

Abstract
High-grade serous ovarian carcinoma (HGSOC) is the most lethal gynecologic malignancy in industrialized countries and has limited treatment options. Targeting ATR/CHK1-mediated S and G2/M cell cycle checkpoints has been a promising therapeutic strategy in HGSOC. To improve the efficacy of CHK1 inhibitor (CHK1i), we conducted a high-throughput drug combination screening in HGSOC cells. PI3K/mTOR pathway inhibitors (PI3K/mTORi) showed supra-additive cytotoxicity with CHK1i. Combined treatment with CHK1i and PI3K/mTORi significantly attenuated cell viability and increased DNA damage, chromosomal breaks and mitotic catastrophe compared to monotherapy. PI3K/mTORi decelerated fork speed by promoting new origin firing via increased CDC45, thus potentiating CHK1i-induced replication stress. PI3K/mTORi also augmented CHK1i-induced DNA damage by attenuating DNA homologous recombination repair activity and RAD51 foci formation. High expression of replication stress markers was associated with poor prognosis in patients with HGSOC. Our findings indicate that combined PI3K/mTORi and CHK1i induces greater cell death in HGSOC cells and in vivo models by causing lethal replication stress and DNA damage. This insight can be translated therapeutically by further developing combinations of PI3K and cell cycle pathway inhibitors in HGSOC.
Keywords: CHK1, PI3K/mTOR pathway, replication stress, mitotic catastrophe, high-grade serous ovarian cancer
Introduction
Ovarian carcinoma is the deadliest gynecologic malignancy and one of the most common causes of cancer mortality among women worldwide (1). High-grade serous ovarian carcinoma (HGSOC) is the most common (~70%) and fatal subtype of ovarian cancer because it typically presents as an advanced disease with a high frequency of recurrence after initial platinum-based chemotherapy (1). Approximately 25-30% of HGSOC are deficient in homologous recombination (HR) repair due to BRCA1 and BRCA2 germline or somatic mutations (2) leading to sensitivity to poly (ADP-ribose) polymerase inhibitor (PARPi). However, the clinical activity of PARPi monotherapy is limited in platinum-resistant ovarian cancer with BRCA wild-type (BRCAwt), a majority of the HGSOC population (3). Therefore, a critical need remains for new effective therapeutic strategies.
Checkpoint signaling is necessary for coordination between DNA damage response and cell cycle control. HGSOC cells are heavily dependent on ataxia-telangiectasia and Rad3-related (ATR)/cell cycle checkpoint kinase1 (CHK1)-mediated G2/M arrest for DNA repair because of its universal TP53 mutation that disrupts the G1/S cell cycle checkpoint. CHK1, which regulates the G2/M checkpoint, is also overexpressed in nearly all HGSOC (4), making it a rational target to induce DNA damage and tumor cell death.
CHK1 is activated by ATR and ataxia-telangiectasia mutated (ATM) kinases in response to DNA damage or replication stress (5). CHK1 suppresses the activity of cyclin-dependent kinases (CDKs) during S phase to keep cells from cell cycle progression without optimal DNA replication (6,7). CHK1 also stabilizes stalled replication forks by decreasing origin firing and promoting DNA repair thereby limiting replication stress (6,7). Replication stress is thus exaggerated in the absence of ATR/CHK1 activation because of persistent stalled replication forks and increased unscheduled new origin firings (7,8). Also, HR DNA double-stranded breaks (DSBs) repair is partly involved in stalled replication fork stabilization, restart and repair via loading RAD51 to the single-stranded DNA (ssDNA) at replication forks (9,10). CHK1 contributes to HR repair by inducing RAD51 phosphorylation and nuclear translocation, as shown in several cancer cell lines including HGSOC (9,10). Hence, targeting CHK1 is a promising therapeutic strategy in HGSOC to augment replication stress while attenuating DNA repair responses. However, only modest clinical activity of the CHK1 inhibitor (CHK1i) monotherapy has been reported thus far in HGSOC patients (11) highlighting the critical unmet need for combination treatment strategies.
In this study, we conducted an unbiased high-throughput drug combination screening of the second generation CHK1i, prexasertib with 1,912 drugs using a panel of TP53 mutant HGSOC cell lines. Among candidates, we identified that phosphatidylinositol-3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway inhibitors have supra-additive cytotoxic effects with a CHK1i. The PI3K/mTOR pathway is frequently (>70%) upregulated in ovarian cancer (1,12) and its activation is associated with aggressive phenotypes, chemoresistance and poor prognosis, therefore making it an important target for treatment (1,12). Moreover, studies suggest that the PI3K/mTOR pathway is involved in DNA repair and replication stress (13). For instance, the active PI3K pathway is required for CHK1 activation following DNA damage for optimal DNA replication (14). Also, mTORC1/2 upregulates the protein levels of CHK1 via inhibiting the translational repressor eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) for protein synthesis upon DNA damage (15). As such, PI3K pathway inhibitors (i.e., mTOR inhibitor PP242 and PI3K/mTOR inhibitor omipalisib) increase replication catastrophe and DNA damage during S and G2 phases as shown in breast cancer cell lines (16,17). Together, these data support the PI3K pathway is a rational target for a CHK1i combination and also highlight the need to further investigate the interactions between the two pathways in HGSOC.
In the present study, we found that PI3K/mTOR blockade potentiated CHK1i-induced replication stress in HGSOC cells via increasing levels of CDC45 which consequently results in excess new origin firing, leading to lethal DNA damage and mitotic catastrophe. Combination treatment also reduced tumor growth in an HGSOC mouse model without significant toxicities. Furthermore, we found that co-expression of high replication stress markers, i.e., high CDC45 with high RPA was associated with poor prognosis in patients with ovarian cancer. Collectively, our results provide initial findings and mechanistic insights to justify combination treatment strategies using PI3K pathway blockade, and cell cycle checkpoint inhibition for recurrent HGSOC.
Materials and Methods
A full description of methods was described in the Supplementary Materials, including drug preparation, cell viability, clonogenic assay, siRNA transfections, immunofluorescence staining, DNA comet assay, caspase 3/7 activity assay, immunoblotting, metaphase spread, cell cycle, DNA fiber assays, HR reporter assay, immunohistochemistry staining and databases for genomic profile, mRNA expression and progression-free survival (PFS) analysis of ovarian cancer.
Cell lines
BRCAwt TP53 mutant HGSOC cell lines OVCAR3, OVCAR5 and OVCAR8 were obtained from NCI-60 collection at the NCI Frederick (MD, USA) and OV90 was purchased from ATCC. BRCA2 mutant (BRCA2m) PEO1 (#10032308) and BRCA2 reversion mutant PEO4 (#10032309) cell lines were purchased from Sigma-Aldrich (Saint Louis, MO). All cell lines were grown in RPMI 1640 medium with (+) L-glutamine supplemented with 10% fetal bovine serum, 0.01 mg/ml insulin, and 1% penicillin/streptomycin.
High-throughput drug combination screening
The screening methods conducted in this study were based on those previously published (18,19). Briefly, we first screened BRCA2m PEO1 cells against 1,912 oncology drugs using a 48-hour cell proliferation assay with an ATP-based readout CellTiterGlo (#G7570, Promega, Madison, WI) to determine activity and potency of compounds in a dose-response manner. We created the second screen moving from 6×6 pilot format to the more robust 10×10 matrix in PEO1 and BRCAwt TP53 mutant (OVCAR5, OVCAR8 and PEO4) HGSOC cell lines. Matrix blocks were dispensed using an acoustic dispenser (EDC Biosystems, Fremont, CA) and 48-hour CellTiterGlo or 8- and 16-hour Caspase-Glo® 3/7 Assay System (#G8090, Promega) readouts were utilized to inform on cell viability and apoptosis induction as described in manufacturers. Signal was measured as median relative luminescence units on a ViewLux (Perkin-Elmer, Waltham, MA) reader. Efficacious compounds from single agent screens were advanced to matrix combinations studies to assess additivity and synergy.
Animal study
All animal experiments were conducted under an approved by Institutional Animal Care and Use Committee of National Cancer Institute (ASP 19-251, NCI, Frederick, MD). OVCAR8 cells (5×106) were counted and prepared as suspensions in 0.1 ml PBS for subcutaneous injections into 5-week-old female NOD-SCID mice purchased from the Jackson Laboratory (Bar Harbor, ME). The volume of tumor was measured three times per week according to the formula V = ½ (length × width2). When tumors reached 100 mm3, mice were randomized into 4 groups (n=6/group). Mice received vehicle or 8 mg/kg CHK1i prexasertib by intraperitoneal (i.p.) injection twice daily (BID) for 3 days, followed by 4 days of no treatment for a total of 3 weeks. For combination studies, 2 study arms were added, and mice received 7.5 mg/kg LY3023414 twice orally (PO, BID) for 5 days, followed by 2 days of no treatment with or without 8 mg/kg prexasertib by i.p. BID for 3 days, followed by 4 days of no treatment for 3 weeks. All mice were sacrificed at the end of treatments, and the xenografted tumors were harvested and assayed for subsequent experiments.
Statistical analysis
All experiments were performed at least in triplicate. Data were analyzed using one-way ANOVA test and shown as mean ± standard deviation (SD) or mean ± standard error of the mean (SEM). All differences were considered statistically significant if p < 0.05. The PFS of ovarian cancer patients was analyzed using Kaplan-Meier curves applying logrank (Mantel-Cox) test, the hazard ratio with 95% confidence intervals (Mantel-Haenszel method), and logrank p values were also calculated. All statistical analyses were done using GraphPad Prism v. 7.1 (GraphPad Software, LA Jolla, CA).
Results
High-throughput drug combination screening identifies PI3K/mTOR pathways as actionable targets for a CHK1i combination
An initial 6×6 pilot screen in PEO1 cells found that CHK1i prexasertib combined synergistically with multiple drugs across multiple pharmacological classes. We then set an arbitrary threshold of −120 to describe synergistic combinations using the ExcessHSA metric. This revealed a set of 192 drugs (10%) of the United States (U.S.) Food and Drug Administration-approved and investigational drugs in our collection (Fig. 1A). There were several enriched signatures including 13 of the 24 drugs listed as mTOR inhibitors (54%), i.e., sirolimus (rank 6) and everolimus (rank 9), and 14 of 42 agents (33%) listed as PI3K inhibitors, i.e., copanlisib (rank 106) (Supplementary Table S1).
Figure 1. Inhibition of PI3K/mTOR induces synergistic cytotoxic effects with a CHK1 inhibitor by causing DNA damage and cell apoptosis.
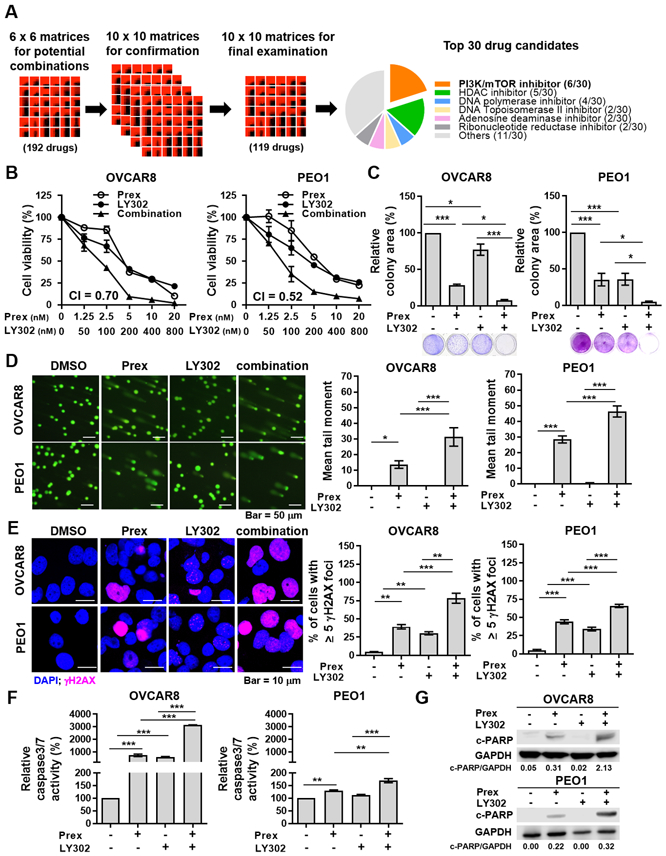
A, The workflow of drug combination screen in BRCA wild-type (BRCAwt, OVCAR5, OVCAR8 and PEO4) and BRCA2 mutant (BRCA2m, PEO1) HGSOC cells. 20% of top 30 drugs that showed synergistic cytotoxic effects with a CHK1 inhibitor prexasertib (Prex) were PI3K/mTOR pathway inhibitors. B, Cell viability was examined using XTT assay. Cells were treated with Prex, PI3K/mTOR inhibitor LY3023414 (LY302) or in combination at indicated doses for 72 hours. The combination index (CI) quantitatively depicts synergism (CI < 1). C, Cells were seeded at low density and treated with Prex (5 nM) and/or LY302 (200 nM) grown for 7 days in normal serum medium. Colonies were visualized by 0.01% (w/v) crystal violet staining. Quantification was performed by ImageJ software with ColonyArea plugin. D, Cells were treated with Prex (5 nM) and/or LY302 (200 nM) for 48 hours. DNA damage was examined by alkaline comet assay. At least 100 cells events in each treatment were quantified using CometScore© software. Representative images are shown (left) and the mean tail moment which considers both the migration of the genetic material as well as the relative amount of DNA is plotted (right). Scale bar is 50 μm. E, Cells were assessed by staining for γH2AX foci (pink) and confocal immunofluorescence imaging. Cell nuclei were stained with DAPI (blue). Representative images were taken at x 63 magnification and scale bar is 10 μm (left). The percentage of cells with ≥ 5 γH2AX foci representing cells with DNA damage is plotted (right). F-G, Cell apoptosis effect was assessed by caspases 3/7 activity assay (F) and Western blotting of cleaved PARP (c-PARP) and GAPDH (G). Densitometric values of c-PARP relative to GAPDH are shown. All data are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Next, we conducted a series of 10×10 screening experiments in both BRCAwt and BRCA2m HGSOC cell lines (Supplementary Tables S2-6). Several of these experiments highlighted strong combination effects when prexasertib was combined with chemotherapeutic drugs (e.g., gemcitabine) (Supplementary Tables S2-5). We deprioritized chemotherapeutic drugs when considering candidates for translation into human clinical trials to avoid overlapping hematological side effects with a CHK1i. A final examination in the PEO1 cell model evaluated 119 drug combinations using each previous round of evaluation to enrich for the most promising agents (Supplementary Table S6). This experiment revealed that the PI3K/mTOR pathway inhibitors were among the top-ranked drug candidates (Fig. 1A). We therefore proceeded with a PI3K/mTOR pathway inhibitor for further in vitro and in vivo studies with CHK1i.
Combined inhibition of CHK1 and PI3K/mTOR pathways shows synergistic cytotoxic effects in multiple HGSOC cell lines independent of BRCA mutation status
LY3023414 (LY302), a dual PI3K/mTOR inhibitor (PI3K/mTORi), was chosen as a test compound for in vitro validation as LY302 has been studied both clinically and preclinically in multiple cancer types (20,21) including gynecologic malignancy (22). IC50 values for prexasertib and LY302 monotherapies ranged from 6.1 nM to 53.2 nM, and 217.8 to 725.7 nM in five BRCA-proficient (OVCAR3, OVCAR5, OVCAR8, OV90, and PEO4) and one BRCA-deficient (PEO1) HGSOC cell lines, respectively (Supplementary Fig. S1A). BRCAwt OVCAR8 and BRCA2m PEO1 cells are shown in the main figures as representative models. Combination therapy using clinically attainable concentrations (20,23) of CHK1i prexasertib (5 nM) and PI3K/mTORi LY302 (200 nM) showed additively or synergistically attenuated cell viability (combination index [CI] < 1) compared with each monotherapy in all 6 HGSOC cells (Fig. 1B and Supplementary Fig. S1B). Similarly, dual inhibition of CHK1 and PI3K/mTOR significantly decreased the colony-forming ability of all HGSOC cell lines compared with each drug alone (Fig. 1C and Supplementary Fig. S1C).
We next examined on-target effects of prexasertib and/or LY302 treatments using clinically attainable concentrations. 5 nM of prexasertib attenuated the expression of active CHK1 (p-CHK1 S296), consistent with previous reports (24), and 200 nM of LY302 decreased the levels of downstream targets of PI3K/mTOR pathway such as phosphorylation of S6 ribosomal protein (p-S6) and 4E-BP1 (p-4E-BP1) (13) (Supplementary Fig. S1D). In addition, LY302 (200 nM) monotherapy did not change the levels of active CHK1 or total CHK1 proteins in both OVCAR8 and PEO1 cells (Supplementary Fig. S1D).
To determine whether the CHK1 and PI3K pathway interactions caused underlying synergistic cytotoxicity, we first examined the cell survival by using other inhibitors that also target CHK1 or PI3K pathways and siRNAs against CHEK1, PIK3CA or MTOR. Consistent with the results of prexasertib and LY302 combination, the combinations of another CHK1/2i (AZD7762) or a highly selective CHK1i (CCT245737) (25) with either LY302, a PI3K inhibitor (AZD8186) or an mTOR inhibitor (AZD2014) (Supplementary Fig. S2A-E) showed synergistic cytotoxic effects (CI < 1) in all HGSOC cell lines regardless of their BRCA mutation status. Moreover, silencing PIK3CA or MTOR in combination with CHEK1 knockdown significantly decreased cell viability compared to CHEK1, PIK3CA or MTOR knockdown alone (Supplementary Fig. S2F) indicating the synergistic cytotoxicity of CHK1i and PI3K/mTORi is a pathway-specific effect rather than drug’s off-target effects. We also knocked down PRKDC which encodes DNA-dependent protein kinase (DNA-PK) in cells transfected with siRNAs against CHEK1 since LY302 has been reported to inhibit the activity of DNA-PK (21). Knockdown of DNA-PK alone reduced the cell viability; however, it did not further increase cytotoxicity when combined with CHEK1 depletion (Supplementary Fig. S2F), indicating that the effect of LY302 on synergistic cytotoxicity is likely driven by PI3K/mTOR inhibition rather than DNA-PK inhibition.
Dual CHK1 and PI3K/mTOR inhibition induces greater DNA damage and apoptosis in HGSOC cells
Next, we performed DNA comet assay and immunofluorescence staining of γH2AX foci to examine if the observed greater cytotoxicity in the combination therapy was caused by DNA damage. Prexasertib alone induced greater DNA damage, while LY302 monotherapy showed no difference compared to the control (Fig. 1D and Supplementary Fig. S3A). We found the mean comet tail moment was further increased in cells treated with the combination, using clinically achievable concentrations of both drugs (prexasertib [5 nM] and LY302 [200 nM]) (20,23), compared with monotherapy (Fig. 1D and Supplementary Fig. S3A). The combination of CHK1i and PI3K/mTORi also showed increased percentage of cells with ≥ 5 γH2AX foci, indicating cells with DNA damage, compared with either drug alone (Fig. 1E and Supplementary Fig. S3B). We also noted the increased percentage of cells with pan-γH2AX staining by the combination treatment, suggesting more apoptotic cells (26) (Supplementary Fig. S3C).
We performed caspase 3/7 activity assay and Western blot analysis of cleaved PARP (c-PARP) to evaluate if the combination therapy caused cell death through apoptosis. Greater activation of caspase 3/7 (Fig. 1F and Supplementary Fig. S3D) and c-PARP expressions (Fig. 1G and Supplementary Fig. S3E) were observed with the combination compared to each monotherapy. Together, our findings indicate the CHK1i and PI3K/mTORi combination induces greater DNA damage leading to apoptosis and cell death relative to monotherapy alone in HGSOC cells.
Dual inhibition of CHK1 and PI3K/mTOR pathways causes massive accumulation of small condensed chromosomal fragments, increased micronuclei and mitotic catastrophe
To investigate whether prexasertib/LY302 combination would cause direct DNA damage on chromosomes, a metaphase spread assay was done given that CHK1 regulates the intra-S and G2/M checkpoints to prevent cells with damaged or incompletely replicated DNA from entering mitosis (6,7). HGSOC cells were treated with monotherapy or combination for 48 hours in the presence of pan-caspase inhibitor zVAD to exclude apoptosis-driven DNA fragmentation. CHK1i and PI3K/mTORi combination induced dramatic fragmentation of chromosomes compared to each monotherapy (Fig. 2A and Supplementary Fig. S4A). Of note, additional caspase inhibition did not alleviate this fragmentation, indicating it was caused by DNA damage but unlikely by the induction of apoptosis (Fig. 2A and Supplementary Fig. S4A). Micronuclei are generally formed from mis-segregated or chromosome fragments indicating replication stress in early S phase (27) and abnormal mitosis (28). Consistently, we noted increased micronuclei by combination treatment (Fig. 2B), suggesting chromosomal breakages were associated with increased replication stress and abnormal mitosis.
Figure 2. Inhibition of PI3K/mTOR pathway increases CHK1 inhibitor-induced chromosome breaks and mitotic cell death in HGSOC cells.
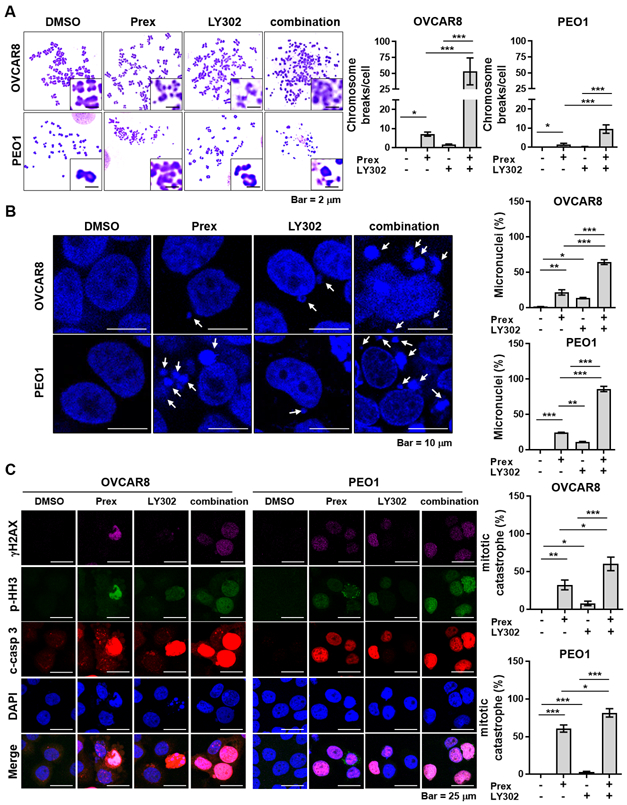
A, Cells were treated with prexasertib (Prex, 5 nM) and/or LY3023414 (LY302, 200 nM) for 48 hours in the presence of 20 μM zVAD pan-caspase inhibitor to exclude apoptosis-driven DNA fragmentation. Metaphase spreads were prepared as described in Supplementary Methods. A total of 35 metaphases were analyzed from each sample. Representative images of metaphase spreads are shown (left). Chromosomal breaks and fusions under each condition are plotted (right). Scale bar is 2 μm. B, Cells were treated with Prex (5 nM) and/or LY302 (200 nM) for 48 hours. Representative images of cells with micronuclei are shown (arrowheads, left). Percentage of cells containing micronuclei are plotted (right). Scale bar is 10 μm. At least ten field images were counted (> 100 cells). C, Cells were treated with Prex (5 nM) and/or LY302 (200 nM) for 48 hours. The DNA double-stranded break marker γH2AX (pink), mitotic marker phosphorylated histone H3 (p-HH3; green) and apoptosis marker cleaved caspase 3 (c-casp 3; red) were analyzed by immunofluorescence (left). Scale bar is 25 μm. Quantification of triple-positive cells as mitotic catastrophe was performed by ImageJ software and plotted (right). All data are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Cells with multiple micronuclei usually result from mitotic catastrophe, which is a type of cell death due to abnormal mitosis (28). A hallmark of mitotic catastrophe is the mitotic entry in the presence of unrepaired damaged DNA (28). We therefore examined the expressions of phosphorylated histone H3 (p-HH3, mitotic cell marker), γH2AX (DSB damage marker) and cleaved caspase 3 (c-casp-3, apoptosis marker) by immunofluorescence staining to investigate whether the chromosome breakages found in combination treatment were associated with increased mitotic catastrophe. Cells with γH2AX/p-HH3/c-casp-3 triple-positive stainings were considered as cells undergoing mitotic catastrophe (29-31). We found that a greater number of γH2AX/p-HH3/c-casp-3 triple-positive populations with combination therapy compared with each monotherapy, (Fig. 2C and Supplementary Fig. S4B), indicating lethal mitotic catastrophe caused by combination treatment.
Inhibition of PI3K/mTOR augments CHK1i-induced replication stress
CHK1i, prexasertib has been reported to increase origin firing, fork degradation and DNA damage in S phase, leading to accumulation of stalled forks, resulting in replication stress (32,33). Although the detailed mechanism is not fully understood, PI3K or mTOR inhibition has also shown increased DNA damage accumulation in S phase in p53-deficient triple-negative breast cancer (TNBC) cells (17). Therefore, we hypothesized that combined inhibition of the CHK1 and PI3K/mTOR pathway would exacerbate replication stress, leading to lethal DNA damage and cell apoptosis in HGSOC.
To test this hypothesis, we studied cell cycle using propidium iodide staining to investigate whether observed micronuclei in combination treatment was caused by increased replication stress in the early S phase. A greater portion of HGSOC cells treated with combination therapy arrested in S phase compared with cells treated with each monotherapy in OVCAR8 and PEO1 cells (Fig. 3A), suggesting possible fork stalling during S phase. Next, HGSOC cells were labeled with 5-Bromo-2’-deoxyuridine (BrdU) treated with drugs for 1 and 4 hours, and immunostained with anti-BrdU and anti-γH2AX antibodies to further determine if exposure to prexasertib and/or LY302 leads to DNA damage in the early S phase. Exposure of OVCAR8 and PEO1 cells to prexasertib and/or LY302 increased levels of γH2AX in S phase (Fig. 3B), suggesting that dual inhibition of CHK1 and PI3K/mTOR pathways may have arrested cells in S phase due to severe DNA damage (Fig. 3A).
Figure 3. PI3K/mTOR inhibitor augments CHK1 blockade-induced replication stress compared with monotherapy in HGSOC cells.
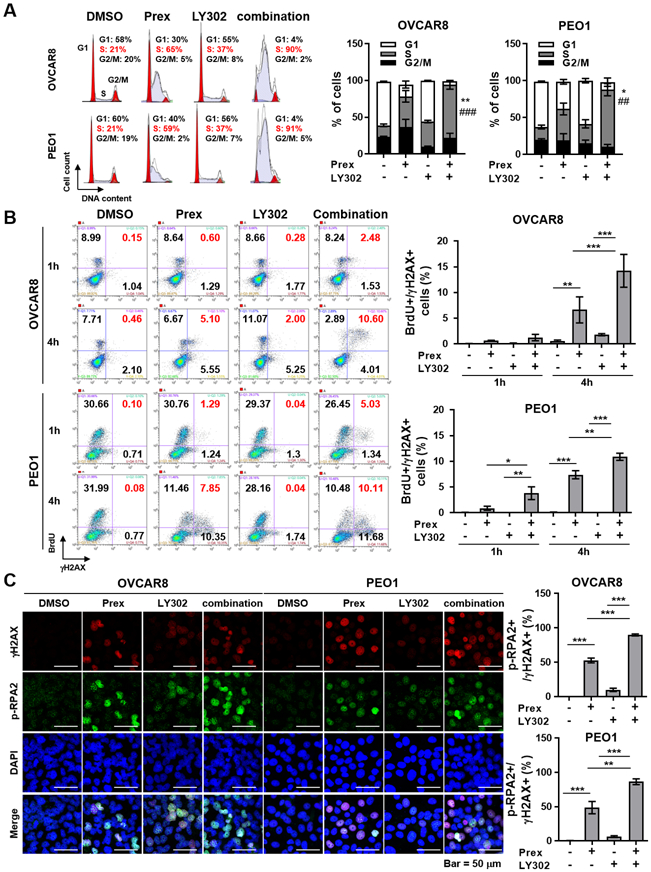
A, Cells were treated with prexasertib (Prex, 5 nM) and/or LY3023414 (LY302, 200nM) for 24 hours. Cells were fixed and stained with propidium iodide for cell cycle distribution analyzed using flow cytometry. Representative images of cell cycle distribution are shown (left). Percentage of cells in different phases is plotted (right). Data are shown as mean ± SD. * compared to Prex; *p < 0.05, **p < 0.01. # compared to LY302; ##, p < 0.01; ###, p < 0.001. B, Cells were treated with Prex (5 nM) and/or LY302 (200 nM) for 1 and 4 hours and incubated with BrdU for 1 hour. Cells were fixed and stained with anti-BrdU and anti-γH2AX using flow cytometry. Representative images are shown (left). Percentage of cells in S phase with DNA damage (γH2AX) is plotted (right). C, Levels of γH2AX (red; DNA double-stranded break marker) and p-RPA2 (S4/S8) (green; stalled replication forks marker) were analyzed by immunofluorescence. Representative images are shown (left) and scale bar is 50 μm. The percentage of double-positive cells suggesting replication stress is plotted (right). Data from B and C are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Excessive replication stress is lethal to cancer cells as it induces mitotic catastrophe (34). ATR/CHK1 signaling activation is therefore important to maintain genomic integrity by preventing the accumulation of excess ssDNAs. Under replication stress, phosphorylated replication protein A (p-RPA) proteins protect ssDNAs at stalled replication forks from nucleases and serve as an indicator of ssDNAs at stalled forks (35). To address whether the greater mitotic catastrophe was caused by increased replication stress, we performed immunofluorescence staining of γH2AX and p-RPA2 (S4/S8). PI3K/mTORi alone increased replication stress compared with control, and further augmented CHK1i-induced replication stress in HGSOC, as examined by increased γH2AX/p-RPA2 (S4/S8) double-positive populations (Fig. 3C). Together, our data show that dual inhibition of CHK1 and PI3K/mTOR augments replication stress, leading to chromosome breakage and mitotic catastrophe.
PI3K/mTORi potentiates CHK1i-induced replication stress by promoting unscheduled origin firing via increasing the levels of CDC45 on chromatin
CHK1 depletion has been shown to decrease the rates of replication fork progression via increased unscheduled origin firing in osteosarcoma (7), however, the role of PI3K/mTOR pathway on replication progression has been poorly understood. We therefore investigated the DNA replication fork dynamics to study whether the addition of PI3K/mTORi to CHK1i can further slow down replication progression. We first treated cells with monotherapy or in combination and then incubated cells with CldU and IdU to determine the replication progression rate. Replication fork progression speed was significantly slower with combination therapy compared to each monotherapy (Fig. 4A).
Figure 4. PI3K/mTOR inhibition enhances CHK1 blockade-induced replication stress by causing new origin firings through increased CDC45.
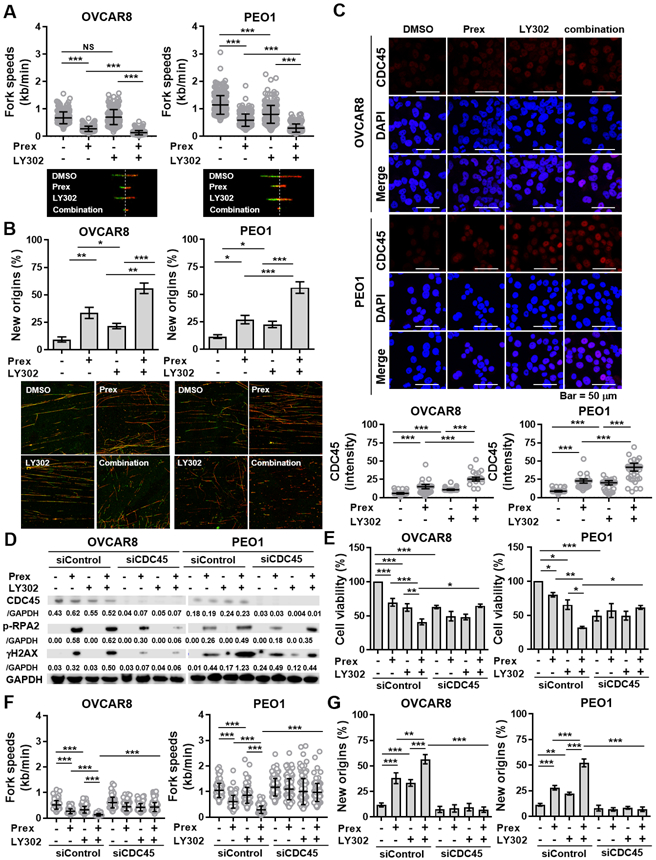
A-B, DNA fiber assays were performed to study alterations in replication fork dynamics, as described in Supplementary Methods. Cells were pretreated with prexasertib (Prex, 5 nM), LY3023414 (LY302, 200 nM) or in combination for 24 hours and then pulse labeled with CldU (green) and IdU (red) for 30 minutes each. Average replication fork speeds (top) and representative images of CldU and IdU replication tracks (bottom) in each group are shown (A). Relative percentage of cells with IdU only represent newly fired origins compared to control group are shown (B). C, Immunofluorescence staining of CDC45 (red) in cells treated with Prex (5 nM) and/or LY302 (200 nM) for 48 hours was conducted. Fluorescence intensity for CDC45 was quantified for at least 200 cells by using ImageJ and plotted. Scale bar is 50 μm. D, Cells were transfected with or without CDC45 siRNA and then treated with prexasertib (Prex, 5 nM) and/or LY3023414 (LY302, 200 nM) for 48 hours. Replication stress was measured by immunoblotting of γH2AX and p-RPA2 (S4/S8). Densitometric values of CDC45, γH2AX and p-RPA2 (S4/S8) relative to GAPDH are shown. E, Cell viability was examined using XTT assay. Cells were treated with Prex, LY302 or in combination at indicated doses with CDC45 knockdown for 72 hours. F-G, DNA fiber assays were performed to study replication speed (F) and new origin firing (G) in cells with CDC45 knockdown and treated with Prex and/or LY302. All data are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Origin firing is the initiation of DNA replication that takes place at the replication origins (36). Increased new origin firings lead to the depletion of nucleotides for DNA synthesis and thus decelerate the fork progression (36). Therefore, this observed effect of CHK1i and PI3K/mTORi combination on delayed S phase and replication progression prompted us to hypothesize that combination treatment would increase new origin firings, thus slowing down DNA replication progression, leading to replication stress. We therefore measured the percentage of IdU-labeled fibers only which represent the newly fired origins. Notably, the PI3K/mTOR inhibition alone increased new origin firings compared to the untreated group (Fig. 4B). Moreover, with combination treatment, HGSOC cells showed significantly augmented unscheduled origin firings compared to monotherapy (Fig. 4B) supporting the concept that PI3K/mTORi can further delay the CHK1i-induced replication progression (Fig. 4A).
CDC45 is a rate-limiting factor for the firing of active replication origins (37). CHK1 inhibition induces CDC45 accumulation at unscheduled fired origins and ongoing replication forks (32). We thus examined the CDC45 expression levels using immunofluorescence to evaluate whether PI3K/mTORi can further enhance CHK1i-induced origin firings via regulating CDC45 loading. We noted increased CDC45 expression in cells treated with CHK1i and PI3K/mTORi combination compared to those treated with monotherapy (Fig. 4C).
CDC45 knockdown rescues cells from CHK1i and PI3K/mTORi combination-induced replication stress and cell death by reducing new origin firings
Next, we transfected the cells with siRNA against CDC45 and then treated them with prexasertib and/or LY302 to investigate the role of CDC45 on this augmented unscheduled origin firings by combination treatment. As shown in Fig. 4D, exposure to either CHK1i or CHK1i and PI3K/mTORi combination induced p-RPA2 (S4/S8) and γH2AX. The effects were partially rescued by CDC45 knockdown in both BRCAwt and BRCA2m cells. Further, cell viabilities were reduced after treated with prexasertib and/or LY302, whereas rescued after CDC45 knockdown (Fig. 4E). We then performed DNA fiber assays in CDC45-knockdown cells treated with prexasertib and/or LY302 to further study whether CDC45 is a key player of increased origin firings and reduced replication progression by combination therapy. While there was no significant change of replication speed between cells transfected with control siRNA and CDC45 siRNA, the observed reduced replication speed and increased new origins by CHK1i and PI3K/mTORi combination were reversed by CDC45 knockdown (Figs. 4F and 4G).
In addition, we preformed DNA fiber assays to investigate whether PI3K/mTORi enhances CHK1i-induced replication stress via increasing both unscheduled origin firing and fork destabilization. PI3K/mTORi did not augment CHK1i-induced fork destabilization when compared to CHK1i treatment alone (Supplementary Fig. S5A), suggesting fork degradation is unlikely the main mechanism underlying CHK1i and PI3K/mTORi combination.
Inhibition of PI3K/mTOR pathway increases CHK1i-induced DNA damage by attenuating HR repair, thus further worsening replication stress
Replication stress is aggravated when DNA repair process is interfered (38). CHK1 inhibition impairs HR repair by attenuating transnuclear localization of RAD51 which subsequently leads to augmented replication stress (10). Similarly, the suppression of PI3K function impairs HR via down-regulating BRCA2 or RAD51 in BRCAwt HGSOC cell lines (39). We therefore assessed whether combination treatment would decrease HR functionality in HGSOC cells. The DRGFP reporter system-based assay was adapted to assess the HR function (40). We co-transfected a DRGFP-expressing and an I-Sce1 expression plasmid to induce DSBs in cells treated with CHK1i and/or PI3K/mTORi. Cells were analyzed for GFP expression by flow cytometry and we observed significant HR deficiency in cells with combination therapy compared to monotherapy (Fig. 5A).
Figure 5. Combined PI3K/mTOR and CHK1 inhibition mitigates RAD51-mediated HR repair in HGSOC cells.
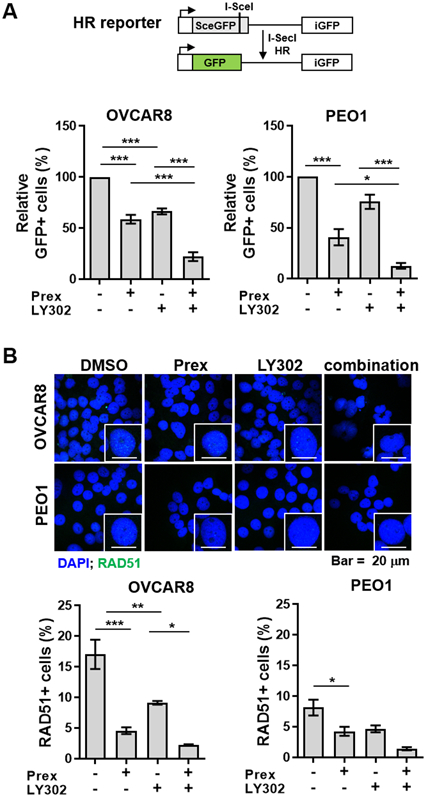
A, Combination of CHK1 and PI3K/mTOR pathway inhibitors attenuated homologous recombination (HR) repair activity in a DRGFP reporter assay in both OVCAR8 and PEO1 cells. GFP-positive cells were analyzed by flow cytometry. B, Cells were treated with prexasertib (Prex, 5 nM) and/or LY3023414 (LY302, 200 nM) following the PARP inhibitor olaparib (5 μM) treatment for 48 hours. Immunofluorescence staining of RAD51 foci (green) was conducted for HR repair activity. Representative images were taken at x 63 magnification and scale bar is 20 μm (up). Cells with ≥ 5 RAD51 foci were counted as RAD51+ cells and the percentage of RAD51+ cells is plotted (bottom). All data are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Also, we evaluated RAD51 foci formation in BRCAwt OVCAR8 cells following DNA damage induced by 5 μM PARP inhibitor olaparib. Notably, we observed a significant reduction in nuclear RAD51 foci formation in OVCAR8 cells with CHK1i and PI3K/mTORi combination compared to each drug alone (Fig. 5B), suggesting attenuated DNA repair through HR. We also found a trend of reduced RAD51 foci in combination therapy in comparison with monotherapy in BRCA2m PEO1 cells despite the relative deficiency of BRCA2 function in this cell line (Fig. 5B). We further confirmed decreased RAD51 protein levels in both BRCAwt and BRCA2m HGSOC cells treated with CHK1i and PI3K/mTORi combination (Supplementary Fig. S5B). Together, PI3K/mTORi accelerates CHK1i-induced DNA damage in part by attenuating HR repair, thus promoting lethal replication stress.
Combined inhibition of both CHK1 and PI3K/mTOR pathway decreases tumor growth and increases replication stress in vivo
Lastly, the NOD-SCID mice were subcutaneously implanted with BRCAwt OVCAR8 cells to confirm the efficacy of CHK1i and PI3K/mTORi in in vivo. Our in vivo data showed that CHK1i and PI3K/mTORi combination reduced tumor growth (Fig. 6A) without significant weight loss (Fig. 6B). Western blotting and IHC staining were performed to study whether combined CHK1i and PI3K/mTORi would increase replication stress and DNA damage via CDC45 in in vivo. Consistent with in vitro findings, the expression levels of p-RPA2 (S4/S8), γH2AX and CDC45 were significantly elevated in tumors treated with combination treatment compared to monotherapy (Figs. 6C and 6D). Together, these findings demonstrate the therapeutic potential of combined inhibition of CHK1 and PI3K/mTOR in HGSOC in vivo.
Figure 6. Dual inhibition of PI3K/mTOR and CHK1 suppresses tumor growth accompanied by high levels of replication stress in vivo and CDC45 combined with replication stress markers shows potential prognostic impacts in patients with ovarian cancer.
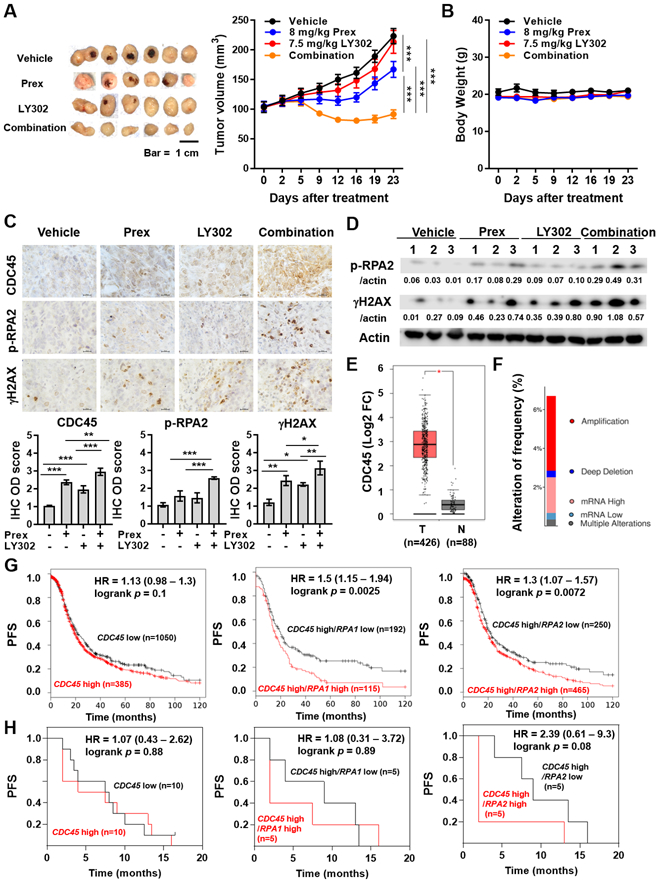
A-B, Combination efficacy of prexasertib (Prex) with LY3023414 (LY302) on tumor growth in OVCAR8 HGSOC xenograft tumors. Dosing schedules are described in Methods. Tumor volume (A) and body weight (B) are plotted. Scale bar is 1 cm. Data are shown as mean ± SEM. ***, p < 0.001. C-D, Tumor tissues from A were collected. C, Representative IHC results for CDC45, p-RPA2 (S4/S8) and γH2AX. All images were taken at x 60 magnification and scale bar is 20 μm (top). Quantification was performed by ImageJ software with IHC Profiler plugin as described in Methods. The IHC optical density (OD) scores of nuclear CDC45, replication stress marker p-RPA2 (S4/S8) and DSBs marker γH2AX are plotted (bottom). Data are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, Immunoblotting results and densitometric values of p-RPA2 (S4/S8) and γH2AX relative to β-actin are shown. E, The boxplots present the expression of CDC45 in serous ovarian cancer tissues and normal ovary tissue were produced by the Gene Expression Profiling Interactive Analysis online platform (GEPIA, http://gepia.cancer-pku.cn). *, p < 0.05. F, The alteration frequency of CDC45 in patients with serous ovarian cancer from The Cancer Genome Atlas (TCGA, Firehose Legacy) was determined using cBioPortal (http://www.cbioportal.org). The alteration frequency included amplification (red), deep deletion (blue), mRNA high (pink), mRNA low (benzo blue) and multiple alterations (gray). G, The prognostic value of CDC45 was obtained from Kaplan-Meier plotter (KM plotter, http://kmplot.com/analysis/) [ovarian cancer] databases. Ovarian cancer patients with high CDC45 levels (n=1,050) and low CDC45 levels (n=385) were divided using auto select best cutoff shown on the website (left). Patients with high CDC45 levels were further divided into high- and low-expression groups based on the median expression of RPA1 (middle) or RPA2 (right) using multiple genes analysis. The PFS of ovarian cancer patients during 120-month follow-up was analyzed by KM plotter website, and the hazard ratio with 95% confidence intervals and logrank p values were calculated. H, RNA-seq datasets were performed using pretreatment biopsy samples from 20 recurrent HGSOC patients (5). According to the median expression of CDC45, HGSOC patients were divided into high- (n=10) and low-expression (n=10) groups (left). HGSOC patients with high CDC45 levels were further divided into high- and low-expression groups of RPA1 (middle) or RPA2 (right) based on their median expression levels.
Co-occurrence of high CDC45 with high RPA is associated with poor prognosis in ovarian cancer patients
To explore whether differential levels of CDC45 expression could be used as a biomarker for patient selection for combination therapy, we analyzed mRNA expression, gene alteration and prognostic impact of CDC45 from cBioPortal, Gene Expression Profiling Interactive Analysis (GEPIA) and Kaplan-Meier plotter [Ovarian Cancer] databases. The expression of CDC45 was upregulated in ovarian tumor tissues compared to those of normal ovary tissues (Fig. 6E) and CDC45 amplification/overexpression was found in approximately 5.6% (34/606) of serous ovarian cancer (Fig. 6F). Furthermore, survival analysis showed a trend of poor prognosis in ovarian cancer patients with high CDC45 levels (n=1,050) compared to those with low CDC45 levels (n=385) using Kaplan-Meier plotter [Ovarian Cancer] databases (median PFS 19.13 vs 21.29 months) (Fig. 6G). We thus investigated other replication factors interacting with CDC45 such as RPA (41,42). RPA1 or RPA2 expression alone did not correlate with prognosis in ovarian cancer (Supplementary Fig. 6). However, ovarian cancer patients with high/high co-expressions of replication stress markers, i.e., CDC45/RPA1 (median PFS 15.01 vs 19.27 months; logrank p = 0.0025) or CDC45/RPA2 (median PFS 19.13 vs 22 months; logrank p = 0.0072) were associated with worse PFS, representing subgroups of patients with unmet medical need (Fig. 6G).
We also analyzed RNA-seq datasets from pretreatment biopsy samples of 20 recurrent HGSOC patients on CHK1i prexasertib monotherapy clinical trial (11). The HGSOC patients were divided into high- (n=10) and low-expression (n=10) groups based on the median expression of CDC45. Similarly, our data also showed a trend of poor prognosis in recurrent HGSOC patients with high CDC45 levels compared to those with low CDC45 expressions (median PFS 5.75 vs 7.75 months) (Fig. 6H). We also found that HGSOC patients with high/high co-expressions of CDC45/RPA1 or CDC45/RPA2 (median PFS 2 vs 9 months and median PFS 2 vs 9 months, respectively) showed a trend of worse PFS (Fig. 6H). Taken together, these data suggest that co-expression patterns of high CDC45 and high RPA might represent a subgroup of ovarian cancers with high replication stress which may benefit from the combination treatment.
Discussion
Here, we demonstrate that inhibition of PI3K/mTOR pathway augments CHK1i-induced replication stress and acts synergistically in HGSOC in vitro and in vivo models, representing a new therapeutic opportunity for HGSOC. Mechanistically, combined inhibition of CHK1 and PI3K/mTOR pathways interferes with successful DNA replication by causing excess new origin firings via increasing CDC45, leading to replication stress, genomic instability and consequent mitotic catastrophe. The combination of CHK1i and PI3K/mTORi also increases DNA damage via impairing RAD51 mediated-HR repair, thus further enhancing lethal replication stress and cell death. Another key strength of the current study lies in the use of clinically attainable concentrations of two inhibitors and commonly used validated in vitro and in vivo ovarian cancer models (21,43).
There are emerging data demonstrating synergistic cytotoxicity of inhibition of cell cycle and PI3K pathways in various tumor types (15,44), although the mechanisms underlying synergistic effect require further investigation. In TNBC, Chopra et al. reported that mTOR inhibition increased an ATR inhibitor AZ20 or CHK1i rabusertib-induced replication stress in PI3K-activated TNBC cell models with underlying PIK3CA mutation (H1047R) or low levels of PTEN and/or INPP4B (16). In the present study, we demonstrated augmented replication stress in HGSOC cell lines that do not have PIK3CA-activating mutation or PTEN loss, thus representing broader applicability of this combination (45,46). Furthermore, we found increased micronuclei by the combination treatment which prompted us to speculate that these micronuclei may be partly associated with immune-response signaling (47), requiring further investigation. Overall, our results in HGSOC models align with those findings in different tumor types, indicating that the synergy between CHK1 and PI3K/mTOR inhibition may be ubiquitous.
In addition, our findings provide novel insights into the mechanisms of the PI3K pathway inhibition on replication stress via CDC45 in HGSOC although CHK1 inhibition-mediated CDC45 overexpression has been previously reported in other tumor types (48,49). CDC45 acts as a proliferation-associated antigen, promoting the rapid division of cancer cells (50). Our findings demonstrated that CDC45 knockdown by siRNAs attenuated DNA damage, RPA phosphorylation and rescued cell death in HGSOC cells from combination therapy, supporting a major role of CDC45 in the highly regulated replication process. We also found that CDC45 gene was upregulated in a subgroup of serous ovarian cancer tissues and co-expression of high CDC45 and high RPA was associated with worse PFS. Hence, we hypothesize that dual inhibition of CHK1 and PI3K/mTOR pathways may benefit this subgroup of HGSOC patients by inducing excess origin firings and lethal replication stress, and may be applied as a biomarker for optimal patient selection, requiring further validation in prospective clinical trials.
It is possible that other factors, i.e., CDKs and CDC7, may have played a role in this unscheduled origin firing resulting in synergistic cytotoxicity by the combination treatment because they also participate in DNA replication by assembling CDC45, MCM2-7 and GINS (CMG complex) at the origins and by activating the replicative helicases (51). As such, the CHK1/2 inhibitor (AZD7762) induces CDK1-mediated RIF1 phosphorylation and disrupts the interaction between RIF1 and PP1 at licensed origins, leading to increased dormant origin firing and replication stress in U2OS osteosarcoma cells (52). In TNBC cell lines, CHK1 inhibition also causes over-activation of CDC7- or CDK2-mediated origin firing, leading to augmented replication stress (7,53). Further mechanistic studies of the interactions between these factors with a CHK1i and PI3K/-based therapy are needed in the context of HGSOC.
In this study, we found that most HGSOC cells treated with prexasertib were arrested in S phase. We speculate that this S phase arrest was likely due to a simultaneously activated DNA replication machinery and decreased HR DNA repair response by CHK1i, all leading to prolonged accumulation of cells in the S phase for optimal DNA replication before entering the mitosis. Similar observations have been made in head and neck cancer cell lines (54) demonstrating that prexasertib induced an increased S phase population and less DNA synthesis. Other groups also noted that the prexasertib causes cells predominantly arrested in the S phase and speculated this S phase arrest was likely related to DNA damage response (32,54-56).
Lastly, HR repair is involved in DNA replication by protecting, restarting and repairing dysfunctional stalled forks through loading RAD51 to the ssDNAs at replication forks (57). Therefore, the unresolved replication intermediates and un-replicated DNA persist and some of them enter into mitosis when HR repair is impaired (57), leading to lethal replication stress and mitotic catastrophe. PI3K indirectly regulates the recruitment of RAD51 to foci of DNA damage via promoting the transcription of BRCA genes (58). Suppression of the PI3K/mTOR pathway increases γH2AX in concert with a decrease of RAD51 in PIK3CA-mutated and wild-type ovarian cancer cell lines as well as in vivo models (39,59). Consistently, we showed that CHK1i and PI3K/mTORi acted synergistically to attenuate HR activity as well as DSB-induced RAD51 foci formation, thus potentiating lethal replication stress in HGSOC.
In summary, we demonstrate the therapeutic potential of combining CHK1i with PI3K/mTORi in HGSOC models and combination treatment is tolerable in mouse tumors. Here, we report the novel mechanistic insights how CHK1i and PI3K/mTORi combination leads to synergistic DNA damage and anti-tumor activity in HGSOC through augmenting replication stress via CDC45-mediated origin firing and reduced HR. Together, our study provides a clear rationale for CHK1i and PI3K/mTORi-based clinical trials, aids the development of new treatment options and biomarkers, and ultimately results in improved clinical outcomes for women with recurrent HGSOC.
Supplementary Material
Significance.
Dual inhibition of CHK1 and PI3K/mTOR pathways yields potent synthetic lethality by causing lethal replication stress and DNA damage in high-grade serous ovarian carcinoma, warranting further clinical development.
Acknowledgments
We are grateful to Dr. Langston Lim (Confocal Microscopy Core Facility, CCR, NCI, NIH) for confocal microscopy analyses; Dr. Keli Agama and Dr. Yves Pommier (NCI/NIH) for their invaluable advice on DNA fiber assay; Dr. Yeajin Song (NINDS/NIH) for advising on tumor tissue sections; Dr. Rajarshi Guha for help with data loading and analysis; Dr. Simone Difilippantonio and Ms. Stacy Stocks (Animal Research Technical Support and Gnotobiotics Facility, Frederick National Laboratory for Cancer Research, NIH) for helping with animal studies; Dr. Seth Steinberg for sharing his expertise on statistical analysis. This research was fully funded by the intramural research program of the NIH, National Cancer Institute (NCI), Center for Cancer Research (CCR; Grant No. ZIA BC011525 [JL]), USA. The drug combination screening work was supported by the intramural research programs of the National Center for Advancing Translational Sciences and the CCR of NCI, USA (Grant No. 1ZIATR000047-05 [CJT]).
Financial support: This work was supported by the Intramural Research Program of the National Cancer Institute (NCI) (JL, #ZIA BC011525), Center for Cancer Research (CCR) and the National Center for Advancing Translational Sciences, CCR, NCI (CJT, #1ZIATR000047-05).
Footnotes
Competing interests: The authors declare no competing interests.
References
- 1.Lheureux S, Gourley C, Vergote I, Oza AM. Epithelial ovarian cancer. Lancet (London, England) 2019;393:1240–53 [DOI] [PubMed] [Google Scholar]
- 2.O'Sullivan CC, Moon DH, Kohn EC, Lee JM. Beyond Breast and Ovarian Cancers: PARP Inhibitors for BRCA Mutation-Associated and BRCA-Like Solid Tumors. Front Oncol 2014;4:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 2011;12:852–61 [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim H, George E, Ragland R, Rafail S, Zhang R, Krepler C, et al. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin Cancer Res 2017;23:3097–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao H, Ji F, Helleday T, Ying S. Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petermann E, Woodcock M, Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci U S A 2010;107:16090–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moiseeva T, Hood B, Schamus S, O'Connor MJ, Conrads TP, Bakkenist CJ. ATR kinase inhibition induces unscheduled origin firing through a Cdc7-dependent association between GINS and And-1. Nat Commun 2017;8:1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 2008;27:3977–85 [DOI] [PubMed] [Google Scholar]
- 10.Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 2005;7:195–201 [DOI] [PubMed] [Google Scholar]
- 11.Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol 2018;19:207–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin Cancer Biol 2019 [DOI] [PubMed] [Google Scholar]
- 13.Huang TT, Lampert EJ, Coots C, Lee JM. Targeting the PI3K pathway and DNA damage response as a therapeutic strategy in ovarian cancer. Cancer Treat Rev 2020;86:102021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selvarajah J, Elia A, Carroll VA, Moumen A. DNA damage-induced S and G2/M cell cycle arrest requires mTORC2-dependent regulation of Chk1. Oncotarget 2015;6:427–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koppenhafer SL, Goss KL, Terry WW, Gordon DJ. mTORC1/2 and Protein Translation Regulate Levels of CHK1 and the Sensitivity to CHK1 Inhibitors in Ewing Sarcoma Cells. Molecular Cancer Therapeutics 2018;17:2676–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chopra SS, Jenney A, Palmer A, Niepel M, Chung M, Mills C, et al. Torin2 Exploits Replication and Checkpoint Vulnerabilities to Cause Death of PI3K-Activated Triple-Negative Breast Cancer Cells. Cell Syst 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silvera D, Ernlund A, Arju R, Connolly E, Volta V, Wang J, et al. mTORC1 and −2 Coordinate Transcriptional and Translational Reprogramming in Resistance to DNA Damage and Replicative Stress in Breast Cancer Cells. Mol Cell Biol 2017;37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ceribelli M, Kelly PN, Shaffer AL, Wright GW, Xiao W, Yang Y, et al. Blockade of oncogenic IkappaB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc Natl Acad Sci U S A 2014;111:11365–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A 2014;111:2349–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bendell JC, Varghese AM, Hyman DM, Bauer TM, Pant S, Callies S, et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin Cancer Res 2018;24:3253–62 [DOI] [PubMed] [Google Scholar]
- 21.Smith MC, Mader MM, Cook JA, Iversen P, Ajamie R, Perkins E, et al. Characterization of LY3023414, a Novel PI3K/mTOR Dual Inhibitor Eliciting Transient Target Modulation to Impede Tumor Growth. Mol Cancer Ther 2016;15:2344–56 [DOI] [PubMed] [Google Scholar]
- 22.Rubinstein MM, Hyman DM, Caird I, Won H, Soldan K, Seier K, et al. Phase 2 study of LY3023414 in patients with advanced endometrial cancer harboring activating mutations in the PI3K pathway. Cancer 2020;n/a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol 2016;34:1764–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nair J, Huang TT, Murai J, Haynes B, Steeg PS, Pommier Y, et al. Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer. Oncogene 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walton MI, Eve PD, Hayes A, Henley AT, Valenti MR, De Haven Brandon AK, et al. The clinical development candidate CCT245737 is an orally active CHK1 inhibitor with preclinical activity in RAS mutant NSCLC and Emicro-MYC driven B-cell lymphoma. Oncotarget 2016;7:2329–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Feraudy S, Revet I, Bezrookove V, Feeney L, Cleaver JE. A minority of foci or pan-nuclear apoptotic staining of gammaH2AX in the S phase after UV damage contain DNA double-strand breaks. Proc Natl Acad Sci U S A 2010;107:6870–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabatinos SA, Ranatunga NS, Yuan JP, Green MD, Forsburg SL. Replication stress in early S phase generates apparent micronuclei and chromosome rearrangement in fission yeast. Mol Biol Cell 2015;26:3439–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat 2001;4:303–13 [DOI] [PubMed] [Google Scholar]
- 29.Dietlein F, Kalb B, Jokic M, Noll EM, Strong A, Tharun L, et al. A Synergistic Interaction between Chk1- and MK2 Inhibitors in KRAS-Mutant Cancer. Cell 2015;162:146–59 [DOI] [PubMed] [Google Scholar]
- 30.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007;11:175–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, et al. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev 2009;23:1895–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, et al. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol Cancer Ther 2015;14:2004–13 [DOI] [PubMed] [Google Scholar]
- 33.Parmar K, Kochupurakkal BS, Lazaro JB, Wang ZC, Palakurthi S, Kirschmeier PT, et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin Cancer Res 2019;25:6127–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 2009;11:753–60 [DOI] [PubMed] [Google Scholar]
- 35.Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D, et al. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol 2014;206:493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prioleau MN, MacAlpine DM. DNA replication origins-where do we begin? Genes Dev 2016;30:1683–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu PY, Nurse P. Establishing the program of origin firing during S phase in fission Yeast. Cell 2009;136:852–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ubhi T, Brown GW. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res 2019;79:1730–9 [DOI] [PubMed] [Google Scholar]
- 39.Wang D, Li C, Zhang Y, Wang M, Jiang N, Xiang L, et al. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol Oncol 2016;142:548–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999;13:2633–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakaya R, Takaya J, Onuki T, Moritani M, Nozaki N, Ishimi Y. Identification of proteins that may directly interact with human RPA. J Biochem 2010;148:539–47 [DOI] [PubMed] [Google Scholar]
- 42.Szambowska A, Tessmer I, Prus P, Schlott B, Pospiech H, Grosse F. Cdc45-induced loading of human RPA onto single-stranded DNA. Nucleic Acids Res 2017;45:3217–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brill E, Yokoyama T, Nair J, Yu M, Ahn YR, Lee JM. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017;8:111026–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Massey AJ, Stephens P, Rawlinson R, McGurk L, Plummer R, Curtin NJ. mTORC1 and DNA-PKcs as novel molecular determinants of sensitivity to Chk1 inhibition. Mol Oncol 2016;10:101–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 2013;4:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanrahan AJ, Schultz N, Westfal ML, Sakr RA, Giri DD, Scarperi S, et al. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov 2012;2:56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017;548:466–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzalez Besteiro MA, Calzetta NL, Loureiro SM, Habif M, Betous R, Pillaire MJ, et al. Chk1 loss creates replication barriers that compromise cell survival independently of excess origin firing. EMBO J 2019;38:e101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo C, Kumagai A, Schlacher K, Shevchenko A, Shevchenko A, Dunphy WG. Interaction of Chk1 with Treslin negatively regulates the initiation of chromosomal DNA replication. Mol Cell 2015;57:492–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pollok S, Bauerschmidt C, Sanger J, Nasheuer HP, Grosse F. Human Cdc45 is a proliferation-associated antigen. FEBS J 2007;274:3669–84 [DOI] [PubMed] [Google Scholar]
- 51.Labib K How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev 2010;24:1208–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moiseeva TN, Yin Y, Calderon MJ, Qian C, Schamus-Haynes S, Sugitani N, et al. An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc Natl Acad Sci U S A 2019;116:13374–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Warren NJH, Eastman A. Inhibition of checkpoint kinase 1 following gemcitabine-mediated S phase arrest results in CDC7- and CDK2-dependent replication catastrophe. J Biol Chem 2019;294:1763–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Harten AM, Buijze M, van der Mast R, Rooimans MA, Martens-de Kemp SR, Bachas C, et al. Targeting the cell cycle in head and neck cancer by Chk1 inhibition: a novel concept of bimodal cell death. Oncogenesis 2019;8:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghelli Luserna Di Rora A, Iacobucci I, Imbrogno E, Papayannidis C, Derenzini E, Ferrari A, et al. Prexasertib, a Chk1/Chk2 inhibitor, increases the effectiveness of conventional therapy in B-/T- cell progenitor acute lymphoblastic leukemia. Oncotarget 2016;7:53377–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mani C, Jonnalagadda S, Lingareddy J, Awasthi S, Gmeiner WH, Palle K. Prexasertib treatment induces homologous recombination deficiency and synergizes with olaparib in triple-negative breast cancer cells. Breast Cancer Res 2019;21:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ait Saada A, Lambert SAE, Carr AM. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair (Amst) 2018;71:135–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmana J, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov 2012;2:1048–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang D, Wang M, Jiang N, Zhang Y, Bian X, Wang X, et al. Effective use of PI3K inhibitor BKM120 and PARP inhibitor Olaparib to treat PIK3CA mutant ovarian cancer. Oncotarget 2016;7:13153–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.