

Abstract
Despite a rapidly growing literature, the role played by the brain in both normal glucose homeostasis and in type 2 diabetes pathogenesis remains poorly understood. In this review, we introduce a framework for understanding the brain’s essential role in these processes based on evidence that the brain, like the pancreas, is equipped to sense and respond to changes in the circulating glucose level. Further, we review evidence that glucose sensing by the brain plays a fundamental role in establishing the defended level of blood glucose, and that defects in this control system contribute to type 2 diabetes pathogenesis. We also consider the possibility that the close association between obesity and type 2 diabetes arises from a shared defect in the highly integrated neurocircuitry governing energy homeostasis and glucose homeostasis. Thus, whereas obesity is characterised by an increase in the defended level of the body’s fuel stores (e.g. adipose mass), type 2 diabetes is characterised by an increase in the defended level of the body’s available fuel (e.g. circulating glucose), with the underlying pathogenesis in each case involving impaired sensing of (or responsiveness to) relevant humoral negative feedback signals. This perspective is strengthened by growing preclinical evidence that in type 2 diabetes the defended level of blood glucose can be restored to normal by therapies that restore the brain’s ability to properly sense the circulating glucose level.
Keywords: Brain, Diabetes, Glucose, Hypothalamus, Obesity, Review
Graphical Abstract
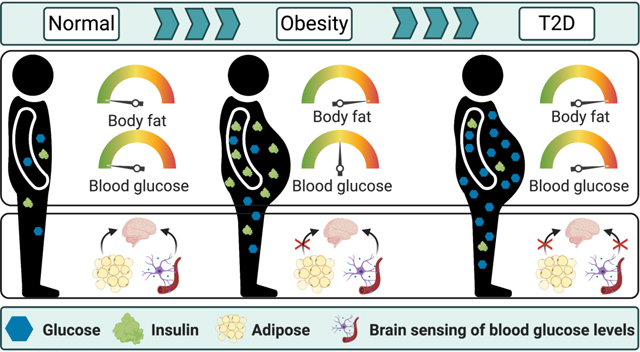
Introduction
Like other homeostatically defended variables, the circulating glucose level in healthy individuals is continuously maintained within narrow physiological limits. The stability of this glucose ‘set point’ arises from an elegant and highly integrated multi-organ control system that dynamically coordinates glucose entry into and removal from the circulation. Although many tissues are involved, the pancreas and brain exert primary control over this process. As ingested nutrients are absorbed into the circulation following a meal, increased insulin secretion promotes glucose disposal into muscle and fat and inhibits endogenous glucose production by the liver, thereby minimising changes in blood glucose levels. The brain helps to coordinate not only the magnitude and timing of the insulin response [1] but also insulin-independent mechanisms that reduce glucose production while enhancing disposal [2]. Here, we review interactions between brain and pancreas that establish the defended blood glucose level and present evidence from humans and animals that both organs must sense the circulating glucose level for this process to function normally. We conclude that impairment of this central control system is fundamental to the pathogenesis of type 2 diabetes.
Evidence linking brain glucose sensing to the glycaemic set point
Evolution of brain glucose sensing
Roughly seven decades before the discovery of insulin in 1921, Claude Bernard invoked a key role for the brain in glucose homeostasis [3], consistent with the view that the brain is responsible for homeostatic control of a broad range of variables upon which survival depends. Since the brain relies almost exclusively on glucose as a fuel, its role in ensuring its availability is in keeping with principles of physiology. While this concept was largely abandoned following the discovery of insulin, recent findings suggest that it warrants a second look.
Work in the fruit fly Drosophila melanogaster [4] reveals that both the systemic glucose level (in haemolymph) and glucose-induced secretion of the fly insulin homologue are governed by a single pair of neurons that sense glucose using cellular machinery analogous to that found in mammalian beta cells. Silencing these neurons causes elevation of systemic glucose levels, demonstrating that their function is essential for normal glucose homeostasis in flies. Interestingly, this effect is associated with reduced insulin secretion even though insulin-secreting cells themselves are not directly impacted [4]. These findings suggest that over the course of evolution, glucose homeostasis originated as a process governed by the brain, as envisioned by Claude Bernard, with insulin secretion lying downstream of brain glucose sensing. Another implication is that cellular machinery for glucose sensing evolved originally in neurons, subsequently being co-opted for use in mammalian beta cells. Although glucose homeostasis in mammals is more complex, evidence suggests that a version of the Drosophila brain control system has been retained over the course of mammalian evolution.
We propose herein a model in which normal glucose homeostasis depends upon afferent information regarding the circulating glucose level provided to the brain by both peripheral and central glucose-sensing mechanisms (Fig. 1a). This information is in turn predicted to modulate glucose-induced insulin secretion (GSIS) and other components of the glucose homeostasis system so as to balance the rates of glucose entering and leaving the circulation. Pancreatic islets function with a high degree of autonomy under usual conditions; minimal input from the brain is required while blood glucose levels remain within their defended physiological range [5]. Accordingly, the role of the brain becomes more apparent when levels deviate from this range.
Fig. 1.
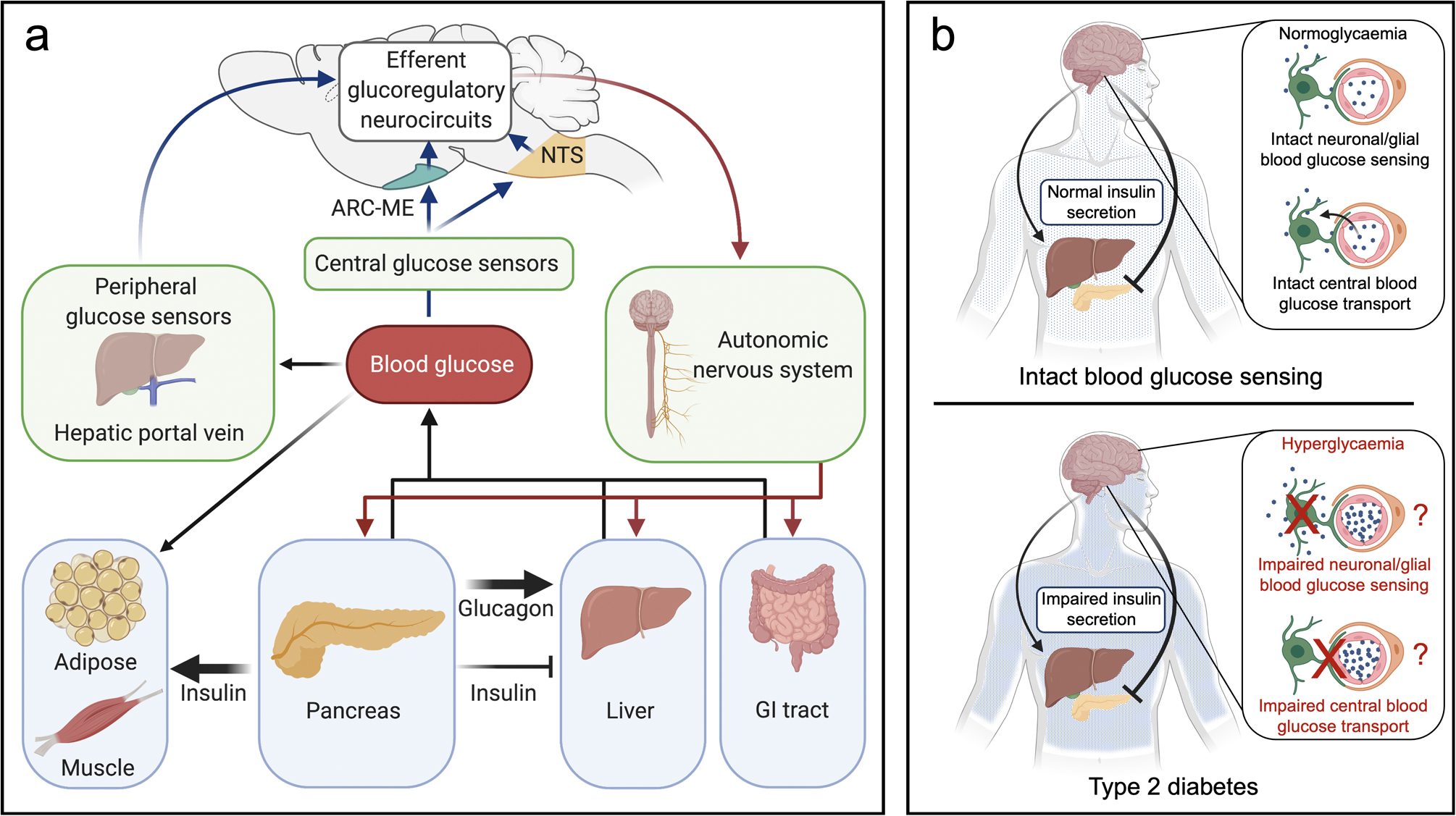
Model describing the role of the brain in glucose homeostasis. (a) Maintenance of blood glucose levels within a narrow physiological range requires balancing of glucose disappearance from and entry into the bloodstream. This balance is achieved via both insulin-dependent and insulin-independent mechanisms that are enhanced when blood glucose deviates from its regulated levels. Secreted by the pancreas in response to rising glucose levels, insulin promotes glucose disposal by inducing glucose uptake into insulin-sensitive tissues (e.g. adipose, muscle), while also reducing glucose appearance by inhibiting hepatic glucose production (black arrows). Glucagon opposes the latter effect by stimulating glucose production by the liver. The blood glucose level is also sensed by the brain via afferent input (blue arrows) to both central (e.g. arcuate nucleus–median eminence, NTS) and peripheral sensing mechanisms. When glucose levels deviate from the defended level, the brain powerfully adjusts both insulin-dependent and insulin-independent determinants of glucose entry into and removal from the circulation (red arrows) (in part via effects on insulin secretion) through the autonomic nervous system that ultimately return the glucose level to normal. (b) Impairment of the brain’s ability to sense blood glucose levels (blue dots) can result from either a genetic or acquired defect. This causes the perceived glucose level to be lower than it truly is and, in response, the brain raises the defended blood glucose level in part by inhibiting GSIS. This pathogenic sequence is proposed to play a major role in type 2 diabetes pathogenesis. ARC, arcuate nucleus; ME, median eminence; NTS, nucleus tractus solitarius.
Acquired or inherited defects in the brain’s ability to sense circulating glucose levels are proposed to result in the perception of the levels being lower than they actually are. While the nature of this glucose-sensing defect remains unclear, several possibilities exist. First, the increase in brain glucose concentration and brain glucose uptake that accompanies the increase in blood glucose is blunted in diabetic rodents [6, 7] and humans [8–11]. Second, neuronal glucose sensing may be dysfunctional [5]. In mice, brain-specific ablation of GLUT2 [12] or glucokinase [13] (key cellular mediators of glucose sensing in both beta cells and neurons) impairs glucose homeostasis. In rats, diabetes is associated with reduced hypothalamic glucokinase activity, and glucose homeostasis is improved by reversing this defect [14, 15]. These observations are consistent with a model in which brain sensing of circulating glucose levels is impaired in type 2 diabetes and, in response, the brain raises the defended blood glucose level, with reduced GSIS being a key component of this response (Fig. 1b). Additional evidence implicating this type of central defect in the pathogenesis of type 2 diabetes is reviewed below.
Brain control of glucose homeostasis during hypoglycaemia
Glucose-sensing neurons are concentrated in brain areas involved in glucose homeostasis [16], and the brain establishes the lower boundary of the blood glucose level in mammalian species. Specifically, the brain mounts ‘counter-regulatory’ responses (CRRs) that restore blood glucose to the normal level should it drop below this boundary [17]. Because this lower boundary is seldom crossed in healthy individuals, the brain’s role in glucose counter-regulation is usually viewed as an emergency response to a pathological state rather than an integral aspect of day-to-day glycaemic control. Nevertheless, specific downstream components of this brain-mediated CRR, such as adrenaline (epinephrine) secretion, can be considered robust biomarkers of what the brain perceives as the lower limit of the defended blood glucose level; the steadiness of this lower boundary suggests continuous homeostatic inputs that involve neural control. This raises the question of whether the brain has the capacity to raise the defended blood glucose level above the normal range in response to a perceived deficit in fuel availability. This capacity is illustrated by experimental induction of ‘neuroglycopenia’, which has been undertaken in many mammalian species, including non-human primates [18]. Neuroglycopenia is induced by administration of 2-deoxyglucose or other non-metabolisable glucose analogues that impair neuronal glucose utilisation. The brain’s response to this stimulus mimics that induced by hypoglycaemia: increased glucose production combined with decreased peripheral glucose utilisation drives blood glucose levels upwards to a stable, hyperglycaemic plateau sufficient to overcome the underlying defect in neuronal glucose sensing. This hyperglycaemia is fully recapitulated in rodents by optogenetic or chemogenetic activation of subsets of neurons in the CRR circuit activated by hypoglycaemia that are situated in the hypothalamic ventromedial nucleus (VMN) [19–21]. The expected increase in insulin secretion in response to this neurocircuit-induced hyperglycaemia is suppressed as part of the brain response to reduced glucose availability. Thus, the brain responds to experimentally reduced glucose availability by raising the defended blood glucose level, in part by inhibiting insulin secretion.
Brain control of glucose homeostasis during diabetic hyperglycaemia
Importantly, the lower limit of the defended blood glucose range increases as mildly abnormal glucose metabolism progresses to the initial phase of type 2 diabetes [22]. Stated differently, the threshold at which adaptive CRRs are recruited increases progressively in parallel with the onset of hyperglycaemia, such that the threshold for CRR activation is higher than normal [22, 23]. This implies that in type 2 diabetes, the elevated glycaemic set point continues to be defended by the brain even as it rises out of the normal range. The brain’s contribution to diabetic hyperglycaemia is also illustrated by studies in rodents wherein subsets of the CRR glucoregulatory neurons in the VMN are inactivated, providing information on what the neurons actually do (as opposed to what they are capable of doing when activated). In a recent study by Flak and colleagues [21], silencing a subset of glutamatergic CRR neurons in the VMN (marked by expression of the cholecystokinin B [CCK-B] receptor) of otherwise normal mice caused a ~25% reduction in blood glucose levels. Implicit in this finding is the intriguing concept that a specific subset of VMN neurons participating in the CRR is also a physiological determinant of the defended blood glucose level. More important is the observation that inactivation of these neurons not only impairs the ability to mount CRRs in response to neuroglycopenia [24, 25] but also ameliorates hyperglycaemia in diabetic mice [21]. Thus, neurons in the circuit responsible for mounting the brain response to reduced glucose availability are key contributors to hyperglycaemia in diabetic animals.
These observations are consistent with a model in which the capacity of the brain to sense blood glucose levels is impaired in diabetes, such that neurocircuits that raise the defended glucose level are activated to compensate for perceived glucose deficiency. Clinical evidence supporting this hypothesis is provided by data from individuals with MODY 2, caused by mutation of the gene encoding glucokinase (MODY2 [also known as GCK]) [22]. Glucokinase is essential for cellular glucose sensing in hypothalamic neurons as well as in beta cells, in both humans and rodents [26] and, although glucose homeostasis in individuals with MODY 2 remains largely intact, their glycaemic range is set somewhat higher than in unaffected people. Furthermore, the threshold for activating central nervous system (CNS)-driven CRR responses to hypoglycaemia (e.g. adrenaline secretion) is increased in MODY 2, mimicking the situation in type 2 diabetes [22, 23]. Although the MODY2 mutation impairs glucose sensing in both beta cells and neurons, the former cannot explain the upward re-setting of the glycaemic threshold for adrenaline secretion, as this response is entirely dependent on the brain. The elevated glycaemic threshold for adrenaline secretion in affected individuals therefore offers prima facie evidence that impaired neuronal glucose sensing is sufficient to raise the lower boundary of the defended blood glucose level in humans with MODY 2, as observed in type 2 diabetes. Full mapping and characterisation of the neurocircuitry involved in this response are a priority.
CNS control of islet function
One obvious way for the brain to maintain glucose homeostasis is by modulation of pancreatic islet function. Five decades of research incorporating a diverse range of human and animal experimentation has demonstrated conclusively that release of insulin and glucagon in response to circulating and sensory stimuli is influenced by neural input under physiological conditions [27, 28]. The neuronal architecture that allows this brain–islet connection involves neurons situated in hypothalamic and brainstem nuclei, many having glucose-sensing properties and some overlapping with neurocircuits that control food intake and body fat mass. Neurons in these brain areas project to the islet via multi-synaptic relays involving both limbs of the autonomic nervous system. Plasma insulin levels are altered rapidly by various perturbations that disrupt normal activity of these neurocircuits. Thus, the case for the existence of a refined system for brain control of islet hormone release is convincing and well supported, and the relevant neurocircuitry, although incompletely understood, overlaps with that involved in energy homeostasis.
Sympathetic and parasympathetic nervous system outflow
Pancreatic islets are innervated by sympathetic nervous system (SNS) and parasympathetic nervous system (PNS) fibres. Recent 3D imaging of cleared human pancreatic tissue shows dense innervation similar to that reported in rodents [29]. The activity of SNS or PNS fibres influences secretion of both insulin and glucagon in ways that can potently impact blood glucose levels. Whereas nutrient-mediated secretion of insulin during a meal is augmented by an associated increase of PNS outflow to the pancreas [1], the hypoglycaemia-induced increase in SNS outflow to the islet [30] stimulates glucagon secretion and potently inhibits GSIS [31]. Sympathetic fibres supplying the liver are also activated as part of the CRR and, together with increased plasma adrenaline (arising from activation of the adrenal medulla) and glucagon, these responses drive increased hepatic glucose production in an effort to restore normoglycaemia [32]. Suppression of insulin secretion in this setting involves activation of alpha-adrenergic receptors on beta cells resulting from either increased SNS outflow directly to the islet or increased circulating adrenaline levels (or both) [33, 34]. Neural control of islet function also plays a physiological role in cephalic-phase insulin release (insulin secretion in response to feeding cues but before nutrient absorption or increase in blood glucose) [35]. The cephalic phase is mediated by vagal cholinergic signals, is amenable to behavioural entrainment, and contributes to glucose tolerance [36]. While cephalic insulin release has become the defining feature of neural regulation of insulin secretion, neural factors also contribute to the postprandial insulin response. Meal consumption triggers parasympathetic outflow to the islet, and pharmacological blockade of these signals reduces prandial insulin in humans and animal models [1]. How CNS signals interact with endocrine control of the pancreas during meal absorption is incompletely understood but a component of brain regulation of prandial islet function seems likely.
A stronger case can be made for CNS control of glucagon secretion [28]. As noted above, hypoglycaemia-induced activation of the SNS triggers glucagon secretion from islet alpha cells [37], which in turn activates hepatic glucose production. This glucagon response is impaired if islets are denervated (e.g. islet/pancreas transplantation or diabetic neuropathy) [38]. The brain is also implicated in the control of postprandial glucagon secretion [39], and growing evidence suggests that this glucagon response plays a key role in meal-induced insulin secretion [40]. Together, these findings support a model in which meal-induced PNS outflow coordinates the islet response to feeding. Ample evidence also points to a role for the brain in control of glucose handling by the liver. Recent work shows that in both humans and rodents, intact brain KATP channel activity is required for the ability of hyperglycaemia to suppress endogenous glucose production (a key component of ‘glucose effectiveness’, the ability of glucose to promote its own disposal independent of insulin action) [2]. In addition to its influence over islet function, discussed below, control of glucose effectiveness is emerging as an important mechanism whereby the brain controls glucose homeostasis.
Neurocircuits linking islet and brain
Efforts to map brain-to-islet circuitry have identified glucoregulatory neuronal populations within brainstem, midbrain and hypothalamus that are linked synaptically to autonomic neurons supplying the pancreas. Recent work is beginning to functionally characterise this circuitry [41–44]. Many glucose-sensing neurons able to affect islet function have been identified, although whether and how they might be integrated into the broader network of neurocircuits involved in glucose homeostasis is unknown. Recently, Rosario and colleagues [41] showed that the hypothalamic arcuate nucleus, lateral hypothalamic area and VMN contain neurons that are connected via multi-synaptic relays to efferent autonomic fibres supplying pancreatic islets in mice. Moreover, experimentally lowering neuronal glucose sensing within each hypothalamic region produces a distinct effect on glucose homeostasis. Additional work is needed to map the neurocircuitry underlying brain control of islet function.
Targeting the brain to restore normoglycaemia in rodent models of diabetes
Transient glucose lowering in diabetes
Perhaps the most clear-cut evidence of the brain’s ability to control blood glucose levels derives from rodent models of diabetes (type 1 and type 2) in which the brain is targeted to ameliorate hyperglycaemia. A recurring theme from these studies is that in diabetic animals, insulin-independent mechanisms play a key role in brain-mediated glucose lowering. One early study involved intracerebroventricular (i.c.v.) administration of the adipocyte hormone leptin to rats or mice with uncontrolled, insulin-deficient diabetes [45]. This work unexpectedly showed not only that hyperglycaemia can be fully normalised by continuous central leptin administration but also that the dose needed to achieve this effect was low enough to have no measurable effect when given systemically. Most leptin actions occur within minutes to hours after i.c.v. administration, so the fact that it took 4–5 days of continuous leptin administration for correction of hyperglycaemia to occur was also unexpected. The i.c.v. administration of leptin normalised both the excessive hepatic glucose production and the reduced tissue glucose utilisation characteristic of uncontrolled diabetes, despite persistent, severe insulin deficiency. This highlights the brain’s capacity to engage glucose-lowering mechanisms resembling those engaged by systemic insulin, even though they are insulin-independent [46]. Hypothalamic areas such as the VMN, crucial to normal glucose homeostasis, are also implicated in leptin’s glucose-lowering effect [47], suggesting an overlap in neurocircuits governing energy homeostasis and glucose homeostasis.
Further evidence stems from investigation into the potent glucose-lowering actions of fibroblast growth factor (FGF) family members. This work initially focused on two hormonal members of this family, FGF21 and FGF19, secreted primarily by the liver and gastrointestinal tract, respectively. When administered systemically at pharmacological doses, both peptides transiently normalised blood glucose in rodent models of type 2 diabetes [48]. Although originally proposed to involve a peripheral mechanism, subsequent work established that at pharmacological doses, both FGF21 and FGF19 promote glucose lowering largely if not exclusively via actions in the brain [49, 50]. Once again, insulin-independent mechanisms appear to be involved [50].
Sustained blood glucose lowering
Extending this work are studies focused on glucose lowering by central administration of the tissue growth factor FGF1. Hyperglycaemia is normalised in rodent models of type 2 diabetes following i.c.v. injection of FGF1 but, unlike the effects of FGF19 or FGF21, the glucose-lowering action of a single i.c.v. injection of FGF1 lasts for weeks or even months [51]. This FGF1 action is mediated in the mediobasal hypothalamus (MBH) [52], largely through insulin-independent mechanisms, although a protective effect on both basal insulin secretion and beta cell mass is also observed [53, 54].
While mechanisms underlying the sustained glucose-lowering action of FGF1 in the MBH await additional study, our recent work points to a role for glia–neuron interactions [55] and associations with FGF1-induced changes in the extracellular matrix [56]. Whatever the mechanism, the data collectively suggest that in rodent models of type 2 diabetes, hyperglycaemia arises from pathological processes that can be corrected (or overridden) through the hypothalamic action of FGF1. Efforts to identify brain mechanisms driving hyperglycaemia in type 2 diabetes, and determine how they are ameliorated by FGF1, are a priority for future work.
Reconciling evidence supporting islet-based models of diabetes pathogenesis
Pancreatic islets are sufficient to control blood glucose without input from the brain
Normoglycaemia can be achieved by islet or pancreas transplantation in diabetic humans and animals, implying that an adequate supply of functional islets is sufficient for normal glucose homeostasis without input from the brain and, therefore, that brain input is dispensable for normal glucose homeostasis. While we concur with the former assertion, the latter is misguided because under physiological conditions, islets are under neural control. Consequently, removing this control creates a non-physiological state that is informative as to what islets can do in the absence of brain control but does little to inform our understanding of how physiological glucose homeostasis normally works.
Accounting for beta cell dysfunction in type 2 diabetes
There is little doubt that abnormal insulin secretion is fundamental to the pathogenesis of type 2 diabetes [57]. In the transition from normal to abnormal glucose tolerance, for example, beta cell dysfunction is detectable even before the onset of frank hyperglycaemia [58]. Although the nature of the underlying beta cell lesion remains uncertain, defective proinsulin processing, amyloid formation and increased cell death are each reported [59]. A causal role for cell-autonomous beta cell dysfunction in the pathogenesis of type 2 diabetes is further strengthened by evidence that beta cells express a majority of gene variants identified as being associated with type 2 diabetes [60] (although many of these variants are also expressed in the brain). That beta cell dysfunction contributes to type 2 diabetes pathogenesis is therefore not in question. What remains to be established is the extent to which this dysfunction originates within the beta cell and/or constitutes the key initiating event in the disease process.
Does beta cell dysfunction in type 2 diabetes involve a primary, cell-autonomous lesion or does it instead reflect an interaction between genetic susceptibility and metabolic consequences of the disease (e.g. ‘glucotoxicity’ and ‘lipotoxicity’ [61])? We favour the latter, and suggest that in genetically susceptible individuals, any pathological process that raises the defended level of blood glucose out of the normal range (including diminished brain sensing of the circulating glucose level) has the potential to trigger the vicious cycle of escalating beta cell dysfunction and metabolic impairment characteristic of type 2 diabetes. Input from the brain may exacerbate beta cell dysfunction in this setting since most endocrine cell types become dysfunctional if they are subjected to either prolonged inhibitory input (e.g. a sustained increase of SNS tone to the islet) or withdrawal of trophic support (e.g. reduced PNS tone). A CNS mechanism that drives hyperglycaemia in part by inhibiting GSIS (as occurs when glucoregulatory VMN neurons are activated), therefore, can potentially set this pathological cascade in motion.
Reconsidering the link between obesity and type 2 diabetes
The obesity to type 2 diabetes transition
The conceptual basis for the model advanced herein has its roots in our current understanding of obesity, a closely related metabolic disorder. Although many factors can contribute to excessive weight gain, body fat mass continues to be biologically defended even as it increases out of the normal range in obese individuals (Fig. 2). Stated differently, obesity is a disorder characterised by a progressive increase in the defended level of the body’s primary stored fuel (triacylglycerol), so weight lost through energy restriction tends to be regained, regardless of whether one starts out being lean or obese [62]. Elevated body fat mass in obesity may be explained by the brain becoming resistant to input from negative feedback signals (e.g. leptin) that inform the brain regarding the amount of body fuel stored as fat [62, 63]. This resistance causes the brain to perceive the amount of body fat to be lower than it actually is and hence activates responses that raise body fat mass to a level sufficient to overcome the resistance. The net effect is a new steady state in which body fat mass and circulating negative feedback signals are both sufficiently increased to overcome the brain’s resistance to this input. While this proposed mechanism is not intended to capture the complexities of obesity pathogenesis in their entirety, it is highly likely that it is involved in the defence of elevated body fat mass characteristic of most obese individuals [62].
Fig. 2.
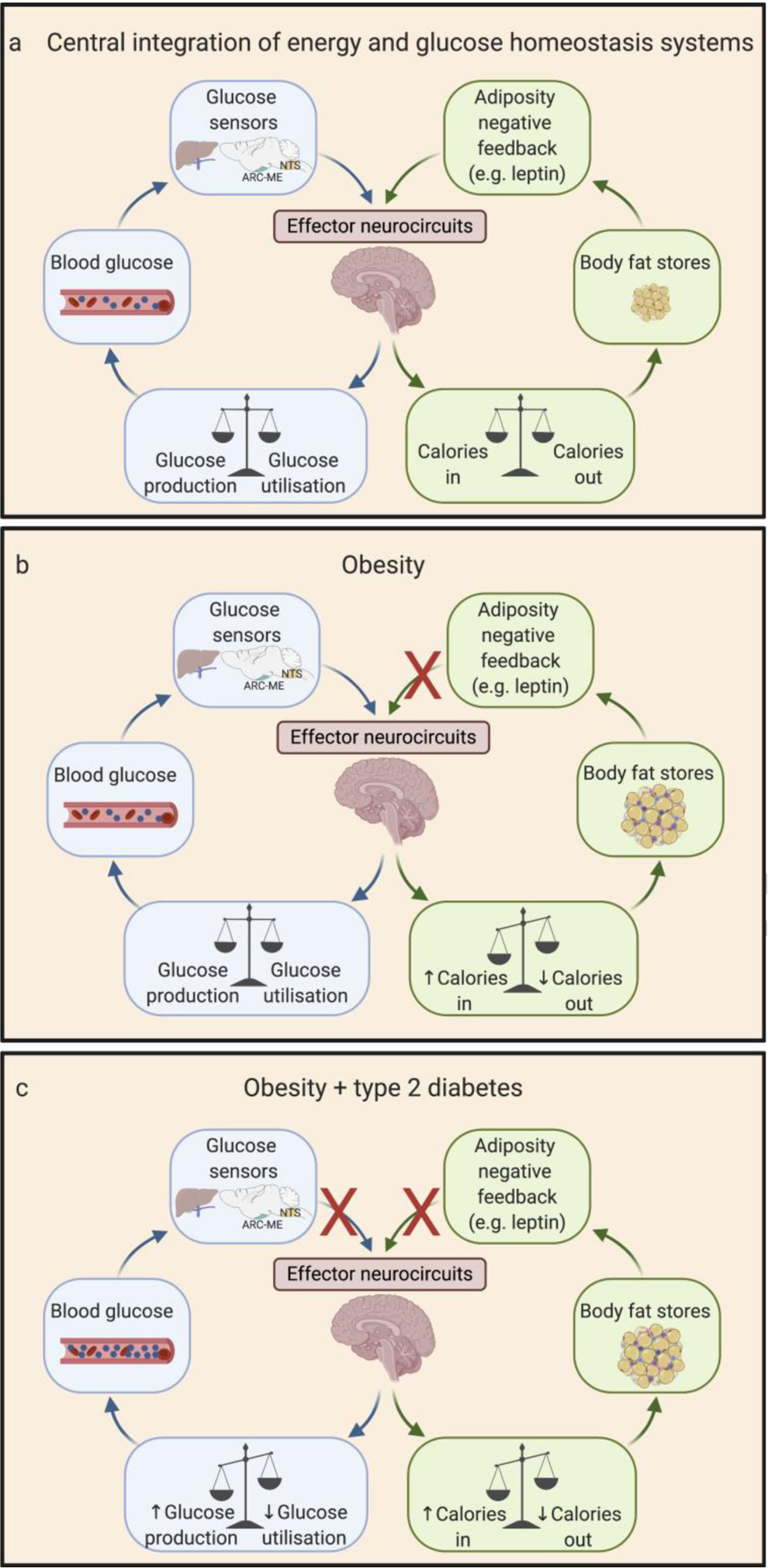
Brain-based model linking obesity to the pathogenesis of type 2 diabetes. Normal energy homeostasis (a) entails brain sensing of circulating adiposity negative feedback signals (such as the hormone leptin). In response to this input, the brain facilitates the matching of energy intake to energy expenditure over time so as to promote stability in the amount of fuel stored as fat. In obese individuals (b), reduced brain sensing of adipose-related negative feedback signals favours the defence of an elevated level of body fat mass. Progression of obesity to type 2 diabetes (c) is proposed to involve an expansion of the underlying brain defect to include impaired sensing of the blood glucose level. This combination of defects causes the defended levels of both blood glucose and adiposity to rise out of the normal range, thus contributing to the close association between obesity and type 2 diabetes. ARC, arcuate nucleus; ME, median eminence; NTS, nucleus tractus solitarius.
By analogy, we hypothesise that type 2 diabetes involves a progressive increase in the defended level of the body’s dominant circulating fuel (glucose). This in turn explains the transient nature of glucose lowering induced by insulin and most other glucose-lowering drugs; once the effect has worn off, the blood glucose level returns to its original defended value, regardless of whether an individual has type 2 diabetes. Furthermore, we postulate that this pathogenic sequence begins with or is exacerbated by an impaired ability of the brain to accurately sense the circulating glucose level (i.e. a form of ‘brain glucose resistance’) [64] (Fig. 1). Although there is currently no direct evidence of a causal role for defective brain sensing of circulating glucose levels in type 2 diabetes pathogenesis, impaired brain responses to glucose are well-documented in humans with type 2 diabetes [8–10]. Additional investigation of this hypothesis is an important priority.
Based on this model, we propose that just as the defence of increased body fat stores in obese individuals can be viewed as a compensatory response [62], the defence of elevated blood glucose levels is the predicted consequence of brain glucose resistance (Fig. 2). Specifically, we propose that in type 2 diabetes, impaired brain sensing of the blood glucose level activates MBH glucoregulatory neurons, which in turn raises the blood glucose ‘set point’ in an effort to compensate for the underlying defect, and that suppression of GSIS is an integral component of this response.
Additional support for the conceptual overlap between the pathogenesis of obesity and type 2 diabetes stems from the close association between these two disorders in humans and the extensive overlap that exists between brain systems involved in energy and glucose homeostasis (including the VMN and adjacent hypothalamic areas) [16, 61]. Thus, we propose that the pathogenesis of obesity and type 2 diabetes involves an overlapping defect in two closely linked brain control systems: energy homeostasis and glucose homeostasis (Fig. 2). Extending this reasoning, just as the defence of an elevated level of stored fuel (triacylglycerol) can be viewed as a consequence of blunted responsiveness to adiposity negative feedback signals in obesity, we propose that in those obese individuals who go on to develop type 2 diabetes, the underlying defect expands to include an impaired ability of the brain to sense the level of circulating fuel (glucose), with the defended level of blood glucose increasing in compensation. Accordingly, beta cell dysfunction leading to type 2 diabetes is proposed to arise at least in part as a secondary consequence of this underlying brain defect, since centrally mediated suppression of GSIS is required for the brain to raise the defended level of blood glucose sufficiently to overcome impaired sensing of the blood glucose level by the brain. In genetically susceptible individuals, a vicious cycle is then created whereby hyperglycaemia and associated metabolic decompensation cause further beta cell dysfunction. Studies to test this hypothesis are a research priority.
A noteworthy and seemingly paradoxical finding is that hyperinsulinaemia is reported to be predictive of the future development of type 2 diabetes [57, 65, 66]. Neither the mechanisms underlying this insulin hypersecretion nor its link to type 2 diabetes pathogenesis are understood. Nevertheless, it occurs in normoglycaemic individuals and since it is not associated with insulin resistance [65], affected individuals may also have reduced glucose effectiveness (otherwise, one expects a lower blood glucose level). Indeed, reduced glucose effectiveness is itself predictive of future development of type 2 diabetes in humans [67]. These considerations raise two questions worthy of future study: (1) is hyperinsulinaemia a biomarker of reduced glucose effectiveness in individuals at risk for type 2 diabetes; and (2) given its unambiguous ability to regulate glucose effectiveness, does the brain play a role in the mechanism underlying this reduced glucose effectiveness?
Conclusions
Herein, we introduce a model in which normal glucose homeostasis hinges on intact brain sensing of the circulating glucose level (Fig. 1) and propose that dysfunction of this sensing process can be acquired in association with obesity and plays a central role in type 2 diabetes pathogenesis (Fig. 2). This model is supported in part by evidence that the brain has the capacity to restore the defended level of blood glucose to normal in animal models of type 2 diabetes [51, 52], implying that a defect fundamental to the pathogenesis of hyperglycaemia must reside in the brain and is targeted by peptides such as FGF1. Despite substantial progress, a mechanistic understanding of the brain’s role in both glucose homeostasis and type 2 diabetes pathogenesis remains in its infancy and opportunities to refine this understanding exist. Key questions include: how does the brain sense the circulating glucose level; to what extent does this process involve neurons vs non-neuronal cell types (e.g. astrocytes, oligodendrocytes, tanycytes); what mechanisms might underly the pathogenesis of brain ‘glucose resistance’ in type 2 diabetes; is this defect linked to mechanisms underlying the defence of elevated body fat stores in obese individuals; and how does the brain regulate glucose effectiveness? Answers to these questions may one day inform type 2 diabetes treatment strategies designed to restore the defended level of blood glucose to normal, rather than to transiently lower blood glucose below its defended level. The latter approach places limits on the efficacy of most current therapies and can also place an unwelcome burden upon beta cells that are ill-prepared to handle it.
Supplementary Material
Acknowledgements
We are grateful to D. Porte Jr (Medicine, University of California San Diego, CA, USA), J. M. Scarlett (Medicine, University of Washington, WA, USA), C. L. Faber (Medicine, University of Washington, WA, USA), G. J. Taborsky (Medicine, University of Washington, WA, USA), G. J. Morton (Medicine, University of Washington, WA, USA) and S. C. Woods (MMPC, University of Cincinnati, OH, USA) for their constructive feedback.
Funding
Work in the authors’ laboratories is supported by National Institute of Diabetes and Digestive and Kidney Diseases grants DK083042 and DK089056 (to MWS) and DK101991 (to DAD). KA is supported by National Institute of Diabetes and Digestive and Kidney Diseases F32 Training Grant DK122662 and Diabetes Research Center P&F Grant DK017047.
Authors’ relationships and activities
MWS has received research support from Novo Nordisk A/C. All other authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Abbreviations
- CCK-B
Cholecystokinin B
- CCR
Counter-regulatory response
- CNS
Central nervous system
- FGF
Fibroblast growth factor
- GSIS
Glucose-induced insulin secretion
- i.c.v.
Intracerebroventricular
- MBH
Mediobasal hypothalamus
- PNS
Parasympathetic nervous system
- SNS
Sympathetic nervous system
- VMN
Ventromedial nucleus
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- [1].DʼAlessio DA, Kieffer TJ, Taborsky GJ Jr, Havel PJ (2001) Activation of the parasympathetic nervous system is necessary for normal meal-induced insulin secretion in rhesus macaques. J Clin Endocrinol Metab 86(3): 1253–1259. 10.1210/jcem.86.3.7367 [DOI] [PubMed] [Google Scholar]
- [2].Carey M, Lontchi-Yimagou E, Mitchell W, et al. (2020) Central KATP channels modulate glucose effectiveness in humans and rodents. Diabetes 69(6): 1140–1148. 10.2337/db19-1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bernard C (1849) Chiens rendus diabetiques. C R SocBio 1: 60 [Google Scholar]
- [4].Oh Y, Lai JS, Mills HJ, et al. (2019) A glucose-sensing neuron pair regulates insulin and glucagon in Drosophila. Nature 574(7779): 559–564. 10.1038/s41586-019-1675-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schwartz MW, Seeley RJ, Tschöp MH, et al. (2013) Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 503(7474): 59–66. 10.1038/nature12709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pardridge WM, Triguero D, Farrell CR (1990) Downregulation of blood-brain barrier glucose transporter in experimental diabetes. Diabetes 39(9): 1040–1044. 10.2337/diab.39.9.1040 [DOI] [PubMed] [Google Scholar]
- [7].Cornford EM, Hyman S, Cornford ME, Clare-Salzler M (1995) Down-regulation of blood-brain glucose transport in the hyperglycemic nonobese diabetic mouse. Neurochem Res 20(7): 869–873. 10.1007/bf00969700 [DOI] [PubMed] [Google Scholar]
- [8].Hwang JJ, Jiang L, Hamza M, et al. (2017) Blunted rise in brain glucose levels during hyperglycemia in adults with obesity and T2DM. JCI insight 2(20): e95913 10.1172/jci.insight.95913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Eskian M, Alavi A, Khorasanizadeh M, et al. (2019) Effect of blood glucose level on standardized uptake value (SUV) in 18F-FDG PET-scan: a systematic review and meta-analysis of 20,807 individual SUV measurements. Eur J Nucl Med Mol Imaging 46(1): 224–237. 10.1007/s00259-018-4194-x [DOI] [PubMed] [Google Scholar]
- [10].Sprinz C, Altmayer S, Zanon M, et al. (2018) Effects of blood glucose level on 18F-FDG uptake for PET/CT in normal organs: A systematic review. PLoS One 13(2): e0193140 10.1371/journal.pone.0193140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li W, Risacher SL, Huang E, Saykin AJ (2016) Type 2 diabetes mellitus is associated with brain atrophy and hypometabolism in the ADNI cohort. Neurology 87(6): 595–600. 10.1212/wnl.0000000000002950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Thorens B (2015) GLUT2, glucose sensing and glucose homeostasis. Diabetologia 58(2): 221–232. 10.1007/s00125-014-3451-1 [DOI] [PubMed] [Google Scholar]
- [13].Ma Y, Ratnasabapathy R, De Backer I, et al. (2020) Glucose in the hypothalamic paraventricular nucleus regulates GLP-1 release. JCI insight 5(8): e132760 10.1172/jci.insight.132760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nishio T, Toyoda Y, Hiramatsu M, Chiba T, Miwa I (2006) Decline in glucokinase activity in the arcuate nucleus of streptozotocin-induced diabetic rats. Biological and Pharmaceutical Bulletin 29(2): 216–219. 10.1248/bpb.29.216 [DOI] [PubMed] [Google Scholar]
- [15].Ma Y, Ratnasabapathy R, Izzi-Engbeaya C, et al. (2018) Hypothalamic arcuate nucleus glucokinase regulates insulin secretion and glucose homeostasis. Diabetes, obesity & metabolism 20(9): 2246–2254. 10.1111/dom.13359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kang L, Dunn-Meynell AA, Routh VH, et al. (2006) Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes 55(2): 412–420. 10.2337/diabetes.55.02.06.db05-1229 [DOI] [PubMed] [Google Scholar]
- [17].Cryer PE (1993) Glucose counterregulation: prevention and correction of hypoglycemia in humans. Am J Physiol 264(2 Pt 1): E149–E155. 10.1152/ajpendo.1993.264.2.E149 [DOI] [PubMed] [Google Scholar]
- [18].Asplin CM, Raghu PK, Koerker DJ, Palmer JP (1985) Glucose counterregulation during recovery from neuroglucopenia: which mechanism is important? Metabolism 34(1): 15–18. 10.1016/0026-0495(85)90053-8 [DOI] [PubMed] [Google Scholar]
- [19].Meek TH, Nelson JT, Matsen ME, et al. (2016) Functional identification of a neurocircuit regulating blood glucose. Proc Natl Acad Sci U S A 113(14): E2073–E2082. 10.1073/pnas.1521160113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Faber CL, Matsen ME, Velasco KR, et al. (2018) Distinct neuronal projections from the hypothalamic ventromedial nucleus mediate glycemic and behavioral effects. Diabetes 67(12): 2518–2529. 10.2337/db18-0380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Flak JN, Goforth PB, DellʼOrco J, et al. (2020) Ventromedial hypothalamic nucleus neuronal subset regulates blood glucose independently of insulin. J Clin Invest 130(6): 2943–2952. 10.1172/jci134135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chakera AJ, Hurst PS, Spyer G, et al. (2018) Molecular reductions in glucokinase activity increase counter-regulatory responses to hypoglycemia in mice and humans with diabetes. Mol Metab 17: 17–27. 10.1016/j.molmet.2018.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Spyer G, Hattersley AT, MacDonald IA, Amiel S, MacLeod KM (2000) Hypoglycaemic counter-regulation at normal blood glucose concentrations in patients with well controlled type-2 diabetes. Lancet 356(9246): 1970–1974. 10.1016/s0140-6736(00)03322-5 [DOI] [PubMed] [Google Scholar]
- [24].Chowdhury GMI, Wang P, Ciardi A, et al. (2017) Impaired glutamatergic neurotransmission in the ventromedial hypothalamus may contribute to defective counterregulation in recurrently hypoglycemic rats. Diabetes 66(7): 1979–1989. 10.2337/db16-1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tong Q, Ye C, McCrimmon RJ, et al. (2007) Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell metabolism 5(5): 383–393. 10.1016/j.cmet.2007.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Roncero I, Alvarez E, Chowen JA, et al. (2004) Expression of glucose transporter isoform GLUT-2 and glucokinase genes in human brain. J Neurochem 88(5): 1203–1210. 10.1046/j.1471-4159.2003.02269.x [DOI] [PubMed] [Google Scholar]
- [27].Woods SC, Porte D Jr (1974) Neural control of the endocrine pancreas. Physiol Rev 54(3): 596–619. 10.1152/physrev.1974.54.3.596 [DOI] [PubMed] [Google Scholar]
- [28].Osundiji MA, Evans ML (2013) Brain control of insulin and glucagon secretion. Endocrinol Metab Clin North Am 42(1): 1–14. 10.1016/j.ecl.2012.11.006 [DOI] [PubMed] [Google Scholar]
- [29].Tang SC, Baeyens L, Shen CN, et al. (2018) Human pancreatic neuro-insular network in health and fatty infiltration. Diabetologia 61(1): 168–181. 10.1007/s00125-017-4409-x [DOI] [PubMed] [Google Scholar]
- [30].Havel PJ, Veith RC, Dunning BE, Taborsky GJ Jr. (1988) Pancreatic noradrenergic nerves are activated by neuroglucopenia but not by hypotension or hypoxia in the dog. Evidence for stress-specific and regionally selective activation of the sympathetic nervous system. The Journal of Clinical Investigation 82(5): 1538–1545. 10.1172/JCI113763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Faber CF, Deem JD, Carlos CA, Taborsky GJ, Morton GJ (2020) CNS Control of the Endocrine Pancreas Diabetologia 10.1007/s00125-020-05204-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Perseghin G, Regalia E, Battezzati A, et al. (1997) Regulation of glucose homeostasis in humans with denervated livers. J Clin Invest 100(4): 931–941. 10.1172/jci119609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Halter JB, Beard JC, Porte D Jr. (1984) Islet function and stress hyperglycemia: plasma glucose and epinephrine interaction. Am J Physiol 247(1 Pt 1): E47–E52. 10.1152/ajpendo.1984.247.1.E47 [DOI] [PubMed] [Google Scholar]
- [34].Lerner RL, Porte D Jr (1971) Epinephrine: selective inhibition of the acute insulin response to glucose. J Clin Invest 50(11): 2453–2457. 10.1172/jci106744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Teff KL (2011) How neural mediation of anticipatory and compensatory insulin release helps us tolerate food. Physiol Behav 103(1): 44–50. 10.1016/j.physbeh.2011.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Thorens B (2014) Neural regulation of pancreatic islet cell mass and function. Diabetes Obes Metab 16(Suppl 1): 87–95. 10.1111/dom.12346 [DOI] [PubMed] [Google Scholar]
- [37].Havel PJ, Mundinger TO, Taborsky GJ (1996) Pancreatic sympathetic nerves contribute to increased glucagon secretion during severe hypoglycemia in dogs. The American journal of physiology 270(1 Pt 1): E20–E26. 10.1152/ajpendo.1996.270.1.e20 [DOI] [PubMed] [Google Scholar]
- [38].Taborsky GJ Jr., Mundinger TO (2012) Minireview: the role of the autonomic nervous system in mediating the glucagon response to hypoglycemia. Endocrinology 153(3): 1055–1062. 10.1210/en.2011-2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jessen L, Smith EP, Ulrich-Lai Y, et al. (2017) Central nervous system GLP-1 receptors regulate islet hormone secretion and glucose homeostasis in male rats. Endocrinology 158(7): 2124–2133. 10.1210/en.2016-1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Capozzi ME, Svendsen B, Encisco SE, et al. (2019) β cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight 4(5): e126742 10.1172/jci.insight.126742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rosario W, Singh I, Wautlet A, et al. (2016) The brain-to-pancreatic islet neuronal map reveals differential glucose regulation from distinct hypothalamic regions. Diabetes 65(9): 2711–2723. 10.2337/db15-0629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jansen AS, Hoffman JL, Loewy AD (1997) CNS sites involved in sympathetic and parasympathetic control of the pancreas: a viral tracing study. Brain Res 766(1–2): 29–38. 10.1016/s0006-8993(97)00532-5 [DOI] [PubMed] [Google Scholar]
- [43].Buijs RM, Chun SJ, Niijima A, Romijn HJ, Nagai K (2001) Parasympathetic and sympathetic control of the pancreas: a role for the suprachiasmatic nucleus and other hypothalamic centers that are involved in the regulation of food intake. J Comp Neurol 431(4): 405–423. [DOI] [PubMed] [Google Scholar]
- [44].Tokunaga K, Fukushima M, Kemnitz JW, Bray GA (1986) Effect of vagotomy on serum insulin in rats with paraventricular or ventromedial hypothalamic lesions. Endocrinology 119(4): 1708–1711. 10.1210/endo-119-4-1708 [DOI] [PubMed] [Google Scholar]
- [45].German JP, Thaler JP, Wisse BE, et al. (2011) Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology 152(2): 394–404. 10.1210/en.2010-0890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Meek TH, Wisse BE, Thaler JP, et al. (2013) BDNF action in the brain attenuates diabetic hyperglycemia via insulin-independent inhibition of hepatic glucose production. Diabetes 62(5): 1512–1518. 10.2337/db12-0837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Meek TH, Matsen ME, Dorfman MD, et al. (2013) Leptin action in the ventromedial hypothalamic nucleus is sufficient, but not necessary, to normalize diabetic hyperglycemia. Endocrinology 154(9): 3067–3076. 10.1210/en.2013-1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Owen BM, Mangelsdorf DJ, Kliewer SA (2015) Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab 26(1): 22–29. 10.1016/j.tem.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Owen BM, Ding X, Morgan DA, et al. (2014) FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab 20(4): 670–677. 10.1016/j.cmet.2014.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Morton GJ, Matsen ME, Bracy DP, et al. (2013) FGF19 action in the brain induces insulin-independent glucose lowering. J Clin Invest 123(11): 4799–4808. 10.1172/jci70710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Scarlett JM, Rojas JM, Matsen ME, et al. (2016) Central injection of fibroblast growth factor 1 induces sustained remission of diabetic hyperglycemia in rodents. Nat Med 22(7): 800–806. 10.1038/nm.4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brown JM, Scarlett JM, Matsen ME, et al. (2019) The hypothalamic arcuate nucleus-median eminence is a target for sustained diabetes remission induced by fibroblast growth factor 1. Diabetes 68(5): 1054–1061. 10.2337/db19-0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Scarlett JM, Muta K, Brown JM, et al. (2019) Peripheral mechanisms mediating the sustained antidiabetic action of FGF1 in the brain. Diabetes 68(3): 654–664. 10.2337/db18-0498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tennant KG, Lindsley SR, Kirigiti MA, True C, Kievit P (2019) Central and peripheral administration of fibroblast growth factor 1 improves pancreatic islet insulin secretion in diabetic mouse models. Diabetes 68(7): 1462–1472. 10.2337/db18-1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bentsen MA, Rausch DM, Mirzadeh Z, et al. (2020) Transcriptomic analysis links diverse hypothalamic cell types to fibroblast growth factor 1-induced sustained diabetes remission. Nature Communications 11(1): 4458 10.1038/s41467-020-17720-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Alonge KM, Mirzadeh Z, Scarlett JM, et al. (2020) Hypothalamic perineuronal net assembly is required for sustained diabetes remission induced by fibroblast growth factor 1. Nature Metabolism. doi: 10.1038/s42255-020-00275-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tabák AG, Jokela M, Akbaraly TN, Brunner EJ, Kivimäki M, Witte DR (2009) Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet 373(9682): 2215–2221. 10.1016/s0140-6736(09)60619-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cnop M, Vidal J, Hull RL, et al. (2007) Progressive loss of β-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care 30(3): 677–682. 10.2337/dc06-1834 [DOI] [PubMed] [Google Scholar]
- [59].Kahn SE, Cooper ME, Del Prato S (2014) Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383(9922): 1068–1083. 10.1016/s0140-6736(13)62154-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Krentz NAJ, Gloyn AL (2020) Insights into pancreatic islet cell dysfunction from type 2 diabetes mellitus genetics. Nat Rev Endocrinol 16(4): 202–212. 10.1038/s41574-020-0325-0 [DOI] [PubMed] [Google Scholar]
- [61].Poitout V, Robertson RP (2008) Glucolipotoxicity: fuel excess and β-cell dysfunction. Endocr Rev 29(3): 351–366. 10.1210/er.2007-0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Schwartz MW, Seeley RJ, Zeltser LM, et al. (2017) Obesity Pathogenesis: an endocrine society scientific statement. Endocr Rev 38(4): 267–296. 10.1210/er.2017-00111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Myers MG Jr, Heymsfield SB, Haft C, et al. (2012) Challenges and opportunities of defining clinical leptin resistance. Cell Metab 15(2): 150–156. 10.1016/j.cmet.2012.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Brown JM, Scarlett JM, Schwartz MW (2019) Rethinking the role of the brain in glucose homeostasis and diabetes pathogenesis. J Clin Invest 129(8): 3035–3037. 10.1172/jci130904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA (2005) β-Cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab 90(1): 493–500. 10.1210/jc.2004-1133 [DOI] [PubMed] [Google Scholar]
- [66].Ferrannini E, Natali A, Bell P, Cavallo-Perin P, Lalic N, Mingrone G (1997) Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). The Journal of clinical investigation 100(5): 1166–1173. 10.1172/JCI119628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Martin BC, Warram JH, Krolewski AS, Bergman RN, Soeldner JS, Kahn CR (1992) Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet 340(8825): 925–929. 10.1016/0140-6736(92)92814-v [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.