

Abstract
Mutations in the calcium-binding protein calsequestrin cause the highly lethal familial arrhythmia catecholaminergic polymorphic ventricular tachycardia (CPVT). In vivo, calsequestrin multimerizes into filaments, but an atomic-resolution structure of a calsequestrin filament is lacking. We report a crystal structure of a human cardiac calsequestrin filament with supporting mutational analysis and in vitro filamentation assays. We identify and characterize a novel disease-associated calsequestrin mutation, S173I, that is located at the filament-forming interface, and further show that a previously reported dominant disease mutation, K180R, maps to the same surface. Both mutations disrupt filamentation, suggesting that disease pathology is due to defects in multimer formation. An ytterbium-derivatized structure pinpoints multiple credible calcium sites at filament-forming interfaces, explaining the atomic basis of calsequestrin filamentation in the presence of calcium. Our study thus provides a unifying molecular mechanism by which dominant-acting calsequestrin mutations provoke lethal arrhythmias.
The Ca2+ ion is a ubiquitous chemical messenger in eukaryotic cells, where transient changes in intracellular calcium trigger a diverse set of signal transduction pathways. In muscle cells, the transduction of an electrical signal at the membrane into calcium flows that activate the contractile apparatus is known as excitation-contraction coupling. Although some of the calcium required for muscle contraction enters the cell through voltage-gated calcium channels in the plasma membrane, most is released into the cytoplasm from the sarcoplasmic reticulum (SR, a muscle-specific extension of the endoplasmic reticulum, ER)1. Myocytes possess enhanced intra-SR calcium buffering near sites of storage and release, permitting a high total SR calcium content with much lower free calcium and maintaining the global SR free calcium level close to a steady state despite large fluxes2,3. The principal SR calcium buffer in cardiac and skeletal muscle is calsequestrin (CSQ), a 44 kD highly acidic protein that stores up to 50 % of SR calcium in a bound state, with each calsequestrin monomer storing up to 40-50 calcium ions4–11. Calsequestrin is complexed to the SR calcium channel, the ryanodine receptor (RyR), thereby ensuring that calcium is stored near the site of release1.
Since their initial identification, in 1971, a substantial field of research has formed around calsequestrins. Calsequestrins (CASQ1 in skeletal muscle and CASQ2 in cardiac muscle) are highly homologous in structure and function, and 64 % identical in sequence, with skeletal muscle calsequestrin appearing to have higher calcium capacity10. Calcium-binding propensity is explained in large part by a remarkable fraction of negatively charged acidic residues (26 % glutamate or aspartate in CASQ2 and 28 % in CASQ1, with corresponding average isoelectric points of 4.2 and 4.0, respectively). In both cardiac and skeletal muscle, calsequestrin localizes to the junctional SR (jSR) of muscle and forms multimers that are anchored to the luminal SR membrane. Low resolution electron micrographs of isolated skeletal muscle fibers reveal the possibility of more than one multimer type: in some studies a filamentous morphotype is clearly discernible, whereas in others a dense, reticular condensate is observed8,12. Multimers are anchored to a complex consisting of RyR and the single-pass transmembrane proteins triadin and junctin1. Extended homo-multimerization provides high density calcium storage, but multimerization appears to be essential for localization, too, in that multimerization-defective mutants are trafficked along the secretory pathway and lost from the SR13–15. Additional insights into calsequestrin biochemistry and cell biology encompass post-translational modifications16,17, calcium-storage capacity10, and interactions with the calcium release unit18–20.
Calsequestrin’s relevance to human disease is well established. Variants in skeletal muscle calsequestrin have been putatively linked to malignant hyperthermia and to vacuolar myopathies21, while variants in cardiac calsequestrin are well known to cause catecholaminergic polymorphic ventricular tachycardia (CPVT), a highly lethal familial arrhythmia. CPVT is caused by a state of cardiac calsequestrin deficiency, whether arising from null or hypomorphic alleles or from point mutants that disrupt calsequestrin multimerization and localization22,23. Most CPVT-causing CASQ2 point mutations have recessive inheritance, which is concordant with a mechanism whereby deficiency leads to disease. For a protein whose function depends on multimerization, there is, at least to date, a surprising paucity of variants with putative dominant negative mechanism. The sole known CASQ2 disease variant with strong evidence for dominant inheritance, K180R, was not reported to cause defects in multimerization and was proposed to act via as-yet undetermined alternative mechanisms24. The biochemical mechanism for dominant-acting CASQ2 disease variants thus remains an open question.
Despite the rich body of work concerning calsequestrin biology, no compelling high-resolution candidate structure for a calsequestrin filament has emerged. Although sixteen crystal structures of calsequestrins have been published9,10,16,21,25–27, none of these contain a convincing filament-like assembly. All 16 prior structures reveal calsequestrin dimers that are nearly identical to one another, but a search for dimer-to-dimer interfaces within and across these crystal unit cells reveals only weak crystallographic packing contacts that appear incompatible with robust biological multimerization. In addition, the observed dimer-to-dimer interfaces vary substantially from one lattice to another. Critically, a lack of mutagenesis studies supporting proposed oligomerization interfaces in the prior published structures calls into question whether the relevant biological multimer has ever been observed. Cumulatively, the prior studies have established that calsequestrins dimerize in a wide variety of conditions, with intra-dimer interactions that are largely the same across published structures. The mechanism by which dimers assemble into higher order multimers (inter-dimer assembly) remains elusive.
A structure of a calsequestrin filament (or other relevant high molecular weight multimer) would advance our understanding of calsequestrin biology and permit a comprehensive mapping of disease-causing point mutations to biologically relevant multimerization interfaces. We report here an investigation of dominant-acting cardiac calsequestrin disease mutants in conjunction with a new X-ray structure of cardiac calsequestrin that reveals a biologically relevant filament-forming interface. We show that known dominant-acting mutants - one previously reported, and one described here for the first time - map to the newly reported multimerization interface. Furthermore, we provide supporting biochemical analysis of likely calcium-binding sites where the protein multimerizes. Finally, we provide a plausible mechanism by which point mutations at different filament-forming interfaces may lead to different disease inheritance patterns. These findings fundamentally advance our understanding of the mechanism by which calsequestrin contributes to calcium homeostasis and provide a missing link in our understanding of how dominant-acting CASQ2 variants cause disease.
Autosomal dominant CASQ2 disease mutations disrupt calsequestrin multimerization.
We encountered a proband from a family with CPVT-like tachy-arrhythmias and multiple cases of sudden, unexplained death at a young age (Fig. 1a). The proband presented at age 33 with a biventricular tachycardia on ECG and underwent an electrophysiologic study that revealed an inducible ventricular tachycardia with focal origination next to the anterior fascicle, and an inducible atypical atrioventricular nodal reentry tachycardia that was successfully ablated. Targeted sequencing of channelopathy genes (KCNQ1, KCNH2, SCN5A, ANK2, KCNE1, KCNE2, KNCJ2, CAV3, RYR2, and CASQ2) revealed only a heterozygously-carried isoleucine-for-serine substitution at position 173 in cardiac calsequestrin. The proband’s son and multiple other family members report a tachycardic phenotype (ranging from self-reported palpitations to diagnosed episodes of tachycardia). The family history is also notable for multiple cases of sudden cardiac death at a young age. The distribution of affected individuals in the pedigree is potentially consistent with dominant inheritance. As the family did not consent to follow-up genetic testing or clinical phenotyping, we were unable to rigorously assess disease segregation with the variant.
Fig. 1: Autosomal dominant CASQ2 disease mutations disrupt calsequestrin multimerization.
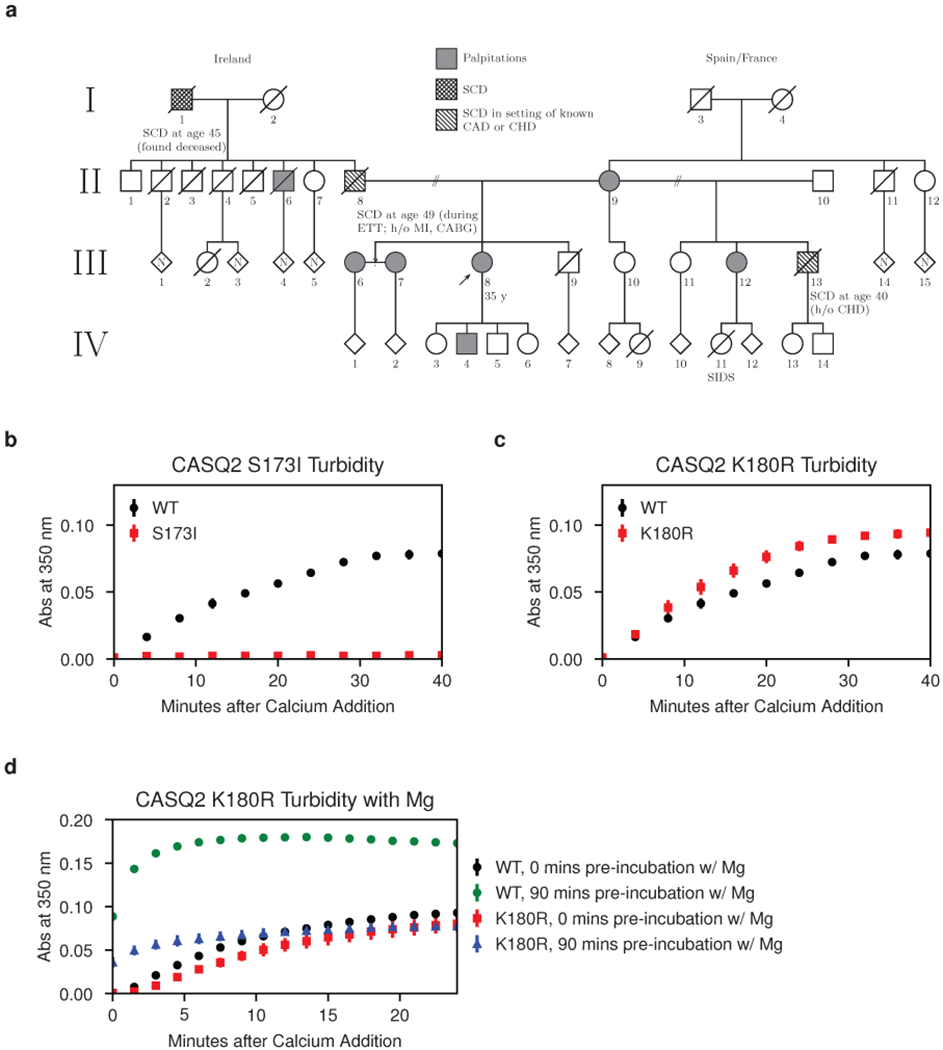
a, Pedigree of a large extended family with the S173I mutation and a CPVT-like phenotype. CABG = coronary artery bypass graft; CAD = coronary artery disease; CHD = congenital heart disease; ETT = exercise treadmill test; MI = myocardial infarction; SCD = sudden cardiac death; SIDS = sudden infant death syndrome. b, Multimerization kinetics of purified S173I mutant protein assessed by turbidity assays following addition of 1 mM CaCl2 (see Methods for details). c, Multimerization kinetics of the K180R mutant determined under the same conditions as in (b). d, Multimerization kinetics of the K180R mutant observed using a turbidity assay (same conditions as in b, but with 2 mM MgCl2 added prior to calcium). Error bars represent the mean ± s.d. of 3 technical replicates.
As turbidimetric monitoring of calsequestrin’s calcium-induced multimerization is a well-established practice22,23, we elected to investigate the S173I variant biochemically in a simple turbidity assay. In this assay, addition of calcium to purified cardiac calsequestrin causes an increase in turbidity that is fully reversible with addition of stoichiometric EDTA, indicating that the calcium-induced light-scattering is due to patterned assembly rather than non-specific aggregation (Extended Data Fig. 1a). Although strong genetic evidence for pathogenicity of the S173I variant is lacking, the turbidity assay for the S173I mutant reveals a profound decrease in multimerization rate (Fig. 1b). Minimal improvement in the multimerization rate is observed in a non-physiologic 0 mM potassium condition (Extended Data Fig. 1b), demonstrating that the mutant protein is intact but defective in multimerization.
The striking effect of S173I on CASQ2 mutimerization prompted us to reexamine the multimerization capacity of the previously reported K180R variant - to date, the only known CASQ2 disease variant with strong evidence for dominant pathogenicity. The turbidity assay for K180R under the same conditions used for S173I shows little difference from the wild type CASQ2 variant (Fig. 1c). However, prior reports suggested that calsequestrin maintains distinct magnesium and calcium binding sites28. Therefore, we investigated multimerization kinetics of the K180R mutant in the presence of magnesium. Strikingly, prolonged incubation of the K180R mutant with magnesium (2 mM MgCl2) prior to addition of calcium yields a substantial multimerization defect (Fig. 1d). Interestingly, neither S173 nor K180 fall at credible, previously identified candidate multimerization interfaces. As the dominant inheritance pattern is classically associated with disease variants that disrupt protein-protein interactions, we elected to pursue a structure of a calsequestrin multimer in the belief that the biologically relevant assembly has not yet been observed.
Helical architecture of the cardiac calsequestrin filament.
We determined a new crystal structure of human cardiac calsequestrin obtained from a full-length construct in very low-pH (3–3.5) crystallization conditions. The previously characterized calsequestrin dimer is again observed, but now in an arrangement that produces a tightly packed filament (Fig. 2a). Crystallographic data collection and processing statistics are summarized in Table 1 for the native structure as well as an ytterbium-soaked condition used to identify probable calcium-binding sites.
Fig. 2: Helical domain architecture of the cardiac calsequestrin filament.
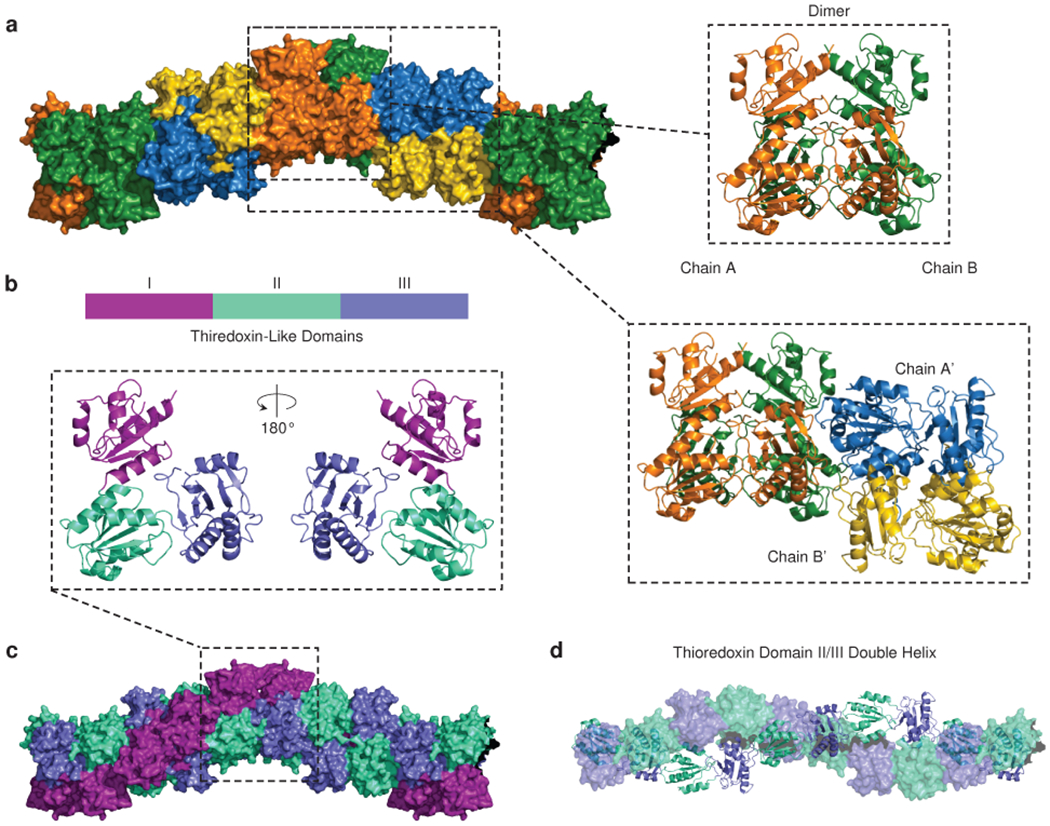
a, A space-filling model of the cardiac calsequestrin candidate filament (PDB ID 6OWV) is shown, along with ribbon diagrams of a dimeric and a tetrameric assembly (boxed images). Dimers are stacked on a screw axis with 90 degrees of rotation per dimer. b, Cardiac calsequestrin monomers are colored by thioredoxin domain. The monomers are translated but remain in their dimer-forming orientation. c, The helical character of the filament is revealed at the domain level. Viewed at the level of its thioredoxin domains (3 per protomer), the filament consists of an inner thioredoxin double helix (domains II and III) with an outer thioredoxin single helix (domain I) wrapping the double helical core. d, The inner double helix of the filament consists of thioredoxin domain II and III. For clarity, one strand is rendered as ribbon (domains from chains A and A’), while the other strand is rendered as surface (domains from chains B and B’).
Table 1:
Data Collection and Refinement Statistics
| Human Cardiac Calsequestrin (PDB 6OWV) | Human Cardiac Calsequestrin, Ytterbium-Complexed (PDB 6OWW) | |
|---|---|---|
| Data Collection | ||
| Space Group | P 43 2 2 | P 1 21 1 |
| Cell Dimensions | ||
| a, b, c (Å) | 62.5329, 62.5329, 213.189 | 85.8318, 86.0152, 214.34 |
| α, β, γ (°) | 90, 90, 90 | 90, 89.9072, 90 |
| Resolution (Å) | 53.94 - 1.88 (1.947 - 1.88)a | 107.2 - 3.84 (3.977 - 3.84) |
| Rpim (%) | 1.8 (194.8) | 16.2 (85.0) |
| I/σ(I) | 14.82 (0.52) | 4.52 (0.96) |
| CC1/2 | 0.999 (0.422) | 0.992 (0.616) |
| Completeness (%) | 97.76 (79.77) | 98.46 (98.05) |
| Redundancy | 22.5 (12.6) | 11.2 (11.5) |
| Refinement | ||
| Resolution (Å) | 53.94 - 1.88 | 107.2 - 3.84 |
| No. Reflections | 35501 (3425) | 29897 (2924) |
| Rwork / Rfree (%) | 21.4 / 25.0 | 29.6 / 35.1 |
| No. atoms | ||
| Protein | 2728 | 21720 |
| Ligand | 35 | 88 |
| Water | 33 | N/A |
| B Factors | ||
| Protein | 82.51 | 98.59 |
| Ligand/ion | 117.84 | 138.64 |
| Water | 55.83 | N/A |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.009 | 0.008 |
| Bond angles (°) | 1.27 | 1.23 |
Values in parentheses are for highest-resolution shell.
The new structure provides a compelling candidate for a biologically plausible higher order multimer. The oligomer-forming contacts that exist between dimers in our structure differ substantially from previously reported calsequestrin crystal structures (Extended Data Fig. 2). The repeating unit of the native crystal resides in a higher-symmetry point group compared to prior calsequestrin structures (Supplementary Table 1), and the candidate filament-forming interfaces collectively encompass significantly greater buried surface area than previously characterized calsequestrin inter-dimer interfaces (Extended Data Fig. 2).
The cardiac calsequestrin monomer, like its skeletal counterpart, consists of an N terminal loop, 3 thioredoxin domains, and a disordered acidic tail. The dimer forms by mutual exchange of N-terminal loops, as previously observed (Fig. 2b). In our structure, the dimers are stacked along a screw axis to form the filament, with each dimer rotated 90° with respect to its neighbors. Although the dimers are positioned at discrete 90° rotations with respect to one another, the underlying architecture of the filament is in fact helical at the level of thioredoxin domains. An outer helix or “collar” of larger radius, consisting only of thioredoxin domain I, winds around a more compact inner double helix consisting of thioredoxin domains II and III (Fig. 2c–d). All helices are left-handed, corresponding to the left-handed screw axis at the level of dimer-stacking.
A 3-Helix configuration of the filament promotes close-packing of thioredoxin domains.
Helical packing permits each domain to contact multiple other domains in multiple other protomers, which is in stark contrast to other reported candidate structures, which lack the helical pitch and have much more limited interacting surface area (Extended Data Fig. 3). The close-packing of the new filament candidate is clearly visible when thioredoxin domains are represented as equal-sized spheres centered at the domain center of mass (Extended Data Fig. 3, right-hand side). Prior putative calsequestrin filament candidates have fewer inter-domain contacts, fewer inter-protomer contacts, and substantially less buried surface area compared to the new candidate.
Notably, our crystallization condition produced crystals only at a low pH (3-3.5, measured by applying a crystallization drop to litmus paper) resulting from the use of aged PEG reagents (presumably having undergone degradation to glycolic acid) or from direct addition of concentrated HCl. This restriction obtained irrespective of calcium in concentrations ranging from trace to 14 mM (higher calcium concentrations led to precipitation, likely due to the presence of sulfate, and were therefore not assessed).
Lanthanide substitution reveals the biochemical basis of cation-driven filament assembly.
To localize candidate calcium sites within the context of the new filament structure, we collected data from an Ytterbium (Yb)-soaked crystal (Table 1). From the anomalous map, we identified approximately 8 sites with strong Yb signal per CASQ2 chain (> 3.8 σ in the anomalous map, and positive difference density in the Fo-Fc map). As prior work to identify calsequestrin’s calcium-binding sites was based largely on the indirect method of inference from metal coordination geometry with nearby side chains and waters26, the use of Yb here provides a direct approach to confirming the prior findings, while extending our understanding to calcium sites at the inter-dimer filament-forming interfaces.
Cation binding leads to conformational shifts in calsequestrin dimers.
We first assessed the presence of Yb at the intra-dimer interface. We identified several high occupancy Yb sites, most of which are clustered in a narrow region between protomers, lined by highly conserved acidic residues (Fig. 3a and Extended Data Fig. 4). Consistent with the location of a putative calcium ion in the prior study, we find a Yb atom coordinated primarily by E147 of chain A and D278 of chain B. In addition, we find a Yb atom coordinated primarily by E143 and E275. In the native structure, the close interaction of E147 and D278 can be explained as a carboxyl-carboxylate bond stabilized by low pH28,29. Anomalous signal is found at this site in the derivative structure, but consistently on just one side of the otherwise symmetric dimer. This subtle asymmetry, with a cation bound on one side and a carboxyl-carboxylate bond on the other, likely reflects a degree difference in occupancy rather than a sharp distinction, but it is a consistent feature of the density map across all 4 dimers in the crystal asymmetric unit and may be conformationally conducive to filament formation.
Fig. 3: Cation binding leads to conformational shifts in calsequestrin dimers.
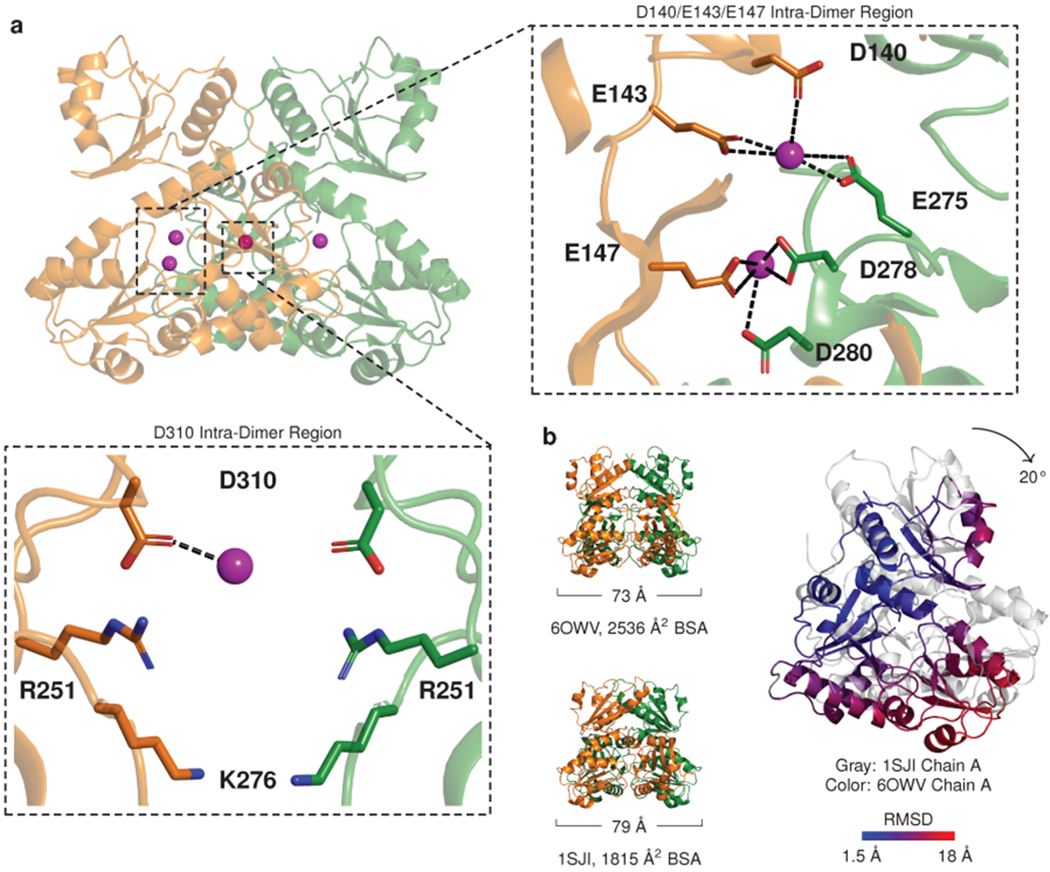
a, Ribbon diagram of a dimer with ytterbium (Yb) sites (magenta spheres) within its interior cavity. Boxed zoom images highlight Yb positions that bridge dimer chains A and B: relatively high occupancy is observed in a narrow intra-dimer cleft (right panel), while a site of weaker interaction and weaker occupancy is identified in the intra-dimer cavity (lower panel). b, Comparison of a previously reported cardiac calsequestrin dimer (1SJI) to the more tightly packed dimer of the present study. The tightly packed dimer results primarily from rigid body rotation of the dimer chains inward (for a single chain, a 20° counterclockwise rotation is observed in the plane of the page when the other chain is fixed to the reference dimer). The inward rotation produces an increase in buried surface area (BSA) in thioredoxin domains II and III.
Another Yb atom is poorly coordinated by D310 and a putative sulfate anion within a solvent cavity enclosed by the dimer (Fig. 3a, lower). The low resolution of the derivative structure precludes identification of waters that could contribute to coordination geometry, but all identified cation sites are supported by electron density and anomalous signal. Together, these Yb bridging sites stabilize the interaction between thioredoxin domain II from protomer A and thioredoxin domain III of protomer B.
In both the native structure and Yb-soaked structure, the dimer subunits have undergone substantial inward rotation in comparison to the other high-resolution cardiac calsequestrin structure (PDB ID 1SJI) (Fig. 3b). The inward domain movement is produced largely by rotation of the monomer as a rigid body, resulting in a conformation where domains II and III are more tightly packed. This conformational shift recapitulates a similar finding from the prior study for skeletal calsequestrin dimers crystallized in a high calcium buffer26. Inspection of all prior published calsequestrin structures reveals that 6 prior calsequestrin structures belong to this “tightly packed dimer” group, while 10 others contain dimers that are more loosely packed (Extended Data Fig. 5). The more tightly packed structures were all crystallized in the presence of multivalent cations (usually calcium), or in one case (PDB ID 2VAF) a monovalent cation at extremely high concentration (2 M NaCl). Dimers from the other group were crystallized with no added multivalent cations, with the exception of PDB ID 3TRP, which contained calcium at much lower concentration. The tightly packed structures consistently exhibit a greater degree of conformational disorder in Domain I (Extended Data Fig. 6), accompanied by a modest loss of intra-dimer contact in Domain I, while multiple hydrophobic side chains from Domains II and III that were not buried now obtain buried surface area (Supplementary Fig. 1). Thus, our data provide additional evidence for conformational change within the dimer upon calcium-binding (likely induced by closer approximation of acidic side chains), independent confirmation of intra-dimer calcium binding sites, and an explanation for why an altered conformation of the dimer becomes energetically tolerable in the presence of calcium.
Cations are trapped at inter-dimer filament-forming interfaces.
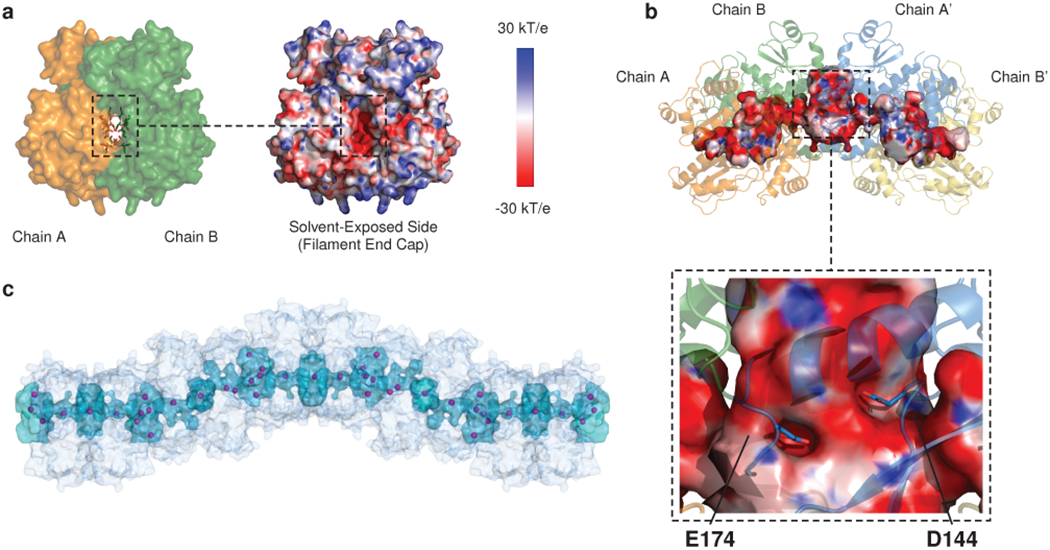
At the inter-dimer interface, acidic surfaces enclose a large solvent cavity with interior and exterior Yb binding sites (Fig. 4a and Extended Data Fig. 7). Whereas the negative charge at the dimer interface is accommodated partly by cation binding and partly by carboxyl-carboxylate bonds, the inter-dimer interface has comparatively higher cation occupancy, measured by magnitude of anomalous signal (Extended Data Fig. 7). Residues E184 and E187 coordinate Yb atoms with very substantial anomalous peaks near the mouth of the cavity, partially shielded from exposure to the bulk solvent. Within the cavity, residues D144 and E174 appear to interact weakly with Yb atoms (the precise geometries of D144 and E174 are difficult to discern, due to lack of side chain density in the electron density maps of both the native and Yb-soaked structures). At the cavity base, residues D348 and D350 are oriented along with their symmetry mates to form a cluster of 4 acidic side chains that coordinate a single cation. Finally, outside the inter-dimer cavity, on the fully solvent-exposed exterior of the filament, residues D351 and E357 and their symmetry mates form bidentate interactions with two Yb atoms, adopting conformations in which opposing acidic rotamers are bent away from one another, thereby alleviating electrostatic repulsion that would otherwise disrupt multimer formation.
Fig. 4: Cations are trapped at inter-dimer filament-forming interfaces.
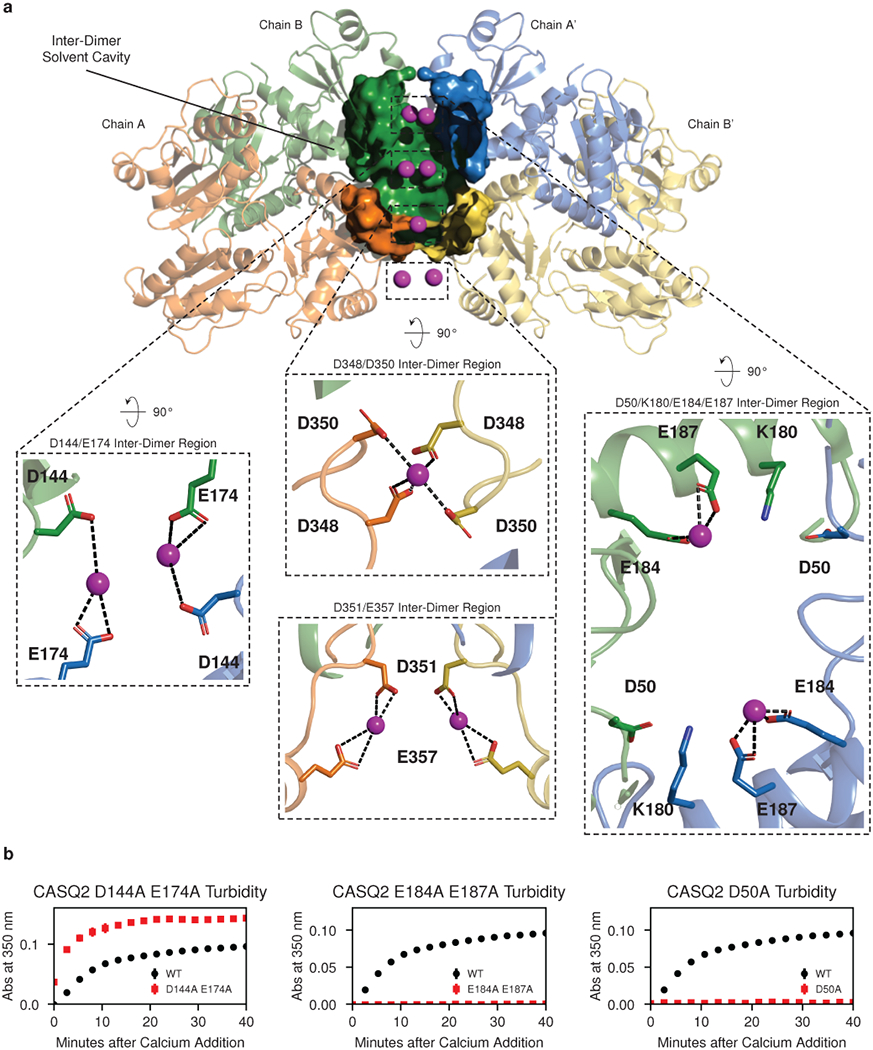
a, Yb (magenta spheres) bound within the walled pocket formed by the inter-dimer interface, with boxed zoomed images highlighting ytterbium sites at D144 and E174, E184 and E187, D348 and D350, and D351 and D357. Thioredoxin domain II of chain A’ (blue) is omitted to allow visualization of the interior of the solvent pocket formed by the inter-dimer interface. b, Turbidity assays after alanine mutagenesis of putative calcium-binding and salt bridge residues. Left: D144A and E174A. Middle: E184A and E187A. Right: D50A. Error bars indicate the mean ± s.d. of 3 technical replicates.
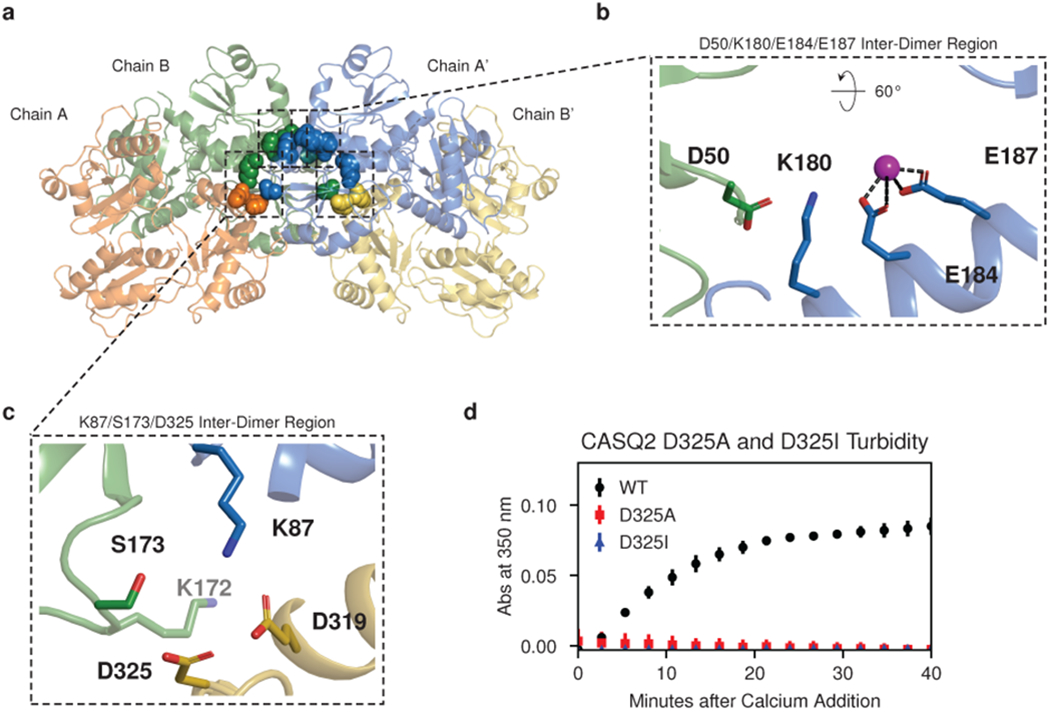
Identification of these Yb occupancy sites at the inter-dimer interface provides a testable model for the biological relevance of the new filament structure. To test our model, we mutated the Yb-binding aspartate or glutamate side chains to alanine and examined the effect of these mutations on multimerization kinetics. Mutation of residues D144 and E174 to alanine results in a dramatic increase in multimerization rate (Fig. 4b, left). Conversely, mutation of residues E184 and E187 to alanine results in a profound multimerization defect (Fig. 4b, middle). Of note, glutamates 184 and 187 belong to an alpha helix that provides a linkage between domains of the outer thioredoxin collar. This helix, belonging to thioredoxin domain II, sits between thioredoxin I domains of different dimers and interacts with inter-dimer salt bridges on either side. Alanine mutagenesis of the D50 residue that participates in a salt bridge with K180 at the N-terminal end of this helix produces a similar defect (Fig. 4b, right). At the remaining two Yb sites (D348 with D350 and D351 with E357), mutating acidic residues to alanine would be expected to relieve mutual repulsion of tightly packed acidic side chains. Consistent with this, alanine mutagenesis of these residues has a largely net neutral effect on multimerization kinetics (Extended Data Fig. 8a–b).
The long axis of the cardiac calsequestrin filament contains a continuous, solvent-accessible lumen.
Intriguingly, the majority of Yb sites that we identify are located within a continuous solvent cavity that winds through the interior of the filament. The calculated electrostatic potential at the protein surface becomes highly electronegative at the entrance to this cavity, which is lined by acidic residues (Fig. 5a). The small size of this cavity, lined by acidic side chains and then surrounded by the bulk of one of the most acidic proteins known to exist, results in a remarkably electronegative character (Fig. 5b). The side chains of D144 and E174 extend far into the high electronegativity region, possibly explaining why mutating these side chains to alanine resulted in improved multimerization kinetics - in contrast to the effect of alanine mutagenesis of the other interfacial acidic residues, which are much closer to or even outside the boundary of the pocket. A continuous lumen results from the fact that each dimer contains a solvent pocket within its interior, while another solvent pocket is formed at each inter-dimer interface. Stacking of dimers thus extends the cavity along the entire length of the filament (Fig. 5c). The calcium ions that would appear to be bound in the dimer’s interior could constitute a separate store of calcium that is more slowly mobilized than the highly accessible pool of ions bound to the surface and the solvated acidic tail.
Fig. 5: The cardiac calsequestrin filament contains a continuous, solvent-accessible lumen along its long axis.
a, Left: space-filling model of the interior cavity of the dimer viewed down its long axis. Residues that interact with Yb atoms within the intra-dimer cleft (Fig. 3) are shown as sticks. All other residues are rendered as surface. Right: APBS-generated electrostatic surface charge distribution of the same region. b, The lumen is continuous down the length of filament because of the large solvent cavity formed at each dimer-dimer interface. The APBS-generated electrostatic surface of the lumen (traced by HOLLOW using a 1.4 Å probe) is shown, with closeup of residues D144 and E174 deep within the electronegative cavity. c, View of the filament and its continuous interior cavity, with Yb sites shown as magenta spheres.
Dominant disease mutations disrupt the filament-forming interface.
The newly observed filament structure permits insight into the disease mechanisms of CPVT-associated calsequestrin variants. Remarkably, both of the variants that exhibit multimerization defect in this study (namely the arrhythmia-associated S173I variant that initiated this investigation, as well as the K180R variant previously implicated in CPVT24) affect residues at the inter-dimer interface (Fig. 6a).
Fig. 6: Dominant disease-associated mutations disrupt the filament-forming interface.
a, Inter-dimer interface residues in the vicinity of S173 and K180 are shown as spheres colored as in Fig 2a. b, Closeup of K180 with the adjacent E184 and E187 cation binding site, with the coordinated Yb atom shown as a magenta sphere. c, Hydrophilic pocket at S173 created by interactions of 3 different thioredoxin domains from 3 distinct chains (K87, S173, D325). K172 (light gray label) is the most proximal of several residues that shield the pocket from bulk solvent. d, Turbidity assay with the D325A and D325I mutations. Error bars indicate the mean ± s.d. of 3 technical replicates.
Despite the conservative nature of the arginine-for-lysine substitution, strong genetic evidence from previously published work implicates the K180R variant as pathogenic. We have shown (in Fig. 1) that the K180R variant results in a multimerization defect, but only in the presence of magnesium. The fact that magnesium is required to observe the multimerization defect of the K180R mutant suggests that rather than disrupting the salt bridge with the adjacent D50 residue, K180R instead alters the electrostatics of the nearby divalent cation-binding site (Fig. 6b). The high conservation of K180 in evolution (Supplementary Fig. 2) and key role of K180 and its interacting partners in supporting calsequestrin multimerization provide additional support for the physiological relevance of our new structure.
The dimer-stacking architecture of the filament likewise explains the disruptive effect of the S173I variant. Remarkably, S173 occupies a critical position at the filament-forming interface: a charged pocket formed by the interaction of K87, S173, and D325 (Fig. 6c and Extended Data Fig. 9). This pocket enforces an interaction between 3 thioredoxin domains from 3 different protomers at a single site. To test the importance of this pocket for multimer formation, we mutated residue D325 to D325I or D325A. Both mutants exhibit similarly depressed multimerization kinetics in the turbidity assay, demonstrating that hydrophobic substitution in this interfacial pocket has profound effects (Fig. 6d).
Discussion
We report a crystal structure of a human calsequestrin filament. Although we do not exclude the possibility that other higher-molecular weight calsequestrin complexes exist, multiple lines of evidence support the biological relevance of the filamentous form that we describe. We also elucidate the biochemical basis of calsequestrin’s divalent cation-induced filamentation. Using Yb substitution, we confirm previous reports of intra-dimer cation binding sites, and we reveal the basis of inter-dimer cation-trapping that appears to govern the rate of multimerization.
The crystal structure of the filament provides insight into subtle conformational changes that may favor filamentation. The prevailing view has been that calsequestrin filamentation is driven by an increase in solvent entropy that occurs as divalent cations lose their hydration shells to bind the protein at filament interfaces28. This hypothesis is complicated, however, by our observation of stable filamentation in the near-absence of ligand (only trace concentrations of divalent cations were present in the low-pH crystallization condition). The low-pH filament is stabilized instead by carboxyl-carboxylate interactions between closely juxtaposed acidic residues. While increased solvent entropy likely contributes to filamentation, we suggest instead that protein conformational entropy is likely to be the major factor. The structure we report is remarkably disordered by conventional B-factor metrics, and especially so in Domain I. In fact, all tightly packed calsequestrin dimers (7 published structures total, including this study) exhibit increased disorder in the solvent-exposed loops of Domain I (Extended Data Fig. 6). The consistency of this observation makes it less likely that conformational disorder within Domain I is a feature of our specific crystallization condition and more likely that it is energetic compensation for the adoption of the more tightly packed conformation favorable for filamentation. This would be consistent with results from studies of another calcium-binding protein, calmodulin, where changes in conformational disorder appear linearly related to the binding energy at its targets30.
Calsequestrin’s pH-sensitivity prompts questions about the possible role of intra-luminal pH changes in the regulation of calcium uptake and release. Carboxyl-carboxylate side chain interactions modulated by prevailing pH are a common feature of calcium-binding proteins, wherein a shared proton provides a stabilizing interaction when the metal cation is unavailable28,31. These carboxyl-carboxylate interactions have a higher pKa than a solitary carboxylate, permitting them to play a significant role at pH ranges closer to physiologic28,29. Studies of calsequestrin and calmodulin have revealed that protons are released upon calcium binding even at neutral pH, consistent with loss of carboxyl-carboxylate bonds28,31. A small decrease in SR luminal pH during calcium release has been observed32, and multiple groups have proposed that proton influx into the SR constitutes a small but important fraction of the counterion flow required to maintain charge neutrality when large calcium fluxes occur. Prior work showed a change in calsequestrin’s intrinsic fluorescence at a pH of 6.033, suggesting that dynamic effects on calsequestrin are not limited to the low pH regime used in our crystallization experiments. It is important to note that regulatory effects of dynamic SR pH are currently speculative and require further elucidation.
Until recently, CASQ2 disease variants were thought to be recessively inherited, with no strong genetic evidence for a dominant disease allele until the report of the K180R variants24. The recessive inheritance group corresponds to variants at or near the intra-dimer interface (e.g. R33Q, D307H, and P308L). Since filamentation is necessary for calsequestrin to remain in the jSR13–15, these intra-dimer interface mutants are assumed to be trafficked out of the jSR, leaving any remaining pool of wild type protein largely unaffected. By this model, filamentation defects overlap mechanistically with a class of calsequestrin-deficient conditions arising from null or hypomorphic alleles, all leading to a final common endpoint of decreased calcium-buffering capacity, and a resulting susceptibility to diastolic calcium leak.
In contrast to the traditional recessive genetic model of CASQ2-associated disease, the variants investigated in this study (S173I, K180R) are associated with dominant disease inheritance. These variants disrupt filamentation at the inter-dimer interface instead of the intra-dimer interface. The apparent discrepancy in inheritance patterns, mapping cleanly to distinct interfaces albeit with limited sampling, is striking and demands explanation. One possibility is that the reduced buffering power of dissociated calsequestrin dimers is sufficient on its own to cause penetrant disease. However, only 7 Yb atoms (a total of 28 per tetrameric assembly) are localized to the inter-dimer interface. If this interfacial/total cation ratio is similar for calcium, then the buffering power of dissociated dimers should remain approximately 75 % of the filament buffering power. Although a number of factors may affect the vivo buffering capacity of the filament, the fact that the unobserved acidic tail of each calsequestrin monomer is thought to bind close to half of the total bound cation pool10 means that in the most likely scenario, the true interfacial calcium fraction is even lower. Alternatively, it is tempting to propose that multimerization is poisoned in classical dominant-negative fashion, whereby dimers that contain a mutant protomer remain in the jSR and interfere with assembly. However, this disregards insights provided by studies of calsequestrin trafficking: unincorporated dimers should be just as susceptible as monomers to jSR export. Instead, we propose a combined mechanism that is more complex than a classical dominant negative effect: for as long as mutant-containing dimers may be present in the jSR, they interfere with multimerization, but crucially, when they leave, they “steal” half of the total WT protein with them. The result is somewhat paradoxical: insufficiency by way of underlying dominant negative biochemistry (Fig. 7). In sum, we can combine the present work with the existing rich body of calsequestrin research to explain the inheritance puzzle associated with CPVT-causing calsequestrin variants. Dimer-defective mutants produce monomers which are trafficked away, but the heterozygous state is rescued because WT protein is unaffected by the pool of defective monomeric protein. In contrast, variants that interfere with the inter-dimer interaction result in depletion of a substantial fraction of WT protein, as unincorporated dimers containing a mix of WT and mutant protein are continually lost. Future work should seek to confirm this hypothetical mechanism in cell-based assays.
Fig. 7: Heterozygous mutations at different calsequestrin interfaces display different disease inheritance patterns.
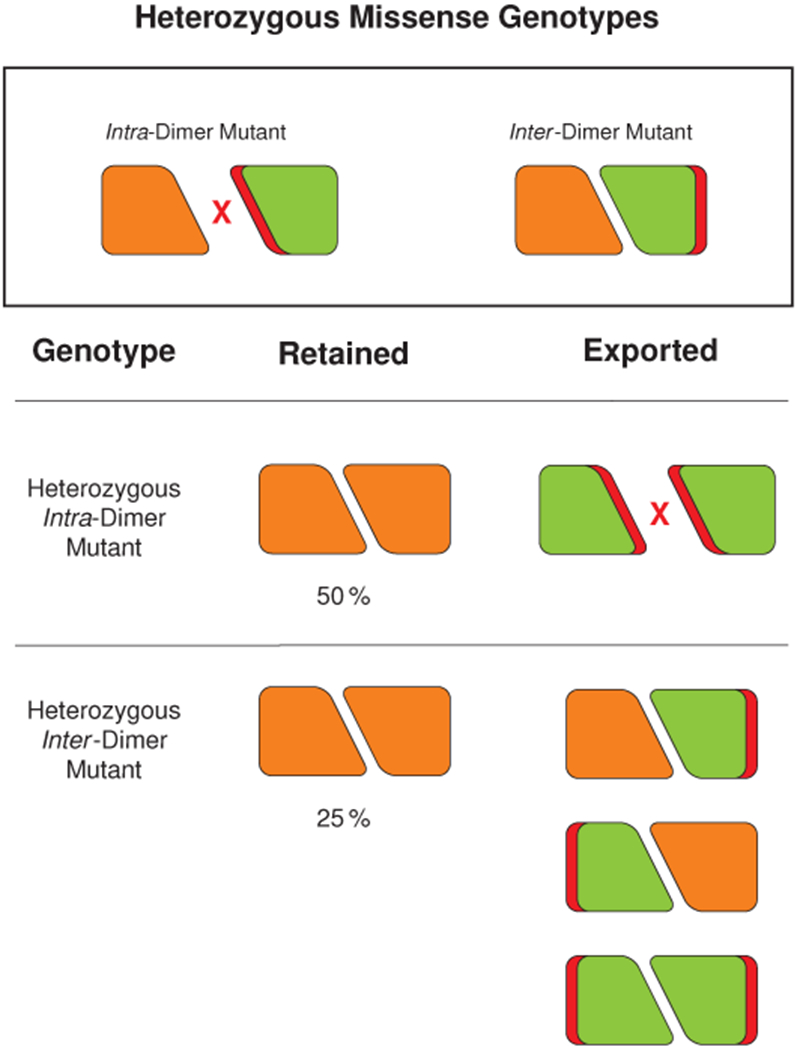
Mutations that inhibit dimerization are likely to cause penetrant disease only when carried recessively - a consequence of export of mutant monomers from the SR. However, mutations that inhibit inter-dimer interaction are likely to have dominant effects - a consequence of the fact that only 25 % of dimers remain able to participate in filament formation.
Materials and Methods
Human Subjects
The patient included in the study provided informed consent as part of a research protocol approved by the University of California, San Francisco Committee on Human Research. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Cloning and Generation of Plasmids
Full-length cardiac calsequestrin was cloned from human cardiac mRNA by reverse transcription, PCR, A-tailing of a PCR product, and TA ligation. A clone lacking the signal peptide sequence was subcloned by PCR and Gibson Assembly into a T7-based bacterial overexpression vector (pET28a) in front of a 6His site and TEV protease cleavage sequence. Point mutants were generated using the protocol from the Q5 Site-Directed Mutagenesis Kit (New England BioLabs), using either the Q5 or Phusion polymerases. All constructs were transformed in NEB Stable or XL-1 Blue E. coli, purified by miniprep, verified by Sanger sequencing, and retransformed into Rosetta (DE3)pLysS E. coli for overexpression. Primers used for cloning and mutagenesis are provided in (Supplementary Table 2). Selection for the pET28a vector was performed using 50 μg/L kanamycin. Selection for pLysS was performed by adding 25 μg/L chloramphenicol. Plasmids are available upon request to the corresponding author.
Expression and Purification of Cardiac Calsequestrin
pET28a-based expression constructs were transformed into Rosetta (DE3)pLysS E. coli. Overnight starter cultures were used to inoculate large cultures (typically 750 mL of broth per 2.8 L flask), which were grown to OD 0.4 and then induced with 0.25 mM IPTG. Upon induction, temperature was reduced from 37 °C to 24 °C. Cultures were grown for 6-9 hours post-induction or overnight and then spun down (optimal yields were observed from shorter durations of growth). All cultures were grown in standard LB in 50 μg/L kanamycin and 25 μg/L chloramphenicol. Pellets were resuspended in lysis buffer (20 mM Tris pH 7.4, 500 mM NaCl, 10 mM imidazole, 1 EDTA-free protease inhibitor tablet per 50 mL) and frozen at −80 °C. Frozen suspensions were thawed, sonicated on ice (5 min at 1 s on/1 s off), and clarified (15,000 g, 45 min, 4 °C). The clarified supernatant was filtered (0.2 μm), and calsequestrin-containing fractions were isolated by IMAC using a 5 mL HisTrap FF column attached to a GE Akta FPLC (IMAC Buffer A: 20 mM Tris pH 7.4, 500 mM NaCl, 10 mM imidazole; IMAC Buffer B: 20 mM Tris pH 7.4, 500 mM NaCl, 300 mM imidazole). Protein was eluted in 10% steps of Buffer B. The first eluted fraction (10% Buffer B) was always discarded (consistently observed to be impure as determined by SDS-PAGE). Remaining protein-containing fractions were pooled. TEV protease was added to the pooled fractions at a concentration of 1:40 by mass, and the protein was dialyzed overnight at 4 °C in TEV protease dialysis buffer (50 mM Tris pH 8.0, 0.5 mM EDTA, 1 mM DTT). The cleaved protein was further dialyzed for several hours in EDTA dialysis buffer (20 mM HEPES pH 7.3, 100 mM NaCl, 5 mM EDTA) and then overnight into Anion Exchange Buffer A (20 mM HEPES pH 7.3, 100 mM NaCl). Anion exchange polishing was performed using a HisTrap FF column in series with 3×1 mL Mono Q columns. (Buffer A: 20 mM HEPES pH 7.3, 100 mM NaCl; Buffer B: 20 mM HEPES pH 7.3, 1 M NaCl). Protein was eluted in a continuous gradient up to 100% Buffer B), with calsequestrin-rich fractions consistently eluting at 40–50% Buffer B. Fractions were collected and analyzed for purity by SDS-PAGE and A260/A280 ratio. Fractions that were optimally pure and free of A260 contamination were pooled, concentrated to 20 mg/mL, and frozen at −80 °C. Alanine mutants were purified as described above, except that phosphate IMAC buffers were employed (Buffer A: 20 mM phosphate buffer at pH 7.4, 500 mM NaCl, 10 mM imidazole; Buffer B: 20 mM phosphate buffer at pH 7.4, 500 mM NaCl, 300 mM imidazole), and an on-column high-salt wash was performed (20 mM phosphate buffer at pH 7.4, 2 M NaCl). In addition, the TEV protease dialysis buffer used for these purifications contained 100 mM NaCl.
Crystallization of Cardiac Calsequestrin
Crystallization screens were carried out in 96-well hanging-drop format and monitored using a Formulatrix Rock Imager automated imaging system. Conditions conducive to crystal growth were optimized and then reproduced in a 24-well format. The best diffraction was obtained by mixing thawed protein (10–20 mg mL−1 in 20 mM HEPES pH 7.3, 400–500 mM NaCl) 1:1 with 15% PEG 4000 and 400 mM Li2SO4. The pH of the PEG 4000 solution used to produce the best-diffracting crystals was tested by litmus paper and found to be approximately 3–3.5. Despite the presence of 20 mM HEPES in the protein reagent, the pH of the drops in which crystals grew was controlled by the PEG and remained 3–3.5. Freshly made PEGs were incompatible with calsequestrin crystal growth except when concentrated HCl was added to the mother liquor, producing crystals similar to those observed with benchtop-aged PEGs. Interestingly, only unbuffered conditions yielded crystals. Multiple attempts to grow crystals at a buffered low pH (using acetate or glycine-based buffers) failed.
Ytterbium Soak of Cardiac Calsequestrin Crystals
We initially attempted to identify calcium sites using anomalous signal from calcium (CaCl2) added to the crystallization condition described above. We were unsuccessful, likely due to a combination of several factors. The calcium absorption edge is unreachable with conventional tunable x-ray sources and in normal atmosphere; thus, it can only be approached, with resulting weakened anomalous signal. In addition, calsequestrin has an average K d for calcium of 1 mM. Thus, occupancy at a typical site would be expected to be lower as compared to other calcium-binding proteins. Presence of sulfate in the crystallization condition limited calcium concentrations to approximately 14 mM and below, above which a precipitate was observed. Although this limit is above the Kd, it was insufficient for robust anomalous signal. Crystals of calsequestrin that formed in trace calcium were therefore soaked in YbCl3. Hanging drops containing calsequestrin crystals were uncovered and an Eppendorf Microloader was used to inject 2 μL crystallization drops (1 μL protein and 1 μL mother liquor) with 200 μL of 2 M YbCl3. Data were collected within 5 minutes with no back-soaking.
Crystal Data Collection and Structure Determination
Hanging drops were uncovered and submerged in a drop of Parabar 10312 (previously known as Paratone). For Yb soaks, Yb was quickly injected prior to application of the oil. Crystals were looped and pulled through the oil. Excess oil was blotted away, and the loop was mounted directly into the cryostream of the Tom-Alber-Tron endstation at ALS beamline 8.3.1. Frames were collected at 1.116 keV (1.386 keV for Yb-soaked crystals) using the endstation’s Pilatus3 S 6M detector with a strategy designed to balance redundancy against radiation damage.
Structure Determination
Diffraction images were processed with xia2 using the DIALS integration pipeline and a resolution cutoff of CC1/2 > 0.334–38. For the native structure, the merged diffraction intensities were used to find a molecular replacement solution in Phaser39 with the previously published canine cardiac calsequestrin structure, 1SJI10, serving as the initial model. This resulted in a solution in space group P4322 containing one calsequestrin chain per AU. This solution was refined in PHENIX40 with PHENIX AutoBuild40–44, with extensive manual model-building in Coot45. For the Yb-complexed dataset, data were processed as above but with preservation of anomalous signal (no merging of Friedel pairs). The refined native structure was used a molecular replacement model, and a solution was found in space group P43212 containing a dimer in the AU. This solution refined poorly. The Yb-complexed dataset was reprocessed in P1, and a molecular replacement solution was found in P1 with 16 chains in the AU. Refinement of this model was tested using Zanuda46, and the best R-free was found to be in space group P1211. The Yb-complexed dataset was reprocessed in space group P1211, and a molecular replacement solution was found with 8 chains in the AU. This solution refined well. The anomalous map was used in refinement to place Yb atoms in the structure.
Turbidity Assays
Recombinant protein samples were thawed, diluted in 2 mL–3 mL of Turbidity Assay Buffer (15 mM Tris pH 7.4, 20 mM NaCl, 85 mM KCl) and dialyzed in Turbidity Assay Buffer plus 10 mM EDTA. Samples were then re-dialyzed overnight in the same buffer without EDTA. Protein A280 was measured in triplicate (Nanodrop) using the appropriate matching buffer as background, and protein was diluted to 2.25 μM in a 140 μL volume in half-area wells of a μClear 96-well plate. The plate was covered, and protein in the wells was allowed to equilibrate on the benchtop for 20 minutes. The turbidity assay was performed using a BioTek Synergy 2 plate reader equipped with reagent injectors. Seven μL of 20 mM CaCl2 solution was injected into each well for a final concentration of approximately 1 mM, and the plate underwent shaking for 20 s. Absorbance at 350 nm was monitored for 45 min. The protocol was performed in plate synchronized mode for consistent well-to-well timing. A 100 mM ion-selective electrode calcium standard (Sigma, cat no. 21059) stock solution was used for all CaCl2 dilutions.
In the assessment of the response of calsequestrin assemblies to EDTA, slightly different conditions were used. The Turbidity Assay Buffer contained Tris at (15 mM Tris pH 7.2 and contained no potassium. This assay was performed in a black plate with μClear bottom. The well-synchronized plate reader mode was used to rapidly measure the effect of EDTA addition. After addition of CaCl2 to a final concentration of 1 mM, absorbance at 350 nm was monitored. Monitoring of absorbance continued as EDTA was then added to a final concentration of 1 mM using the plate reader’s reagent injector module.
Continuum Electrostatics
Atomic coordinates of a calsequestrin dimer-dimer complex were prepared using PDB2PQR47. The Assisted Model Building with Energy Refinement (AMBER) force field was used48. Electrostatic calculations were performed by The Adaptive Poisson-Boltzmann Solver (APBS)49 using the nonlinear Poisson-Boltzmann equation. Relevant conditions were a protein relative dielectric of 2, a solvent dielectric of 78.5, 150 mM monovalent cation/anion, and a temperature of 25 °C.
Visualization and Statistical Analysis
Protein structure figures were generated using PyMOL50. The interior cavity of the calsequestrin filament was traced using HOLLOW51. Sequence alignments were generated using TEXshade52. Plots were generated using python matplotlib53. Data points in figures represent mean values, with error bars representing standard deviation. All turbidity assay data points are mean of 3 technical replicates.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data Availability
The structures determined as part of this work are deposited in the Protein Data Bank (PDB) under identifiers 6OWV (native) and 6OWW (ytterbium-soaked). The raw diffraction dataset for the native structure is deposited in Zenodo under doi:10.5281/zenodo.2941360. The raw diffraction dataset for the ytterbium-complexed structure is likewise deposited in Zenodo under doi:10.5281/zenodo.2943248. Source data for all plots are provided with the manuscript online. All data presented in the manuscript are also available at https://github.com/errontitus/casq2-structure-function.
Code Availability
Code to generate all results and figures from the manuscript is available at https://github.com/errontitus/casq2-structure-function. The manuscript and all figure layouts were constructed entirely in LATEX using PGF/TikZ.
Extended Data
Extended Data Figure 1: Turbidity assay controls; related to Fig. 1.
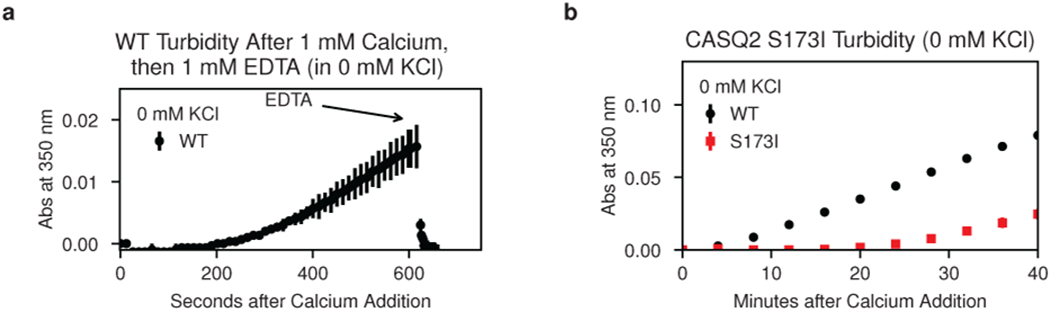
a, Stoichiometric addition of EDTA demonstrates immediate reversal of calcium-induced turbidity. b, Turbidity assay for the S173I mutant in 0 mM KCl. Error bars represent the mean ± s.d of n=3 technical replicates.
Extended Data Figure 2: The inter-dimer interface of the new candidate cardiac calsequestrin filament exhibits all-by-all contacts and greater buried surface area (BSA) compared to all other published calsequestrin structures; related to Fig. 2.
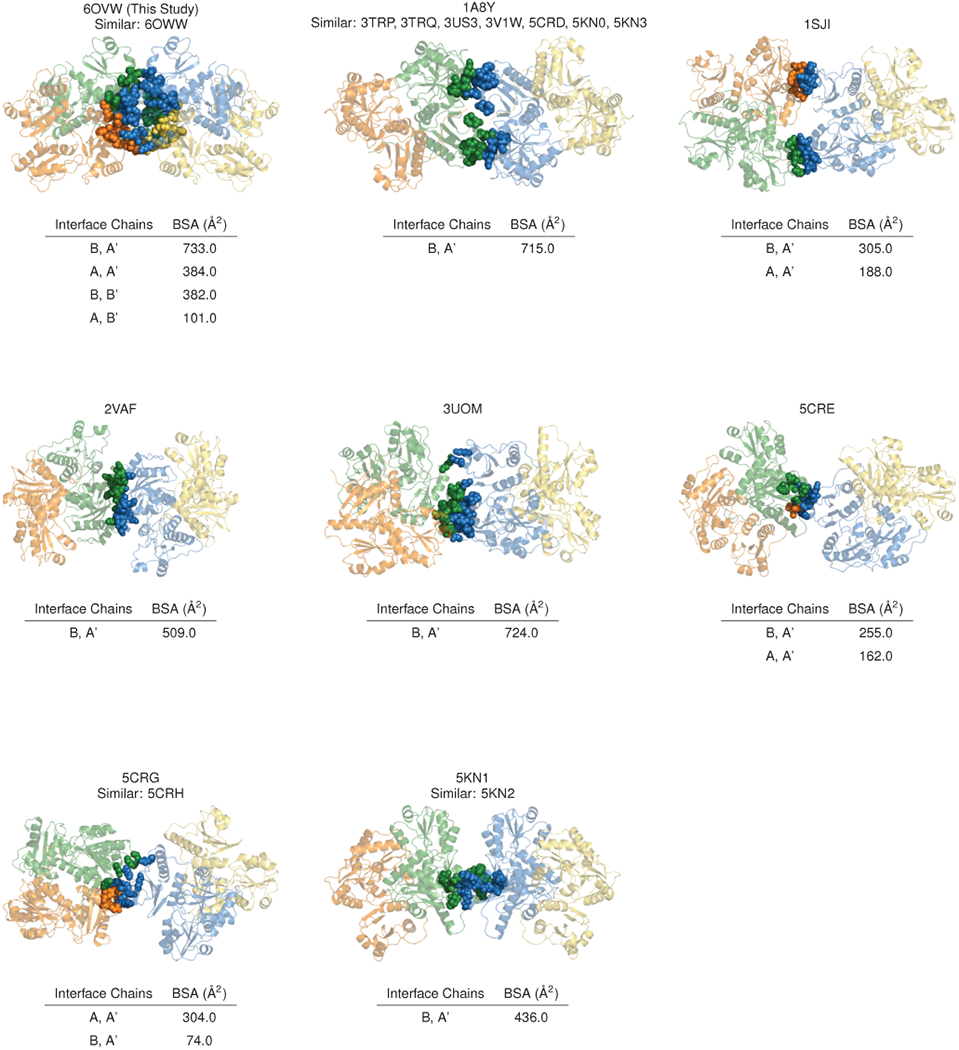
For each published calsequestrin structure, the inter-dimer interface with the greatest buried surface area is shown. Residues with buried surface area at the interface are rendered as spheres. Where similar PDB codes are listed, inter-dimer interfaces are roughly isomorphous with the example structure shown, although the space group and unit cell used to determine the structure sometimes differ.
Extended Data Figure 3: The 3-Helix configuration of the new filament candidate promotes close packing of thioredoxin domains.
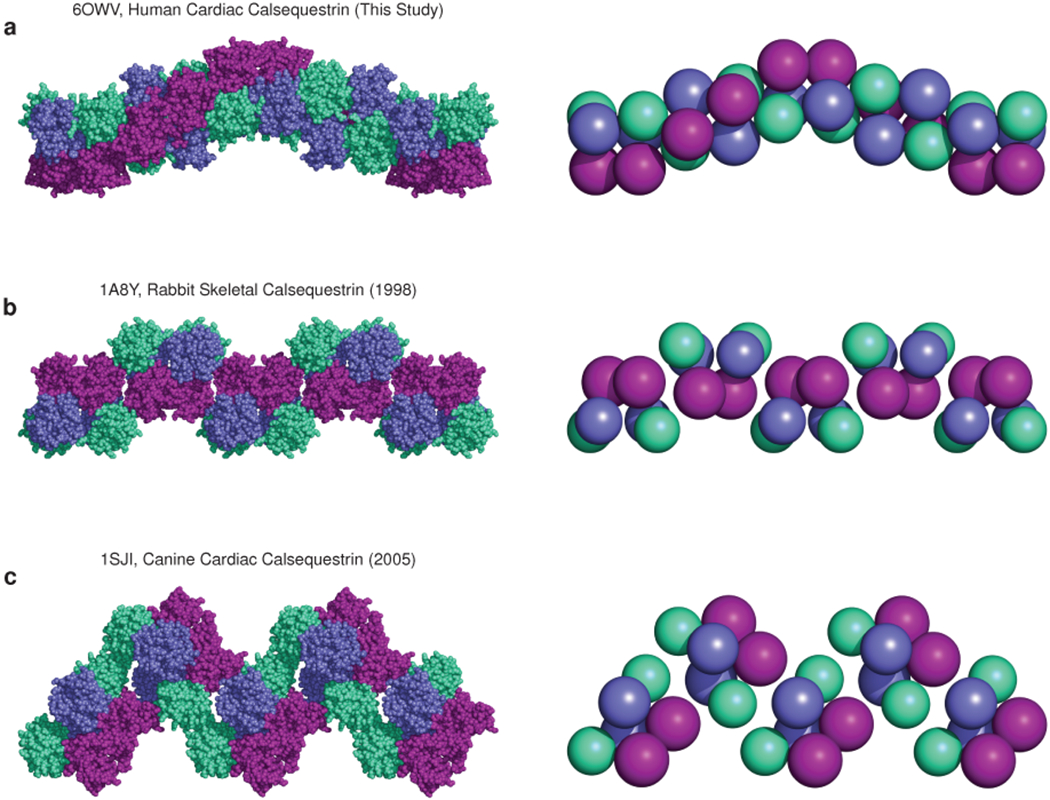
a, The candidate cardiac calsequestrin filament assembled from crystallographic symmetry operations on PDB ID 6OWV (human CASQ2, this study). The filament exhibits tight packing of protomers and thioredoxin domains (shown on the right using equal-size spheres placed at the center of mass of each thioredoxin domain). b, A putative skeletal calsequestrin filament assembled from crystallographic symmetry operations on PDB ID 1A8Y (rabbit CASQ1, 1998). Right-side: equal-size spheres represent thioredoxin domains. c, A putative skeletal calsequestrin filament assembled from crystallographic symmetry operations on PDB ID 1SJI (canine CASQ2, 2005). Right-side: equal-size spheres represent thioredoxin domains.
Extended Data Figure 4: Electron density and anomalous difference maps for Yb-binding sites at the cardiac calsequestrin intra-dimer interface; related to Fig. 3.
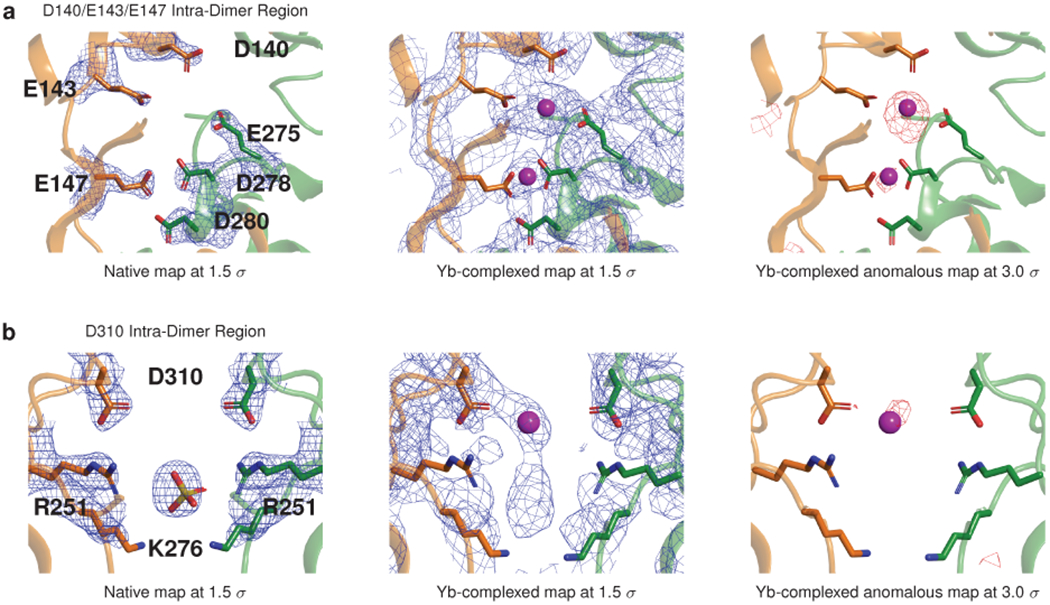
a, Electron density (blue mesh) and anomalous difference maps for the D140-E143-E147 region. b, Electron density and anomalous difference maps for the D310 region.
Extended Data Figure 5: Dimer overlays reveal that calsequestrin structures can be classified into tightly packed or loosely packed dimers; related to Fig. 3.
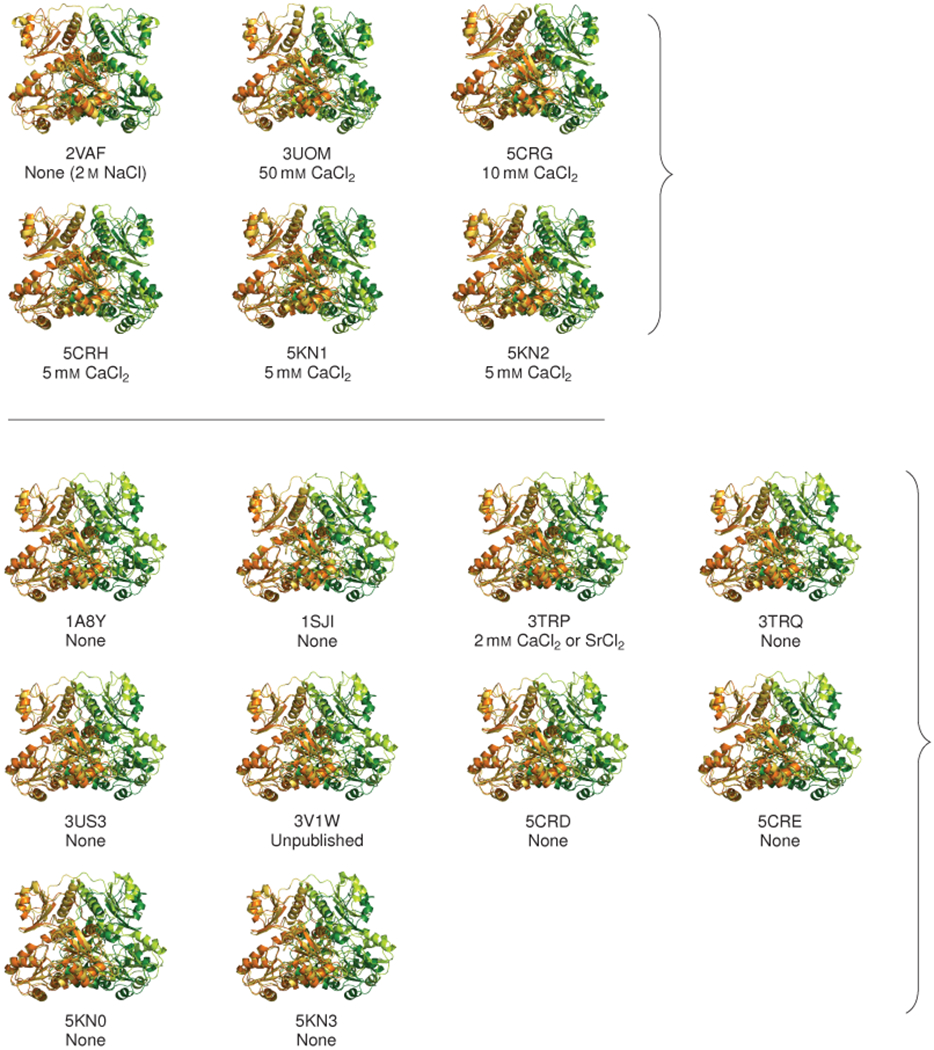
Dimers from published calsequestrin structures (lighter orange and green) are overlaid onto the tightly packed dimer from the present study (6OVW, darker orange and green). In each dimer pair, chain A is aligned to chain A to illustrate the relative displacement of chain B. The concentration of divalent cations used in the crystallization conditions is noted below. The overlays reveal two distinct conformational groupings. The more tightly packed conformation with inwardly rotated chains resembles the dimer in this study and appears to form at low pH or in the presence of neutralizing divalents.
Extended Data Figure 6: Tightly packed calsequestrin dimers consistently exhibit increased conformational disorder in domain I; related to Fig. 3.

The top panel shows tightly packed calsequestrin dimers (i.e. dimers of calsequestrin crystallized in low pH or with high concentration of multivalent cations). In these structures, solvent-exposed loops in domain I are consistently disordered. In PDB 6OVW, the disordered loop region is omitted due to the high level of disorder. This same region (boxed, residues 58-68) is highly disordered in similar structures. The bottom panel shows loosely packed calsequestrin dimers (i.e. dimers of calsequestrin crystallized at neutral pH with low or trace concentrations of multivalent cations). The resolution for each structure is indicated, and several structures of non-comparable resolution are excluded (2VAF, 5CRE, 5KN0).
Extended Data Figure 7: Electron density and anomalous difference maps for Yb-binding Sites at the cardiac calsequestrin filament’s inter-dimer interface; related to Fig. 4.
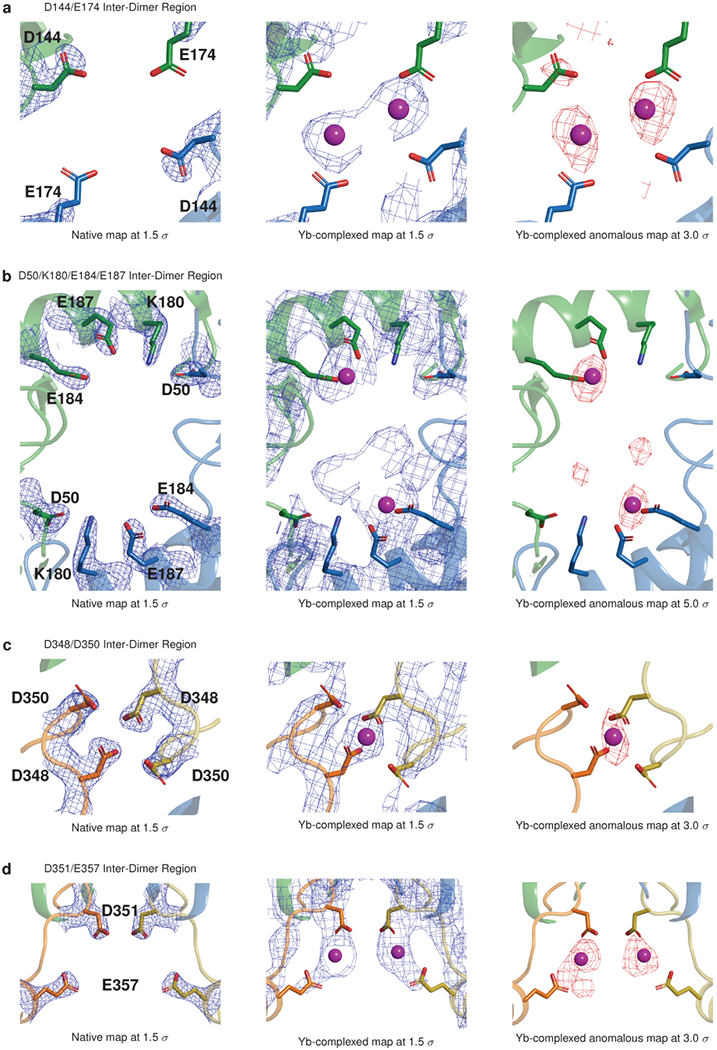
a, Electron density and anomalous difference maps for the D144-E174 region of interest. b, Electron density and anomalous difference maps for the D50-K180-E184-E187 region. c, Electron density and anomalous difference maps for the D348-D350 region. d, Electron density and anomalous difference maps for the D351-E357 region.
Extended Data Figure 8: Turbidity assays showing effect of alanine mutagenesis of additional Yb-binding sites at the cardiac calsequestrin inter-dimer interface; related to Fig. 4.
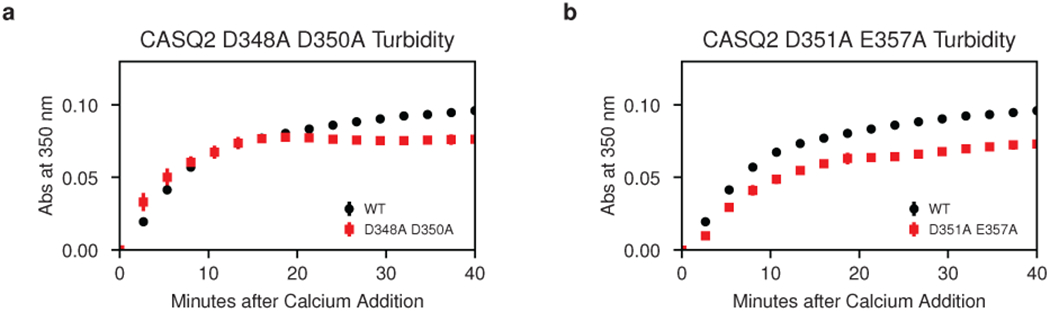
a, Turbidity assay after alanine mutagenesis of the putative calcium-coordinating residues D348 and D350. b, Turbidity assay after alanine mutagenesis of the putative calcium-coordinating residues D351 and E357. Error bars represent the mean ± s.d of n=3 technical replicates.
Extended Data Figure 9: Electron density map for a hydrophilic pocket at the cardiac calsequestrin filament’s inter-dimer interface; related to Fig. 6.
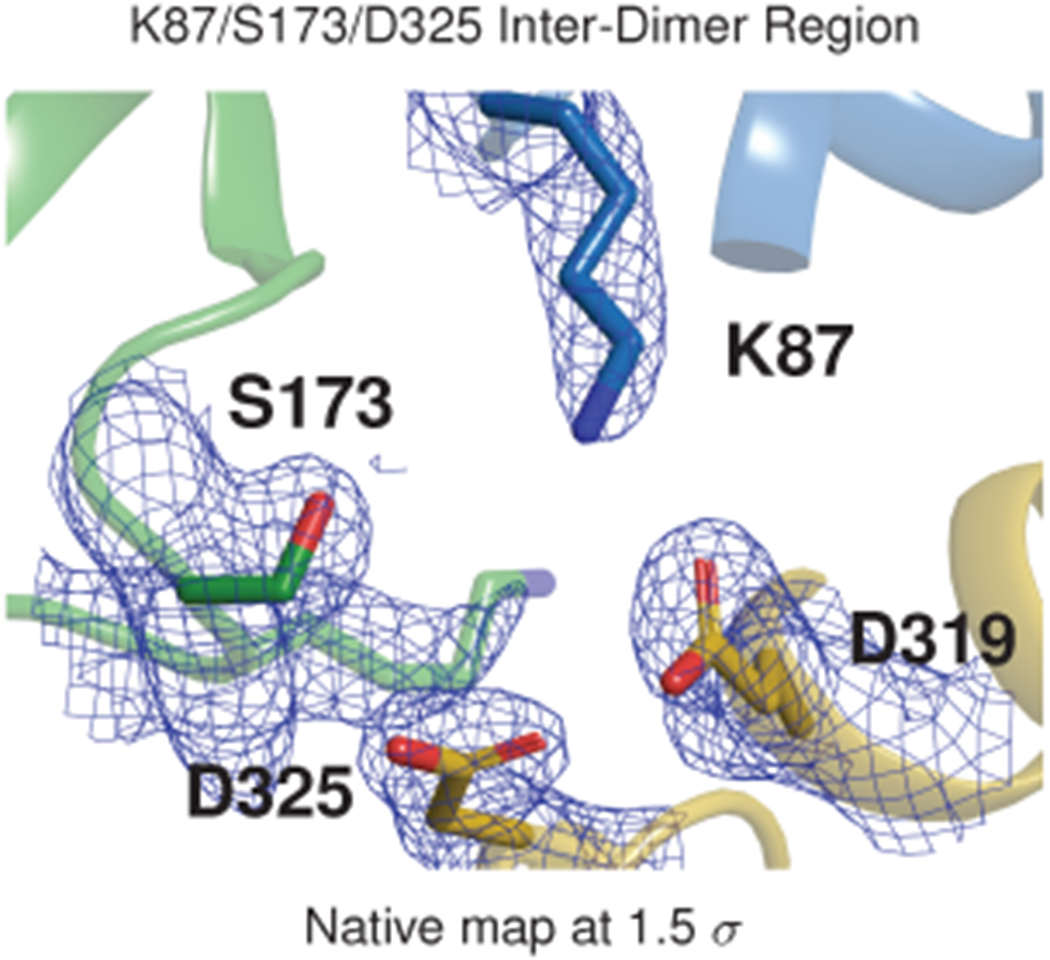
The S173 inter-dimer region of the calsequestrin filament with electron density shown as a blue mesh.
Supplementary Material
Acknowledgments
We thank J. Roberts for expert clinical review. We thank C. Agnew, J. Fraser, J. Holton, L. Liu, M. Lolicato, B. Mensa, and T. Thaker for technical assistance with crystallographic data collection and processing. We thank L. Liu and T. Thaker for technical assistance with protein preparation and crystallization screens. We thank M. Grabe, A. Kao, and O. Rosenberg for helpful discussions.
This work was supported by NIH/NHLBI grant F30HL137329 (E.W.T.), a Sarnoff Foundation Fellowship (E.W.T), NIH/NIGMS grant T32GM007618 to the UCSF Medical Scientist Training Program (E.W.T), NIH/NHLBI grant DP2HL123228 (R.C.D.), and American Heart Association grant 17IRG33460152 (R.C.D.).
Beamline 8.3.1 at the Advanced Light Source is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15-328599, the National Institutes of Health (R01 GM124149 and P30 GM124169), Plexxikon Inc., and the Integrated Diffraction Analysis Technologies program of the US Department of Energy Office of Biological and Environmental Research. The Advanced Light Source (Berkeley, CA) is a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy under contract number DEAC02-05CH11231, Office of Basic Energy Sciences.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- 1.Bers DM Macromolecular complexes regulating cardiac ryanodine receptor function. J. Mol. Cell. Cardiol 37, 417–429 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Royer L & Ríos E Deconstructing calsequestrin. Complex buffering in the calcium store of skeletal muscle. J. Physiol 587, 3101–3111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guerrero-Hernández A et al. in Calcium Signaling (ed Islam MS) 337–370 (Springer International Publishing, 2020). [Google Scholar]
- 4.MacLennan DH & Wong PT Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc. Natl. Acad. Sci. U. S. A 68, 1231–1235 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacLennan DH Isolation of a second form of calsequestrin. J. Biol. Chem 249, 980–984 (1974). [PubMed] [Google Scholar]
- 6.Ostwald TJ & MacLennan DH Isolation of a high affinity calcium-binding protein from sarcoplasmic reticulum. J. Biol. Chem 249, 974–979 (1974). [PubMed] [Google Scholar]
- 7.Costello B et al. Characterization of the junctional face membrane from terminal cisternae of sarcoplasmic reticulum. J. Cell Biol 103, 741–753 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franzini-Armstrong C, Kenney LJ & Varriano-Marston E The structure of calsequestrin in triads of vertebrate skeletal muscle: a deep-etch study. J. Cell Biol 105, 49–56 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang S et al. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat. Struct. Biol 5, 476–483 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Park H et al. Comparing skeletal and cardiac calsequestrin structures and their calcium binding: a proposed mechanism for coupled calcium binding and protein polymerization. J. Biol. Chem 279, 18026–18033 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Knollmann BC et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Invest 116, 2510–2520 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perni S, Close M & Franzini-Armstrong C Novel details of calsequestrin gel conformation in situ. J. Biol. Chem 288, 31358–31362 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milstein ML, Houle TD & Cala SE Calsequestrin isoforms localize to different ER sub-compartments: evidence for polymer and heteropolymer-dependent localization. Exp. Cell Res 315, 523–534 (2009). [DOI] [PubMed] [Google Scholar]
- 14.McFarland TP, Milstein ML & Cala SE Rough endoplasmic reticulum to junctional sarcoplasmic reticulum trafficking of calsequestrin in adult cardiomyocytes. J. Mol. Cell. Cardiol 49, 556–564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knollmann BCA “rough” journey to the sarcoplasmic reticulum–implications of altered calsequestrin trafficking for cardiac arrhythmia. J. Mol. Cell. Cardiol 49, 554–555 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanchez EJ, Lewis KM, Munske GR, Nissen MS & Kang C Glycosylation of skeletal calsequestrin: implications for its function. J. Biol. Chem 287, 3042–3050 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirchhefer U et al. The human CASQ2 mutation K206N is associated with hyperglycosylation and altered cellular calcium handling. J. Mol. Cell. Cardiol 49, 95–105 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, Kelley J, Schmeisser G, Kobayashi YM & Jones LR Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem 272, 23389–23397 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Rani S, Park CS, Sreenivasaiah PK & Kim DH Characterization of Ca2+-Dependent Protein-Protein Interactions within the Ca2+ Release Units of Cardiac Sarcoplasmic Reticulum. Mol. Cells 39, 149–155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Handhle A et al. Calsequestrin interacts directly with the cardiac ryanodine receptor luminal domain. J. Cell Sci 129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis KM, Ronish LA, Ríos E & Kang C Characterization of Two Human Skeletal Calsequestrin Mutants Implicated in Malignant Hyperthermia and Vacuolar Aggregate Myopathy. J. Biol. Chem 290, 28665–28674 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bal NC et al. The catecholaminergic polymorphic ventricular tachycardia mutation R33Q disrupts the N-terminal structural motif that regulates reversible calsequestrin polymerization. J. Biol. Chem 285, 17188–17196 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bal NC et al. Probing cationic selectivity of cardiac calsequestrin and its CPVT mutants. Biochem. J 435, 391–399 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Gray B et al. A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 13, 1652–1660 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim E et al. Characterization of human cardiac calsequestrin and its deleterious mutants. J. Mol. Biol 373, 1047–1057 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Sanchez EJ, Lewis KM, Danna BR & Kang C High-capacity Ca2+ binding of human skeletal calsequestrin. J. Biol. Chem 287, 11592–11601 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis KM et al. Characterization of Post-Translational Modifications to Calsequestrins of Cardiac and Skeletal Muscle. Int. J. Mol. Sci 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krause KH, Milos M, Luan-Rilliet Y, Lew DP & Cox JA Thermodynamics of cation binding to rabbit skeletal muscle calsequestrin. Evidence for distinct Ca2+- and Mg2+-binding sites. J. Biol. Chem 266, 9453–9459 (1991). [PubMed] [Google Scholar]
- 29.Sawyer L & James MNG Carboxyl–carboxylate interactions in proteins. Nature 295, 79–80 (1982). [DOI] [PubMed] [Google Scholar]
- 30.Frederick KK, Marlow MS, Valentine KG & Wand AJ Conformational entropy in molecular recognition by proteins. Nature 448, 325–329 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milos M, Schaer JJ, Comte M & Cox JA Calcium-proton and calcium-magnesium antagonisms in calmodulin: microcalorimetric and potentiometric analyses. Biochemistry 25, 6279–6287 (1986). [DOI] [PubMed] [Google Scholar]
- 32.Kamp F, Donoso P & Hidalgo C Changes in luminal pH caused by calcium release in sarcoplasmic reticulum vesicles. Biophys. J 74, 290–296 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hidalgo C, Donoso P & Rodriguez PH Protons induce calsequestrin conformational changes. Biophys. J 71, 2130–2137 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 34.Winter G et al. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr D Struct Biol 74, 85–97 (February 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winter G xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr 43, 186–190 (2010). [Google Scholar]
- 36.Evans PR & Murshudov GN How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans P Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr 62, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (August 2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (February 2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Afonine PV et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terwilliger T SOLVE and RESOLVE: automated structure solution, density modification and model building. Journal of Synchrotron Radiation 11, 49–52 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Terwilliger TC et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr 64, 61–69 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zwart PH, Grosse-Kunstleve RW & Adams PD Xtriage and Fest: automatic assessment of X-ray data and substructure structure factor estimation. CCP4 Newsl 43, 27–35 (2005). [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (April 2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lebedev AA & Isupov MN Space-group and origin ambiguity in macromolecular structures with pseudo-symmetry and its treatment with the program Zanuda. Acta Crystallogr. D Biol. Crystallogr 70, 2430–2443 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Dolinsky TJ, Nielsen JE, McCammon JA & Baker NA PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–7 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cornell WD et al. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc 117, 5179–5197 (1995). [Google Scholar]
- 49.Jurrus E et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schrödinger LLC. The PyMOL Molecular Graphics System, Version 2.2.3 (2019). [Google Scholar]
- 51.Ho BK & Gruswitz F HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct. Biol 8, 49 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beitz E TeXshade: shading and labeling of multiple sequence alignments using LaTeX2e. Bioinformatics 16, 135–139 (2000). [DOI] [PubMed] [Google Scholar]
- 53.Hunter JD Matplotlib: A 2D graphics environment. Computing In Science & Engineering 9, 90–95 (2007). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structures determined as part of this work are deposited in the Protein Data Bank (PDB) under identifiers 6OWV (native) and 6OWW (ytterbium-soaked). The raw diffraction dataset for the native structure is deposited in Zenodo under doi:10.5281/zenodo.2941360. The raw diffraction dataset for the ytterbium-complexed structure is likewise deposited in Zenodo under doi:10.5281/zenodo.2943248. Source data for all plots are provided with the manuscript online. All data presented in the manuscript are also available at https://github.com/errontitus/casq2-structure-function.