 2012, 30 (10), 918–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Eng JK; Jahan TA; Hoopmann MR Comet: an open-source MS/MS sequence database search tool. Proteomics 2013, 13 (1), 22–4. [DOI] [PubMed] [Google Scholar]
- (36).Eng JK; Deutsch EW Extending Comet for global amino acid variant and post-translational modification analysis using the PSI extended FASTA format. Proteomics 2020, No. e1900362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Deutsch EW; Sun Z; Campbell DS; Binz PA; Farrah T; Shteynberg D; Mendoza L; Omenn GS; Moritz RL Tiered human integrated sequence search databases for shotgun proteomics. J. Proteome Res 2016, 15 (11), 4091–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lam H; Deutsch EW; Eddes JS; Eng JK; King N; Stein SE; Aebersold R Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 2007, 7 (5), 655–67. [DOI] [PubMed] [Google Scholar]
- (39).Keller A; Nesvizhskii AI; Kolker E; Aebersold R Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem 2002, 74 (20), 5383–92. [DOI] [PubMed] [Google Scholar]
- (40).Shteynberg D; Deutsch EW; Lam H; Eng JK; Sun Z; Tasman N; Mendoza L; Moritz RL; Aebersold R; Nesvizhskii AI iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 2011, 10 (12), M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Keller A; Eng J; Zhang N; Li XJ; Aebersold R A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol 2005, DOI: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Deutsch EW; Mendoza L; Shteynberg D; Farrah T; Lam H; Tasman N; Sun Z; Nilsson E; Pratt B; Prazen B; Eng JK; Martin DB; Nesvizhskii AI; Aebersold R A guided tour of the Trans-Proteomic Pipeline. Proteomics 2010, 10 (6), 1150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shteynberg DD; Deutsch EW; Campbell DS; Hoopmann MR; Kusebauch U; Lee D; Mendoza L; Midha MK; Sun Z; Whetton AD; Moritz RL PTMProphet: Fast and accurate mass modification localization for the Trans-Proteomic Pipeline. J. Proteome Res 2019, 18 (12), 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Thompson A; Schafer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem 2003, 75 (8), 1895–904. [DOI] [PubMed] [Google Scholar]
- (45).Deutsch EW; Bandeira N; Sharma V; Perez-Riverol Y; Carver JJ; Kundu DJ; Garcia-Seisdedos D; Jarnuczak AF; Hewapathirana S; Pullman BS; Wertz J; Sun Z; Kawano S; Okuda S; Watanabe Y; Hermjakob H; MacLean B; MacCoss MJ; Zhu Y; Ishihama Y; Vizcaino JA The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2019, 48 (D1), D1145–D1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wang M; Wang J; Carver J; Pullman BS; Cha SW; Bandeira N Assembling the Community-Scale Discoverable Human Proteome. Cell Syst 2018, 7 (4), 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Pullman BS; Wertz J; Carver J; Bandeira N ProteinExplorer: A repository-scale resource for exploration of protein detection in public mass spectrometry data sets. J. Proteome Res 2018, 17 (12), 4227–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Martens L; Hermjakob H; Jones P; Adamski M; Taylor C; States D; Gevaert K; Vandekerckhove J; Apweiler R PRIDE: the proteomics identifications database. Proteomics 2005, 5 (13), 3537–45. [DOI] [PubMed] [Google Scholar]
- (49).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Perez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yilmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaino JA The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47 (D1), D442–d450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Desiere F; Deutsch EW; Nesvizhskii AI; Mallick P; King NL; Eng JK; Aderem A; Boyle R; Brunner E; Donohoe S; Fausto N; Hafen E; Hood L; Katze MG; Kennedy KA; Kregenow F; Lee H; Lin B; Martin D; Ranish JA; Rawlings DJ; Samelson LE; Shiio Y; Watts JD; Wollscheid B; Wright ME; Yan W; Yang L; Yi EC; Zhang H; Aebersold R Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol. 2004, 6 (1), R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Deutsch EW; Sun Z; Campbell D; Kusebauch U; Chu CS; Mendoza L; Shteynberg D; Omenn GS; Moritz RL State of the human proteome in 2014/2015 as viewed through PeptideAtlas: Enhancing accuracy and coverage through the AtlasProphet. J. Proteome Res 2015, 14 (9), 3461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Moriya Y; Kawano S; Okuda S; Watanabe Y; Matsumoto M; Takami T; Kobayashi D; Yamanouchi Y; Araki N; Yoshizawa AC; Tabata T; Iwasaki M; Sugiyama N; Tanaka S; Goto S; Ishihama Y The jPOST environment: an integrated proteomics data repository and database. Nucleic Acids Res. 2019, 47 (D1), D1218–d1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Ma J; Chen T; Wu S; Yang C; Bai M; Shu K; Li K; Zhang G; Jin Z; He F; Hermjakob H; Zhu Y iProX: an integrated proteome resource. Nucleic Acids Res. 2019, 47 (D1), D1211–d1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Sharma V; Eckels J; Schilling B; Ludwig C; Jaffe JD; MacCoss MJ; MacLean B Panorama Public: A public repository for quantitative data sets processed in Skyline. Mol. Cell. Proteomics 2018, 17 (6), 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Bruderer R; Muntel J; Muller S; Bernhardt OM; Gandhi T; Cominetti O; Macron C; Carayol J; Rinner O; Astrup A; Saris WHM; Hager J; Valsesia A; Dayon L; Reiter L Analysis of 1508 plasma samples by capillary-flow data-independent acquisition profiles proteomics of weight loss and maintenance. Mol. Cell. Proteomics 2019, 18 (6), 1242–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ruiz-Romero C; Lam MPY; Nilsson P; Onnerfjord P; Utz PJ; Van Eyk JE; Venkatraman V; Fert-Bober J; Watt FE; Blanco FJ Mining the proteome associated with rheumatic and autoimmune diseases. J. Proteome Res 2019, 18 (12), 4231–4239. [DOI] [PubMed] [Google Scholar]
- (57).Ubaida-Mohien C; Lyashkov A; Gonzalez-Freire M; Tharakan R; Shardell M; Moaddel R; Semba RD; Chia CW; Gorospe M; Sen R; Ferrucci L Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. eLife 2019, DOI: 10.7554/eLife.49874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Vasaikar S; Huang C; Wang X; Petyuk VA; Savage SR; Wen B; Dou Y; Zhang Y; Shi Z; Arshad OA; Gritsenko MA; Zimmerman LJ; McDermott JE; Clauss TR; Moore RJ; Zhao R; Monroe ME; Wang YT; Chambers MC; Slebos RJC; Lau KS; Mo Q; Ding L; Ellis M; Thiagarajan M; Kinsinger CR; Rodriguez H; Smith RD; Rodland KD; Liebler DC; Liu T; Zhang B; et al. Clinical proteomic tumor analysis consortium, Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 2019, 177 (4), 1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Clark DJ; Dhanasekaran SM; Petralia F; Pan J; Song X; Hu Y; da Veiga Leprevost F; Reva B; Lih TM; Chang HY; Ma W; Huang C; Ricketts CJ; Chen L; Krek A; Li Y; Rykunov D; Li QK; Chen LS; Ozbek U; Vasaikar S; Wu Y; Yoo S; Chowdhury S; Wyczalkowski MA; Ji J; Schnaubelt M; Kong A; Sethuraman S; Avtonomov DM; Ao M; Colaprico A; Cao S; Cho KC; Kalayci S; Ma S; Liu W; Ruggles K; Calinawan A; Gumus ZH; Geiszler D; Kawaler E; Teo GC; Wen B; Zhang Y; Keegan S; Li K; Chen F; Edwards N; Pierorazio PM; Chen XS; Pavlovich CP; Hakimi AA; Brominski G; Hsieh JJ; Antczak A; Omelchenko T; Lubinski J; Wiznerowicz M; Linehan WM; Kinsinger CR; Thiagarajan M; Boja ES; Mesri M; Hiltke T; Robles AI; Rodriguez H; Qian J; Fenyo D; Zhang B; Ding L; Schadt E; Chinnaiyan AM; Zhang Z; Omenn GS; Cieslik M; Chan DW; Nesvizhskii AI; Wang P; Zhang H; et al. Clinical Proteomic Tumor Analysis Consortium, Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 2019, 179 (4), 964–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Pflughoeft KJ; Mash M; Hasenkampf NR; Jacobs MB; Tardo AC; Magee DM; Song L; LaBaer J; Philipp MT; Embers ME; AuCoin DP Multi-platform approach for microbial biomarker identification using Borrelia burgdorferi as a model. Front. Cell. Infect. Microbiol 2019, 9, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Laferriere F; Maniecka Z; Perez-Berlanga M; Hruska-Plochan M; Gilhespy L; Hock EM; Wagner U; Afroz T; Boersema PJ; Barmettler G; Foti SC; Asi YT; Isaacs AM; Al-Amoudi A; Lewis A; Stahlberg H; Ravits J; De Giorgi F; Ichas F; Bezard E; Picotti P; Lashley T; Polymenidou M TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci 2019, 22 (1), 65–77. [DOI] [PubMed] [Google Scholar]
- (62).Gerez JA; Prymaczok NC; Rockenstein E; Herrmann US; Schwarz P; Adame A; Enchev RI; Courtheoux T; Boersema PJ; Riek R; Peter M; Aguzzi A; Masliah E; Picotti P A cullin-RING ubiquitin ligase targets exogenous alpha-synuclein and inhibits Lewy body-like pathology. Sci. Transl. Med 2019, 11 (495), eaau6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Soste M; Charmpi K; Lampert F; Gerez JA; van Oostrum M; Malinovska L; Boersema PJ; Prymaczok NC; Riek R; Peter M; Vanni S; Beyer A; Picotti P Proteomics-based monitoring of pathway activity reveals that blocking diacylglycerol biosynthesis rescues from alpha-synuclein toxicity. Cell Syst 2019, 9 (3), 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Lum KK; Howard TR; Pan C; Cristea IM Charge-mediated pyrin oligomerization nucleates antiviral IFI16 sensing of herpesvirus DNA. mBio 2019, DOI: 10.1128/mBio.01428-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kumar S; Murray D; Dasari S; Milani P; Barnidge D; Madden B; Kourelis T; Arendt B; Merlini G; Ramirez-Alvarado M; Dispenzieri A Assay to rapidly screen for immunoglobulin light chain glycosylation: a potential path to earlier AL diagnosis for a subset of patients. Leukemia 2019, 33 (1), 254–257. [DOI] [PubMed] [Google Scholar]
- (66).Rius-Perez S; Perez S; Torres-Cuevas I; Marti-Andres P; Talens-Visconti R; Paradela A; Guerrero L; Franco L; Lopez-Rodas G; Torres L; Corrales F; Sastre J Blockade of the trans-sulfuration pathway in acute pancreatitis due to nitration of cystathionine beta-synthase. Redox Biol. 2020, 28, 101324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Jin Y; Diffee GM; Colman RJ; Anderson RM; Ge Y Top-down mass spectrometry of sarcomeric protein post-translational modifications from non-human primate skeletal muscle. J. Am. Soc. Mass Spectrom 2019, 30 (12), 2460–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Santo-Domingo J; Dayon L; Wiederkehr A Protein lysine acetylation: Grease or sand in the gears of beta-cell mitochondria? J. Mol. Biol 2020, 432 (5), 1446–1460. [DOI] [PubMed] [Google Scholar]
- (69).Beekhof R; van Alphen C; Henneman AA; Knol JC; Pham TV; Rolfs F; Labots M; Henneberry E; Le Large TY; de Haas RR; Piersma SR; Vurchio V; Bertotti A; Trusolino L; Verheul HM; Jimenez CR INKA, an integrative data analysis pipeline for phosphoproteomic inference of active kinases. Mol. Syst. Biol 2019, 15 (4), e8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Waas M; Pereckas M; Jones Lipinski RA; Ashwood C; Gundry RL SP2: Rapid and automatable contaminant removal from peptide samples for proteomic analyses. J. Proteome Res 2019, 18 (4), 1644–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Pfister N; Williams EG; Peters J; Aebersold R; Bühlmann P Stabilizing variable selection and regression. arXiv, November 5, 2019, arXiv:1911.01850v1. [Google Scholar]
- (72).Cristea IM; Lilley KS Editorial overview: Untangling proteome organization in space and time. Curr. Opin. Chem. Biol 2019, 48, A1–A4. [DOI] [PubMed] [Google Scholar]
- (73).Uhlen M; Karlsson MJ; Zhong W; Tebani A; Pou C; Mikes J; Lakshmikanth T; Forsstrom B; Edfors F; Odeberg J; Mardinoglu A; Zhang C; von Feilitzen K; Mulder J; Sjostedt E; Hober A; Oksvold P; Zwahlen M; Ponten F; Lindskog C; Sivertsson A; Fagerberg L; Brodin P A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 2019, 366 (6472), eaax9198. [DOI] [PubMed] [Google Scholar]
- (74).Sjostedt E; Zhong W; Fagerberg L; Karlsson M; Mitsios N; Adori C; Oksvold P; Edfors F; Limiszewska A; Hikmet F; Huang J; Du Y; Lin L; Dong Z; Yang L; Liu X; Jiang H; Xu X; Wang J; Yang H; Bolund L; Mardinoglu A; Zhang C; von Feilitzen K; Lindskog C; Ponten F; Luo Y; Hokfelt T; Uhlen M; Mulder J An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 2020, 367 (6482), eaay5947. [DOI] [PubMed] [Google Scholar]
- (75).Robinson JL; Kocabas P; Wang H; Cholley PE; Cook D; Nilsson A; Anton M; Ferreira R; Domenzain I; Billa V; Limeta A; Hedin A; Gustafsson J; Kerkhoven EJ; Svensson LT; Palsson BO; Mardinoglu A; Hansson L; Uhlen M; Nielsen J An atlas of human metabolism. Sci. Signaling 2020, 13 (624), eaaz1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Uhlen M; Karlsson MJ; Hober A; Svensson AS; Scheffel J; Kotol D; Zhong W; Tebani A; Strandberg L; Edfors F; Sjostedt E; Mulder J; Mardinoglu A; Berling A; Ekblad S; Dannemeyer M; Kanje S; Rockberg J; Lundqvist M; Malm M; Volk AL; Nilsson P; Manberg A; Dodig-Crnkovic T; Pin E; Zwahlen M; Oksvold P; von Feilitzen K; Haussler RS; Hong MG; Lindskog C; Ponten F; Katona B; Vuu J; Lindstrom E; Nielsen J; Robinson J; Ayoglu B; Mahdessian D; Sullivan D; Thul P; Danielsson F; Stadler C; Lundberg E; Bergstrom G; Gummesson A; Voldborg BG; Tegel H; Hober S; Forsstrom B; Schwenk JM; Fagerberg L; Sivertsson A The human secretome. Sci. Signaling 2019, 12 (609), eaaz0274. [DOI] [PubMed] [Google Scholar]
- (77).Hikmet F; Mear L; Edvinsson A; Micke P; Uhlen M; Lindskog C The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol 2020, 16 (7), e9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Ziegler CGK; Allon SJ; Nyquist SK; Mbano IM; Miao VN; Tzouanas CN; Cao Y; Yousif AS; Bals J; Hauser BM; Feldman J; Muus C; Wadsworth MH 2nd; Kazer SW; Hughes TK; Doran B; Gatter GJ; Vukovic M; Taliaferro F; Mead BE; Guo Z; Wang JP; Gras D; Plaisant M; Ansari M; Angelidis I; Adler H; Sucre JMS; Taylor CJ; Lin B; Waghray A; Mitsialis V; Dwyer DF; Buchheit KM; Boyce JA; Barrett NA; Laidlaw TM; Carroll SL; Colonna L; Tkachev V; Peterson CW; Yu A; Zheng HB; Gideon HP; Winchell CG; Lin PL; Bingle CD; Snapper SB; Kropski JA; Theis FJ; Schiller HB; Zaragosi LE; Barbry P; Leslie A; Kiem HP; Flynn JL; Fortune SM; Berger B; Finberg RW; Kean LS; Garber M; Schmidt AG; Lingwood D; Shalek AK; Ordovas-Montanes J; et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020, 181 (5), 1016–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Uhlen M; Bandrowski A; Carr S; Edwards A; Ellenberg J; Lundberg E; Rimm DL; Rodriguez H; Hiltke T; Snyder M; Yamamoto T A proposal for validation of antibodies. Nat. Methods 2016, 13 (10), 823–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Sivertsson Å; Lindström E; Oksvold P; Katona B; Hikmet F; Vuu J; Gustavsson J; Sjöstedt E; von Feilitzen K; Kampf C; Schwenk J; Uhlén M; Lindskog C, Enhanced validation of antibodies for discovery of missing proteins. J. Proteome Res 2020, in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Pineau C; Hikmet F; Zhang C; Oksvold P; Chen S; Fagerberg L; Uhlen M; Lindskog C Cell type-specific expression of testis elevated genes based on transcriptomics and antibody-based proteomics. J. Proteome Res 2019, 18 (12), 4215–4230. [DOI] [PubMed] [Google Scholar]
- (82).Hwang H; Jeong JE; Lee HK; Yun KN; An HJ; Lee B; Paik YK; Jeong TS; Yee GT; Kim JY; Yoo JS Identification of missing proteins in human olfactory epithelial tissue by liquid chromatography-tandem mass spectrometry. J. Proteome Res 2018, 17 (12), 4320–4324. [DOI] [PubMed] [Google Scholar]
- (83).Li S; He Y; Lin Z; Xu S; Zhou R; Liang F; Wang J; Yang H; Liu S; Ren Y Digging more missing proteins using an enrichment approach with ProteoMiner. J. Proteome Res 2017, 16 (12), 4330–4339. [DOI] [PubMed] [Google Scholar]
- (84).Wang Y; Chen Y; Zhang Y; Wei W; Li Y; Zhang T; He F; Gao Y; Xu P Multi-protease strategy identifies three PE2 missing proteins in human testis tissue. J. Proteome Res 2017, 16 (12), 4352–4363. [DOI] [PubMed] [Google Scholar]
- (85).Siddiqui O; Zhang H; Guan Y; Omenn GS Chromosome 17 missing proteins: Recent progress and future directions as part of the neXt-MP50 challenge. J. Proteome Res 2018, 17 (12), 4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Chen LC; Liu MY; Hsiao YC; Choong WK; Wu HY; Hsu WL; Liao PC; Sung TY; Tsai SF; Yu JS; Chen YJ Decoding the disease-associated proteins encoded in the human chromosome 4. J. Proteome Res 2013, 12 (1), 33–44. [DOI] [PubMed] [Google Scholar]
- (87).Weldemariam MM; Han CL; Shekari F; Kitata RB; Chuang CY; Hsu WT; Kuo HC; Choong WK; Sung TY; He FC; Chung MCM; Salekdeh GH; Chen YJ Subcellular proteome landscape of human embryonic stem cells revealed missing membrane proteins. J. Proteome Res 2018, 17 (12), 4138–4151. [DOI] [PubMed] [Google Scholar]
- (88).Baker MS; Ahn SB; Mohamedali A; Islam MT; Cantor D; Verhaert PD; Fanayan S; Sharma S; Nice EC; Connor M; Ranganathan S Accelerating the search for the missing proteins in the human proteome. Nat. Commun 2017, 8, 14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Adhikari S; Sharma S; Ahn SB; Baker MS In silico peptide repertoire of human olfactory receptor proteomes on high-stringency mass spectrometry. J. Proteome Res 2019, 18 (12), 4117–4123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.