

SUMMARY
Cancer-associated mutations that stabilize NRF2, an oxidant defense transcription factor, are predicted to promote tumor development. Here, utilizing 3D cancer spheroid models coupled with CRISPR-Cas9 screens, we investigate the molecular pathogenesis mediated by NRF2 hyperactivation. NRF2 hyperactivation was necessary for proliferation and survival in lung tumor spheroids. Antioxidant treatment rescued survival but not proliferation, suggesting the presence of distinct mechanisms. CRISPR screens revealed that spheroids are differentially dependent on mTOR for proliferation and the lipid peroxidase GPX4 for protection from ferroptosis of inner, matrix-deprived cells. Ferroptosis inhibitors blocked death from NRF2 downregulation, demonstrating a critical role for NRF2 in protecting matrix-deprived cells from ferroptosis. Interestingly, proteomic analyses show global enrichment of selenoproteins, including GPX4, by NRF2 downregulation, and targeting both NRF2 and GPX4 killed spheroids overall. These results illustrate the value of spheroid culture in revealing environment- or spatial-dependent differential dependencies on NRF2, and reveal exploitable vulnerabilities of NRF2-hyperactivated tumors.
Graphical Abstract
eTOC Blurb
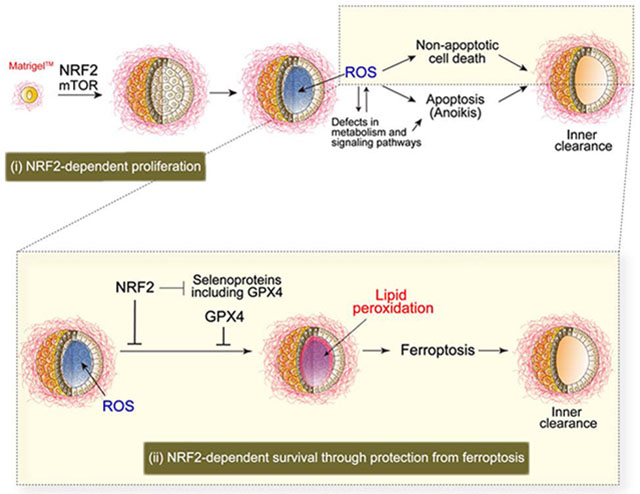
Takahashi and Cho et al. demonstrate that NRF2-hyperactivation is necessary for tumor spheroid formation through regulation of proliferation and protection from ferroptosis, the lipid peroxidation-induced non-apoptotic cell death. Loss of NRF2 upregulates most selenoproteins including the lipid peroxidase GPX4. Targeting both NRF2 and GPX4 kills cells throughout the spheroid.
INTRODUCTION
Cancer cells rewire metabolic networks to provide a steady source of energy and biosynthetic intermediates needed for cell division and rapid growth (Possemato et al., 2011; Vander Heiden et al., 2009). However, this causes the production of toxic metabolic by-products, including reactive oxygen species (ROS), that can promote oxidative stress and impair cancer cell viability (DeBerardinis and Chandel, 2016; Gorrini et al., 2013; Schafer et al., 2009; Tennant et al., 2010). In particular, conditions associated with tumorigenesis and metastasis, such as detachment from extracellular matrix (ECM), induce marked metabolic changes and impose further oxidative stress on tumor cells (Cheung et al., 2020; DeBerardinis and Chandel, 2016; Gorrini et al., 2013; Schafer et al., 2009). Thus, there is a selection for tumor cells that upregulate oxidative-stress defense programs in order to overcome the toxicity of oxidative stress, which represents a significant hurdle that impedes tumor initiation and progression (Chandel and Tuveson, 2014; Chio and Tuveson, 2017; Glasauer et al., 2014).
The transcription factor NRF2 is a master regulator of cellular redox homeostasis in response to oxidative stress. NRF2 activation induces the expression of antioxidants as well as enzymes involved in GSH synthesis and promotes NADPH production by regulating enzymes in the pentose phosphate and serine biosynthesis pathways (Kitamura and Motohashi, 2018; Rojo De La Vega et al., 2018). Regulation of NRF2 in homeostatic conditions is tightly controlled at the protein level by direct binding to KEAP1, which acts as a substrate adaptor for the cullin-3 (CUL3) E3 ubiquitin ligase complex. This complex maintains NRF2 in the cytosol where it is continually targeted for proteasomal degradation. Under conditions of oxidative stress, key cysteine residues of KEAP1 are oxidatively modified to block interaction with NRF2, stabilizing the transcription factor. Notably, mutations in NFE2L2 (the gene encoding NRF2), KEAP1, or CUL3 that induce constitutive activation of NRF2 are frequently observed in multiple types of human cancer. These mutations are particularly prevalent in non-small cell lung cancer (Collisson et al., 2014; Hammerman et al., 2012), where mutations occur in ~30% of lung squamous cell carcinoma (LUSC) patients and in ~25% of lung adenocarcinoma (LUAD) patients, as well as in other squamous cell carcinomas (Sanchez-Vega et al., 2018). NRF2 is also transcriptionally upregulated by oncogenes, such as K-RASG12D, B-RafV619E, or MycERT2 (Denicola et al., 2011). Hyperactivation of NRF2 is associated with a worse clinical prognosis (Cescon et al., 2015; Romero et al., 2017; Solis et al., 2010). These lines of evidence suggest that cancer cells with NRF2 hyperactivation have a competitive fitness advantage (Cescon et al., 2015; Homma et al., 2009; Rojo De La Vega et al., 2018; Singh et al., 2008; Tao et al., 2014). However, the molecular mechanisms by which NRF2 hyperactivation imparts a survival and proliferative advantage to tumor cells remain poorly defined. In particular, it is controversial whether NRF2 promotes cancer exclusively through maintenance of redox homeostasis based on differences in the extent to which antioxidant treatment rescues NRF2 deficiency-induced phenotypes (Chio et al., 2016; Denicola et al., 2011; Reddy et al., 2007; Vartanian et al., 2019). As such, it is important to dissect the diverse and complex nature of NRF2 hyperactivation in order to understand how NRF2 hyperactivation leads to tumor progression as well as present new vulnerabilities for therapeutic intervention in patients with NRF2-hyperactivated tumors.
Epithelial cells are dependent on interactions with specific ECM components for survival, proliferation, and differentiation functions. However, during tumor initiation and progression, the normal epithelial organization is disrupted, and malignant cells proliferate and survive outside their normal niches (Chiarugi and Giannoni, 2008). This process is not well recapitulated in two-dimensional (2D) monolayer cultures, and thus findings from such cultures may obfuscate aspects of the mechanisms underlying molecular pathogenesis of cancer and potentially explain, in part, the failure of many therapeutic approaches in clinical trials.
Three-dimensional (3D) spheroid models recapitulate in vivo morphologies, such as cell polarization, organization of cell layers, and interactions with ECM, to regulate proliferation and survival. Survival of non-transformed epithelial cells in 3D culture is dependent on attachment to ECM – inner cells, lacking ECM attachment, undergo both apoptotic, namely anoikis, and non-apoptotic cell death, generating a hollow lumen (Chiarugi and Giannoni, 2008; Debnath and Brugge, 2005; Hawk and Schafer, 2018). While numerous studies have unveiled multi-faceted strategies that cancer cells utilize to evade anoikis, the mechanisms that contribute to non-apoptotic cell death remain poorly understood. We have previously shown that the death of centrally localized, matrix-deprived cells is preceded by an elevation in ROS levels due to metabolic alterations and that treatment of spheroids with exogenous antioxidants is sufficient to reduce this cell death (Schafer et al., 2009). Tumor cells, on the other hand, are able to survive in the high oxidative stress inner spheroid space (Jiang et al., 2016; Takahashi et al., 2018), raising the possibility that tumor cells upregulate oxidative-stress defense programs to prevent inner clearance in spheroids.
Here, we investigated the molecular pathogenesis mediated by NRF2 hyperactivation through utilization of 3D cancer spheroid models coupled with CRISPR-Cas9 screens. The findings provide critical insights into NRF2-induced phenotypic changes that could not be assayed in standard 2D cultures and reveal exploitable vulnerabilities of NRF2-hyperactivated tumors.
RESULTS
3D Spheroids of NRF2-Hyperactivated Lung Cancer Cells Are More Dependent on NRF2 for Survival and Proliferation, Compared with 2D Monolayer Cells
To examine the effects of downregulation of NRF2 in both conventional 2D monolayer and 3D spheroid cultures, we generated A549 and H1437 non-small cell lung tumor cell lines that express two distinct, inducible shRNAs targeting NRF2 mRNA under the control of doxycycline. These tumor cell lines exhibit constitutive activation (hyperactivation) of NRF2 due to inactivating genetic alterations in KEAP1 that result in stabilization of NRF2 (Denicola et al., 2015). Notably, NRF2 downregulation caused a much more significant reduction in cell number in 3D cultures compared to 2D cultures (Figure 1A). Moreover, in co-culture of parental cells with either RFP-positive shControl or shNRF2 cells, shNRF2 cells were more significantly out-competed in 3D cultures compared to 2D cultures (Figure S1A), suggesting that these tumor cells are more dependent on NRF2 for survival or proliferation under 3D culture conditions. Differences in the extent of reduction in cell numbers in H1437 and A549 cells correlate with the reduction in both expression of NRF2 and NRF2 activity as measured by transcription of its target NQO1 (Figure 1B).
Figure 1. Downregulation of NRF2 Significantly Impacts Cell Growth in 3D Culture.

(A) Growth of the indicated cell lines in 2D and 3D culture conditions. For A549 cells, data shown as mean ± SEM from three independent experiments. For H1437 cells, data shown as mean ± SD from one representative experiment out of two independent experiments performed in duplicate–quadruplicate. (B) mRNA expression of NRF2 and NQO1 in the indicated cell lines after 72 hours of shRNA induction in 2D culture from three independent experiments. Data were normalized to shControl. (C) Representative confocal images of the indicated spheroids from three independent experiments. (D) Quantification of spheroid size from experiments described in (C) (n = 55–112). (E) Quantification of Ki67 signal relative to shControl from experiments described in (C) (n = 22–46). (F) Representative confocal images of the indicated spheroids from three independent experiments. (G) Timeline of doxycycline treatment (1 μg/ml) in (F). (H) Definition of inner space and calculation of % filled. White line represents the boundary of inner space. (I) Percentage of filled inner space from experiments described in (F) (n = 45–77). (J) Representative confocal images of the indicated spheroids from two independent experiments. (K) Quantification of spheroid size in the indicated day-8 spheroids (n = 59–150). (L) Quantification of Ki67 signal relative to spheroids transduced with both shControl and an empty vector in the indicated day-8 spheroids (n = 59–150). (M) Representative confocal images of the indicated spheroids from two independent experiments. (N) Percentage of filled inner space from experiments described in (M) (n = 14–22). Unless otherwise noted, the cells were treated with 1 μg/ml doxycycline throughout the experiments. In (B), (D), (E), and (I), unpaired two-tailed t-test was used to determine statistical significance (*p < 0.05, **p < 0.01, and ***p < 0.001 compared to shControl). In (K), (L), and (N), one-way ANOVA was used to determine statistical significance (*p < 0.05, **p < 0.01, and ***p < 0.001). Scale bar represents 100 μm. All data shown as mean ± SEM unless otherwise indicated.
Growth of spheroids can be separated into two stages: an early, highly proliferative stage in which single cells begin to form spheroids (approximately the first week of culture) followed by a proliferation arrest stage in the second week of culture (Debnath and Brugge, 2005). After eight days (i.e. early stage) of growth in 3D culture, spheroids with NRF2 knockdown were smaller than spheroids without NRF2 knockdown (Figures 1C and 1D), with the strongest effect observed in H1437 spheroids. Furthermore, immunofluorescent staining for the cell proliferation marker Ki67 was also decreased in spheroids with NRF2 knockdown (Figures 1C and 1E). These data suggest that NRF2 regulates proliferation during the early, proliferative stage of spheroid growth in 3D culture.
After twelve days (i.e. late stage) of growth in 3D culture, NRF2 knockdown induced clearing of inner, matrix-deprived cells in A549 spheroids (Figures 1F–1I). Since NRF2 knockdown from the start of 3D culture greatly suppressed spheroid growth in H1437 cells, we cultured the H1437 spheroids for one week prior to addition of doxycycline so that the spheroids would become large enough to assess the effect of NRF2 knockdown on inner clearance (Figure 1G). Consistent with A549 spheroids, induction of NRF2 knockdown after formation of H1437 spheroids induced clearing of the inner cells (Figures 1F–1I). These results indicate that NRF2 controls survival of the inner matrix-deprived cells during the late stage of spheroid formation. The observed phenotypes induced by NRF2 knockdown are on-target, as transduction of NFE2L2 mutants (NRF2* and NRF2**) that are not targetable by the shNRF2 hairpins (Figures S1B–S1D) rescued both the reduction in proliferation and survival of inner cells (Figures 1J–1N).
High NRF2 Activity Is Required for Efficient Lung Cancer Spheroid Formation
To further evaluate the impact of NRF2 on spheroid growth, we examined the relationship between the status of NRF2 activation and spheroid growth in a large panel of lung cancer cell lines. Given that NRF2 hyperactivation is induced not only by genetic alterations in the NRF2 pathway but also by other factors, including oncogenes and unknown mechanisms, we first derived an NRF2 gene signature to quantify the extent of NRF2 activation in each cell line.
To derive an NRF2 gene signature, we mined primary patient data from four cancer types with frequent genetic alterations in the NRF2 pathway [LUSC, LUAD, head and neck squamous cell carcinoma (HNSC), and cervical squamous cell carcinoma (CESC)] in The Cancer Genome Atlas (TCGA) project. We identified 1,466 genes that have significantly higher or lower expression in tumors with NRF2-hyperactivating genetic alterations in the NRF2 pathway (mutation/amplification of NFE2L2 or mutation/deletion of KEAP1 or CUL3) than tumors without NRF2-hyperactivating genetic alterations (p < 0.05, Figure 2A and Table S1). We derived a 55-gene NRF2 signature by selecting genes that were more highly expressed in altered tumors compared to unaltered tumors in LUSC and at least two out of the other three tumor types (Figures 2B and 2C and Table S1). The median normalized expression value of the NRF2 signature genes was used as an “NRF2 score” which serves as an indication of NRF2-hyperactivation for a panel of cancer cell lines (Figure 2C and Table S2).
Figure 2. High NRF2 Activity Is Required for Efficient Spheroid Formation.

(A) Genes associated with NRF2 hyperactivation in TCGA LUSC samples (n = 198). Heatmap shows log2 median-centered RNA-seq gene expression data. Status of genetic alterations in the NRF2 pathway for each tumor is displayed on the right. (B) Venn diagram showing the number of genes associated with NRF2 hyperactivation in TCGA cancer types. The 1,466 genes associated with NRF2 hyperactivation were further limited to a 55-gene NRF2 signature using the outlined criteria. (C) Expression of the 55 NRF2 signature genes in 675 cancer cell lines. Heatmap shows log2 median-centered RNA-seq gene expression data. Copy number (CN) alteration and mutation status of KEAP1, CUL3, and NFE2L2 as well as tissue of origin are displayed. (D) Representative images of day-16 spheroids of lung cancer cell lines. Asterisks indicate the existence of genetic alterations in the NRF2 pathway. (E) Correlation between NRF2 score and spheroid size from experiments described in (D). H522, H1975, and H23 cells were excluded from the correlation analysis because they do not form spheroids but just clumps of cells. (F) mRNA expression of NQO1 in the indicated cell lines from three independent experiments. Data were normalized to sgControl. (G) Representative confocal images of the indicated day-14 spheroids from two independent experiments. (H) Quantification of spheroid size from experiments described in (G) (n = 65–97). (I) Representative confocal images of the indicated day-12 spheroids from two independent experiments. (J) Quantification of spheroid size and percentage of filled inner space from experiments described in (I) (n = 53–91). In (F), (H), and (J), one-way ANOVA (F) or unpaired two-tailed t-test (H and J) was used to determine statistical significance. **p < 0.01 and ***p < 0.001 compared to sgControl. Scale bar represents 100 μm. All data shown as mean ± SEM.
Consistent with the NRF2 knockdown data in 3D cultures, lung cancer cell lines with lower NRF2 scores did not form spheroids efficiently, and NRF2 score of lung cancer cell lines correlated with spheroid size (Figures 2D and 2E; Pearson’s r = 0.73, p = 0.0031). Notably, cell lines that do not have genetic alterations in the NRF2 pathway but exhibit higher NRF2 scores (i.e. H520 and SK-MES-1 cells) formed spheroids, and cell lines that have genetic alterations in the NRF2 pathway but exhibit lower NRF2 score (i.e. H23 cells) did not form spheroids. NRF2 knockdown decreased spheroid size and increased clearing of inner cells in H520 and SK-MES-1 cells, and overexpression of NRF2** rescued spheroid growth and survival of inner cells (Figures S1E–S1P). Interestingly, a small proportion of H596 cells, which exhibit an intermediate NRF2 score, formed spheroids, but they exclusively showed strong inner clearance (Figure 2D, arrowheads). Thus, these results suggest that high NRF2 activity is necessary for efficient spheroid formation, at least in lung cancer cell lines.
To further investigate the effect of NRF2-hyperactivation on spheroid formation, we examined whether forced stabilization of NRF2 by CRISPR-Cas9-mediated KEAP1 knockout could enhance spheroid size and survival of inner cells in lung cancer cell lines that do not have genetic alterations in the NRF2 pathway and exhibit low or intermediate NRF2 scores: H226 and H596. We confirmed that KEAP1 knockout increased NRF2 protein expression and activity as determined by increased expression of NQO1 (Figures 2F and S1Q). KEAP1 knockout increased the size of spheroids in both cell lines (Figures 2G–2J) and mitigated the death of inner spheroid cells in H596 cells (Figures 2I and 2J). These data provide additional evidence that high NRF2 activity can promote efficient spheroid formation in lung cancer cells.
The NRF2 score of LUSC and LUAD primary patient tumors positively correlated with proliferation signature scores (Pearson’s r = 0.24, p = 2.6 × 10−14) (Selfors et al., 2017) (Figures S2A and S2B), supporting a potential association between NRF2 activity and proliferation in human tumors. Interestingly, NRF2 score did not correlate with 2D cell growth in a panel of lung cancer cell lines (Pearson’s r = −0.145, p = 0.565; Figures S2C and S2D), suggesting that NRF2 hyperactivation does not account for differences in 2D proliferation in lung cancer cell lines.
Antioxidants Rescue Survival, but Not Proliferation Defects, Caused by NRF2 Downregulation
Given that NRF2 regulates a cellular antioxidant program, we investigated cellular redox status in 2D versus 3D culture conditions. 3D cultures displayed enhanced ROS levels and decreased GSH/GSSG levels (Figures 3A and 3B) relative to 2D cultures, indicating that spheroid cells are subjected to more oxidative stress compared to monolayer cells. The induction of oxidative stress was more prominent at Day 10, potentially due to strong oxidative stress in inner spheroid cells at late stage of 3D culture (Schafer et al., 2009; Takahashi et al., 2018). While NRF2 mRNA expression in 3D cultures was comparable to those in 2D cultures at any time point except for Day 10, NQO1 expression was higher in 3D culture (Figure S2E), suggesting both that NRF2 still has capacity for being further activated by ROS despite inactivating genetic alterations in KEAP1 in A549 and H1437 cells and that cells carrying extremely hyperactivated NRF2 are selected in 3D cultures.
Figure 3. Antioxidant Treatment Rescues Survival of Inner Cells but Not Loss of Proliferation Caused by NRF2 Downregulation in Spheroids.

(A) ROS levels relative to monolayer cells in the indicated spheroids from three independent experiments. (B) GSH/GSSG ratio in the indicated cells cultured in 2D or 3D from three independent experiments. (C and D) Representative confocal images of the indicated spheroids from two independent experiments. (E) Percentage of filled inner space from experiments described in (C) and (D) (n = 10–64). (F) Representative confocal images of the indicated spheroids from two independent experiments. (G) Quantification of spheroid size from experiments described in (F) (n = 4–24). (H) Quantification of Ki67 signal relative to NAC- and GSH-EE-untreated spheroids with shControl from experiments described in (F) (n = 4–24). In (A) and (B), unpaired two-tailed t-test was used to determine statistical significance (*p < 0.05, **p < 0.01, and ***p < 0.001 compared to 2D cells). In (C), (D), and (F), the spheroids were treated with 1 μg/ml doxycycline and either vehicle, 1 mM NAC, or 1 mM GSH-EE for the indicated time period. In (E), (G), and (H), one-way ANOVA was used to determine statistical significance (***p < 0.001). Scale bar represents 100 μm. All data shown as mean ± SEM.
We next examined whether antioxidant treatment rescues NRF2 knockdown-induced clearance of inner spheroid cells and reduced proliferation. Survival of A549 and H1437 cells in the inner luminal space following NRF2 knockdown was substantially increased by treatment with the antioxidants N-acetylcysteine (NAC) or glutathione ethyl ester (GSH-EE), a membrane-permeable derivative of glutathione (Figures 3C–3E). In contrast, these antioxidants did not rescue proliferation of NRF2-depleted spheroids based on the Ki67 signal or spheroid size in H1437 spheroids (Figures 3F–3H), indicating that antioxidant responses are not sufficient for NRF2-mediated proliferation in spheroids. Thus, these results show that NRF2 regulates survival of inner matrix-deprived cells and proliferation in lung cancer spheroids by distinct mechanisms.
CRISPR-Cas9 Screens in 2D vs 3D Cultures Reveal Dependencies of Spheroid Growth in NRF2-Hyperactivated Cancer Cells
To identify critical genes and/or programs that mediate NRF2 hyperactivation-induced spheroid growth, and to define candidate dependencies of NRF2-hyperactivated tumors, we performed pooled CRISPR-Cas9 screens in A549 and H1437 cells. Because of issues associated with scaling to full genome screens using spheroids plated sparsely in reconstituted basement membrane (Matrigel™), we chose to screen genes most likely to play a role in NRF2-hyperactivated tumors – that is, genes that significantly correlated with genetic alterations that hyperactivate NRF2 in human tumors (Figures 2A, 2B, and 4A). Since antioxidants have been implicated in tumorigenesis and survival in 3D models, we also included a set of redox regulatory genes and other NRF2-regulated genes curated from the literature. In addition, we included a number of genes identified through NRF2 network analysis (GeneGO) or analysis of existing functional genomic screening data (Project Achilles: https://depmap.org/portal/achilles/). The list was pared by removing genes expressed at low levels in lung tumors or cell lines with alterations in KEAP1/CUL3/NFE2L2 and genes that were either coamplified with NFE2L2 or co-deleted with KEAP1/CUL3 (Figure 4B). Additionally, a number of non-targeting and positive control sgRNAs were included in the library for a total of 14,058 sgRNAs targeting ~1,500 genes (Figure 4B and Table S3).
Figure 4. CRISPR Screening Approach to Identify Dependencies in Spheroids of NRF2-Hyperactivated Cell Lines.

(A) Schematic for the identification of genes for the focused CRISPR library. (B) Distribution of source of genes in the focused CRISPR library. The most common sources of genes are highlighted. Non-identified groups include genes derived from more than one of the common sources or through Project Achilles. Expression filters were applied to further narrow down the list to ~1,500 genes. (C) Schematic for the pooled CRISPR screening approach. (D) Hit genes common to both A549 and H1437 cell lines. For each cell line, the difference in β-score between 3D and 2D growth culture conditions is plotted.
Given that the effects of NRF2 knockdown on 2D cell growth were not as robust as on 3D spheroid growth (Figures 1A and S1A), our screening approach was to simultaneously perform pooled CRISPR-Cas9 screens in A549 and H1437 cells under 2D and 3D culture conditions, in order to identify genes that are important particularly for spheroid growth (Figure 4C). Figure 4D shows those hits that were either enriched (positive 3D–2D β-score) or depleted (negative 3D–2D β-score) in both cell lines in 3D compared to 2D culture conditions. In total, sgRNAs targeting 41 genes were positively selected, and sgRNAs targeting 23 genes were negatively selected for during spheroid growth in both cell lines (all CRISPR screening results are available in Table S4). The evidence that both NFE2L2 and its binding partner MAFG were drop-out hits in both cell lines, with greater depletion in the 3D than in the 2D culture condition, serves as a validation of the CRISPR screening approach. Moreover, the dropout hits included three genes that encode for glycolytic and pentose phosphate pathway enzymes, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), glucose-6-phosphate isomerase (GPI), and transketolase (TKT), all of which are at important branchpoints in glucose metabolism for entry into/exit out of the pentose phosphate pathway (PPP). The dependency on these enzymes is consistent with previous reports suggesting that NRF2 supports cellular proliferation by redirecting glucose metabolism into the PPP (Best et al., 2018; Mitsuishi et al., 2012; Singh et al., 2013; Zhao et al., 2018). Notably, the most consistent and significant hits that were greater dependencies under 3D culture conditions include enrichment of sgRNAs targeting TSC1 and depletion of sgRNAs targeting GPX4.
Loss of TSC1 and GPX4 Enhances Proliferation and Inner Clearance, respectively, in Spheroids
TSC1, in complex with TSC2, serves as an upstream repressor of mTOR signaling. The enrichment of sgRNAs targeting TSC1 in the CRISPR-Cas9 screens suggests that activation of mTOR signaling provides a fitness advantage for growth in 3D culture conditions. Interestingly, analysis of drug sensitivity data from the Genomics of Drug Sensitivity in Cancer (GDSC; Release 7.0) (Yang et al., 2012) shows that lung cancer cell lines with high NRF2 scores are correlated with enhanced sensitivity to the PI3K/mTOR inhibitor BEZ235 (Figure S3A) and the rapamycin analog Temsirolimus (Figure S3B), suggesting that NRF2-hyperactivated tumors are more dependent on mTOR signaling.
To examine the effect of TSC1 loss on spheroid growth in detail, we knocked out TSC1 by CRISPR-Cas9 in the previously generated A549 and H1437 cells with or without NRF2 knockdown (Figure S3C). In both cell lines, loss of TSC1 increased the size of spheroids in cells expressing shControl, but not in cells expressing shNRF2 (Figures S3D–S3G). The observed phenotypes induced by TSC1 loss in spheroids with shControl are through the mTOR signaling, as Torin1, a specific mTOR inhibitor, abolished sgTSC1-induced increase in spheroid size (Figures S3H and S3I). These results suggest that proliferation in 3D culture is enhanced by activation of the mTOR signaling pathway, but only when NRF2 signaling is intact. Furthermore, while loss of TSC1 increased spheroid size, it did not increase survival of the inner cells of spheroids in NRF2-depleted cells (Figures S3F and S3G), suggesting that enhancement of proliferation in cancer cells is not sufficient to promote survival of inner spheroid cells.
The most prominent dropout hit in both A549 and H1437 cells was GPX4, which is protective against a form of non-apoptotic cell death known as ferroptosis by converting lipid hydroperoxides into non-toxic lipid alcohol (Stockwell et al., 2017; Wan et al., 2014). Given our previous studies demonstrating that the levels of ROS are elevated in the inner core of cancer spheroids (Schafer et al., 2009; Takahashi et al., 2018), these results raised the possibility that inner matrix-deprived cells are vulnerable to ROS-dependent ferroptosis.
Spheroids treated with the either of two GPX4 inhibitors, ML210 or RSL3, after formation of spheroids displayed decreased survival of cells in the inner luminal space in A549, H1437, and H520 cells (Figures 5A, 5B, and S4A–S4D). RSL3-induced inner clearance of spheroids was abolished by treatment with Ferrostatin-1 (Fer-1) (Figures S4C and S4D), a potent and selective inhibitor of ferroptosis (Dixon et al., 2012), suggesting that inner spheroid cells are vulnerable to ferroptosis. This is supported by the observation that treatment with linoleic acid, a precursor of ω6 polyunsaturated fatty acid-containing phospholipids (PUFA-PLs) which is a substrate for the reaction of lipid peroxidation (Doll et al., 2017; Kagan et al., 2017; Yang et al., 2016), induces clearing of the inner cells in A549 and H1437 spheroids (Figures S4E and S4F). These results phenocopied the increased clearance observed with downregulation of NRF2 in spheroids and implicate ferroptosis as the cell death mechanism responsible for inner cell clearance of spheroids.
Figure 5. NRF2 Controls Survival of Inner Spheroid Cells through Protection from Ferroptosis.

(A) Representative confocal images of the indicated spheroids from three independent experiments. Spheroids were treated with or without 10 μM ML210 for the last three days. (B) Percentage of filled inner space from experiments described in (A) (n = 35–77). (C) Representative confocal images of the indicated spheroids from three independent experiments. Spheroids were treated with 1 μg/ml doxycycline and either vehicle or 3 μM Fer-1 for 12 days. (D) Percentage of filled inner space from experiments described in (C) (A549) and Figure S4G (H1437) (n = 11–77). (E) Representative confocal images of the indicated spheroids from three independent experiments. (F) Quantification of spheroid size from experiments described in (E) (n = 18–112). (G) Quantification of Ki67 signal relative to Fer-1-untreated spheroids with shControl from experiments described in (E) (n = 4–24). (H) H2O2 levels (arbitrary units) in the indicated day-10 spheroids from three independent experiments. (I) Representative C11-Bodipy ratiometric images of the indicated day-10 A549 spheroids from two independent experiments. Spheroids were treated with or without 1 μM ML210 for the last three days. (J) Quantification of C11-Bodipy ratio in inner region relative to outer region from experiments described in (I) (n = 12–20). (K) Fe2+ levels in the indicated day-10 spheroids from three independent experiments. (L) Immunoblot analysis of NRF2 and ACSL4 in H1437 spheroids. In (E–G), H1437 spheroids were treated with 1 μg/ml doxycycline and either vehicle or 3 μM Fer-1 throughout the 3D culture. In (H), (K), and (L), the spheroids were treated with 1 μg/ml doxycycline throughout the experiments. In (B), (H), and (J), unpaired two-tailed t-test was used to determine statistical significance. In (D), (F), and (G), one-way ANOVA was used to determine statistical significance. In (K), paired two-tailed t-test was used to determine statistical significance. *p < 0.05, **p < 0.01, and ***p < 0.001. All data shown as mean ± SEM. Scale bar represents 100 μm.
NRF2 Regulates Survival of Inner Matrix-Deprived Cells through Protection from Ferroptosis
To address whether the inner clearance in spheroids associated with NRF2 knockdown is mediated by ferroptosis, we cultured A549 and H1437 spheroids in the presence of Fer-1. Fer-1 treatment prevented inner clearance in spheroids with NRF2 knockdown (Figures 5C, 5D, and S4G), suggesting that NRF2 regulates inner cell survival through protection from ferroptosis. Treatment of spheroids with Fer-1 did not rescue spheroid size or Ki67 staining (Figures 5E–5G), suggesting that NRF2-mediated regulation of proliferation does not involve protection from ferroptosis.
ROS as well as ω6 PUFA-PLs are substrates for the reaction of lipid peroxidation (Doll et al., 2017; Kagan et al., 2017). NRF2 knockdown increased levels of hydrogen peroxide (H2O2) in both A549 and H1437 spheroids (Figure 5H), suggesting that NRF2 suppresses lipid peroxidation through reduction of cellular ROS levels. Notably, NRF2 knockdown as well as ML210 treatment substantially increased the levels of lipid peroxides in the core of spheroids, as determined by the ratiometric lipid-peroxidation probe C11-BODIPY (Pap et al., 1999) (Figures 5I, 5J, S4H, and S4I), indicating both that inner spheroid cells are vulnerable to lipid peroxidation and that NRF2 regulates lipid peroxidation in spheroids. NRF2 knockdown did not increase intracellular levels of Fe2+, which can produce hydroxyl radical through the Fenton reaction and promote ferroptosis (Dixon et al., 2012), or expression of ACSL4, an enzyme responsible for ω6 PUFA-PL generation (Doll et al., 2017) (Figures 5K and 5L), suggesting that the increased sensitivity to ferroptosis in cells with downregulation of NRF2 is due to neither Fe2+ nor ACSL4. These results support our observation that NRF2 regulates survival of inner cells through antioxidant-dependent pathways in spheroids.
We next investigated the relationship between NRF2 and GPX4. Given that previous papers have suggested induction of GPX4 expression by NRF2 (Dodson et al., 2019; Osburn et al., 2006; Rojo De La Vega et al., 2018; Stockwell et al., 2017; Wu et al., 2011), we interrogated GPX4 levels in cells with NRF2 downregulation. Surprisingly, NRF2 downregulation dramatically increased GPX4 levels in both 2D and 3D culture conditions (Figures 6A and 6B). One possible explanation for this is that enhanced oxidative stress caused by NRF2 downregulation can activate other cytoprotective programs, such as NF-κB, that upregulate transcription of antioxidant enzymes. However, NRF2 knockdown did not increase GPX4 mRNA levels (Figure S5A), and oxidative stress induced by treatment with tert-butyl hydroperoxide (TBHP), a relatively stable alkyl hydroperoxide, or a GPX4 inhibitor ML210 did not enhance GPX4 protein levels in A549 and H1437 cells (Figure S5B). Furthermore, treatment with the antioxidants NAC or Trolox did not abolish NRF2 knockdown-induced enhancement of GPX4 expression in A549 and H1437 cells (Figure S5C). While NAC treatment suppressed GPX4 expression in H1437 cells with shNRF2, the level of expression was still higher compared to H1437 cells with shControl, and Trolox did not reduce GPX4 expression in H1437 cells with shNRF2. Thus, these data indicate that GPX4 mRNA expression is not induced by NRF2 knockdown and that enhanced oxidative stress is not sufficient to stabilize GPX4 protein.
Figure 6. Downregulation of NRF2 Induces Global Enrichment of Selenoproteins Including GPX4.

(A and B) Immunoblot analysis of NRF2 and GPX4 in the indicated cells. In (B), the blots for NRF2 and β-actin in 3D are from Figure 5L as these experiments were performed concurrently as the experiments presented in Figure 6B. (C) Schematic of proteomics approach. For H1437 cells with shControl, two samples were used for the experiment because the plex was limited to 11 for each TMT experiment. (D) All selenoproteins or proteins required for selenoprotein biosynthesis detected by proteomics in A549 cells transduced with shControl or shNRF2-#2. Data presented are mean log2 (fold change) of shNRF2-#2 compared to shControl cells from three technical replicates. (E) Intensity of all selenoproteins detected in the indicated A549 cells. Data presented are mean ± SD from three technical replicates. Uncorrected Student’s t-test was used to determine statistical significance. *p < 0.05, **p < 0.01, and ***p < 0.001. Unless otherwise noted, the cells were cultured for 72 hours upon treatment with 1 μg/ml doxycycline.
GPX4 is a selenoprotein that contains selenocysteine (Sec), the 21st amino acid. Therefore, a possible explanation for the induction of GPX4 protein following NRF2 downregulation could be a change in translation of selenoproteins. Interestingly, analysis of The Cancer Dependency Map (Tsherniak et al., 2017) (DepMap) data shows that the top five co-dependencies of GPX4 are genes required for the biosynthesis of Sec (Figure S5D). To interrogate whether translation of all selenoproteins or just GPX4 specifically is affected by NRF2 downregulation, we used isobaric tandem mass tag (TMT) labeling combined with LC-MS/MS analysis (McAlister et al., 2012) (Figure 6C). From the proteomics data (Figure S5E and Table S5), 11 of the 25 described selenoproteins in humans were detected (Table S6), and surprisingly, most of the detected selenoproteins exhibited increased expression following NRF2 downregulation (Figures 6D, 6E, and S5F). This suggests that the induction of GPX4 after NRF2 knockdown is not specific for this selenoprotein, rather most selenoproteins are affected by a global modulation to selenoprotein biosynthesis. In addition, of the three (out of five) selenoprotein biosynthesis genes that were co-dependencies of GPX4 (Figure S5D) and were detected by proteomics (Table S6), only SEPHS2, which itself is a selenoprotein, was changed (increased) after NRF2 downregulation. In contrast, TXNRD1, a bona fide NRF2 target gene (Malhotra et al., 2010), is strongly decreased after NRF2 knockdown even though TXNRD1 is also a selenoprotein (Figures 6E, S5F, and S5G). Given that TXNRD1 is highly abundant, up to orders of magnitudes higher than the other detectable selenoproteins (Figures 6E and S5F), the decrease in TXNRD1 protein levels in cells with NRF2 knockdown could release a large fraction of the total Sec pool, which could then be incorporated into other selenoproteins, resulting in increased expression of the selenoproteins. Indeed, sodium selenite treatment increased GPX4 protein expression in both cells with shControl and cells with shNRF2 (Figure S5G), indicating that selenium availability is a critical factor for GPX4 expression. Interestingly, selenium supplementation did not induce TXNRD1 expression. This could be at least in part due to the fact that selenium sufficiency induces an upregulation of the methylated isoform of the tRNA carrying selenocysteine mcm5Um tRNA[Ser]Sec. Since the unmethylated mcm5U isoform is specifically incorporated into TXNRD1 while both are incorporated in GPX4, an increase in TXNRD1 would not be predicted (Howard et al., 2013; Labunskyy et al., 2014). It has been also shown that expression level of TXNRD1 protein is unaffected by selenium availability because cysteine can substitute Sec in TXNRD1 under selenium deficiency conditions (Xu et al., 2010).
Targeting both NRF2 and GPX4 Leads to Substantial Cell Death in Lung Cancer Spheroids
Given that inner spheroid cells exhibit higher levels of lipid peroxidation, together with our observation that NRF2 downregulation increases GPX4 expression, it is possible that inner and outer cells of spheroids have differential oxidative stress capacities or that outer cells are able to adapt to increased oxidative stress in response to reduction of NRF2 or GPX4. Notably, treatment of spheroids with ML210 in combination with NRF2 downregulation resulted in death of both inner and outer cells (Figures 7A and 7B).
Figure 7. Combined Loss of NRF2 and Inhibition of GPX4 Leads to Death of both Inner and Outer Cells of Spheroids.

(A and B) Representative confocal images of the indicated spheroids from three independent experiments. Spheroids were treated with 10 μM ML210 and 1 μg/ml doxycycline for the indicated time periods. (C–E) Total area of cells/spheroids relative to ML210-untreated controls in the indicated cells treated with ML210 for the last three days. A549 and H1437 cells were cultured in 2D for four days or 3D for 12 days in the presence of either 1 μg/ml doxycycline (C) or both 1 μg/ml doxycycline and 3 μM Fer-1 (D and E). H520 and SK-MES-1 cells were cultured in either 2D for four days or 3D for 15 days in the presence of 1 μg/ml doxycycline. (F) Representative C11-Bodipy ratiometric images of the indicated day-10 A549 spheroids upon treatment with 1 μM ML210 for the last three days from two independent experiments. (G) Quantification of C11-Bodipy ratio in the indicated day-10 A549 spheroids treated with or without 1 μM ML210 for the last three days. The data for both ML210-untreated spheroids and ML210-treated spheroids with shControl are from Figures 5I, 5J, and S4I as these experiments were performed concurrently as the experiment presented in Figure 7G. Data shown as mean ± SEM (n = 8–20). One-way ANOVA was used to determine statistical significance. *p < 0.05 and ***p < 0.001. In (C), (D), and (E), data shown as mean ± SD from four independent experiments, and two-way ANOVA was used to determine statistical significance (*p < 0.05, **p < 0.01, and ***p < 0.001 compared to shControl). Scale bar represents 100 μm.
Next, we investigated whether combined reduction of NRF2 and inhibition of GPX4 activity was specifically lethal in 3D culture or whether this combination was also lethal to cells in 2D culture conditions. Interestingly, A549 cells were sensitive to ML210 even in the absence of genetic manipulations in 2D culture conditions, whereas H1437, H520, and SK-MES-1 cells were less or not sensitive (Figure 7C, left panels). This could be due to the difference in KRAS mutation state since ferroptosis inducers, such as ML210, were initially identified to be selectively lethal in RAS-mutant tumor cells and mesenchymal cell states (Dixon et al., 2012; Viswanathan et al., 2017; Weïwer et al., 2012; Yang and Stockwell, 2008) – A549 cells have an activating KRAS mutation, whereas the others contain wild-type KRAS. In 2D cultures, downregulation of NRF2 marginally sensitized A549, H1437, H520, and SK-MES-1 cells to ML210 (Figure 7C, left panels). In 3D cultures, induction of shNRF2 more strongly sensitized cells to ML210 (Figures 7C, right panels, and S6A). In both 2D and 3D culture conditions, sensitivity to ML210 was completely rescued by co-treatment with Fer-1 (Figures 7D and 7E). KEAP1 knockout enhanced survival of spheroid cells treated with ML210 in H596 cells, a cell line exhibiting an intermediate NRF2 score (Figure 2D), and ML210 induced inner clearance in H596 spheroids with sgKEAP1 (Figures S6B–S6E). This phenocopied the effect of NRF2 in NRF2-hyperactivated lung cancer cell lines.
Selenium supplementation did not affect survival of inner cells and proliferation but reduced sensitivity to ML210 in both spheroids with shControl and spheroids with shNRF2 (Figures S6F–S6K). However, NRF2 knockdown still substantially sensitized spheroid cells to ML210 even after selenium supplementation (Figure S6K), suggesting that silencing of NRF2 allows spheroid cells to be highly vulnerable to ferroptosis even when GPX4 expression is upregulated.
We next assessed whether the combined loss of NRF2 and GPX4 exacerbated lipid peroxidation to potentiate cell death (Figures 7F and 7G). Cells were treated with one-tenth the amount of ML210 previously used to avoid total cell death. In shGFP-transduced control A549 spheroids, ML210 treatment increased lipid peroxidation specifically in the inner region of spheroids. In A549 spheroids with NRF2 knockdown, ML210 treatment increased lipid peroxidation in both the inner and outer regions of the spheroids although the induction was more prominent in the inner region compared with the outer region of the spheroids. Interestingly, lipid peroxidation levels in the outer region of NRF2-knockdown spheroids with ML210 were comparable to those in the inner region of shNRF2-transduced spheroids without ML210 and shGFP-transduced spheroids with ML210 (Figure 7G). Both shNRF2-transduced spheroids without ML210 and shGFP-transduced spheroids with ML210 exhibited inner clearance. Given these results, the observed lipid peroxidation levels in the outer region of NRF2-knockdown spheroids with ML210 could be higher than the threshold level of lipid peroxidation that causes ferroptosis. Thus, the combined targeting of multiple oxidative stress defense programs is required to kill tumor cells efficiently.
DISCUSSION
Use of spheroid models enabled us to reveal that high NRF2 activity is necessary for lung cancer spheroid formation and that loss of NRF2 has significant effects on two distinct processes during the formation of spheroids: proliferation (an effect not rescued by antioxidants) and differential survival of inner spheroid cells (rescued by antioxidants) (Figure S7A).
Very recently, Han et al. carried out CRISPR screens in cells that form 3D cell colonies in medium without exogenous ECM, a condition that requires anchorage-independent proliferation and survival (Han et al., 2020). Their findings provided evidence that dependencies of tumor cells under these 3D conditions more closely resemble in vivo dependencies than those derived from monolayer culture. Importantly, their culture condition was significantly different from ours since we utilized a reconstituted basement membrane ECM (Matrigel™)-based 3D culture, which is typically used in organoid and other 3D models in order to better model in vivo cell-to-ECM interaction (Figure S7B). Interestingly, of our 1,500 CRISPR gene list, 93 genes overlap with their top 911 gene list, and phenotypes of the 93 genes from our screen correlated with those from their screen – in particular, GPX4 and TSC1 were among the strongest hits in A549 and H1437 cells in their screen (Figure S7C). The strong correlation may be attributed to the fact that ECM-free 3D colony cells may in part resemble the inner cells of ECM-based 3D spheroids. Moreover, A549 and H1437 cells could produce ECM by themselves in ECM-free 3D conditions.
Our study suggests that ferroptosis is involved in the non-apoptotic cell death associated with the loss of natural matrix interactions. The interplay between lipid peroxidation and ECM detachment is supported by the previous observation that in mice lacking the proapoptotic factor Bim, significant levels of 4-HNE, an indirect marker of lipid peroxidation, are detected in dying ECM-detached cells (Mailleux et al., 2007), which fail to undergo apoptosis due to Bim knockout, within mammary ducts. Consistent with our results in 2D cultures, it has been shown that NRF2 downregulation marginally sensitizes cells cultured in monolayers to ferroptosis-inducing compounds (Cao et al., 2019; Fan et al., 2017), except for cisplatin-resistant cancer cells (Shin et al., 2018). Moreover, NRF2 expression levels are shown to be a weak predictor of sensitivity to ferroptosis-inducing compounds across hundreds of cancer cell lines cultured as monolayers, based on the Cancer Therapeutics Response Portal database (https://portals.broadinstitute.org/ctrp/) (Cao et al., 2019; Rees et al., 2016). It is possible that the strong ECM-matrix complexes allow 2D monolayer cells to tolerate ferroptosis, as we observed in outer versus inner spheroid cells.
Our results suggest that NRF2 downregulation upregulates GPX4 expression through enhanced availability of selenium. This is supported by the recent study showing that loss of TXNRD1 increases GPX4 protein expression potentially by influencing availability of selenium (Cai et al., 2020). While selenium supplementation enhances GPX4 expression, it does not affect survival of inner cells and proliferation in spheroids, which could be consistent with the previous report demonstrating that selenium treatment does not affect colony formation in the presence of 10% FBS (Vande Voorde et al., 2019). Interestingly, selenium supplementation shifts dose-response curves for ML210 to the right; however, NRF2 knockdown still substantially sensitizes spheroid cells to ML210 even after selenium supplementation. Given that ROS are substrates for the reaction of lipid peroxidation and that GPX4 uses GSH as a cofactor for its enzymatic activity, these results suggest that silencing of NRF2, a key regulator of ROS and GSH, allows spheroid cells to be highly vulnerable to ferroptosis even when GPX4 expression is upregulated. A complete understanding of the relationship between NRF2 and selenoproteins requires further investigation.
The relevance of NRF2 effects on cell proliferation in human lung cancer is supported by our finding that the NRF2 score of primary patient lung tumors is correlated with the expression of a proliferation signature. Intriguingly, most other types of cancer cell lines formed spheroids effectively without NRF2 hyperactivation. For example, we have reported that breast cancer cell lines, T-47D, MDA-MB468, and HCC1569, as well as ovarian cancer cell lines, MCAS, SK.OV.3, and OvCA432, are able to form spheroids (Muranen et al., 2012; Takahashi et al., 2018); all of these cells lines have low NRF2 scores (Table S2). Moreover, NRF2 disruption did not alter spheroid growth in ductal carcinoma in situ (DCIS) and triple-negative breast cancer (TNBC) cells (Pereira et al., 2020). These lines of evidence suggest a unique dependence of lung cancer cell lines on NRF2 hyperactivation for spheroid formation. Indeed, the frequency of tumors with alterations in KEAP1, CUL3, or NFE2L2 varies from tissue to tissue. In good agreement with the clinical observation, Kras:Tp53:Keap1 triple mutations in the lung cause cancers with aggressive proliferation (Romero et al., 2017), whereas these triple mutations in the pancreas do not cause cancers but result in fibrosis instead (Hamada et al., 2018) in mice. Tissue-specific environmental factors are likely to determine the prerequisites for NRF2-addicted cancer development.
In general, metabolic reprogramming greatly contributes to the rapid proliferation of cancer largely through supporting biosynthetic needs. Indeed, most cytosolic NADPH (> 80%) is devoted to biosynthesis rather than to redox defense (Ye et al., 2014). One hypothesis for the NRF2-regulation of cell proliferation is through the involvement of NADPH or NADH-producing enzymes, including glycolytic and PPP enzymes that we obtained as dropout hits from the screen. Recent works have also implicated PPP enzymes as mediators for NRF2-induced proliferation since loss of NRF2 strongly represses PPP activities (Best et al., 2018; Mitsuishi et al., 2012; Zhao et al., 2018). It remains challenging to demonstrate this hypothesis as NRF2 induces numerous metabolic enzymes to carry out the appropriate rescue experiments.
The findings from our study have several important implications for cancer therapeutics. Our study has provided further support for the use of 3D culture models to identify therapeutic targets that may be masked in conventional 2D assays. Moreover, our findings provide further support for targeting the antioxidant capacity of cells as a therapeutic strategy in cancer. Importantly, we have observed that inner and outer cells are differentially vulnerable to ferroptosis, which suggests that different regions of tumors are likely to have differential capacities for oxidative stresses and sensitivity to ferroptosis-inducing compounds. Therefore, targeting multiple oxidative stress defense programs may be required for maximal killing of all cells within a tumor. In this context, a therapeutic strategy may be through induction of ferroptosis either by targeting NRF2 directly, targeting GPX4 directly, targeting NRF2 transcriptional targets that impact ferroptosis (such as SLC7A11), or through targeting other pathways that impact sensitivity to lipid peroxidation (such as ACSL4). The development of compounds targeting anti-ferroptotic regulators may be a promising approach and could be particularly effective in combination treatments akin to combination strategies including inhibitors of anti-apoptotic proteins which are being tested in clinical trials (Montero and Letai, 2018; Sun et al., 2016).
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents may be directed to and will be fulfilled by the Lead Contact, Joan S. Brugge (joan_brugge@hms.harvard.edu).
Materials Availability
All unique/stable reagents generated in this study will be made available upon request to the Lead Contact.
Data and Code Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD018634.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
All cell lines were obtained from, and authenticated by, ATCC. A549 and H1437 cells were further authenticated by STR profiling. The cells were cultured in RPMI 1640 (Life Technologies) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% penicillin/streptomycin (Life Technologies) (referred to as ‘complete media’ hereafter) at 37°C with 5% CO2. For cell counts and growth of A549 and H1437 spheroids shown in Figure 1A, cells were plated on 24-well ultra-low attachment plates (Corning) in complete media supplemented with 4% Matrigel™ (Corning). Media was refreshed every four days by carefully aspirating 50% of the well volume and replacing with fresh complete media containing 1% Matrigel™. For confocal images of spheroids, cells were plated on Matrigel™-coated glass bottom 24-well plates in complete media supplemented with 2% Matrigel™. The cells were re-fed with fresh media every two or three days. Cells were negative for mycoplasma contamination.
METHOD DETAILS
Plasmids, shRNAs, CRISPR/Cas9, and Virus Production
TRIPZ-inducible lentiviral NRF2 shRNAs were obtained from Dharmacon (Clone ID: V3THS_306092 for shNRF2-#1 and V3THS_306096 for shNRF2-#2). Inducible lentiviral non-silencing shRNA control was obtained from Dharmacon (RHS4743). TRC lentiviral NRF2 shRNAs (non-inducible) were obtained from Dharmacon (Clone ID: TRCN0000007555 for shNRF2-#1*and TRCN0000007558 for shNRF2-#2*). shRNA against GFP was purchased from Addgene (plasmid #: 30323) (Sancak et al., 2008). NFE2L2 mutants were constructed using NFE2L2-pLX304 (Dharmacon, Clone ID: OHS6085-213573755) according to the protocol of Q5® Site-Directed Mutagenesis (NEB) and QuikChange II XL Site-Directed Mutagenesis Kit (Agilent). Primers used for the construction are shown in Table S7. The nucleotide sequences of the mutants were verified by sequencing the corresponding cDNA. LentiCRISPR plasmids were constructed using lentiCRISPRv2-puro or lentiCRISPRv2-blast containing two expression cassettes, hSpCas9 and the chimeric guide RNA, according to the standard protocol (Sanjana et al., 2014; Shalem et al., 2014). All constructs were sequence verified, and primer sequence information is shown in Table S7. Lentiviruses for shRNAs, CRISPR/Cas9, and NFE2L2-pLX304 were made in 293T cells according to standard protocol, and transduced cells were selected with puromycin or blasticidin for at least 1 week.
Cell Viability Assays in 2D and 3D culture
Since cells expressing doxycycline-inducible shRNAs also express RFP driven by the same tetracycline response element, RFP-positive areas were measured using an Acumen Cellista (TTPLabTech) or ImageXpress Pico (Molecular Devices) in order to determine relative cell viability. For 2D culture of cells expressing doxycycline-inducible shRNAs, the cells were cultured on 96 black well plates in complete media supplemented with 1 μg/mL doxycycline. For 3D culture of cells expressing doxycycline-inducible shRNAs, the cells were plated on Matrigel™-coated 96 black well plates in complete media supplemented with 2% Matrigel™ and 1 μg/mL doxycycline. The spheroid cells were re-fed with fresh media every two or three days and were fixed with 4% paraformaldehyde at the end point. For competition assays shown in Figure S1A, the cells were treated with 1 μg/ml doxycycline from two-day before the initiation of co-culture experiments. RFP-positive shControl or shNRF2 cells were then co-cultured with parental cells at a 1:1 ratio in the presence of 1 μg/ml doxycycline. For cell counts and growth of a panel of lung cancer cell lines shown in Figure S2C, cells were cultured with complete media on 96 black well plates, fixed with 4% paraformaldehyde, and then stained with DAPI (Sigma). DAPI-positive cells were counted using ImageXpress Pico (Molecular Devices).
Quantitative PCR
RNA prepared from cell extracts using PureLink™ RNA Mini Kit (Life Technologies) was reverse-transcribed into cDNA using the qScript cDNA synthesis kit (Quanta Biosciences). Real-time qPCR was performed on a QuantStudio 7 Flex Real Time PCR System (Thermo Fisher) or StepOnePlus™ Real Time PCR Systems (Thermo Fisher) using Power SYBR Green PCR Mix (Life Technologies). The qPCR primer sequences used are listed in Table S7. Expression was normalized to RPS9 or β-actin.
Immunofluorescence Staining and Confocal Imaging
Cells were plated on Matrigel™-coated, glass-bottom, 24-well plates in complete media supplemented with 2% Matrigel™. Media was replaced every two or three days with fresh complete media containing 2% Matrigel™. Complete media was supplemented with doxycycline, NAC, GSH-EE, ML210, RSL3, Torin1, or Ferrostatin-1 at the concentrations and durations indicated in figure legends. Spheroids were fixed with 4% paraformaldehyde and stained as previously described (http://brugge.hms.harvard.edu). Ki67 antibody (Dako Cat. No. M724029-2) was used for cell proliferation analysis. Nuclei were counterstained with DAPI (Sigma). Fluorescent images were acquired using the A1R point scanning confocal microscope (Nikon) or the LSM 700 laser scanning confocal microscope (Carl Zeiss) and are representative of at least two independent experiments where indicated. Data was processed with NIS Elements software (Nikon) or ZEN microscope software (Carl Zeiss).
Bioinformatic Analyses to Derive NRF2 Signature and NRF2 Score
LUSC, LUAD, HNSC, CESC patient data (RNAseqV2, mutation and copy number) was retrieved from The Cancer Genome Atlas (TCGA) Research Network (https://www.cancer.gov/tcga). Within each type of cancer, patient data was divided into groups: those with hyperactivating genetic alterations in the NRF2 pathway (mutation/amplification of NFE2L2 or mutation/deletion of KEAP1 or CUL3) and those without genetic alterations in the NRF2 pathway. Differentially expressed genes were identified between these groups using a Student’s t-test with a Bonferroni correction. In total, 1,466 genes from all four cancer types were found to be associated with hyperactivating genetic alterations in the NRF2 pathway. From the large number of genes that were associated with NRF2-hyperactivating alterations, a 55 gene “NRF2 signature” was selected by limiting to those genes that were more highly expressed in altered tumors compared to unaltered tumors and associated with genetic alterations in LUSC and at least two out of the other three tumor types. The “NRF2 score” was defined as the median value for the 55 NRF2 signature genes in cell lines and patient data.
Pooled CRISPR-Cas9 Screen in 2D and 3D culture
A custom, pooled lentiCRISPR library was generated by Sigma-Aldrich. Identification of approximately 1,500 genes for the custom library are described in the Results. For 1,400 of these genes (including 5 essential genes that were specifically included as positive controls), the coverage was 10 sgRNAs per gene. For the remaining genes of interest, only those genes in which at least 3 sgRNAs were designed were included in the production of the library. Additionally, 100 non-targeting sgRNAs were included in the library for negative controls. Cloning of sgRNAs into the lentiGuide-puro backbone, verification of library content by deep sequencing, and production and titering of virus were performed by Sigma. A549 and H1437 cells were transduced with lentiCas9-blast and then with the lentiGuide-puro library at an expected MOI of 0.25 and coverage of 500 cells per sgRNA. Immediately after puromycin selection, one aliquot of cells was saved as the reference sample (T0 sample). The remaining cells were plated for 2D and 3D culture in complete media supplemented with 1% Matrigel™. Cells cultured in 2D were passaged approximately every four days or as needed, with aliquots of cells saved at each passage. Cells in 3D culture were maintained in ultra-low attachment flasks (Corning), and media was replenished every four days by carefully aspirating 50% of the media and replacing with fresh complete media supplemented with 1% Matrigel™. After 20 days in culture, the number of population doublings in 3D was determined and cells were collected for gDNA isolation (3D sample). The saved aliquot of cells cultured in 2D with the most closely matched number of population doublings was submitted as the 2D sample. gDNA from the T0, 2D, and 3D samples were isolated using the QIAamp DNA Mini Kit (Qiagen). A nested PCR amplification protocol was used to amplify sgRNA sequences. PCR using outer primers was conducted for 12 cycles under the following conditions: initial denaturation was 2 min at 95°C, then 20 sec at 95°C, followed by a 30-sec annealing step at 56°C and 18-sec elongation at 72°C, and a final elongation of 2 min at 72°C. PCR using inner primers was conducted for 18 cycles under the following conditions: initial denaturation was 2 min at 95°C, then 20 sec at 95°C, following by a 30-sec annealing step at 56°C and 18-sec elongation at 72°C, and a final elongation of 2 min at 72°C. The primers used are listed in Table S7. Amplified PCR product was purified and gel extracted using the QIAquick PCR Purification Kit (Qiagen) and QIAquick Gel Extraction Kit (Qiagen), respectively. Quality control and quantification of PCR product was determined by TapeStation and qPCR analysis at the Bauer Core (Harvard University). Samples were pooled 1:1 and a 50% PhiX DNA spike-in was included and sequenced on an Illumina NexSeq at the Bauer Core. Quality control, mapping, normalization, and analysis of sequencing data was performed using MAGeCK-VISPR (Li et al., 2015).
Western Blots
For analysis of NRF2, cells were treated with MG132 (20 μM) for the last 1.5 hours of culture. Cells were lysed in RIPA lysis buffer (Boston BioProducts) containing protease and phosphatase inhibitor cocktails (Roche) and MG132 (Sigma-Aldrich). Protein concentration was quantified by BCA assay (Thermo Fisher). Equal amounts of protein were run on 4–20% Tris-Glycine gels (Life Technologies) and transferred to PVDF membranes. Membranes were blocked with 5% w/v bovine serum albumin or 5% w/v non-fat dry milk in Tris-buffered saline with 0.1% Tween-20. The following primary antibodies were used: β-actin (Sigma Cat. No. A1978), NRF2 (Cell Signaling Cat. No. 12721), GPX4 (Abcam Cat. No. ab125066), TSC1 (Cell Signaling Cat. No. 6935), ACSL4 (Santa Cruz Cat. No. sc-271800), TXNRD1 (Cell Signaling Cat. No. 15140), RPS6 (Cell Signaling Cat. No. 2217). Blots were imaged using Luminata Western HRP substrate (EMD Millipore) or IRdye secondary antibodies (LI-COR) and are representative of at least two independent experiments where indicated.
ROS, Fe2+, and GSH/GSSG Measurement
For ROS measurement in 2D cells vs 3D spheroids shown in Figure 3A, cells were cultured in either complete media using 6 well plates (2D) or complete media supplemented with 1% Matrigel™ using ultra-low attachment 6 well plates (3D). The cells were treated with trypsin in order to dissociate cells and to remove Matrigel and then cultured with complete media for 30 min at 37°C. The cells were then subjected to ROS assay using OxiSelect™ Hydrogen Peroxide Assay Kit (Cell Biolabs) according to the manufacturer’s protocol. For GSH/GSSG measurement in 2D cells vs 3D spheroids shown in Figure 3B, cells were cultured in either complete media using 96 white well plates (2D) or complete media supplemented with 2% Matrigel™ using Matrigel™-coated 96 white well plates (3D). The cells were then subjected to GSH/GSSG assay using GSH/GSSG-Glo™ Assay kit (Promega) according to the manufacturer’s protocol. For ROS and Fe2+ measurements in shControl vs shNRF2 spheroids shown in Figures 5H and 5K, cells were cultured in complete media supplemented with 1% Matrigel™ using ultra-low attachment flasks. ROS levels were assessed using the ROS-Glo™ H2O2 Assay (Promega) according to the manufacturer’s protocol. Ferrous (Fe2+) and ferric (Fe3+) iron were assayed using an Iron Assay Kit (Abcam) according to the manufacturer’s protocol.
C11-BODIPY Lipid Peroxidation Assay in Spheroids
Cells were transduced with TRC non-inducible lentiviral shGFP, shNRF2-#1*, or shNRF2-#2* as TRIPZ-inducible lentiviral NRF2 shRNAs have the tetracycline-dependent promoter that induces expression of both shRNA and RFP, which disturbs C11-BODIPY™ 581/591 signals. The cells were cultured on Matrigel™-coated, glass-bottom, 24-well plates in complete media supplemented with 2% Matrigel™ and 1 μM C11-BODIPY™ 581/591 (Invitrogen) for 10 days. Media was refreshed every two or three days. Ratiometric images of C11-BODIPY 581/591 were obtained by simultaneous acquisition of fluorescent images (excitation: 488 nm, emission: 520 nm and 595 nm) using the Eclipse Ti-E microscopes with A1R point scanning confocal (Nikon). Data was processed using the NIS Elements software (Nikon).
Proteomic Mass Spectrometry, Data Processing and Analysis
(i). Sample Preparation
Cells were plated in complete media and allowed to adhere to plates for 24 hours. shRNAs were induced by replacing media with fresh complete media containing doxycycline (1 μg/mL). Three replicates of shControl and shNRF2 of A549 cells and two replicates of shControl and three replicates of shNRF2 of H1437 cells were conducted. Seventy-two hours after induction of shRNAs, cells were trypsinized, washed with PBS, and snapped frozen. Cells were syringe-lysed in 8 M urea and 200 mM EPPS pH 8.5 with protease inhibitor. BCA assay was performed to determine protein concentration of each sample. Samples were reduced in 5 mM TCEP, alkylated with 10 mM iodoacetamide, and quenched with 15 mM DTT. One hundred μg protein was chloroform-methanol precipitated and re-suspended in 100 μL 200 mM EPPS pH 8.5. Protein was digested by Lys-C at a 1:100 protease-to-peptide ratio overnight at room temperature with gentle shaking. Trypsin was used for further digestion for 6 hours at 37°C at the same ratio with Lys-C. After digestion, 30 μL acetonitrile (ACN) was added into each sample to 30% final volume. Two hundred μg TMT reagent (126C, 127N, 127C, 128N, 128C, 129N, 129C, 130N, 130C, 131N or 131C) in 10 μL ACN was added to each sample. After 1 hour of labeling, 2 μL of each sample was combined, desalted, and analyzed using mass spectrometry. Total intensities were determined in each channel to calculate normalization factors. After quenching using 0.3% hydroxylamine, eleven samples were combined in 1:1 ratio of peptides based on normalization factors. The mixture was desalted by solid-phase extraction and fractionated with basic pH reversed phase (BPRP) high performance liquid chromatography (HPLC), collected onto a 96 six well plate and combined for 24 fractions in total. Twelve fractions were desalted and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Navarrete-Perea et al., 2018).
(ii). Liquid chromatography and tandem mass spectrometry
Mass spectrometric data were collected on an Orbitrap Fusion mass spectrometer coupled to a Proxeon NanoLC-1200 UHPLC. The 100 μm capillary column was packed with 35 cm of Accucore 50 resin (2.6 μm, 150Å; ThermoFisher Scientific). The scan sequence began with an MS1 spectrum (Orbitrap analysis, resolution 120,000, 350–1400 Th, automatic gain control (AGC) target 4E5, maximum injection time 50 ms). SPS-MS3 analysis was used to reduce ion interference (Gygi et al., 2019; Paulo et al., 2016). The top ten precursors were then selected for MS2/MS3 analysis. MS2 analysis consisted of collision-induced dissociation (CID), quadrupole ion trap analysis, automatic gain control (AGC) 1E4, NCE (normalized collision energy) 35, q-value 0.25, maximum injection time 60 ms), and isolation window at 0.7. Following acquisition of each MS2 spectrum, we collected an MS3 spectrum in which multiple MS2 fragment ions are captured in the MS3 precursor population using isolation waveforms with multiple frequency notches. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap (NCE 65, AGC 3E5, maximum injection time 150 ms, resolution was 50,000 at 400 Th).
(iii). Data analysis
Mass spectra were processed using a Sequest-based pipeline (Eng et al., 1994; Huttlin et al., 2010). Spectra were converted to mzXML using a modified version of ReAdW.exe. Database searching included all entries from the Human UniProt database (downloaded: 2014-02-04). This database was concatenated with one composed of all protein sequences in the reversed order. Searches were performed using a 50 ppm precursor ion tolerance for total protein level analysis. The product ion tolerance was set to 0.9 Da. TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification.
Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR) (Elias and Gygi, 2007, 2010). PSM filtering was performed using a linear discriminant analysis (LDA), as described previously (Huttlin et al., 2010), while considering the following parameters: XCorr, ΔCn, missed cleavages, peptide length, charge state, and precursor mass accuracy. For TMT-based reporter ion quantitation, we extracted the summed signal-to-noise (S:N) ratio for each TMT channel and found the closest matching centroid to the expected mass of the TMT reporter ion. For protein-level comparisons, PSMs were identified, quantified, and collapsed to a 1% peptide false discovery rate (FDR) and then collapsed further to a final protein-level FDR of 1%, which resulted in a final peptide level FDR of <0.1%. Moreover, protein assembly was guided by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides.
Proteins were quantified by summing reporter ion counts across all matching PSMs, as described previously (Huttlin et al., 2010). PSMs with poor quality, MS3 spectra with more than eight TMT reporter ion channels missing, MS3 spectra with TMT reporter summed signal-to-noise of less than 100, or having no MS3 spectra were excluded from quantification (McAlister et al., 2012). Each reporter ion channel was summed across all quantified proteins and normalized assuming equal protein loading of all 11 samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
Error bars represent either the SD or SEM, as described in the figure legends. The sample size for each experiment, n, is included in the associated figure legend. Statistical significance was determined by unpaired or paired two-tailed t-test and the one-way or two-way ANOVA followed by Tukey’s or Dunnett’s test using GraphPad Prism 7.0 or RStudio. p-values < 0.05 were considered significant. p-values for each experiment are also included in the associated figure legends.
Supplementary Material
Table S1. 1,466 Genes That Significantly Correlate with NRF2-Hyperactivating Genetic Alterations and 55 NRF2 Signature Genes, Related to Figure 2.
Table S2. NRF2 Score of 675 Cancer Cell Lines, Related to Figure 2.
Table S3. 1,500 Genes Selected for CRISPR-Cas9 Screens, Related to Figure 4.
Table S4. Summary of Results from CRISPR-Cas9 Screens in 2D and 3D for A549 and H1437 Cell Lines, Related to Figure 4. Data on all the genes and hit genes common to both cell lines are shown.
Table S5. LS-MS/MS Data (Sum Date), Related to Figure 6.
Table S6. Abundance of Selenoproteins and Proteins Required for Selenoprotein Biosynthesis by Proteomics in A549 and H1437 Cells, Related to Figure 6.
Table S7. Primer Sequences, Related to STAR METHODS. Primer sequences used for the construction of NFE2L2 mutants and LentiCRISPR plasmids, quantitative PCR, and amplification of sgRNA are shown.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal antibody anti-Ki-67 | Dako | Cat# M7240, RRID:AB_2142367 |
| Mouse monoclonal antibody anti-beta-Actin | Sigma-Aldrich | Cat# A1978, RRID:AB_476692 |
| Rabbit monoclonal antibody anti-NRF2 | Cell Signaling Technology | Cat# 12721, RRID: AB_2715528 |
| Rabbit monoclonal antibody anti-GPX4 | Abcam | Cat# ab125066, RRID: AB_10973901 |
| Rabbit monoclonal antibody anti-TSC1 | Cell Signaling Technology | Cat# 6935, RRID: AB_10860420 |
| Mouse monoclonal antibody anti-ACSL4 | Santa Cruz Biotechnology | Cat# sc-271800, RRID: AB_10715092 |
| Rabbit monoclonal antibody anti-TRXR1 | Cell Signaling Technology | Cat# 15140, RRID: AB_2798725 |
| Rabbit monoclonal antibody anti-S6 Ribosomal Protein (RPS6) | Cell Signaling Technology | Cat# 2217, RRID: AB_331355 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DAPI | Sigma-Aldrich | Cat# D9542, CAS: 28718-90-3 |
| Doxycycline hydrochloride | Sigma-Aldrich | Cat# D3447, CAS: 10592-13-9 |
| ML210 | Sigma-Aldrich | Cat# SML0521, CAS: 1360705-96-9 |
| RSL3 | Sigma-Aldrich | Cat# SML2234, CAS: 1219810-16-8 |
| Torin1 | Tocris | Cat# 4247 CAS: 1222998-36-8 |
| Ferrostatin-1 | Sigma-Aldrich | Cat# SML0583, CAS: 347174-05-4 |
| BODIPY™ 581/591 C11 | Invitrogen | Cat# D3861 |
| Roche cOmplete, EDTA-free | Sigma-Aldrich | Cat#11873580001 |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher | Cat# 90058 |
| Lys-C, Mass Spectrometry Grade | Wako Chemicals | Barcode#4987481427648 |
| TMT10plex Isobaric Label Reagent Set plus TMT11-131C Label Reagent | Thermo Fisher | Cat# A34808 |
| Critical Commercial Assays | ||
| Q5® Site-Directed Mutagenesis Kit | NEB | Cat# E0554S |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent | Cat# 200522 |
| ROS-Glo™ H2O2 Assay | Promega | Cat# G8821 |
| OxiSelect™ Hydrogen Peroxide/Peroxidase Assay Kit | Cell Biolabs | Cat# STA-344 |
| GSH/GSSG-Glo™ Assay | Promega | Cat# V6611 |
| Iron Assay Kit | Abcam | Cat# ab83366 |
| Deposited Data | ||
| RNAseqV2, mutation and copy number | The Cancer Genome Atlas | https://www.cancer.gov/tcga |
| Functional genomic screening data | Project Achilles | https://depmap.org/portal/achilles/ |
| Small-molecule sensitivity data | Genomics of Drug Sensitivity in Cancer | https://www.cancerrxgene.org/ |
| Small-molecule sensitivity data | The Cancer Therapeutics Response Portal | https://portals.broadinstitute.org/ctrp/ |
| Mass spectrometry proteomics data for A549 and H1437 cells with or without NRF2 knockdown | This paper | ProteomeXchange Consortium (https://www.proteomexchange.org), Identifier: PXD018634 |
| Experimental Models: Cell Lines | ||
| H522 | ATCC | Cat# CRL-5810, RRID: CVCL_1567 |
| H1975 | ATCC | Cat# CRL-5908, RRID: CVCL_1511 |
| H226 | ATCC | Cat# CRL-5826, RRID: CVCL_1544 |
| SW 900 | ATCC | Cat# HTB-59, RRID: CVCL_1731 |
| H1568 | ATCC | Cat# CRL-5876, RRID: CVCL_1476 |
| H1838 | ATCC | Cat# CRL-5899, RRID: CVCL_1499 |
| H23 | ATCC | Cat# CRL-5800, RRID: CVCL_1547 |
| H358 | ATCC | Cat# CRL-5807 RRID: CVCL_1559 |
| H596 | ATCC | Cat# HTB-178 RRID: CVCL_1571 |
| H520 | ATCC | Cat# HTB-182 RRID: CVCL_1566 |
| SK-MES-1 | ATCC | Cat# HTB-58 RRID: CVCL_0630 |
| H1792 | ATCC | Cat# CRL-5895, RRID: CVCL_1495 |
| H2228 | ATCC | Cat# CRL-5935, RRID: CVCL_1543 |
| A549 | ATCC | Cat# CCL-185, RRID: CVCL_0023 |
| H1437 | ATCC | Cat# CRL-5872, RRID: CVCL_1472 |
| H460 | ATCC | Cat# HTB-177, RRID: CVCL_0459 |
| H1299 | ATCC | Cat# CRL-5803, RRID: CVCL_0060 |
| H2030 | ATCC | Cat# CRL-5914, RRID: CVCL_1517 |
| H2122 | ATCC | Cat# CRL-5985, RRID: CVCL_1531 |
| H1944 | ATCC | Cat# CRL-5907, RRID: CVCL_1508 |
| Oligonucleotides | ||
| Primers for the construction of NFE2L2 mutants: listed in Table S7 | This paper | N/A |
| Primers for the construction of LentiCRISPR plasmids: listed in Table S7 | This paper | N/A |
| Primers for quantitative PCR: listed in Table S7 | This paper | N/A |
| Primers for amplification of sgRNA: listed in Table S7 | This paper | N/A |
| Recombinant DNA | ||
| TRIPZ-inducible lentiviral shNRF2-#1 | Dharmacon | V3THS_306092 |
| TRIPZ-inducible lentiviral shNRF2-#2 | Dharmacon | V3THS_306096 |
| Inducible lentiviral non-silencing shRNA | Dharmacon | RHS4743 |
| TRC lentiviral shNRF2-#1* | Dharmacon | TRCN0000007555 |
| TRC lentiviral shNRF2-#2* | Dharmacon | TRCN0000007558 |
| shRNA against GFP | Addgene (Sancak et al., 2008) | Plasmid #30323 |
| NFE2L2-pLX304 | Dharmacon | OHS6085-213573755 |
| NFE2L2-pLX304 mutants | This paper | N/A |
| LentiCRISPR plasmids | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7.0 | GraphPad Prism Software | N/A |
| RStudio | RStudio | N/A |
| Database searching and reporter ion quantitation in-house software | Huttlin et al., 2010 | NA |
| Sequest algorithm | Eng et al., 1994 | NA |
| R | https://cran.r-project.org/ | Version 3.5.1 |
| MAGeCK-VISPR | Li et al., 2015 | Version 0.5.6 |
| Other | ||
| Waters 200mg Sep-Pak | Waters | Prod#WAT054945 |
| Orbitrap Fusion | Thermo Fisher | IQLAAEGAAPFADBMBCX |
Highlights.
Proliferation and survival of lung tumor spheroid cells require NRF2 hyperactivation
NRF2 prevents ferroptotic death of inner, matrix-deprived spheroid cells
NRF2 regulates selenoprotein expression through alteration of selenium availability
Targeting both NRF2 and GPX4 efficiently kills all cells within spheroids
ACKNOWLEDGMENTS
We are grateful to Angie Martinez-Gakidis for scientific editing and to Stephan Gysin, Daniel Rohle, and Astrid Ruefli-Brasse for helpful discussions. We also thank Yasuo Mori and Itaru Hamachi as well as the staff of the Nikon Imaging Center and the ICCB-Longwood Screening Facility at Harvard Medical School for providing support and access to instruments. This work was supported by a grant from F. Hoffmann-La Roche Ltd (J.S.B.), Japan Agency for Medical Research and Development (N.T.), and National Institutes of Health GM97645 (S.P.G.).
DECLARATION OF INTERESTS
J.S.B. receives funding for this project from F. Hoffmann-La Roche Ltd. J.S.B. is a consultant for Agios Pharmaceuticals, eFFECTOR Therapeutics, and Frontier Medicines. I.S.H. is a consultant for ONO Pharmaceuticals US.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Best SA, De Souza DP, Kersbergen A, Policheni AN, Dayalan S, Tull D, Rathi V, Gray DH, Ritchie ME, McConville MJ, et al. (2018). Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. Cell Metab. 27, 935–943. [DOI] [PubMed] [Google Scholar]
- Cai LL, Ruberto RA, Ryan MJ, Eaton JK, Schreiber SL, and Viswanathan VS (2020). Modulation of ferroptosis sensitivity by TXNRD1 in pancreatic cancer cells. bioRxiv. 2020: 10.1101/2020.06.25.165647. [DOI] [Google Scholar]
- Cao JY, Poddar A, Magtanong L, Lumb JH, Mileur TR, Reid MA, Dovey CM, Wang J, Locasale JW, Stone E, et al. (2019). A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep. 26, 1544–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cescon DW, She D, Sakashita S, Zhu CQ, Pintilie M, Shepherd FA, and Tsao MS (2015). NRF2 pathway activation and adjuvant chemotherapy benefit in lung squamous cell carcinoma. Clin. Cancer Res. 27, 2499–2505. [DOI] [PubMed] [Google Scholar]
- Chandel NS, and Tuveson DA (2014). The promise and perils of antioxidants for cancer patients. N. Engl. J. Med 377, 177–178. [DOI] [PubMed] [Google Scholar]
- Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, and Vousden KH (2020). Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 37, 168–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi P, and Giannoni E (2008). Anoikis: A necessary death program for anchorage-dependent cells. Biochem. Pharmacol 76, 1352–1364. [DOI] [PubMed] [Google Scholar]
- Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Öhlund D, et al. (2016). NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 766, 963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio IIC, and Tuveson DA (2017). ROS in cancer: the burning question. Trends Mol. Med 23, 411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, Beer DG, Cope L, Creighton CJ, Danilova L, et al. (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci. Adv 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, and Brugge JS (2005). Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 5, 675–688. [DOI] [PubMed] [Google Scholar]
- Denicola GM, Chen P-H, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. (2015). NRF2 regulates serine biosynthesis in non–small cell lung cancer. Nat. Genet 47, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson M, Castro-Portuguez R, and Zhang DD (2019). NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2010). Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol 604, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, and Savaskan N (2017). Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasauer A, Sena LA, Diebold LP, Mazar AP, and Chandel NS (2014). Targeting SOD1 reduces experimental non–small-cell lung cancer. J. Clin. Invest 124, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, and Mak TW (2013). Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov 12, 931–947. [DOI] [PubMed] [Google Scholar]
- Gygi JP, Yu Q, Navarrete-Perea J, Rad R, Gygi SP, and Paulo JA (2019). Web-based search tool for visualizing instrument performance using the triple knockout (TKO) proteome standard. J. Proteome Res 18, 687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada S, Shimosegawa T, Taguchi K, Nabeshima T, Yamamoto M, and Masamune A (2018). Simultaneous K-ras activation and Keap1 deletion cause atrophy of pancreatic parenchyma. Am. J. Physiol. Gastrointest. Liver Physiol 314, G65–G74. [DOI] [PubMed] [Google Scholar]
- Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, Stojanov P, McKenna A, Lander ES, Gabriel S, et al. (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Pierce SE, Li A, Spees K, Anderson GR, Seoane JA, Lo YH, Dubreuil M, Olivas M, Kamber RA, et al. (2020). CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 580, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawk MA, and Schafer ZT (2018). RIPK1-dependent mitophagy: A novel mechanism to eliminate cells detached from the extracellular matrix. Mol. Cell. Oncol 5, e1465015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma S, Ishii Y, Morishima Y, Yamadori T, Matsuno Y, Haraguchi N, Kikuchi N, Satoh H, Sakamoto T, Hizawa N, et al. (2009). Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res 15, 3423–3432. [DOI] [PubMed] [Google Scholar]
- Howard MT, Carlson BA, Anderson CB, and Hatfield DL (2013). Translational redefinition of UGA codons is regulated by selenium availability. J. Biol. Chem 288, 19401–19413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, et al. (2016). Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura H, and Motohashi H (2018). NRF2 addiction in cancer cells. Cancer Sci. 109, 900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labunskyy VM, Hatfield DL, and Gladyshev VN (2014). Selenoproteins: molecular pathways and physiological roles. Physiol. Rev 94, 739–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Köster J, Xu H, Chen C-H, Xiao T, Liu JS, Brown M, and Liu XS (2015). Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 16, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, and Brugge JS (2007). BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev. Cell 12, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, et al. (2010). Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, et al. (2012). Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem 84, 7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, and Motohashi H (2012). Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79. [DOI] [PubMed] [Google Scholar]
- Montero J, and Letai A (2018). Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 25, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, and Brugge JS (2012). Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 21, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete-Perea J, Yu Q, Gygi SP, and Paulo JA (2018). Streamlined Tandem Mass Tag (SL-TMT) protocol: an efficient strategy for quantitative (phospho)proteome profiling using tandem mass tag-synchronous precursor selection-MS3. J. Proteome Res 17, 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osburn WO, Wakabayashi N, Misra V, Nilles T, Biswal S, Trush MA, and Kensler TW (2006). Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch. Biochem. Biophys 454, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pap EHW, Drummen GPC, Winter VJ, Kooij TWA, Rijken P, Wirtz KWA, Op Den Kamp JAF, Hage WJ, and Post JA (1999). Ratio-fluorescence microscopy of lipid oxidation in living cells using C11-BODIPY 581/591. FEBS Lett. 453, 278–282. [DOI] [PubMed] [Google Scholar]
- Paulo JA, O’Connell JD, and Gygi SP (2016). A triple knockout (TKO) proteomics standard for diagnosing ion interference in isobaric labeling experiments. J. Am. Soc. Mass Spectrom 27, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira EJ, Burns JS, Lee CY, Marohl T, Calderon D, Wang L, Atkins KA, Wang CC, and Janes KA (2020). Sporadic activation of an oxidative stress-dependent NRF2-p53 signaling network in breast epithelial spheroids and premalignancies. Sci. Signal 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo H-K, Jang HG, Jha AK, et al. (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy NM, Kleeberger SR, Cho H-Y, Yamamoto M, Kensler TW, Biswal S, and Reddy SP (2007). Deficiency in Nrf2-GSH signaling impairs type II cell growth and enhances sensitivity to oxidants. Am. J. Respir. Cell Mol. Biol 37, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, Javaid S, Coletti ME, Jones VL, Bodycombe NE, et al. (2016). Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol 12, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo De La Vega M, Chapman E, and Zhang DD (2018). NRF2 and the hallmarks of cancer. Cancer Cell 34, 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, Leboeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sanchez-Rivera FJ, et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med 23, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, and Sabatini DM (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, and Brugge JS (2009). Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selfors LM, Stover DG, Harris IS, Brugge JS, and Coloff JL (2017). Identification of cancer genes that are independent of dominant proliferation and lineage programs. Proc. Natl. Acad. Sci. USA 114, E11276–E11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D, Kim EH, Lee J, and Roh J-L (2018). Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med 129, 454–462. [DOI] [PubMed] [Google Scholar]
- Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J, Blackford A, Goodman SN, Bunz F, Watson WH, et al. (2008). RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 68, 7975–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, Nasipuri P, Krausz KW, Wakabayashi N, Dewi R, et al. (2013). Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Invest 123, 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, Corvalan AH, Biswal S, Swisher SG, Bekele BN, et al. (2010). Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res 16, 3743–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, and Tang D (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Chen H-Y, Harris IS, Stover DG, Selfors LM, Bronson RT, Deraedt T, Cichowski K, Welm AL, Mori Y, et al. (2018). Cancer cells co-opt the neuronal redox-sensing channel TRPA1 to promote oxidative-stress tolerance. Cancer Cell 33, 985–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK, and Zhang DD (2014). Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 74, 7430–7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant DA, Durán RV, and Gottlieb E (2010). Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 10, 267–277. [DOI] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a cancer dependency map. Cell 170, 564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Voorde J, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, Nixon C, Blyth K, Gottlieb E, and Tardito S (2019). Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv 5, eaau7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian S, Lee J, Klijn C, Gnad F, Bagniewska M, Schaefer G, Zhang D, Tan J, Watson SA, Liu L, et al. (2019). ERBB3 and IGF1R signaling are required for Nrf2-dependent growth in KEAP1-mutant lung cancer. Cancer Res. 79, 4828–4839. [DOI] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, Sriramaratnam R, Matthew, Shimada K, Skouta R, Vasanthi, Jaime, Paul, Alykhan, Clary, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weïwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, Macpherson L, Dandapani S, Palmer M, Stockwell BR, Schreiber SL, et al. (2012). Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett 22, 1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KC, Cui JY, and Klaassen CD (2011). Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci 123, 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XM, Turanov AA, Carlson BA, Yoo MH, Everley RA, Nandakumar R, Sorokina I, Gygi SP, Gladyshev VN, and Hatfield DL (2010). Targeted insertion of cysteine by decoding UGA codons with mammalian selenocysteine machinery. Proc. Natl. Acad. Sci. USA 107, 21430–21434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et al. (2012). Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, and Stockwell BR (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 113, E4966–E4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, and Stockwell BR (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol 15, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J, Finley LWS, Lu C, Lindsten T, Cross JR, et al. (2014). Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 4, 1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Badur MG, Luebeck J, Magaña JH, Birmingham A, Sasik R, Ahn CS, Ideker T, Metallo CM, and Mali P (2018). Combinatorial CRISPR-Cas9 metabolic screens reveal critical redox control points dependent on the KEAP1-NRF2 regulatory axis. Mol. Cell 69, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. 1,466 Genes That Significantly Correlate with NRF2-Hyperactivating Genetic Alterations and 55 NRF2 Signature Genes, Related to Figure 2.
Table S2. NRF2 Score of 675 Cancer Cell Lines, Related to Figure 2.
Table S3. 1,500 Genes Selected for CRISPR-Cas9 Screens, Related to Figure 4.
Table S4. Summary of Results from CRISPR-Cas9 Screens in 2D and 3D for A549 and H1437 Cell Lines, Related to Figure 4. Data on all the genes and hit genes common to both cell lines are shown.
Table S5. LS-MS/MS Data (Sum Date), Related to Figure 6.
Table S6. Abundance of Selenoproteins and Proteins Required for Selenoprotein Biosynthesis by Proteomics in A549 and H1437 Cells, Related to Figure 6.
Table S7. Primer Sequences, Related to STAR METHODS. Primer sequences used for the construction of NFE2L2 mutants and LentiCRISPR plasmids, quantitative PCR, and amplification of sgRNA are shown.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD018634.