

Abstract
Over the past decade, large-scale genetic studies have successfully identified hundreds of genetic variants robustly associated with risk for psychiatric disorders. However, mechanistic insight -- and clinical translation -- continues to lag the pace of risk variant identification, hindered by the sheer number of targets, their predominant non-coding localization, as well as pervasive pleiotropy and incomplete penetrance. Successful next steps require identification of ‘causal’ genetic variants and their proximal biological consequences; placing variants within biologically-defined functional contexts, reflecting specific molecular pathways, cell-types, circuits, and developmental windows; and characterizing the downstream, convergent neurobiological impact of polygenicity within an individual. Here, we discuss opportunities and challenges of high-throughput transcriptomic profiling in human brain, and how transcriptomics can help pinpoint mechanisms underlying genetic risk for psychiatric disorders at a scale necessary to tackle daunting levels of polygenicity. These include transcriptome-wide association studies (TWAS) and related approaches for candidate risk gene prioritization through integration of GWAS with expression quantitative trait loci (eQTL). We outline transcriptomic results informing our understanding of the brain-level molecular pathology of psychiatric disorders, including autism (ASD), bipolar disorder (BD), major depression (MDD), and schizophrenia (SCZ). Finally, we discuss systems-level approaches for integration of distinct genetic, genomic and phenotypic levels, including combining spatially-resolved gene expression and human neuroimaging maps. Results highlight the importance of understanding gene expression (dys)regulation across human brain development as a major contributor to psychiatric disease pathogenesis, from common variants acting as expression quantitative trait loci (eQTL) to rare variants enriched for gene expression regulatory pathways.
Keywords: transcriptomics, genomics, GWAS, brain, coexpression, Schizophrenia, autism
Introduction
A major barrier to the treatment of psychiatric disorders is our limited understanding of pathogenic mechanisms across molecular, cellular, and systems levels. For most disorders, the majority of liability is mediated by heritable genetic variation, thereby providing a tractable framework for gaining neurobiological, mechanistic insights (1). Accordingly, large-scale genetic studies have made tremendous gains identifying thousands of risk variants. However, biological interpretation of these variants is challenged by predominant localization in non-coding regions (3), substantial linkage disequilibrium (LD), pleiotropy, incomplete penetrance, and daunting polygenicity. Whereas brain non-coding regions were previously poorly annotated, now the opposite problem is true--there are many functional annotations for a given variant. These challenges culminate in ~1000s of small effect-size risk alleles with unknown function which can only be confidently localized to a given LD block (5).
As variant discovery continues on its exponential trajectory, biological interpretation is now the major obstacle impeding translation. Mechanistic insight requires finding robust ‘causal’ variant(s), identifying the biological effect of a variant (e.g., putative target gene), understanding how multiple variants converge onto specific downstream molecular pathways, and finally understanding how an individual’s variants aggregate to mediate phenotypic risk. Here, we discuss how the transcriptome--the collection of RNAs expressed in a given cell/tissue--represents a proximal, quantitative readout enabling mechanistic interrogation of the biological impact of genetic variation, individually or in aggregate, across both clinical and experimental settings (Figure 1).
Figure 1 --. A transcriptomic framework for mechanistic dissection of complex psychiatric traits.
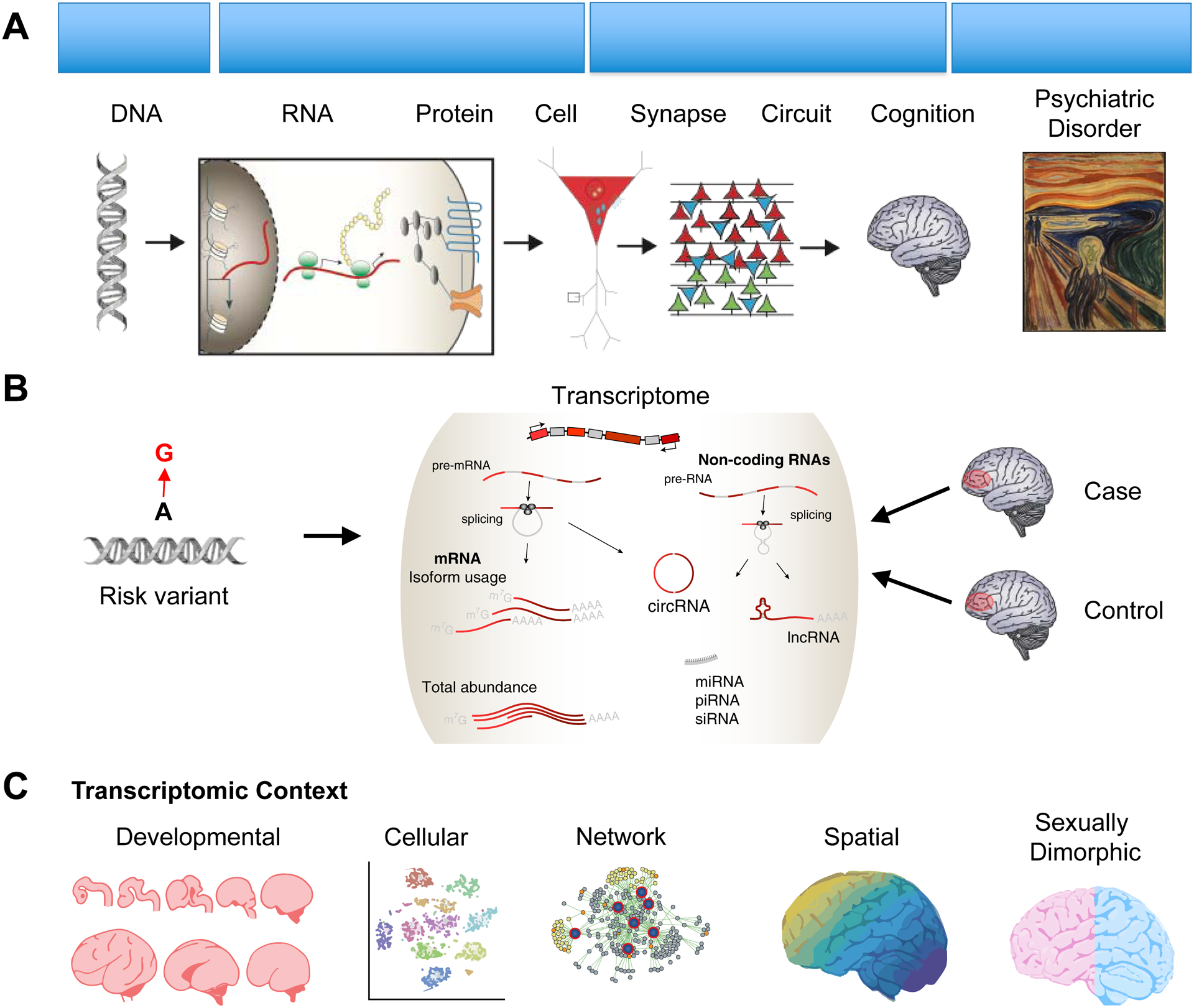
A) A hierarchical neurobiological organization linking genotype to phenotype. B) Two main approaches are highlighted. Left, disease-associated genetic risk variants can be integrated with gene expression measures through transcriptome-wide association studies (TWAS) and related approaches to prioritize proximal biological mechanisms, particularly for non-coding variants. Right, differential gene expression analyses of brain tissue from subjects with psychiatric disorders compared with controls can be used to identify a reactive, brain-level molecular pathology of disease. C) Gene expression patterns are highly dynamic and interpretation requires understanding the relevant biological context.
Tissue Considerations
Transcriptomics has received renewed focus in psychiatry, particularly in the post-GWAS era. The etiologically relevant tissue for psychiatric traits is the human brain (7), which shows complex gene expression patterns due to the large number of expressed genes, including non-coding RNAs, and abundant alternative splice isoforms (4,10). More accessible tissues, such as blood, fail to recapitulate these complexities (6); only ~2/3 of brain-expressed genes are detected in blood, which undergo less splicing to yield fewer isoforms (7). Post-mortem brain samples have been used to fill this gap, but are not without challenges, including limited access, ascertainment, agonal and degradation effects (13). Interpretation is further complicated by potential reverse causality, hidden confounding, and pleiotropic effects. Nevertheless, the transcriptomic architecture of the human brain has demonstrated remarkable stability across cortical regions, individuals, and studies, including its genetic regulation (4,15,16). Further, such challenges can be mitigated, in part, by associating expression changes with germline variation, across large numbers of individuals, providing a directional anchor. Finally, methods for generating neurons and 3D cortical organoids from patient-derived human induced pluripotent stem cells (hiPSCs) can now be performed at the necessary scale to fill some of these gaps, although there are still key biological differences between these models and true brain development (9,11).
Transcriptomic Methods
RNA-sequencing enables accurate, quantitative transcriptomic profiling with wide dynamic range, including across protein-coding and non-coding genes, splicing events, and unannotated genomic regions (19,21,23). Recent developments in single-cell and long-read sequencing enable interrogation of individual cells and full-length isoforms, respectively (21). RNA-seq quantifications, however, are relative and often biased by technical factors (Figure 2), including PCR amplification, GC content, RNA quality, gene and mitochondrial mapping rates, gene body coverage uniformity, among others (25,27). The importance of experimental batch correction was recognized for microarrays (29), and with RNA-seq, these can be introduced at many steps. To enable direct comparison, libraries should be processed together in parallel, and multiplexed in random groups where possible (31). RNA quality of most human brains is lower than cell lines or tissues from experimental models. Proper detection, visualization, and correction for such factors should therefore be a critical aspect of any workflow (12,25). Achieving an appropriate balance between under- and over-correction remains a challenge, as many commonly used latent variable approaches are effective but also prone to capture true biological signal (34,36,38).
Figure 2. Factors influencing gene expression patterns across cortical brain samples in GTEx.
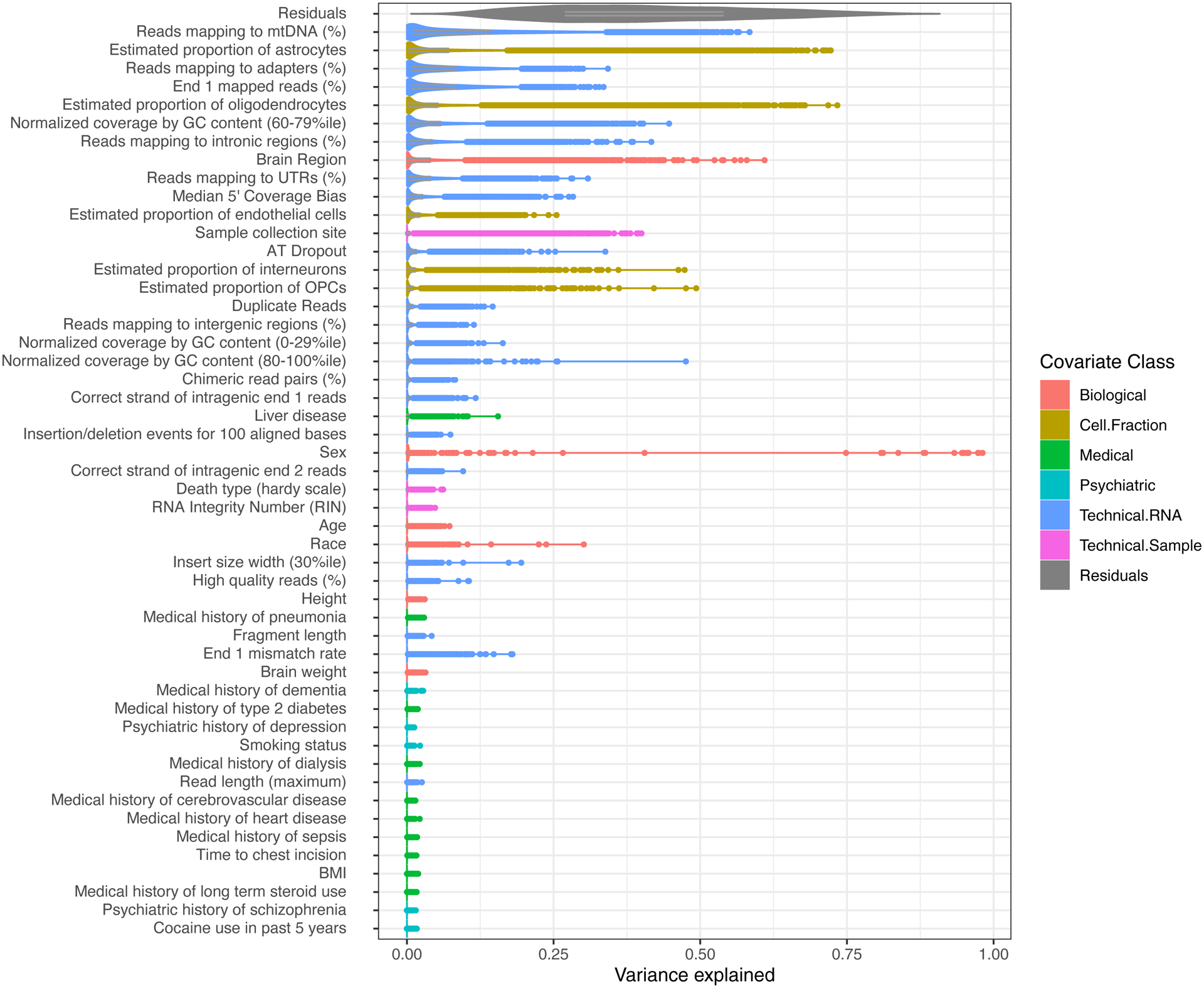
Sequencing related technical factors, along with estimated cell proportions, are top drivers of expression variance across anterior cingulate and frontal cortex brain samples in GTEx (v8; n=629 samples). Sequencing-related technical covariates were computed using PicardTools (“collectMultipleMetrics”) and combined with sample and subject-level metadata provided GTEx. Collinear covariates were removed and gene expression variance explained by each covariate was calculated using a linear mixed effects model using the variancePartition package (28). Sample specific proportions for major cortical cell-types were estimated with Bisque using single-nucleus RNA-seq data as a reference (56,57)
The human brain transcriptome exhibits a robust, hierarchical organization of co-expression patterns, reflecting cell-types, subcellular organelles and region-, sex-, and age-specific processes (15,16,40,42). Dimensionality reduction techniques can capture co-expression ‘modules’ reflecting these processes, boosting power and facilitating interpretability (44). Weighted gene correlation network analysis (WGCNA) is a popular unsupervised approach (46), although many methods are available (48,50). As subtle shifts in cellular proportion contribute are among the largest source of expression variation across samples (Figure 2), particularly in brain (24), the most connected (‘hub’) genes of a module often show strong, selective enrichment for cell-type-specific markers (25). Modules can be further functionally annotated by assessing overlap with protein-protein interaction (PPI) databases, brain-relevant gene ontology pathways (52), transcription-factor or miRNA targets, among others. Finally, additional insight is provided by gene connectivity within a module, as hubs are more likely to act as drivers or regulators, making them useful targets for experimental validation (53–55).
Prioritization of target genes and proximal mechanisms underlying GWAS loci
GWAS loci are largely localized within non-coding, regulatory regions (3,5), that control gene expression and/or splicing often in a species and cell-type-specific manner. However, regulatory elements exhibit temporally dynamic and pleiotropic biological effects, necessitating high-throughput unbiased interrogation of their impact across distinct contexts (58). Along these lines, large-scale efforts have been undertaken to map expression and splicing-QTLs across development (14,59–64), facilitated by the high-throughput and systematic nature of transcriptomic phenotyping. For QTL discovery, sample size and tissue/cell-type are critical (65). PsychENCODE has compiled the largest panel to date (64), with >30,000 eGenes identified with a cis-eQTL, but results are restricted to bulk frontal cortex (35). Although cortical regions exhibit relatively homogenous transcriptomic patterns, distinct ‘patterning’ differences have been identified and non-cortical regions are highly distinct (37). Such differences are driven by unique cell-type compositions (56), which are obscured when profiling bulk tissue. Further, QTL mapping is typically performed after removing dozens of large expression variance components (e.g., ‘PEER’ factors), which maximizes identification of cis-eQTLs but also removes distinct cell type signals and other trans-acting factors (34). Temporal eQTL variability also remains underexplored, but is likely to uncover additional hidden signal, particularly during fetal and early postnatal timepoints (60,63,66).
Under the assumption that gene expression mediates the effect of genetic variation on a complex trait, several methods integrate GWAS and cis-eQTL signals to prioritize candidate risk genes (67). Colocalization-based methods estimate the probability of a shared causal variant between expression and trait, given observed marginal association statistics (68,69). Summary mendelian randomization (SMR) estimates the mediating effect of gene expression on a trait using top eQTLs as instrumental variables (70). Under multivariate predictive models, transcriptomic imputation methods (e.g., TWAS or S-PrediXcan) test for genetic correlation between the cis-regulated component of gene expression and a trait (71,72). Here, a sparse set of local SNP predictors is trained for each gene, using a large tissue-specific reference panel, followed by imputation into an association cohort to prioritize candidate risk genes and their direction of dysregulation. By aggregating effects of multiple SNPs onto specific features, these methods increase power for detection of associations potentially even outside GWAS loci. However, as these are association tests, they remain susceptible to potential confounds of linkage and pleiotropy (67). Recent frameworks have been developed to identify potential associations driven by linkage (70,73), to control for pleiotropy (74), and to provide probabilistic interpretation for locus-specific associations (75). Ultimately, this is an active research area which will continue to improve with incorporation of additional annotations from specific cell-types, tissues (76), and other (e.g., epigenetic) regulatory mechanisms (77).
These methods have prioritized several high-confidence psychiatric risk genes. For example, in SCZ, increased expression of C4A can (partially) explain the top GWAS-associated locus in Europeans (59,67,78–80). The CommonMind Consortium (CMC) performed colocalization of frontal cortex cis-eQTL, prioritizing several SCZ risk genes, including SNAP91, FURIN, and TSNARE1 (61), and SMR and tWaS have concordantly prioritized SNX19,among others (70,79). PsychENCODE, with its larger frontal cortex reference panel (80), prioritized many of these candidate genes and several more, including RERE, SETD6, SETD8, MCHR1, JKAMP, and AKT3--all of which were also concordantly differentially expressed (DE) in SCZ. Finally, incorporating splicing into the TWAS framework uncovered a number of candidates not identified through expression (79). Highlighting the importance of the reference panel tissue, TWAS-prioritized genes from fetal brain (60) or within distinct GTEx brain tissues (76) showed only modest overlap.
Such discrepancies highlight the need for validation, although true experimental replication remains challenging. For GWAS loci with a single high probability credible/causal variant and a specific eQTL colocalization, CRISPR/Cas9 genome editing can potentially be used for target validation (81). Yet, such strategies cannot distinguish between pleiotropic associations, if a given variant shows a functional effect across multiple genes. Integration of orthogonal annotations, such as chromosomal interactions with Hi-C or chromatin accessibility with ATACseq (63), can provide additional evidence of functionality and connect enhancers to genes, but remains susceptible to pleiotropy and generally lacks the resolution to pinpoint effects of individual variants. Integration with robustly-associated rare coding variants for the same trait may provide validation if available (82,83). In ASD, for example, the lysine methyltransferase KMT2E shows genome-wide significant associations across both common and rare variant studies (84,85). We recommend a probabilistic interpretation of prioritized candidate genes (75), including a potential null result, and hypothesize that integration of biologically-informed pathway-level priors will significantly boost performance.
Finally, these methods assume that genetic risk for disease is mediated through regulation of gene expression. Yet, recent evidence suggests that the overall proportion of disease heritability mediated by cis-eQTLs is much lower than previously thought, around ~10% in SCZ at least with current bulk tissue reference panels (86). The missing signal may be explained by distal QTLs, missing cis-eQTLs for lowly expressed genes (e.g., ncRNAs), those not captured in bulk tissue (cell-type-specific QTLs) (37,38), or distinct biological contexts, such as fetal brain (60). Effects other than expression regulation, including SNPs tagging structural variants, splicing or isoform-QTLs, methylation- or chromatin-accessibility QTLs, coding variants or even variants within ncRNA exons may explain additional missing variance.
Identifying Biological Convergence Through Transcriptomics
Due to the overwhelming polygenicity of psychiatric disorders, two unrelated affected individuals likely possess distinct combinations of risk variants. As such, the next challenge is to characterize whether the effects of multiple risk variants converge onto ‘key’ downstream molecular pathways. Early examples came from ASD-associated copy-number variants (CNVs), found to be enriched for neuronal and synaptic cell-adhesion genes (87). Exome sequencing studies found enrichment for genes harboring ASD-associated rare, de novo, protein-disrupting variants (RDNVs) among synaptic, chromatin, and gene regulation pathways (88). Further, although the overall RDNV burden in SCZ is smaller, similar pathways including synaptic genes, glutamate signaling, and activity-regulated cytoskeletal pathways are implicated (89,90). Common-variants across SCZ, MDD, and BD, in aggregate, show similar enrichments across synaptic and gene regulatory pathways (91).
Such enrichments are based on manually annotated pathways, which are unlikely to capture the full genomic complexity of the human brain (22). Transcriptomics provides a natural, bottom-up framework for systematically extending such annotations. The molecular underpinnings of human brain development are under exquisite spatiotemporal regulation, prompting several efforts to map expression across brain regions throughout development (40,92,93). Regional “patterning” of gene expression has been well captured in fetal (94) and adult human brain (9), a trend which will only be amplified with the advent of spatial transcriptomic profiling (95). These data enable systematic characterization of potential spatio-temporal, regional, and cell-type-specific convergence of the molecular genetic underpinnings of psychiatric disease. Along these lines, nearly all well-powered psychiatric genetic studies show enrichment for brain-expressed genes (7,84,85,96–98). Temporally, RDNVs associated with ASD and SCZ show enrichment among mid-fetal cortex gene networks (99–101). Experimentally-defined genomic targets of the RNA-binding proteins FMRP, RBFOX1, and CELF4 exhibit among the strongest enrichments for cross-disorder genetic risk (80,89,97,102,103).
In addition to RNA-binding proteins, non-coding RNAs such as microRNAs (miRNAs) are known to play an important role in human brain development and are implicated in psychiatric disease risk, as reviewed (87). miRNAs fine-tune gene expression by binding to the 3’ untranslated region (UTR) of specific target genes, inhibiting translation or promoting degradation. Several established genetic loci associated with psychiatric disorders harbor miRNAs, notably miR-137 as one of the top SCZ GWAS hits (88) as well as miR-130B, miR-185, and DGCR8 in 22q11.2 region. The exact nature and extent of dysregulation of miRNAs or other short non-coding RNAs (e.g. snoRNAs) in psychiatric disorders needs to be further explored.
Transcriptomic Insight into the Molecular Pathology Associated with Psychiatric Disorders
Psychiatric disorders lack a clearly defined anatomic pathology. Yet, given strong genetic roots, the question becomes -- what are the downstream biological consequences of these genetic risk factors? This question has fueled hundreds of case-control studies attempting to characterize group-level differences in molecular biomarkers, such as gene expression (13,44). Here, transcriptomics can provide a strategy for high-throughput, comprehensive molecular phenotyping of affected neural systems to characterize the current reactive state of a biological sample. Tissue and sample size are again critical, particularly given substantial levels of heterogeneity.
Initial seminal studies employed expression microarrays or in-situ hybridization in human postmortem prefrontal cortex from matched pairs of subjects with SCZ and controls. These studies identified notable dysregulation in synaptic machinery (89), interneuron markers (90), mitochondrial processes (91), and myelin-related genes (92), among others, as reviewed (13). Following on this initial work, post-mortem brain transcriptomics became widely performed across many disorders, fueled by the growth of brain banks (93) and advances in high-throughput profiling. Decreased expression of interneuron markers was also reported in BD and major depression (MDD; (104) as well as mitochondrial dysfunction in BD (95,96). Synaptic dysregulation was also observed in ASD cortex, as were elevated neuroinflammatory markers and notable splicing changes (105). Cross-disorder comparisons have been conducted, with layer-specific pyramidal cell changes observed in SCZ, but not BD or MDD (106). Further, when directly compared, transcriptomic changes were substantially larger in ASD than SCZ or BD (107). However, results have been variable across studies, particularly for individual DE genes-discrepancies which have been attributed to methodological, analytic, and/or cohort specific effects. Consistency tends to be greater at the level of pathways, cell-types, and networks, for example, and with meta- and mega-analytic approaches (107–109).
To address these challenges, consortia such as the Common Mind (61) and BrainSeq (63) were formed to aggregate and uniformly profile large numbers of psychiatric brain samples. Common Mind profiled more than 500 prefrontal cortex brain samples from SCZ and control subjects (>250 per group), prioritizing several new candidate risk genes through eQTL and GWAS colocalization (61). Several hundred DE genes were identified and, although effect sizes were modest, modeling estimated >40% of the transcriptome is perturbed in SCZ--a level of polygenicity that parallels GWAS findings to date. A neuronal co-expression module, associated with SCZ DE genes, was enriched for common and rare variation as well as postsynaptic density and synaptic signaling pathways, pointing to a convergent disease biology. More recently, PsychENCODE has compiled and meta-analyzed transcriptomic data from >2000 samples, including hundreds of individuals across SCZ, ASD, and BD as well as ~1000 controls for frontal cortex (64,80). Concordant results include interneuron marker downregulation, especially PVALB and SST, across SCZ and ASD (104,107,110). Many neuronal processes were dysregulated, particularly in ASD and SCZ, including those related to synaptic signaling and/or regulated by RBFOX1 (63,80,105,107). Notably, neuronal isoform-level changes showed the greatest effect-size changes in ASD and SCZ as well as the largest genetic enrichments (80). Mitochondrial and metabolic processes were broadly disrupted (106,107,111) as were blood-brain-barrier markers (80). In contrast, a number of processes were concordantly upregulated, such as NFkB and interferon response pathways along with astrocyte genes in SCZ and ASD (80,112,113). Microglia genes exhibited more distinct changes, with upregulation in ASD and downregulation in SCZ and BD (80). Many of these ASD results were recapitulated by a recent sn-RNAseq study (114). Importantly, regional differences need to be characterized, particularly for non-cortical brain structures, which can exhibit highly distinctive changes (115,116).
Interpretation of such changes is challenging, however, as they may reflect a compensatory or reactive consequence of disease, rather than a true causal pathophysiology, as argued (116). Integration of established genetic risk factors, imparted at birth, with identified expression changes can provide a directional framework for interpretation, although pleiotropy can still confound observed associations. DE genes or co-expression modules can be assessed for GWAS enrichment with methods such as LD-score regression (117) and MAGMA (110).
Cell-type specificity
Interpretation of bulk tissue transcriptomic results is further challenged due to its component mixture of individual cell types. Indeed, as much as ~85% of tissue-level RNA-seq data in human brain is driven by cell-type proportional differences (64). Computational approaches, including unsupervised co-expression network analysis or supervised cell-type deconvolution methods, can be used to gain inference into specific cell classes at a population level (38,57,64,107,118). However, individual cellular-level variability is lost and these methods are often limited to common cell-types. Initial targeted experimental methods, such as laser capture microdissection, flow cytometry or immunopanning, used cell-type specific visualization or labeling to enrich for a given population followed by transcriptomic profiling (119), although these approaches can be labor intensive and limited in throughput.
Advances in high-throughput single-cell/nuclei RNA-seq now provide cellular-level resolution to the aforementioned approaches (120,121). Microfluidic technologies capture individual cells in an oil droplet along with a barcoded bead and enzymes. Cells are lysed and polyA+ mRNAs are reverse transcribed into cDNA, which is amplified, fragmented, and sequenced. Barcoded cDNAs are pooled for PCR amplification, library construction, and fragmentation. Libraries are typically only sequenced at the 3’ end of a transcript, which is sufficient to quantify overall gene abundance. Transcripts from the same cell contain the same barcode, whereas a unique molecular index controls for biases due to PCR amplification. To avoid artifacts from tissue dissociation, single-nuclei RNA-seq using frozen tissue samples has become popular, as nuclei are more resistant to the stresses of freeze-thaw during isolation (122). snRNA-seq is generally comparable to scRNA-seq, although it detects more intronic reads and fewer total genes (122,123). Resulting datasets are large but sparse, with substantial gene dropout and noisy quantification of at most a few thousand genes per cell. Analytic methods are rapidly evolving, including quality control, normalization, batch correction, and multimodal integration (121,124,125). Non-linear dimensionality reduction methods identify unique cell-type clusters, which can then be contrasted across experimental conditions (121). Emerging methods can infer cellular lineage and “pseudo-temporal” trajectories, to characterize cell-type transition states and branch points (124,126). Nevertheless, as most methods rely on polyA+ 3’ sequencing, these data generally fail to capture the full transcriptome, missing many non-coding genes and splicing changes which are likely important contributors to psychiatric pathophysiology (80,115,127,128).
Large-scale sc/sn-RNAseq efforts are underway to fully elucidate the ‘parts-list’ of the human brain across development, including mid-gestational human fetal brain (129,130) and adult cortex across multiple regions (56,131), as well as hippocampus, striatum (132), and substantia nigra (125). Results highlight notable species-specific differences, including primate-specific striatal interneuron populations (132) and human-specific cell-type expression patterns for several serotonin and glutamate receptors (131). GWAS enrichment analyses have identified a set of genetically “vulnerable” cell-types in schizophrenia, including cortical pyramidal cells, interneurons, and DRD2+ medium spiny neurons (133). Rare variants associated with ASD, which are most highly expressed early during brain development, show particular enrichment among excitatory and inhibitory neuronal lineages (84,129,130). Concordantly, snRNAseq profiling of ASD cortex found notable alterations in synaptic gene expression in upper layer excitatory neurons (114). Finally, there is hope that cell-type-specific eQTL mapping will provide critical missing annotations for psychiatric GWAS loci and uncover hidden disease mechanisms. Some evidence already supports this (69), and while cell-type-specific analyses will undoubtedly identify new eQTLs, many non-expression-based mechanisms (e.g,. splicing) will remain hidden.
Model Systems
Transcriptomics can provide an important mechanistic readout in experimental settings, such as with animal models, hiPSC-derived neurons, or 3D cortical organoids. hiPSC-based methods provide an exciting opportunity to interrogate subject-specific neurobiology in an otherwise inaccessible context (127). To date, these approaches have been most fruitful characterizing effects of rare, large-effect-size mutations. For example, hiPSC-derived neurons from SCZ subjects harboring rare, pathogenic deletions in NRXN1 show strong allele-specific accumulation of ‘mutant’ isoforms and concomitant reduction in neuronal activity (128). hiPSC-derived neurons from subjects with MECP2mutations associated with Rett Syndrome show transcriptomic signatures of increased stress and senescence (129). In the context of common variation, genetic background effects can be difficult to control, although strategies enriching for subjects with strong polygenic burden have been successfully used (130). High-throughput genome-editing approaches coupled with transcriptional readouts are likely to provide important insights (134), for example, identifying convergent patterns of neuronal differentiation delay or acceleration associated with repression of distinct groups of ASD risk genes (132). Similar approaches taken in the context of animal models have begun to elucidate convergent biological mechanisms underlying psychiatric risk mutations. For example, overlapping transcriptomic changes in mouse models of 3 distinct CNVs associated with SCZ and ASD pinpointed convergent dysregulation of neuronal mitochondrial function (133). However, species differences in genomic regulation and brain cell-type organization (131) poses notable challenges for translational investigation of brain-relevant traits (134).
Integrative Approaches: Linking brain imaging & transcriptomics
Magnetic resonance imaging (MRI) is an important tool to investigate changes in brain structure, connectivity, and function that are associated with psychiatric traits at the macroscale. Due to a lack of resources integrating human neuroimaging data with gene expression patterns across cortical regions, previous studies have primarily examined associations between polygenic risk scores and brain structure/function. However, the recent availability of whole-brain gene expression atlases has enabled investigation into how regional gene expression relates to spatial patterns of in vivo neuroimaging phenotypes (9). The most widely used technique for integrating neuroimaging and transcriptomics links regional patterning of gene expression with spatial variation in brain structure using the Allen Human Brain Atlas (AhBA). A detailed description of this approach is provided elsewhere (135,136). The AHBA consists of 6 adult human brains from which T1-weighted MRI images were collected and co-registered with gene expression profiled from ~900 neuroanatomically defined regions. The resulting dataset provides an index of gene expression by brain region, which is then related to spatial patterns of neuroimaging phenotypes. This integrative method has the potential to inform our understanding of how gene networks relate to the hierarchical organizing principles of brain structural topography (137) and cortico-cortical connectivity patterns (138) relevant to behavioral symptoms associated with psychiatric disorders, and may also elucidate the genetic mechanisms by which particular brain regions are susceptible to disease-associated pathology as indexed in vivo by MRI. Indeed, work in this area has already shown that regional gene expression relates to atypical structural connectivity in SCZ (139,140) and regional variability of cortical thickness in ASD (141). Importantly, as the AHBA is derived from 6 neurotypical adults, future research integrating neurodevelopmental patterns of gene expression with age-appropriate neuroimaging data, as well as disease-specific expression profiles, will undoubtedly shed light on how polygenic risk for mental illness relates to the emergence of psychiatric illness across the lifespan. Overall, these methods have the potential to inform our understanding of how individual variability in gene expression affects both behavioral and brain-based phenotypes of psychiatric dysfunction, as well as how genetic control of transcription relates to symptomatology and neuro-endophenotypes across time.
Conclusions and Future Directions
Altogether, transcriptomics provides a rich resource for informing our understanding of the mechanisms underlying genetic risk for psychiatric disorders. Yet, we emphasize this is not the only approach, nor is it without limitations. Indeed, RNA-seq provides only a static snapshot of the transcriptional state of a biological system, and RNA abundance is highly dynamic, undergoing concomitant synthesis, splicing, and degradation. Integration of orthogonal measures, including epigenetic and proteomic annotations, including PPIs, will be highly complementary (142,143). Future approaches to non-invasively interrogate the genomic architecture of the human brain--whether through 3D organoids or by exosomes--will be important. Nevertheless, there is much more to be gained through investigation of postmortem human brains, including fully elucidating the component cell-types and their genetic regulation (113,115), characterization of spatial transcriptomic patterning (78), and detailed investigation of the full complexity of local splicing and isoform-level regulation (6,40,128,144). Finally, the notable enrichment of psychiatric genetic risk among gene regulatory pathways, particularly during fetal timepoints, highlights a strong, continued need for basic research into the complex molecular genetic principles orchestrating human brain development.
Acknowledgments
This work was supported by the Simons Foundation Bridge to Independence Award (MJG), the National Institute of Mental Health (R01MH121521 to MJG; K00MH119663 to LMH; T32MH073526 to MK), and the UCLA Medical Scientist Training Program (T32GM008042 to MK). The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, nHgRI, nHlBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from: the GTEx Portal and dbGaP accession number phs000424.v8.p2.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures
All authors declare no biomedical financial interests or potential conflicts of interest.
References
- 1.Gandal MJ, Leppa V, Won H, Parikshak NN, Geschwind DH (2016): The road to precision psychiatry: translating genetics into disease mechanisms. Nat Neurosci 19: 1397–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. (2012): Systematic localization of common disease-associated variation in regulatory DNA. Science 337: 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Claussnitzer M, Cho JH, Collins R, Cox NJ, Dermitzakis ET, Hurles ME, et al. (2020): A brief history of human disease genetics. Nature 577: 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, et al. (2018): Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet 50: 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, et al. (2015): Human genomics. The human transcriptome across tissues and individuals. Science 348: 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merkin J, Russell C, Chen P, Burge CB (2012): Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 338: 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai C, Langfelder P, Fuller TF, Oldham MC, Luo R, van den Berg LH, et al. (2010): Is human blood a good surrogate for brain tissue in transcriptional studies? BMC Genomics, vol. 11 p 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horváth S, Mirnics K (2015): Schizophrenia as a disorder of molecular pathways. Biol Psychiatry 77: 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. (2012): An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489: 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH (2008): Functional organization of the transcriptome in human brain. Nat Neurosci 11: 1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, et al. (2020): Cell stress in cortical organoids impairs molecular subtype specification. Nature 578: 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.SEQC/MAQC-III Consortium, Su Z, Łabaj PP, Li S, Thierry-Mieg J, Thierry-Mieg D, et al. (2014): A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat Biotechnol 32: 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stark R, Grzelak M, Hadfield J (2019): RNA sequencing: the teenage years. Nat Rev Genet 20: 631–656. [DOI] [PubMed] [Google Scholar]
- 14.Wang Z, Gerstein M, Snyder M (2009): RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li S, Łabaj PP, Zumbo P, Sykacek P, Shi W, Shi L, et al. (2014): Detecting and correcting systematic variation in large-scale RNA sequencing data. Nat Biotechnol 32: 888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng H, Zhang X, Zhang C (2015): mRIN for direct assessment of genome-wide and gene-specific mRNA integrity from large-scale RNA-sequencing data. Nat Commun 6: 7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, et al. (2010): Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet 11: 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Auer PL, Doerge RW (2010): Statistical design and analysis of RNA sequencing data. Genetics 185: 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman GE, Schadt EE (2016): variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinformatics 17: 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stegle O, Parts L, Piipari M, Winn J, Durbin R (2012): Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc 7: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stegle O, Parts L, Durbin R, Winn J (2010): A Bayesian Framework to Account for Complex Non-Genetic Factors in Gene Expression Levels Greatly Increases Power in eQTL Studies. PLoS Comput Biol 6: e1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim-Hellmuth S, Aguet F, Oliva M, Munoz-Aguirre M, Wucher V, Kasela S, et al. (2019, October 17): Cell type specific genetic regulation of gene expression across human tissues. BioRxiv. p 806117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. (2011): Spatio-temporal transcriptome of the human brain. Nature 478: 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawrylycz M, Miller JA, Menon V, Feng D, Dolbeare T, Guillozet-Bongaarts AL, et al. (2015): Canonical genetic signatures of the adult human brain. Nat Neurosci 18: 1832–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parikshak NN, Gandal MJ, Geschwind DH (2015): Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet 16: 441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B, Horvath S (2005): A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4: Article17. [DOI] [PubMed] [Google Scholar]
- 27.Saelens W, Cannoodt R, Saeys Y (2018): A comprehensive evaluation of module detection methods for gene expression data. Nat Commun 9: 1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ballouz S, Verleyen W, Gillis J (2015): Guidance for RNA-seq co-expression network construction and analysis: safety in numbers. Bioinformatics 31: 2123–2130. [DOI] [PubMed] [Google Scholar]
- 29.Kelley KW, Nakao-Inoue H, Molofsky AV, Oldham MC (2018): Variation among intact tissue samples reveals the core transcriptional features of human CNS cell classes. Nat Neurosci 21: 1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu X, Wells AB, O’Brien DR, Nehorai A, Dougherty JD (2014): Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J Neurosci 34: 1420–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skene NG, Grant SGN (2016): Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Front Neurosci 10: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koopmans F, van Nierop P, Andres-Alonso M, Byrnes A, Cijsouw T, Coba MP, et al. (2019): SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron 0 10.1016/j.neuron.2019.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, et al. (2006): Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A 103: 17402–17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, et al. (2013): Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153: 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swarup V, Hinz FI, Rexach JE, Noguchi K-I, Toyoshiba H, Oda A, et al. (2019): Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat Med 25: 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, et al. (2018): Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36: 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jew B, Alvarez M, Rahmani E, Miao Z, Ko A, Garske KM, et al. (2020): Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat Commun 11: 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strober BJ, Elorbany R, Rhodes K, Krishnan N, Tayeb K, Battle A, Gilad Y (2019): Dynamic genetic regulation of gene expression during cellular differentiation. Science 364: 1287–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Brien HE, Hannon E, Hill MJ, Toste CC, Robertson MJ, Morgan JE, et al. (2018): Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders. Genome Biol 19: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker RL, Ramaswami G, Hartl C, Mancuso N, Gandal MJ, de la Torre-Ubieta L, et al. (2019): Genetic Control of Expression and Splicing in Developing Human Brain Informs Disease Mechanisms. Cell 179: 750–771 .e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. (2016): Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci 19: 14421453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng B, White CC, Klein H-U, Sieberts SK, McCabe C, Patrick E, et al. (2017): An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci 20: 1418–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaffe AE, Straub RE, Shin JH, Tao R, Gao Y, Collado-Torres L, et al. (2018): Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat Neurosci 21: 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, et al. (2018): Comprehensive functional genomic resource and integrative model for the human brain. Science 362 10.1126/science.aat8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aguet F, Barbeira AN, Bonazzola R, Brown A, Castel SE, Jo B, et al. (2019, October 3): The GTEx Consortium atlas of genetic regulatory effects across human tissues. BioRxiv. p 787903. [Google Scholar]
- 46.GTEx Consortium (2017): Genetic effects on gene expression across human tissues. Nature 550: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Werling DM, Pochareddy S, Choi J, An J-Y, Sheppard B, Peng M, et al. (2020): Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell Rep 31: 107489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wainberg M, Sinnott-Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D, et al. (2019): Opportunities and challenges for transcriptome-wide association studies. Nat Genet 51: 592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V (2014): Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10: e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hormozdiari F, van de Bunt M, Segré AV, Li X, Joo JWJ, Bilow M, et al. (2016): Colocalization of GWAS and eQTL Signals Detects Target Genes. Am J Hum Genet 99: 1245–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. (2016): Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48: 481–487. [DOI] [PubMed] [Google Scholar]
- 52.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, et al. (2016): Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 48: 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, et al. (2018): Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun 9: 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbeira AN, Pividori M, Zheng J, Wheeler HE, Nicolae DL, Im HK (2019): Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet 15: e1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Porcu E, Rüeger S, Lepik K, eQTLGen Consortium, BIOS Consortium, Santoni FA, et al. (2019): Mendelian randomization integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits. Nat Commun 10: 3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mancuso N, Freund MK, Johnson R, Shi H, Kichaev G, Gusev A, Pasaniuc B (2019): Probabilistic fine-mapping of transcriptome-wide association studies. Nat Genet 51: 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huckins LM, Dobbyn A, Ruderfer DM, Hoffman G, Wang W, Pardinas AF, et al. (2019): Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nat Genet. 10.1038/s41588-019-0364-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang W, Voloudakis G, Rajagopal VM, Readhead B, Dudley JT, Schadt EE, et al. (2019): Integrative transcriptome imputation reveals tissue-specific and shared biological mechanisms mediating susceptibility to complex traits. Nat Commun 10: 3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. (2016): Schizophrenia risk from complex variation of complement component 4. Nature 530: 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gusev A, Mancuso N, Won H, Kousi M, Finucane HK, Reshef Y, et al. (2018): Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat Genet 50: 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, et al. (2018): Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362 10.1126/science.aat8127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schrode N, Ho S-M, Yamamuro K, Dobbyn A, Huckins L, Matos MR, et al. (2019): Synergistic effects of common schizophrenia risk variants. Nat Genet. 10.1038/s41588-019-0497-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, Geschwind DH (2018): The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172: 289–304.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Freund MK, Burch KS, Shi H, Mancuso N, Kichaev G, Garske KM, et al. (2018): Phenotype-Specific Enrichment of Mendelian Disorder Genes near GWAS Regions across 62 Complex Traits. Am J Hum Genet 103: 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barbeira AN, Bonazzola R, Gamazon ER, Liang Y, Park Y, Kim-Hellmuth S, et al. (2019, October 22): Widespread dose-dependent effects of RNA expression and splicing on complex diseases and traits. BioRxiv. p 814350. [Google Scholar]
- 66.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, et al. (2020): Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 10.1016/j.cell.2019.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. (2019): Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 10.1038/s41588-019-0344-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yao DW, O’Connor LJ, Price AL, Gusev A (2019, August 9): Quantifying genetic effects on disease mediated by assayed gene expression levels. BioRxiv. p 730549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jaffe AE, Hoeppner DJ, Saito T, Blanpain L, Ukaigwe J, Burke EE, et al. (2020): Profiling gene expression in the human dentate gyrus granule cell layer reveals insights into schizophrenia and its genetic risk. Nat Neurosci 23: 510–519. [DOI] [PubMed] [Google Scholar]
- 70.Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, et al. (2009): Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459: 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. (2014): Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515: 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. (2014): De novo mutations in schizophrenia implicate synaptic networks. Nature 506: 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. (2014): A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506: 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium (2015): Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 18: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, et al. (2011): Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478: 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, et al. (2018): Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362 10.1126/science.aat7615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A, et al. (2014): Transcriptional landscape of the prenatal human brain. Nature 508: 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. (2019): Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363: 1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landén M, et al. (2016): Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci 19: 1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. (2018): Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 50: 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. (2019): Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet 51: 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, et al. (2013): Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155: 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. (2013): Integrative functional genom analyses implicate specific molecular pathways and circuits in autism. Cell 155: 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, et al. (2013): Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154: 518–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pardinas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. (2018): Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong backgroun selection. Nat Genet 50: 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cross-Disorder Group of the Psychiatric Genomics Consortium. Electronic address: plee0@mgh.harvard.edu, Cross-Disorder Group of the Psychiatric Genomics Consortium (2019): Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 179: 1469–1482.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Issler O, Chen A (2015): Determining the role of microRNAs in psychiatric disorders. Nat Rev Neurosci 16 201–212. [DOI] [PubMed] [Google Scholar]
- 88.Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014): Biological insights from 1C schizophrenia-associated genetic loci. Nature 511: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mirnics K, Middleton FA, Lewis DA, Levitt P (2001): Analysis of complex brain disorders with gene expression microarrays: schizophrenia as a disease of the synapse. Trends Neurosci 24: 479–486. [DOI] [PubMed] [Google Scholar]
- 90.Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, et al. (2003): Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 23: 6315–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P (2002): Gene Expression Profiling Reveals Alterations of Specific Metabolic Pathways in Schizophrenia. J Neurosci 22: 2718–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, et al. (2001): Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A 98: 4746–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Deep-Soboslay A, Benes FM, Haroutunian V, Ellis JK, Kleinman JE, Hyde TM (2011): Psychiatric brain banking: three perspectives on current trends and future directions. Biol Psychiatry 69: 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sibille E, Morris HM, Kota RS, Lewis DA (2011): GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int J Neuropsychopharmacol 14: 721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Iwamoto K, Bundo M, Kato T (2005): Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet 14: 241–253. [DOI] [PubMed] [Google Scholar]
- 96.Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, Heckers S (2004): Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry 61: 300–308. [DOI] [PubMed] [Google Scholar]
- 97.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. (2011): Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474: 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arion D, Huo Z, Enwright JF, Corradi JP, Tseng G, Lewis DA (2017): Transcriptome Alterations in Prefrontal Pyramidal Cells Distinguish Schizophrenia From Bipolar and Major Depressive Disorders. Biol Psychiatry 82: 594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, et al. (2018): Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359: 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Elashoff M, Higgs BW, Yolken RH, Knable MB, Weis S, Webster MJ, et al. (2007): Meta-analysis of 12 genomic studies in bipolar disorder. J Mol Neurosci 31: 221–243. [DOI] [PubMed] [Google Scholar]
- 101.Toker L, Mancarci BO, Tripathy S, Pavlidis P (2018): Transcriptomic Evidence for Alterations in Astrocytes and Parvalbumin Interneurons in Subjects With Bipolar Disorder and Schizophrenia. Biol Psychiatry 84: 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chung DW, Chung Y, Bazmi HH, Lewis DA (2018): Altered ErbB4 splicing and cortical parvalbumin interneuron dysfunction in schizophrenia and mood disorders. Neuropsychopharmacology 43: 2478–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pacifico R, Davis RL (2017): Transcriptome sequencing implicates dorsal striatum-specific gene network, immune response and energy metabolism pathways in bipolar disorder. Mol Psychiatry 22: 441–449. [DOI] [PubMed] [Google Scholar]
- 104.Ramaker RC, Bowling KM, Lasseigne BN, Hagenauer MH, Hardigan AA, Davis NS, et al. (2017): Post mortem molecular profiling of three psychiatric disorders. Genome Med 9: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Volk DW, Moroco AE, Roman KM, Edelson JR, Lewis DA (2019): The Role of the Nuclear Factor-KB Transcriptional Complex in Cortical Immune Activation in Schizophrenia. Biol Psychiatry 85: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, et al. (2019): Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364: 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, et al. (2016): Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540: 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Collado-Torres L, Burke EE, Peterson A, Shin J, Straub RE, Rajpurohit A, et al. (2019): Regional Heterogeneity in Gene Expression, Regulation, and Coherence in the Frontal Cortex and Hippocampus across Development and Schizophrenia. Neuron. https://doi.org/10.1016Zj.neuron.2019.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, et al. (2015): Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47: 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.de Leeuw CA, Mooij JM, Heskes T, Posthuma D (2015): MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11: e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Miller JA, Cai C, Langfelder P, Geschwind DH, Kurian SM, Salomon DR, Horvath S (2011): Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinformatics 12: 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Poulin J-F, Tasic B, Hjerling-Leffler J, Trimarchi JM, Awatramani R (2016): Disentangling neural cell diversity using single-cell transcriptomics. Nat Neurosci 19: 1131–1141. [DOI] [PubMed] [Google Scholar]
- 113.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. (2015): Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161: 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kulkarni A, Anderson AG, Merullo DP, Konopka G (2019): Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr Opin Biotechnol 58: 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, et al. (2017): Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 14: 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, et al. (2018): Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One 13: e0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stuart T, Satija R (2019): Integrative single-cell analysis. Nat Rev Genet 20: 257–272. [DOI] [PubMed] [Google Scholar]
- 118.Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ (2019): Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 0 10.1016/j.cell.2019.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. (2018): RNA velocity of single cells. Nature. 10.1038/s41586-018-0414-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, et al. (2016): RNA splicing is a primary link between genetic variation and disease. Science 352: 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Takata A, Matsumoto N, Kato T (2017): Genome-wide identification of splicing QTLs in the human brain and their enrichment among schizophrenia-associated loci. Nat Commun 8: 14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Polioudakis D, de la Torre-Ubieta L, Langerman J, Elkins AG, Shi X, Stein JL, et al. (2019): A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron. 10.1016/j.neuron.2019.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, et al. (2017): Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358: 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, et al. (2019): Conserved cell types with divergent features in human versus mouse cortex. Nature 573: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Krienen FM, Goldman M, Zhang Q, del Rosario R, Florio M, Machold R, et al. (2019, July 23): Innovations in Primate Interneuron Repertoire. BioRxiv. p 709501. [Google Scholar]
- 126.Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, et al. (2018): Genetic identification of brain cell types underlying schizophrenia. Nat Genet 50: 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Marton RM, Paşca SP (2019): Organoid and assembloid technologies for investigating cellular crosstalk in human brain development and disease. Trends Cell Biol 30: 133–143. [DOI] [PubMed] [Google Scholar]
- 128.Flaherty E, Zhu S, Barretto N, Cheng E, Deans PJM, Fernando MB, et al. (2019): Neuronal impact of patient-specific aberrant NRXN1a splicing. Nat Genet 51: 1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ohashi M, Korsakova E, Allen D, Lee P, Fu K, Vargas BS, et al. (2018): Loss of MECP2 Leads to Activation of P53 and Neuronal Senescence. Stem Cell Reports 10: 1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hoffman GE, Hartley BJ, Flaherty E, Ladran I, Gochman P, Ruderfer DM, et al. (2017): Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nat Commun 8: 2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, et al. (2019): A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 176: 377–390.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lalli MA, Avey D, Dougherty JD, Milbrandt J, Mitra RD (2020, February 11): High-throughput single-cell functional elucidation of neurodevelopmental disease-associated genes reveals convergent mechanisms altering neuronal differentiation. BioRxiv. p 862680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gordon A, Forsingdal A, Klewe IV, Nielsen J, Didriksen M, Werge T, Geschwind DH (2019): Transcriptomic networks implicate neuronal energetic abnormalities in three mouse models harboring autism and schizophrenia-associated mutations. Mol Psychiatry 10.1038/s41380-019-0576-0 [DOI] [PubMed] [Google Scholar]
- 134.Miller JA, Horvath S, Geschwind DH (2010): Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A 107: 12698–12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fornito A, Arnatkevičiūtė A, Fulcher BD (2019): Bridging the Gap between Connectome and Transcriptome. Trends Cogn Sci 23: 34–50. [DOI] [PubMed] [Google Scholar]
- 136.Arnatkevic Iūtė A, Fulcher BD, Fornito A (2019): A practical guide to linking brain-wide gene expression and neuroimaging data. Neuroimage 189: 353–367. [DOI] [PubMed] [Google Scholar]
- 137.Burt JB, Demirtaş M, Eckner WJ, Navejar NM, Ji JL, Martin WJ, et al. (2018): Hierarchy of transcriptomic specialization across human cortex captured by structural neuroimaging topography. Nat Neurosci 21: 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Krienen FM, Yeo BTT, Ge T, Buckner RL, Sherwood CC (2016): Transcriptional profiles of supragranular-enriched genes associate with corticocortical network architecture in the human brain. Proc Natl Acad Sci U S A 113: E469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Romme IAC, de Reus MA, Ophoff RA, Kahn RS, van den Heuvel MP (2017): Connectome Disconnectivity and Cortical Gene Expression in Patients With Schizophrenia. Biol Psychiatry 81: 495–502. [DOI] [PubMed] [Google Scholar]
- 140.Morgan SE, Seidlitz J, Whitaker KJ, Romero-Garcia R, Clifton NE, Scarpazza C, et al. (2019): Cortical patterning of abnormal morphometric similarity in psychosis is associated with brain expression of schizophrenia-related genes. Proc Natl Acad Sci U S A 116: 9604–9609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Romero-Garcia R, Warrier V, Bullmore ET, Baron-Cohen S, Bethlehem RAI (2019): Synaptic and transcriptionally downregulated genes are associated with cortical thickness differences in autism. Mol Psychiatry 24: 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. (2012): Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485: 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Horn H, Lawrence MS, Chouinard CR, Shrestha Y, Hu JX, Worstell E, et al. (2018): NetSig: network-based discovery from cancer genomes. Nat Methods 15: 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, et al. (2016): Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 166: 1147–1162.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]