

Abstract
To investigate the role of placental growth factor/vascular endothelial growth factor (PlGF-VEGF) heterodimers are involved in the blood-retinal barrier (BRB) breakdown and the associated mechanism, human retinal endothelial cells (HRECs) were treated with recombinant human (rh)PlGF-VEGF heterodimers and rhPlGF and studied in normal and high-glucose conditions. HREC barrier function was evaluated by the measurement of trans-endothelial electrical resistance (TEER). Adeno-Associated Virus Type 5 (AAV5) vectors overexpressed PlGF in the retina by intravitreal injection into the C57BL6 mouse eye. AAV5-GFP vector and naïve animals were used as controls. Immunofluorescence (IF) and western blots examined the protein expression of PlGF-VEGF heterodimers, VEGF, PlGF, NFκB, p-IκBα, ZO-1, and VE-cadherin in HREC and mouse retina. PlGF-VEGF heterodimers were detected predominantly in the HREC cell nuclei based on IF and cytoplasmic and nuclear fractionation experiments. High glucose treatment increased PlGF-VEGF nuclear abundance. Dot immunoblotting demonstrated a strong affinity of the 5D11D4 antibody to PlGF-VEGF heterodimers. rhPlGF-VEGF disrupted the barrier function of HREC, which was prevented by the neutralization of PlGF-VEGF by the 5D11D4 antibody. Stimulation of HRECs with rhPlGF also led to an increase in the nuclear signals for PlGF-VEGF, p-IκBα, and colocalization of NFκB p65 and PlGF-VEGF in the nuclei. The selective IKK2 inhibitor IMD0354 disrupted the nuclear colocalization. Treatment with IMD0354 restored the barrier function of HREC, as indicated by the ZO-1 and VE-cadherin expression. In the mouse retinas, PlGF overexpression by AAV5 vector reduced ZO-1 expression and increased abundance of pIκBα. PIGF/VEGF heterodimers mediate BRB breakdown potentially through the canonical NFκB activation.
Keywords: PlGF-VEGF, PlGF, NFκB, IKK2, IMD0354, Blood-retinal barrier
Introduction
Placental growth factor (PlGF, also known as PGF), a member of the vascular endothelial growth factor (VEGF) family and a homolog of VEGF-A, is a multifunctional peptide associated with angiogenesis and implicated in pathological angiogenesis in the eye and non-ocular conditions 1,2 In the pathophysiology of diabetic retinopathy (DR), one of the leading causes of vision loss is characterized by the breakdown of the blood-retinal barrier (BRB), and PlGF was shown to be expressed in the vitreous of these patients 3-6 In proliferative DR, PIGF colocalizes with endothelial cells (ECs) 7 and has been shown to mediate proangiogenic properties in this cell type by stimulating endothelial cell migration and recruitment of pericytes and other inflammatory cells, such as microglia and macrophages 8-10 PlGF is also a critical promoter of vascular leakage under ischemic conditions 11-13, and its overexpression can lead to an early onset of DR 5,6 Previous reports have indicated heterodimerization between PIGF and VEGF can induce VEGFR1 homodimerization or VEGFR1/VEGFR2 heterodimerization to modulate angiogenesis 14-16, but cannot directly bind with VEGFR2. Synergistic interactions of PlGF and VEGF have been observed in ischemia 8,17. PlGF has been shown to promote tumor angiogenesis and metastasis, positioning it as a potential target for anticancer-directed therapy 18. Although VEGF and PlGF are upregulated and critical for angiogenesis in part through heterodimerization, little is known how these proteins mediate their function in DR's pathophysiology.
In our previous work, we have demonstrated that the ablation of PlGF in mice suppresses diabetes-induced degradation of the junctional proteins ZO-1 and VE-cadherin, whose degradation is an early sign of the pathophysiology of diabetic retinopathies 1. Furthermore, a deficiency of PlGF in Akita diabetic mice results in the inhibition of hypoxia-inducible factor (HIF)1α–VEGF signaling, as indicated by the downregulation of HIF1α, VEGF, and VEGFR1–3 and by the reduction of phosphor-(p)-VEGFR1, p-VEGFR2, and p–endothelial nitric oxide synthase. Whole proteome analysis revealed that increased neuron survival and antioxidant defense, along with reduced insulin resistance, may contribute to the retinal protection against diabetes-induced retinal damage in the diabetic Akita mice with PlGF ablation 19.
Recently, we demonstrated that not only does high glucose increase the level of PlGF-VEGF heterodimers, it also increases their binding capacity to ECs with the decreased levels of secreted PlGF-VEGF heterodimers and increases in cell lysates. In addition, recombinant human (rh)PlGF-VEGF heterodimers have been shown to decrease the barrier function of human retinal microvascular endothelial cells (HREC) in vitro by disrupting ZO-1 and VE-cadherin protein expression and diminishing their cell-to-cell distribution. 20However, the exact mechanism mediating these effects is not entirely understood. Here, we demonstrate a nuclear translocation of PlGF-VEGF heterodimers and its potential role in modulating the pathophysiology of early DR.
Materials and methods
Primary human retinal endothelial cells (HREC) culture
Primary HRECs were procured from Cell Systems (ACBR1 181, Kirkland, WA, USA). HRECs were seeded on fibronectin-coated (10 μg/ml, overnight, 33016015, Gibco) plastic culture vessels and grown with the EBM2-MV medium (cc-4176, Lonza, Walkersville, MD, USA), supplemented with 10% fetal bovine serum (FBS), 1% of penicillin/streptomycin (P/S), and EGM MV SingleQuots growth supplement kit (cc-4147, Lonza, Walkersville, MD, USA).
Cell treatments
Primary HRECs were cultured, as described above. After reaching confluence, culture media were replaced with a fresh media containing either of the following treatments: Mouse Isotype Control (Clone B-Z1; Cell Sciences); 5D11D4 antibody 25 μg/ml; D-glucose (25 mM), recombinant human (rh) PlGF protein (264-PGB-010/CF, R&D Systems, Minneapolis, MN, USA), IKK2 activation inhibitor IMD-0354 (I3159-5MG, solid, Sigma-Aldrich, St Louis, MO, USA) 10 ng/ml Dimethyl Sulfoxide (DMSO). HRECs were evaluated 48 h after the start of the incubation.
Dot immunoblotting assay
Previously we reported that the 5D11D4 antibody interacts with human and bovine PlGF. 20 However, it was unknown if 5D11D4 has an affinity to PlGF as part of PlGF-VEGF heterodimers. Dot immunoblotting assay was performed as described previously. 20 Briefly PlGF-VEGF heterodimers protein (200 ng; Cat# 297-VP/CF, R&D Systems), rhPlGF protein (200 ng; 264-PGB-010/CF, R&D Systems); VEGF165 (200 ng, 293-VE-010/CF, R&D Systems) and 500 ng of Bovine serum albumin Pierce™ Bovine Serum Albumin Standard (23209; Thermo Fisher Scientific, Waltham, MA, USA) was deposited in the volume of 2 μl to the dry nitrocellulose membrane (Bio-Rad) and immobilized by 15 minutes incubation at the RT. The total protein deposition was visualized by incubation with Pierce™ Reversible Protein Stain Kit (24580; Thermo Fisher Scientific, Waltham, MA, USA). The membranes were then washed with PBS Then, after phosphate-buffered saline (1x, Fisher Bioreagents, Fair Lawn, NJ, US, 187672, BP399-1L). Blocked with 2.5% bovine serum albumin (BSA; A7096, Sigma-Aldrich, St Louis, MO, USA) in PBS at room temperature (RT) for 1 h and then incubated overnight at 4°C with mouse 5D11D4 antibody 0.5 μg/ml. The membrane was washed 3 times for 5 minutes with PBS-Tween 20 (0.02%, Amresco Tween-20, Lot:1392C184, Cat:0777-1L) (PBS-T) and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody Goat anti-Mouse IgG 172-1011 (1:1000; Bio-Rad) for 1 h at room temperature. Following additional washing, signals were developed with enhanced chemiluminescence (ECL) using a Super Signal West Pico kit (Thermo Fisher Scientific, Waltham, MA, USA) and detected with ImageQuant LAS500 (GE HealthCare Life Science, Pittsburgh, PA, USA).
Cytoplasmic and nuclear protein isolation
Nuclear proteins were isolated using NE-PER™ nuclear and cytoplasmic extraction reagents according to the kit's instructions (Thermo Fisher Scientific, Waltham, MA, USA). Briefly, cell pellets of ~1x106 were treated with ice-cold cytoplasmic extraction reagent (CER) (100 ul) and supplemented with 1:100 Halt™ Protease Inhibitor Cocktail (78425). Following pellet resuspension, 10 min of incubation CER II 5.5 ul was added. After another 1 min of incubation, the lysates were centrifuged at 17,000 g for 5 min. The resulting soluble cytoplasmic fraction was transferred to precooled sample tubes and stored at −80 C. An insoluble nuclear pellet was washed with PBS and resuspended in ice-cold nuclear extraction reagent (NER, 50 ul). Nuclear lysis was performed over 40 min of incubation on ice and with vigorous agitation every 10 min. Finally, the nuclear lysates were centrifuged at 17000 g for 10 min, and the supernatants transferred to precooled sample tubes and stored at −80 C until use.
Western blotting (WB) analysis
WB was performed as previously noted 9,21. HRECs were grown in six-well plates, washed with Dulbecco's-PBS (DPBS; Gibco, Paisley, UK, Lot:2085148, Cat:14190-144) three times, detached with a cell scraper, and collected by centrifugation. Mouse retinas were carefully isolated on ice immediately after euthanasia. Cell pellets or mouse retinas were sonicated in a cold RIPA buffer supplemented with a 1:100 FAST protease inhibitor (S8830, Sigma-Aldrich, St Louis, MO, USA). Disruption of the material was performed by a Q55 Sonicator (Qsonica, NY, USA) with four pulses for 22 kHz, 5 s each at 30% power output, and on ice. The lysates were centrifuged at 17,000 for 10 min. The total Protein concentration of resulting supernatants was determined using the Qubit 4 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). The 30 μg total protein loaded per lane were separated by SDS-PAGE (4–20% polyacrylamide gel; Biorad) before electrophoretic transfer to 0.45-μm pore nitrocellulose membranes. The membranes were blocked with 2.5% BSA (A7096, Sigma-Aldrich, St Louis, MO, USA) at RT for 1 h and then incubated overnight at 4 °C with the primary antibodies (Table 1). After washing with PBS-T buffer, the blots were incubated with horseradish peroxidase (HRP)-conjugated 1:1000 secondary antibody in 1% BSA in PBS-T (Goat anti-Rabbit IgG, 170-6515; Goat anti-Mouse IgG 172-1011; Goat anti-Rat IgG 5204-2504 Bio-Rad) for 1 h at RT. Signals were developed with ECL using a Super Signal West Pico kit (Thermo Fisher Scientific, Waltham, MA, USA) and detected with ImageQuant LAS500 (GE HealthCare Life Science, Pittsburgh, PA, USA). The densitometric analysis of WBs was performed with ImageJ software (National Institute of Health, Bethesda, MD, USA) using 8-bit grayscale positive chemiluminescent membrane images. All the quantification results were averaged from three protein blots and expressed as the mean ratio of the values (target protein/housekeeping protein) ± standard deviation (SD).
Table 1:
Primary antibodies and dilutions used in the study
| Target protein | Produced by | Catalog number |
Host | IHC dilution |
WB dilution |
|---|---|---|---|---|---|
| PLGF-VEGF Heterodimers1 | R&D Systems, Minneapolis, MN, USA | MAB297-SP | Mouse | 1:100 | 1:1000 |
| β-actin | Thermo Fisher Scientific, Waltham, MA, USA | MA5-15739 | Mouse | 1:200 | 1:2000 |
| β-tubulin | Thermo Fisher Scientific, Waltham, MA, USA | MA5-16308 | Mouse | 1:200 | 1:1000 |
| ZO-1 | Thermo Fisher Scientific, Waltham, MA, USA | 61-7300 | Rabbit | 1:100 | 1:1000 |
| VE-cadherin | Thermo Fisher Scientific, Waltham, MA, USA | 50-128-96 | Mouse | 1:100 | 1:1000 |
| VEGF-a | Santa Cruz Biotechnology, Dallas, TX, US | A-20, sc-152 | Rabbit | 1:100 | 1:1000 |
| PlGF | Novus Biologicals, Centennial, CO, US | NBP2-67067 | Rabbit | 1:100 | 1:1000 |
| p-IκB-α | Santa Cruz Biotechnology, Dallas, TX, US | B-9, sc-8404 | Mouse | 1:100 | 1:500 |
| NFκB p65 | Thermo Fisher Scientific, Waltham, MA, USA | SL259361 | Rabbit | 1:100 | 1:1000 |
| GFP | Abcam, Cambridge, MA, USA | ab6556 | Rabbit | 1:100 | 1:1000 |
| Adenovirus type 5 | Abcam, Cambridge, MA, USA | ab6982 | Rabbit | 1:500 | 1:5000 |
| Neural/glial antigen 2 | Cell Signaling Technology, Danvers, MA, USA | 4235S | Rabbit | 1:50 | 1:500 |
MAB297-SP antibody does not cross-react with VEGF or PlGF proteins when not as part of heterodimers. The antibody specificity was validated by enzyme-linked immunosorbent assay by the vendor (R&D Systems; MAB297-SP datasheet), as well in our WB experiments, where MAB297-SP produces a single specific band at 52 kDa, consistent with the combined molecular weight of the individual heterodimer fragments (PlGF 30 kDa+VEGF 22 kDa).
Trans-endothelial electrical resistance measurement by an electrical cell-impedance sensing system
The primary HRECs were seeded on two 8-well electrical cell-impedance sensing (ECIS) arrays (8W10E; (Applied BioPhysics) and cultured as described above. Trans-endothelial electrical resistance (TEER) was monitored with the ECIS system (Applied BioPhysics) in real-time. The changes in TEER have been monitored automatically every 300 s at 4 kHz AC frequency and recorded with ECIS software.22 After achieving stable readings of approximately 1300 Ohm, signifying the formation of the HREC monolayer, that was also confirmed by microscopy, cultures were treated with: control PBS, PlGF-VEGF heterodimers (200 ng/ml; Cat# 297-VP/CF, R&D Systems) or PlGF-VEGF heterodimers (200 ng/ml), preincubated with 5D11D4 antibody (25 μg/ml) for 30 min at RT. The unreacted 5D11D4 antibody was further neutralized by 30 minutes incubation with 25 μg/ml of Goat-anti-mouse IgG (AF007, Novusbio; Centennial, CO, USA) at RT. 5D11D4 antibody (25 μg/ml) without neutralization was used as control. Mouse Isotype Control (25 μg/ml; Clone B-Z1; Cell Sciences; Newburyport, MA, USA) was added to PBS, 5D11D4, PlGF-VEGF treated groups.
Tube formation
5 × 105 HREC were seeded onto a Matrigel (Geltrex; Thermo Fisher Scientific, Waltham, MA, USA) pre-coated 24-well-plate and incubated in (EBM-2 with 10% FCS, 1% Pen/Strep) with 5D11D4 antibody (25 μg/ml) or Mouse Isotype Control (25 μg/ml; Clone B-Z1; Cell Sciences; Newburyport, MA, USA). Cells were incubated for 12 hours and visualized by Calcein-AM (Thermo Fisher; C1430; 1 μg/ml) in culture for 30 min before imaging. Digital images were acquired using the EVOS Fl fluorescent microscope. Quantification of the number of junctions, nodes, and segments was performed by Angiogenesis Analyzer for ImageJ (http://image.bio.methods.free.fr/ImageJ/?Angiogenesis-Analyzer-for-ImageJ).
Immunocytofluorescence analysis
HRECs were seeded into Millicell EZ slides (Millipore, Billerica, MA, USA). At the experimental endpoint, the samples were fixed mildly in HistoChoice Molecular Biology fixative (H120-1L 1x; VWR Life Science, Radnor, PA, US) for 10 min, permeabilized with 0.05% Triton-X100, and then blocked with 5% BSA (A7096, Sigma-Aldrich, St Louis, MO, USA) for 1 h at room temperature (RT). The samples were then incubated with primary antibodies (Table 1). Then, after phosphate-buffered saline (1x, Fisher Bioreagents, Fair Lawn, NJ, US, 187672, BP399-1L)-Tween 20 (0.02%, Amresco Tween-20, Lot:1392C184, Cat:0777-1L) (PBS-T) washing, the samples were incubated with Goat anti-Rabbit IgG (H+L), Cyanine5 (A10523, 1:1000; Thermo Fisher Scientific, Waltham, MA, USA), Goat anti-Rabbit IgG (H+L), Alexa Fluor 488 (A-11034, 1:1000, Thermo Fisher Scientific, Waltham, MA, USA), Goat anti-Mouse IgG (H+L), Cyanine 5 (A10524, 1:1000, Thermo Fisher Scientific, Waltham, MA, USA). The cell nuclei were stained by incubation with 4',6-diamidino-2-phenylindole (DAPI) 1:5000 (Sigma-Aldrich, St Louis, MO, USA). The slides were mounted with the ProLong Diamond antifade reagent (Thermo Fisher Scientific, Waltham, MA, USA) and imaged with a Leica TCS SP8 inverted laser confocal microscope (Leica Camera AG, Wetzlar, Germany). Samples incubated with the blocking buffer (primary antibody was omitted), followed by secondary antibody incubation, were used as the background control. The colocalization analysis between PlGF-VEGF and NFκB signals was performed using EzColocalization plug-in for ImageJ. 23 Individual fluorescent channel images were converted to 8-bit grayscale and analyzed using default parameters.
Evaluation of 5D11D4 fluorescence after incubation with live HREC
To evaluate the fluorescence component of 5D11D4 incubation with live HREC cells and the immunostainings' specificity, HRECs were seeded into Millicell EZ slides (Millipore, Billerica, MA, USA). Live HREC cultures were incubated with 5D11D4 antibody 25 μg/ml for 24 hours then mildly fixed with HistoChoice Molecular Biology fixative (H120-1L 1x; VWR Life Science, Radnor, PA, US) for 10 min, permeabilized with 0.05% Triton-X100 for 10 min. Samples were blocked with 5% BSA (A7096, Sigma-Aldrich, St Louis, MO, USA) for 1 h at room temperature (RT) and then incubated with Cyanine 5 (A10524, 1:1000, Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature (RT). The cell nuclei were stained by incubation with 4',6-diamidino-2-phenylindole (DAPI) 1:5000 (Sigma-Aldrich, St Louis, MO, USA). The slides were mounted with the ProLong Diamond antifade reagent (Thermo Fisher Scientific, Waltham, MA, USA) and imaged with a Leica TCS SP8 inverted laser confocal microscope (Leica Camera AG, Wetzlar, Germany).
Experimental animals and housing
Eight-week-old C57BL/6 male mice were purchased from Jackson Laboratory and housed in a specific pathogen-free facility at the University of Missouri Department of Biomedical Sciences. All mice were fed with normal chow diets and provided with water ad libitum. During experimentation, the mice were anesthetized with ketamine hydrochloride (100 mg/kg body weight) and xylazine (4 mg/kg body weight). All experiments were approved by the Institutional Animal Care and Use Committee of the University of Missouri School of Medicine (protocol number: 9520) and were in accordance with the guidelines of the Association for Research in Vision and Ophthalmology Statement for the use of animals in ophthalmic and vision research.
Viral vectors and intravitreal injections and experimental endpoint
Viral vectors AAV5-PlGF or AAV5-GFP were designed by Dr. Sandro De Falco (Angiogenesis Lab, Institute of Genetics and Biophysics, CNR, Naples, Italy). The vectors were stored at −80°C until use. Intravitreal injections of adenovirus constructs were performed under the Olympus SZSTB1 (Olympus) dissection microscope. The mice were anesthetized, and an incision was made through the conjunctiva with micro scissors to expose approximately 0.2–0.3 mm of the sclera. The sclera was then punctured with a 28-G needle, and successful access to the vitreous cavity was verified by the presence of intravitreal fluid in the puncture. Using the volume displacement Pico-Injector (PLI-100; Harvard Apparatus, Holliston, MA, USA), 1 μl of AAV5-GFP or AAV5-PlGF vector material (approximately 2.8 × 108 vgc/μl) was then delivered to the mouse vitreous through the microinjection needle via scleral puncture. The animals were then evaluated for one week for posttraumatic cataract and injection site recovery, and animals with signs of cataract or unresolved inflammation at the injection site were excluded from the study. Successful infection of retinal cells by the AAV vectors was confirmed by AAV5 and PlGF proteins in the retinal samples and the presence of GFP signal in the control eyes. Retinal lysates from naïve C57BL/6 mice were used as a negative control for AAV5 and ZO-1 protein detection. Animals were sacrificed by i.p. injection of ketamine hydrochloride (300 mg/kg body weight) 4 weeks after intravitreal injection of the AAV5 vectors.
Histological analysis
The eyeballs were fixed with HistoChoice Molecular Biology fixative (VWR Life Science) for 12 hours at 4C and stored in PBS until being processed for paraffin embedding and sectioning (5 μm thick). As the GFP fluorescence was reduced significantly by tissue fixation and processing, sections were rehydrated, and the GFP signal was enhanced by incubation overnight at 4 °C with GFP antibody (Table 1), following by incubation with Goat anti-Rabbit IgG (H+L), Cyanine5 (A10523, 1:1000; Thermo Fisher Scientific, Waltham, MA, USA), secondary antibody for 1 h at RT, and counterstained with DAPI 1:5000 (Sigma-Aldrich, St Louis, MO, USA) and mounted with the ProLong Diamond antifade reagent (Thermo Fisher Scientific, Waltham, MA, USA). Sections were imaged using the EVOS FL fluorescent microscope (Thermo Fisher Scientific, Waltham, MA, USA).
Retinal flat mounts
The retinal flatmounts were prepared, as described previously 24. Briefly under an SZ-STB1 (Olympus) dissection microscope, fixed (HistoChoice Molecular Biology fixative, 12 hours at 4C) anterior segment tissues and vitreous were removed the retinas were carefully separated from retinal pigment epithelial/choroidal complexes. The retinas was permeabilized within a blocking solution composed of 5% Normal Goat Serum (NGS; Thermo Fisher Scientific, Waltham, MA, USA) in PBS overnight with 0.01% Triton-X; the samples were removed from a blocking solution and washed with PBS and then incubated with primary antibody (Table 1) for 24 h, followed by washing with PBS-T three times for 10 min and then incubation for 24 h with Goat anti-Mouse IgG H&L (Cy5 ®) preadsorbed (ab6563) 1:1000; Goat anti-Rabbit IgG H&L (Cy5 ®) preadsorbed (ab97077) 1:1000 and Pacific Blue Goat-anti Rabbit IgG H&L (P10994, Thermo Fisher Scientific, Waltham, MA, USA) secondary antibody in 2.5% NGS and DAPI 1:5000 (Sigma-Aldrich, St Louis, MO, USA). After another PBS-T washing, the samples were mounted on slides, photoreceptor side down, with ProLong Diamond antifade reagent (Thermo Fisher Scientific, Waltham, MA, USA) and imaged with a Leica TCS SP8 inverted laser confocal microscope (Leica Camera AG, Wetzlar, Germany). Samples incubated with the blocking buffer (primary antibody was omitted), followed by secondary antibody incubation, were used as the background control.
Statistical analysis
All experiments were performed in triplicates. Experimental values were expressed as the mean ± standard deviation (SD) for the respective groups. Statistical analyses were performed with GraphPad Prism software (https://www.graphpad.com/scientific-software/prism/). A one-way ANOVA with Tukey multiple comparisons was used whenever comparing multiple groups. A p-value of less than 0.05 was considered significant. The following designations for the p-value were used in the manuscript figures: n.s. p>0.05; * p<0.05; ** p<0.01; *** p<0.001.
Results
Nuclear PlGF-VEGF heterodimerization is enhanced by high glucose
We first examined the cellular localization of PlGF-VEGF heterodimers in HREC using immunostaining; we unexpectedly found that PlGF-VEGF heterodimers were detected primarily in HREC nuclei (Fig. 1A). It is worth noting that the primary antibody (MAB297, R&D Systems) binds only to the PlGF-VEGF heterodimer isoform and has no cross-reactivity with either subunit alone. Incubation of HREC cultures with 5D11D4 mouse PlGF antibody (25 μg/ml) for 24 hours decreased the heterodimer's presence, which suggests that the 5D11D4 antibody can bind with and neutralize not only PlGF protein but also the PlGF-VEGF heterodimers. (Fig. 1B). The likelihood of the 5D11D4 antibody affinity with PlGF-VEGF heterodimers and PlGF was investigated by dot immunoblot assay. As shown in Fig. 1C, the results showed that this antibody had a strong affinity with PlGF-VEGF heterodimer, less degree with PlGF, but lack of affinity with VEGF. The fluorescent component of incubation HREC with 5D11D4 (25 μg/ml) antibody for 24 hours was evaluated by immunofluorescence (Fig S1). The faint fluorescent signals were detected in the cell membrane surface, but not in the cell nuclei, further confirming the specificity of the PlGF-VEGF heterodimers staining.
Figure 1.

Detection of PlGF-VEGF heterodimers in the endothelial cells. PlGF-VEGF heterodimers are expressed in HREC with predominantly nuclear localization (A), 5D11D4 antibody 25 μg/ml markedly decreases the PlGF-VEGF heterodimers signals (B). Dot immunoassay demonstrating affinity of 5D11D4 antibody to rhPlGF-VEGF heterodimers and rhPlGF, but not rhVEGF or BSA control. (C)
Next, we investigated the influence of high glucose on PlGF-VEGF's nuclear localization. Western blots (WB) analysis was performed for the cytoplasmic and nuclear extracts from HRECs incubated in normal and high glucose conditions (Fig. 2A). The quantitative results indicated decreased cytoplasmic (Fig 2B; p<0.05) and increased nuclear presence (Fig 2C; p<0.05) of PIGF/VEGF heterodimers in high glucose condition. The purity of nuclear extracts was confirmed by the absence of the β-Tubulin protein, which was abundant in cytoplasmic extracts. Total protein loading was confirmed by β-actin protein. Consistent with the WB results, immunofluorescent staining further confirmed that stimulation with high glucose promoted the nuclear presence of PlGF-VEGF heterodimers (Fig. 2). As expected, the expression of VEGF-A (Fig. S2) and PlGF (Fig. 2D-E) was also promoted by high glucose stimulation. However, neither VEGF-A nor PlGF colocalized within the cell nuclei in normal or high glucose conditions. Loss of barrier function proteins VE-cadherin and ZO-1 in high glucose conditions was confirmed by immunofluorescence (Fig. S3).
Figure 2.

Western blot analysis PlGF-VEGF heterodimers, in cytoplasmic and nuclear extracts from HREC in normal and high glucose conditions (A). β-actin is used as loading control; β-tubulin is used to confirm the purity of the nuclear extracts. Quantitative analysis of PlGF-VEGF relative abundance in cytoplasmic (B) and nuclear (C) extracts of HREC in normal and high glucose conditions. The student's t-test was used to determine statistical significance. *p < 0.05; n=3. Immunofluorescent analysis of PlGF-VEGF heterodimerization and PlGF expression in normal (D) high-glucose (E) conditions. Nuclei counterstained by DAPI.
PlGF-VEGF heterodimers decrease the barrier function of the HREC monolayer
The effect of PlGF-VEGF heterodimers on the barrier function of HREC monolayer was first assessed by the transendothelial electrical resistance (TER) measured with the electrical cell-impedance system (ECIS) in a real-time manner. Previously we have reported that the addition of rhPlGF or rhVEGF165 reduces the resistance of monolayer.20 The addition of PlGF-VEGF heterodimers (200 ng/ml; Fig. 3A; black chart) rapidly decreased the TER of HREC monolayer. Neutralization of the PlGF-VEGF heterodimers by preincubation with mouse PlGF antibody (5D11D4; Fig. 3A; cyan chart) resulted in a resistance curve similar to control (Fig. 3A; magenta chart) in which HREC cultures were incubated with isotype control. Addition of 5D11D4 that inhibited both PlGF and PlGF-VEGF heterodimers determined an increase of the HREC monolayer barrier function (Fig. 3A; red chart), which is consistent with our previous observations of PlGF blockade contributing to increase if ZO-1 and VE-cadherin along with pentose phosphate pathways proteins in HREC20
Figure 3.

PlGF-VEGF heterodimers negatively regulate the barrier function. (A). Trans-endothelial electrical resistance measurement in HREC treated with 5D11D4 antibody 25 μg/ml (Red); Igg control (Magenta); PlGF-VEGF heterodimers (200 ng/ml, Black); PlGF-VEGF heterodimers (200 ng/ml) + 5D11D4 antibody 25 μg/ml (Cyan). A one-way ANOVA test with Tukey multiple comparisons was used to determine statistical significance between the experimental groups after 15 hours post treatment and till the experimental endpoint at 48 hours. n.s. p > 0.05; ***p < 0.001; n = 4 Immunofluorescent analysis of VE-cadherin and ZO-1 in the Igg control (B), PlGF-VEGF heterodimers 200 ng/ml (C); PlGF-VEGF heterodimers (200 ng/ml) + 5D11D4 antibody 25 μg/ml (D).
Consistently, PlGF-VEGF protein decreased the abundance level of two barrier-forming proteins VE-cadherin and ZO-1 in the cell-cell interaction sites, which were recovered by the preincubation of PlGF-VEGF and 5D11D4 antibody (Fig. 3B-D). Consistent with our previous observation, the addition of the 5D11D4 antibody increased both ZO-1 and VE-cadherin abundance at cell-to-cell interaction sites (Fig. 3E).
5D11D4 antibody disrupts the tube formation of HREC.
We next investigated whether the 5D11D4 antibody, which neutralizes PlGF-VEGF and PlGF based on our immunofluorescent and dot immunoassay findings above, inhibits tube formation in vitro. HREC grown on Geltrex were treated with either with 5D11D4 antibody (25 μg/ml) or isotype control for 24 hours. Tube formation was visualized Calcein AM staining (Fig 4A, B). As expected, isotype control-treated HREC formed a prominent tubule network when cultured on Geltrex support. However, 5D11D4 antibody-treated HREC demonstrated partially disintegrated tubule structures. Quantitative analysis of the images indicated a significant reduction in junction formation (Fig. 4C; p < 0.05), the numbers of nodes (Fig. 4D), and the numbers of segments (Fig. 4E; p < 0.01), relative to controls.
Figure 4.

Inhibition of PlGF and PlGF-VEGF heterodimers decreases tube formation in HREC. Tube formation assay in Igg control (A) and 5D11D4 antibody 25 μg/ml treated (B) HREC cultures 12-hours after seeding to geltrex substrate and visualized by calcein-AM. Quantitive analysis of the number of junctions (C), nodes (D), and segments (E) in digital images of the tube formation assay. The student's t-test was used to determine statistical significance. *p < 0.05; **p < 0.01; n=8.
Recombinant human PlGF promotes PlGF-VEGF heterodimerization and NFκB activation, with a blockade of IKK2 dissociating this interaction and suppression of NFκB activation
The question of whether PlGF and PlGF-VEGF regulate NFκB canonical activation was further addressed in HREC by rhPlGF stimulation. Double labeling of PlGF-VEGF heterodimers and NFκB in control HREC culture showed that NFκB p65 was detected primarily in the cytoplasm, with minimal nuclear presence, which was in contrast to the nuclear localization of PlGF-VEGF (Fig. 5A). However, stimulation of HREC with 100 ng/ml of recombinant human PlGF resulted in increased nuclear translocation of NFκB p65 (Fig. 5B) and increased cytoplasmic detection p-IκBα (Fig 5E). PBS (Fig. 5F) and LPS 1 μg/ml treated HREC (Fig. 5G) were negative and positive controls for p-IκBα staining. PlGF-VEGF heterodimers were colocalized with NFκB p65 upon rhPlGF stimulation (Fig 5B). The pretreatment of HREC with the selective IKK2 inhibitor: IMD-0354 (10 ng/ml) disrupted this colocalization (Fig. 5C). These results were further confirmed by quantifying cell numbers with the colocalization of PlGF-VEGF heterodimers and NFκB p65 (Fig 5D), and consistent with a recent study demonstrating anti-PlGF treatment prevents HREC damages caused by high glucose through the inhibition of PlGF/VEGFR1/ERK/NFκB/TNFa inflammatory signaling pathways25
Figure 5.

Immunofluorescence analysis of rhPlGF-induced NFκB activation and PlGF-VEGF heterodimerization. The PlGF-VEGF heterodimers and NFκB expression in control (A). Expression of NFκB increased following induction with rhPIGF, with increased activation shown by a stronger colocalization NFκB signal with PlGF-VEGF heterodimers than the control (B). Treatment with an IKK2 inhibitor, IMD0354, following stimulation with rhPIGF, suppressed the activation of NFκB signaling and colocalization of PlGF-VEGF heterodimers (C). Quantitative analysis of the number of cells with PlGF-VEGF heterodimers and NFκB colocalization (D). A one-way ANOVA test with Tukey multiple comparisons was used to determine statistical significance. n.s. p > 0.05; ***p < 0.001; n = 5 Increased p-IκBα presence in rhPlGF treated (100 ng/ml) HREC (E). Background activity of p-IκBα in the PBS treated negative control (F). Positive control HREC treated with 1 μg/ml of LPS shows a dramatic increase in p-IκBα signals (G).
IKK2 inhibition prevents rhPlGF-induced loss of barrier function proteins ZO-1, VE-cadherin
In agreement with our previous experiments 20, treatment of HREC with rhPlGF (100 ng/ml) resulted in a decrease of the barrier function, as indicated by the suppression of ZO-1 and VE-cadherin (Fig. 6A). However, pretreatment with IMD-0354 10 ng/ml prevented PlGF-induced loss of barrier function proteins. The quantification of protein bands in western blots indicated a significant decrease of ZO-1 (p<0.01) and VE-cadherin (p<0.01) in rhPlGF treated HREC lysates when compared to the control. Pretreatment with IMD0354 has restored barrier function proteins' expression to normal (p >0.05). Interestingly IMD0354 treatment by itself did not affect the expression of barrier function proteins (p>0.05) when compared to the control group (Fig. 6B, C). These results support that PlGF and PlGF-VEGF negatively regulate HREC barrier function through activation of canonic NFκB signaling.
Figure 6.
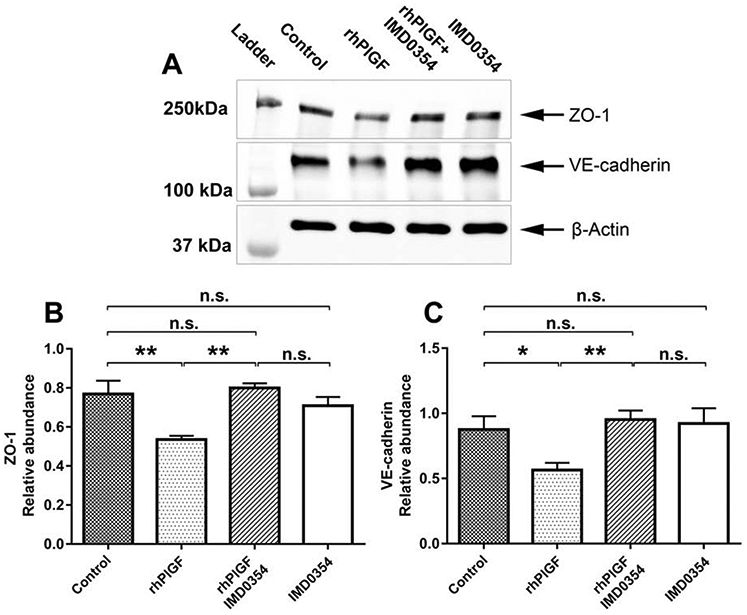
WB analysis of rhPlGF-induced ZO-1 and VE-cadherin expression in HREC (A). Expression of ZO-1 and VE-cadherin in control cells. The stimulation of HREC cells with rhPIGF (200 ng/ml) suppressed the expression of ZO-1 and VE-cadherin when compared with the control. Suppression of NFκB activation by pretreatment with IMD0354 (10 ng/ml) followed by rhPIGF (200 ng/ml) preserved the barrier function by maintaining the expression of the junctional proteins ZO-1 and VE-cadherin. Quantitative analysis of ZO-1 (B) and VE-cadherin (C) relative abundance in HREC lysates treated with PBS, rhPlGF 200 ng/ml; IMD0354 (10 ng/ml) and rhPIGF (200 ng/ml); IMD0354 (10 ng/ml). A one-way ANOVA test with Tukey multiple comparisons was used to determine statistical significance. n.s. p > 0.05; *p < 0.05; **p < 0.01; n=3.
Overexpression of PlGF in the retina by adeno-associated virus type 5 (AAV5) vector resulted in NFκB activation and decreased ZO-1
To investigate the role of PlGF-VEGF heterodimers in vivo, adult C57BL/6 mice retinas were transduced with PlGF-AAV5 vectors by intravitreal injection, and the GFP-AAV5 vector was used as control. Effective transduction of the retina by AAV5-GFP vector as indicated by the presence of the strong GFP signals in all retinal layers (Fig. 7A), but no GFP signals were detected in AAV5-PlGF transduced retinas (Fig. 7B). Transduction of the AAV5-PlGF vectors without the GFP-tag was confirmed by western blot analysis, which showed the presence of AAV5 capsid proteins in all AAV5 injected eyes by the absence of this protein in naïve control retina (Fig. 7C). Immunostaining to PlGF indicated massive PlGF signals from the AAV5-PlGF transduced eyes, but not the AAV5-GFP treated eyes (Fig. 7D, E).
Figure 7.

Analysis of AAV5-mediated transduction of retinal cells with GFP and PlGF in C57BL/6 mice. AAV5-GFP vector effectively transduced retinal cells, as indicated by the presence of GFP across the retinal cell layers (A); no GFP signals were observed in AAV5-PlGF transduced retinas (B). The presence of AAV5 capsid proteins bands for AAV5-GFP and AAV5-PlGF vectors by WB analysis; naïve retina lysate were used as the negative control (C). Immunofluorescent analysis of PlGF expression in AAV5-GFP transduced retinas (D) and AAV5-PlGF (E). Immunofluorescent p-IκBα signals from control animals transduced with AAV5-GFP vector (F), with an increase of p-IκBα presence in AAV5-PlGF transduced retinas (G). Western blot analysis of the junctional protein ZO-1 in naïve, AAV5-GFP, and AAV5-PlGF transduced retinal lysates, β-actin used as the loading control. (I) The relative abundance of ZO-1 in ZO-1 in naïve, AAV5-GFP, and AAV5-PlGF transduced retinal lysates PlGF overexpression leads to a decrease in the junctional protein ZO-1. A one-way ANOVA test with Tukey multiple comparisons was used to determine statistical significance. n.s. p > 0.05; **p < 0.01; n=3. Detection of PLGF-VEGF heterodimers AAV5-GFP (J) and AAV5-PLGF (K) transduced retinas counterstained with NG2 marker. White squares indicate the enlarged areas of the image. PLGF-VEGF heterodimers in AAV5-GFP control retinas are present in NG2 positive cells (magenta, double-positive for NG2 and PLGF-VEGF; white arrows). Overexpression of PlGF in the retina by AAV5-PLGF indicates heterodimers' presence in ganglion cells (consistent with PlGF signals in E; yellow arrows); visualized vasculature and NG2 double-positive signals (magenta, white arrows).
The effects of AAV5-mediated PlGF overexpression on NFκB signaling and ZO-1 protein expression were further examined. Minimal p-IκBα signals were observed in control retinas transduced with AAV5-GFP (Fig 7F). However, p-IκBα signals were increased in the retina of mice transduced with AAV5-PlGF (Fig 7G). Overexpression of PlGF by the AAV5 vector resulted in a suppression of ZO-1 in the retina (Fig. 7H). Western blot results were further confirmed by the quantitative analysis of ZO-1 bands (Fig 7I) and immunofluorescence in retinal flat mounts (Fig S4). The ZO-1 was prominently expressed in the vessel wall of retinal microvasculature in naïve and AAV5-GFP-transduced retinas (Fig S4A-B). However, this expression was reduced in AAV5-PlGF transduced eyes (Fig S4C).
Finally, we examined the potential cell sources of the endogenously and exogenously expressed PlGF-VEGF in the retina. Double immunostaining PlGF-VEGF heterodimers staining with Neuron-glial antigen 2 (NG2) indicated the presence of PlGF-VEGF heterodimers predominantly in NG2 positive cells (Fig. 7J) across the vasculature with the cell morphology consistent with pericytes in AAV5-GFP control retinas (Fig. 7K). The overexpression of PlGF demonstrated strong PlGF-VEGF heterodimers signals in the NG2 positive cells (white arrows), ganglion (yellow arrows), consistent with PlGF overexpression (Fig 7E).
Discussion
Ocular diseases such as DR are a major clinical challenge because of the lack of efficient treatments and a limited understanding of these conditions' pathophysiology. The standard treatment for PDR—photocoagulation—has well-documented limitations, such as requiring patient cooperation and the possible permanent loss of peripheral vision, nyctalopia, among other issues 26 On the other hand, the VEGF signaling pathway has been implicated in the pathological angiogenesis in PDR 27,28; however, antiangiogenic treatments for pathological angiogenesis in the eye, in general, are of limited efficacy 29.
PlGF-VEGF heterodimerizes naturally in certain pathophysiological conditions and can also be reconstituted by recombinant proteins in vitro 30. We recently showed that both PlGF and VEGF were expressed and secreted by the cultured HREC and formed a heterodimer in culture supernatants 20. We also demonstrated that high glucose levels could induce an interaction between PlGF-VEGF heterodimers and membrane receptors; however, the further fate of these complexes has remained unclear at the time. 20
Similarly, diabetes upregulated the levels of PlGF-VEGF heterodimers in the retina. These new results from the current study indicate a potential functional role of PlGF-VEGF in high glucose conditions. Because of the inclusion of both PlGF and VEGF-A components and the differential affinity capacity of VEGFR1 and VEGFR2, PlGF-VEGF heterodimer has been proposed as playing regulatory roles in angiogenesis and vascular permeability.
PIGF, a member of the VEGF family of proteins, is involved in angiogenesis and can heterodimerize with VEGF-A 30,31. For example, PlGF-VEGF dimerization has been shown to modulate choroidal neovascularization 32. PlGF was previously thought to be expressed in tissues primed for angiogenesis, such as the placenta, but not in normal adult tissue 33,34. However, the genetic ablation of PlGF in mice does not produce any reproductive abnormalities, producing healthy animals 35. In a previous study, PIGF was shown to mediate cutaneous inflammation and vasodilation, it was postulated that VEGF-driven angiogenesis depends on the local expression of PIGF 36, further supporting the importance of PlGF-VEGF dimerization under pathological conditions. In one in-vitro study, it was observed that PlGF-VEGF heterodimers showed less mitogenic and chemotactic properties compared with VEGF165 homodimers 30. PlGF-VEGF heterodimers have a strong affinity with VEGFR1 and can induce VEGFR1/VEGFR2 dimerization, indicating that PlGF-VEGF heterodimerization may be essential for VEGF-induced angiogenesis 14. So far, the PIGF/VEGF complex's exact mode of action has not been sufficiently studied and hence still poorly understood. In the current study, we demonstrated a potential regulatory mechanism involving PlGF-VEGF heterodimers colocalize with NFκB p65 in the cell nuclei to suppress the expression of junctional proteins ZO-1 and VE-cadherin, whose expression was rescued by IKK2 inhibition, thereby suggesting the involvement in BRB function.
Previously, we reported IMD-0354 non-ATP-binding competitive selective IKK-2 inhibitor to ameliorate DR in mice 37. NFκB signaling is a well-described pathway essential for cell survival; however, its activation in certain conditions has been shown to promote inflammation and angiogenesis, for instance, in corneal neovascularization 38,39. DeNiro et al. showed a nexus between VEGF and NFκB signaling under hypoxia in driving pathological angiogenesis in vitro 40. Our current findings indicate the increase of PlGF-VEGF heterodimer's nuclear presence and colocalization with active NFκB in high glucose conditions and when stimulated by rhPlGF. Furthermore, blockade of NFκB activation has prevented PlGF induced loss of barrier proteins ZO-1 and VE-cadherin in HREC.
In one of our recently completed studies, we showed that PlGF and PlGF-VEGF heterodimers negatively regulate the EC barrier function through the activation of VEGFR1 and VEGFR2 and suppression of G6PD activity and pentose phosphate pathway (PPP) flux in vitro. In the same work, we also demonstrated a negative regulation of barrier function by PIGF/VEGF heterodimers and increased signals in the endothelial cells in high glucose conditions. 20 Our current findings, especially the fact that the 5D11D4 antibody has an affinity with both PlGF and PlGF-VEGF heterodimers, suggest that some of our previous findings may be attributed to the combined effects of PlGF and PlGF-VEGF heterodimers being inhibited by 5D11D4 antibody treatment.
In the present study, we further demonstrated that NFκB is involved in nuclei's regulatory networks. Cramer et al. 41 demonstrated NFκB regulatory role on PlGF expression in vitro, through NFκB interaction with Sp1 binding site, metal responsive element (MRE), hypoxia-responsive element (HRE) 42, and HNF3b (BF-2) binding sites 43 in the promoter region of PlGF. Mirjam et al. reported that overexpression of NFκB in HEK293 cells caused increased numbers of PlGF transcripts. While PlGF and VEGF are not present in the nuclei, several putative binding sites for NFκB in the PlGF promoter/enhancer region of the DNA were identified.41 PlGF overexpression can induce the formation of the retina's heterodimer and disrupt the BRB integrity, as shown by the reduced level of ZO-1 protein in the retina. Both in vitro and in vivo data support the interactions of PlGF, PlGF-VEGF, and their receptors with NFκB components. In a healthy retina, PlGF-VEGF heterodimers are predominantly detected in NG2 positive cells that include pericytes as indicated by double staining with NG2 pericytes marker with AAV5 induced PlGF overexpression. It is known that diabetic conditions can cause pericyte loss in the retina, although the exact mechanism of induction of pericyte loss remains unclear.44,45 Further in-vitro and in-vivo studies are required to understand whether the PlGF-VEGF heterodimer formation potentially plays a role in the acellular-occluded capillaries lacking pericytes.
Although several studies showed that VEGF variants, such as VEGF-165b46, VEGF-D 47, and long-VEGF (with a 188aa extra extension compared to normal VEGF)48, had nuclear localization via endocytosis under certain developmental, physiological, and pathological conditions, the classical signaling pathways for VEGF and PlGF are to bind with their transmembrane tyrosine kinase receptors (VEGFR1 and VEGFR2) and then activate the intracellular transduction cascade. We expected that VEGF-PlGF would cause hetero-dimerization of VEGFR1 and VEGFR2 and amplify the effects on HRECs permeability. However, we unexpectedly found that VEGF-PlGF heterodimer had a nuclear localization associated with high glucose and NFκB signaling pathways. We hypothesize that these dimers in the nucleus, together with NFκB and human antigen R49, act as trans-regulatory factors and regulate inflammatory genes (e.g., TNFa) concerning diabetic retinopathy.
The present study's findings are of the potential therapeutic implications in providing new therapeutic target sites for diabetic retinopathy, such as PlGF and VEGF interaction. Using anti-PlGF/VEGF therapy to treat vascular disorders associated with NFkB activation and inflammation, preventing pericytes loss or dysfunction by the inhibition of VEGF and PlGF hetero-dimerization.
Statements of limitation
1. The amount of 100 ng/ml of PlGF-VEGF heterodimers to decrease TEER and barrier function proteins was established in vitro experimentally. However, the exact amount of the PlGF-VEGF heterodimers produced in vivo by the AAV5-PlGF vector transduction remains undetermined.
2. While the morphology of NG2 positive cells presented in Fig. 7J-K is consistent with pericytes, NG2 is also expressed in microglia cells.50 Thus, we can not entirely exclude potential double staining of PlGF-VEGF heterodimers and NG2 positive microglia cells.
Conclusion
Our results support that PlGF-VEGF heterodimers negatively regulate EC barrier function in a high-glucose environment both in vivo and in vitro, acting as an independent property that may play an essential role in vasculopathy and can be considered a new therapeutic target. Further studies focused on PlGF-VEGF formation. Its role in DR and the creation of small molecules, specifically disrupting heterodimers' formation in the retina, are needed to elucidate its therapeutic potential.
Supplementary Material
Highlights.
Placental growth factor/vascular endothelial growth factor (PlGF-VEGF) heterodimers were detected predominantly in the HREC cell nuclei
High glucose and recombinant human PlGF treatment increased PlGF-VEGF nuclear abundance.
PlGF-VEGF heterodimers are involved in the blood-retinal barrier (BRB) breakdown.
Neutralization of PlGF-VEGF heterodimers or inhibition of NFkB activation restored the HREC barrier function proteins
Overexpression of PlGF in mouse retina by AAV5 vector resulted in an increase of heterodimers formation, NFκB activation, and a decrease of ZO-1 in vivo.
Acknowledgments
The authors would like to acknowledge the following contributors: Allen Raye (University of Missouri Department of Biomedical Sciences, Columbia, Missouri, USA) for assistance with animal resources; Molecular Cytology core (University of Missouri, Columbia, Missouri, USA) for technical assistance with confocal imaging.
Funding
This work was supported by the following grants: National Eye Institute R01 EY027824 (Hu Huang); Missouri University start-up fund (Hu Huang); Missouri University Postdoctoral Research Grant (Anton Lennikov), and Project SATIN-POR Campania FESR 2014/2020 (Sandro De Falco).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Availability of data and materials
All other data generated and analyzed in the current study are included in this published article and its supplementary information.
Ethics approval and consent to participate
All experiments were approved by the Institutional Animal Care and Use Committee of the University of Missouri School of Medicine (protocol number: 9520) and were in accordance with the guidelines of the Association for Research in Vision and Ophthalmology Statement for the use of animals in ophthalmic and vision research.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
References
- 1.Huang H et al. Deletion of placental growth factor prevents diabetic retinopathy and is associated with Akt activation and HIF1α-VEGF pathway inhibition. Diabetes 64, 200–212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen QD et al. Placental growth factor and its potential role in diabetic retinopathy and other ocular neovascular diseases. Acta ophthalmologica 96, e1–e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dewerchin M & Carmeliet P PlGF: a multitasking cytokine with disease-restricted activity. Cold Spring Harbor perspectives in medicine 2, a011056 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al Kahtani E et al. Vitreous levels of placental growth factor correlate with activity of proliferative diabetic retinopathy and are not influenced by bevacizumab treatment. Eye (Lond) 31, 529–536, doi: 10.1038/eye.2016.246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyamoto N et al. Placental growth factor-1 and epithelial haemato-retinal barrier breakdown: potential implication in the pathogenesis of diabetic retinopathy. Diabetologia 50, 461–470, doi: 10.1007/s00125-006-0539-2 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Kowalczuk L et al. Placental growth factor contributes to micro-vascular abnormalization and blood-retinal barrier breakdown in diabetic retinopathy. PloS one 6, e17462, doi: 10.1371/journal.pone.0017462 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khaliq A et al. Increased expression of placenta growth factor in proliferative diabetic retinopathy. Laboratory investigation; a journal of technical methods and pathology 78, 109–116 (1998). [PubMed] [Google Scholar]
- 8.Carmeliet P et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med 7, 575–583, doi: 10.1038/87904 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Huang H, Shen J & Vinores SA Blockade of VEGFR1 and 2 suppresses pathological angiogenesis and vascular leakage in the eye. PLoS One 6, e21411, doi: 10.1371/journal.pone.0021411 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang H, Parlier R, Shen JK, Lutty GA & Vinores SA VEGF receptor blockade markedly reduces retinal microglia/macrophage infiltration into laser-induced CNV. PLoS One 8, e71808, doi: 10.1371/journal.pone.0071808 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joussen AM et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 16, 438–440, doi: 10.1096/fj.01-0707fje (2002). [DOI] [PubMed] [Google Scholar]
- 12.Koizumi K et al. Contribution of TNF-alpha to leukocyte adhesion, vascular leakage, and apoptotic cell death in endotoxin-induced uveitis in vivo. Investigative ophthalmology & visual science 44, 2184–2191 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Gardiner TA et al. Inhibition of tumor necrosis factor-alpha improves physiological angiogenesis and reduces pathological neovascularization in ischemic retinopathy. The American journal of pathology 166, 637–644 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao Y et al. Heterodimers of Placenta growth factor/vascular endothelial growth factor endothelial activity, tumor cell expression, and high affinity binding to Flk-1/KDR. Journal of Biological Chemistry 271, 3154–3162 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Boulton M, Cai J, Jiang W & Ahmed A Role of Placenta Growth Factor (PlGF) and PlGF/VEGF Heterodimer in the Regulation of In Vitro Angiogenesis. Investigative Ophthalmology & Visual Science 43, 2762–2762 (2002). [Google Scholar]
- 16.Apicella I, Cicatiello V, Acampora D, Tarallo V & De Falco S Full Functional Knockout of Placental Growth Factor by Knockin with an Inactive Variant Able to Heterodimerize with VEGF-A. Cell reports 23, 3635–3646 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Autiero M et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nature medicine 9, 936–943, doi: 10.1038/nm884 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Snuderl M et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 152, 1065–1076, doi: 10.1016/j.cell.2013.01.036 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saddala MS et al. Proteomics reveals ablation of PlGF increases antioxidant and neuroprotective proteins in the diabetic mouse retina. Sci Rep 8, 16728, doi: 10.1038/s41598-018-34955-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang H et al. Placental growth factor negatively regulates retinal endothelial cell barrier function through suppression of glucose-6-phosphate dehydrogenase and antioxidant defense systems. The FASEB Journal 0, fj.201901353R, doi: 10.1096/fj.201901353R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H et al. Reduced retinal neovascularization, vascular permeability, and apoptosis in ischemic retinopathy in the absence of prolyl hydroxylase-1 due to the prevention of hyperoxia-induced vascular obliteration. Investigative ophthalmology & visual science 52, 7565–7573, doi: 10.1167/iovs.11-8002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giaever I & Keese CR Micromotion of mammalian cells measured electrically. Proceedings of the National Academy of Sciences of the United States of America 88, 7896–7900 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stauffer W, Sheng H & Lim HN EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Scientific reports 8, 15764, doi: 10.1038/s41598-018-33592-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saddala MS et al. Discovery of novel L-type voltage-gated calcium channel blockers and application for the prevention of inflammation and angiogenesis. Journal of neuroinflammation 17, 132, doi: 10.1186/s12974-020-01801-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazzara F et al. Aflibercept regulates retinal inflammation elicited by high glucose via the PlGF/ERK pathway. Biochem Pharmacol 168, 341–351, doi: 10.1016/j.bcp.2019.07.021 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Shimura M et al. Quantifying alterations of macular thickness before and after panretinal photocoagulation in patients with severe diabetic retinopathy and good vision. Ophthalmology 110, 2386–2394 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Wang G & Wang Y Intravitreous vascular endothelial growth factor and hypoxia-inducible factor 1a in patients with proliferative diabetic retinopathy. American journal of ophthalmology 148, 883–889 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Witmer A, Vrensen G, Van Noorden C & Schlingemann R Vascular endothelial growth factors and angiogenesis in eye disease. Progress in retinal and eye research 22, 1–29 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Wells JA et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema: two-year results from a comparative effectiveness randomized clinical trial. Ophthalmology 123, 1351–1359 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao Y et al. Heterodimers of placenta growth factor/vascular endothelial growth factor. Endothelial activity, tumor cell expression, and high affinity binding to Flk-1/KDR. J Biol Chem 271, 3154–3162, doi: 10.1074/jbc.271.6.3154 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Cao Y, Linden P, Shima D, Browne F & Folkman J In vivo angiogenic activity and hypoxia induction of heterodimers of placenta growth factor/vascular endothelial growth factor. J Clin Invest 98, 2507–2511, doi: 10.1172/JCI119069 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Apicella I, Cicatiello V, Acampora D, Tarallo V & De Falco S Full Functional Knockout of Placental Growth Factor by Knockin with an Inactive Variant Able to Heterodimerize with VEGF-A. Cell Rep 23, 3635–3646, doi: 10.1016/j.celrep.2018.05.067 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Shalaby F et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Takahashi A et al. Markedly increased amounts of messenger RNAs for vascular endothelial growth factor and placenta growth factor in renal cell carcinoma associated with angiogenesis. Cancer Research 54, 4233–4237 (1994). [PubMed] [Google Scholar]
- 35.Carmeliet P & Jain RK Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307, doi: 10.1038/nature10144 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carmeliet P et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nature medicine 7, 575 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Lennikov A et al. IkappaB kinase-beta inhibitor IMD-0354 beneficially suppresses retinal vascular permeability in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci 55, 6365–6373, doi: 10.1167/iovs.14-14671 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Lan W, Petznick A, Heryati S, Rifada M & Tong L Nuclear Factor-κB: central regulator in ocular surface inflammation and diseases. The ocular surface 10, 137–148 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Lennikov A et al. Selective IKK2 inhibitor IMD0354 disrupts NF-κB signaling to suppress corneal inflammation and angiogenesis. Angiogenesis 21, 267–285 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeNiro M, Al-Mohanna FH, Alsmadi O & Al-Mohanna FA The nexus between VEGF and NFκB orchestrates a hypoxia-independent neovasculogenesis. PLoS One 8, e59021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Cramer M et al. NF-kappaB contributes to transcription of placenta growth factor and interacts with metal responsive transcription factor-1 in hypoxic human cells. Biol Chem 386, 865–872, doi: 10.1515/BC.2005.101 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Green CJ et al. Placenta growth factor gene expression is induced by hypoxia in fibroblasts: a central role for metal transcription factor-1. Cancer Res 61, 2696–2703 (2001). [PubMed] [Google Scholar]
- 43.Zhang H et al. Transcriptional activation of placental growth factor by the forkhead/winged helix transcription factor FoxD1. Curr Biol 13, 1625–1629, doi: 10.1016/j.cub.2003.08.054 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Beltramo E & Porta M Pericyte loss in diabetic retinopathy: mechanisms and consequences. Curr Med Chem 20, 3218–3225, doi: 10.2174/09298673113209990022 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Pfister F, Przybyt E, Harmsen MC & Hammes HP Pericytes in the eye. Pflugers Arch 465, 789–796, doi: 10.1007/s00424-013-1272-6 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Baba T et al. VEGF 165 b in the developing vasculatures of the fetal human eye. Developmental dynamics : an official publication of the American Association of Anatomists 241, 595–607, doi: 10.1002/dvdy.23743 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El-Chemaly S et al. Nuclear localization of vascular endothelial growth factor-D and regulation of c-Myc-dependent transcripts in human lung fibroblasts. Am J Respir Cell Mol Biol 51, 34–42, doi: 10.1165/rcmb.2013-0417OC (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenbaum-Dekel Y et al. Nuclear localization of long-VEGF is associated with hypoxia and tumor angiogenesis. Biochem Biophys Res Commun 332, 271–278, doi: 10.1016/j.bbrc.2005.04.123 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Lejbkowicz F, Goldberg-Cohen I & Levy AP New horizons for VEGF. Is there a role for nuclear localization? Acta histochemica 106, 405–411, doi: 10.1016/j.acthis.2004.11.003 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Zhu L et al. NG2 expression in microglial cells affects the expression of neurotrophic and proinflammatory factors by regulating FAK phosphorylation. Scientific reports 6, 27983, doi: 10.1038/srep27983 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.