

Abstract
Objective.
Pristane-induced lupus is associated with nonresolving inflammation and deficiency of proresolving macrophages. Proresolving non-classical macrophages (NCM) are less responsive to type I interferon than classical (proinflammatory) macrophages (CM) reflecting their relative expression levels of the type I interferon receptor (IFNAR). This study focused on the regulation of IFNAR expression in macrophages.
Methods.
We carried out gene expression profiling of purified CM and NCM from mice treated with pristane (develop lupus) or mineral oil (MO, non-lupus control). Macrophage differentiation and IFNAR expression were examined in mice treated with Nrf2 activators and inhibitors and in Nrf2-deficient mice. Nrf2 activity also was assessed in blood cells from SLE patients.
Results.
RNA-sequencing analysis revealed increased expression of genes regulated by the transcription factor Nrf2 in NCM from MO- vs. pristane-treated mice and in NCM vs. CM. The Nrf2 activator CDDO-imidazole (CDDO-Im) decreased CM (P < 0.0001) and promoted the development of proresolving NCM (p=0.08) (Student t-test), whereas the Nrf2 inhibitor brusatol increased CM (P < 0.05) and decreased NCM (P < 0.001) (Student t-test). CDDO-Im decreased Ifnar1(P < 0.001 by Student t-test). and interferon-stimulated gene (ISG) expression in macrophages and alleviated oxidative stress (P < 0.05 by Student t-test) , whereas brusatol had the opposite effect(P < 0.01 by Student t-test). Moreover, Ifnar1 and ISG expression was higher in Nrf2-knockout mice than controls (P < 0.05 by Student t-test). As seen in lupus mice, SLE patients showed evidence of low Nrf2 activity.
Conclusion.
We conclude that Nrf2 activation favors the resolution of chronic inflammation in lupus. Since autoantibody production and lupus nephritis depend on IFNAR signaling, the ability of Nrf2 activators to repolarize macrophages and reduce the interferon signature suggests that these agents may warrant consideration for treating lupus.
Introduction
Peripheral blood mononuclear cells (PBMCs) in most lupus patients express high levels of gene transcripts regulated by type I interferons (IFN-I) (1). In both humans (2, 3) and mice (4, 5), the “interferon signature” is related to the pathogenesis of organ involvement (e.g. nephritis) and the serological abnormalities (e.g. anti-Sm/RNP, dsDNA autoantibodies) of lupus. Consistent with that possibility, patients with interferonopathies often develop lupus-like disease (6) and mice deficient in the Ifnar1 subunit of the type I interferon receptor (IFNAR) (7), are resistant to the induction of lupus nephritis and autoantibodies by pristane (8).
Abnormal monocyte and macrophage (Mϕ) function also contributes to the pathogenesis of lupus due to impaired clearance of apoptotic cells and over-production of proinflammatory cytokines (9–11). Activated monocytes/Mϕ infiltrate the kidneys of patients with lupus nephritis and monocyte/Mϕ depletion protects pristane-treated mice from diffuse alveolar hemorrhage (DAH) and pulmonary vasculitis (12, 13). An unusual non-classical Mϕ (NCM) subset bearing the cell surface marker CD138 is associated with resolution of sterile peritoneal inflammation and protection from lupus in mineral oil (MO)-treated mice and is deficient in mice with nonresolving inflammation due to pristane (10). In contrast, peritoneal exudate cells (PEC) from pristane-treated mice consist primarily of proinflammatory Ly6Chi classical monocytes/Mϕ (CM).
CM are more responsive to IFN-I than NCM (14). The ability of monocyte/Mϕ to respond to IFN-I signaling correlates with IFNAR expression, but the mechanism(s) regulating IFNAR expression are unclear. We report that the transcription factor Nrf2, a key regulator of the cellular response to oxidative stress, also regulates IFNAR mRNA/protein expression.
Methods
Mice.
C57BL/6 (B6), B6.129X1-Nfe2l2 tm1Ywk/J (B6 Nrf2 KO), and B6.129S2-Ifnar1tm1Agt/Mmjax (B6 IFNARKO) mice (Jackson Laboratory) maintained under specific pathogen free conditions were injected i.p. with 0.5 ml of pristane or MO as described (14). There were 5-15 mice per group unless otherwise noted. Experiments were repeated at least twice. PEC were collected by lavage 3-14 days later (15). DAH was assessed as described (13). This study followed the recommendations of the Animal Welfare Act and US Government Principles for the Utilization and Care of Vertebrate Animals and was approved by the UF IACUC.
Patients.
Peripheral blood was collected in PAXgene tubes (BD Biosciences) from 14 patients with SLE and 10 healthy controls and RNA was isolated as described (15). These studies were approved by the University of Florida IRB.
Flow cytometry, cell sorting, and RNA isolation.
Flow cytometry was performed using anti-mouse CD16/32 (Fc Block; BD Biosciences) before staining with primary antibody or isotype controls. Cells were surface-stained, then fixed/permeabilized (Fix-Perm buffer, eBioscience) before intracellular staining for HSP70 and IRF7. Monoclonal antibodies are listed in Table S1. For flow-sorting, PEC were incubated with anti-CD11b-BV421, CD138-APC, Ly6C-Alexa Fluor 488, and Ly6G-APC-Cy7 antibodies (15). NCM (CD11b+CD138+Ly6C−Ly6G−) and CM (CD11b+CD138−Ly6ChiLy6G−) (10) were flow-sorted using a FACSAria cell sorter (3 mice/group, 30,000 cells/mouse). Cell purity was verified and RNA was isolated (RNeasy Microkit, Qiagen).
Gene expression profiling.
RNA sequencing (RNA-seq) was performed at Broad Institute using the Smart-Seq2 platform as described (14). Gene set enrichment analysis (GSEA) was performed using the Broad Institute GSEA Desktop v2.2.4 software and hallmark gene sets from the Molecular Signature Database (MSigDB v.6.2) (14). The RNA-seq data discussed in this publication are available from the corresponding author upon request.
Quantitative PCR (qPCR).
qPCR was performed as described (14) using RNA isolated from 106 mouse PEC or from human blood collected in PAXgene tubes. Gene expression was normalized to 18S RNA and expression was calculated using the 2−ΔΔCt method. Primer sequences are in Table S2.
Nrf2 binding to target sequence.
Nrf2 interacts with antioxidant response elements (AREs) of target genes (16, 17). To measure ARE binding activity, B6 mice were treated with pristane or MO and PEC were collected 7-d later. Nuclear extracts were purified using a Nuclear Extraction Kit (Cayman Chemical). DNA binding activity was determined using the Cayman Nrf2 Transcription Factor Assay Kit.
Mitochondrial superoxide and membrane potential.
Superoxide anion is the predominant reactive oxygen species (ROS) in mitochondria (18). To assess mitochondrial superoxide, PEC obtained 14-d after pristane- or MO- treatment were stained with the cell-permeant mitochondrial superoxide-specific indicator dye MitoSOX Red (ThermoFisher). MitoSOX staining was quantified as mean fluorescence intensity (MFI). Mitochondrial membrane potential (ΔΨm), a measure of mitochondrial and cell health, was assessed by staining with tetramethylrhodamine methyl ester (TMRM, ThermoFisher). After staining with MitoSOX or TMRM, cells were co-stained anti-CD11b, -Ly6C and -CD138 antibodies.
In vivo Nrf2 activator and inhibitor treatment.
The synthetic oleanane triterpenoid 1-[2-cyano-3, 12-dioxooleana-1,9(11)-dien-28-oyl]imidazole (CDDO-Im) (Tocris) modifies Cys151 of Keap1 and potently inhibits binding of Cul3 to Keap1, stabilizing Nrf2 (19, 20). The methyl ester (CDDO-Me, Bardoloxone methyl) also was tested. B6 mice were treated with pristane and received either CDDO-Im (2.5 mg/kg in DMSO-PBS i.p. every other day), CDDO-Me (2.5 mg/kg body weight in DMSO-PBS, i.p. daily), or vehicle. PEC were assessed at 14-d by flow cytometry and qPCR. Expression of IFN-I regulated proteins (CD169, PDCA-1, Ly6C, IRF7) was evaluated by flow cytometry. Expression of IFN-I stimulated genes (ISGs) Mx1, Isg15, and Irf7 was measured by qPCR. DAH was assessed at 14-d.
Brusatol (11β,12α,15β)-13,20-Epoxy-3,11,12-trihydroxy-15-[(3-methyl-1-oxo-2-buten-1-yl)oxy]-2,16-dioxo-picras-3-en-21-oic acid methyl ester) is a plant-derived quassinoid that inhibits ARE-driven gene expression by promoting the ubiquitination and degradation of Nrf2 (21). B6 mice were treated with pristane and received either brusatol (Sigma, 2.0 mg/kg in DMSO-PBS, i.p. every other day) or vehicle. PEC were assessed at 9-d by flow cytometry. DAH was assessed at 9-d.
In vivo treatment with MitoTEMPO.
To assess the role of mitochondrial superoxide on peritoneal Mϕ, pristane-treated mice were given MitoTEMPO, an antioxidant that accumulates in the mitochondria and scavenges mitochondrial superoxide (22). Mice were injected with pristane plus MitoTEMPO (1.5 mg/kg in PBS i.p. daily) or vehicle for 9 or 14-d. PEC were assessed by flow cytometry. DAH was determined at 9 and 14-d.
IFNAR expression.
Pristane-treated B6 mice received either CDDO-Im or vehicle. At 3 or 6-d, PEC stained with anti-CD11b, Ly6C, CD138, and Ifnar1 for flow cytometry or lysed in TRIzol for qPCR. Ifnar1 and Ifnar2 mRNA levels were normalized to 18S RNA. Surface expression of the IFN-I regulated proteins PDCA-1 (CD317, BST2), CD169 (Siglec-1), and Ly6C on CD11b+CD138+ cells was determined by flow cytometry 6-d after pristane treatment in B6 mice, B6 mice treated with CDDO-Im, or B6 IFNARKO mice.
IFNα-stimulated Stat1 phosphorylation.
Pristane-treated B6 mice received either CDDO-Im or vehicle every other day. At 8-d, PEC were cultured for 15-min in AIM-V medium in the presence/absence of IFNα15 (200 ng/ml, Biolegend). Cells were fixed, permeabilized, and stained with anti-CD11b-BV-421, Ly6G-APC-Cy7, and anti-phospho-Stat1-PE antibodies. The MFI of phospho-Stat1 staining in CD11b+Ly6G− cells was determined.
Treatment of Mϕ cell line with Nrf2 activators and inhibitors.
RAW264.7 cells (ATCC) were cultured with or without 100 nM brusatol. After 1-h, CDDO-Im (0.5 μM) or dimethyl fumarate (DMF, 10 μM, Sigma) was added. After culturing an additional 6-h, Gclc, Gpx4, Nqo1, and Sod2 expression was measured (qPCR). In other experiments, RAW264.7 cells were cultured with CDDO-Im (0.1, 1.0, or 2.0 μM) or vehicle (DMSO). After 1-h, IFNα4 (100 U/ml in PBS, R&D Systems) or PBS alone, was added and the cells were cultured an additional 6-h (gene expression) or 24-h (flow cytometry). ISG expression (Isg15, Irf7, Mx1) relative to 18S RNA was determined by qPCR. Nrf2-regulated gene expression (Nqo1) relative to 18S RNA also was determined by qPCR. For flow cytometry, cells were stained with anti-CD169-APC and anti-PDCA1-PE.
Statistical analysis.
Statistical analyses were performed using Prism 6.0 (GraphPad Software). Differences between groups were analyzed by two-sided unpaired Student t test unless otherwise indicated. Data were expressed as mean ± SD. p < 0.05 was considered significant. Experiments were repeated at least twice.
Results
Both IFN-I production and IFNAR expression on monocytes/Mϕ influence the interferon signature in SLE (14). The high IFN-I responsiveness of CM vs. anti-inflammatory NCM reflects their relative IFNAR expression. However, the mechanism(s) controlling differential IFNAR expression has not been determined. We explored the regulation of IFNAR expression.
NCM from MO-treated mice express Nrf2-regulated genes.
NCM from MO-treated mice exhibit an anti-inflammatory phenotype promoting the resolution of inflammation whereas NCM from pristane-treated mice are more proinflammatory (15). In addition to higher ISG expression (14), GSEA suggested that genes involved in the response to ROS (23, 24) were up-regulated in NCM from 14-d MO- vs. pristane-treated mice (Fig. 1A). In contrast, expression of these genes was not significantly different in CM (not shown). Gclc (glutamate-cysteine ligase, catalytic subunit, the rate-limiting enzyme for intracellular glutathione synthesis), was the top ROS pathway transcript over-expressed in NCM from MO-treated mice (Fig. 1A). As this key mitochondrial anti-oxidant enzyme is regulated by Nrf2 (16), we asked whether other Nrf2-regulated transcripts were up-regulated. Seven of the 19 enriched genes in the GSEA (Gclc, Nqo1, Sod2, Gsr, Gpx4, Srxn, and Prdx1) were Nrf2-regulated (Fig. 1A). By qPCR, expression of Nfe2l2 (encoding Nrf2), was substantially higher in flow-sorted NCM and CM from MO- vs. pristane-treated mice (Fig. 1B). In MO-treated mice, Nfe2l2 expression was higher in NCM vs. CM. Consistent with Nfe2l2 levels, expression of two representative Nrf2-regulated genes (Gpx4 and Prdx1) was higher in NCM vs. CM from MO-treated mice (Fig. 1C). Although Gpx4 and Prdx1 expression was lower in pristane-treated mice, Nrf2-regulated gene expression generally was higher in NCM vs. CM from pristane-treated mice. Intracellular staining of the Nrf2-regulated protein HSP70 (25) exhibited a similar pattern: higher in NCM from MO- vs. pristane-treated mice and lower in CM vs. NCM (Fig. 1D). The Nrf2-regulated proteins Marco and CD36 also are over-expressed in NCM (14). Finally, Nrf2 protein binding to its target sequence was higher in PEC nuclear extracts from MO- vs. pristane-treated mice (Fig. 1E). Together, these data indicate that 1) Nrf2 activity is higher in NCM vs. CM and 2) NCM from MO-treated mice exhibit higher activity than NCM from pristane-treated mice. Similarly, expression of a subset of Nrf2-regulated genes, exemplified by SOD2, was lower in leukocytes from lupus patients vs. healthy controls (Fig. 1F). As expected, SOD2 and NEF2L2 expression correlated positively.
Figure 1. Transcriptional profiling of Mϕ.

A, NCM and CM from pristane and mineral oil (MO)-treated mice (d-14, 3 mice/group) were flow-sorted and analyzed by RNA-Seq. GSEA of RNA from NCM, MO vs. pristane treated mice, is shown on left. Nrf2-regulated genes are indicated by a red dot. Right, GSEA enrichment plot, hallmark reactive oxygen species pathway. B, Expression of Nfe2l2 relative to 18S rRNA by qPCR in flow-sorted NCM and CM from pristane (Pris) and MO-treated mice (4/group). C, Expression of Nrf2-regulated genes Gpx4 (left) and Prdx1 (right) relative to 18S rRNA in flow-sorted NCM and CM from pristane- and MO- treated mice. D, Intracellular staining (flow cytometry) for the Nrf2-regulated protein HSP70 (heat shock protein 70) in NCM and CM 14-days after pristane or MO treatment (MFI, mean fluorescence intensity). E, Nrf2 binding ARE-motif binding activity in 7-day pristane- vs. MO-treated B6 mice (Cayman Nrf2 transcription factor assay). F, Expression of SOD2 and NFE2L2 in the whole blood of SLE patients and healthy donors (HD, by qPCR). Left SOD2 expression relative to 18S rRNA. Right, bivariate analysis of SOD2 vs. NFE2L2 expression. *P < 0.05; **P < 0.01; ***P < 0.001 vs. control (Student t-test).
Effect of Nrf2 on myeloid subsets.
Pristane-injected mice were treated with Nrf2 activators (CDDO-Im, CDDO-Me). After 14-d peritoneal myeloid cell subsets were identified by flow cytometry (Fig. 2A, left). Neutrophils were defined as CD11b+Ly6G+ cells (R1). The CD11b+Ly6G− population was sub-divided into Ly6ChiCD138− CM (R2) and Ly6Clo/negCD138+ NCM (R3) subsets. CDDO-Im treatment did not significantly affect the total PEC count or absolute numbers of CM or NCM, whereas neutrophils increased (Fig. 2B). Surface IFNAR staining of PEC from pristane-treated mice exhibited marked differences over time (Fig. 2C). In CM, staining was highest 3-6 days after pristane treatment and decreased by 14-d. In contrast, IFNAR staining initially was low on NCM, but increased by 14-d. CDDO-Im treatment reduced IFNAR staining in both CM and NCM (Fig. 2C).
Figure 2. Nrf2 activation decreases CM and increases NCM.
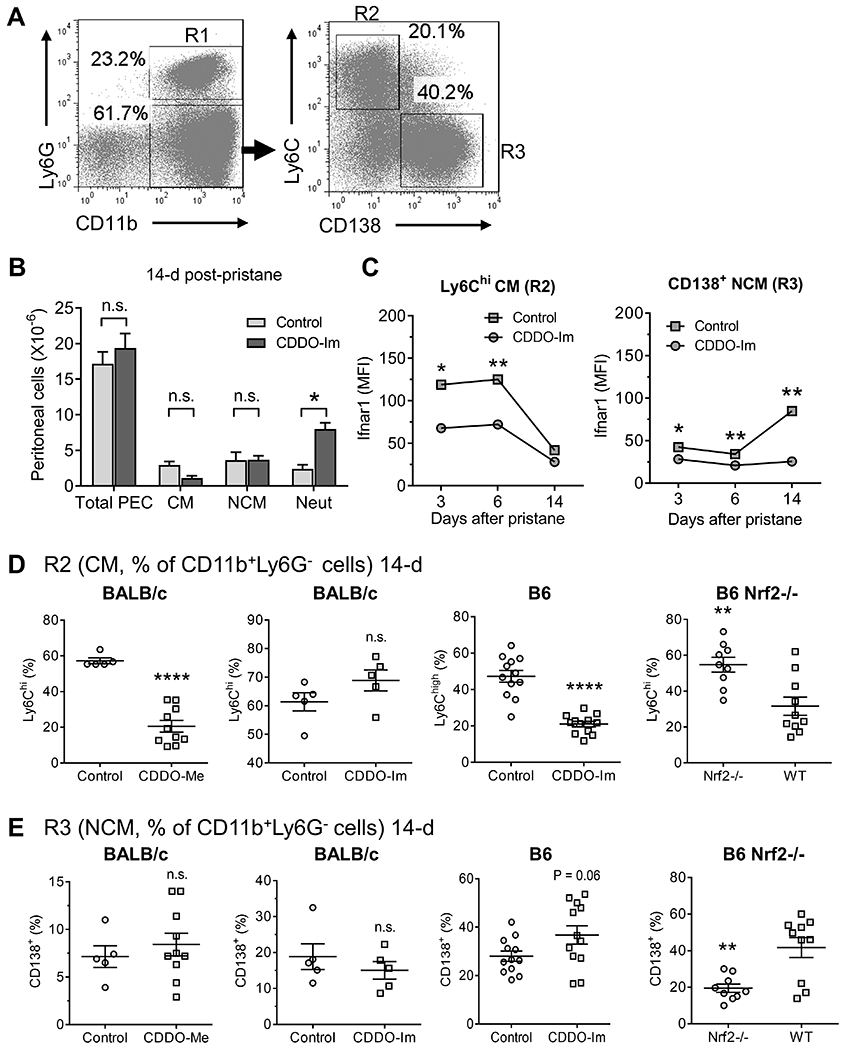
Mice were injected with pristane plus either CDDO-Im (2.5 mg/kg in PBS i.p. every other day), CDDO-Me (2.5 mg/kg in PBS i.p. daily) or vehicle (Control). Day-14 PEC were analyzed by flow cytometry. A, Gating strategy for CD11b+Ly6G+ neutrophils (R1), CD11b+Ly6G−Ly6Chigh (CM) (R2) and CD11b+Ly6G−CD138+ (NCM) (R3). B, Total peritoneal (PEC), CM, NCM and neutrophil (Neut) cell numbers (5/group). C, Ifnar1 surface staining (mean fluorescence intensity, MFI) of CM (left) and NCM (right) in pristane-treated B6 mice that received CDDO-IM or vehicle 3, 6, or 14 days. D, Left two panels: percentage of CM (R2) in pristane-treated BALB/c mice that received CDDO-Me, CDDO-Im, or vehicle. Third panel, percentage of CM in pristane-treated wild-type B6 mice with or without CDDO-Im treatment. Far right panel, CM in pristane-treated B6 Nrf2−/− mice. E, Percentage of NCM (R3) in pristane-treated BALB/c that received CDDO-Me, CDDO-Im, or vehicle. Third panel, percentage of NCM in pristane-treated wild-type B6 mice with or without CDDO-Im treatment. Far right panel, NCM in pristane-treated B6 Nrf2−/− mice. * P < 0.05, ** P < 0.01, **** P < 0.0001 (Student t-test); n.s., not significant.
As shown in Fig. 2D, CM (R2) as a % of CD11b+Ly6G− cells decreased in B6 mice receiving CDDO-Im. This was not apparent in BALB/c mice treated with CDDO-Im, but was seen in BALB/c mice treated with a more potent activator (CDDO-Me, Fig. 2D, far left). The percentage of CM also correlated negatively with Nrf2 activity in wild-type B6 vs. B6 Nfe2l2−/− (Nrf2-deficient) mice (Fig. 2D, far right). CDDO-Im and CDDO-Me had little effect on NCM (R3) as a % of CD11b+Ly6G− cells, although there was a trend toward increased numbers in CDDO-Im treated B6 mice (Fig. 2E). Wild-type mice had a higher percentage of NCM than Nrf2-deficient mice (Fig. 2E, far right). Neutrophils (R1) expressed as % of CD11b+ cells were not consistently affected by Nrf2 activity (not shown).
Mitochondrial dysfunction in pristane-treated mice.
Nrf2 is a master regulator of genes involved in cellular defense against oxidative and electrophilic stress (16, 26). Expression of the Nrf2-regulated genes glutathione peroxidase 4 (Gpx4), which protects against oxidative stress (16), peroxiredoxin-1 (Prdx1), and superoxide dismutase 2 (Sod2), which encode mitochondrial matrix proteins (16, 26), was higher in peritoneal Mϕ from MO vs. pristane treated mice (Fig. 1C; data not shown). We looked for evidence of mitochondrial dysfunction by examining mitochondrial superoxide (MitoSox Red) and mitochondrial membrane potential (TMRM). In both NCM (left) and CM (right), MitoSox Red staining was higher in pristane- vs. MO-treated B6 mice, whereas TMRM staining was lower in pristane-treated mice (Fig. 3A).
Figure 3. Nrf2 activation opposes mitochondrial dysfunction.
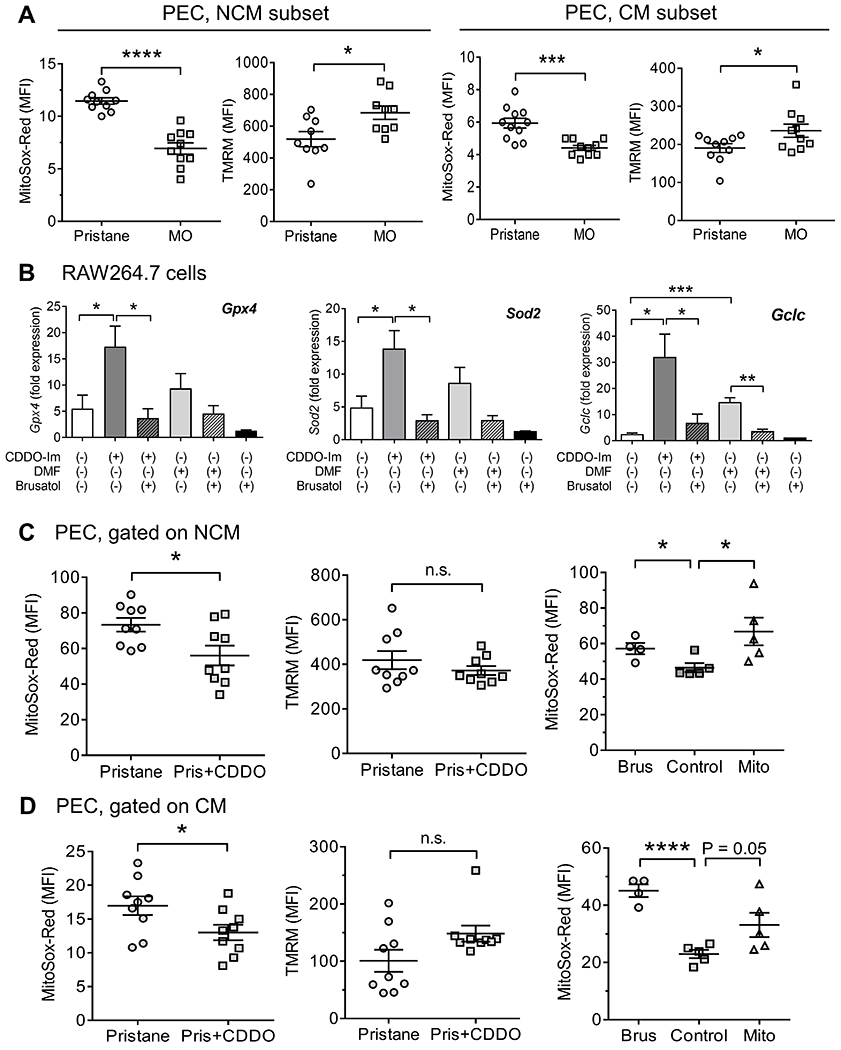
A, PEC from 14-d pristane or MO treated mice were analyzed by flow cytometry. Mitochondrial superoxide (MitoSox Red staining) and mitochondrial membrane potential (Ψ, tetramethylrhodamine, TMRM, staining) in NCM (left two panels) or CM (right two panels) from pristane and MO mice. B, RAW 264.7 cells were pretreated with/without brusatol (0.1 μM) for 1-h, followed by CDDO-Im (0.5 μM) or DMF (10 μM) for 6-h. Expression of Gpx4, Sod2, and Gclc were quantified by qPCR (4/group). C and D, Mice were treated with pristane plus either CDDO-Im or vehicle for 14-d. Right panel, mice were treated with pristane plus brusatol (2 mg/kg in PBS i.p. every other day), MitoTEMPO (1.5 mg/kg in PBS i.p.), or vehicle (Control) for 9-d. PEC were analyzed for mitochondrial superoxide (MitoSox Red staining) and mitochondrial membrane potential (TMRM, staining) by flow cytometry. C, PEC were gated on NCM; D, PEC were gated on CM. * P < 0.05, *** P < 0.001, **** P < 0.0001 (Student t-test). n.s., not significant.
As in primary Mϕ, CDDO-Im treatment of RAW 264.7 cells enhanced Gpx4, Sod2, and Gclc expression (Fig. 3B). The Nrf2 inhibitor brusatol reduced CDDO-enhanced transcription of Gpx4, Sod2, and Gclc to baseline. A weaker Nrf2 activator, DMF, did not stimulate Gpx4 or Sod2 transcription, but did activate Gclc (Fig. 3B).
To examine whether mitochondrial dysfunction was associated with altered Nrf2 activity, we measured mitochondrial superoxide and membrane potential in PEC from mice treated with CDDO-Im, brusatol, or MitoTEMPO. In pristane-treated mice, Mito-Sox Red staining in both NCM (Fig. 3C) and CM (Fig. 3D) was lower in mice receiving CDDO-Im vs. controls, whereas TMRM staining was unaffected, suggesting that Nrf2-regulated gene expression down-regulates mitochondrial superoxide. Conversely, brusatol treatment in vivo increased MitoSox Red staining in both NCM and CM (Fig. 3C, D). MitoTEMPO paradoxically increased mitochondrial superoxide (MitoSox staining).
Nrf2 alters the phenotype of NCM.
IFNAR signaling induces the interferon signature, which is strongly associated with lupus in humans (1, 27) and pristane-treated mice (5). Although CDDO-Im reduced IFNAR staining in CM (Fig. 2C), it had only modest effects on the IFN-regulated proteins Ly6C, PDCA-1, and CD169 (Fig. 4A). In contrast, CDDO-Im reduced surface expression of Ly6C, PDCA-1, and CD169 on NCM, while increasing CD138. Similarly, Ly6C and PCDA-1 staining was lower and CD138 was higher in wild-type vs. Nrf2-deficient mice (Fig. 4B). However, CD169 surface staining was comparable. As expected, NCM from brusatol-treated mice had higher Ly6C and lower CD138 staining than controls (Fig. 4C). Brusatol-treated mice also had more peritoneal CM and fewer NCM than controls.
Figure 4. Nrf2 activation changes the phenotype of NCM.
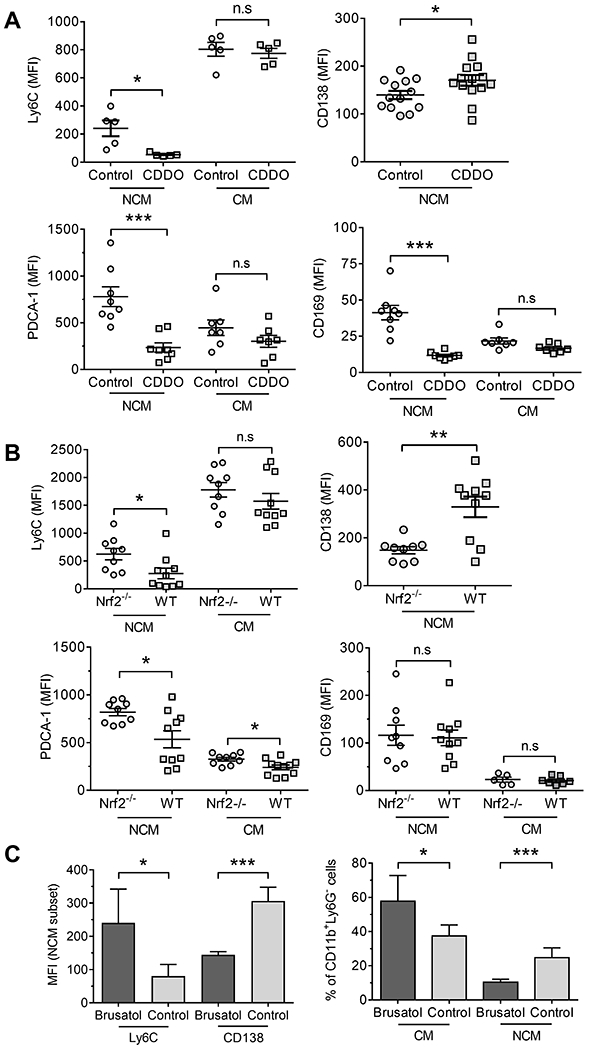
A, Mice were injected with pristane plus CDDO-Im as in figure 2, or vehicle (Control) for 14-d. Surface staining (flow cytometry) of PEC for Ly6C, PDCA-1 and CD169 on CM and NCM and for CD138 on NCM. B, Wild-type B6 (WT) and B6 Nrf2−/− mice were injected with pristane and 14-d later PEC were analyzed by flow cytometry. Surface staining for Ly6C, PDCA-1 and CD169 on CM and NCM and CD138 expression level on NCM. C, Mice (4-5/group) were injected with pristane plus brusatol as in figure 3 or vehicle (Control) for 9-d. Left, surface staining of PEC for Ly6C and CD138 on NCM (left). Right, percentages of CM and NCM in CD11b+Ly6G− cells (right). * P < 0.05, ** P < 0.01, *** P < 0.001 (Student t-test).
Nrf2 activity influences Ifnar expression.
CM have higher Ifnar expression than NCM and respond more robustly to IFN-I (14). Nrf2 activation with CDDO-Im decreased Ifnar1 (but not Ifnar2) gene expression in pristane-treated mice (Fig. 5A). Flow cytometry confirmed that Ifnar1 surface staining was lower on NCM vs. CM 6-d after pristane treatment (Fig. 5B, “Con”). CDDO-Im reduced Ifnar1 staining on both CM and NCM (Fig. 5B), but expression was still lower on NCM vs. CM. There was little Ifnar1 staining of Ly6G+ neutrophils or “Other” cells (primarily lymphocytes) and CDDO-Im treatment had no effect. Although CDDO-Im reduced Ifnar1 staining on NCM 8-d after pristane treatment, it was higher than on NCM from pristane-treated Ifnar1-deficient (IFNARKO) mice (Fig. 5C). The residual Ifnar protein is likely to be functional, as surface staining of the IFN-I regulated proteins PDCA-1 (Fig. 5C) and Ly6C (not shown) on NCM was above the background in IFNARKO mice. In the CM subset, CDDO-Im had only a modest effect on Ifnar1 and PDCA-1 staining. However, brusatol enhanced Ifnar1 staining in both NCM and CM (Fig. 5D). Consistent with the low levels of ISGs/proteins in Mϕ from CDDO-Im treated mice, CD11b+Ly6G− PEC from mice treated with pristane+CDDO-Im for 8-d did not increase phospho-Stat1 staining following 15-min culture with IFNα4 (Fig. 5E).
Figure 5. High Nrf2 activity is associated with low Ifnar1 expression.
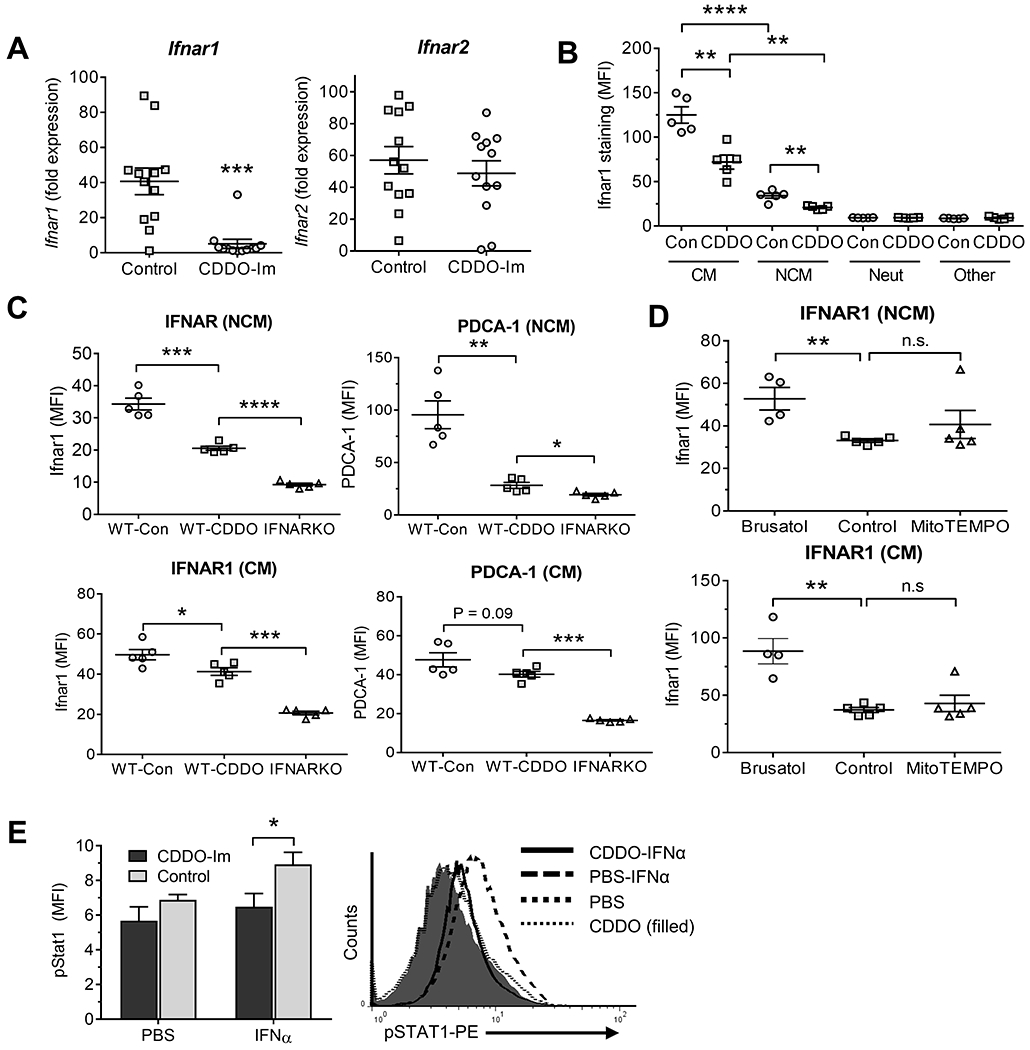
Pristane-treated B6 mice received either CDDO-Im or vehicle (Control) as in Fig. 2. PEC were analyzed at 6-d. A, Expression of Ifnar1 and Ifnar2 mRNA relative to 18S RNA in total PEC (qPCR). B, Flow cytometry of Ifnar1 expression in PEC gated on CD11b+Ly6G−CD138+ cells (NCM), CD11b+Ly6G−Ly6Chi cells (CM), CD11b+Ly6G+CD138− neutrophils (Neut), and CD11b−Ly6G−CD138− (Other) cells, mainly lymphocytes. C, wild-type B6 (WT) and B6-Ifnar knockout (IFNARKO) mice injected with pristane received CDDO-Im or vehicle (Control, Con) as above. PEC gated on NCM (top) and CM (bottom) were analyzed by flow cytometry at 8-d for IFNAR1 (left) and the IFN-regulated protein PDCA-1 (right). D, Pristane-treated B6 mice received brusatol, MitoTEMPO, or vehicle (Control) as in Fig. 3 for 9-d. IFNAR1 surface staining of peritoneal NCM (Top) and CM (Bottom) was determined. E, pristane-treated B6 mice received CDDO-Im or PBS for 8-d. PEC were treated in vitro with IFNα15 (200 ng/ml) or PBS for 15-min and phosphorylated Stat1 (pStat1) was measured in CD11b+Ly6G− cells by flow cytometry (5/group). Right, representative histograms of IFNAR surface staining in mice treated with IFNα15 vs. PBS from mice treated with pristane + CDDO-Im or pristane alone. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 (Student t-test). n.s., not significant. (MFI, mean fluorescence intensity).
Nrf2 activation inhibits ISG expression.
Surface staining for the IFNAR and interferon-regulated proteins (Ly6C, PDCA-1, CD169) decreased in NCM from mice treated with CDDO-Im + pristane (14-d) vs. pristane-treated controls (Figs. 4–5). mRNA transcripts for the ubiquitously expressed ISGs Mx1, Isg15, and Irf7 also were expressed at lower levels in 14-d PEC from CDDO-Im treated B6 mice vs. controls (Fig. 6A). Expression of the IFN-I regulated proteins PDCA-1 and IRF7 following pristane injection was decreased in both CM and NCM from CDDO-Im-treated mice vs. untreated controls (Fig. 6B). Consistent with the results in CDDO-Im treated mice, Mx1, Isg15, and Irf7 expression was higher in PEC from Nrf2-deficient mice vs. controls, whereas Nfe2l2 expression was absent as expected (Fig. 6C).
Figure 6. High Nrf2 is associated with low interferon-stimulated gene (ISG) expression.
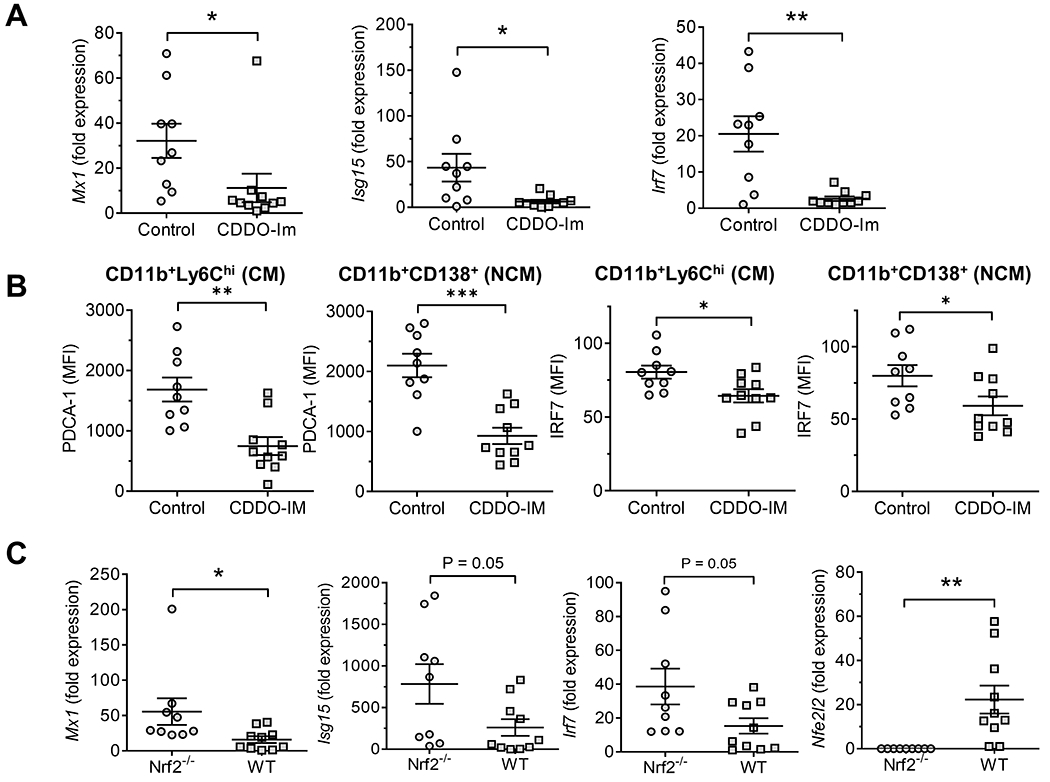
Pristane-treated B6 mice received either CDDO-Im or vehicle (Control) as in Fig. 2. B6 wild-type (WT) mice and Nrf2−/− mice were treated with pristane. PEC were analyzed at 14-d. A, ISG expression (Mx1, Isg15, Irf7) relative to 18S RNA (qPCR) in total PEC from CDDO-IM and control mice. B, Flow cytometry of IFN-inducible proteins (PDCA-1 and IRF7) in CD11b+Ly6ChiCD138− conventional macrophages (CM) and CD11b+CD138+Ly6C− non-conventional macrophages (NCM) from CDDO-IM treated vs. control mice. C, Mx1, Isg15, Irf7, and Nfe2l2 expression in Nrf2−/− and WT mice. * P < 0.05, ** P < 0.01, *** P < 0.001 (Student t-test).
Nrf2 activation had similar effects on RAW264.7 cells. When cultured for 24-h with IFNα4 (100 U/ml), CD169 staining increased (Fig. 7A). CDDO-Im reduced IFNα4-stimulated CD169 staining in a dose-dependent manner, which was near maximal at 1 μM. A similar result was obtained with a second IFN-I regulated protein, PDCA-1, the expression of which was even more strongly inhibited by CDDO-Im, also with a maximal inhibition at about 1 μM (Fig. 7B). As expected, expression of the Nrf-2 regulated gene Nqo1 was stimulated by 0.5 μM CDDO-Im treatment in both IFNα and PBS treated RAW264.7 cells (Fig. 7C). In contrast, IFNα4-stimulated Isg15, Irf7, and Mx1 expression was inhibited by CDDO-Im. CDDO-Im (0.5 μM) (Fig. 7D). also inhibited Ifnar1 expression in RAW264.7 cells (not shown).
Figure 7. CDDO-Im inhibits interferon stimulated gene expression in RAW264.7 cells.
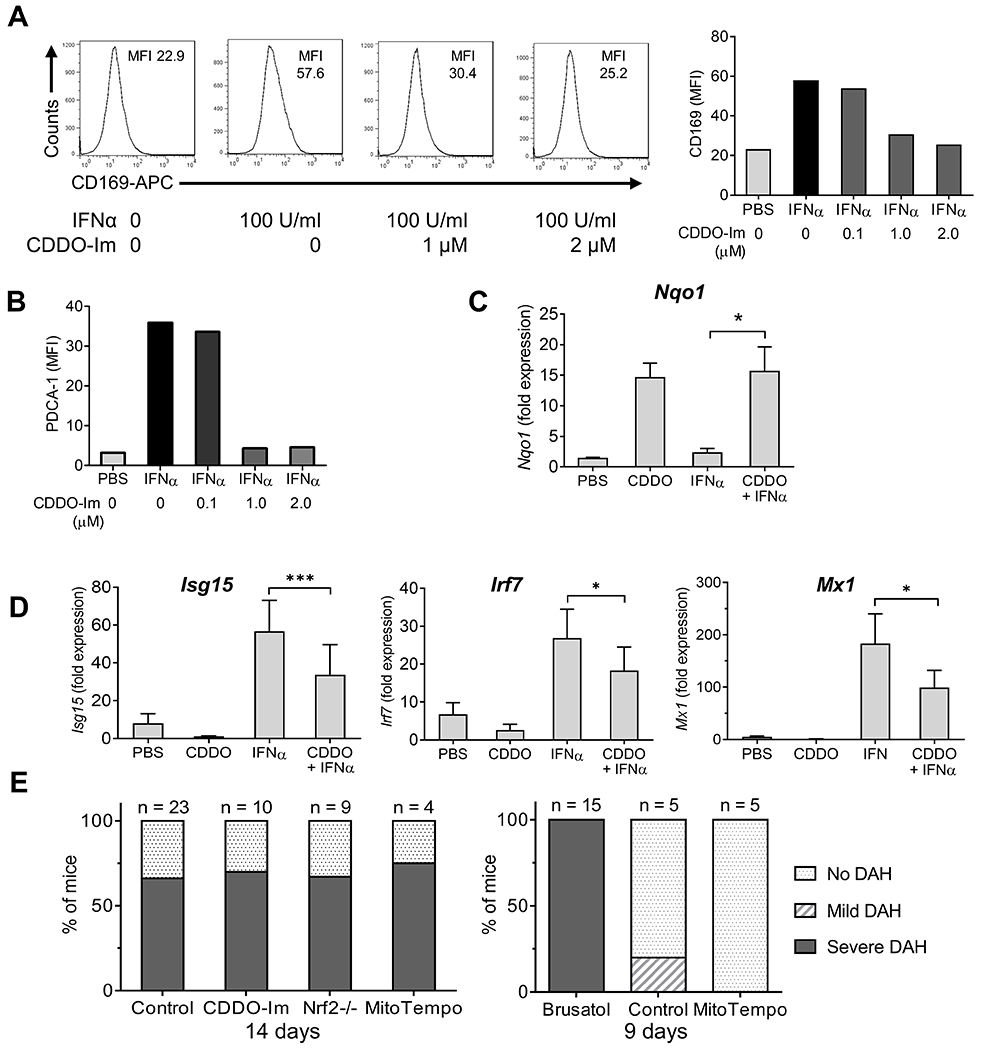
A, RAW264.7 cells were pretreated with CDDO-Im (0.1, 1.0, or 2.0 μM in DMSO) or vehicle alone for 1-h, and then challenged with IFNα4 (100 U/ml) for 24-h. Surface staining of the IFN-I regulated protein CD169 (MFI) was analyzed by flow cytometry. Left, representative histograms of CD169 surface staining; right, quantification. B, Quantification of surface staining (flow cytometry) for a second IFN-inducible protein (PDCA-1) showing a pattern similar to CD169. C-D, RAW264.7 cells were pretreated with CDDO-Im (0.5 μM) or PBC and then challenged with IFNα4 (100 U/ml in PBS) or PBS alone for 6-h. C, Nrf2-regulated gene expression (Nqo1) relative to 18S RNA (qPCR) was determined (qPCR). D, ISG expression (Isg15, Irf7, Mx1) relative to 18S RNA (qPCR) was determined (qPCR). E, Mice were injected with pristane plus CDDO-Im (2.5 mg/kg in PBS i.p. every other day), brusatol (2 mg/kg in PBS i.p. every other day), MitoTEMPO (1.5 mg/kg in PBS i.p.), or vehicle. Left, DAH was assessed at 14-d in B6 mice receiving CDDO-Im, MitoTEMPO, or vehicle (Control) and in B6 Nrf2−/− mice. Right, DAH was assessed at 9-d in B6 mice receiving brusatol, MitoTEMPO, or vehicle. * P < 0.05, ** P < 0.01, *** P < 0.001 (Student t-test).
Autoantibody production and renal disease in pristane-induced lupus require IFNAR signaling, whereas DAH is IFNAR-independent (8, 13). Consistent with the interferon-independence of DAH, neither CDDO-Im treatment nor Nrf2 deficiency affected the induction of DAH in B6 mice (Fig. 7E, left). However, brusatol unexpectedly induced the rapid onset of severe DAH requiring termination of the experiment at d-9 (Fig. 7E, right). Pristane-treated mice that received MitoTEMPO did not develop DAH by d-9, but it developed at d-14 (Fig. 7E, left).
Discussion
Chronic peritoneal inflammation induced by pristane is associated with a high interferon signature, persistence of Ly6Chi (inflammatory) CM, and lupus (5). The initial peritoneal inflammatory response to MO is similar, but the interferon signature is absent, pro-resolving CD138+ NCM replace the CM after ~2 weeks, and lupus does not develop (10). The induction of lupus autoantibodies and nephritis by pristane is greatly impaired in mice lacking the IFNAR (5) and SLE patients with a high interferon signature have an increased frequency of lupus autoantibodies and nephritis (2, 3). In pristane-treated mice, CM exhibit a stronger interferon signature than NCM exposed to same concentration of IFN-I reflecting their relative IFNAR expression (14). We show that activation of Nrf2 shifts Mϕ toward an M2-like phenotype and down-regulates IFNAR expression on CM and NCM.
Nrf2 activity regulates the phenotype of NCM.
GSEA revealed that Nrf2-regulated genes were upregulated in NCM (but not CM) from MO- vs. pristane- treated mice (Fig. 1). Expression of Nfe2l2 and Nrf2-regulated gene and protein expression also was higher in MO- vs. pristane-treated mice. CM expressed only low levels of Nfe2l2 and Nrf2-regulated genes. But although Nrf2-regulated gene expression was low in pristane-treated mice, it was higher in NCM vs. CM.
Nrf2 regulates the cellular response to oxidative stress by forming a cytoplasmic complex with Keap1, a thiol-rich electrophile sensor, and the ubiquitin ligase Cullin3 (Cul3) (17, 26). Under unstressed conditions, Keap1 promotes ubiquitin-mediated degradation of Nrf2 (16, 26). Modification of the reactive cysteine residues of Keap1 by electrophiles reduces the ubiquitin ligase activity of Keap1/Cul3, resulting in Nrf2 stabilization, translocation to the nucleus, dimerization with small musculoaponeurotic fibrosarcoma (sMAF) proteins, and binding to AREs of target genes (16, 17). Nrf2 regulates over 100 genes involved in cellular defense against oxidative and electrophilic stress, of which about a third are involved in cellular redox homeostasis or stress responses (26, 28). Interestingly, some Nrf2-dependent genes do not contain AREs (28). Notably, Nrf2-regulated downregulation of the transcription of several proinflammatory cytokines is independent of AREs and ROS (29).
PECs from pristane- and MO- treated mice contain pro- and anti-inflammatory Mϕ subsets (10). Proinflammatory CM predominate in pristane-treated mice, whereas anti-inflammatory/proresolving NCM predominate in MO-treated mice. Pristane-treated Nrf2-deficient mice had more CM and fewer NCM than wild-type controls (Fig. 2D,E). Similarly, Nrf2 activators decreased CM in pristane-treated wild-type mice, although the effect on NCM was small. Conversely, brusatol increased CM and decreased NCM (Fig. 4C). These data suggest that Nrf2 inhibits M1-like and promotes M2-like Mϕ polarization. Altered Mϕ recruitment to the peritoneum is an alternative explanation, but we feel it is less likely because Ly6Chi monocyte recruitment is CCR2-dependent and subsequently these cells develop into both Ly6Chi CM and Ly6Clo/− NCM (30).
Consistent with this model, Nrf2 activation promotes M2 polarization and the resolution of acute respiratory distress syndrome (ARDS) (31). However, in contrast to its beneficial effect in ARDS, Nrf2 activation did not improve DAH in pristane-induced lupus (Fig. 7E). Nrf2 also promotes the development of Mox Mϕ, a proinflammatory subset distinct from M1 and M2 Mϕ that is weakly phagocytic, expresses high levels of heme oxygenase I, Nr4A2 (Nurr1) and other Nrf2-regulated anti-oxidative genes, and may be involved in atherogenesis (32). Although CD138+ NCM have increased Nrf2-regulated gene expression, they are strongly phagocytic with an M2-like phenotype (10), suggesting that they are distinct from Mox Mϕ.
Nrf2 downregulates Ifnar expression.
In view of the importance of IFNAR signaling in lupus, Nrf2 regulation of Ifnar1 expression may influence disease pathogenesis. Ifnar1 expression varied with time after pristane exposure (Fig. 2C). Acutely, Ifnar1 staining was higher in CM than NCM. However, at 14-d, staining decreased in CM but increased in NCM. Nrf2 activation reduced Ifnar1 protein and mRNA levels in both subsets and reduced ISG expression (Figs. 2, 4–6). The opposite was seen in mice treated with a Nrf2 inhibitor. Consistent with the inhibition by Nrf2 activators, ISG expression was higher in Nrf2-deficient mice vs. controls. However, although CDDO decreased Ifnar1 and ISG expression, it was higher than in Ifnar1-deficient mice (Fig. 5), suggesting that basal Ifnar1 expression is Nrf2-independent.
Regulation of IFNAR expression by Nrf2 has not been reported previously. Although CDDO potently activates Nrf2/Keap1-regulated genes, there also are Keap-1 independent effects. At high doses, CDDO inhibits IKKβ, blocking NFκB activation (33, 34), raising the possibility that it might alter Ifnar1 expression via an IKKβ/NFκB-dependent mechanism. In ARDS, Nrf2 blocks M1 polarization by interfering with the NFκB pathway (31). But several lines of evidence suggest that decreased IFNAR expression in CDDO-treated mice is a direct effect of Nrf2. Three Nrf2 activators, CDDO-Im, CDDO-Me, and DMF, all reduced Ifnar1 expression (Figs. 2–3) and brusatol, which enhances the degradation of Nrf2 (21), increased it (Fig. 5). In addition, Nrf2-deficient mice have higher IFNAR expression than wild-type mice (SH unpublished data). Nrf2-deficient mice are more susceptible than controls to endotoxic shock and produce more TNFα, IL-1α, and IL-6 due to ROS-mediated upregulation of NFκB (35). However, neither the mitochondria-targeted antioxidant Mito-TEMPO (Fig. 5D) nor the NFκB inhibitor JSG-23 (SH, unpublished) down-regulated Ifnar1 expression.
Nrf2 binds near promoter regions of the IL6, IL1a, and IL1b genes in M1 Mϕ (29). Although Nrf2-dependent, inhibition of LPS-driven transcriptional upregulation of these cytokines is independent of ARE motifs and ROS. Like the IL1a and IL1b genes (36), the Ifnar1 gene is not known to have a functional ARE. Thus, negative regulation of Ifnar1 by Nrf2 may be another example of ARE-independent negative transcriptional regulation, analogous to Il1a, Il1b, and Il6 (29). Further studies are necessary to define the mechanisms.
Exacerbation of DAH by brusatol.
Brusatol depletes Nrf2 via a Keap1-independent post-transcriptional mechanism, sensitizing cells to oxidative stress (37). As expected, the effects of brusatol generally were opposite those of CDDO-Im. However, Nrf2 deficiency had little effect on DAH, whereas brusatol unexpectedly exacerbated it (Fig. 7E). We suspect that this is due to brusatol’s Nrf2-independent effects on protein synthesis (38).
CDDO-Im alleviates oxidative stress in pristane-treated mice.
Pristane causes oxidative stress in Mϕ, as suggested by increased lipid peroxidation and decreased serum superoxide dismutase, glutathione, and catalase activity (39). The increased mitochondrial superoxide and decreased mitochondrial membrane potential in pristane-treated mice (Fig. 3) further support the idea that pristane induces mitochondrial oxidative stress. Consistent with the downregulation of mitochondrial ROS (40), Nrf2 activation decreased mitochondrial oxidative stress in pristane-treated mice (Fig. 3C–D), which may promote the resolution of inflammation.
Clinical significance.
Autoantibody production and nephritis are less severe in lupus mice lacking the IFNAR (8, 41). Similarly, the interferon signature is associated with autoantibodies and nephritis in SLE patients (2, 3). Consistent with the possibility that Nrf2-regulated changes in IFN-I responsiveness are pathogenic, Nrf2-deficient mice have greater renal damage than controls following pristane treatment and develop spontaneous lupus-like disease with aging (42, 43). Moreover, DMF reduces autoantibody production and renal disease in pristane-induced lupus (44) and CDDO-Me decreases proteinuria, BUN, anti-dsDNA autoantibodies, and renal pathology in B6.Sle1.Sle3 and MRL/lpr mice (45). The suppression of IFNAR responsiveness by Nrf2 provides a plausible explanation for the beneficial effect of Nrf2 activators in lupus.
A recent phase II clinical trial suggests that anti-IFNAR monoclonal antibody treatment improves disease in SLE patients with high interferon signatures (46). Small molecule Nrf2 activators may be an alternative approach to block interferon signaling. DMF is approved for multiple sclerosis (47). CDDO-Im and CDDO-Me are 100-fold more potent than DMF and target somewhat different sets of genes and signaling pathways (33, 48). Our preliminary studies suggest that, as in murine lupus, Nrf2 activity is low in leukocytes from SLE patients vs. healthy controls (Fig. 1F). Based on their relative safety, efficacy in preclinical lupus models, and ability to attenuate IFNAR signaling, Nrf2 activators may warrant further study for treating human SLE.
Supplementary Material
Acknowledgments
Supported by research grants R01-AR44731 (WR), P30-AR070253 (Joint Biology Consortium, Brigham and Women’s Hospital, PAN), R01 AR065538 (PAN), and K08-AR074562 (PYL) from NIH/NIAMS and a Lupus Research Alliance Target Identification in Lupus Grant (PAN). PYL was the recipient of a Rheumatology Research Foundation Investigator Award from the American College of Rheumatology. We are grateful for the assistance provided by the Broad Institute Technology Labs. We thank Dr. Shi-Wu Li for assisting with promoter analysis and Annie Chan, R.N. for assisting with the human studies.
Footnotes
Conflict of Interest Disclosures: The authors declare no competing financial interests
References
- 1.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100(5):2610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhuang H, Narain S, Sobel E, Lee PY, Nacionales DC, Kelly KM, et al. Association of anti-nucleoprotein autoantibodies with upregulation of Type I interferon-inducible gene transcripts and dendritic cell maturation in systemic lupus erythematosus. Clin Immunol. 2005;117(3):238–50. [DOI] [PubMed] [Google Scholar]
- 3.Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52(5):1491–503. [DOI] [PubMed] [Google Scholar]
- 4.Nacionales DC, Kelly KM, Lee PY, Zhuang H, Weinstein JS, Sobel E, et al. Type I interferon production by tertiary lymphoid tissue developing in response to 2, 6, 10, 14 tetramethylpentadecane (pristane). Am J Pathol. 2006;168:1227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30(9):455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alperin JM, Ortiz-Fernandez L, Sawalha AH. Monogenic Lupus: A Developing Paradigm of Disease. Front Immunol. 2018;9:2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Weerd NA, Samarajiwa SA, Hertzog PJ. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. 2007;282(28):20053–7. [DOI] [PubMed] [Google Scholar]
- 8.Nacionales DC, Kelly-Scumpia KM, Lee PY, Weinstein JS, Sobel E, Satoh M, et al. Deficiency of the Type I interferon receptor protects mice from experimental lupus. Arthritis Rheum. 2007;56:3770–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinbach F, Henke F, Krause B, Thiele B, Burmester GR, Hiepe F. Monocytes from systemic lupus erythematous patients are severely altered in phenotype and lineage flexibility. Ann Rheum Dis. 2000;59(4):283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han S, Zhuang H, Shumyak S, Wu J, Li H, Yang LJ, et al. A Novel Subset of Anti-Inflammatory CD138(+) Macrophages Is Deficient in Mice with Experimental Lupus. J Immunol. 2017;199(4):1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Sanchez C, Barbarroja N, Messineo S, Ruiz-Limon P, Rodriguez-Ariza A, Jimenez-Gomez Y, et al. Gene profiling reveals specific molecular pathways in the pathogenesis of atherosclerosis and cardiovascular disease in antiphospholipid syndrome, systemic lupus erythematosus and antiphospholipid syndrome with lupus. Ann Rheum Dis. 2015;74(7):1441–9. [DOI] [PubMed] [Google Scholar]
- 12.Bethunaickan R, Berthier CC, Ramanujam M, Sahu R, Zhang W, Sun Y, et al. A unique hybrid renal mononuclear phagocyte activation phenotype in murine systemic lupus erythematosus nephritis. J Immunol. 2011;186(8):4994–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhuang H, Han S, Lee PY, Khaybullin R, Shumyak S, Lu L, et al. Pathogenesis of diffuse alveolar hemorrhage in murine lupus. Arthritis Rheumatol. 2017;69:1280–93 (see commentary). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han S, Zhuang H, Lee PY, Li M, Yang L, Nigrovic PA, et al. Differential Responsiveness of Monocyte and Macrophage Subsets to Interferon. Arthritis Rheumatol. 2019;Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han S, Zhuang H, Shumyak S, Wu J, Xie C, Li H, et al. Liver X Receptor Agonist Therapy Prevents Diffuse Alveolar Hemorrhage in Murine Lupus by Repolarizing Macrophages. Front Immunol. 2018;9:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39(4):199–218. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 System: a Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol Rev. 2018;98(3):1169–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Batandier C, Fontaine E, Keriel C, Leverve XM. Determination of mitochondrial reactive oxygen species: methodological aspects. J Cell Mol Med. 2002;6(2):175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA, et al. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PloS one. 2014;9(6):e98896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petronelli A, Pannitteri G, Testa U. Triterpenoids as new promising anticancer drugs. Anticancer Drugs. 2009;20(10):880–92. [DOI] [PubMed] [Google Scholar]
- 21.Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci USA. 2011;108(4):1433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trnka J, Blaikie FH, Smith RA, Murphy MP. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic Biol Med. 2008;44(7):1406–19. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glory A, Averill-Bates DA. The antioxidant transcription factor Nrf2 contributes to the protective effect of mild thermotolerance (40 degrees C) against heat shock-induced apoptosis. Free Radic Biol Med. 2016;99:485–97. [DOI] [PubMed] [Google Scholar]
- 26.Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88(Pt B):179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197(6):711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal. 2010;13(11):1713–48. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee PY, Li Y, Kumagai Y, Xu Y, Weinstein JS, Kellner ES, et al. Type-I interferon modulates monocyte recruitment and maturation in chronic inflammation. Am J Pathol. 2009;175:2023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei J, Chen G, Shi X, Zhou H, Liu M, Chen Y, et al. Nrf2 activation protects against intratracheal LPS induced mouse/murine acute respiratory distress syndrome by regulating macrophage polarization. Biochem Biophys Res Commun. 2018;500(3):790–6. [DOI] [PubMed] [Google Scholar]
- 32.Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107(6):737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liby KT, Sporn MB. Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol Rev. 2012;64(4):972–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7(5):357–69. [DOI] [PubMed] [Google Scholar]
- 35.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116(4):984–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu P, Rojo de la Vega M, Sammani S, Mascarenhas JB, Kerins M, Dodson M, et al. RPA1 binding to NRF2 switches ARE-dependent transcriptional activation to ARE-NRE-dependent repression. Proc Natl Acad Sci USA. 2018;115(44):E10352–E61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olayanju A, Copple IM, Bryan HK, Edge GT, Sison RL, Wong MW, et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic Biol Med. 2015;78:202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vartanian S, Ma TP, Lee J, Haverty PM, Kirkpatrick DS, Yu K, et al. Application of Mass Spectrometry Profiling to Establish Brusatol as an Inhibitor of Global Protein Synthesis. Mol Cell Proteomics. 2016;15(4):1220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ouyang Q, Huang Z, Wang Z, Chen X, Ni J, Lin L. Effects of pristane alone or combined with chloroquine on macrophage activation, oxidative stress, and TH1/TH2 skewness. J Immunol Res. 2014;2014:613136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kovac S, Angelova PR, Holmstrom KM, Zhang Y, Dinkova-Kostova AT, Abramov AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta. 2015;1850(4):794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, et al. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197(6):777–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang T, Tian F, Zheng H, Whitman SA, Lin Y, Zhang Z, et al. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-kappaB-mediated inflammatory response. Kidney Int. 2014;85(2):333–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, et al. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001;60(4):1343–53. [DOI] [PubMed] [Google Scholar]
- 44.Ebihara S, Tajima H, Ono M. Nuclear factor erythroid 2-related factor 2 is a critical target for the treatment of glucocorticoid-resistant lupus nephritis. Arthritis Res Ther. 2016;18(1):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu T, Ye Y, Min SY, Zhu J, Khobahy E, Zhou J, et al. Prevention of murine lupus nephritis by targeting multiple signaling axes and oxidative stress using a synthetic triterpenoid. Arthritis Rheumatol. 2014;66(11):3129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017;69(2):376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–97. [DOI] [PubMed] [Google Scholar]
- 48.To C, Ringelberg CS, Royce DB, Williams CR, Risingsong R, Sporn MB, et al. Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis. 2015;36(7):769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.