

Abstract
Macrophages within the tumor microenvironment (TME) exhibit a spectrum of pro-tumor and anti-tumor functions, yet it is unclear how the TME regulates this macrophage heterogeneity. Standard methods to measure macrophage heterogeneity require destructive processing, limiting spatiotemporal studies of function within the live, intact 3D TME. Here we demonstrate two-photon autofluorescence imaging of NAD(P)H and FAD to non-destructively resolve spatiotemporal metabolic heterogeneity of individual macrophages within 3D microscale TME models. Fluorescence lifetimes and intensities of NAD(P)H and FAD were acquired at 24, 48, and 72 hours post-stimulation for mouse macrophages (RAW 264.7) stimulated with IFN-γ or IL-4 plus IL-13 in 2D culture, confirming that autofluorescence measurements capture known metabolic phenotypes. To quantify metabolic dynamics of macrophages within the TME, mouse macrophages or human monocytes (RAW264.7 or THP-1) were cultured alone or with breast cancer cells (mouse PyVMT or primary human IDC) in 3D microfluidic platforms. Human monocytes and mouse macrophages in tumor co-cultures exhibited significantly different FAD mean lifetimes and greater migration than monocultures at 24, 48, and 72 hours post-seeding. In co-cultures with primary human cancer cells, actively-migrating monocyte-derived macrophages had greater redox ratios (NAD(P)H/FAD intensity) compared to passively-migrating monocytes at 24 and 48 hours post-seeding, reflecting metabolic heterogeneity in this sub-population of monocytes. Genetic analyses further confirmed this metabolic heterogeneity. These results establish label-free autofluorescence imaging to quantify dynamic metabolism, polarization, and migration of macrophages at single-cell resolution within 3D microscale models. This combined culture and imaging system provides unique insights into spatiotemporal tumor-immune crosstalk within the 3D TME.
INTRODUCTION
Immune cells are a critical component of the tumor microenvironment (TME) and regulate tumor initiation, progression, and therapeutic response(1). The phenotype and function of immune cells surrounding or infiltrating the tumor are crucial to tumor fate(1). Macrophages are a subset of immune cells with diverse function that are abundant within numerous tumor types, especially breast and pancreatic tumors(1,2). This prevalence suggests that macrophages may serve as important immune mediators of tumor behavior, yet macrophage activity is complex and not well understood. For example, multiple macrophage phenotypes have been identified, each with unique functional and stimulatory activity that influence the surrounding cellular environment(3,4). Two well-characterized classes of macrophages, M1-like and M2-like macrophages, exhibit distinct responses to tumors(4). M1-like macrophages promote anti-tumor activity through apoptotic or phagocytic signaling, secondary immune stimulation via cytokine secretion and antigen presentation, and nutrient deprivation of tumors(4,5). M2-like macrophages support tumor growth by suppressing immune recognition and promoting cancer cell proliferation, motility, and vessel growth(4,5). However, intermediate macrophage states have also been observed within tumors, suggesting a more complex model of macrophage heterogeneity than this M1-M2 dichotomy(6). This heterogeneity within macrophage populations presents a substantial challenge for effective treatments, emphasizing the need to characterize cell-level macrophage function within the 3D TME.
Cell function has been correlated with metabolism across numerous cell types, including macrophages(4,6,7). Therefore, monitoring metabolic preferences within macrophages could reveal heterogeneity in macrophage phenotype and function. Metabolic demands greatly differ between M1-like and M2-like macrophage classes(4). M1-like macrophages upregulate glycolysis to sustain viability, induce tumor cell death and recruit additional monocytes to the tumor. This increases the production of the metabolic co-enzyme, NADH, via glycolysis and sustains the viability of M1-like macrophages within tumors(6,7). Conversely, elevated fatty acid oxidation and oxidative phosphorylation in M2-like macrophages support angiogenesis and tumor growth(6–8). Microenvironmental changes can regulate these distinct metabolic demands and produce diverse macrophage phenotypes(6,9). This highlights a continuum of metabolic activity related to macrophage function that is provoked by complex stimuli from the TME(5,6). Therefore, monitoring macrophage metabolic heterogeneity may improve understanding of tumor-macrophage crosstalk.
3D tumor models provide reliable portrayal of tumor structure and TME conditions that influence immune cell function. The key advantages of microscale models include controllable features that can be easily tuned to simulate the TME and designs enabling various combinations of culture conditions that support high-throughput assessment(10). These microscale platforms also provide a simple approach to recapitulate paracrine signaling, protein-protein interactions, and in vivo tumor-associated environmental conditions (e.g., hypoxia, nutrient availability) that regulate both macrophage phenotype and function(10–12). These representative 3D tumor models provide attractive systems to evaluate macrophage heterogeneity in response to spatiotemporal changes in the microenvironment.
Limitations of destructive, population-level functional assays (e.g., flow cytometry, ELISA) and low-resolution whole body imaging (e.g., PET/CT, MRI) prohibit monitoring spatiotemporal macrophage heterogeneity within the complex 3D TME(13,14). Non-invasive autofluorescence imaging has previously quantified cellular heterogeneity in vivo and in 3D cultures using label-free two-photon microscopy(15–19). Specifically, autofluorescence imaging can quantify the endogenous fluorescence of metabolic coenzymes NADH and FAD, which are both used across several cellular metabolic processes(20). NADH and NADPH have overlapping fluorescence properties, and are collectively referred to as NAD(P)H(21). Fluorescence intensity measurements can inform on the intra-cellular concentrations of NAD(P)H and FAD. The optical redox ratio, defined as the ratio of NAD(P)H intensity to FAD intensity, provides a measure of the relative oxidation-reduction state of the cell(15,20). Fluorescence lifetime imaging microscopy (FLIM) of NAD(P)H and FAD provide additional information specific to protein binding activity(19). Lifetime measurements can distinguish between free and protein-bound forms of NAD(P)H and FAD, characterized by distinct molecular conformations that affect fluorescence quenching(19). Previous studies have shown that metabolic autofluorescence imaging detects spatial and temporal changes in stromal cells across in vivo and 3D in vitro models(17,22–24). This study aims to validate whether autofluorescence imaging of NAD(P)H and FAD can monitor changes in macrophage metabolism with polarization and migration. A novel technology combining metabolic autofluorescence imaging and microscale 3D models was established to quantify metabolic activity and visualize macrophage heterogeneity within the 3D TME using primary human cancer, human cell lines, and mouse cell lines. These technologies could provide valuable insights for future studies of tumor-associated macrophage function.
MATERIALS AND METHODS
2D cell culture
RAW 264.7 murine macrophages (ATCC TIB-71) were maintained in culture medium composed from standard RPMI 1640 (Gibco), 10% fetal bovine serum, and 1% penicillin:streptomycin. RAW macrophages were seeded in 35 mm glass bottom dishes (MatTek) for imaging experiments. All imaging samples were plated at a density of 1×105 cells per dish and incubated at 37 ⁰C and 5% CO2, for 24 hours to allow cell adhesion. Separate cultures of macrophages polarized with M1-like (IFN-γ) or M2-like (IL-4/IL-13) cytokines were generated by standard media substitution with 2 mL cytokine-supplemented media and incubated between 24–72 hours. Media for M(IFN-γ) stimulation consisted of RPMI 1640 supplemented with 10 ng/ml interferon-γ (IFN-γ; R&D systems), and media for M(IL4/IL13) stimulation consisted of RPMI 1640 with 20 ng/ml interleukin 4 (IL-4; Invitrogen) and 20 ng/ml interleukin 13 (IL-13; Gibco).
Microdevice design and fabrication
Description of the Stacks microfluidic platform is previously described in detail(11). 24-well polystyrene microdevices (dimensions: 75 mm x 50 mm) were fabricated by injection molding and sterilized via sonication in isopropanol. Collagen hydrogels (2.0 mg/ml) were prepared and a 4.5 µL volume was suspended within each microwell of the device (thickness ~1.2 mm), creating an open-air culture system compatible for two-photon imaging. The hydrogel was generated from a mixture of 6 µL 10X PBS, 14 µL sterile water, 6 µL NaOH (Sigma), 160 µL Bovine collagen type I (PureCol, Advanced BioMatrix), 3 µL fibronectin (Sigma) and 50 µL cell type-specific medium (RPMI1640 – mouse; DMEM/complete DMEM/F12 (Gibco) – human). Collagen hydrogels were polymerized by incubation at 37⁰C for at least 6 hours, prior to sequential seeding of breast carcinoma cells and macrophages (~1000 cells/μL each) on the opposing ends of the collagen layer.
3D microdevice culture
The 3D microfluidic platform was used for co-cultures of primary and immortalized macrophages and breast carcinoma of both mouse and human origin. PyVMT (established from FVB MMTV-PyVmT mouse tumors) and MDA-MB-231 (ATCC HTB-26) human breast cancer cells were cultured in medium composed from standard Dulbecco’s Modified Eagle Medium (DMEM, Gibco), 10% fetal bovine serum, and 1% penicillin:streptomycin. RAW 264.7 macrophages (ATCC TIB-71) and THP-1 human monocytes (ATCC TIB-202) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin:streptomycin. All immortalized lines were used less than 15 passages from thawing. Patient-derived invasive breast carcinoma cells (IDC; 171881–019-R-J1-PDC, Passages 7–12) were obtained upon request from the NCI Patient-Derived Models Repository (NCI PDMR)(25). Culture of patient-derived cells required specialized media consisting of 1X Advanced DMEM/F12 (Gibco) supplemented with 5% Fetal Bovine Serum, 1.1 µM Hydrocortisone (Sigma), 1.61 nM EGF Recombinant Human Protein (Invitrogen), 0.178 mM Adenine (Sigma), 1% penicillin:streptomycin, 2 mM L-Glutamine (Invitrogen), and 0.01 mM Y-27632 dihydrochloride (Tocris)(25). All cell lines were tested negative for mycoplasma by the University of Wisconsin-Madison or NCI PDMR and verified with short-tandem repeat (STR) profiling by ATCC and NCI PDMR. 3D microwell conditions included macrophages incubated with and without tumor cells over 72 hours. For gene expression analysis, tumor cells and macrophages were cultured on separate Stacks layers to prevent cell type cross-contamination. An additional Stacks layer with wells only containing 10 μL culture medium was placed on top of each Stacks layer with cells. These layers were then overlaid forming a four-layer Stacks setup (media layer-macrophage layer-media layer-tumor layer). Media changes were performed daily by aspirating old media from each microwell and replacing with 10 µL of fresh culture media during incubation between imaging experiments.
Autofluorescence imaging
FLIM images were acquired with an Ultima two-photon imaging system (Bruker) composed of an ultrafast tunable laser source (Insight DS+, Spectra Physics) coupled to a Nikon Ti-E inverted microscope equipped with time correlated single photon counting electronics (SPC 150, Becker & Hickl GmbH). The ultrafast tunable laser source enabled excitation of NAD(P)H (750 nm) and FAD (890 nm) fluorescence. For 2D macrophage imaging, NAD(P)H and FAD fluorescence were excited sequentially, and their respective emission was isolated using 440/80 and 550/100 bandpass filters (Chroma). Laser power at the sample for NAD(P)H and FAD excitation was approximately 11.3 mW and 11.5 mW, respectively. Simultaneous excitation of NAD(P)H (750 nm) and FAD (895 nm) was performed for all 3D samples by wavelength mixing of 750 nm and 1041 nm laser lines to virtually generate a 895 nm excitation effect as described previously(26,27). Wavelength mixing was implemented by delaying the tunable laser line and matching the collimation of both the fixed 1041 nm and tunable lasers with a single beam telescope to align both laser lines spatially and temporally within the focal volume (Supplementary Fig 2A). Efficient 895 nm excitation was confirmed with excitation of fixed GFP-labeled cells across a range of 750 nm and 1041 nm laser powers (0 – 1.5 mW) relative to single-color 895 nm excitation, supporting minimal excitation at the single 750 or 1041 nm wavelengths compared with dual excitation. Co-localization of GFP signal with one- (1041 nm) and two-laser (750 & 1041 nm) excitation was also used to regularly correct alignment of the laser lines. For wavelength mixing experiments, all samples were illuminated through a 40X/1.15 NA objective (Nikon) with an optical zoom parameter of 2, decreasing the scan angle and resulting field of view to capture fluorescence emission only from the lateral area of overlap between the two laser lines. Approximate laser power at the sample during wavelength mixing was 10.7 mW at 750 nm and 16.3 mW at 1041nm. NAD(P)H and FAD emission during wavelength mixing were isolated using a 466/40 and a 540/24 nm bandpass filter (Chroma), respectively. Wavelength mixing results were further validated by comparing two-color sequential imaging and wavelength mixing autofluorescence trends in standard culture of a breast carcinoma cell line (BT474, ATCC), as well as following perturbation with 4 mM NaCN for 5 min in BT474 monolayers(27). Emission was detected with GaAsP photomultiplier tubes (Hamamatsu 7422PA-40). Per-pixel lifetime decay curves were collected by scanning the field of view (256×256 pixels; 300µm X 300 µm) over 60 seconds with a 4.8 microsecond pixel dwell time. To prevent photobleaching within the sample, photon count rates were maintained at ~1–3×105 photons/second. The instrument response function was generated from second harmonic signal of urea crystals excited at 900 nm, with a full width at half maximum of 240 picoseconds. A fluorescence lifetime standard measurement was collected daily by imaging a YG fluorescent bead (Polysciences Inc), measured as 2.1±0.09 nanoseconds consistent with reported lifetime values(28). Autofluorescence images were captured for each 2D polarization condition at 24, 48, and 72 hours post-stimulation, with a minimum of four representative fields of view per sample (~1000–2000 cells). For 3D cultures, autofluorescence lifetime volumes were acquired across 2–3 microwells for each macrophage monoculture and co-culture condition for at least 3 separate devices (i.e., 6 to 9 microwells per condition). Image volumes captured images at 3 µm z-steps starting at the macrophage seeding plane and ending at the leading edge of the macrophage layer (~1000–3000 cells). Z-stack image depths ranged from 24 to 170 µm.
Image analysis
Per-pixel fluorescence lifetimes of free and protein-bound NAD(P)H and FAD were calculated from fitting fluorescence decays to the following bi-exponential model: (19). Fluorescence intensity images were generated by integrating photon counts over the per-pixel fluorescence decays. The per-pixel ratio of NAD(P)H fluorescence intensity to FAD intensity was calculated to determine optical redox ratio. A customized CellProfiler pipeline was used to segment individual cell cytoplasms(19). Cytoplasm masks were applied to all images to determine single-cell redox ratio and NAD(P)H and FAD fluorescence lifetime variables. Fluorescence lifetime variables consist of the mean lifetime (τm = τ1α1 + τ2α2), free- and protein-bound lifetime components (τ1, τ2), and their fractional contributions (α1 and α2; where α1 + α2 = 1) for each cell cytoplasm. Eleven total variables were analyzed for each cell cytoplasm: NAD(P)H τm, FAD τm, NAD(P)H τ1, FAD τ1, NAD(P)H τ2, FAD τ2, NAD(P)H α1, FAD α1, NAD(P)H intensity, FAD intensity, and optical redox ratio.
Metabolic Inhibition
To validate that metabolic changes were specific to macrophage polarization, 2D polarized macrophages were treated with a panel of metabolic inhibitors with known activity and imaged post-treatment. RAW264.7 macrophages were stimulated to glycolytic M(IFN-γ) or oxidative M(IL4/IL13) phenotype for 72 hours(29,30). Next, glycolysis was inhibited with media + 10 mM 2-deoxyglucose (2DG; Sigma), FAO was inhibited with media + 100 nM etomoxir (ETO; Sigma), or OXPHOS was inhibited with media + 4mM sodium cyanide (NACN; Sigma)(31). Autofluorescence imaging was performed immediately before adding inhibitor and after treatment for 5 mins (NaCN), 1 hour (2DG), or 24 hours (ETO).
Quantification of macrophage migratory activity
Macrophage migration was plotted as the percentage of macrophages within each z-plane versus z-plane distance from the macrophage seeding plane. Specifically, the percent of macrophages at each z-plane was calculated as the number of macrophages at each z-plane divided by the total number of macrophages in each microwell. This percent of macrophages for each z-plane was plotted versus z-plane distance from the macrophage seeding plane for histograms of macrophage migration distance, to further define migratory behavior (i.e., “passive” and “active” migration). The maximum z-plane distance for the monoculture condition at each timepoint was used as the threshold to discriminate passively- (closest to macrophage seeding plane) vs. actively-migrating (farthest from macrophage seeding plane) macrophages in co-cultures. Passive migration is defined as random, undirected migratory activity, while active migration represented a directional chemotactic response(32).
Immunofluorescence staining and imaging
Following autofluorescence imaging, unstimulated and cytokine-stimulated 2D cultures of RAW264.7 macrophages were stained according to supplier instructions with both PE-conjugated CD86 antibody (Tonbo Biosciences) and FITC-conjugated CD206 antibody (BioRad) to confirm macrophage polarization. Conjugated antibodies were diluted to 1:30 in PBS+goat serum. 2D immunofluorescence images were acquired using the two-photon microscope setup described above. CD206-FITC fluorescence intensity was excited at 890 nm and collected with a 550/100 nm bandpass filter, while CD86-PE fluorescence was excited at 1050 nm and collected with a 650/47 nm filter.
mRNA Isolation, cDNA Synthesis and real time-quantitative PCR (RT-qPCR)
To assess genetic heterogeneity in macrophage polarization, expression levels of genes specific to broad macrophage classes (i.e., M1-like, M2-like, mixed phenotypes) were analyzed by qPCR. For 2D cultures, separate cultures of M0, M(IFN-γ), M(IL4/IL13) RAW 264.7 macrophages were maintained in 6-well plates prior to mRNA extraction. For 3D cultures, RAW264.7 macrophages or THP-1 monocytes were cultured alone or with breast cancer cells in the 3D Stacks microfluidic platform. For all experiments, mRNA was extracted at 24, 48 and 72 hours using a Dynabeads™ mRNA DIRECT™ Purification Kit (Thermo Fisher, 61012). Purified mRNA was quantified using a Qubit fluorometer (Thermo Fisher) and a Qubit™ RNA BR Assay Kit (Thermo Fisher, Q10210). mRNA was reverse transcribed using RT² PreAMP cDNA synthesis kit (Qiagen, 330451). The prepared cDNA was preamplified using the RT² PreAMP Primer Mix for Human and Mouse PCR Array (Qiagen, PBH-181Z). cDNA was analyzed by RT-qPCR using a Qiagen RT2 profiler custom panel (Qiagen, PAHS-181Z, CLAM3313, CLAH36077) following supplier instructions. Gene lists for mouse and human experiments are reported in Supplementary Table 1.
Population density modeling
Population density modeling of single-cell metabolism was used to observe differences in macrophage heterogeneity across 2D polarized cultures and 3D tumor-macrophage cultures over time. Frequency histograms were generated based on single-cell metabolic autofluorescence measurements across samples. Multiple Gaussian probability density functions were fit to each distribution histogram to identify the presence of sub-populations with distinct metabolic activity(33). Accurate sub-population identification was evaluated by iteratively increasing the number of fitted Gaussian curves and calculating the Akaike Information Criterion (AIC) for each iteration(33). The number of fitted Gaussians yielding the lowest AIC value indicate the optimal fit conditions and number of sub-populations per sample(33).
Random Forest Classification
Multi-class Random Forest classifiers were trained to distinguish 1) macrophage polarization conditions (M0, M(IFNγ) and M(IL4/IL13)) pooled across the stimulation timecourse (24, 48, and 72 hours) and passively- and actively-migrating cells in 2) mouse PyVMT+RAW264 co-cultures and 3) human IDC+THP co-cultures across timepoints. Conditions were pooled across timepoints to allow robust classification of each group without overfitting. Autofluorescence measurements for all 2D macrophage conditions (cell number = 14504), mouse 3D macrophage conditions (cell number = 360), and human 3D macrophage conditions (cell number = 4079), respectively, were randomly sampled from each condition and timepoint to generate balanced classification datasets before assignment to training and test sets. Classification accuracy was assessed as a function of varying proportions of training/test data: 12.5%/87.5%, 25%/75%, 37.5%/62.5%, 50%/50%, 62.5%/37.5%, 75%/25%, & 87.5%/12.5%.
Statistics
Tukey’s tests for non-parametric, unpaired comparisons were performed to assess differences in autofluorescence variables between 2D cytokine-stimulated cultures and 3D monoculture and co-culture conditions over 72 hours. Multiple student’s t-tests were performed to assess significance of fold change differences for redox ratio changes in 2D macrophage sub-population analysis and in response to metabolic inhibitors and gene expression changes with respect to M0 conditions for 2D mouse macrophages or monoculture for 3D mouse and human macrophages. Coefficient of variation was calculated to evaluate variability across 2D polarized mouse cultures and 3D mouse monocultures and co-cultures over 72 hours. The non-parametric, squared-ranks test was performed to assess the equality of coefficients of variation between macrophage groups (MATLAB, RRID:SCR_001622, SquaredRanksTest)(34). Metabolic autofluorescence results are represented as line plots, dot plots, or bar graphs plotted in GraphPad (GraphPad Prism, RRID:SCR_002798). Heatmaps of autofluorescence variables with respect to depth were generated in MATLAB.
RESULTS
Microscale 3D co-culture and metabolic autofluorescence imaging provide innovative and complementary tools to monitor 3D changes in macrophage metabolism within the TME
Autofluorescence imaging can non-destructively monitor live, single-cell metabolism and 3D migration, while 3D microscale co-cultures reliably model tumor complexity with selective environmental control and high-throughput capabilities. Specifically, the Stacks 3D microfluidic system enables multi-cellular cultures with minimal biological material, flexible configuration, and well-characterized environmental gradients(11). The Stacks system was combined with metabolic autofluorescence imaging to monitor tumor-mediated changes in macrophage metabolism and migration (Fig 1A)(11). Microscale 3D co-cultures of tumor cells and macrophages were established in the Stacks 3D system using primary human cancer cells, and cell lines from mouse and human origin (Fig 1A). A collagen-based ECM was polymerized within each microwell, then monocytes/macrophages were seeded with (co-culture) or without (monoculture) breast cancer cells at the opposing end of the ECM layer (Fig 1A). Both the metabolic autofluorescence and microfluidic culture technologies were validated by imaging macrophages in 2D culture with standard stimulation techniques and in 3D co-cultures of macrophages alone in the Stacks 3D system (Fig 1B). Finally, the metabolic dynamics of tumor-stimulated macrophages were characterized by collecting NAD(P)H and FAD autofluorescence image volumes of monocytes/macrophages migrating across the ECM at 24, 48, and 72 hours (Fig 1C). Overall, this approach provides unique, quantitative imaging of macrophage spatiotemporal dynamics in the TME that cannot be achieved with conventional assessments that lack single-cell resolution, 3D complexity, and/or non-destructive monitoring capability.
Figure 1: Metabolic autofluorescence imaging of macrophages in the Stacks 3D microscale co-culture system.
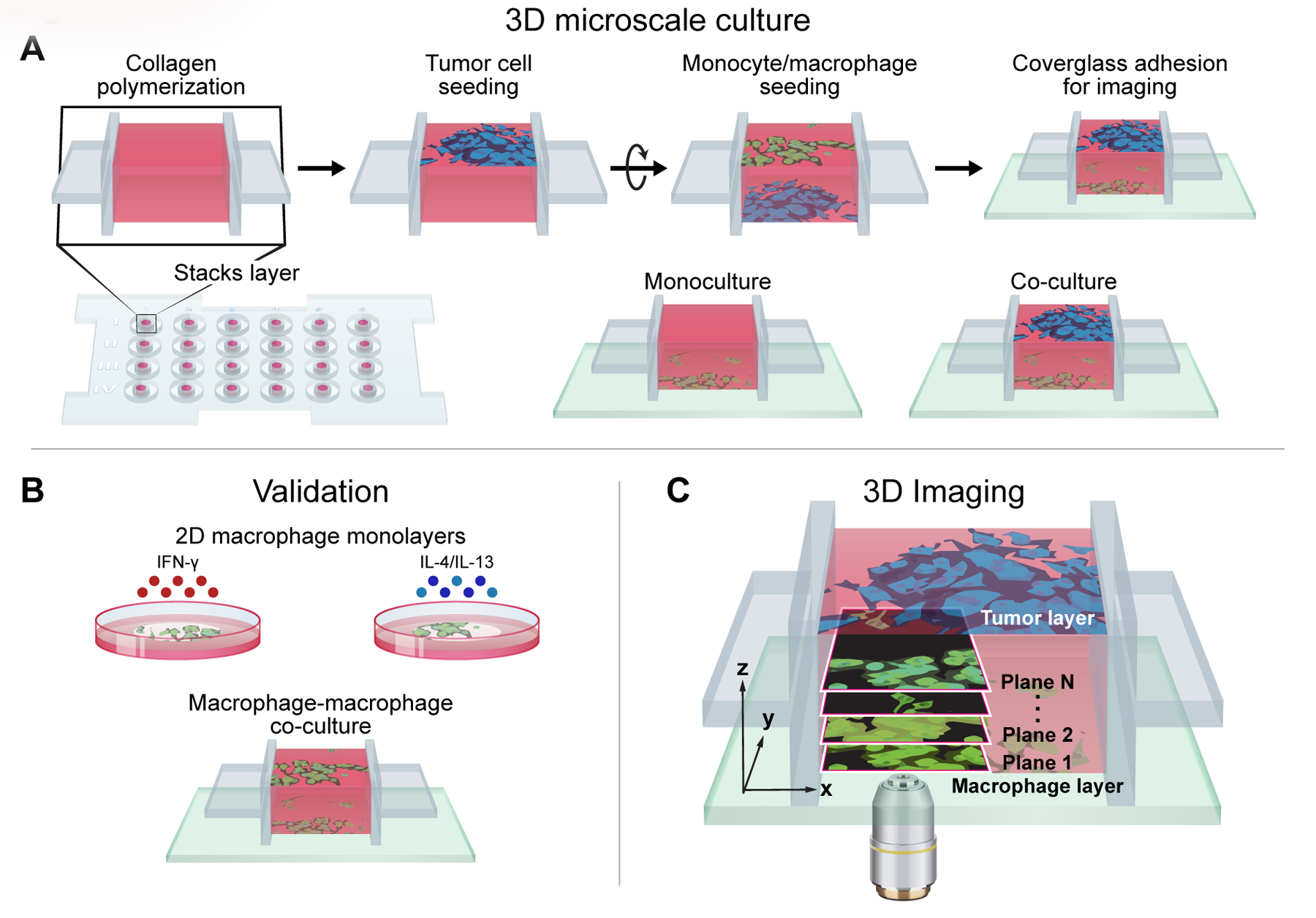
Illustration of experimental workflow showing (A) design of the Stacks 3D co-culture of macrophages/monocytes and tumor cells from primary patient samples, along with cell lines of mouse and human origin, (B) validation of metabolic autofluorescence imaging in 2D culture with standard stimulations and in Stacks 3D microscale system with macrophages alone, and (C) metabolic autofluorescence imaging of 3D migration and single-cell metabolism for macrophages in the Stacks 3D microscale co-culture system.
Metabolic imaging validation: macrophage stimulation in 2D in vitro culture
To establish the sensitivity of metabolic autofluorescence imaging to distinct macrophage phenotypes, metabolic autofluorescence measurements were first validated in 2D cultures of RAW264.7 mouse macrophages stimulated with IFN-γ (M(IFN-γ): anti-tumor M1-like phenotype), IL-4/IL-13 (M(IL4/IL13): pro-tumor M2-like phenotype), or without cytokine stimulation (M0: naïve macrophages). Macrophages were maintained in cytokine-supplemented media and imaged over 24–72 hours. Autofluorescence imaging of NAD(P)H and FAD demonstrates time-dependent differences in M(IFN-γ) and M(IL4/IL13) macrophages (Fig 2A). M(IFN-γ) macrophages have increased redox ratio compared to M(IL4/IL13) macrophages, with the greatest redox ratio differences at 72 hours post-polarization (Fig 2A). Individual M(IFN-γ) and M(IL4/IL13) macrophages have heterogeneous NAD(P)H and FAD lifetimes across fields-of-view at all timepoints. A flattened, cuboid structure was observed for M(IFN-γ) macrophages, whereas M(IL4/IL13) macrophages extended outward with spindle projections, consistent with reported morphology of polarized macrophages (Fig 2A)(35). These known subset-specific morphological changes become more pronounced over time (Fig 2A). Staining for common M1-like and M2-like surface proteins (CD86 and CD206, respectively) show increasing CD86 expression in M(IFN-γ) macrophages across the 72 hour time course, while M(IL4/IL13) macrophages have stronger CD206 expression (Fig 2A).
Figure 2: Metabolic autofluorescence imaging is sensitive to temporal changes in macrophage metabolism in 2D culture.
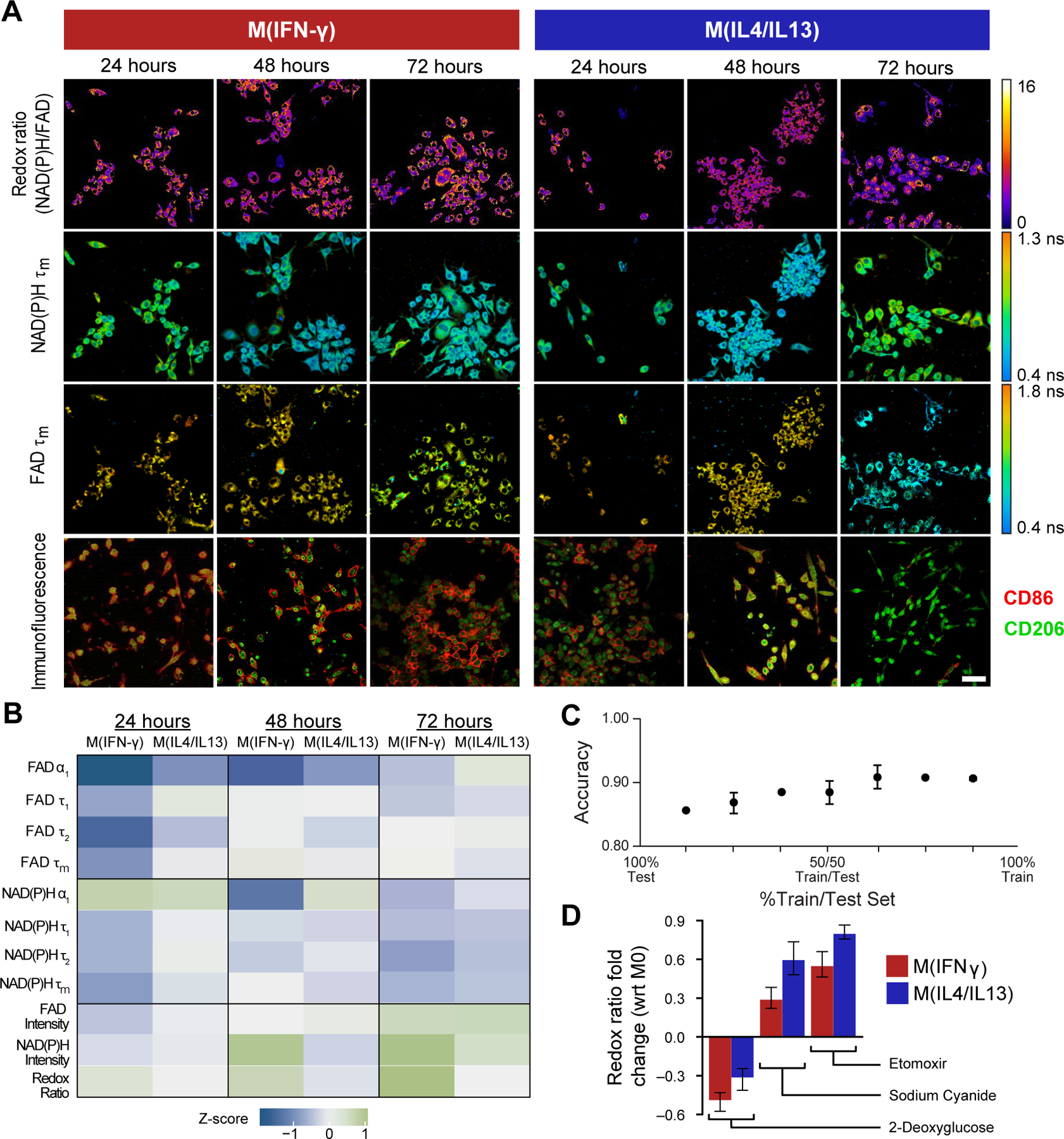
A) Representative optical redox ratio, NAD(P)H and FAD mean lifetime (τm) images of RAW 264.7 macrophages cytokine-stimulated to M(IFN-γ) (anti-tumor M1-like phenotype), M(IL4/IL13) (pro-tumor M2-like phenotype), or unstimulated (M0 naïve phenotype). Representative immunofluorescence images show surface expression of known M1-like and M2-like macrophage markers (CD86, CD206). Scale Bar = 50 μm. B) Z-score heatmaps representing metabolic autofluorescence changes of M(IFN-γ) and M(IL4/IL13) RAW264.7 mouse macrophages relative to M0 macrophages at 24, 48, or 72 hours. Z-scores are calculated as the difference between variable mean per condition and variable mean of the monoculture condition, divided by the monoculture standard deviation. C) Random forest classification accuracy of M0, M(IFNγ), and M(IL4/IL13) RAW264.7 mouse macrophages at across all timepoints (3 groups). Metabolic characterization was performed via inhibition with D) 2-deoxyglucose (2DG, glycolysis inhibitor), etomoxir (ETO, fatty acid oxidation inhibitor), and sodium cyanide (NaCN, oxidative phosphorylation inhibitor). Reported values are fold change of redox ratio with respect to M0 control under the same polarization conditions.
Quantitative autofluorescence measurements were then compared between M0, M(IFN-γ) and M(IL4/IL13) macrophages over the 72 hour time course to determine whether metabolic autofluorescence can distinguish macrophage polarization states. Z-score heatmaps of all 11 metabolic variables show the average value of each polarization state with respect to unpolarized, M0 macrophages (Fig 2B). The full distributions of each metabolic variable are observed in Supplementary Figure 1. The redox ratio was significantly different (p<0.0001) between M0, M(IFN-γ) and M(IL4/IL13) macrophages at 48 and 72 hours post-polarization (Fig 2B, Supplementary Fig 1A). NAD(P)H and FAD lifetimes further demonstrate dynamic metabolic behavior across all three macrophage conditions, consistently exhibiting significant differences 24 hours post-stimulation (p<0.05,0.001) (Fig 2B, Supplementary Fig 1B–C). The short (τ1) and long lifetimes (τ2) and their relative contribution (α1) for NAD(P)H and FAD influence changes in τm over the time course (Fig 2B, Supplementary Fig 1D–I). Multi-class models of random forest classification were then generated to classify macrophage polarization states ((M0, M(IFNγ) and M(IL4/IL13)) across each of the 3 experimental timepoints (24, 48, and 72 hours). Classification accuracy of test data predictions for all 3 groups was highest (90.6%) upon training with 75% and testing on 25% of the total data set (Fig 2C). Accuracies of >84% were observed for test data predictions regardless of training/test proportions (Fig 2C, Supplementary Fig 1L).
Metabolic inhibitors were used to confirm observed autofluorescence differences between M(IFN-γ) and M(IL4/IL13) macrophages. Glycolysis, oxidative phosphorylation, and fatty acid oxidation were inhibited with 2-deoxyglucose, sodium cyanide, and etomoxir, respectively. Fold changes in redox ratio with respect to untreated controls with the same stimulation are shown in Fig. 2D. Inhibition of glycolysis decreased the redox ratio of M(IFN-γ) macrophages to a greater degree than M(IL4/IL13) macrophages (Fig 2D), consistent with published studies that show increased reliance on glycolysis for these anti-tumor, M1-like macrophages compared to pro-tumor, M2-like macrophages(7). Conversely, M(IL4/IL13) macrophages showed greater increases in redox ratio than M(IFN-γ) macrophages with inhibition of oxidative phosphorylation and fatty acid oxidation (Fig 2D). This is consistent with prior studies that show increased reliance on oxidative phosphorylation and fatty acid oxidation in M2-like macrophages compared to M1-like macrophages(7,8). Fold changes are statistically significant between control and inhibitor-treated conditions across all treatments (Supplementary Table 2). Fold change of both NAD(P)H and FAD mean lifetimes with inhibitors are shown in Supplementary Fig 1J–K. These findings agree with previous studies reporting upregulation of glycolysis in M1-like macrophages, while M2-like macrophages preferentially shift towards a more oxidative metabolic state(3,6,8). Collectively, these results demonstrate that metabolic autofluorescence imaging can assess macrophage phenotype and metabolism.
The variable metabolic behavior observed between polarization states over the time course prompted further analysis of heterogeneity within the aggregated single-cell data. Population density modeling of single-cell redox ratios was performed to determine whether metabolic autofluorescence imaging can resolve macrophage heterogeneity. Population density curves revealed heterogeneous metabolic sub-populations of macrophages across conditions and time points (Fig 3A–C). All stimulation conditions and time points have a narrow population of cells with similarly low redox ratio along with a second population of cells with a broad range of higher redox ratios (Fig 3A–C). Note that the proportion of the high redox ratio sub-population (p2) is a substantial proportion of all cells for the M(IFN-γ) condition (red) at 48 and 72 hours (Fig 3B–C), which contributes to its higher average redox ratio at 48 and 72 hours (Supplementary Fig 1A). Additionally, shifts within the low redox population (p1) and within the high redox population (p2) between conditions are significant at each time point (Fig 3A–C).These distributions illustrate the heterogeneous metabolic state of macrophages on a single-cell level. Population distributions overlap between M0, M(IFN-γ), and M(IL4/IL13) conditions, which highlights a continuum of metabolic activity between conditions. M(IFN-γ) and M(IL4/IL13) macrophages also exhibited greater variability compared to M0 macrophages at 24 and 72 hours (Supplementary Table 3). Increasing numbers of sub-populations are observed from population distribution analysis of single-cell NAD(P)H and FAD τm during macrophage stimulation, highlighting substantial heterogeneity in enzyme binding activity (Supplementary Fig 1M–R).
Figure 3: Metabolic autofluorescence measurements resolve metabolic heterogeneity linked to heterogeneous 2D cytokine-stimulated macrophage polarization.
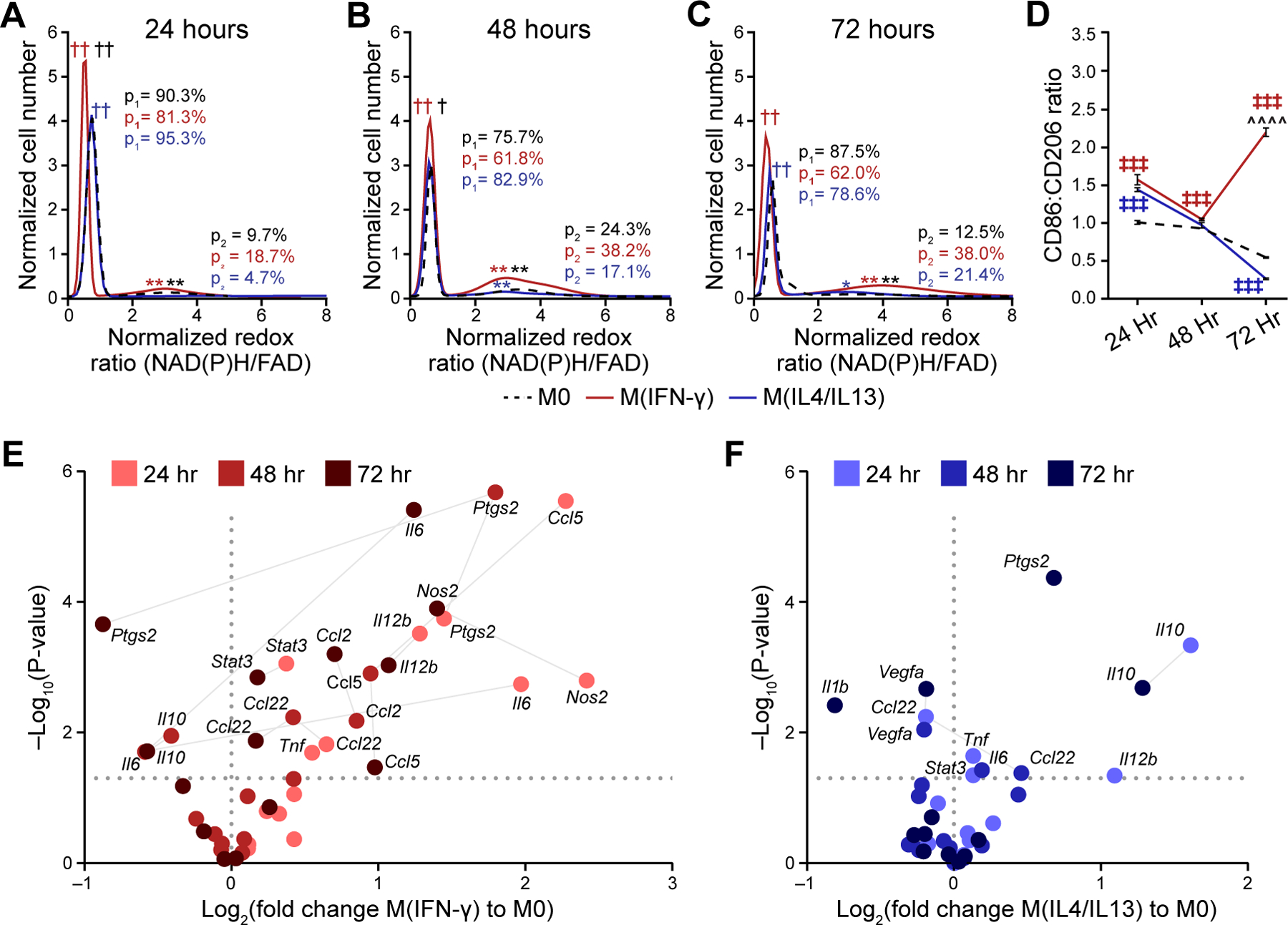
Population density modeling of redox ratios per cell illustrates heterogeneous macrophage metabolism at A) 24 hours, B) 48 hours, and C) 72 hours of stimulation. Two distinct sub-populations of cell metabolism are present for each time point and stimulation condition. The proportions of the low (p1) and high (p2) redox ratio sub-populations were calculated to represent the contribution of each sub-population to the overall distribution. Cell number per time point (24/48/72 hours) – M0:1008/2153/2263 cells; M(IFN-γ):1289/999/1100 cells; M(IL4/IL13):1443/1821/2157 cells. †,†† p<0.05, 0.01 – low population; *,**p<0.05, 0.01 – high population; red – M0 vs. M(IFN-γ), blue – M0 vs. M(IL4/IL13), black – M(IFN-γ) vs. M(IL4/IL13). D) Expression levels of known M1-like and M2-like macrophage markers (CD86, CD206) were quantified for M0, M(IFN-γ), and M(IL4/IL13) macrophages at 24 hours, 48 hours and 72 hours; ╪╪╪ p<0.001, ^^^^p<0.0001. Expression levels of M1-like and M2-like genetic markers for E) M(IFN-γ), and F) M(IL4/IL13) conditions were evaluated with qPCR at 24 hours, 48 hours, and 72 hours following stimulation.
Heterogeneity in macrophage function within stimulation conditions (M(IFN-γ) or M(IL4/IL13)) was further assessed from gene and protein expression. Immunofluorescence was quantified from images of M(IFN-γ) and M(IL4/IL13) macrophages stained for CD86 and CD206 expression (Fig 3D). Both stimulation conditions have basal expression of these markers that fluctuate during polarization(36). Thus, the ratio of CD86 to CD206 expression was used to assess protein changes associated with macrophage polarization(37). The CD86:CD206 ratio increased significantly in M(IFN-γ) compared with M(IL4/IL13) conditions by 72 hours, while heterogenous expression was observed at earlier time points (Fig 3D). Gene expression of cytokines and other mediators of inflammation (Supplementary Table 1) were also measured with qPCR as a parallel measure of heterogeneity in macrophage function across 24–72 hours of stimulation (Fig 3E–F). Ccl2 and Ccl5 expression and downregulation of Il10 and Ptgs2 in M(IFN-γ) conditions at 72 hours indicates polarization towards M1-like phenotype (Fig 3E). IFN-γ stimulation at 72 hours also upregulated mixed phenotype Il12b, IL6, Ccl22 and Nos2 and M2-like gene, Stat3. Accordingly, IL-4/IL-13 stimulation gradually upregulated Il10 and Ptgs2 while Il1b and Vegfa were significantly downregulated by 72 hours, confirming M2-like polarization (Fig 3F). Variability in expression of M1-like, M2-like, and mixed phenotype genes at 24- and 48-hours of stimulation suggests incomplete polarization at earlier time points (Fig 3E–F).
Evaluating non-specific effects of 3D Stacks co-culture system on macrophage metabolism and migration
The Stacks system was previously characterized for diffusion gradients and cell viability, survival, and recruitment(11). To determine non-specific changes in metabolism, metabolic activity was assessed in 2D breast cancer cultures with and without wavelength mixing (Supplementary Fig 2A–D) and in macrophages 1 hours after seeding for RAW264.7 macrophage monocultures and RAW264.7+PyVMT mouse breast carcinoma co-cultures within the Stacks system (Supplementary Fig 2E–G). Minimal differences in autofluorescence measurements were observed between both imaging methods (Supplementary Fig 2B–D). Autofluorescence measurements were not significantly different between monocultured macrophages and co-cultured macrophages, confirming that macrophages were similar for both conditions at the time of seeding (Supplementary Fig 2E–G). Additionally, no appreciable depth-dependent attenuation of NAD(P)H and FAD intensities were observed in these studies of optically transparent collagen I gels imaged within ~170 µm depths (Supplementary Fig 2H–I), ensuring accurate redox ratio trends without corrections for depth-dependent light attenuation. Additionally, RAW264.7 macrophages were seeded on both ends of the ECM layer to account for non-specific macrophage migration in the 3D Stacks system without tumor stimulation. Minimal change in macrophage migration was detected over 72 hours within macrophage-macrophage cultures (Supplementary Fig 2J–L). Overall, this data suggests that changes in macrophage migration and metabolism in co-culture at later time-points are due to tumor-specific signaling.
Mouse macrophage metabolism and migration using 3D metabolic imaging and tumor microscale Stacks system
Next, metabolic autofluorescence was characterized in microfluidic cultures of mouse Polyoma-Middle T Virus (PyVMT) mammary carcinoma and mouse RAW264.7 macrophages over a 72 hour time-course. Redox ratio, NAD(P)H τm and FAD τm of co-cultured macrophages from a representative z-plane (migration distance = 6 µm) are visually distinct from monocultured macrophages at the same z-plane as early as 24 hours post-seeding (Fig 4A). Morphological differences also emerge between monocultured and co-cultured macrophages over the 72 hour time course. Co-cultured macrophages were larger than monocultured macrophages and adopted a mixture of flat, cuboidal or elongated, spindle-like morphologies, commonly observed for polarized macrophage states (Fig 4A)(35). Genetic heterogeneity between macrophages within 3D co-cultures was evaluated by qPCR for a gene panel associated with M1-like, M2-like, and mixed macrophage phenotypes (Supplementary Table 1)(38). Minimal changes in gene expression of select M1-like, M2-like, and mixed phenotype genes were observed between monocultured and co-cultured macrophages at early time points (Fig 4B). However, 72 hours of co-culture resulted in significantly increased expression across Ccl2, Ccl5, and Il23a (M1-like), Vegfa and Stat3 (M2-like), and Il6 (mixed phenotype) (Fig 4B). Additionally, Il12b was significantly downregulated in co-cultured macrophages at 72 hours (Fig 4B).
Figure 4: Metabolic autofluorescence imaging of mouse macrophages within 3D Stacks tumor co-cultures.
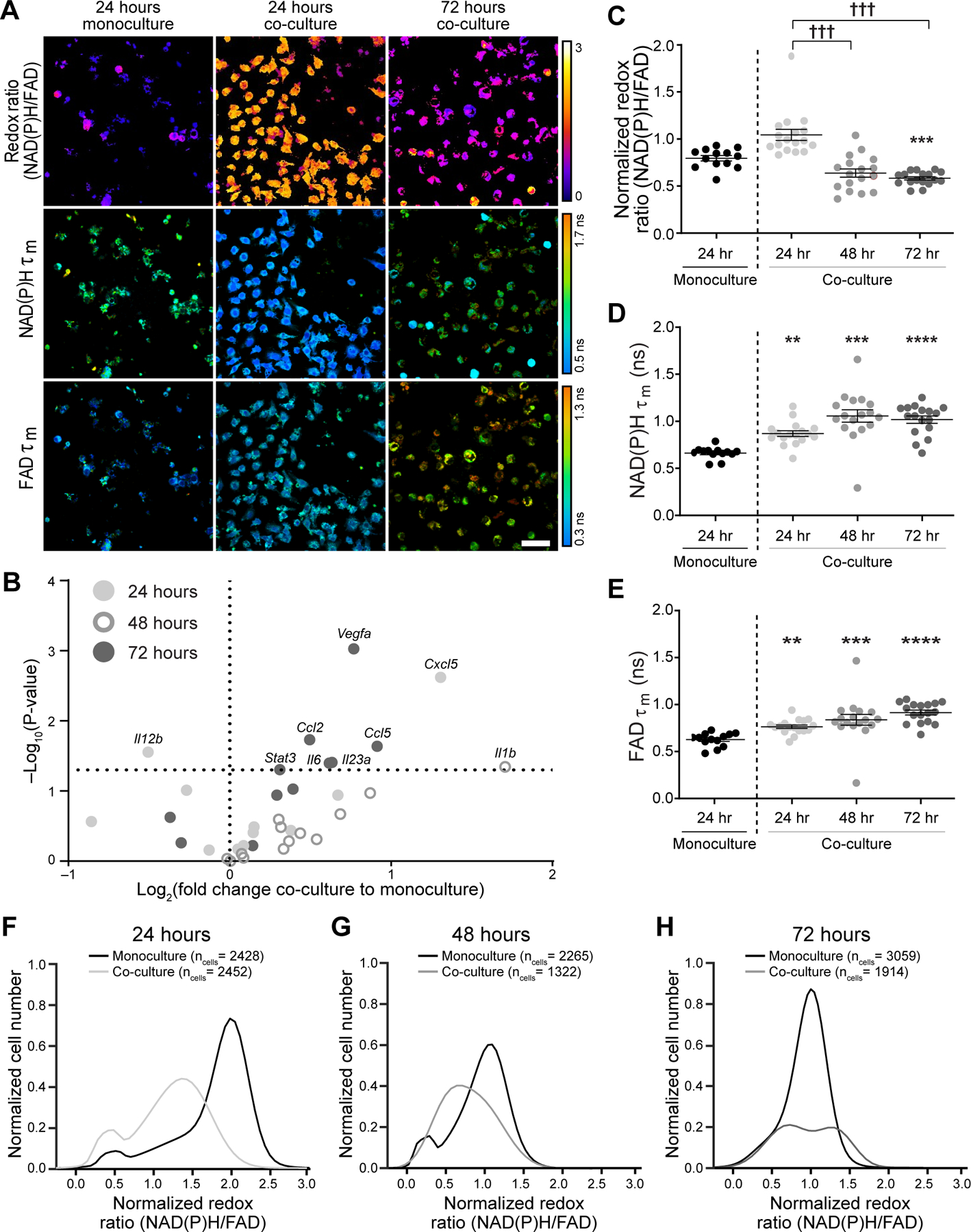
A) Representative autofluorescence images (z-plane distance: 6 µm) demonstrate qualitative differences in the redox ratio, NAD(P)H and FAD mean lifetimes (τm) of RAW264.7 macrophages in co-culture with PyVMT mouse mammary tumor cells. Scale bar = 50 μm. B) Gene expression levels of M1-like and M2-like markers were evaluated for monocultured and co-cultured macrophages at 24, 48, and 72 hours using qPCR. Upregulation of both M1-like and M2-like genes were observed in co-cultures by 72 hours, demonstrating heterogeneity in macrophage polarization in response to tumor stimulation. Quantitative analysis of C) redox ratio, D) NAD(P)H τm, and E) FAD τm highlight dynamic metabolic changes in macrophages during prolonged tumor-macrophage co-culture. Monocultured macrophages were not significantly different (p>0.05) across time points, so only the 24 hour monoculture is shown above. Significant differences in NAD(P)H and FAD τm are observed between monocultures and co-cultures, as early as 24 hours post-seeding. Additionally, macrophage co-cultures exhibit a gradual decline in redox ratio over time, consistent with transition to a more oxidized state; **,***,**** p<0.01,0.001,0.001 vs. monoculture, ††† p<0.001. Population density curves of single-cell redox ratios in monocultures and co-cultures at F) 24 hours, G) 48 hours, and H) 72 hours demonstrate variable cellular-level heterogeneity in co-cultured macrophages over time in monocultures.
Quantitative autofluorescence measurements more clearly capture metabolic changes across 3D macrophage cultures. Significant differences in NAD(P)H τm and FAD τm were observed between monocultured and co-cultured macrophages at 24, 48 and 72 hours, while the redox ratio of co-cultured macrophages significantly decreased only at 72 hours compared to monoculture conditions (Fig 4C–E). The redox ratio gradually decreased over 72 hours of co-culture with tumor cells (Fig 4C), while NAD(P)H and FAD τm did not significantly change over time (Fig 4D&E). Further changes in NAD(P)H and FAD lifetime components were also observed over the co-culture time course (Supplementary Fig 3A–F). Heterogeneity during RAW264.7 macrophage polarization in response to PyVMT tumor cell co-culture was also quantified with population density modeling of cell-level metabolic autofluorescence over the 72-hour imaging time course (Fig 4F–H). Two distinct redox ratio sub-populations of macrophages are present in co-culture at the 24 hour and 72 hour time points, and in monoculture at 24 and 48 hours (Fig 4F&H). Variability in redox ratio of macrophage monocultures is less than co-cultures at all time points (Supplementary Table 3). Heterogeneity in enzyme binding activity is reflected in single-cell NAD(P)H and FAD τm distributions, which also detect macrophage sub-populations during stimulation (Supplementary Fig 3G–L). These results demonstrate that this 3D tumor co-culture can stimulate heterogeneous changes in macrophage gene expression (Fig 4B) that may contribute to the metabolic heterogeneity between individual macrophages that is exclusively captured with metabolic autofluorescence measurements (Fig 4F–H).
The 3D culture and imaging approach enabled an analysis of migration as a source of metabolic heterogeneity within 3D mouse co-cultured macrophages. Specifically, mouse macrophage metabolism was monitored during migration toward the tumor layer within 3D co-cultures by acquiring volumes of metabolic autofluorescence images spanning the macrophage layer. Heatmaps of optical redox ratio at each z-plane show that macrophage metabolism and migratory distance vary over the 72 hour time course (Fig 5A). Heatmaps of NAD(P)H τm and FAD τm with migration distance are represented in Supplementary Fig 3M–N. The number of macrophages within each z-plane (depth increments of 3 µm) was divided by the total number of macrophages within each image volume to quantify the migration distance of macrophages over time (Fig 5B–D). Monocultured macrophages exhibited minimal migration from the seeding plane, whereas co-cultured macrophages migrated further into the ECM toward the tumor layer especially at 24 and 48 hours (Fig 5B&C). Therefore, the maximum migration distance for the monoculture condition was used to threshold passive from active macrophage migration at each time point (Fig 5B–D)(32). Actively-migrating macrophages in co-culture have lower redox ratio at all time points, suggesting that these macrophages shift towards oxidative metabolism to enhance migration in the TME (Fig 5E–F). Additionally, NAD(P)H τm decreases in actively-migrating macrophages at 24 and 48 hours, while FAD τm increases significantly at 72 hours (Fig 5F, Supplementary Fig 3O&P). Z-scores for each metabolic autofluorescence variable were calculated for passively- and actively-migrating mouse macrophages in co-culture by subtracting the variable mean of the monoculture condition from the variable mean per condition, then dividing by the monoculture standard deviation at each time point (Fig 5F). Multi-class random forest models were generated from all 11 autofluorescence variables to classify passive and active migration in 3D mouse co-cultures across all timepoints (24, 48, and 72 hours). High classification accuracy was observed for test data predictions for passive vs. active migration, with an accuracy of at least 85% for all test data predictions (Fig 5G, Supplementary Fig 3Q). These results highlight the combination of metabolic autofluorescence imaging and 3D Stacks co-culture to reveal novel relationships between metabolic activity and macrophage migration.
Figure 5: Microscale tumor co-cultures regulate mouse macrophage metabolism and migration over time.
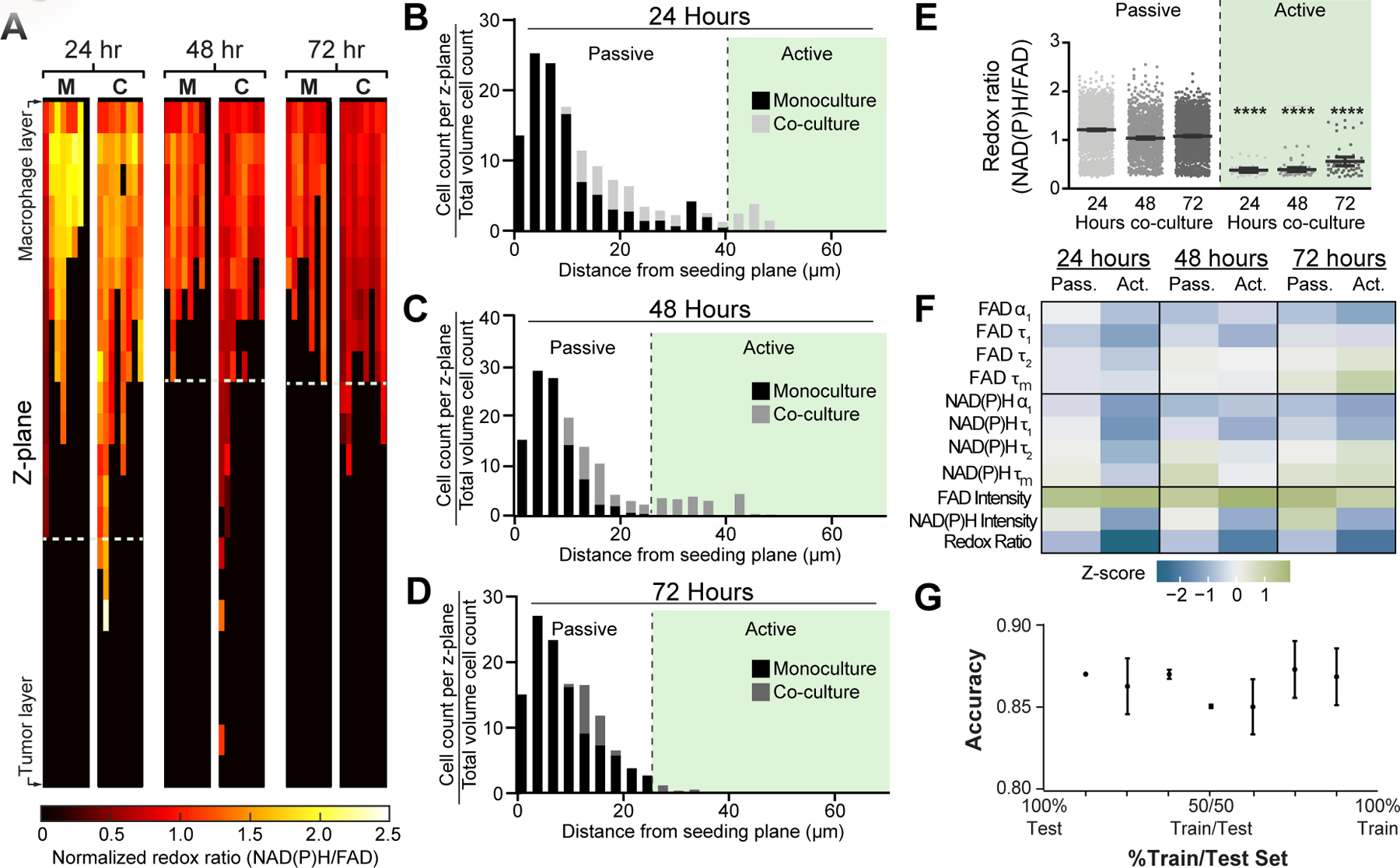
A) Representative heatmaps show changes in optical redox ratio with z-plane and time within monocultured, “M”, and co-cultured, “C”, macrophages. Cell density distributions of migration distance identify a distinct subpopulation of actively-migrating RAW264.7 macrophages in the PyVMT mouse mammary tumor co-culture condition at B) 24 hours, C) 48 hours, and D) 72 hours. E) Passively- and actively- migrating macrophage populations were defined in co-cultures over 72 hours to observe time-dependent relationships between metabolic autofluorescence and migratory activity. Actively-migrating populations exhibit lower redox ratio than passive-migrating populations at all time points, suggesting macrophages undergo a metabolic switch during migration;****p<0.0001 vs passive migration. F) Z-score heatmaps representing metabolic autofluorescence changes for passive and actively-migrating RAW264.7 mouse macrophages in co-culture relative to monocultured macrophages. Z-scores are calculated as the variable mean per condition minus variable mean of the monoculture condition, divided by the monoculture standard deviation. G) Random forest classification accuracy of passive and active migration of RAW264.7 mouse macrophages in co-culture at 24, 48 and 72 hours (2 groups – Passive vs. Active).
Human macrophage metabolism and 3D migration in response to patient-derived breast cancer cells
To further observe human tumor-mediated macrophage polarization and migration, THP-1 human monocytes were cultured alone or with patient-derived IDC cells, and metabolic autofluorescence changes were measured over 72 hours. Qualitative redox images from a representative z-plane (migration distance = 12 µm) highlight cell-level heterogeneity in redox ratio and lifetimes for co-cultures compared to monoculture (Fig 6A). Increased cell and nucleus size is observed in co-cultured cells, consistent with reported morphological differences between human monocytes and activated macrophages (Fig 6A)(39). Expression of M1-like and M2-like macrophage signature genes demonstrate heterogeneity in macrophage phenotype for human co-cultures(40,41). 24 hours of co-culture significantly upregulated only IL1B, while expression of TNF and PTGS2 significantly decreased in 48 hour co-cultures (Fig 6B). Mixed phenotype genes were variably expressed at both 24 and 48 hours (Fig 6B). Conversely, mixed phenotype IL12B, TGFB1, CSF1, NOS2, IL10 was upregulated after 72 hours co-culture with patient-derived tumor cells (Fig 6B). IL1B, TNF (M1-like) and CCL22, VEGFA, PTGS2 (M2-like) were also significantly increased by 72 hours (Fig 6B). Quantitative autofluorescence measurement showed monocyte-derived macrophages in co-culture have significantly higher redox ratio at 48 hours (Fig 6C), no change in NAD(P)H τm at any time point (Fig 6D), and significantly lower FAD τm at all time points compared to monoculture (Fig 6E). NAD(P)H and FAD lifetime components were also affected by tumor co-culture (Supplementary Fig 4A–F). These parallel metabolic and genetic findings demonstrate variation in tumor-mediated monocyte activation and macrophage polarization across species, culture in 2D vs. 3D, and breast tumor sub-types, which highlights the unique insights enabled by combining 3D Stacks and autofluorescence imaging technologies.
Figure 6: Metabolic autofluorescence imaging captures human monocyte-derived macrophage metabolism in 3D Stacks co-cultures with primary human invasive ductal carcinoma.
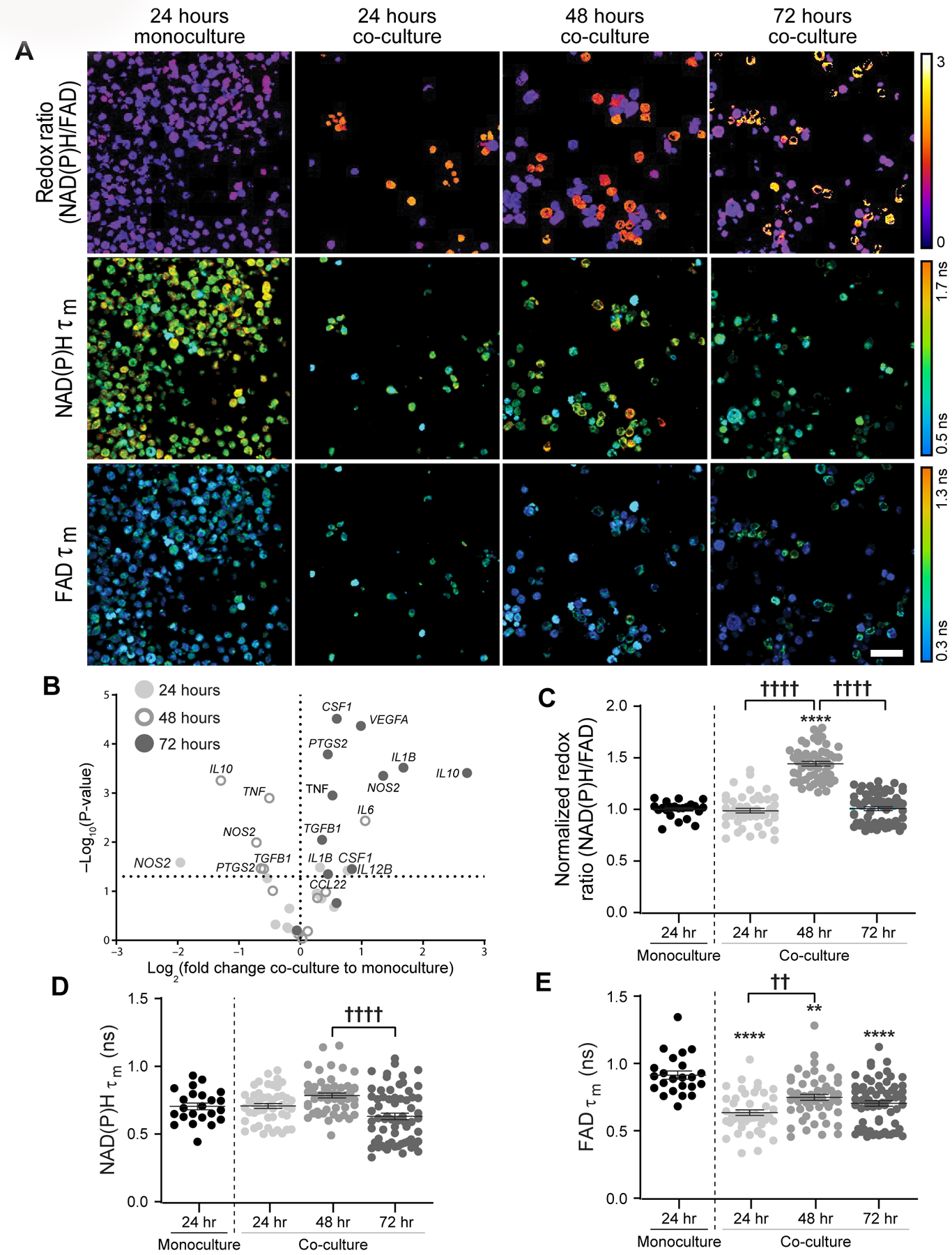
A) Representative images (z-plane distance: 12µm) display qualitative changes in redox ratio, NAD(P)H τm, and FAD τm between monoculture and co-cultures over 72 hours. Scale bar = 50 µm. B) Gene expression changes in co-cultured monocyte-derived macrophages over 24, 48, and 72 hours measured from qPCR. Changes are reported as log fold change with respect to the average of the monoculture condition for each measurement and time point. Metabolic changes in human THP-1 monocytes were quantified following co-culture with primary human IDC: C) Redox ratio, D) NAD(P)H τm, and E) FAD τm. **,**** p<0.01,0.0001 vs. monoculture; ††, †††† p<0.01,0.0001.
The considerable heterogeneity observed in metabolic autofluorescence across 2D and 3D models of mouse macrophage polarization prompted an investigation of heterogeneity due to migration within 3D human tumor-macrophage co-cultures. Here, tumor-mediated migration was also assessed in 3D co-cultures of primary IDC and THP-1 monocytes. Heatmaps of redox ratio changes with z-plane show increased migration in co-culture conditions and decreased redox ratio in macrophages localized closer to the tumor at 72 hours (Fig 7A), similar to the behavior of RAW264.7 cells (Fig 5A). NAD(P)H and FAD τm of macrophages also vary with time and z-plane (Supplementary Fig 4G–H). Monocyte-derived macrophages stimulated in co-culture exhibited substantial migration towards the tumor layer at all time points, in contrast to monocytes in monoculture (Fig 7B–D). Passively- and actively-migrating macrophages were then defined based on monoculture migration distances at each time point (Fig 7B–D). Both the redox ratio and FAD τm of actively-migrating macrophages co-culture were higher at early time points compared to passively-migrating macrophages, but gradually declined over 72 hours (Fig 7E–F, Supplementary Fig 4J). Actively-migrating macrophages in co-culture also have decreased NAD(P)H τm at 24 and 72 hours (Supplementary Fig 4I). Z-scores for metabolic autofluorescence of passively- and actively-migrating human macrophage co-cultures were calculated relative to the monoculture condition at each time point, demonstrating substantial differences between monocultured and actively-migrating co-cultured macrophages, while passively-migrating co-cultured macrophages minimally changed relative to monoculture (Fig 7F). Random forest classification with all 11 autofluorescence variables was also performed to distinguish passively- and actively-migrating macrophages at 24, 48, and 72 hours of co-culture with human primary invasive ductal carcinoma. Classification accuracy for passive and active migration was > 83% across all test data predictions (Fig 7G, Supplementary Fig 4K). Furthermore, the versatility of this platform was demonstrated from a time course of autofluorescence changes in THP-1 human monocytes in co-culture with a triple negative breast cancer cell line (MDA-MB-231, Supplementary Fig 5A–J) in contrast with patient-derived IDC co-cultures (Figs 6–7), highlighting breast cancer cell origin as a source of variability in monocyte-derived macrophage metabolism. Overall, these results establish the utility and versatility of the platform to address the challenge of evaluating dynamic macrophage function across human TME models.
Figure 7: Spatial and temporal metabolic changes of human monocyte-derived macrophages in co-culture with primary human invasive ductal carcinoma.
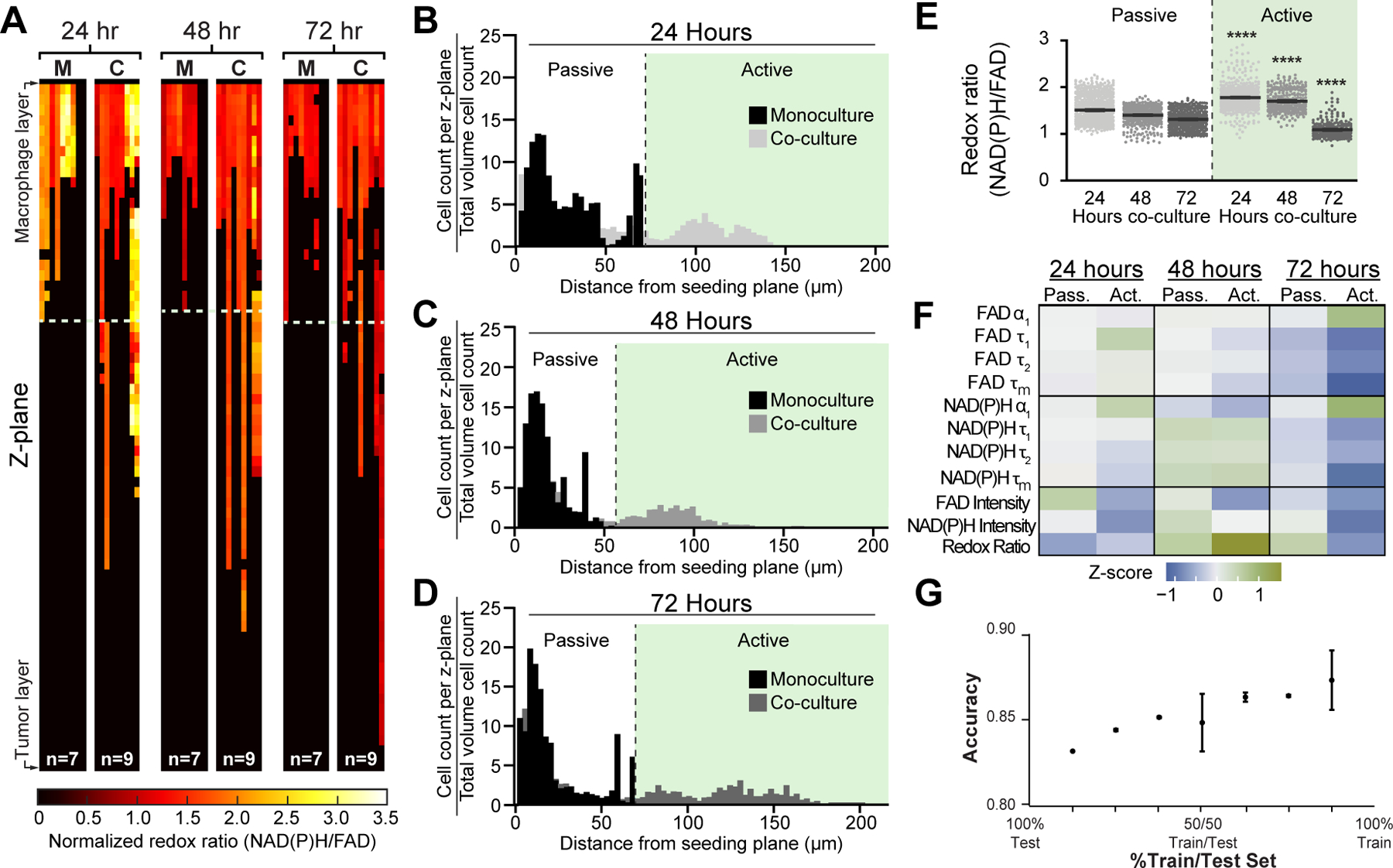
A) Representative heatmaps depicting changes in redox ratio with z-plane. Co-cultured macrophages, “C”, exhibit greater migratory activity and gradual decreases in redox ratio over time compared to monocultured macrophages, “M”. Representative cell density distributions reveal significant monocyte-derived macrophage migration in response to tumor co-culture, with a large population of actively-migrating macrophages at B) 24 hours, C) 48 hours, and D) 72 hours. E) Differences in redox ratio between actively- and passively-migrating populations were quantified at each time point. ***,**** p<0.001,0.0001 vs. passive migration. F) Z-score heatmaps representing metabolic autofluorescence changes for passive and actively-migrating THP-1 human macrophages in co-culture relative to monocultured macrophages. G) Random forest classification accuracy of passive and active migration of THP-1 human macrophages in co-culture at 24, 48 and 72 hours (2 groups – Passive vs. Active).
DISCUSSION
Macrophage plasticity leads to frequent shifts in function and metabolism in the TME, resulting in heterogeneous tumor-associated macrophage populations(9). Here, we show that metabolic autofluorescence imaging can monitor macrophage heterogeneity during polarization and migration within a 3D microfluidic model of the TME. Metabolic autofluorescence imaging enables non-destructive monitoring of metabolic changes in individual, live cells within 3D in vitro cultures and in vivo tumors(16–18,22). Additionally, 3D microfluidic culture systems, such as the Stacks microfluidic device used in this study, provide simple platforms to recapitulate dynamic environmental conditions that characterize in vivo tumors (e.g., hypoxia, acidosis, nutrient starvation) and are powerful to study primary human cells in an in vivo-like environment(42). Previous studies have shown that metabolic autofluorescence can identify macrophages in 2D cultures and in vivo but have not explored spatiotemporal heterogeneity in macrophage metabolism within the TME(16–18,24). This is the first study to monitor cell-level macrophage metabolism in microfluidic models of intact TME. Here, we quantify metabolic heterogeneity of macrophages in the Stacks 3D microdevice platform using autofluorescence imaging to provide novel insights into spatiotemporal heterogeneity of macrophages in the TME.
Temporal changes in microenvironment conditions affect macrophage metabolism and function, so this study quantified time-dependent changes in macrophage autofluorescence within microfluidic 3D tumor co-cultures. This study first confirmed that metabolic autofluorescence imaging can quantify changes in macrophage metabolism and distinguish macrophage sub-populations within intact, living samples. Differences in NAD(P)H and FAD autofluorescence were first observed in 2D cytokine-stimulated macrophages over 72 hours and reflected the expected metabolic shifts for M(IFN-γ) and M(IL4/IL13) macrophages (Fig 2)(43–45). This result is consistent with previous metabolic flux and metabolite accumulation studies showing increased glycolysis in LPS- or IFN-γ+TNF-α-stimulated (M1-like) macrophages and increased TCA cycle in IL-4-stimulated (M2-like) macrophages, as well as NAD(P)H lifetime studies distinguishing IFN-γ/LPS-treated and IL-4/IL-13 treated mouse bone marrow-derived macrophages (BMDMs)(16). Generation of a multiparametric classification model incorporating all 11 metabolic autofluorescence measurements further improved discrimination of these 2D macrophage polarization states (Fig 2C, Supplementary Fig 1L). The classifier performed well for identifying M0, M(IFN-γ), and M(IL4/IL13) macrophage conditions regardless of stimulation period, indicating that this classifier could accurately identify polarization in samples where the period of stimulation is unknown. Additionally, metabolic heterogeneity was observed across population density distributions of single-cell redox ratios for 2D mouse macrophages over 24–72 hours of polarization, revealing distinct populations of low and high redox ratio (Fig 3A–C). This metabolic heterogeneity is consistent with heterogeneous gene and protein expression profiles (Fig 3D–F), but autofluorescence measurements resolve cell-level heterogeneity unlike these bulk gene and protein measurements. This analysis also demonstrates the sensitivity of autofluorescence measurements to temporal functional plasticity commonly reported during macrophage polarization. For example, the bimodal populations observed in redox ratio of polarized macrophages may reflect early- and late-phase metabolic reprogramming shown in recent studies to promote initial upregulation of glycolytic or oxidative processes prior to reestablishing a resting metabolic state(43,46). Notably, autofluorescence imaging provides a unique tool to monitor metabolically-distinct cell sub-populations, unlike standard metabolic flux or metabolite production measurements that monitor bulk changes within a pooled population of cells.
Next, RAW264.7 macrophages co-cultured in 3D with PyVMT breast cancer displayed changes in redox ratio consistent with metabolic changes from tumor stimulation in previous studies (Fig 4A&C)(47). Differences in redox ratio and fluorescence lifetime measurements were observed between RAW264.7 macrophages in cytokine-stimulated 2D cultures (Fig 2B) and tumor-stimulated 3D cultures (Fig 4C–E), consistent with reported differences in intracellular metabolite concentration and metabolic flux based on stimulation condition (e.g., cytokines, tumor conditioned media) and culture in 2D vs. 3D(48,49). Additionally, 3D co-cultured RAW264s exhibited heterogeneous cell-level autofluorescence and pooled gene expression (Fig 4B, F–H). This is consistent with reported time courses of metabolic and genetic changes in co-cultures of tumor cells and monocytes or macrophages(50). Concentrations of metabolites, including NAD(P)H and FAD, can be replenished or depleted more rapidly than the downstream gene expression initiated by these metabolic changes, so correlations between gene expression and autofluorescence changes are difficult to interpret(50). However, the genetic and metabolic heterogeneity observed here is supported by recent studies that similarly show tumor-associated macrophages adopt a mixture of M1-like and M2-like characteristics and functions(51). More importantly, autofluorescence measurements capture early metabolic changes that are followed by these late changes in gene and protein expression(52). These different timescales of metabolic, gene, and protein expression changes in macrophages are likely responsible for differences between autofluorescence measurements and standard genomic/proteomic readouts. This reinforces a model of tumor-associated macrophages with diverse phenotypes(53), which can be further explored using the methods demonstrated here. Ultimately, these studies show that metabolic autofluorescence imaging is sensitive to heterogeneity in macrophage function and metabolism within 3D models of the TME.
Microfluidic culture is also attractive for mimicking the 3D spatial structure of the TME. Accordingly, multi-photon excitation of metabolic autofluorescence enables cellular-resolution imaging in thick, scattering samples to monitor environmental gradients and spatial heterogeneity in 3D tumor models(54). Therefore, we measured metabolic heterogeneity of RAW264 macrophages with 3D migration during co-culture with PyVMT tumor cells (Fig 5). Macrophages in co-culture exhibited greater migration across the ECM layer over 72 hours compared to monocultured macrophages, which was shown to be a tumor-specific behavior (Fig 5A–D, Supplementary Fig 2J–L,3M–N). Previous studies suggest that mouse macrophages often exhibit random migration during chemotaxis(55). This is consistent with our observations of mouse macrophage migration (Fig 5A–D). Prior studies have also shown that M2-like macrophages are typically more migratory, which is consistent with the oxidative metabolic shift in mouse macrophages localized closest to the tumor layer (actively-migrating) (Fig 5E)(56,57). However, variability in individual metabolic autofluorescence measures (e.g., redox ratio, NAD(P)H/FAD lifetimes) limits their classification accuracy when used alone. Thus, classification models based on all 11 metabolic autofluorescence variables were generated to collectively describe metabolic changes in macrophages with tumor stimulation in 3D and improve discrimination of passive and active migratory phenotypes (Figs 5G, 7G, Supplementary Figs 3Q, 4K). Notably, this 3D imaging platform enables further exploration of cell-level metabolic heterogeneity as a function of migration, not resolved from the genetic profile pooled across all migratory depths. Overall, metabolic autofluorescence imaging and microfluidic models of 3D microenvironment illustrate tumor-driven spatiotemporal changes in macrophage metabolism.
An important advantage of the Stacks system is the use of primary human cells for studies of human tumor-immune interactions. We highlighted this advantage with 3D co-cultures of primary IDC and THP-1 monocytes (Fig 6). Previous studies have shown that human THP-1 monocytes will differentiate and preferentially polarize to M2-like macrophages as early as 48 hours after treatment with tumor-conditioned media(58,59). This is consistent with redox ratio changes of THP-1s in co-culture with primary tumor cells (Fig 6C). The late oxidative, M2-like switch captured by the redox ratio is also consistent with previous reports of 2D differentiated THP-1 cells exposed to IL-4 (M2-like) cytokine stimulation(18). Although autofluorescence changes were less pronounced in human co-cultures when all macrophages were pooled, spatial analyses revealed more substantial autofluorescence changes due to macrophage migration in co-culture. Co-cultured THP-1s also migrated farther across the ECM than monocultures over 72 hours (Fig 7A–D). Human macrophages and monocytes exhibit strong directional migration toward chemotactic gradients (Fig 7B–D), consistent with our observations of human macrophage and monocyte migration(12). The redox ratio of actively-migrating human macrophages increased at 24 hours and gradually declined over 72 hours (Fig 7E–F), consistent with reports of upregulated glycolytic metabolism prior to oxidative shifts in human macrophages during chemoattractant-induced migration(60). These studies indicate that metabolic autofluorescence imaging in human tumor-macrophage microfluidic cultures provides a valuable platform to characterize novel spatiotemporal dynamics of macrophage metabolism within the human TME.
Cell function is variable across species and individuals. This variability is reflected in metabolic autofluorescence differences between mouse PyVMT/RAW264.7 and human primary invasive ductal carcinoma/THP-1 co-cultures (Figs 4&6, Supplementary Figs 3&4). Our results are consistent with previous reports of genetic and metabolic differences between mouse and human macrophage lines, including RAW264.7 and THP-1s(40,41,43,44,61). These changes are likely due to distinct profiles of cytokine secretion and other polarizing stimuli(36,40). Differences in macrophage response to tumor co-culture are also likely due to unique tumor cell origin and functional activity. The human primary invasive ductal carcinoma line was derived from resected tissue of a moderately differentiated and hormone receptor-positive breast cancer, while PyVMT mouse breast carcinoma originates from a transgenic mouse model with spontaneous oncoprotein-controlled tumor growth that more closely mimics human triple-negative breast cancer(25,62). Furthermore, genetic and transcriptional profiles of human breast cancer are only partially conserved in PyVMT mouse mammary cancer(62). Therefore, variations in macrophage metabolism in response to these breast cancer co-cultures are likely due to differences in cell phenotype and function (e.g., mutation/receptor status, secretory profiles) across species.
The combined metabolic autofluorescence imaging and 3D Stacks microscale platform provides a unique system to evaluate tumor-macrophage crosstalk within the TME. Though this model system mimics key components of the TME, limitations exist for fully recapitulating the cellular and architectural diversity of the TME(10). One limitation in our studies is the use of macrophage cell lines rather than primary macrophages. These macrophage cell lines (RAW264.7 and THP-1) were previously characterized under conditions similar to those tested in this study(11,43,47), so these cell lines were chosen to provide a validated model for autofluorescence imaging in 3D Stacks. However, RAW264.7 and THP-1 lines originated from monocytic leukemias, which introduce phenotypic and functional differences related to the origin site and malignant background compared to primary macrophages derived from peripheral blood or bone marrow(63,64). The autofluorescence imaging and 3D Stacks system demonstrated here is compatible with primary macrophages, which should be investigated in the future to provide deeper insights into functional dynamics in macrophage migration and metabolism. Furthermore, environmental factors (e.g., oxygen and pH gradients) are another source of contrast for autofluorescence lifetimes(19,42). Decreased FAD autofluorescence lifetimes have been previously correlated with increased pH, which suggests that our observed decreased FAD mean lifetimes with prolonged co-culture and increased migration in human monocyte-derived macrophages may be due increased local pH levels(65). An additional consideration for this platform is the efficiency of excitation using the wavelength mixing approach. This method has been shown to excite FAD more efficiently than two-color sequential imaging(27). However, wavelength mixing and two-color sequential imaging provide comparable excitation efficiency for NAD(P)H and comparable fluorescence lifetime values for both fluorophores(27). In our study, all redox ratio values are referenced to control groups, and relative changes in redox ratio are conserved between wavelength mixing and two-color sequential imaging (Supplementary Fig 2). Considering these constraints for future adaptations of this 3D imaging and microscale culture platform should improve its versatility for characterizing cellular dynamics in the TME.
Overall, we have established a novel, single-cell imaging and 3D microfluidic culture platform to monitor the spatial and temporal dynamics of macrophage metabolism within the TME. Autofluorescence imaging of 3D tumor-macrophage co-cultures characterized metabolic heterogeneity with tumor-mediated macrophage polarization and migration. This approach could be used to evaluate metabolic heterogeneity in more complex 3D microscale cultures with additional immune cells, stromal cells, and vasculature. Additionally, this technology supports live, high-throughput biological studies to probe mechanisms of macrophage response to microenvironmental stimulation in parallel to imaging studies. Direct manipulation of environmental gradients (e.g., oxygen, nutrients, pH) during culture and imaging could assess the relationship between environmental pressures and macrophage metabolism, function, and organization. Ultimately, these tools may improve our understanding of cellular heterogeneity and metabolism in the TME, and their effects on tumor progression and treatment response.
Supplementary Material
SIGNIFICANCE.
Label-free metabolic imaging and microscale culture technologies enable monitoring of single-cell macrophage metabolism, migration, and function in the 3D tumor microenvironment.
ACKNOWLEDGMENTS
We would like to thank Rupsa Datta and Jose Ayuso for their valuable input on the experimental design and manuscript composition. We acknowledge Jens Eickoff for his guidance in statistical analysis of the reported data. We also acknowledge Kayvan Samimi and Matthew Stefely for input on design of scientific diagrams and data representation. M.C. Skala acknowledges support from the NCI (R01 CA211082, R01 CA205101, R01 CA185747), the NSF (CBET-1642287), Stand Up to Cancer (SU2C-AACR-IRG-08–16, SU2C-AACR-PS-18), the Morgridge Institute for Research, and the University of Wisconsin Carbone Cancer Center. Stand Up To Cancer is a division of the Entertainment Industry Foundation. The cited SU2C grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. T.M. Heaster acknowledges the support of the NSF Graduate Research Fellowship (DGE-1256259). M. Humayun acknowledges support by NIH Fellowship F31 CA247248.
Footnotes
Conflicts of interest statement:
David J. Beebe holds equity in Bellbrook Labs LLC, Tasso Inc., Salus Discovery LLC, Lynx Biosciences Inc., Stacks to the Future LLC, Turba LLC, and Onexio Biosystems LLC. David J. Beebe is also a consultant for Abbott Laboratories. Jiaquan Yu holds equity in Stacks to the Future LLC and Turba LLC.
REFERENCES
- 1.Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nature Immunol. 2013;14(10):1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panni RZ, Linehan DC, DeNardo DG. Targeting tumor-infiltrating macrophages to combat cancer. Immunotherapy. 2013;5(10):1075–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray PJ. Macrophage Polarization. Annu Rev Physiol. 2017;79(1):541–66. [DOI] [PubMed] [Google Scholar]
- 4.Jagannathan N, Ramanathan S. Tumor Associated Macrophage: A Review on the Phenotypes, Traits and Functions. Iran J Cancer Prev Iran J Cancer Prev. 2014;7(71):1–81. [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verdeguer F, Aouadi M. Macrophage heterogeneity and energy metabolism. Exp Cell Res. 2017;360(1):35–40. [DOI] [PubMed] [Google Scholar]
- 7.Pearce EL, Pearce EJ. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity. 2013;38(4):633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019;30(1):36–50. [DOI] [PubMed] [Google Scholar]
- 10.Montanez-Sauri SI, Beebe DJ, Sung KE. Microscale screening systems for 3D cellular microenvironments: platforms, advances, and challenges. Cell Mol Life Sci. 2015;72(2):237–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu J, Berthier E, Craig A, de Groot TE, Sparks S, Ingram PN, et al. Reconfigurable open microfluidics for studying the spatiotemporal dynamics of paracrine signalling. Nat Biomed Eng. 2019;3(10):830–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bzymek R, Horsthemke M, Isfort K, Mohr S, Tjaden K, Müller-Tidow C, et al. Real-time two- and three-dimensional imaging of monocyte motility and navigation on planar surfaces and in collagen matrices: roles of Rho. Sci Rep. 2016;6(1):25016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chattopadhyay PK, Gierahn TM, Roederer M, Love JC. Single-cell technologies for monitoring immune systems. Nat Immunol. 2014;15(2):128–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ottobrini L, Martelli C, Trabattoni DL, Clerici M, Lucignani G. In vivo imaging of immune cell trafficking in cancer. Eur J Nucl Med Mol Imaging. 2011;38(5):949–68. [DOI] [PubMed] [Google Scholar]
- 15.Georgakoudi I, Quinn KP. Optical Imaging Using Endogenous Contrast to Assess Metabolic State. Annu Rev Biomed Eng. 2012;14(1):351–67. [DOI] [PubMed] [Google Scholar]
- 16.Alfonso-García A, Smith TD, Datta R, Luu TU, Gratton E, Potm EO, et al. Label-free identification of macrophage phenotype by fluorescence lifetime imaging microscopy. J Biomed Opt. 2016;21(4):46005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szulczewski JM, Inman DR, Entenberg D, Ponik SM, Aguirre-Ghiso J, Castracane J, et al. In Vivo Visualization of Stromal Macrophages via label-free FLIM-based metabolite imaging. Sci Rep. 2016;6(1):25086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smokelin IS, Mizzoni C, Erndt-Marino J, Kaplan D, Georgakoudi I. Optical changes in THP-1 macrophage metabolism in response to pro- and anti-inflammatory stimuli reported by label-free two-photon imaging. J Biomed Opt. 2020;25(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Datta R, Heaster TM, Sharick JT, Gillette AA, Skala MC. Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications. J Biomed Opt. 2020;25(7):1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chance B, Schoener B, Oshino R, Itshak F, Nakase Y. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J Biol Chem. 1979;254(11):4764–71. [PubMed] [Google Scholar]
- 21.Blacker TS, Duchen MR. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic Biol Med. 2016;100:53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharick JT, Jeffery JJ, Karim MR, Walsh CM, Esbona K, Cook RS, et al. Cellular Metabolic Heterogeneity In Vivo Is Recapitulated in Tumor Organoids. Neoplasia. 2019;21(6):615–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ayuso JM, Gillette A, Lugo-Cintrón K, Acevedo-Acevedo S, Gomez I, Morgan M, et al. Organotypic microfluidic breast cancer model reveals starvation-induced spatial-temporal metabolic adaptations. EBioMedicine. 2018;37:144–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Liu T-M. Discovering Macrophage Functions Using In Vivo Optical Imaging Techniques. Front Immunol. 2018;9:502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.NCI-Frederick, Frederick National Laboratory for Cancer Research, Frederick M The NCI Patient-Derived Models Repository (PDMR).
- 26.Mahou P, Zimmerley M, Loulier K, Labroille G, Morin X, Supatto W, et al. Multicolor two-photon tissue imaging by wavelength mixing. Nat Methods. 2012;9(8):815. [DOI] [PubMed] [Google Scholar]
- 27.Stringari C, Abdeladim L, Malkinson G, Mahou P, Solinas X, Lamarre I, et al. Multicolor two-photon imaging of endogenous fluorophores in living tissues by wavelength mixing. Sci Rep. 2017;7(1):3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bird DK, Yan L, Vrotsos KM, Eliceiri KW, Vaughan EM, Keely PJ, et al. Metabolic mapping of MCF10A human breast cells via multiphoton fluorescence lifetime imaging of the coenzyme NADH. Cancer Res. 2005;65(19):8766–73. [DOI] [PubMed] [Google Scholar]
- 29.He C, Carter AB. The Metabolic Prospective and Redox Regulation of Macrophage Polarization. J Clin Cell Immunol. 2015;6(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galván-Peña S, O’Neill LAJ. Metabolic reprograming in macrophage polarization. 2014;5:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pålsson-McDermott EM, O’Neill LAJ. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020;30:300–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pakshir P, Alizadehgiashi M, Wong B, Coelho NM, Chen X, Gong Z, et al. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat Commun. 2019;10(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walsh AJ, Skala MC. Optical metabolic imaging quantifies heterogeneous cell populations. Biomed Opt Express. 2015;6(2):559–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edward Miller G Use of the squared ranks test to test for the equality of the coefficients of variation. Commun Stat Comput. 1991;20(2–3):743–50. [Google Scholar]
- 35.Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2:578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith TD, Tse MJ, Read EL, Liu WF. Regulation of macrophage polarization and plasticity by complex activation signals. Integr Biol. 2016;8(9):946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z, Zhao M, Li T, Zheng J, Liu X, Jiang Y, et al. Decidual macrophage functional polarization during abnormal pregnancy due to Toxoplasma gondii: role for LILRB4. Front Immunol. 2017;8:1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bardi GT, Smith MA, Hood JL. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine. 2018;105:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rostam HM, Reynolds PM, Alexander MR, Gadegaard N, Ghaemmaghami AM. Image based machine learning for identification of macrophage subsets. Sci Rep. 2017;7(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spiller KL, Wrona EA, Romero-Torres S, et al. Differential gene expression in human, murine, and cell line-derived macrophages upon polarization. Exp Cell Res. 2016;347(1):1–13. [DOI] [PubMed] [Google Scholar]
- 41.Surdziel E, Clay I, Nigsch F, Pallotta I, Graney PL, Witherel CE, et al. Multidimensional pooled shRNA screens in human THP-1 cells identify candidate modulators of macrophage polarization. PLoS One. 2017;12(8):e0183679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Damgaci S, Ibrahim-Hashim A, Enriquez-Navas PM, Pilon-Thomas S, Guvenis A, Gillies RJ. Hypoxia and acidosis: immune suppressors and therapeutic targets. Immunology. 2018;154(3):354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seim GL, Britt EC, John SV, Yeo FJ, Johnson AR, Eisenstein RS, et al. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nat Metab. 2019;1(7):731–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol. 2019;10:1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tavakoli S, Downs K, Short JD, Nguyen HN, Lai Y, Jerabek PA, et al. Characterization of macrophage polarization states using combined measurement of 2-deoxyglucose and glutamine accumulation: implications for imaging of atherosclerosis. Arterioscler Thromb Vasc Biol. 2017;37(10):1840–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi L, Jiang Q, Bushkin Y, Subbian S, Tyagi S. Biphasic Dynamics of Macrophage Immunometabolism during Mycobacterium tuberculosis Infection. MBio. 2019;10(2):e02550–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khabipov A, Kaeding A, Liedtke KR, Freund E, Partecke L-I, Bekeschus S. RAW 264.7 Macrophage Polarization by Pancreatic Cancer Cells–A Model for Studying Tumour-promoting Macrophages. Anticancer Res. 2019;39(6):2871–82. [DOI] [PubMed] [Google Scholar]
- 48.Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee H-J, et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019;29(6):1390–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caires HR, Esteves T, Quelhas P, Barbosa MA, Navarro M, Almeida CR. Macrophage interactions with polylactic acid and chitosan scaffolds lead to improved recruitment of human mesenchymal stem/stromal cells: a comprehensive study with different immune cells. J R Soc Interface. 2016;13(122):20160570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Plüddemann A, et al. Ovarian Cancer Cells Polarize Macrophages Toward A Tumor-Associated Phenotype. J Immunol. 2006;176(8):5023–32. [DOI] [PubMed] [Google Scholar]
- 51.Hassanzadeh Ghassabeh G, De Baetselier P, Brys L, Noël W, Van Ginderachter JA, Meerschaut S, et al. Identification of a common gene signature for type II cytokine–associated myeloid cells elicited in vivo in different pathologic conditions. Blood. 2006;108(2):575–83. [DOI] [PubMed] [Google Scholar]
- 52.Shamir M, Bar-On Y, Phillips R, Milo R. SnapShot: timescales in cell biology. Cell. 2016;164(6):1302. [DOI] [PubMed] [Google Scholar]
- 53.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mulligan SJ, Macvicar BA, Méndez-Vilas A, Diaz J. Two-Photon Fluorescence Microscopy: Basic Principles, Advantages and Risks. Modern Research and Educational Topics in Microscopy. 2007; 1:881–99 [Google Scholar]
- 55.Hind LE, Mackay JL, Cox D, Hammer DA. Two-dimensional motility of a macrophage cell line on microcontact-printed fibronectin. Cytoskeleton. 2014;71(9):542–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vogel DYS, Heijnen PDAM, Breur M, de Vries HE, Tool ATJ, Amor S, et al. Macrophages migrate in an activation-dependent manner to chemokines involved in neuroinflammation. J Neuroinflammation. 2014;11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hind LE, Lurier EB, Dembo M, Spiller KL, Hammer DA. Effect of M1–M2 polarization on the motility and traction stresses of primary human macrophages. Cell Mol Bioeng. 2016;9(3):455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sawa-Wejksza K, Dudek A, Lemieszek M, Kaławaj K, Kandefer-Szerszeń M. Colon cancer–derived conditioned medium induces differentiation of THP-1 monocytes into a mixed population of M1/M2 cells. Tumor Biol. 2018;40(9):1010428318797880. [DOI] [PubMed] [Google Scholar]
- 59.Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giasjuddin S, et al. Inflammatory Breast Cancer Promotes Development of M2 Tumor-associated Macrophages and Cancer Mesenchymal Cells Through a Complex Cytokine Network. Cancer Res. 2019; 79(13):3360–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. 2016;7(1):11635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thapa B, Lee K. Metabolic influence on macrophage polarization and pathogenesis. BMB Rep. 2019;52(6):360–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rennhack JP, To B, Swiatnicki M, Dulak C, Ogrodzinski MP, Zhang Y, et al. Integrated analyses of murine breast cancer models reveal critical parallels with human disease. Nat Commun. 2019;10(1):3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tedesco S, De Majo F, Kim J, Trenti A, Trevisi L, Fadini GP, et al. Convenience versus Biological Significance: Are PMA-Differentiated THP-1 Cells a Reliable Substitute for Blood-Derived Macrophages When Studying in Vitro Polarization? Front Pharmacol. 2018;9:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berghaus LJ, Moore JN, Hurley DJ, Vandenplas ML, Fortes BP, Wolfert MA, et al. Innate immune responses of primary murine macrophage-lineage cells and RAW 264.7 cells to ligands of Toll-like receptors 2, 3, and 4. Comp Immunol Microbiol Infect Dis. 2010;33(5):443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Islam MS, Honma M, Nakabayashi T, Kinjo M, Ohta N. pH dependence of the fluorescence lifetime of FAD in solution and in cells. Int J Mol Sci. 2013;14(1):1952–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.