

Abstract
Complexities in treating breast cancer with bone metastasis are enhanced by a vicious protumorigenic pathology, involving a shift in skeletal homeostasis towards aggressive osteoclast activity and polarization of immune cells supporting tumor growth and immune suppression. Recent studies signify the role of receptor activator of nuclear factor-κB ligand (RANKL) beyond skeletal pathology in breast cancer; including tumor growth and immunosuppression. By using an osteoprotegerin (OPG) variant we developed recently through protein engineering to uncouple TNF-related apoptosis-inducing ligand (TRAIL) binding, the present study established the potential of a cell-based OPGY49R therapy for both bone damage and immunosuppression in an immunocompetent mouse model of orthotopic and metastatic breast cancers. In combination with agonistic death receptor (DR5) activation, the OPGY49R therapy significantly increased both bone remolding and long-term anti-tumor immunity, protecting mice from breast cancer relapse and osteolytic pathology. With limitations, cost, and toxicity issues associated with the use of denosumab, bisphosphonates, and chemotherapy for bone metastatic disease, use of OPGY49R combination could offer a viable alternate therapeutic approach.
Keywords: Breast cancer, Bone metastasis, Macrophage, Osteoprotegerin, RANK signaling
INTRODUCTION
Osteolytic bone metastasis is a major event in patients with breast cancer, accounting for about 70% with skeletal pathology (1). A key signaling event that triggers aggressive osteoclast activity involves receptor activator of nuclear factor κB ligand (RANKL; TNFSF11), produced by osteoblast, bone marrow stromal cells, monocytes, T-helper cells, and disseminated cancer cells, and its cognate receptor RANK (TFNRSF11A) on osteoclast precursors (2). In normal skeletal homeostasis, a balance between osteoclast function and bone remodeling is controlled by osteoprotegerin (OPG; TNFRSF11B), a soluble decoy receptor for RANK with high-affinity binding to RANKL thereby blocking osteoclast activation (2). However, OPG also binds to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL; TNFSF10) of the tumor necrosis factor superfamily, enabling tumor cells to evade apoptosis (3). As skeletal dissemination of breast cancer progresses, the checkpoint effect of OPG is compromised, which leads to aggressive osteoclast activity (4). This effect is further compounded by the production of an inhibitor of Wnt signaling pathway, Dickkopf-related protein 1 (DKK1) that blocks osteoblast differentiation (5). Studies have also shown that RANKL-RANK axis promotes tumor cell development and mammary tumorigenesis. RANK expression leads to the spontaneous development of mammary tumor and MMTV-RANK mice showed higher incidence of adenocarcinomas (6), but a lack of RANK showed reduced mammary tumorigenesis (7). Importantly, treatment with soluble RANK-Fc attenuated mammary tumorigenesis in MMTV-RANK mice (6). Recent studies have provided direct proof that RANKL-RANK pathway plays a vital role in the progression of BRAC1 mutation-associated breast cancer (8). Moreover, high RANK expression was found to be significantly associated with poor disease-free survival in patients with breast cancer, especially the triple-negative breast cancer (TNBC) (9). Taken together, RANKL targeting is promising for breast cancer with bone metastasis, beyond its effects on skeletal pathology.
In addition to its role in bone remodeling, RANKL-RANK signaling has been implicated in the immune regulation (10). In the context of bone metastasis, tumor-promoting immunosuppressive M2 macrophages have been identified to express high surface RANK (10–12), prompting the importance of RANKL-RANK signaling in macrophage polarization (13,14). Further, expression of RANK has also been reported in breast cancer cells (15,16). Recent clinical and preclinical studies have revealed a potential role for blockade of RANKL-RANK interaction in patients with cancer and in animal models to potentiate anticancer immunotherapy (17–20). These studies showed that the combination of anti-RANKL antibody with anti-CTLA and/or anti-PD-1 antibodies significantly reduced human melanoma and experimental mouse melanoma lung metastases via CD8+ T cells and natural killer (NK) cells, compared to individual treatment (18,19). Thus, targeting RANKL-RANK signaling is emerging as a promising approach to treat pleomorphic tumor-associated pathologies.
In this context, the present study investigated the potential of individual and combination cell therapy, using a novel OPG variant (OPGY49R), devoid of TRAIL binding with an agonistic antibody (MD5-1) targeting the TRAIL receptor DR5 (21) in an immunocompetent mouse model of osteolytic breast cancer. Results of the study indicated targeting excess RANKL with OPGY49R without affecting endogenous TRAIL function not only restored bone remodeling against breast cancer-induced osteolytic damage, but also improved antitumor immune response by directly affecting M2 macrophages polarization and production of immunosuppressive cytokines and chemokines. Further, in combination with MD5-1 antibody, OPG cell therapy approach significantly increased long-term anti-tumor immunity, leading to long-term survival and protection against tumor relapse.
MATERIALS AND METHODS
Cell lines and reagents
The human embryonic kidney cell line, HEK 293T, human breast cancer cell lines, MDA-MB-231 (TNBC), MCF-7 [estrogen receptor (ER)-positive], and the murine macrophage cell line, RAW 264.7 (a kind gift from Dr. Xu Feng, The University of Alabama at Birmingham, UAB), were maintained in DMEM (Gibco, 11965-092), supplemented with 10% FBS (Omega Scientific, FB-02) and 1% Pen-Strep (Gibco, 15140-122). BALB/c mouse mesenchymal stem cells (MSCs) were provided by Dr. Donald G. Phinney, Scripps Research, Florida, and maintained in alpha-MEM (Gibco, 32561-037) supplemented with 10% FBS and 1% Pen-Strep. The human prostate cancer cell line, PC3 (a kind gift from Dr. Kenneth J. Pienta, University of Michigan), and murine TNBC cell line, 4T1.2 (kindly provided by Dr. Robin L. Anderson, Olivia Newton-John Cancer Research Institute, Melbourne, Australia), were maintained in RPMI-1640 medium (Gibco, 11875-093), supplemented with 10% FBS, and 1% Pen-Strep.
Recombinant TRAIL (GF092) and chemiluminescent HRP substrate (WBKLS0500) were purchased from MilliporeSigma, and bacterially purified recombinant RANKL was a kind gift from Dr. Xu Feng, UAB (22). Recombinant OPG (459-MO) and M-CSF (216-MC) proteins were purchased from R&D Systems. Recombinant murine IL-4 (214-14) and IL-13 (210-13) were purchased from Peprotech. Mouse IL-2 (130-120-662) was purchased from Miltenyi Biotec. Details of all antibodies used in this study were listed in the Supplementary Table S1.
Development of 4T1.2 cell line (4T1.2Luc), constitutively expressing firefly luciferase
4T1.2 cells were transduced with a recombinant lentiviral vector expressing luciferase and stable clones were selected by puromycin. Luciferase expression was confirmed by in vitro luciferase assay and in vivo non-invasive luciferase imaging. Identical in vivo growth kinetics of 4T1.2Luc to the parental 4T1.2 cell line was verified in female BALB/c mice. For further details, see Supplementary Methods.
Transduction of MSCs with AAV-OPGWT/Y49R
MSCs were plated in 60 mm dishes at a density of 4×105. Next day, the cells were infected with rAAV-OPGWT or rAAV-OPGY49R variant at an MOI of 1×105 vector particles/cell and cultured for 2 days. On day-3, OPG-expressing MSCs were harvested for in vivo injection. Prior to in vivo application, lysates and culture supernatants from rAAV-OPGWT/Y49R transduced MSCs were confirmed by Western blotting for expression and extracellular secretion of the transgenic protein. For cloning, packaging, and purification of rAAV-OPGWT and rAAV-OPGY49R variant, see Supplementary Methods.
In vitro assays
TRAIL-induced apoptosis assay was performed using PC3 cells by culturing them in media containing recombinant TRAIL alone or in combination with conditioned media from OPGWT or OPGY49R plasmid-transfected HEK 293T cells. Osteoclast differentiation assay was performed using RAW264.7 cells by culturing cells in medium containing recombinant RANKL alone or in combination with conditioned media containing OPGWT or OPGY49R. 4T1.2Luc cell proliferation was performed by culturing cells in medium containing recombinant RANKL ± recombinant OPG. Further details are provided in Supplementary Methods.
Bone marrow-derived macrophage differentiation
Femurs and tibiae were obtained from 8-week-old female BALB/c mice and bone marrow cells (BMCs) were flushed out with a syringe filled with RPMI-1640 medium. Next, cells were treated with ACK buffer (Lonza, 10-548E) to lyse red blood cells, then resuspended in FACS buffer and stained with CD11b-VioBlue antibody. CD11b+ BMCs were flow-sorted and plated in 6-well plate (1×106 cells/well). To generate bone marrow-derived macrophages, cells were cultured in RPMI 1640, supplemented with 10% FBS, 1% Pen-Strep and 20 ng/ml M-CSF for 7 days, then differentiated to M2 macrophages with IL-4 (50 ng/ml) and IL-13 (20 ng/ml) in the presence of RANKL ± OPG. M2 differentiation was confirmed by flow cytometry using M2 macrophage markers and by qRT-PCR for M2 macrophage-specific gene transcripts.
Orthotopic and metastasis models of breast cancer and treatment strategies
4T1.2Luc cells (1×105) in PBS were injected into the 4th mammary fat pad of eight-week-old female BALB/c mice. Mice were divided into four groups and treated with OPGY49R, MD5-1, OPGY49R and MD5-1 combination; untreated group was used as the control. MD5-1 antibody (100 μg per mouse) was injected intraperitoneally three times: on day-2, day-5, and day-9. Approximately, 2×105 of MSC-OPGY49R was injected by tail vein into mice on day-3 and day-10. Tumor progression was monitored using a digital caliper every other day, starting 1 week post-implantation and tumor volume was calculated as: volume = (length × width2)/2. For the metastasis model, primary tumors developed as above were surgically removed when tumors reached a volume of ~100 mm3, following which OPGY49R and MD5-1 combination treatment was given. One cohort was maintained without treatment as control. Metastasis development was monitored using non-invasive IVIS imaging. All animal studies were conducted in accordance with an approved protocol of the Institutional Animal Care and Use Committee (IACUC) of UAB.
Micro-CT and histology of bone tissue
For determination of the 3-dimensional (3D) architecture of the trabecular bones, mice were sacrificed, tibiae were harvested and scanned using a SCANCO µCT40 instrument (Scanco Medical). For histology, bone tissues were decalcified and sections of the bones were prepared. Hematoxylin and eosin (H&E) staining, Goldner’s trichrome staining and TRAP staining were performed as detailed in Supplementary Methods.
Analysis of cytokine and chemokine in the tumor microenvironment (TME)
Soluble proteins were extracted from primary tumors of untreated and OPGY49R-treated mice and analyzed using Proteome Profiler Mouse Cytokine Array Kit (R&D Systems, ARY006) following manufacturer’s instructions. For further details, see Supplementary Methods.
Adoptive CD8+ T cell transfer and tumor cell re-challenge studies
For adoptive T cell transfer, CD8+ T cells were flow-sorted from treated mice on day-20. Normal BALB/c mice were used as control. Sorted CD8+ T cells were plated in anti-CD3 (10 μg/ml)-coated 12-well tissue culture plates and cultured in RPMI-1640 medium plus IL-2 (50 U/ml) and anti-CD28 antibody (2 μg/ml) for 3 days. Later, CD8+ T (1×106) cells were harvested, and injected through tail vein into recipient mice with pre-established orthotopic tumors. Tumor progression was monitored using digital caliper every other day, starting 5 days post-tumor challenge. For tumor cell re-challenge studies, 1×105 4T1.2Luc cells were inoculated at the opposite 4th mammary fat pad of the BALB/c mice that were originally implanted with tumor cells and treated with MD5-1 or a combination of OPGY49R and MD5-1 antibody. A new cohort of 4T1.2Luc-challenged female BALB/c mice were used as control for tumor growth kinetics. Tumor progression was monitored using a digital caliper every other day, starting 1 week post-implantation.
Statistical Analysis
Results consisting of three groups or more were analyzed using two way ANOVA with Tukey’s multiple comparisons test by GraphPad Prism 8. Analysis of results containing two groups was performed using Student’s t test. All data are presented as mean ± SD. A p values < 0.05 was considered statistically significant.
Additional details on Materials and Methods are provided in Supplementary Methods.
RESULTS
Development and characterization of murine OPG variant retaining RANKL binding, but abolishing TRAIL binding
We recently identified a TRAIL-binding aromatic residue on human OPG and established that removal of this side-chain abolished interaction with TRAIL, without affecting RANKL binding (23,24). Interestingly, comparison of OPG orthologs revealed that this motif is highly conserved across species (Supplementary Figure S1). To utilize this OPG variant (OPGY49R) for therapy effects on bone remodeling without affecting immune cell-produced TRAIL effects on breast cancer cells in an immunocompetent mouse model, we first created a mouse OPGY49R variant by site-directed mutagenesis and tested its effects, first in a TRAIL assay. Conditioned media from mouse wild-type OPG (OPGWT) or OPGY49R variant plasmids transfected HEK 293T cells were tested on PC3 cells in the presence of recombinant TRAIL protein. TRAIL-induced apoptosis was analyzed by cell viability and Western blot, using anti-caspase-3 antibody. We observed that TRAIL treatment induced significant apoptosis in both recombinant TRAIL treated group and in combination with conditioned medium containing OPGY49R (Fig. 1A). However, when conditioned medium from OPGWT transfected HEK 293T cells were added with TRAIL, there was a significant protection from apoptosis. Further, Western blot analyses corroborated the above results with increased levels of cleaved caspase-3 and a concomitant decrease in total caspase-3 when conditioned medium from OPGY49R-transfected cells was used, but not with medium from OPGWT-transfected cells (Fig.1B). This result confirmed that OPGY49R does not bind to TRAIL. In parallel, we also confirmed by osteoclast assay that substitution of an aromatic side chain in OPGY49R did not compromise RANKL binding. In an osteoclast assay using mouse macrophage cell line, RAW 264.7, addition of conditioned-medium containing OPGY49R in combination with RANKL significantly prevented osteoclast differentiation. This effect was found to be comparable to the OPGWT treated group (Fig. 1C). These data confirmed that murine OPGY49R abolished TRAIL binding, but retained RANKL binding.
Figure 1. Development and characterization of OPG variant devoid of TRAIL binding.
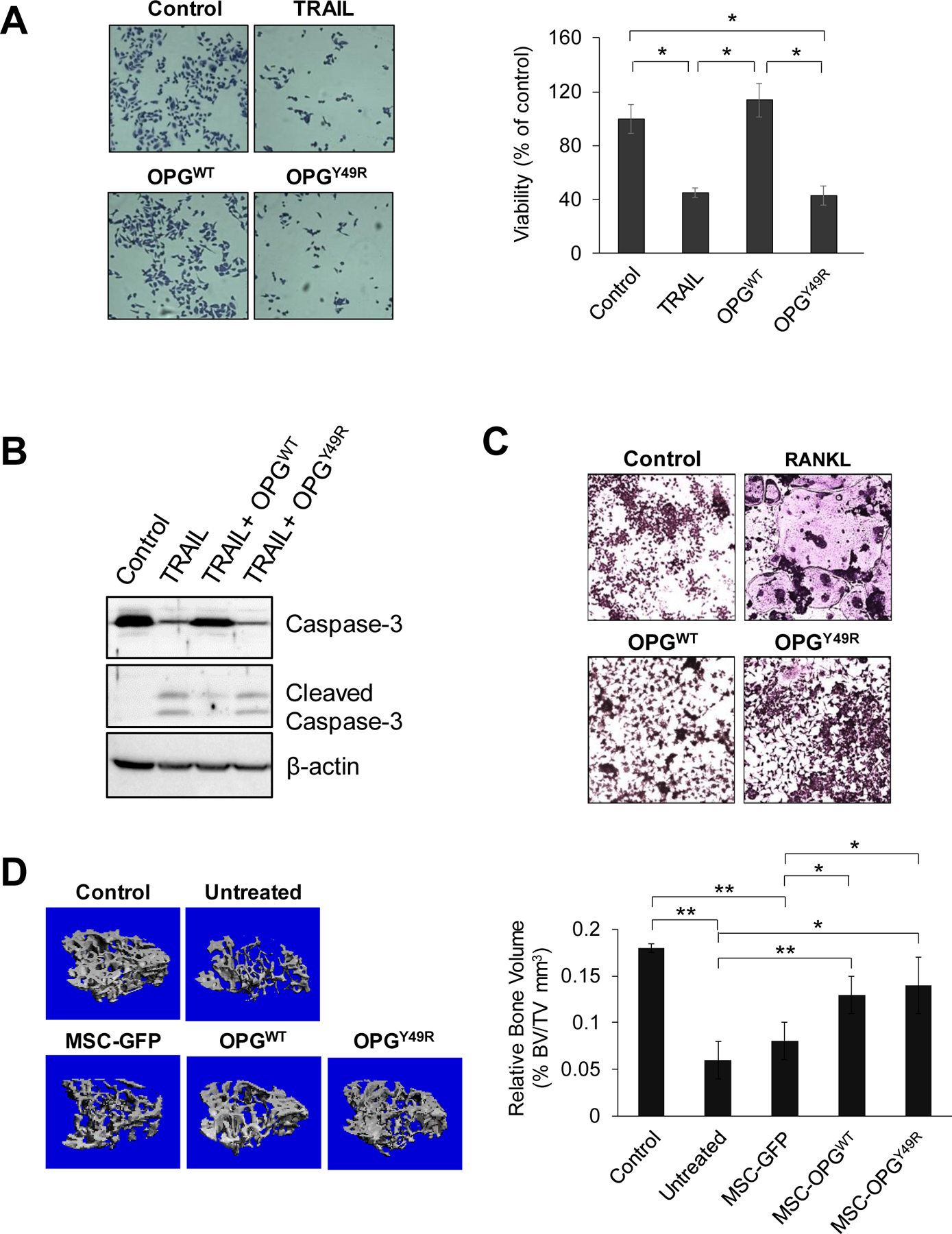
A and B, For TRAIL assay, PC3 cells were maintained in duplicate cultures in medium containing recombinant TRAIL with or without conditioned medium from HEK 293T cells, transfected with vectors expressing OPGWT or OPGY49R. The cells were fixed after 24 hrs using methanol and stained with 0.05% Crystal violet. Images were taken using a light microscope (10x) and cells were enumerated using ImageJ software by counting 5 random fields in each sample (A). Identically-treated PC3 cells were harvested after 5 hrs, and analyzed by Western blotting using anti-caspase-3 and cleaved caspase-3 antibodies. Beta actin was used as loading control (B). C, For osteoclast formation assay, the macrophage cell line, RAW264.7, was cultured for 10 days in medium containing recombinant RANKL with or without conditioned medium from HEK 293T cells, transfected with plasmids expressing OPGWT or OPGY49R. Cells were stained for Tartrate-resistant acid phosphatase (TRAP) and images were taken using a light microscope (10x). D, To determine the potential of OPGY49R in protecting skeletal homeostasis from breast cancer-induced bone loss, female BALB/c mice were inoculated with 4T1.2Luc cells at the 4th mammary fat pad for orthotopic tumor development, following which MSCs, transduced with rAAV-GFP, rAAV-OPGWT or rAAV-OPGY49R were systemically delivered by tail vein injection. Normal, age-matched female BALB/c mice were used as control. Tibiae were collected after 20 days and analyzed by micro-CT. Data from micro-CT were used in 3D reconstruction images (left panel) and quantitative analysis of respective bones for bone volume (right panel). N = 5 per group; *P < 0.05; **P < 0.01.
Osteoprotegerin variant devoid of TRAIL binding restores bone architecture
To utilize OPGY49R for therapy effects on bone remodeling in breast cancer-induced osteolytic damage, without compromising immune cell-produced TRAIL effects in vivo, we adopted a cell therapy approach using mouse MSCs. MSCs were genetically engineered by infection of recombinant adeno-associated virus expressing OPGWT (rAAV-OPGWT) or OPGY49R (rAAV-OPGY49R). Prior to producing rAAV expressing OPG, we tested different AAV serotypes for their ability to transduce MSCs from BALB/c mouse using rAAV-GFP and established that AAV serotype-4 capsid possesses highest transduction efficiency in mouse MSCs (Supplementary Figure S2A). Based on this observation, we cloned, packaged and purified rAAV4, encoding OPGWT or OPGY49R. Prior to tumor challenge studies in vivo, high level expression and extracellular secretion of OPG was confirmed by transducing mouse MSCs with rAAV-OPGWT or rAAV-OPGY49R and analyzing both cell lysates and supernatants by Western blotting (Supplementary Figure S2B). Next, we examined the potential of mouse OPGY49R in protecting tumor-induced bone loss in vivo. To monitor the bone metastasis non-invasively, we established a clonal derivative of 4T1.2 cells by stably transduction with lentiviral vector expressing luciferase (4T1.2Luc) and confirmed the identical in vivo growth kinetics to that of parental 4T1.2 cells (Supplementary Figure S3A). Using the 4T1.2Luc cell line, an osteolytic breast cancer model was established by inoculating this syngeneic mouse breast cancer cell line into 4th mammary fat pad of female BALB/c mice. It was interesting to note that the 4T1.2/BALB/c displayed bone damage by systemic effects without visible bone metastasis (Supplementary Figure S3B). In order to first verify the potential of OPGY49R against tumor-induced bone loss, cohorts of female BALB/c mice with pre-established orthotopic breast tumors were untreated or treated with MSCs producing OPGWT or OPGY49R, or MSC infected with rAAV-GFP. Normal BALB/c mice were used as control. Bone damage was examined on day-18 by micro-CT. Tumor challenge caused significant bone damage in untreated and MSC-GFP treated groups, as shown by decreased trabeculae and bone volume compared to normal BALB/c mice (Fig. 1D). However, treatment with MSCs producing either OPGWT or OPGY49R displayed a significant decrease in osteolytic bone damage compared to untreated and MSC-GFP treated groups (Fig. 1D), suggesting therapy effects with OPGY49R is effective in restoring skeletal homeostasis against cancer-induced osteolysis.
OPG inhibits 4T1.2Luc cell proliferation by blocking RANKL-RANK signaling
Prior to testing the effects of blocking RANKL-RANK signaling directly on tumor growth in vivo, we evaluated the significance of RANKL in 4T1.2Luc cell growth and downstream signaling on tumor cell proliferation. We identified by flow cytometry (Fig. 2A) and Western blotting (Fig. 2B) using anti-RANK (R12-31) antibody (25) that RANK expression is explicit in 4T1.2Luc cells as in human breast cancer cell lines, MBA-MB-231 and MCF 7. Next, we tested direct effects of RANKL on 4T1.2Luc proliferation and activation of downstream signaling pathways, and how OPG influences RANKL-induced effects in these cellular processes. To determine this, 4T1.2Luc cells were cultured in the presence or absence of RANKL. Recombinant OPG was added to the cultures, either alone or with RANKL. Results indicated a significant increase in 4T1.2Luc cell proliferation when RANKL was added to the culture. Interestingly, when OPG was added to the culture in the presence of RANKL, the growth rate of 4T1.2Luc cells decreased to the level without RANKL (Fig. 2C). Addition of OPG alone to 4T1.2Luc cells did not influence rate of cell proliferation (Fig. 2C). These data provide key evidence that RANKL-RANK axis promotes breast cancer proliferation. A major activation pathway following RANKL-RANK ligation involves the recruitment of an adaptor protein TNF Receptor Associated Factor 6 (TRAF6) that relays downstream signaling via IκB to activate transcription factor NF-κB and NFATc1 or via the MAPK cascade involving p38, ERK, and JNK. RANKL-RANK interaction also relays downstream signaling via the PI3K pathway through c-Src, which leads to AKT signaling (26). To investigate which signaling pathway is involved in enhanced breast cancer cell proliferation upon RANKL stimulation, we examined the changes in phosphorylation status of NF-κB p65, ERK1/2, and AKT. We observed a significant increase in NF-κB and MAPK activation, whereas there was no change in AKT phosphorylation (Fig. 2D). Interestingly, when OPG was added to 4T1.2Luc cells in the presence of RANKL, both NF-κB and ERK1/2 phosphorylation restored to levels in cultures without RANKL (Fig. 2D). These data indicated that OPG inhibited RANKL-induced 4T1.2Luc cell proliferation by blocking RANKL-RANK signaling pathway through NF-κB and ERK1/2 MAPK.
Figure 2. RANK expression in human and mouse breast cancer cells and effects of RANKL stimulation on proliferation and downstream signal transduction pathways.
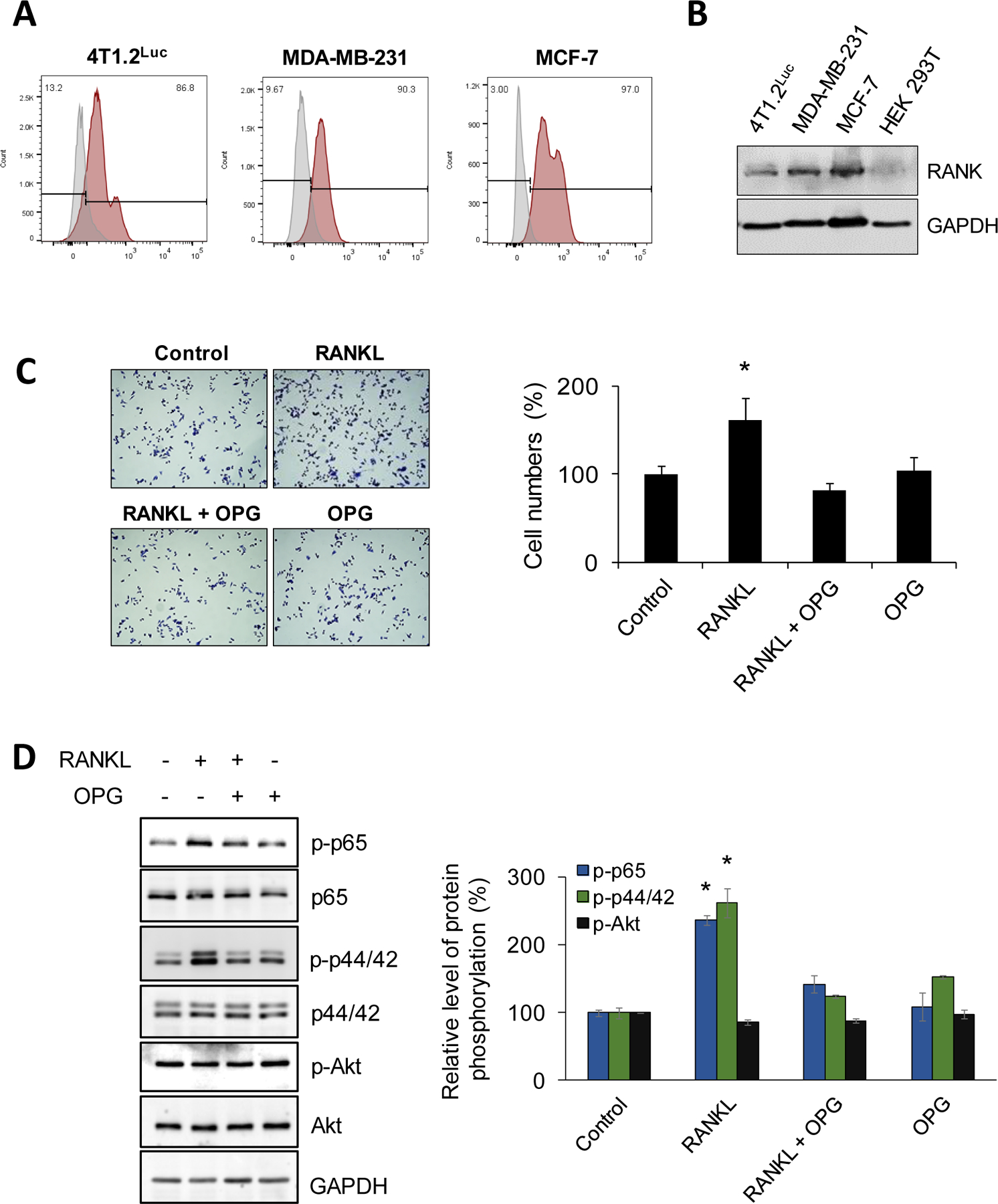
A and B, Human breast cancer cell lines, MDA-MB-231 (TNBC), MCF7 (ER+), and a mouse breast cancer cell line, 4T1.2Luc, were tested for membrane-bound RANK expression by flow cytometry and from cell lysates by Western blotting. For flow cytometry, single cell suspension was prepared and stained with RANK-PE antibody (red) or isotype control antibody (gray) (A). Protein expression was analyzed by immunoblotting using anti-RANK antibody. GAPDH was used as loading control (B). C, To determine the effect of RANKL stimulation on cell proliferation and the impact of OPG on this, 4T1.2Luc cells were treated with RANKL ± OPG and cultured for 48 hrs in DMEM containing 2% FBS. The cells were fixed and stained using crystal violet. Images were taken using light microscope (10x). Cell proliferation was enumerated using ImageJ software by counting 5 random fields in each sample. D, To determine major RANK/RANKL signal transduction pathways on breast cancer cells, 4T1.2Luc cells were serum-starved for 1 hr and then treated with RANKL ± OPG for 10 min. Cells were lysed with RIPA buffer and equal amount of protein were resolved by SDS-PAGE. Protein phosphorylation status of indicated signaling molecules were examined by immunoblotting using phospho-p65, phospho-ERK, phospho-Akt antibodies, and their respective total forms; GAPDH was used as loading control. Protein phosphorylation levels were quantitated by densitometry analysis using ImageJ and normalized to total forms of respective proteins. *P < 0.05.
Systemic cell therapy with OPGY49R suppresses breast cancer growth beyond bone restoration
From recent studies it is becoming increasingly clear that RANKL-RANK pathway is involved in promoting mammary tumorigenesis and protumorigenic immune cell functions. Together with our observations on the potential of OPGY49R in skeletal remodeling in osteolytic malignancy, we hypothesized that systemically stable, genetically engineered cell therapy to produce OPGY49R in physiologically required concentration, without interfering in TRAIL-induced apoptosis of breast cancer cells, would provide therapeutic benefits for osteolytic bone damage by minimizing tumor growth directly and by improving antitumor immunity through dampening tumor-associated M2 macrophage function. We tested this possibility in female BALB/c mice with pre-established orthotopic 4T1.2Luc tumors. To further examine if antibody targeting of TRAIL receptor, DR5, as in clinically tested agonistic monoclonal antibody (mAb) approach (27), would enhance antitumor effects, we included cohorts of mice that received only MD5-1 antibody (mouse DR5-targeting agonistic mAb) or in combination with cell based OPGY49R therapy. MD5-1 antibody was applied intraperitoneally and MSC-OPGY49R was delivered by tail vein injection. Overall experimental schema is shown in Supplementary Figure S4A. Results of the study demonstrated a significant decrease in tumor growth in both MD5-1 and MSC-OPGY49R individual treatments. However, the combination of MD5-1 and OPGY49R resulted in a significantly greater decrease in tumor growth kinetics (Fig. 3A). Tibiae from cohorts of mice from control, MD5-1 and/or MSC-OPGY49R treatment groups were further analyzed by micro-CT and histology to distinguish therapy effects on bone remodeling. Data obtained from micro-CT were analyzed for bone volume, trabecular thickness, trabecular number and trabecular spacing. Results from Micro-CT data, H&E staining, and trichrome staining showed a significant destruction of the trabecular bone in the untreated tumor group, compared to age-matched control (Fig. 3B and C; Supplementary Figure S4B). Increased osteoclast activity was also observed in the untreated tumor group by TRAP staining (Fig. 3D). Interestingly, although MD5-1 standalone treatment decreased tumor growth, there was significant bone damage and osteoclast activity in this group (Fig. 3B–D; Supplementary Figure S4). However, treatment with MSC-OPGY49R alone or in combination with MD5-1 restored bone integrity and decreased osteoclast activity, similar to the control group (Fig. 3B–D; Supplementary Figure S4), confirming superior effects of this combination in simultaneously restoring skeletal integrity and decreasing tumor growth.
Figure 3. Antitumor effects of OPGY49R as standalone therapy and in combination with DR5-targeted agonistic antibody therapy.
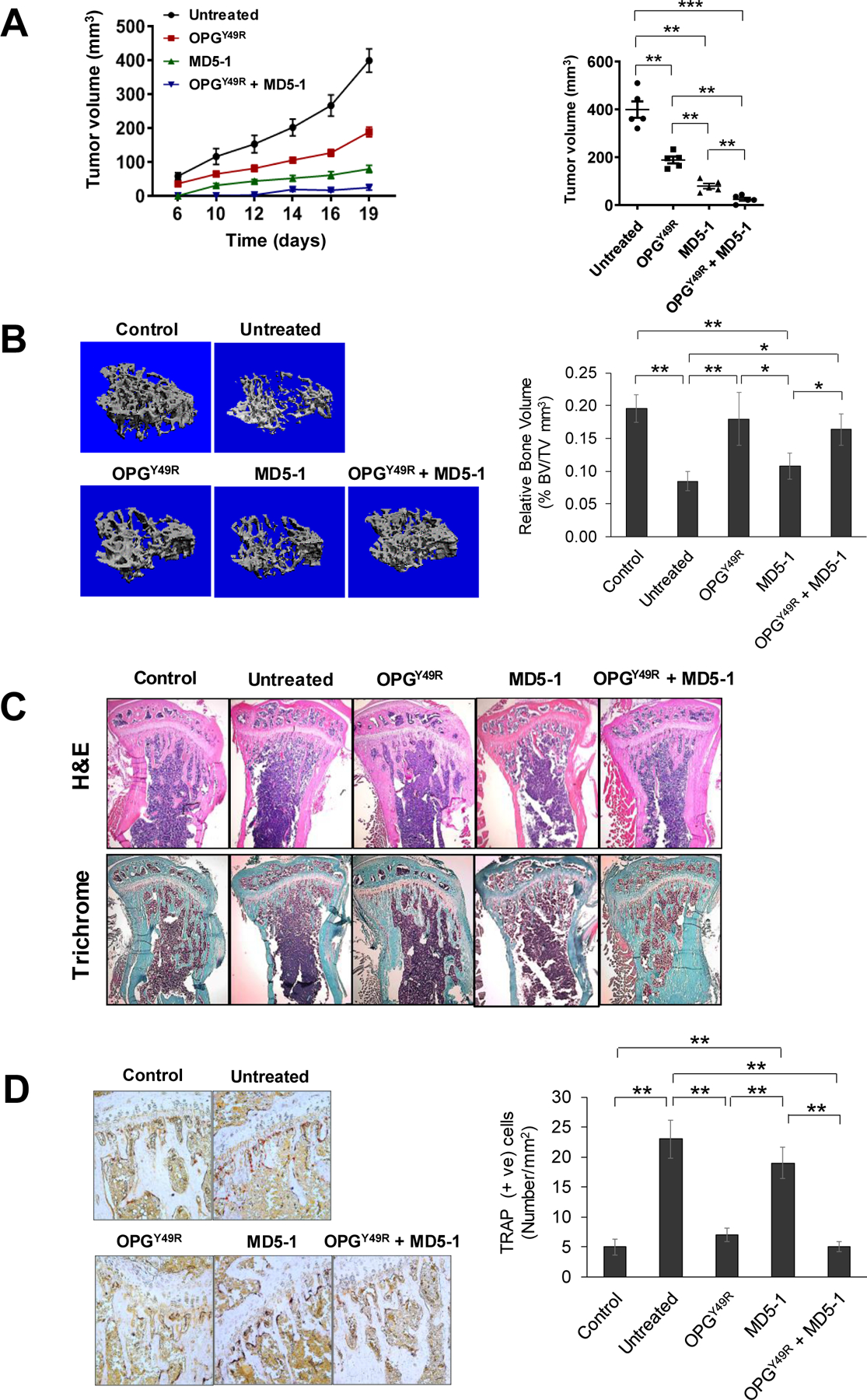
A, Female BALB/c mice were implanted with 4T1.2Luc cells, followed by individual and combination treatments with OPGY49R and MD5-1. Tumor volumes were measured on indicated days using a digital caliper. Data was analyzed by two-way ANOVA and statistical comparisons were made between tumors in mice that received no treatment and those, which received individual or combination treatments. Growth kinetics and tumor volumes at the endpoint are shown from each group. B, Mice were sacrificed on day-20 following initiation of therapy and tibiae were processed for evaluation of bone architecture by micro-CT. Data from micro-CT scans were used in 3D reconstruction images (left panel) and quantitative analysis of bone volume (right panel). C and D, Following micro-CT, tibiae were decalcified and processed for histology by H&E and Trichrome staining. Images were taken using light microscope (5x) (C). TRAP staining was performed to determine osteoclast activity. Images were taken using light microscope (20x, left panel) and TRAP positive osteoclasts were enumerated using ImageJ software (right panel) (D). N = 5; *P < 0.05; **P < 0.01; ***P < 0.001.
Systemic RANKL-targeted OPGY49R cell therapy directly affects protumorigenic macrophages and cytokine milieu at the TME
Recent studies suggest that RANKL-RANK signaling promotes cancer-associated inflammation through M2 macrophage activation, and RANKL induces M2 macrophage polarization (10,14). Based on significant therapy effects of MSC-OPGY49R on both bone remodeling and tumor growth, indicating benefits of targeting RANK expression on tumor cells, we further established higher RANK expression in M2 macrophages and monocytic MDSCs (M-MDSCs), compared to M1 macrophages and polymorphonuclear MDSCs (PMN-MDSCs), respectively. Further there was no significant difference in RANK expression between myeloid DCs (mDCs) and plasmacytoid DCs [pDCs; (Fig. 4A; Supplementary Figure S5A and B)]. Since M2 macrophages displayed highest RANK expression, next, we examined the impact of dampening RANKL signaling systemically with MSC-OPGY49R on M2 macrophages. Spleen and tumors were isolated from control and 4T1.2Luc tumor challenged mice with/without OPGY49R treatment and macrophages were analyzed by flow cytometry. Results of this study showed tumor challenge significantly increased total macrophages (Fig. 4B). Interestingly, there was neither a significant decrease in macrophage numbers nor in M2/M1 ratio based on M1 and M2 macrophage surface markers following OPGY49R treatment (Fig. 4B and C). Based on this, we sought to understand possible effect of OPG on M2 macrophage polarization and/or function. CD11b+ monocytes from bone marrow of BALB/c mice were differentiated with IL-4 and IL-13 for M2 macrophage in the presence of RANKL ± OPG, following which cells were analyzed for M2 polarization markers by flow cytometry and gene expression signatures by qRT-PCR. Results indicated that addition of RANKL didn’t significantly increase M2 macrophage numbers (Fig. 4D, left panel). However, both arginase 1 (28,29) gene expression and enzyme levels were significantly increased following exposure to RANKL, but addition of OPG blocked RANKL-induced arginase 1 production (Fig. 4D, right panel, and E, left panel). We also observed that RANKL increased CCL17 (30,31) expression and addition of OPG significantly blocked it (Fig. 4E, right panel).
Figure 4. Effects of sequestering RANKL by OPGY49R on macrophages at the TME.
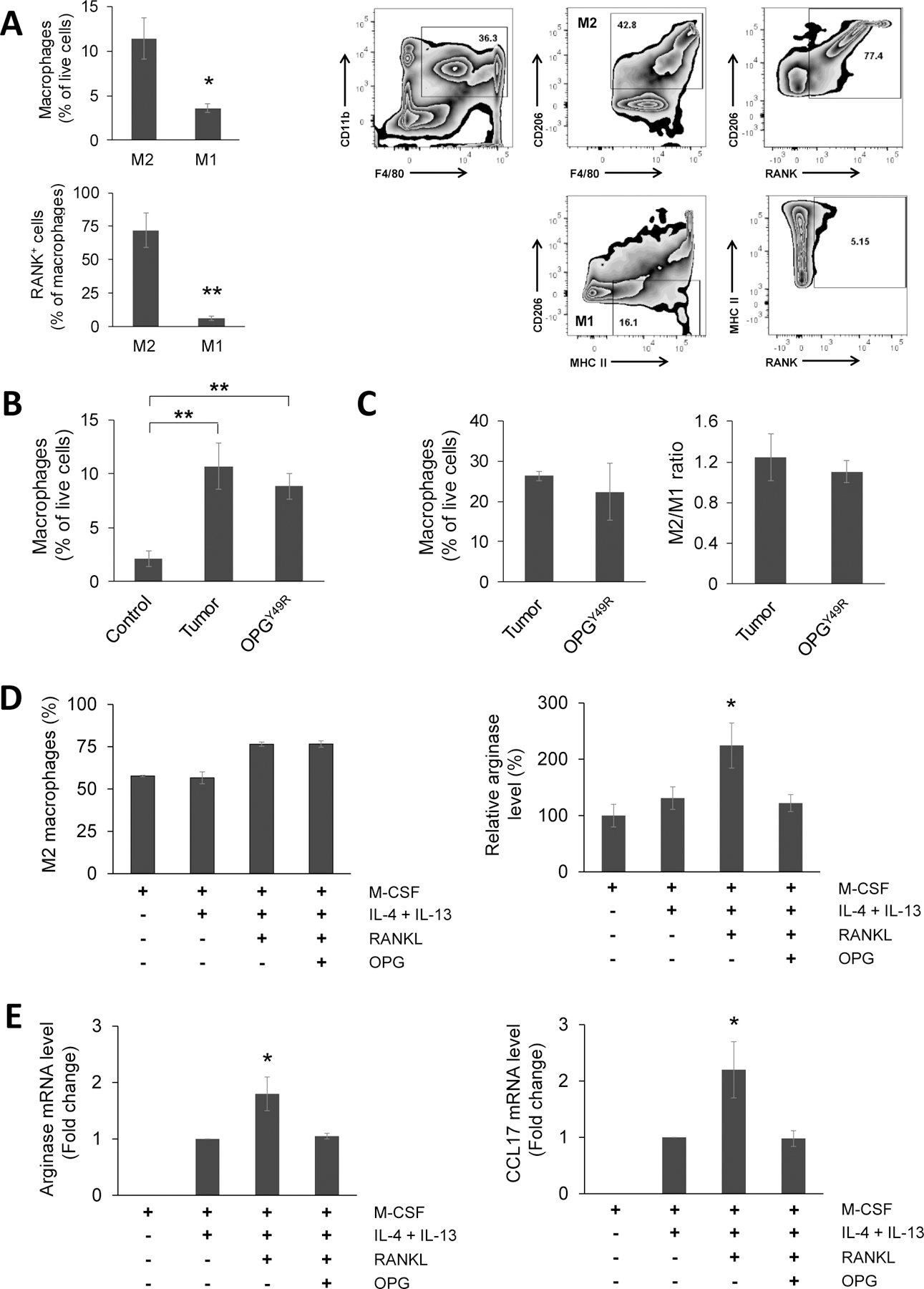
A, Twenty days after orthotopic 4T1.2Luc tumor inoculation, mice were sacrificed and tumor tissues explanted. Single-cell suspensions of explanted tumors were analyzed for M1 and M2 macrophages by flow cytometry, M1, CD11b+F4/80+CD206−MHC-II+; M2, CD11b+F4/80+CD206+. RANK+ macrophages within the M1 and M2 population were analyzed by staining with anti-RANK antibody. Relative percentages of M1 and M2 macrophages and their RANK expression (left panel), and gating strategy (right panel) are shown. B and C, Percentage of macrophages were determined by flow cytometry in spleens from control and 4T1.2Luc tumor-challenged mice that received no treatment or following treatment with OPGY49R (B), and in tumors tissues explanted from mice that received no treatment or OPGY49R treatment (C). Right panel shows the M2/M1 ratio in tumor tissues. D and E, To determine effects of blocking RANKL-induced downstream effects with OPG on M2 macrophages, CD11b+ cells were purified by FACS from BALB/c mouse bone marrow and cultured in medium containing M-CSF for 7 days, then differentiated to M2 macrophages using IL-4 and IL-13 in the presence of RANKL ± OPG in duplicate cultures. One batch of cells was analyzed by flow cytometry to determine arginase-expressing M2 macrophages (CD11b+ F4/80+CD206+Arg+) (D). M2 macrophages from duplicate cultures were analyzed for arginase and CCL17 gene transcript levels by qRT-PCR. Fold-change in gene expression was analyzed using ΔΔCt method, compared to untreated M2 macrophage group (E). *P < 0.05; **P < 0.01.
To further examine if dampening RANKL influences macrophage- and protumorigenic immune cell-produced cytokines and chemokines in vivo, we investigated cytokine/chemokine signals in the TME. Primary tumors from untreated and OPGY49R treated 4T1.2Luc tumor-challenged mice were explanted and soluble proteins were extracted and analyzed using Proteome Profiler Mouse Cytokine Array Kit. Result showed that treatment with MSC-OPGY49R induced remarkable changes in cytokine/chemokine profiles (Fig. 5A and B). Following OPGY49R treatment, there was upregulation of Th1 cytokines: IL-1α, IL-1β, IL-2, IL-12, IL-16, IL-17, IL-23, INFγ, and TNFα (Fig. 5C). In addition, OPGY49R treatment also resulted in an increase in IL-1ra, CCL5 and a decrease in chemokine CXCL1 and CXCL2 (Fig. 5A–C), indicating the potential of OPGY49R in blocking RANKL-mediated protumorigenic functions.
Figure 5. Effects of sequestering RANKL by OPGY49R on soluble cytokine and chemokine mediators at the TME.
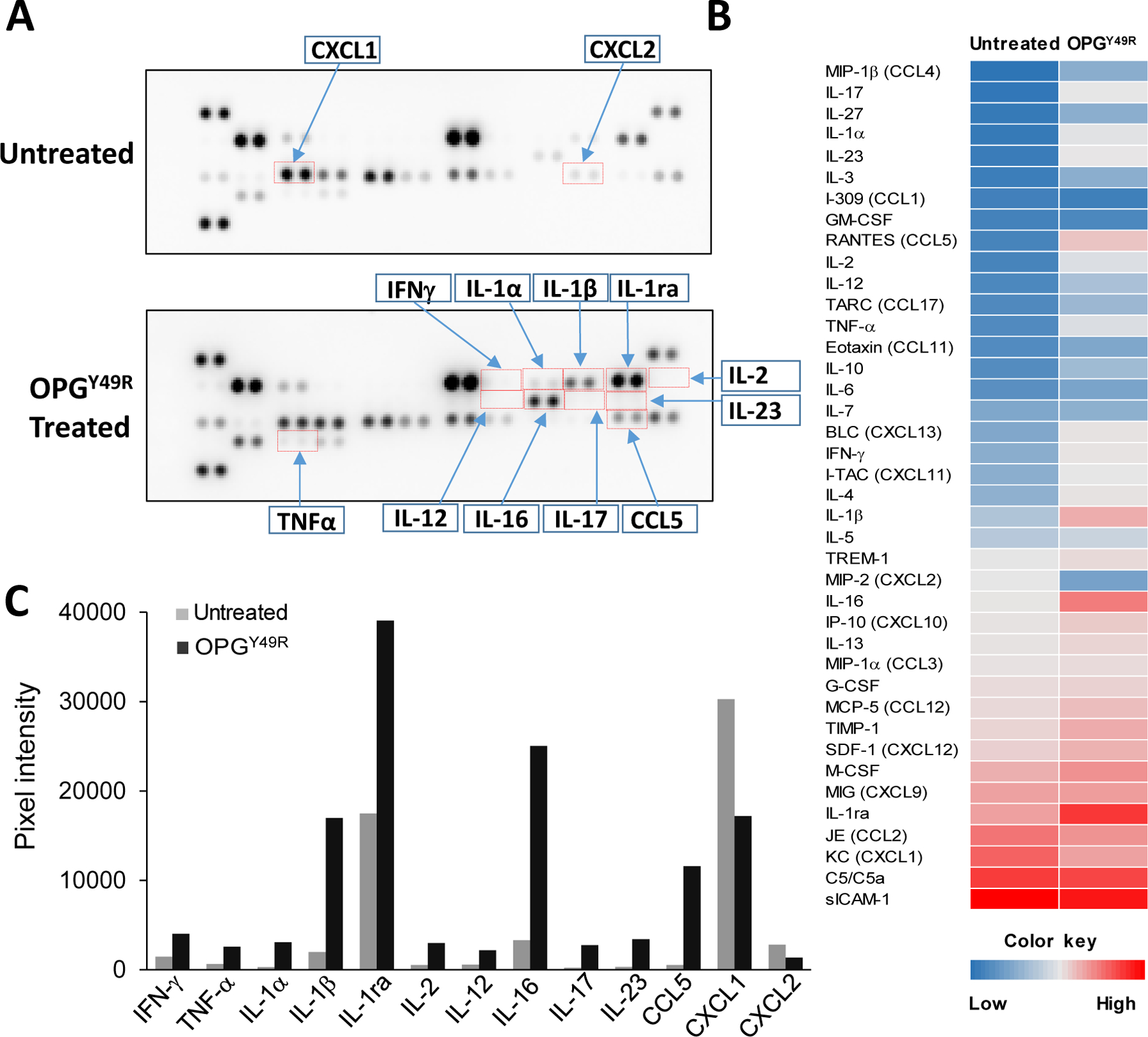
Twenty days after 4T1.2Luc tumor inoculation, mice were sacrificed and tumor tissues explanted. To determine effects of OPGY49R in altering the immune milieu at the TME, lysates were prepared from explanted tumors of untreated and OPGY49R treated mice (N=5). Equal amount of protein from individual samples were pooled together, then analyzed using a Proteome Profiler Mouse Cytokine Assay Kit and relative changes in individual cytokine/chemokine was determined by quantitation of acquired signal intensity using HLImage++ software. The representative images are shown in (A). Heatmap was created based on the mean pixel intensity of duplicate spots of individual cytokine/chemokine (B). The fold change in notable cytokine/chemokine levels in untreated and OPGY49R treated mice TME is shown (C).
Systemic OPGY49R and MD5-1 combination therapy improves tumor-specific T cell memory functions
Upon establishing the dual therapeutic effects of OPGY49R in restoring skeletal homeostasis and reducing M2 macrophage functional mediators, we reasoned that simultaneous targeting of tumor cells and the TRAIL signaling pathway would maximize anti-tumor effects and effector/memory T cell functions. To determine the effect of OPGY49R and MD5-1 combination therapy on long-term T-cell function, we performed adoptive transfer studies wherein splenetic CD8+ T cells from control mice, tumor-challenged untreated mice and tumor-challenged MSC-OPGY49R, MD5-1 and MSC-OPGY49R + MD5-1 treated mice were sorted in flow cytometry and adoptively transferred to female BALB/c mice with pre-established tumors in mammary fat pad and the tumor growth kinetics was monitored. Results of this study indicated a significant decrease in tumor growth following administration of CD8+ T cells from both MD5-1 and MD5-1 and OPG Y49R combination treatment received mice, but the effect was more pronounced in the group of mice that received CD8+ T cells from OPGY49R and MD5-1 combination treatment (Fig. 6A). However, there was no protection against the growth of tumors following adoptive transfer of T cells from control, untreated, or OPGY49R alone treated mice. In addition, cohorts of tumor-free mice following MD5-1 ± OPDY49R treatment were re-challenged with 4T1.2Luc cells to determine anti-tumor memory cell reactivation. Results of this study showed a significant protection against re-growth of secondary tumors in mice that were tumor-free following OPGY49R and MD5-1 combination therapy. Mice that were treated initially with MD5-1, showed a significant decrease in the growth of re-challenged tumors, but in mice that received combination therapy, tumor re-growth was completely blocked (Fig. 6B). These data indicate that OPGY49R and MD5-1 combination therapy improves tumor-specific T cell memory functions.
Figure 6. Effects of OPGY49R and MD5-1 combination therapy in long-term anti-tumor immunity against tumor re-challenge, and on metastasis.
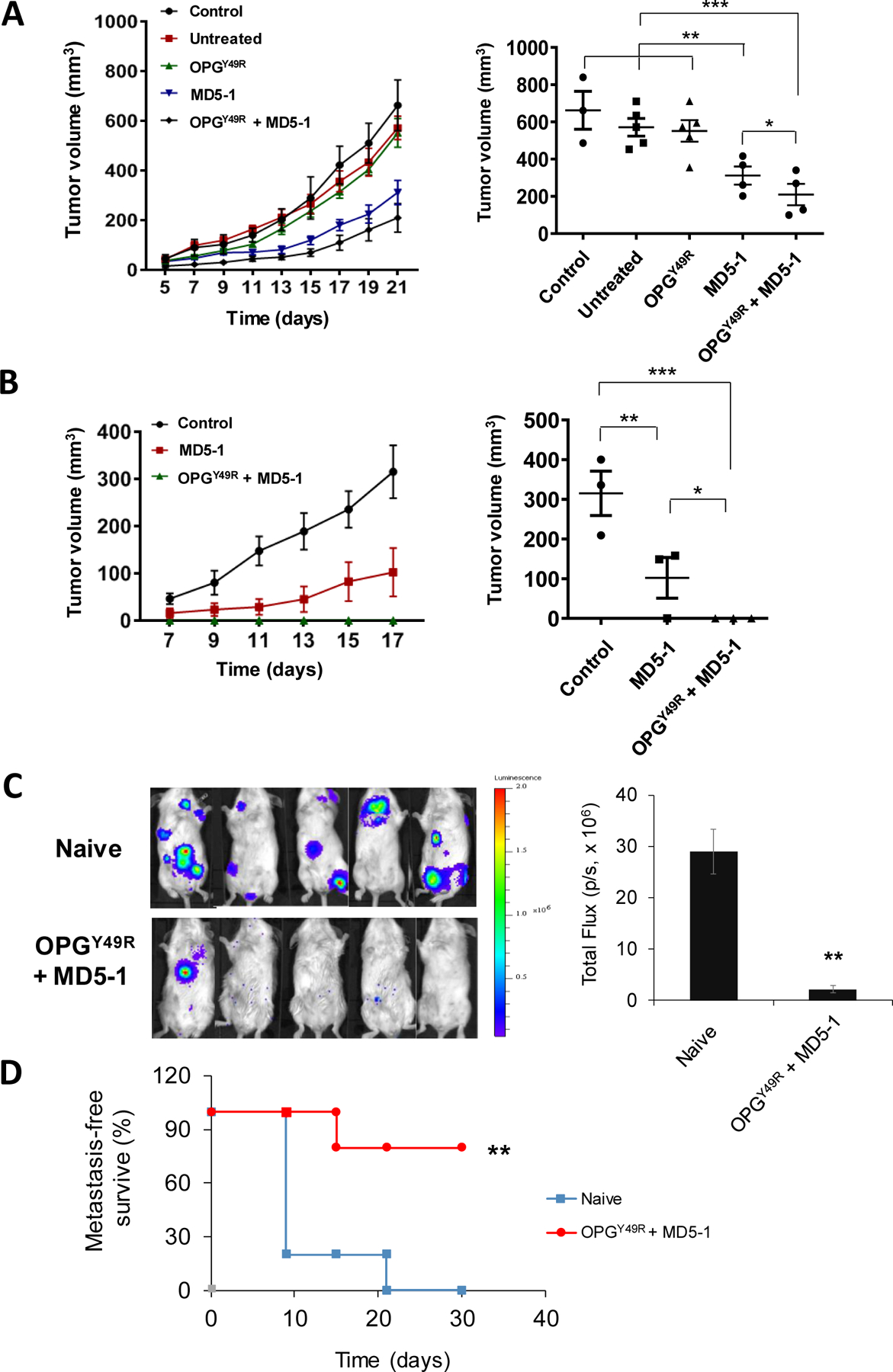
A, To establish the significance of RANKL-targeted OPGY49R and DR5-targeted MD5-1 therapy, either individually or in combination, splenic CD8+ T cells from control mice without tumor, or mice bearing 4T1.2Luc tumors that were either untreated or treated with OPGY49R, MD5-1 or both OPGY49R and MD5-1 were purified by flow sorting. Approximately 1×106 CD8+ T cells were adoptively transferred to female BALB/c mice that were previously challenged orthotopically with 4T1.2Luc tumors. B, To determine long-term anti-tumor immunity against re-growth of secondary tumors, groups of mice that received MD5-1 or MD5-1 and OPGY49R that remained tumor-free were re-challenged with 4T1.2Luc tumors in the opposite 4th mammary fat pad. To compare growth kinetics of re-challenged tumors, one group of control mice was inoculated with the same number of 4T1.2Luc cells. Tumor growth was determined on indicated days using a digital caliper and tumor volumes were compared between the groups by two-way ANOVA. Growth kinetics and tumor volumes at the endpoint are shown from each group. C and D, To determine the effects of OPGY49R and MD5-1 combination on metastasis, primary 4T1.2Luc tumors were established in 4th mammary fat pad of female BALB/c mice. Once tumors reached a volume of ~100 mm3, they were surgically excised. Following excision of the primary tumors, MD5-1 and OPGY49R treatments were given as described earlier. Control mice did not receive any treatment (Naïve). Non-invasive luciferase imaging was performed on indicated days as a measure of tumor re-growth at the primary tumor site and at secondary sites of metastasis. Representative luciferase imaging is shown in the left panel and quantitation of tumor growth at primary and secondary sites from luciferase imaging is shown in the right panel (C). Metastasis-free survival is shown (D). N = 3–5 per group; *P < 0.05; **P < 0.01; ***P<0.001.
Cell-based OPGY49R therapy with MD5-1 antibody combination confers protection against metastasis
Finally, we tested if application of the combination therapy would protect against visceral metastasis, extending its therapeutic benefit when given during resection of the primary tumors. Breast cancer disseminates to both skeletal and non-skeletal tissues. In addition to bone, common sites of breast cancer metastasis are liver, lung, and brain. To determine this, we developed 4T1.2luc tumors orthotopically in 4th mammary fat pad of female BALB/c mice. When the primary tumors reached a volume of ~100 mm3, they were surgically resected, followed by either no treatment (naïve) or application of MSC-OPGY49R and MD5-1 combination therapy Recurrent tumor growth at the primary site and/or visceral metastasis was monitored by non-invasive luciferase imaging. Results of this study demonstrated a significant decrease, both in the incidence of recurrent tumors at the primary site and in visceral metastasis, compared to naïve group without treatment (Fig. 6C). Metastasis-free survival rate was significantly increased in mice that received the combination therapy (Fig. 6D). These data suggest that OPGY49R and MD5-1 combination therapy can also be used in conjunction with debulking surgery to prevent/reduce metastasis.
DISCUSSION
Over the past decade, it has become clear that RANKL-RANK signaling axis is not only involved in bone homeostasis and remodeling, but also in mammary gland development, immune function, and tumorigenesis (32,33). Especially in breast cancer pathophysiology, RANKL-RANK pathway seems to play a vital role in aggressive growth and metastasis, as supported by clinical data correlating RANK and RANKL expression with poor disease-free survival in patients with TNBC (9), higher RANKL levels in estrogen receptor (ER)-positive breast cancers correlating with higher risk (34), and RANKL as a regulator of BRCA1 mutation-driven breast cancer (8). Therefore, targeting overt RANKL-RANK signaling is emerging as a viable option for antitumor functions. Using an immunocompetent mouse TNBC model, recapitulating breast cancer-associated pathologies as in the human disease, we established the potential of a variant OPG, devoid of TRAIL inhibitory function, both as a standalone therapy and in combination with agonistic activation of the death receptor pathway in tumor cells.
In corroboration with published reports that human breast cancer cells express RANK (15,16), we found that 4T1.2Luc cells express RANK and addition of RANKL to the culture significantly increases cell proliferation (Fig. 2A–C). Binding of RANKL to membrane-bound RANK triggers intracellular signaling through recruitment of adaptor proteins: TNF receptor-associated factors (TRAFs), Gab2, Cbl, and c-Src (26). In a cell-specific context, this pleomorphic signaling leads to activation of the NF-κB, PI3K-AKT, and MAPK cascades (26). Although a key activation pathway of RANK signaling during osteoclastogenesis involves NF-κB, NFATC1, and downstream osteoclast-specific genes, including cathepsin K, we found RANKL stimulation of 4T1.2Luc cells resulting in the activation of both the NF-κB and MAPK pathways, involved in aggressive tumor growth (35,36). However, treatment with OPG in the presence of RANKL reverted to basal proliferation rate and protumorigenic signaling molecules in breast cancer cells (Fig. 2C and D). Further, application of systemic MSC-OPGY49R as a standalone therapy not only reverted osteolytic bone damage, but also decreased tumor growth (Fig. 3A–C), indicating the potential of OPGY49R in directly targeting RANKL-mediated tumor growth. Recent reports also identified that RANK-positive tumor cells of luminal progenitors are highly proliferative, compared to RANK-negative tumors (8). Further, in three distinct datasets, including 684 breast cancer patient samples, RANK mRNA expression was higher in primary tumor tissues of TNBC patients, compared to other molecular subtypes, and more interestingly, TNBC patients who exhibited both RANK and RANKL had significantly worse relapse-free survival (9), signifying the use of OPGY49R in co-adjuvant setting for TNBC. Considering the lack of receptors for therapeutic targeting in TNBC and this molecular subtype disease associated with poor prognosis with least 5-year survival rate among all breast cancer subtypes, the use of OPGY49R as a biological molecule for dual therapy implications on skeletal lesions and against tumor proliferation without interfering in TRAIL-mediated apoptosis of tumor cells further augments the relevance of its potential application.
Although cell-based OPGY49R therapy significantly reversed bone damage and established bone architecture to that seen in normal bone anatomy, and decreased tumor growth in vivo, the latter effect was significantly improved when combined with agonistic activation of DR5, using MD5-1 antibody (Fig. 3A–C). This superior combinatorial effect was found to be due to induction of tumor apoptosis and activation of antitumor immune functions. In particular, following combination treatment with OPGY49R and MD5-1, there was a significant increase in tumor-specific T cell memory functions, the potency of which was further verified by adoptive transfer and tumor re-challenge studies (Fig. 6A and B). Though there was no effect in total M2 macrophage numbers (Fig. 4B–D), arginase 1 and CCL17 production induced by RANKL was blocked by OPG (Fig. 4D and E). Both arginase 1 and CCL17 are related to immunosuppression of tumor-associated macrophages (TAMs) (28–31). Superimposing our findings with published studies, which indicated immunosuppressive effects of RANKL on M2 macrophges (11,12) and on the existence of two different populations of M2 macrophages [Arg+ and Arg− (29)], our data suggests RANKL plays a major role in converting Arg− M2 macrophages to Arg+ highly immunosuppressive cells. This data supports a direct therapeutic potential of blocking RANKL-RANK signaling in M2 macrophages by OPGY49R to decrease immunosuppressive effects. Differential analysis of soluble factors in the TME by immune array in mice treated with OPGY49R alone identified upregulation of key Th1 cytokines IL-2, IL-12, INFγ, and downregulation of chemokines CXCL1 and CXCL2 (Fig. 5A–C). Although a precise source of these cytokines and chemokines is not established in this study, most of these soluble immune mediators are known to be produced by cells of both innate and adaptive immune system (37). Interestingly, we also found a stark upregulation of IL-1ra, a biological antagonist of IL-1 signaling (38). IL-1ra has been reported to inhibit tumor growth and metastasis by decreasing MDSCs and TAMs accumulation (39). Thus, blocking the RANKL-RANK axis with OPGY49R at the TME would decrease TAM function and consequently metastasis.
We also observed upregulation of IL-16 following treatment with OPGY49R. IL-16, a ligand for CD4 and functions as chemoattractant for T cells, has been reported to be expressed in high concentrations in M1 macrophages compared to M2 macrophages (40). Data from cytokine array, identifying increased IL-1ra and IL-16, together with a decrease in arginase production by M2 macrophages following OPG treatment is suggestive of beneficial effects of blocking RANKL-RANK signaling to promote effector T cell infiltration at the TME for antitumor effects. CXCL1 and CXCL2 have been reported to enhance MDSCs and promote breast cancer metastasis through NF-κB/SOX4 signaling (41,42). Decrease in chemokine ligand CXCL1 and CXCL2 and increase Th1 cytokines corroborate the significance of OPGY49R combination therapy, possibly by decreasing immunosuppression and promoting infiltration of Th1 cells to the TME as we found an increase in CD8+ T cells following OPGY49R and MD5-1 combination therapy (Supplementary Figure S6). Immunotherapy approaches using immune checkpoint inhibitors (ICIs) are emerging as more powerful alternatives for many cancers, including carcinomas of the breast (43). However, only a minority of patients respond effectively to ICIs, including monoclonal antibodies to CTLA-4, PD1, and PD-L1. Multiple reasons account for this insufficiency including mutation burden, bulk and clonality of neoantigens, levels of immune checkpoints, IFNγ responsiveness, and composition of the TME itself, which enables evasion of immune surveillance through production of additional suppressive mediator such as VEGF, IKK2 (44,45). Hence, combining OPGY49R with other immune modulating agents would be beneficial for patients refractory to ICIs. In this context, targeting the RANKL-RANK axis with OPGY49R, capable of reverting and preventing bone damage, to simultaneously exert antitumor effects directly through arresting RANK-mediated signaling cascades, without interfering in endogenous and/or exogenous TRAIL-induced apoptosis of tumor cells, will be a promising new alternate treatment modality. RANK signaling is also known to play an important role in generating regulatory T cells (Treg) that are immune suppressive (46,47). Hence, blocking augmented RANKL-RANK signaling in both innate and adaptive protumorigenic immune cells will be highly beneficial.
The most prevalent adjuvant therapy for breast cancer with bone dissemination is denosumab, a monoclonal RANKL antibody, used commonly in combination with aromatase inhibitors, especially in patients with malignancy-related hypercalcemia after bisphosphonate therapy (48). From a recently-concluded large ABCSG-18 trial, although denosumab treatment reduced clinical features, there was only a modest disease-free survival (49,50). As encountered with use of monoclonal antibodies in clinical regimen, common side effects with denosumab include infection of abdomen and urinary tract, pancreatitis, musculoskeletal pain, and hypocalcemia (51). Further, controversy exists regarding osteonecrosis of the jaw (52), prompting a careful follow up for patients prescribed denosumab. It was interesting to note in our studies that use of MD5-1 alone although resulted in a significant reduction in tumor growth kinetics in vivo, there was no therapy effect on bone damage. Hence, OPGY49R combination with tumor targeted therapies should extend dual benefits on both skeletal damage and tumor growth. The cell therapy approach we have adopted using MSCs offers additional advantages such as an immune privileged cell source as genetically engineered MSCs can be used in allogenic context to produce systemically stable levels of OPGY49R in physiologically relevant concentrations. Further characterization of this approach from pharmacokinetics and long-term standpoints should result in clinical application of this dual targeting for not only patients with osteolytic malignancy in carcinoma of the breast, but also other osteolytic cancers including multiple myeloma, kidney cancer, and prostate cancer with apparent skeletal pathology.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH grants: R01 AR060948 and R01CA184770 to S.P and R01HL128502 to J.S.D, and grants from the Breast Cancer Research Foundation of Alabama to S.P. We thank the Heflin Center Genomics Core, the Pathology Research Core, the Small Animal Bone Phenotyping Core, the Preclinical Shared Imaging Facility, and the Comprehensive Flow Cytometry Core at UAB for support with DNA sequencing, histology, μCT scan, IVIS imaging, and flow cytometry analyses, respectively.
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- 1.D’Oronzo S, Coleman R, Brown J, Silvestris F. Metastatic bone disease: Pathogenesis and therapeutic options: Up-date on bone metastasis management. J Bone Oncol 2019;15:004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature 2003;423:337–42. [DOI] [PubMed] [Google Scholar]
- 3.Shipman CM, Croucher PI. Osteoprotegerin is a soluble decoy receptor for tumor necrosis factor-related apoptosis-inducing ligand/Apo2 ligand and can function as a paracrine survival factor for human myeloma cells. Cancer Res 2003;63:912–6. [PubMed] [Google Scholar]
- 4.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer 2011;11:411–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinzone JJ, Hall BM, Thudi NK, Vonau M, Qiang YW, Rosol TJ, et al. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 2009;113:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010;468:103–7. [DOI] [PubMed] [Google Scholar]
- 7.Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010;468:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nolan E, Vaillant F, Branstetter D, Pal B, Giner G, Whitehead L, et al. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat Med 2016;22:933–9. [DOI] [PubMed] [Google Scholar]
- 9.Reyes ME, Fujii T, Branstetter D, Krishnamurthy S, Masuda H, Wang X, et al. Poor prognosis of patients with triple-negative breast cancer can be stratified by RANK and RANKL dual expression. Breast Cancer Res Treat 2017;164:57–67. [DOI] [PubMed] [Google Scholar]
- 10.Ahern E, Smyth MJ, Dougall WC, Teng MWL. Roles of the RANKL-RANK axis in antitumour immunity - implications for therapy. Nat Rev Clin Oncol 2018;15:676–93. [DOI] [PubMed] [Google Scholar]
- 11.Fujimura T, Kambayashi Y, Furudate S, Asano M, Kakizaki A, Aiba S. Receptor Activator of NF-kappaB Ligand Promotes the Production of CCL17 from RANK+ M2 Macrophages. J Invest Dermatol 2015;135:2884–7. [DOI] [PubMed] [Google Scholar]
- 12.Fujimura T, Kambayashi Y, Furudate S, Kakizaki A, Hidaka T, Asano M, et al. Receptor activator of nuclear factor kappa-B ligand (RANKL)/RANK signaling promotes cancer-related inflammation through M2 macrophages. Exp Dermatol 2016;25:397–9. [DOI] [PubMed] [Google Scholar]
- 13.Dou C, Ding N, Zhao C, Hou T, Kang F, Cao Z, et al. Estrogen Deficiency-Mediated M2 Macrophage Osteoclastogenesis Contributes to M1/M2 Ratio Alteration in Ovariectomized Osteoporotic Mice. J Bone Miner Res 2018;33:899–908. [DOI] [PubMed] [Google Scholar]
- 14.Meng YH, Zhou WJ, Jin LP, Liu LB, Chang KK, Mei J, et al. RANKL-mediated harmonious dialogue between fetus and mother guarantees smooth gestation by inducing decidual M2 macrophage polarization. Cell Death Dis 2017;8:e3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas RJ, Guise TA, Yin JJ, Elliott J, Horwood NJ, Martin TJ, et al. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology 1999;140:4451–8. [DOI] [PubMed] [Google Scholar]
- 16.Bhatia P, Sanders MM, Hansen MF. Expression of receptor activator of nuclear factor-kappaB is inversely correlated with metastatic phenotype in breast carcinoma. Clin Cancer Res 2005;11:162–5. [PubMed] [Google Scholar]
- 17.Smyth MJ, Yagita H, McArthur GA. Combination Anti-CTLA-4 and Anti-RANKL in Metastatic Melanoma. J Clin Oncol 2016;34:e104–6. [DOI] [PubMed] [Google Scholar]
- 18.Ahern E, Harjunpaa H, Barkauskas D, Allen S, Takeda K, Yagita H, et al. Co-administration of RANKL and CTLA4 Antibodies Enhances Lymphocyte-Mediated Antitumor Immunity in Mice. Clin Cancer Res 2017;23:5789–801. [DOI] [PubMed] [Google Scholar]
- 19.Ahern E, Harjunpaa H, O’Donnell JS, Allen S, Dougall WC, Teng MWL, et al. RANKL blockade improves efficacy of PD1-PD-L1 blockade or dual PD1-PD-L1 and CTLA4 blockade in mouse models of cancer. Oncoimmunology 2018;7:e1431088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Dam PA, Verhoeven Y, Trinh XB, Wouters A, Lardon F, Prenen H, et al. RANK/RANKL signaling inhibition may improve the effectiveness of checkpoint blockade in cancer treatment. Crit Rev Oncol Hematol 2019;133:85–91. [DOI] [PubMed] [Google Scholar]
- 21.Takeda K, Yamaguchi N, Akiba H, Kojima Y, Hayakawa Y, Tanner JE, et al. Induction of tumor-specific T cell immunity by anti-DR5 antibody therapy. J Exp Med 2004;199:437–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McHugh KP, Hodivala-Dilke K, Zheng MH, Namba N, Lam J, Novack D, et al. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest 2000;105:433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higgs JT, Jarboe JS, Lee JH, Chanda D, Lee CM, Deivanayagam C, et al. Variants of Osteoprotegerin Lacking TRAIL Binding for Therapeutic Bone Remodeling in Osteolytic Malignancies. Mol Cancer Res 2015;13:819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higgs JT, Lee JH, Wang H, Ramani VC, Chanda D, Hardy CY, et al. Mesenchymal stem cells expressing osteoprotegerin variants inhibit osteolysis in a murine model of multiple myeloma. Blood Adv 2017;1:2375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chypre M, Seaman J, Cordeiro OG, Willen L, Knoop KA, Buchanan A, et al. Characterization and application of two RANK-specific antibodies with different biological activities. Immunol Lett 2016;171:5–14. [DOI] [PubMed] [Google Scholar]
- 26.Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med 2006;12:17–25. [DOI] [PubMed] [Google Scholar]
- 27.Reck M, Krzakowski M, Chmielowska E, Sebastian M, Hadler D, Fox T, et al. A randomized, double-blind, placebo-controlled phase 2 study of tigatuzumab (CS-1008) in combination with carboplatin/paclitaxel in patients with chemotherapy-naive metastatic/unresectable non-small cell lung cancer. Lung Cancer 2013;82:441–8. [DOI] [PubMed] [Google Scholar]
- 28.Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res 2001;61:1100–6. [PubMed] [Google Scholar]
- 29.Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ, et al. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics 2018;8:5842–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer 2008;122:2286–93. [DOI] [PubMed] [Google Scholar]
- 31.Maruyama T, Kono K, Izawa S, Mizukami Y, Kawaguchi Y, Mimura K, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to infiltration of regulatory T cells in esophageal squamous cell carcinoma. Dis Esophagus 2010;23:422–9. [DOI] [PubMed] [Google Scholar]
- 32.Rao S, Cronin SJF, Sigl V, Penninger JM. RANKL and RANK: From Mammalian Physiology to Cancer Treatment. Trends Cell Biol 2018;28:213–23. [DOI] [PubMed] [Google Scholar]
- 33.Infante M, Fabi A, Cognetti F, Gorini S, Caprio M, Fabbri A. RANKL/RANK/OPG system beyond bone remodeling: involvement in breast cancer and clinical perspectives. J Exp Clin Cancer Res 2019;38:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarink D, Schock H, Johnson T, Overvad K, Holm M, Tjonneland A, et al. Circulating RANKL and RANKL/OPG and Breast Cancer Risk by ER and PR Subtype: Results from the EPIC Cohort. Cancer Prev Res (Phila) 2017;10:525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia L, Tan S, Zhou Y, Lin J, Wang H, Oyang L, et al. Role of the NFkappaB-signaling pathway in cancer. Onco Targets Ther 2018;11:2063–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene 2007;26:3279–90. [DOI] [PubMed] [Google Scholar]
- 37.Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta 2014;1843:2563–82. [DOI] [PubMed] [Google Scholar]
- 38.Voronov E, Apte RN. Targeting the Tumor Microenvironment by Intervention in Interleukin-1 Biology. Curr Pharm Des 2017;23:4893–905. [DOI] [PubMed] [Google Scholar]
- 39.Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep 2016;6:36107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Y, Du KL, Guo PY, Zhao RM, Wang B, Zhao XL, et al. IL-16 regulates macrophage polarization as a target gene of mir-145-3p. Mol Immunol 2019;107:1–9. [DOI] [PubMed] [Google Scholar]
- 41.Wang N, Liu W, Zheng Y, Wang S, Yang B, Li M, et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-kappaB/SOX4 signaling. Cell Death Dis 2018;9:880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi H, Han X, Sun Y, Shang C, Wei M, Ba X, et al. Chemokine (C-X-C motif) ligand 1 and CXCL2 produced by tumor promote the generation of monocytic myeloid-derived suppressor cells. Cancer Sci 2018;109:3826–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Force J, Leal JHS, McArthur HL. Checkpoint Blockade Strategies in the Treatment of Breast Cancer: Where We Are and Where We Are Heading. Curr Treat Options Oncol 2019;20:35. [DOI] [PubMed] [Google Scholar]
- 44.Gil Del Alcazar CR, Aleckovic M, Polyak K. Immune Escape during Breast Tumor Progression. Cancer Immunol Res 2020;8:422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barriga V, Kuol N, Nurgali K, Apostolopoulos V. The Complex Interaction between the Tumor Micro-Environment and Immune Checkpoints in Breast Cancer. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin J, Yang L, Silva HM, Trzeciak A, Choi Y, Schwab SR, et al. Increased generation of Foxp3(+) regulatory T cells by manipulating antigen presentation in the thymus. Nat Commun 2016;7:10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Francisconi CF, Vieira AE, Azevedo MCS, Tabanez AP, Fonseca AC, Trombone APF, et al. RANKL Triggers Treg-Mediated Immunoregulation in Inflammatory Osteolysis. J Dent Res 2018;97:917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thosani S, Hu MI. Denosumab: a new agent in the management of hypercalcemia of malignancy. Future Oncol 2015;11:2865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gnant M, Pfeiler G, Steger GG, Egle D, Greil R, Fitzal F, et al. Adjuvant denosumab in postmenopausal patients with hormone receptor-positive breast cancer (ABCSG-18): disease-free survival results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2019;20:339–51. [DOI] [PubMed] [Google Scholar]
- 50.Gnant M, Pfeiler G, Dubsky PC, Hubalek M, Greil R, Jakesz R, et al. Adjuvant denosumab in breast cancer (ABCSG-18): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015;386:433–43. [DOI] [PubMed] [Google Scholar]
- 51.Adler RA, Gill RS. Clinical utility of denosumab for treatment of bone loss in men and women. Clin Interv Aging 2011;6:119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khan AA, Morrison A, Kendler DL, Rizzoli R, Hanley DA, Felsenberg D, et al. Case-Based Review of Osteonecrosis of the Jaw (ONJ) and Application of the International Recommendations for Management From the International Task Force on ONJ. J Clin Densitom 2017;20:8–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.