

SUMMARY
The linkage, length, and architecture of ubiquitin (Ub) chains are all important variables in providing tight control over many biological paradigms. There are clear roles for branched architectures in regulating proteasome-mediated degradation, however the proteins that selectively recognize and process these atypical chains are unknown. Here, using synthetic and enzyme-derived ubiquitin chains along with intact mass spectrometry, we report that UCH37/UCHL5, a proteasome-associated deubiquitinase, cleaves K48 branched chains. The activity and selectivity toward branched chains is markedly enhanced by the proteasomal Ub receptor RPN13/ADRM1. Using reconstituted proteasome complexes, we find that chain debranching promotes degradation of substrates modified with branched chains under multi-turnover conditions. These results are further supported by proteome-wide pulse-chase experiments, which show that the loss of UCH37 activity impairs global protein turnover. Our work therefore defines UCH37 as a debranching deubiquitinase important for promoting proteasomal degradation.
IN BRIEF
Using designer ubiquitin chains and intact mass spectrometry to characterize heterogeneous ubiquitin conjugates, Deol et. al. discovered that the ubiquitin C-terminal hydrolase UCH37/UCHL5 cleaves branched chains bearing K48 linkages. The debranching activity of UCH37 is enhanced by the proteasome subunit RPN13. Loss of UCH37 activity on the proteasome impedes the degradation of proteins modified with branched chains.
Graphical Abstract
INTRODUCTION
Ubiquitin (Ub) chains are a class of non-template-derived biopolymers with diverse structures that provide precise control over many biological pathways (Oh et al., 2018). The eight amino groups of Ub (M1, K6, K11, K27, K29, K33, K48, and K63) give rise to a wide assortment of chain types (Swatek and Komander, 2016; Yau and Rape, 2016). Each subunit can be conjugated to the C-terminus of another Ub molecule to afford single-linkage (homotypic) and mixed-linkage (heterotypic) chains. Further expanding the diversity, heterotypic chains exist in different configurations: linear/unbranched and branched, with the latter composed of subunits that are modified at more than one amino group with other Ub molecules. Failure to build and remove specific chains at the appropriate times can be detrimental to cellular function (Damgaard et al., 2016; Heger et al., 2018; Popovic et al., 2014; Zhou et al., 2016).
While efforts have largely focused on defining the functions of homotypic chains, heterotypic chains are emerging as important regulators of cellular pathways (Haakonsen and Rape, 2019). K11/K48 chains, for instance, increase in abundance during mitosis and proteotoxic stress to prioritize substrates for degradation by the 26S proteasome (Meyer and Rape, 2014; Samant et al., 2018; Yau et al., 2017). K29/K48 and K48/K63 chains have also been implicated in targeting proteins for degradation (Leto et al., 2019; Liu et al., 2017; Ohtake et al., 2018). M1/K63 chains, on the other hand, function independently of the proteasome to regulate the immune response (Emmerich et al., 2013; Wertz et al., 2015). Together, these results raise the possibility that heterotypic chains can be selectively recognized by Ub-binding domains (UBDs) (Husnjak and Dikic, 2012) and processed by deubiquitinases (DUBs) (Mevissen and Komander, 2017). While there is evidence some UBDs may prefer branched conjugates (Boughton et al., 2020), a DUB selective for branched chains has not yet been identified.
UCH37/UCHL5 is a cysteine protease and member of the small family of DUBs referred to as the Ub C-terminal hydrolases (UCHs). Deficiencies in UCH37 lead to embryonic lethality in mice (Al-shami et al., 2010) and overexpression has been found in several human cancers (Fang et al., 2012, 2013). At the cellular level, UCH37 has been implicated in TGF-β signaling (Wicks et al., 2005, 2006), Wnt signaling (Han et al., 2017), DNA double-strand break repair (Nishi et al., 2014), cell cycle progression (Randles et al., 2016), NF-κB activation (Mazumdar et al., 2010), and adipogenesis (Beekum et al., 2012). Most of these functions have been attributed to UCH37’s association with either the proteasome (Hamazaki et al., 2006; Jørgensen et al., 2006; Qiu et al., 2006; Yao et al., 2006) or INO80 chromatin remodeling complex (Yao et al., 2008). In the case of the proteasome, the dogma is that UCH37 trims homotypic K48 chains to rescue proteins from degradation (Lam et al., 1997). The kinetics of homotypic K48 chain cleavage, however, are exceedingly slow (Bett et al., 2015; Lu et al., 2017; Yao et al., 2006). Moreover, purified human proteasomes with UCH37 as the only non-essential DUB are unable to cleave K48 chains (Lu et al., 2015b). Thus, the function of UCH37 on the proteasome remains poorly understood.
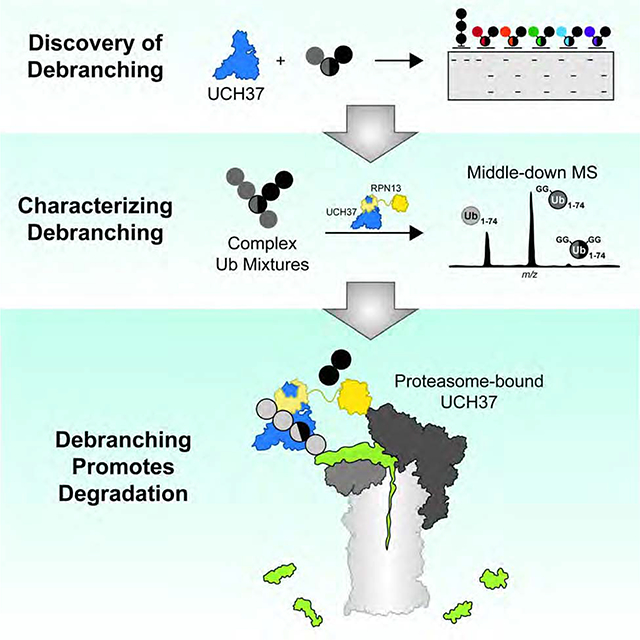
In the present study, we demonstrate that UCH37 exclusively cleaves branched Ub chains. Using a library of designer Ub chains, we show that K48 linkages are readily removed in branched chains. Middle-down mass spectrometry combined with quantitative linkage analysis reveals that the K48-specific debranching activity extends to complex chain mixtures. The proteasomal subunit RPN13/ADRM1 markedly enhances both the activity and selectivity of UCH37. In the context of the proteasome, debranching activity is not only retained but also important for the degradation of substrate proteins modified with K48-containing branched chains. Proteome-wide analysis of global protein turnover further substantiates these findings by demonstrating that proteins are turned over more rapidly in cells when active UCH37 is present. Our work therefore identifies UCH37 as a chain debranching enzyme and uncovers the mechanism by which UCH37 regulates proteasomal degradation.
RESULTS
UCH37 Cleaves K48 Branched Ub Trimers
Several years ago, our lab developed a method to synthesize a diverse set of Ub chains (Figure 1A) (Trang et al., 2012a; Valkevich et al., 2012a). Using this library, we identified branched conjugates bearing K48 linkages as potential targets of UCH37 (Figure 1B). To confirm these results, we also generated native branched trimers. We found that UCH37 cleaves the native K/K48 branched trimer in a time- and concentration-dependent manner (Figures 1C–D), suggesting branched Ub homooligomers could be the principal target of UCH37.
FIGURE 1. UCH37 Cleaves K48 Linkages in Branched Ubiquitin Trimers.

(A) Structures of native and thiol-ene coupling (TEC)-derived isopeptide bonds.
(B) SDS-PAGE analysis of cleavage reactions with TEC-derived branch tri-Ub derivatives. Concentrations used: tri-Ub (10 μM); UCH37 (1 μM). Linkages in branched tri-Ub are represented by X/Y, where X and Y denote the positions of the isopeptide bonds.
(C) SDS-PAGE analysis of K6/K48 branch tri-Ub (10 μM) cleavage with varying concentrations of UCH37 (0, 0.1, 0.25, 0.5, 1, 5, and 10 μM) at 1 h.
(D) SDS-PAGE analysis of the cleavage reaction time course. Concentrations used: native tri-Ub (10 μM); UCH37 (1 μM).
(E) Sortagging native K6/K48 branch tri-Ub with different fluorophores to report on the linkage specificity of UCH37.
(F) Fluorescence analysis of cleavage reactions with either OTUB1 (5 μM) or UCH37 (5 μM) and fluorophore-labeled native K6/K48 branch tri-Ub (10 μM).
See also Figure S1.
To further explore this unique reactivity, we wanted to determine whether UCH37 also exhibits linkage specificity. We used a sortagging approach to selectively label individual Ub subunits of a native chain with different fluorophores (Crowe et al., 2016a). When UCH37 encounters a branch point, there are two possible linkages that can be cleaved. The logic was that K48 cleavage should furnish a fluorescein-labeled di-Ub and TAMRA-labeled mono-Ub (Figure 1E). By contrast, scission of the K6 linkage should yield a TAMRA-labeled di-Ub and fluorescein-labeled mono-Ub. The results are consistent with K48 linkage specificity, as the products from UCH37-mediated cleavage mirror those generated by the K48 linkage specific DUB OTUB1 (Figures 1F, S1A–B). Replacing the non-K48 distal subunit with either GFP or SUMO abrogates cleavage, indicating that all three subunits in the chain must be Ub (Figures S1C–D). Together, our results show that UCH37 has the unprecedented ability to preferentially target K48 branch points.
UCH37 Removes K48 Branch Points in Complex Mixtures of Chains
Although Ub trimers are good model systems, they do not reflect the heterogeneity of ubiquitination observed in cells or in vitro. Thus, we sought to analyze the debranching activity of UCH37 in the context of heterogeneous chain populations where there is considerable variability in chain length and frequency of branch points. Identifying branch points in complex mixtures of chains, however, is challenging. We turned to middle-down mass spectrometry (MS), as this has proven to be a powerful method for identifying and characterizing branched conjugates (Swatek et al., 2019; Valkevich et al., 2014; Xu et al., 2008).
A series of enzymatic reactions were performed based on their ability to generate chains with a specific mixture of linkages (Figure 2A). In each case, high molecular weight (HMW) conjugates were isolated and minimally digested with trypsin to generate a mixture of Ub species representing mono-Ub or the end caps of a chain (Ub1–74), the linear portion of a chain (diGly-Ub1–74), and branch points (2xdiGly-Ub1–74). Analysis of the resulting Ub1–74 derivatives by middle-down MS reveals that the extent of branching varies from 4–14% of the total Ub population (Figures 2B–D; top spectra). According to electron transfer dissociation (ETD) analysis of the 2xdiGly-Ub1–74 peak, NleL generates K6/K48 branch points (Hospenthal et al., 2013), the combination of UBE2S (Bremm et al., 2010) and UBE2R1 forms K11/K48 branches, and UBE2N/UBE2V2 with UBE2R1 builds K48/K63 bifurcations (Nakasone et al., 2013) (Figures S2B–D).
FIGURE 2. UCH37 Removes K48 Branch Points in Complex Chain Mixtures.

(A) Assembly of high molecular weight Ub chain mixtures used in this study.
(B-D) Ub middle-down (Ub MiD) MS analysis of HMW K6/K48, K11/K48, and K48/K63 chains (top) treated with either OTUB1 (3 μM, middle) or UCH37 (3 μM, bottom) for 1 h. Percentages correspond to the relative quantification values of the 11+ charge state for each Ub species: Ub1–74, 1xdiGly-Ub1–74, and 2xdiGly-Ub1–74.
(E-G) Ub-AQUA analysis of HMW K6/K48, K11/K48, and K48/K63 chains before and after a 1 h treatment with OTUB1 (15 μM, middle) or UCH37 (3 μM, last). For all points, *P<0.025, **P<0.01 (Student’s T-test)
All MS spectra are representative traces and quantification values (E-G) are derived from averaging fits of 2 independent experiments shown with SEM. See also Figure S2 and Table S1A–C.
After the addition of UCH37 to HMW chains, middle-down MS shows a complete loss of the 2xdiGly-Ub1–74 species indicative of debranching (Figures 2B–D; bottom spectra). With K6/K48 chains, the disappearance of branched chains (2xdiGly-Ub1–74) coincides with an increase in the linear/unbranched chains (diGly-Ub1–74) (Figure 2B; bottom spectrum). Subjecting the same chains to OTUB1, which should be immune to chain architecture and cleave any K48 linkage, decreases the branch point in addition to the unbranched segments (Figure 2B; middle spectrum). With K11/K48 and K48/K63 chains, both UCH37-catalyzed debranching and the global removal of K48 linkages by OTUB1 afford an increase in Ub1–74 (Figures 2C–D; middle and bottom spectra). Chains built with non-K48 linkages, e.g., K11 and K63, are not targeted for cleavage by UCH37 (Figures S2E–F).
To determine whether UCH37 retains specificity toward K48 linkages in these complex chains, we used isotopically labeled, absolute quantitation (AQUA) peptide standards for each linkage of Ub (Kirkpatrick et al., 2006). HMW conjugates were analyzed both prior to and after the addition of OTUB1 and UCH37. The most significant changes occur in the K48 levels for each set of chains, indicating this linkage is the primary target of UCH37 (Figures 2E–G). The K48-specific debranching activity observed with model Ub trimers thus holds true for more complex chains.
RPN13 Enhances the Debranching Activity of UCH37
Our data with free UCH37 suggest it could function as a chain debranching enzyme; however, there is little evidence that UCH37 acts alone. One of its primary binding partners is the proteasomal Ub receptor RPN13 (Figure 3A) (Hamazaki et al., 2006; Jørgensen et al., 2006; Qiu et al., 2006; Yao et al., 2006). Previous studies have shown that RPN13 enhances UCH37’s ability to cleave the fluorogenic substrate Ub-AMC (Sahtoe et al., 2015; VanderLinden et al., 2015). Thus, we wanted to evaluate the effects of RPN13 on chain debranching. We first attempted to reconstitute the UCH37•RPN13 complex by mixing the purified recombinant proteins; however, we did not observe activity. We then co-purified UCH37 and full length RPN13 (Figure S3A). The resulting UCH37•RPN13 complex exhibits higher activity toward Ub-AMC, branched trimers, and HMW chains relative to free UCH37 (Figures 3B, 3D, S3B). Analysis of cleavage reactions with HMW chains confirmed that the UCH37•RPN13 complex displays the same reactivity toward K48 branch points as free UCH37 (Figures 3C, S3D–E).
FIGURE 3. Steady-State Kinetic Analysis of Chain Debranching.

(A) Constructs used in this study.
(B) Western blot analysis of cleavage reactions with HMW K6/K48 chains. Concentrations used: UCH37 (1 μM) or UCH37•RPN13 (1 μM). Immunoblotting with the α-Ub P4D1 antibody.
(C) Ub MiD MS analysis of HMW K6/K48 chains (top) followed by treatment with UCH37•RPN13 (1 μM, bottom) for 1 h. Percentages correspond to the relative quantification values of the 11+ charge state for each Ub species: Ub1–74, 1xdiGly-Ub1–74, and 2xdiGly-Ub1–74.
(D) SDS-PAGE analysis of debranching reactions with UCH37 (1 μM) or UCH37•RPN13 (1 μM).
(E) Michaelis-Menten plot for the hydrolysis of native K6/K48 branched tri-Ub (solid line) or K48 tri-Ub (dashed line) by either free UCH37 (black) or UCH37•RPN13 (red). Enzyme concentrations are 0.5 μM for native K6/K48 branched tri-Ub and 1 μM for K48 tri-Ub.
(F) Table of kinetic parameters measured for all experiments following the initial rates of di-Ub formation.
All kinetic curves are representative traces and constants (E-F) derived from averaging fits of 3 independent experiments shown with SD. See also Figure S3.
Encouraged by these results, we sought to obtain more quantitative information. With the native K6/K48 branched trimer as a substrate, we measured initial rates using a gel-based assay. Formation of di-Ub was monitored and the resulting data were fit to the Michaelis-Menten equation (Figure 3E). The steady-state parameters show that the presence of RPN13 confers a 3-fold decrease in Km and a 4-fold increase in kcat (Figure 3F). RPN13 therefore boosts the debranching activity of UCH37 by an order of magnitude from 1200 M−1•s−1 to 12000 M−1•s−1. Interestingly, RPN13 has little effect on the cleavage efficiency of homotypic K48 chains. In fact, comparing kcat/Km for the cleavage of K6/K48 tri-Ub and K48 tri-Ub reveals that selectivity for the branched trimer increases from 12- to 90-fold in the presence of RPN13.
The heightened activity bestowed on UCH37 by RPN13 has largely been attributed to the DEUBAD (DEUBiquitinase ADaptor) domain. The C-terminal DEUBAD domain is conserved among other UCH regulatory proteins (Sanchez-pulido et al., 2012) and is necessary and sufficient for enhancing UCH37-catalyzed hydrolysis of an artificial Ub-AMC substrate (Sahtoe et al., 2015; VanderLinden et al., 2015). Congruent with these results, we observed a ~6-fold rate enhancement in Ub-AMC hydrolysis in the presence of RPN13DEUBAD (Figure S3B). For comparison, full-length RPN13 stimulates hydrolysis by ~4-fold. In the chain debranching reaction, however, UCH37•RPN13DEUBAD exhibits ~2-fold lower catalytic efficiency relative to the full-length complex. This reduction can be ascribed to the absence of the Ub-binding PRU (Pleckstrin-like Receptor for Ub) domain since a similar drop in activity is observed when UCH37 is bound to a mutant form of RPN13 incapable of binding Ub (RPN13L56A/F76R/D79N; Lu et al., 2017; Schreiner et al., 2008) (Figure S3C).
Homotypic K48 Chains Inhibit Debranching
That UCH37•RPN13 cleaves homotypic K48 chains at all suggests unbranched chains might interfere with debranching activity. To test this, we first assessed whether UCH37 exhibits a binding preference for branched chains. Pulldown experiments were conducted with a panel of designer homotypic chains. We observed that inactive UCH37 C88A•RPN13 only interacts with K48 tri-Ub (Figure 4A). We then probed the binding to HMW chains using immobilized UCH37 C88A•RPN13 (Figure S4A). As shown by western blot analysis, UCH37 C88A•RPN13 binds both K6/K48 and K48/K63 chains (Figures S4B–C). Eliminating all K48 linkages prior to the pulldown abolishes binding. The removal of branch points with UCH37, however, has little effect on chain enrichment. Together, these results suggest UCH37•RPN13 exhibits a strong binding preference for K48 chains independent of the overall chain architecture.
FIGURE 4. UCH37 Preferentially Binds K48 Chains.

(A) SDS-PAGE analysis of pull-downs with M1-, K6-, K11-, K29-, K33-, K48-, and K63-linked Ub chains. Each chain (50 pmol) was captured using immobilized His-RPN13•UCH37 C88A (5 nmol).
(B) Schematic for measuring the hydrolysis of native K6/K48 branched tri-Ub in the presence of K48 tetra-Ub added in trans.
(C) Michaelis-Menten plot for the hydrolysis of native K6/K48 branched tri-Ub by UCH37•RPN13 (0.5 μM) in the presence of K48 tetra-Ub.
(D-E) ITC analysis of active UCH37•RPN13 binding to K48-linked tri-Ub (C) and tetra-Ub (D).
(F) Schematic for measuring the hydrolysis of native K6/K48 branched tetra-Ub in cis.
(G) Michaelis-Menten plot for the hydrolysis of native K6/K48 branched tetra-Ub by UCH37•RPN13 (0.5 μM).
All ITC curves are representative traces and reported Kds (B, C) are derived from averaging fits of 2 independent experiments. All kinetic curves (A, D) are representative traces and constants derived from averaging fits of 2 independent experiments with SD. See also Figure S4 and Table S1D.
Next, K48 tetra-Ub chains were added in trans at varying concentrations to reactions with K6/K48 tri-Ub as the substrate (Figure 4B). Global fitting of the steady-state kinetic data shows that K48 tetra-Ub acts as a competitive inhibitor (Figures 4C, S4D). The Ki of 0.7 μM for K48 tetra-Ub is approximately 31-fold lower than the Km for K6/K48 tri-Ub with no inhibitor. Analysis of binding by isothermal titration calorimetry (ITC) confirms that the active UCH37•RPN13 complex binds K48 tri- and tetra-Ub with rather high affinity (Kd = 0.33 μM for tri-Ub and 0.15 μM for tetra-Ub) (Figures 4D–E). Similar binding strengths are observed for the inactive UCH37 C88A•RPN13 complex, as evidenced by both ITC (Figures S4E–J) and fluorescence polarization (Du and Strieter, 2018). These results argue that homotypic K48 chains are potent inhibitors of debranching. Interestingly, when the K6/K48 tri-Ub is extended by one unit to generate another K48 linkage (Figure 4F), the steady-state kinetics remain similar to the trimer (Figure 4G), suggesting that a homotypic segment presented in cis does not inhibit debranching. Together, these findings suggest that homotypic K48 chains might act as regulators of UCH37-dependent chain debranching.
Proteasome-Bound UCH37 Debranches Chains
Since RPN13 recruits UCH37 to the proteasome, we wondered whether proteasome-bound UCH37 would also act as a debranching enzyme. Proteasomes (Ptsms) were purified from a HEK293 cell line stably expressing an affinity-tagged version of the proteasomal DUB RPN11 (HEK293RPN11-HTBH) (Figure 5A) (Wang et al., 2007). The other non-essential proteasomal DUB USP14 was removed under high salt conditions (Besche et al., 2009; Lee et al., 2010) to ensure that the observed DUB activity can be ascribed to UCH37 (Figures 5B, S5A). Adding purified Ptsms deficient in USP14, but replete with UCH37 (herein referred to as wild-type (WT) Ptsm), to HMW K6/K48 chains results in a concentration-dependent formation of shorter chains along with mono-Ub (Figure 5C). As evidenced by intact MS, the formation of lower MW species is due to the removal of branch points (Figure 5D). A similar loss of branch points is observed with HMW K11/K48, and K48/K63 chains, indicating that Ptsm-catalyzed debranching is not idiosyncratic to K6/K48 chains (Figures S5C–E).
FIGURE 5. Proteasome-Bound UCH37 Debranches Ub Chains.

(A) Purification scheme for the isolation of various human proteasome (Ptsm) complexes.
*Ptsms are labeled as wild-type (WT); displaced by RPN2 peptide (UCH37•RPN13-depleted); UCH37 deficient (ΔUCH37); RPN13 deficient (ΔUCH37/ΔRPN13); UCH37•RPN13-replenished = WT-r (active UCH37•RPN13) and C88A-r (inactive UCH37 C88A•RPN13)
(B) Western blot and Native PAGE (26S) analysis of purified Ptsm complexes (2 μg).
(C) Western blot analysis of debranching reactions with HMW K6/K48 chain at 1 h. Reactions were performed with two different concentrations of Ptsms (2 and 5 μg). Α-Ub P4D1 antibody used for visualization.
(D) Ub MiD MS analysis of HMW K6/K48 chains subjected to each of the indicated Ptsm complexes (10 μg) for 4 h. Percentages correspond to the relative quantification values of the 11+ charge state for each Ub species: Ub1–74, 1xdiGly-Ub1–74, and 2xdiGly-Ub1–74.
(E) Time course analysis of the removal of branch points from HMW K6/K48 chains by WT Ptsm (10 μg, black) and UCH37•RPN13-replenished Ptsm (10 μg, blue) (top). The 11+ charge state of the 2xdiGly-Ub1–74 species was used to measure the relative abundance of 2xdiGly-Ub1–74 at each time point.
All curves are averaged representative traces from 2 independent experiments with SD. Table of kinetic parameters (kobs) obtained from fits to a first-order decay (bottom). Observed rate constants are normalized based on the concentration of UCH37. See also Figure S5 and Table S1E.
To then ascertain whether the debranching activity of purified Ptsms is due to UCH37, we used two orthogonal approaches. First, we displaced UCH37 and RPN13 from WT Ptsm complexes using a 38-mer peptide from the scaffolding subunit RPN2 (RPN2916−953) (Figure 5A) (Lu et al., 2015a). Subjecting the resulting UCH37•RPN13-depleted Ptsms to the fluorogenic Suc-LLVY-AMC peptide shows cleavage activity similar to WT Ptsms (Figure S5B; bottom). Ub-AMC hydrolysis, however, is severely compromised, supporting the notion that UCH37 is the only non-essential DUB on purified WT Ptsms (Figure S5B; top). Second, we used CRISPR/Cas9 genome editing to remove UCH37 from HEK293RPN11-HTBH cells (Figure 5A). Ptsms purified from these cells are devoid of UCH37 (ΔUCH37 Ptsm), but still capable of cleaving Suc-LLVY-AMC. Like UCH37•RPN13-depleted Ptsms, ΔUCH37 Ptsms display little activity toward HMW chains according to both western blot and intact MS (Figures 5C–D, S5C–E), suggesting UCH37 is responsible for the debranching activity.
For further corroboration, recombinant UCH37•RPN13 complexes were added back to ΔUCH37/ΔRPN13 Ptsms during the purification (Figure 5A). As shown by Native PAGE and western blot, ΔUCH37/ΔRPN13 Ptsms can indeed be reconstituted with either active or inactive forms of UCH37•RPN13 (Figure 5B). Interrogating either UCH37•RPN13-replenished Ptsms (WT-r Ptsms) or UCH37 C88A•RPN13-replenished Ptsms (C88A-r Ptsms) with HMW chains shows that chain debranching occurs with catalytically active UCH37, but not inactive UCH37 C88A (Figure 5C–D, S5C–E). Thus, we conclude that Ptsm-bound UCH37 debranches Ub chains.
Kinetics of Proteasome-Bound UCH37 Resemble the UCH37•RPN13 Complex
Steady-state kinetic data with the UCH37•RPN13 complex showed that the rate of debranching occurs with a turnover number of 16–19 min−1, which means a K48 branch point can be removed within 3–4 s (1/kcat). Assuming the activity of the UCH37•RPN13 complex reflects the Ptsm-bound form, we would expect the kinetics of debranching to be commensurate with degradation rates, as several studies suggest it takes the proteasome tens of seconds to clear a ubiquitinated protein (Bard et al., 2019; Lu et al., 2015b; Peth et al., 2013). We tested this by determining the relative rates of debranching by UCH37•RPN13 and Ptsm-bound UCH37.
Middle-down MS was used to monitor the loss of branch points from HMW chains at different time points using UCH37•RPN13, WT Ptsms and active WT-r Ptsms. These data show that branch points in K6/K48 and K48/K63 chains are rapidly depleted upon addition of UCH37•RPN13, WT Ptsm, and active WT-r Ptsm (Figures 5E, S3F, S5H). After normalizing for differences in the concentration of UCH37, we find the observed rate constants are similar for UCH37•RPN13 and the Ptsm complexes (Figures 5E, S3F, S5F), suggesting the turnover number measured with the complex could reflect the activity of UCH37 on the proteasome.
While the overall reactivity patterns between UCH37•RPN13 and the proteasome complexes are largely the same with K6/K48 and K48/K63 chains, this is not true for K11/K48 chains. In the presence of UCH37•RPN13, the K11/K48 branch point is consumed within 15 min and there is little change in the unbranched portion of the chain even after 2 h (Figures S3F–G). With Ptsm complexes, the K11/K48 branch point is consumed rapidly, similar to reactions with UCH37•RPN13. After a slight delay, however, the unbranched portion also appears to decrease in abundance (Figure S5I; Table S1E). Cleavage of both segments of the chain is dependent on the catalytic activity of UCH37. These results indicate that while UCH37•RPN13 and Ptsm-bound UCH37 display similar debranching kinetics, K11/K48 chains are dismantled to a greater extent in the presence of the Ptsm. As discussed in more detail below, this additional activity could be the result of proteasomal degradation.
Chain Debranching Regulates Degradation
With data suggesting chain debranching occurs on a timescale relevant to proteasome-mediated degradation, we tested the impact of debranching on protein turnover. To do this, we generated a substrate modified with branched chains. While synthesizing K11/K48 chains, we found that the E2 fusion protein UBE2S-UBD (Bremm et al., 2010) modifies itself at three different positions: K117, K178, and K184 (Figure 6A; Table S1F). To determine whether branched chains are tethered to one or more of these sites, we appended a FLAG tag to UBE2S-UBD to separate the autoubiquitinated conjugates from unanchored chains. Middle-down MS analysis shows the extent of branching on UBE2S-UBD is nearly the same as in the bulk (~9%), heterogenous mixture of HMW K11/K48 conjugates (Figure 6A; bottom spectrum). Thus, we decided to use polyubiquitinated UBE2S-UBD (K11/K48 Ubn-UBE2S-UBD) as a substrate for the Ptsm to measure the effect of debranching on degradation.
FIGURE 6. Ubiquitin Chain Debranching Promotes Proteasomal Degradation.

(A) UBE2S-UBD construct (top) showing ubiquitination sites (K117, K201, and K207) and the proteasome-derived UBE2S-UBD peptides detected by parallel reaction monitoring (PRM) MS. Ub MiD MS analysis of K11/K48-Ubn-UBE2S-UBD-FLAG (bottom).
(B) Degradation of K11/K48-Ubn-UBE2S-UBD after 1 h using the indicated Ptsm complexes (5 μg) under multi-turnover conditions in the presence and absence of bortezomib (1 μM).
(C) Schematic for tracking the formation of specific transition ions of UBE2S-UBD-derived proteolytic peptides. All reactions are completed as a two-plex experiment using ubiquitinated light and heavy (15N) UBE2S-UBD with different Ptsm complexes.
(D-E) PRM MS analysis of UBE2S-UBD peptides formed by Ptsms. The UBE2S peptide FPASPPKGY is shown on the left and the UBD peptide FDGSGGNNHAVE is on the right. (D) Light and heavy UBE2S-UBD were mixed with either WT Ptsm (5 μg) or ΔUCH37 Ptsm (5 μg), respectively, in the presence and absence of MG132 (10 μM). (E) Light and heavy UBE2S-UBD were mixed with either WT-r Ptsm (5 μg) or C88A-r Ptsm (5 μg).
(F) K48/K63-Ubn-titin-I27V15P-23-K-35 construct (top) used in this study. Ub MiD MS analysis of K48/K63-Ubn-titin-I27V15P-23-K-35 (middle) followed by treatment with WT Ptsm (10 μg, bottom).
(G) Degradation of K48/K63-Ubn-titin-I27V15P-23-K-35 after 1 h using the indicated Ptsm complexes (5 μg) under multi-turnover conditions in the presence and absence of bortezomib (1 μM). * indicates non-ubiquitinated titin-I27V15P-23-K-35 fragments Ub MiD MS percentages correspond to the relative quantification values of the 11+ charge state for each Ub species: Ub1–74, 1xdiGly-Ub1–74, and 2xdiGly-Ub1–74. Multiple-turnover assays were tracked by tricine-SDS-PAGE and visualized by cy5 fluorescence.
All MS curves are representative depictions from the sum of the transition ions of each monitored peptide (D-E) with a dashed line connecting the averaging of 4 independent experiments with SD. See also Figure S6 and Table S1F–G.
K11/K48 Ubn-UBE2S-UBD was fluorescently labeled using a cysteine-reactive Cy5 dye and degradation by different Ptsm complexes was monitored by SDS-PAGE under multi-turnover conditions. The HMW bands corresponding to K11/K48 Ubn-UBE2S-UBD disappear in the presence of both WT Ptsm and UCH37•RPN13-replenished Ptsm, but not with ΔUCH37 or UCH37 C88A•RPN13-replenished Ptsms (Figures 6B, S6A). The appearance of peptides also depends on the activity of UCH37.
To confirm the formation of UBE2S-UBD-derived peptides, we used parallel reaction monitoring (PRM) MS (Figure 6C). Two peptides from separate regions of UBE2S-UBD–one from UBE2S (FPASPPKGY) and another from the UBD (FDGSGGNNHAVE)–could be detected reproducibly. Both peptides are products of the Ptsm, as inhibition with MG132 abolishes their formation. We then directly compared the abundance of peptides at various time points with different Ptsm complexes by treating the ‘light’ form of K11/K48 Ubn-UBE2S-UBD with one Ptsm complex and the ‘heavy’ form (K11/K48 Ubn-15N-UBE2S-UBD) with another and mixing the resulting peptides. Labeling UBE2S-UBD with 15N does not affect the nature of chains it attaches to itself (Figure S6B). The PRM results show that peptide formation is more robust with WT Ptsms compared to reactions with ΔUCH37 Ptsms (Figures 6D, S6C). This difference can be ascribed to the activity of UCH37 since peptides accumulate to a greater extent when Ptsms contain active UCH37, but not inactive UCH37 C88A (Figures 6E, S6D).
While these data indicate that UCH37 is important for degradation, they do not specifically implicate the debranching activity. We sought to address this by first treating K11/K48 Ubn-UBE2S-UBD with UCH37•RPN13 for one hour to remove branch points (Figure S3F). Unbranched chains remain largely intact during this time period (Figure S3G). UCH37•RPN13 was then inactivated through multiple freeze/thaw cycles before adding ΔUCH37 Ptsms. PRM data shows that prior removal of branch points is sufficient to rescue the activity of ΔUCH37 Ptsms (Figures S6E–F). Together, these results indicate that chain debranching is important for promoting degradation.
To assess whether the degradation of other substrates depends on the activity of UCH37, we availed a well-established model system for Ptsm degradation—titin-I27V15P-23-K-35 (Bard et al., 2019). K48/K63 branched chains were attached to the single C-terminal lysine of titin-I27V15P-23-K-35 using a combination of Rsp5 and UBE2R1 (Figures S6G). Since WT Ptsms are able to debranch the titin-I27-anchored chains (Figure 6F), degradation was then monitored by SDS-PAGE and visualized by labeling cysteine-135 of titin-I27V15P-23-K-35 with Cy5. Like degradation reactions with K11/K48 Ubn-UBE2S-UBD, WT Ptsms breakdown K48/K63 Ubn-titin-I27V15P-23-K-35 conjugates and catalyze robust peptide formation (Figures 6G; S6H). ΔUCH37 Ptsms, by contrast, do not decrease the polyubiquitinated species nor generate peptide products. A comparison of the reactions with replenished Ptsms shows that the polyubiquitinated species are consumed and peptides are produced to a greater extent with active UCH37. Thus, the ability of UCH37 to promote degradation by removing branch points applies to different substrates and different K48 branched chains.
UCH37 Potentiates Proteasomal Degradation In Cells
The abundance of Ub conjugates is often used to monitor the status of Ptsm activity in cells (Bennett et al., 2007). We reasoned that if UCH37 potentiates degradation by removing branch points, Ptsm activity would be muted in UCH37KO cells relative to WT, resulting in the accumulation of Ub conjugates. To test this, the abundance of Ub conjugates was probed using a K11/K48 bispecific antibody (Yau et al., 2017), a K48-specific antibody, and a pan Ub antibody. We find that the levels of Ub conjugates are slightly elevated in UCH37KO cells compared to WT (Figures 7A, S7A), consistent with a recent report in HCT116 cells (Osei-Amponsa et al., 2020). Overexpressing active UCH37 in UCH37KO cells does not completely restore conjugates to WT levels (Figure 7A), but this result likely does not reflect the absence of a rescue. Due to the chain binding properties, supernumerary levels of UCH37 could sequester Ub chains, leading to an observed accumulation. In accordance with this idea, higher levels of conjugates are also observed in WT cells overexpressing UCH37 compared to the empty vector control (Figure 7A). What is important to compare is the abundance of Ub conjugates in UCH37KO cells overexpressing either inactive (C88A or C88S) or active UCH37. Since conjugates accumulate to a much greater extent with the inactive forms (Figure 7A), we assert that the buildup of chains in UCH37KO cells is due to the loss of UCH37 activity.
FIGURE 7. UCH37 Potentiates Proteasomal Degradation in Cells.

(A) Steady-state levels of Ub conjugates were assessed in WT and UCH37KO cells using the indicated antibodies. WT and UCH37KO cells were transfected with inactive (C88A and C88S) and active forms of UCH37 and the levels of Ub conjugates were also probed.
(B) Schematic for GFPu turnover in cells.
(C) Fluorescence histograms of WT HEK293 GFPu cells, UCH37KO GFPu cells, and UCH37KO GFPu cells expressing either WT UCH37 (UCH37KO + WT UCH37) or inactive UCH37 (UCH37KO + UCH37 C88S). Mean GFP fluorescence measured in 2 independent experiments.
(D) Schematic for measuring global protein turnover using azidohomoalanine (AHA) labeling of newly synthesized proteins in cells. AHA incorporated proteins were labeled with DBCO-Cy5, separated by SDS-PAGE and visualized by Cy5 fluorescence.
(E) Cy5 fluorescence analysis of the turnover of AHA-labeled proteins at different chase times in WT, UCH37KO, and UCH37KO HEK293 FT cells expressing WT UCH37 or inactive UCH37 C88S (top) with corresponding Sypro total protein stain (bottom).
(F) Densitometric quantification of AHA labeled proteins with molecular weights ≤ 50 kDa. Values at each time point are adjusted relative to total protein. Results are shown as mean ± SEM from 3 independent experiments.
(G) Model showing UCH37-catalyzed Ub chain debranching promotes proteasomal degradation.
To further assess the impact of UCH37 on Ptsm-mediated degradation in cells, we turned to a well-established model substrate that is known to be modified with branched chains. GFPu was developed as a cellular reporter of Ptsm activity (Bence et al., 2001) and is polyubiquitinated with K29/K48 branched chains to promote degradation through the endoplasmic reticulum-associated degradation (ERAD) pathway (Leto et al., 2019). Cells stably expressing GFPu were edited with CRISPR/Cas9 to remove UCH37. The levels of GFPu in the resulting UCH37KO cells were measured by flow cytometry and compared to WT (Figure 7B). The data reveal the stability of GFPu is enhanced in the absence of UCH37 (Figures 7C and S7B). GFPu-expressing UCH37KO cells were then complemented with either active or inactive UCH37. Only the expression of active UCH37 is able to shift GFPu levels closer to WT (Figure 7C). These results suggest that UCH37 promotes the degradation of a model substrate in cells.
We then wanted to measure the turnover of endogenous proteins in the presence and absence of UCH37. Pulse-chase experiments were performed on a proteome-wide scale using the unnatural amino acid azidohomoalanine (AHA). AHA is activated by tRNAMet-synthetase and incorporated into newly synthesized proteins (NSPs). Previous studies have shown that AHA labeling does not affect protein stability nor does it induce ribosomal fall-off (Dieterich, et al., 2006; McShane et al., 2016; Howden, et al., 2013). After a one-hour pulse with AHA, cells were chased in AHA-free medium containing excess methionine to prevent reincorporation of AHA into new proteins (Figure 7D). NSPs were visualized after conjugation to a Cy5 dye using strain-promoted click chemistry and quantified by densitometry (Figures 7E; top). The resulting values were adjusted relative to total protein (Figures 7E; bottom). As shown with WT HEK293 and UCH37KO cells complemented with active UCH37, AHA-labeled proteins with molecular weights ≤50 kDa are depleted quite rapidly within the first two hours when short-lived proteins are turned over (Figure 7F; Table S1H). Similar behavior is not observed in cells deficient of UCH37 or expressing the inactive form. The difference between WT and knockout/inactive-expressing cells at the later time points–during which the bulk of protein degradation occurs (Zhao et al., 2015)–is even more pronounced (Figure 7F). Together, the biochemical and cellular data have led us to conclude that UCH37 acts as an important regulator of proteasomal degradation by debranching Ub chains (Figure 7G).
DISCUSSION
In this study, we identify UCH37 as a DUB capable of debranching Ub chains. The remarkable topological specificity of UCH37 occurs in conjunction with K48 linkage selectivity. When UCH37 is bound to RPN13 alone or in the context of the proteasome, the rate of debranching is dramatically enhanced. This activity has surprising consequences on protein turnover, as our work reveals that UCH37 removes branch points to enhance proteasomal degradation.
The prevailing view is that UCH37 contributes to the substrate selection process by trimming Ub chains to reduce the residence time on the proteasome (Collins and Goldberg, 2017; de Poot et al., 2017). This supposition has been based on the premise that UCH37 cleaves homotypic K48 chains; however, cleavage of homotypic K48 chains is quite slow (Bett et al., 2015; Lu et al., 2017; Yao et al., 2006). If the purpose is to rescue poorly ubiquitinated proteins from the fate of the proteasome, then UCH37 would have to act quickly. Take for example, the other non-essential cysteine-dependent DUB that associates with the proteasome—USP14. Evidence suggests that USP14 functions before the commitment step (Lee et al., 2016), which does not occur until an unstructured region of the substrate is inserted into the AAA+ ATPase motor of the 19S regulatory particle and the proteasome adopts a conformation conducive to proteolysis (Bard et al., 2019; Lu et al., 2015b). These steps are estimated to take only a few seconds, which means USP14 operates on a much faster timescale and UCH37 would have to as well in order to oppose the degradation process.
By identifying K48 branched chains as targets, we can develop a better picture of how UCH37 fits into the kinetic scheme of degradation. Steady-state kinetic analyses suggest that UCH37 in complex with RPN13 selectively removes K48 branch points every ~3 s (1/kcat) and the level of activity on the proteasome is similar. Thus, in light of the detailed kinetics that have been reported for degradation (Bard et al., 2019; Lu et al., 2015b), Ub chain debranching would occur around the same time as either the commitment step or deubiquitination by the intrinsic metalloprotease RPN11/POH1. Based on this logic, UCH37 would not have a chance to debranch chains prior to the commitment step.
How does Ub chain debranching stimulate degradation? The lid of the proteasome houses multiple Ub receptors (RPN13, RPN10, and RPN1) providing a versatile recognition platform for substrates modified with different Ub chains (Deveraux et al., 1994; Husnjak et al., 2008; Schreiner et al., 2008; Shi et al., 2016). Given the multivalency and high local concentration of Ub in branched chains, cooperation between receptors could promote high avidity interactions. According to a recent study, it may not be necessary for multiple receptors to engage branched chains since RPN1 binds K11/K48 branched chains with higher affinity than the linear counterparts (Boughton et al., 2020). Branched chains could thus prioritize substrates for the proteasome at the initial binding stage. However, affinity does not always correlate with degradation efficiency. Longer chains, for example, bind more tightly to the proteasome than shorter chains, yet degradation rates for substrates modified with either long or short chains are nearly the same (Martinez-Fonts et al., 2020). Debranching could be necessary to take full advantage of the avidity afforded by branched chains yet still ensure efficient degradation. It is possible that branched chains bind in a configuration that impedes the proteasome from transitioning into a catalytically competent conformation or RPN11-mediated deubiquitination (Hyoung et al., 2007; Kim et al., 2009). The removal of branch points could mitigate this problem. Another possibility is that branched chains could slow down product release. Debranching would then free the proteasome for additional rounds of proteolysis.
The ability of a DUB to selectively target branch points for removal is unprecedented. How UCH37 selects K48 branch points from a sea of unbranched chains is unclear. Considering UCH37 engages both branched and unbranched K48 chains, there must be a mechanism for UCH37 to interrogate subtle differences in chain topology. Understanding the molecular details of debranching could provide insight into the design of effective inhibitors of UCH37, the development of which is even more pressing given its role in promoting proteasomal degradation—a process many cancer cells rely on for survival (Deshaies, 2014).
LIMITATIONS
Characterizing the precise architecture of Ub chains attached to cellular proteins remains a formidable challenge. Thus, we cannot rule out the possibility that protein turnover defects in UCH37 deficient cells are due to the loss of other chain editing activities, e.g., the cleavage of unbranched chains, instead of debranching. Our biochemical data do show, however, that pre-treatment with UCH37•RPN13, which results primarily in the loss of branch points, rescues substrate degradation by ΔUCH37 Ptsms. Because the loss of UCH37 function has similar effects in vitro and in cells, our data support a model in which UCH37 debranches Ub chains to promote degradation. For further validation, it will be necessary to characterize the chains that accumulate on cellular substrates whose stability is regulated by UCH37. We also cannot dismiss the possibility that the observed turnover defects are due to the activity of UCH37 in the INO80 complex instead of the proteasome.
STAR*METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Eric R. Strieter (estrieter@umass.edu)
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
Original/source data are available on Mendeley data (https://data.mendeley.com/datasets/pv8t3hbg4t/1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian Cell Culture
HEK293 cells stably expressing His-biotin affinity tagged human RPN11 (RPN11-HTBH) (Wang et al., 2007), HEK293FT, and HEK293 GFPu cells were cultured at 37 °C under 5% CO2 using high glucose DMEM supplemented with 10% FBS, 1xGlutamax (Gibco), and 1xPen/Strep.
Generation of CRISPR KO Cell Lines
Guide RNAs were designed for UCH37 and RPN13 using design tools from Harvard and the Broad Institute. The gRNAs were purchased from IDT to be used in their Alr-R® CRISPR-Cas9 system. CRISPR reactions were performed according to the protocol provided by IDT. Briefly, a 1:1 annealed complex of gRNA: tracrRNA was prepared by mixing 1 μM of each RNA in nuclease-free duplex buffer and warming to 95 °C for 5 min before cooling to room temperature. Once the annealed complex was prepared, a ribonucleotide-protein complex (RNP) was prepared by mixing the RNA complex with Alt-R® S.p. Cas9 Nuclease in Opti-MEM media and incubating at room temp for 5 min. A transfection containing the RNP mixture was then prepared by diluting the RNP in Opti-MEM, adding Lipofectamine RNAimax, briefly vortexing, and allowing the mixture to incubate for 20 min at room temperature. The transfection mixture was then added to a 48 well plate followed by 80,000 cells in antibiotic free media. These cells were allowed to grow for 2 days before trypsinization and dilution to single cell per well density in 96 well plates. Single colonies were identified and screened for protein expression by western blot using appropriate antibodies. Colonies that showed loss of UCH37 or RPN13 were further screened by sequencing and analysis using TIDE software. Colonies with confirmed Indels were used in all subsequent experiments.
Lentivirus Packaging, Infection and Cell Line Creation
HEK293 FT packaging cells were seeded in 60 mm dishes four days prior to transfection at a density of 200,000 cells. HEK293 FT cells were transiently transfected with a mix of 3 μg packaging vector (psPAX2), 2 μg envelope vector (pMD2.G), and 3 μg pINDUCER21 vector containing the ORF corresponding to either WT UCH37 or UCH37 C88S in Opti-MEM (Gibco) using Lipofectamine 3000 following the recommended protocol. At 48 h post-transfection, the cell culture media was collected and filtered through a 0.45 μm filter. 9 mL of lentivirus-containing supernatant was combined with 9 mL of DMEM to reach a final concentration of 0.4μg/mL polybrene. UCH37KO HEK293 FT cells were seeded in T25 flasks at a density of 420,000 cells and incubated under standard cell culture conditions until 60% confluency was reached. A total of 18 mL of lentivirus was used for infection; 6 mL of lentivirus was added to cells every 12 h for a total of 36 h. 24 h after the last infection, the supernatant was removed, and cells were grown in fresh DMEM for another 24 h. Infected cells were selected by flow cytometry using the BD FACSAria II SORP cell sorter (BD Biosciences) equipped with 355 (UV), 405, 488, 561, and 640 nm lasers with 16 color analysis capabilities for the detection of high eGFP expression in the target cells.
Minor modifications were made in the transduction of UCH37KO GFPu cells. Packaging cells were plated at 640,000 cells 2 days prior to transfection. Target cells were seeded at 560,000 cells, 24 h prior to infection. Polybrene was used at 4 μg/mL. Two infections were performed, 12 h apart. The pCW57-MCS1-P2A-MCS2 (RFP) transfer plasmid containing the ORF corresponding to WT UCH37 or UCH37 C88S was used, allowing for selection of RFP positive cells without affecting the GFPu readout. The top 0.5–3% of RFP positive cells were sorted.
METHOD DETAILS
Protein Purification
All plasmids used for purification are described in the key resources table. Proteins were purified at 4 °C unless otherwise indicated. After the final chromatography step, all proteins were concentrated in Amicon Ultra spin concentrators, aliquoted, flash frozen in liquid N2 and stored at −80 °C. Concentrations of all proteins were determined by BCA assay.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti UCH37 | Abcam | ab124931 |
| Anti RPN11 | Abcam | ab109130 |
| Anti RPN13 | Cell Signaling Technology | D9Z1U |
| Anti RPT2 | Abcam | ab3317 |
| Anti PSMB7 | R&D Systems | MAB7590 |
| Anti USP14 | Abcam | ab56210 |
| Anti beta Actin | Abcam | ab8227 |
| Anti Ub | Enzo Lifesciences | BML-PW0930 |
| Anti K48-linkage Specific | Cell Signaling Technology | D9D5 |
| Anti Ub, K11/K48 Bispecific | Genentech | N/A |
| Goat Anti Mouse IR Dye 800CW | LI-COR Biosciences | 926-32210 |
| Goat Anti Rabbit IR Dye 680RD | LI-COR Biosciences | 926-68071 |
| Goat Anti Rabbit IR Dye 800CW | LI-COR Biosciences | 926-32211 |
| Goat Anti Human IR Dye 680LT | LI-COR Biosciences | 926-68032 |
| Bacterial and Virus Strains | ||
| Rosetta 2(DE3)pLysS | EMD Millipore Novagen | 71403-3 |
| BL21(DE3)pLysS | Promega | L1191 |
| One Shot™ TOP10 | Fisher Scientific | C404003 |
| One Shot™ Stbl3 | Fisher Scientific | C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ProBlock Gold Mammalian Protease Inhibitor Cocktail | Gold Biotechnology | GB-331-5 |
| Simple Stop™ 2 Phosphatase Inhibitor Cocktail | Gold Biotechnology | GB-451 |
| Ammonium Chloride (15N, 99%) | Cambridge Isotope Laboratories | NLM-467 |
| SYPRO Ruby Stain | Fisher Scientific | S12000 |
| Cy5 maleimide | Lumiprobe | 23080 |
| DBCO-Cy5 | Sigma-Aldrich | 777374 |
| Click-iT™ AHA (L-Azidohomoalanine) | Fisher Scientific | C10102 |
| Trypsin | Promega | V5113 |
| Chymotrypsin | Promega | V1061 |
| Formic Acid | Sigma-Aldrich | 399388 |
| Acetic Acid | Fisher Scientific | 351269-4 |
| Creatine phosphate disodium salt | Abcam | ab146255 |
| Creatine Kinase | Sigma-Aldrich | 10127566001 |
| Adenosine-5’-triphosphate | Gold Biotechnology | A-081-5 |
| Ub-AMC | Boston Biochem | U-550 |
| Suc-LLVY-AMC | Boston Biochem | S-280 |
| MG132 | Fisher Scientific | 508339 |
| Bortezimib | Selleck Chemicals | S1013 |
| Polybrene | Sigma-Aldrich | TR-1003-G |
| Lipofectamine 3000™ | Fisher Scientific | L3000008 |
| Doxycycline hyclate | Sigma-Aldrich | D9891 |
| L-Methionine | Sigma-Aldrich | 64319-25G-F |
| Carfilzomib (PR-171) | Selleck Chemicals | S2853 |
| AQUA peptides | Cell Signaling Technology | see Table S1 |
| Critical Commercial Assays | ||
| Alt-R® S.p. Cas9 Nuclease 3NLS | Integrated DNA Technologies | 1081058 |
| PTMScan® Ubiquitin Remnant Motif | Cell Signaling Technology | 14482 |
| pENTR™/SD/D-TOPO™ Cloning Kit | Fisher Scientific | K242020 |
| Gateway LR Clonase II Enzyme Mix | Fisher Scientific | 11-791-020 |
| Deposited Data | ||
| Raw and analyzed data | This study; Mendeley Data | http://dx.doi.org/10.17632/pv8t3hbg4t.1 |
| Experimental Models: Cell Lines | ||
| HEK293 Expressing Rpn11-HTBH | Applied Biological Materials | T6007 |
| HEK293 Expressing Rpn11-HTBH UCH37 KO | This study | N/A |
| HEK293 Expressing Rpn11-HTBH RPN13 KO | This study | N/A |
| HEK293 FT | ATCC | CRL-3216 |
| HEK293 FT UCH37 KO | This study | N/A |
| HEK293 GFPu | ATCC | CRL-2794 |
| HEK293 GFPu UCH37 KO | This study | N/A |
| Oligonucleotides | ||
| CRISPR KO sgDNA sequence UCH37 (1) | Integrated DNA Technologies | GTTACTGAACTGTACCCACC |
| CRISPR KO sgDNA sequence UCH37 (2) | Integrated DNA Technologies | CGCCTAAATGGACATCCTGG |
| CRISPR KO sgDNA sequence RPN13 | Integrated DNA Technologies | CACGAACTCTCTGCGCTAGG |
| Recombinant DNA | ||
| pMCSG20: NleL (aa 170-782) | Valkevich et al., 2014 | N/A |
| pQE30: SortaseΔN25 (SrtA) | Crowe et al., 2016 | gifted from O. Schneewind |
| pVP16: UCH37 | DNASU | HsCD00084019 |
| pET19: RPN13 | Yao et al., 2006 | Addgene, Plasmid #19423 |
| pGEX-6P1: hRPN2 (aa 916-953) | Lu et al., 2015a | gifted from K. Walters |
| pET28b: E1 | Trang et al., 2012 | N/A |
| pGEX-4T2: UBE2D3 | Valkevich et al., 2014 | N/A |
| pGEX-6P1: UBE2S-UBD | Bremm et al., 2010 | Addgene, Plasmid #66713 |
| pGEX-6P1: AMSH | Trang et al., 2012 | N/A |
| pOPINK: OTUD1 | Mevissen et al., 2013 | Addgene, Plasmid #61405 |
| pVP16: OTUB1 | Pham et al., 2015 | N/A |
| pOPINS: UBE3C | Michel et al., 2015 | Addgene, Plasmid #66711 |
| pDEST17: UBE2R1 | Pham et al., 2015 | N/A |
| pST39: UBE2N/UBE2V2 | Pham et al., 2015 | N/A |
| pET28b: UBC1 | DNASU | ScCD00009212 |
| pOPINK: RSP5 | DNASU | ScCD00008707 |
| pOPINS: Titin I27V15P 23-K-35 | Bard et al., 2019 | N/A |
| pET22b: Ub and Ub variants | Valkevich et al., 2012 | N/A |
| pET28b: SUMO2 | Mikolajczyk et al., 2007 | Addgene, Plasmid #25102 |
| pET22b: GFP | Dantuma et al., 2000 | Addgene, Plasmid #11938 |
| pMD2.G | gifted from Didier Trono | Addgene, Plasmid #12259 |
| psPAX2 | gifted from Didier Trono | Addgene, Plasmid #12260 |
| pINDUCER21 | Meerbrey et al., 2011 | Addgene, Plasmid #46948 |
| pCW57-MCS1-P2A-MCS2 (RFP) | Barger et al., 2019 | Addgene, Plasmid #80923 |
| Software and Algorithms | ||
| Typhoon FLA 9500 | GE Healthcare | N/A |
| Odyssey CLx Imager | LICOR Biosciences | N/A |
| Image Studio software | LICOR Biosciences | N/A |
| Prism 8 | Graphpad Software | N/A |
| OriginLab 7 SR4 | OriginLab Corporation | N/A |
| Xcalibur 3.0 | Thermo Fisher Scientific | N/A |
| Pinpoint 1.4 | Thermo Fisher Scientific | N/A |
| Proteome Discoverer 2.3 | Thermo Fisher Scientific | N/A |
| Mash Suite | Guner et al., 2014 | N/A |
| FlowJo 10.4 | FlowJo, LLC | N/A |
| Other | ||
| His60 Ni Superflow resin | Clontech | 635660 |
| Glutathione resin | GenScript | L00206 |
| Amylose resin | NEB | E8021S |
| Streptavidin resin | GenScript | L00353 |
| Anti-Flag M2 Affinity Gel | Sigma-Aldrich | A2220 |
| Slide-A-lyzer MINI dialysis units (3.5 kDa MWCO) | Thermo Scientific | PI69552 |
| 100mg SEP-PAK C18 column | Waters | wat043395 |
| C18 StageTips | Thermo Scientific | SP301 |
| Zeba Spin Desalting Column | Thermo Scientific | 89889 |
| NuPAGE Novex 12% Bis-Tris Protein Gels | Fisher Scientific | NPO343BOX |
| 4–20% Mini-PROTEAN Gels | Bio-Rad | 4561096 |
| Syringe Filters, PES, 0.45 μm | Genesee Scientific | 25-246 |
Purification of Ubiquitination machinery
E1, UBE2D3, UBE2R1, UBE2N/UBE2V2, UBC1, and UBE3C were purified as previously described (Bashore et al., 2015; Michel et al., 2015; Pham et al., 2016; Trang et al., 2012). Briefly, E1, UBE2D3, UBE2R1, and UBE2N/UBE2V2 constructs were expressed in Rosetta 2(DE3)pLysS E.coli cells in LB media supplemented with appropriate antibiotics at 37 °C to OD600 ~0.6–0.8 and transferred to 18 °C for 16 h after induction with IPTG. Cultures were harvested, resuspended in lysis buffer A (50 mM Tris pH 7.5, 300 mM NaCl, 1 mM EDTA and 10 mM imidazole), lysed by sonication, and clarified by centrifugation. Clarified lysate was then incubated with Ni-NTA resin for 1 h, washed with lysis buffer A, and eluted into Ni-NTA elution buffer (lysis buffer A plus 300 mM imidazole).
NleL (aa 170–782) were purified as previously (Valkevich et al., 2014). Briefly, NleL was expressed in BL21(DE3)pLysS E.coli cells in LB media supplemented with appropriate antibiotics at 37 °C to OD600 ~0.6 and transferred to 16 °C for 16 h after induction with IPTG. Cultures were harvested, resuspended in lysis buffer B (50 mM Tris pH 8.0, 200 mM NaCl, 1 mM EDTA and 1 mM DTT), lysed by sonication, and clarified by centrifugation. Clarified lysate was then incubated with GST resin for 1 h, washed with lysis buffer B, and eluted into GST elution buffer (lysis buffer B plus 10 mM reduced glutathione). Eluate was concentrated in TEV protease buffer (50 mM Tris pH 8.0, 150 mM NaCl, and 0.5 mM TCEP), cleaved overnight with TEV protease, and further purified using anion exchange chromatography.
UBE2S-UBD was purified as previously described (Bremm et al., 2010). Briefly, UBE2S-UBD constructs were expressed in Rosetta 2(DE3)pLysS E.coli cells in LB media supplemented with appropriate antibiotics at 37 °C to OD600 ~0.6–0.8 and transferred to 16 °C for 16 h after induction with IPTG. Cultures were harvested, resuspended in lysis buffer C (270 mM sucrose, 50 mM Tris pH 8.0, 50 mM NaF, and 1 mM DTT), lysed by sonication, and clarified by centrifugation. Clarified lysate was then incubated with GST resin for 1 h, washed with high salt buffer A (25 mM Tris pH 8.0, 500 mM NaCl, and 5 mM DTT), followed by low salt buffer A (25 mM Tris pH 8.0, 150 mM NaCl, and 5 mM DTT), and resuspended in 3C protease buffer (50 mM Tris pH 8.0 and 150 mM NaCl) for on-resin cleavage with HRV 3C protease overnight.
Minor modifications were made for the expression of 15N-UBE2S-UBD. Using the high-cell-density expression method, cultures were grown to an OD600 of 0.8 at 37 °C, concentrated (4x), and transferred into M9 media (Marley et al., 2001). Cells were incubated for 1 h to allow for discharge of unlabeled metabolites and then supplemented with 20% w/v glucose and 0.5 g 15NH4Cl, induced with IPTG, and grown overnight as described above.
RSP5 were purified as previously described (Worden et al., 2017). Briefly, RSP5 was expressed in Rosetta 2(DE3)pLysS E.coli cells in 2xYT media supplemented with appropriate antibiotics at 37 °C to OD600 ~0.8 and transferred to 18 °C for 16 h after induction with IPTG. Cultures were harvested, resuspended in lysis buffer D (50 mM HEPES pH 7.5, 250 mM NaCl, 1 mM EDTA, 1 mg/mL lysozyme, 1X protease cocktail and 5 mM PMSF), lysed by sonication, and clarified by centrifugation. Clarified lysate was then incubated with GST resin for 1 h, washed with high salt buffer B (25 mM HEPES pH 8.0 and 500 mM NaCl), followed by low salt buffer B (25 mM HEPES pH 8.0 and 150 mM NaCl), and resuspended in 3C protease buffer for on-resin cleavage with HRV 3C protease overnight. Eluate was concentrated and run on a Superdex 200 (GE) gel filtration column in 50 mM HEPES pH 7.5, 50 mM NaCl, and 5% glycerol.
Purification of Deubiquitinating Enzymes
OTUB1 was purified as previously described (Pham et al., 2016). Briefly, OTUB1 was expressed in Rosetta 2(DE3)pLysS E.coli cells in LB media supplemented with appropriate antibiotics at 37 °C to OD600 ~0.6–0.8 and transferred to 18 °C for 16 h after induction with IPTG. Cultures were harvested, resuspended in lysis buffer A, lysed by sonication, and clarified by centrifugation. Clarified lysate was then incubated with Ni-NTA resin for 1 h, washed with lysis buffer A, and eluted into Ni-NTA elution buffer.
AMSH was purified as previously described (Trang et al., 2012). Briefly, AMSH was expressed in BL21(DE3)pLysS E.coli cells in LB media supplemented with appropriate antibiotics at 37 °C to OD600 ~0.6 and transferred to 16 °C for 16 h after induction with IPTG and addition of ZnCl2. Cultures were harvested, resuspended in lysis buffer E (50 mM Tris pH 7.5, 300 mM NaCl and 2 mM DTT), lysed by sonication, and clarified by centrifugation. Clarified lysate was then incubated with GST resin for 1 h, washed with high salt buffer A, followed by low salt buffer A, and resuspended in 3C protease buffer for on-resin cleavage with HRV 3C protease overnight.
OTUD1 was purified as previously described (Mevissen et al., 2013). OTUD1 was expressed in Rosetta 2(DE3)pLysS E.coli cells in LB media supplemented with appropriate antibiotics at 37 °C to OD600 ~0.6–0.8 and transferred to 20 °C for 16 h after induction with IPTG. Cultures were harvested, resuspended in lysis buffer C, lysed by sonication, and clarified by centrifugation. Clarified lysate was then incubated with GST resin for 1 h, washed with high salt buffer A, followed by low salt buffer A, and resuspended in 3C protease buffer for on-resin cleavage with HRV 3C protease overnight. Eluate was concentrated and purified using anion exchange chromatography.
Purification of hRPN2 peptide
hRPN2 (aa 916–953) construct was expressed in BL21(DE3)pLysS E.coli cells in LB media supplemented with appropriate antibiotics at 37 °C to OD600 ~0.5 and transferred to 18 °C for 16 h after induction with IPTG. Cultures were harvested, resuspended in lysis buffer E containing 1 mM PMSF, lysed by French press, and clarified by centrifugation. Clarified lysate was then incubated with GST resin for 2 h, washed with high salt buffer A, followed by low salt buffer A, and eluted into GST elution buffer (lysis buffer E plus 10 mM reduced glutathione). Eluate was concentrated into 3C protease buffer, cleaved overnight with HRV 3C protease, and quenched with 10% (v/v) acetic acid. The acidified solution was poured over a 100 mg SEP-PAK C18 column (Waters) and washed with 3 mL 0.1% TFA in water followed by 1.5 mL each 10, 20, 30, 40, 50, 60, and 70% ACN in 0.1% TFA at room temperature. Fractions containing RPN2 peptide were identified by MALDI-TOF MS, lyophilized, and then reconstituted in water.
Co-Purification of UCH37 and RPN13
UCH37 and RPN13 constructs were expressed in BL21(DE3)pLysS E.coli cells in LB media supplemented with appropriate antibiotics at 37 °C to OD600 ~0.6 and transferred to 20 °C for 16 h after induction with IPTG. Cultures were harvested and stored at −80 °C with UCH37•RPN13 complexes mixed 1:1 (volume) prior to lysis. Cell pellets were resuspended in lysis buffer F (50 mM HEPES pH 7.5, 200 mM NaCl, 1 mM EDTA, and 1 mM TCEP), lysed by sonication, and clarified by centrifugation.
UCH37•RPN13 was purified in three chromatographic steps. (1) Clarified lysate was incubated with amylose resin for 1 h, washed with lysis buffer F, followed by low salt buffer C (50 mM HEPES pH 7.5, 50 mM NaCl, and 1 mM TCEP), eluted into amylose elution buffer (low salt buffer C plus 10 mM maltose), and incubated overnight with TEV protease. (2) Eluate was incubated over Ni-NTA resin for 1 h, washed with low salt buffer C, and eluted with Ni-NTA elution buffer (lysis buffer F plus 300 mM imidazole). (3) Eluate was concentrated and run on a Superdex 200 (GE) gel filtration column in 50 mM HEPES pH 7.5, 50 mM NaCl, 1 mM EDTA, and 1 mM TCEP.
Minor modifications were made for the purification of isolated UCH37 and RPN13. For UCH37, step 2 was omitted and further purified using anion exchange chromatography. For RPN13, step 1 was omitted and further purified using a Superdex 75 (GE) gel filtration column.
Purification of Proteasomes (Ptsms)
WT, UCH37KO, and RPN13KO cells stably expressing RPN11-HTBH were grown, harvested and lysed in Ptsm buffer (40 mM HEPES pH 7.4, 40 mM NaCl, 10 mM MgCl2, 2 mM ATP, 1 mM DTT, and 10% glycerol). The lysates were clarified at 20,000 xg for 20 min and the supernatant was incubated with streptavidin resin overnight with rocking. The resin was washed and incubated for 10 min intervals with high salt wash buffer (Ptsm buffer containing 200mM NaCl) on ice with rocking. The resin was then resuspended in low salt wash buffer (Ptsm buffer without DTT) and incubated with TEV protease for 1.5 h at room temperature.
Minor modifications were made for the purification of UCH37•RPN13-depleted, UCH37•RPN13-replenished (WT-r), and UCH37 C88A•RPN13-replenished (C88A-r) Ptsms. For UCH37•RPN13-depleted Ptsms, clarified lysates derived from WT cells were incubated with streptavidin resin overnight in the presence of 10 μM RPN2 peptide. For UCH37•RPN13-replenish and UCH37 C88A•RPN13-replenished Ptsms, clarified lysates derived from RPN13KO cells were incubated with streptavidin resin overnight with rocking. The resin was pelleted, resuspended in Ptsm buffer containing 10 μM WT or C88A recombinant UCH37•RPN13 complexes, and further incubated for 4 h with rocking prior to high salt washes.
To characterize purified proteasomes, western blots were used to assess individual components of the proteasome, native PAGE was used to assess intact proteasomes, Ub-AMC was used to assess deubiquitinase activity, and suc-LLVY-AMC was used to assess the chymotryptic-like peptidase activity of the proteasomes.
Synthesis of Defined Ub chains
Thiol-ene Trimers:
Reactions were performed as previously described (Valkevich et al., 2012).
Fluorescently labeled Native Trimers:
Reactions were performed as previously described (Crowe et al., 2016).
Native Homotypic Chains:
2 mM Ub, 300 nM E1, 3 μM UBE2R1 (K48) or 3 μM UBE2N/UBE2V2 (K63) were mixed in reaction buffer A (20 mM ATP, 10 mM MgCl2, 40 mM Tris-HCl pH 7.5, 50 mM NaCl, and 6 mM DTT) overnight at 37 °C.
Native K6/K48 Branched Trimer:
2 mM K6/48R Ub, 1 mM UbD77, 300 nM E1, 10 μM UBE2D3, and 1 μM NleL were mixed in reaction buffer A overnight at 37 °C.
Native K6/K48 Branched Tetramer:
Branched tetramer was generated using three reaction steps. (1) 2 mM K6/K48 branched tri-Ub was first cleaved by 1 μM OTUB1 in DUB buffer (50 mM Tris pH 7.5, 150 mM NaCl and 2 mM DTT) at 37 °C for 1 h to generate K6 di-Ub where the proximal subunit is a UbD77 molecule. (2) 2 mM K48R Ub, 1 mM UbD77, 300 nM E1, and 3 μM UBE2R1 were mixed in reaction buffer A overnight at 37 °C to generate K48 di-Ub. This K48-di Ub was then treated with 0.5 μM Yuh1 cleavage in DUB buffer at 25 °C for 4 h to expose its proximal C-terminus. (3) 500 μM K6 di-Ub, 500 μM K48 di-Ub, 300 nM E1, and 3 μM UBE2R1 were mixed in reaction buffer A overnight at 37 °C.
All reactions for native chains were quenched by lowering the pH to <5 via the addition of 5 M ammonium acetate pH 4.4. Enzymes were then precipitated through multiple freeze thaw cycles and further purified using cation exchange chromatography.
Generation of High Molecular Weight (HMW) Ub chains
K6/K48 Ub chains were assembled in a reaction buffer A containing 1 mM Ub, 150 nM E1, 5 μM UBE2D3, and 3 μM NleL. K11/48 Ub chains were assembled in a reaction buffer B (10 mM ATP, 10 mM MgCl2, 40 mM Tris pH 8.5, 100 mM NaCl, 0.6 mM DTT, and 10% (v/v) glycerol) containing 0.6 μM Ub, 150 nM E1, and 5 μM UBE2S-UBD. 3 μM AMSH and 0.5 μM OTUD1 were added after 3 h and the mixture was left overnight at 37 °C. Prior to purification, an additional bolus of AMSH and OTUD1 was added, the mixture was incubated for 3 h at 37 °C and subjected to size exclusion chromatography to isolate products with a mass >35 kDa. These HMW K11-linked chains were then added to 0.6 μM Ub, 150 nM E1, and 3 μM UBE2R1 in reaction buffer B. K48/K63 Ub chains were assembled using 1 mM Ub, 150 nM E1, 5 μM UBE2R1, and 5 μM UBE2N/UBE2V2 in reaction buffer A. K11/K63 Ub chains were assembled from 1 mM Ub, 150 nM E1, 5 μM UBE2S-UBD as described above for K11/K48 chains. Finally, K29/K48 Ub chains were generated from 1 mM Ub, 150 nM E1, 2 μM UBE2D3, and 3 μM UBE3C as previously described (Michel et al., 2015).
All Ub chains were purified using size exclusion chromatography (Superdex 75) to isolate high molecular weight chains of >35kDa used in these studies.
Purification and ubiquitination of titin I27V15P 23-K-35
Titin I27V15P 23-K-35 was purified as previously described (De la Peña et al., 2018). Briefly, titin I27V15P 23-K-35 was expressed in BL21(DE3)pLysS E.coli cells in 2xYT media containing 1% glycerol and supplemented with appropriate antibiotics at 30 °C to OD600 ~1.2–1.5, induced with IPTG, and grown for an additional 5 h. Cultures were harvested, flash frozen, resuspended in lysis buffer G (60 mM HEPES pH 7.5, 100 mM NaCl, 100 mM KCl, 10 mM MgCl2, 0.5 mM EDTA, 1 mg/mL lysozyme, 2 mM PMSF, 20 mM imidazole, and 10% glycerol), lysed by sonication, and clarified by centrifugation. Clarified lysate was then incubated with Ni-NTA resin for 1 h, washed with high salt buffer B, followed by low salt buffer B, and eluted with low salt buffer B containing 300 mM imidazole. Eluate was concentrated into Ulp1 protease buffer (60 mM HEPES pH 7.5 and 150 mM NaCl), cleaved overnight with Ulp1, and further purified using a Superdex 200 (GE) gel filtration column in 50 mM HEPES pH 7.5 and 5% glycerol.
100 μM substrate was modified with 5 μM E1, 5 μM Ubc1, 20 μM Rsp5, and 2 mM Ub in labeling buffer (60 mM HEPES pH 7.5, 20 mM NaCl, 20 mM KCl, 10 mM MgCl2, and 2.5% glycerol) containing 1X ATP regeneration mix for 3 h at room temperature followed by the addition of 5 μM UBE2R1 and incubation overnight at 4 °C.
Fluorescent labeling of Ubiquitinated substrates
Substrates (2 mg/mL) were fluorescently labeled using cyanine5 maleimide at pH 7.2 for 2 h at room temperature and quenched with excess DTT. Free dye was separated from the substrate using a Zeba spin desalting column and buffer exchanged into labeling buffer.
Steady-State Measurements with Defined Ub chains
Stock solutions of enzymes and ubiquitin chains were prepared in assay buffer A (50 mM HEPES pH 7.5, 50 mM NaCl, and 2 mM DTT). All reactions were performed at 37 °C with the exception of the K48 tetra-Ub inhibition kinetics being performed at room temperature. Each sample along with a Ub and di-Ub standard were then separated on a 15% SDS-PAGE gel and followed by SYPRO® Ruby staining. Gels were visualized on a Typhoon FLA 9500 (GE) and densitometry was analyzed on the Ub standards using Image Studio™. Initial velocities of Ub and di-Ub formation were calculated by plotting concentration as a function of time. These values were then fit to the Michealis-Menten equation using nonlinear regression in Prism 8. Error bars represent the standard deviation of three trials for each reaction performed using UCH37 and UCH37•RPN13 complexes.
Pulldown Assays
Ub chains:
His-tagged RPN13•UCH37C88A (5 nmol) was incubated with Ni-NTA resin (20 μL) in pulldown buffer (50 mM HEPES pH 7.4, 150 mM NaCl, and 0.05% IGEPAL) for 2 h. Ni-bound RPN13•UCH37C88A was then incubated with either defined Ub chains (50 pmol) or HMW Ub chains (12 μg) for an additional 2 h, washed three times with pulldown buffer, and eluted with 6x Laemmli loading buffer. For DUB-treated Ub chains, HMW Ub chains were first incubated with OTUB1 or UCH37 for 1 h at 37 °C then added to the Ni-bound His-RPN13•UCH37C88A.
UBE2S-UBD-FLAG:
Anti-FLAG M2 Affinity gel (50 μL) was incubated with ubiquitinated UBE2S-UBD-FLAG (250 ng/μL) in pulldown buffer for 2 h. The resin was washed with pulldown buffer (3x) followed by minimal buffer (50 mM HEPES pH 7.4 and 150 mM NaCl, 2x). Captured UBE2S-UBD-FLAG was resuspended in minimal buffer, trypsin was then added to a 1:150 (w/w) ratio and minimal proteolysis was allowed to proceed for 2.5 h at 37 °C.
K-ε-GG peptides:
Pulldown was performed according to the protocol provided by CST. Briefly, 1 mg of ubiquitinated UBE2S-UBD was resuspended in urea lysis buffer (50 mM HEPES pH 8.0, 50 mM NaCl, and 8 M urea), reduced with 1.25 M DTT at 55 °C for 30 min, alkylated with 1:10 (v/v) iodoacetamide for 15 min, and diluted with 20 mM HEPES pH 8.0 to a final concentration of 2 M urea. Trypsin (2 μg) was added to the diluted solution and digestion was allowed to proceed overnight at 37 °C. Trypsin was quenched was 10% (v/v) formic acid and peptides were purified using a SEP-PAK C18 column where peptides were washed with 2 mL 0.1% TFA in water and eluted with 50% acetonitrile with 0.1% TFA. Eluate was dried using a speed vac, resuspended in IAP buffer with added K-ε-GG antibody bead slurry (40 μL), and incubated for 2 h at 4 °C with rocking. The beads were washed with IAP buffer (2x), followed by washes with water (3x), eluted with 0.15% TFA (2x), and desalted using C18 StageTips then dried with a speed vac.
All pulldown assays were done at room temperature unless otherwise indicated.
Isothermal Titration Calorimetry (ITC) Analysis
ITC measurements were performed on a MicroCal Auto-ITC200 (Malvern) at 25 °C with a setting of 20 × 2 μL injections. UCH37C88A•RPN13 and Ub chains were all buffer exchanged into dialysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, and 500 μM TCEP). For UCH37•RPN13 and UCH37C88A•RPN13 measurements, the syringe contained a concentration of Ub chains at 45 μM, and the cell contained UCH37•RPN13 or UCH37C88A•RPN13 at a concentration of 3 μM. Manufactured supplied Origin software (OriginLab 7 SR4) was used to fit the data to a single-site binding model and to determine the stoichiometry (N), ΔH, ΔS, and the association constant Ka. The dissociation constant, Kd, was calculated from Ka.
Native Gel Electrophoresis
Native gel electrophoresis was performed as previously described (Elsasser et al., 2005). In brief, 2.5 μg purified proteasomes were separated on 4% acrylamide gels at 100V for 3 h at 4 °C. The gel was incubated in developing buffer (50 mM HEPES pH 7.4, 5 mM MgCl2, 1 mM ATP and 50 μM suc-LLVY-AMC) without agitation for 30 min at 30 °C. The gel was then imaged using a UV transilluminator (Bio-Rad) with the excitation for AMC at 360 nm.
Ub-AMC and suc-LLVY-AMC Assays
UCH37, UCH37•RPN13, and Ptsms were assayed for their DUB or proteolytic activity using either Ub-AMC or suc-LLVY-AMC quenched fluorescent reporter substrates respectively. Both assays were performed in black clear bottom 96 well plates. Reactions were performed by prewarming the AMC reagent (250 nM for Ub-AMC or 50 μM suc-LLVY-AMC) dissolved in assay buffer A for UCH37 and UCH37•RPN13 and assay buffer B (40 mM HEPES pH 7.4, 40 mM NaCl, 10 mM MgCl2, 2 mM ATP, and 1 mM DTT) for Ptsms at 37 °C for 20 min. At this point, UCH37 and UCH37•RPN13 (20 nM) or Ptsms (1 μg) in their respective assay buffer were added to the appropriate wells and hydrolysis was monitored continuously for 30 min at 37 °C on a fluorescence plate reader (BioTek Synergy 2, λex = 360 nm, λem = 460 nm).
Debranching and Degradation Assays
Debranching:
Stock solutions of all DUBs and Ptsms along with HMW Ub chains (250 ng/μL) were warmed to 37 °C in assay buffer A (DUBs) or assay buffer B (Ptsms). Reactions were initiated by the addition of DUB or Ptsm. Time points were taken at 1 h for DUBs and 4 h for Ptsms unless otherwise indicated and quenched with either 6x Laemmli loading buffer for immunoblotting and Ub-AQUA analysis or trypsin (1:200 w/w ratio) for middle-down mass spectrometry. For Ub-AQUA analysis, samples were separated by SDS-PAGE on 12% NuPAGE Bis-Tris gels and prepared as previously described (Ries et al., 2019). For middle-down mass spectrometry, minimal proteolysis using trypsin (1:150 w/w) was allowed to proceed for 2.5 h at 37 °C, samples were then acidified to pH 2 with acetic acid to deactivate trypsin and either (1) dialyzed into water for DUB-treated samples as previously described (Valkevich et al., 2014) or (2) separated using a Sep-pak C18 column for Ptsm-treated samples as previously described (Crowe et al., 2017).
Degradation:
Stock solutions of Ptsms along with ubiquitinated substrates (250 ng/μL) were warmed to 37 °C in assay buffer B supplemented with 1X ATP-regeneration mix. For pre-treatment of ubiquitinated substrates with UCH37•RPN13 (1 μM), branch points were removed for 1 h at 37 °C and UCH37•RPN13 was inactivated by multiple freeze/thaw cycles prior to the addition of Ptsms. Time points were taken at 1 h unless otherwise indicated and quenched with either 6x Laemmli loading buffer for gel-based degradation monitoring or 10% (v/v) formic acid for parallel reaction monitoring. For gel-based degradation, samples were separated by SDS-PAGE on homemade 10–16% tricine gels (Schägger, 2006). Cy5 fluorescence was measured on a Typhoon FLA 9500 (GE) using a pixel density of 50 μm per pixel, while total protein staining was performed using SYPRO® Ruby and imaged with a pixel density of 50 μm per pixel. For parallel reaction monitoring, corresponding light and heavy samples were mixed 1:1 (volume) and samples were desalted using C18 StageTips then dried with a speed vac. All mass spectrometry samples were resuspended in 1% formic acid.
Flow Cytometry using GFPu Reporter System
WT and UCH37KO GFPu cells were grown in 96-well plates until cells reached ~70% confluency. HEK293 FT cells were used as a negative control. Cells were treated with either 10 μM MG132 or 0.1% DMSO for 24 h before trypsinization and dilution in PBS with 10% FBS and 1xPen/Strep. At least 10,000 events per sample were analyzed at a flow rate of 200 μL/min on an Attune NxT Acoustic Focusing Flow Cytometer (Life Technologies) equipped with 488 nm laser (530/30 nm emission filter). Data was analyzed using FlowJo version 10.4 (FlowJo, LLC). Statistical analysis was performed using Prism 8 and are represented as the mean fluorescence in BL1-A with standard deviation of three independent experiments. ***P<0.0005 (student’s t-test).
Minor modifications were made for UCH37KO cells expressing either WT or UCH37 C88S. Cells were grown in 12-well plates in the presence of 0.1 μg/ml doxycycline for 48 h. Cell were then harvested, washed with PBS, and pipetted into 1% BSA and 1 mM EDTA in PBS for flow cytometry. At least 30,000 events were collected on a BD LSR Fortessa equipped with 488 nm and 561 nm lasers for excitation of GFP and RFP, respectively.
Pulse-Chase Experiments with L-Azidohomoalanine (AHA) labeling
WT, UCH37KO, or UCH37KO cells expressing either WT or UCH37 C88S were seeded in 6-well plates. Once cells reached ~60% confluency, cells were washed once with PBS and incubated for 1 h in methionine free media supplemented with or without 25 μM AHA. Cells were then either harvested or chased in complete DMEM (cold chase) supplemented with 2 mM excess methionine for 4 h. Negative controls were not treated with AHA and were harvested without chase. Carfilzomib controls were treated with 1 μM carfilzomib during both AHA incubation and 4 h chase. Cells were harvested by adding PBS supplemented with 1 mM EDTA to each well of the plate. Cells were washed once with PBS and resuspended in RIPA buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 0.5% Triton X-100, 140 mM NaCl, 0.1% SDS, and 0.1% sodium deoxycholate supplemented with 1X protease inhibitor cocktail, 1X phosphatase inhibitor cocktail and 10 μM MG132) before flash freezing on dry ice and storing at −80 °C. Pellets were lysed via freeze-thaw cycles and sonicated before centrifugation at 20,000 xg for 20 min at 4 °C. Total protein concentration was quantified using Bradford assay and then labeled with 10 μM DBCO-Cy5 for 30 min. Labeling was quenched with 6x Laemmli loading buffer without bromophenol blue or DTT and samples were separated by SDS-PAGE on 4–20% Mini-PROTEAN gels. Cy5 fluorescence and total protein staining using SYPRO® Ruby was measured on a Typhoon FLA 9500 (GE). For UCH37KO cells expressing either WT or UCH37 C88S, cells were seeded in media containing 0.1 μg/mL doxycycline for 48 h. Doxycycline remained present for the rescue cell lines after each media change.
Ub Middle-down Mass Spectrometry (Ub MiD MS) Analysis
Minimal tryptic fragments of HMW Ub chains were either separated using an Ultimate 3000 UHPLC (Thermo Scientific) prior to analysis using Orbitrap Fusion Tribid mass spectrometer (Thermo Scientific). For separation, the UHPLC was equipped with a MASSPrep™ Micro Desalting VanGuard Pre-Column (2.1 × 5 mm, Waters). Fragments were then separated using a linear gradient of 5% to 70% B over 18 min and 70% to 95% B over 5 min (solvent A: 0.1% formic acid (FA) in water, solvent B: 0.1% FA in ACN) using a flow rate of 10 μL/min. The resolving power of the mass analyzer on the spectrometer was set at 120000. For tandem mass spectrometry (MS/MS) using ETD, individual charge states of protein molecular ions were isolated and dissociated by ETD using a 10 ms reaction time, a 2.0e5 reagent ion target, and 10% supplemental collisionally induced dissociation (CID). All spectra were processed with in-house software (MASH Suite) using a signal-to-noise (S/N) threshold of 3 and a fit factor of 70% and then validated manually (Guner et al., 2014). Percentages correspond to the relative quantification values of the 11+ charge state for all three species: Ub1–74, 1xdiGly-Ub1–74, and 2xdiGly-Ub1–74. For measuring the relative rates of debranching for HMW chains, the relative quantification values were fit to Eq 1 using Prism 8. The reported kobs and error bars represent the standard deviation (SD) of two replicates for UCH37•RPN13 and varying Ptsms.
| (Eq 1) |
Ubiquitin-AQUA High Resolution and Accurate Mass MS (HR/AM MS) Analysis
Full tryptic digests of HMW Ub chains were separated on an Easy nLC 1000 UHPLC equipped with a homemade 15cm nanoLC column (ProntoSIL C4 5 um 300A, Custom PN-81003). Using a flow rate of 300 nL/min, the linear gradient was 0% to 50% over B for 20 min, 50% to 95% over B for 3 min, and 95% hold over B for 7 min (solvent A: 0.1% formic acid (FA) in water, solvent B: 0.1% FA in ACN). The LC system was coupled to the Orbitrap with a resolving power set at 60000. Spectra were recorded over a range 300 to 1500 m/z. For data-dependent MS/MS, the top four most intense ions with charge state of 2–5 were selected using an isolation window of 2 m/z. Fragmentation was achieved by CID at 35% nominal energy with product ion detection in the linear ion-trap. Ion chromatograms were extracted for each peptide of interest with an extraction window of 20 ppm. Chromatograms were smoothed using the Boxcar algorithm with a 7-point window. Integration was then performed using default parameters with manual adjustment as deemed appropriate. Results are normalized against total amount of Ub for each linkage type detected and are represented as means ± SEM of two replicates. For all points, asterisks represented are as follows: *P<0.025, **P<0.01 (student’s t-test).
UBE2S-UBD Degradation using Parallel Reaction Monitoring (PRM) Analysis
Proteolytic peptides from UBE2S-UBD degradation were separated on an Easy nLC 1000 UHPLC equipped with a homemade 15 cm nanoLC column (ProntoSIL C4 5 um 300A, Custom PN-81003). Using a flow rate of 300 nL/min, the linear gradient was 5% to 50% over B for 35 min, 50% to 95% over B for 3 min, and 95% hold over B for 6 min (solvent A: 0.1% formic acid (FA) in water, solvent B: 0.1% FA in ACN). The LC system was coupled to the Orbitrap with a resolving power set at 50000. Spectra were recorded over a range 200 to 1300 m/z. For data-dependent MS/MS, the parent ions for the heavy (15N) and light (14N) UBE2S and UBD peptides (FPASPPKGY and FDGSGGNNHAVE) were selected using an isolation window of 2 m/z and a resolving power set at 15000. Fragmentation was achieved by HCD at a stepped 24, 27, and 30% nominal energy with transition ion detection in the orbitrap. Proteolytic peptides were first identified with an extraction window of 20 ppm using Proteome Discoverer 2.3 and ion chromatograms were then extracted each transition ion of interest with an extraction window of 10 ppm using Pinpoint 1.4. Results are represented as sum ± SD of four replicates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
All statistical data were calculated using GraphPad Prism 8. Detailed parameters used for each experiment are indicated in every figure and table when applicable.
Reproducibility
Unless stated otherwise all quantitative experiments were performed in triplicate and averaged with SEM reported.
Supplementary Material
TABLE S1. Data for Ub AQUA, ITC, Middle-down MS, PRM and AHA Analysis.
(1A) Ub peptides analyzed in this study (related to Figure 2E–G & S3E). Peptide name, peptide sequence, accurate m/z of precursor ion are shown. SDS-PAGE analysis of samples analyzed in this study.
(1B) Standard curves of Ub AQUA peptides. Raw and analyzed data of ten accurate mass AQUA peptides are shown (related to Figure 2E–G & S3E).
(1C) Ub AQUA HR/AM analysis of HMW chains analyzed in this study. Raw and analyzed data are shown for duplicate runs (related to Figure 2E–G & S3E).
(1D) ITC analysis of UCH37•RPN13 and UCH37 C88A•RPN13 complexes with K48-linked and K6/K48-linked Ub chains (related to Figure 4 & S4).
(1E) Middle-down MS analysis of HMW K6/K48, K11/K48, and K48/K63 chains with UCH37•RPN13, WT Ptsm, and UCH37•RPN13-replenish Ptsm (related to Figure 5E & S5F–H).
(1F) MS2 analysis of K-ε-GG pulldown for K11/K48-Ubn-UBE2S-UBD and list of transition ions used for PRM analysis (related to Figure 6A).
(1G) PRM analysis for the degradation of K11/K48-Ubn-UBE2S-UBD with each indicated Ptsm (related to Figure 6D–E & S6C–F).
(1H) AHA labeling densitometry. Raw and analyzed data of 3 individual bioreps are shown (related to Figure 7E–F).
HIGHLIGHTS.
UCH37 cleaves branched ubiquitin chains at the K48 position
The proteasomal subunit RPN13 enhances the activity of UCH37
Proteasome-bound UCH37 retains selectivity for branched chains
UCH37-mediated chain editing potentiates proteasome-mediated degradation
ACKNOWLEDGEMENTS
We thank Dr. Steve Eyles (UMass Amherst) for assistance with high-resolution mass spectrometry, Dr. Amy S. Burnside (UMass Amherst) for assistance with flow cytometry, and Marissa Matsumoto (Genentech) for technical guidance with topology-specific antibodies. This work was funded by the NIH (RO1GM110543 to E.R.S.), a NSF Graduate Research Fellowship (GRFP1451512 to K.K.D.), and a NIH Chemistry and Biology Training Grant (T32GM008515 to J.D. and H.A.B.). The data described herein were acquired on an Orbitrap Fusion mass spectrometer funded by National Institutes of Health grant 1S10OD010645-01A1.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Al-shami A, Jhaver KG, Vogel P, Wilkins C, Humphries J, Davis JJ, Xu N, Potter DG, Gerhardt B, Mullinax R, et al. (2010). Regulators of the Proteasome Pathway, Uch37 and Rpn13, Play Distinct Roles in Mouse Development. PLoS One 5, e13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JAM, Bashore C, Dong KC, and Martin A (2019). The 26S Proteasome Utilizes a Kinetic Gateway to Prioritize Substrate Degradation. Cell 177, 286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger CJ, Branick C, Chee L, and Karpf AR (2019). Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer . Cancers. 11, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashore C, Dambacher CM, Goodall EA, Matyskiela ME, Lander GC, and Martin A (2015). Ubp6 deubiquitinase controls conformational dynamics and substrate degradation of the 26S proteasome. Nat. Struct. Mol. Biol. 22, 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beekum O, Gao Y, Berger R, Koppen A, and Kalkhoven E (2012). A Novel RNAi Lethality Rescue Screen to Identify Regulators of Adipogenesis. PLoS One 7, e37680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, and Kopito RR (2001). Impairment of the Ubiquitin-Proteasome System by Protein Aggregation. Science 292, 1552–1555. [DOI] [PubMed] [Google Scholar]
- Bennett EJ, Shaler TA, Woodman B, Ryu K-Y, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR (2007). Global changes to the ubiquitin system in Huntington’s disease. Nature 448, 704–708. [DOI] [PubMed] [Google Scholar]
- Besche HC, Haas W, Gygi SP, and Goldberg AL (2009). Isolation of mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry 48, 2538–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett JS, Ritorto MS, Ewan R, Jaffray EG, Virdee S, Chin JW, Knebel A, Kurz T, Trost M, Tatham MH, et al. (2015). Ubiquitin C-terminal hydrolases cleave isopeptide- and peptide-linked ubiquitin from structured proteins but do not edit ubiquitin homopolymers. Biochem. J. 466, 489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boughton AJ, Krueger S, and Fushman D (2020). Branching via K11 and K48 Bestows Ubiquitin Chains with a Unique Interdomain Interface and Enhanced Affinity for Proteasomal Subunit Rpn1. Structure 28, 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremm A, Freund SMV, and Komander D (2010). Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat. Struct. Mol. Biol. 17, 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GA, and Goldberg AL (2017). The Logic of the 26S Proteasome. Cell 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe SO, Pham GH, Ziegler JC, Deol KK, Guenette RG, Ge Y, and Strieter ER (2016). Subunit-Specific Labeling of Ubiquitin Chains by Using Sortase: Insights into the Selectivity of Deubiquitinases. ChemBioChem 17, 1525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe SO, Rana ASJB, Deol KK, Ge Y, and Strieter ER (2017). Ubiquitin Chain Enrichment Middle-Down Mass Spectrometry Enables Characterization of Branched Ubiquitin Chains in Cellulo. Anal. Chem. 89, 4428–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, McHale D, Maher ER, McKenzie ANJ, and Komander D (2016). The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 166, 1215–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, and Masucci MG (2000). Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 18, 528–543. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ (2014). Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 12, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux Q, Ustrell V, Pickart C, and Rechsteiner M (1994). A 26 S Protease Subunit That Binds Ubiquitin Conjugates. J. Biol. Chem. 269, 7059–7061. [PubMed] [Google Scholar]
- Du J, and Strieter ER (2018). A fluorescence polarization-based competition assay for measuring interactions between unlabeled ubiquitin chains and UCH37•RPN13. Anal. Biochem. 550, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser S, Schmidt M, and Finley D (2005). Characterization of the proteasome using native gel electrophoresis. Methods Enzymol. 398, 353–363. [DOI] [PubMed] [Google Scholar]
- Emmerich CH, Ordureau A, Strickson S, Arthur JSC, Pedrioli PGA, Komander D, and Cohen P (2013). Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. U. S. A. 110, 15247–15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Mu J, Ma Y, Ma D, Fu D, and Shen X (2012). The interaction between ubiquitin C-terminal hydrolase 37 and glucose-regulated protein 78 in hepatocellular carcinoma. Mol. Cell. Biochem. 359, 59–66. [DOI] [PubMed] [Google Scholar]
- Fang Y, Fu D, Tang W, Cai Y, Ma D, Wang H, Xue R, Liu T, Huang X, Dong L, et al. (2013). Ubiquitin C-terminal Hydrolase 37, a novel predictor for hepatocellular carcinoma recurrence, promotes cell migration and invasion via interacting and deubiquitinating PRP19. Biochim. Biophys. Acta - Mol. Cell Res 1833, 559–572. [DOI] [PubMed] [Google Scholar]
- Guner H, Close PL, Cai W, Zhang H, Peng Y, Gregorich ZR, and Ge Y (2014). MASH Suite: A user-friendly and versatile software interface for high-resolution mass spectrometry data interpretation and visulation. J. Am. Soc. Mass Spectrom. 25, 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haakonsen DL, and Rape M (2019). Branching Out: Improved Signaling by Heterotypic Ubiquitin Chains. Trends Cell Biol. 29, 704–716. [DOI] [PubMed] [Google Scholar]
- Hamazaki J, Iemura SI, Natsume T, Yashiroda H, Tanaka K, and Murata S (2006). A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO J. 25, 4524–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Lee H, and Han J (2017). Ubiquitin C-terminal hydrolase37 regulates Tcf7 DNA binding for the activation of Wnt signalling. Sci. Rep. 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger K, Wickliffe KE, Ndoja A, Zhang J, Murthy A, Dugger DL, Maltzman A, De Sousa E Melo F., Hung J, Zeng Y, et al. (2018). OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 559, 120–124. [DOI] [PubMed] [Google Scholar]
- Hospenthal MK, Freund SMV, and Komander D (2013). Assembly, analysis and architecture of atypical ubiquitin chains. Nat. Struct. Mol. Biol. 20, 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnjak K, and Dikic I (2012). Ubiquitin-binding proteins: Decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 81, 291–322. [DOI] [PubMed] [Google Scholar]
- Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, and Dikic I (2008). Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyoung TK, Kwang PK, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, Gygi SP, and Goldberg AL (2007). Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem. 282, 17375–17386. [DOI] [PubMed] [Google Scholar]
- Jørgensen JP, Lauridsen AM, Kristensen P, Dissing K, Johnsen AH, Hendil KB, and Hartmann-Petersen R (2006). Adrm1, a Putative Cell Adhesion Regulating Protein, is a Novel Proteasome-associated Factor. J. Mol. Biol. 360, 1043–1052. [DOI] [PubMed] [Google Scholar]
- Kim HT, Kim KP, Uchiki T, Gygi SP, and Goldberg AL (2009). S5a promotes protein degradation by blocking synthesis of nondegradable forked ubiquitin chains. EMBO J. 28, 1867–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick DS, Hathaway NA, Hanna J, Elsasser S, Rush J, Finley D, King RW, and Gygi SP (2006). Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat. Cell Biol. 8, 700–710. [DOI] [PubMed] [Google Scholar]
- De la Peña AH, Goodall EA, Gates SN, Lander GC, and Martin A (2018). Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis–driven translocation. Science. 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YA, Xu W, DeMartino GN, and Cohen RE (1997). Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature 385, 737–740. [DOI] [PubMed] [Google Scholar]
- Lee B, Lee MJ, Park S, Oh D, Elsasser S, Chen P, Gartner C, Dimova N, Hanna J, Gygi SP, et al. (2010). Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Lu Y, Prado MA, Shi Y, Tian G, Sun S, Elsasser S, Gygi SP, King RW, and Finley D (2016). USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 532, 398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto DE, Morgens DW, Zhang L, Walczak CP, Elias JE, Bassik MC, and Kopito RR (2019). Genome-wide CRISPR Analysis Identifies Substrate-Specific Conjugation Modules in ER-Associated Degradation. Mol. Cell 73, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Liu W, Ye Y, and Li W (2017). Ufd2p synthesizes branched ubiquitin chains to promote the degradation of substrates modified with atypical chains. Nat. Commun. 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Liu F, Durham SE, Tarasov SG, and Walters KJ (2015a). A High Affinity hRpn2-Derived Peptide That Displaces Human Rpn13 from Proteasome in 293T Cells. PLoS One 10, e0140518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Nowicka U, Sridharan V, Liu F, Randles L, Hymel D, Dyba M, Tarasov SG, Tarasova NI, Zhao XZ, et al. (2017). Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets. Nat. Commun 8, 15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Lee B, King RW, Finley D, and Kirschner MW (2015b). Substrate degradation by the proteasome: A single-molecule kinetic analysis. Science. 348, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marley J, Lu M, and Bracken C (2001). A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75. [DOI] [PubMed] [Google Scholar]
- Martinez-Fonts K, Davis C, Tomita T, Elsasser S, Nager AR, Shi Y, Finley D, and Matouschek A (2020). The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat. Commun. 11, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumdar T, Gorgun FM, Sha Y, Tyryshkin A, Zeng S, Hartmann-Petersen R, Jørgensen JP, Hendil KB, and Eissa NT (2010). Regulation of NF-kappaB activity and inducible nitric oxide synthase by regulatory particle non-ATPase subunit 13 (Rpn13). Proc. Natl. Acad. Sci. U. S. A. 107, 13854–13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al. (2011). The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 108, 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen TET, and Komander D (2017). Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 86, 159–192. [DOI] [PubMed] [Google Scholar]
- Mevissen TET, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F, et al. (2013). OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 154, 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HJ, and Rape M (2014). Enhanced protein degradation by branched ubiquitin chains. Cell 157, 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MA, Elliott PR, Swatek KN, Simicek M, Pruneda JN, Wagstaff JL, Freund SMV, and Komander D (2015). Assembly and specific recognition of K29- and K33-linked polyubiquitin. Mol. Cell 58, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolajczyk J, Drag M, Békés M, Cao JT, Ronai Z, and Salvesen GS (2007). Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs. J. Biol. Chem. 282, 26217–26224. [DOI] [PubMed] [Google Scholar]
- Nakasone MA, Livnat-Levanon N, Glickman MH, Cohen RE, and Fushman D (2013). Mixed-linkage ubiquitin chains send mixed messages. Structure 21, 727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi R, Wijnhoven P, Le Sage C, Tjeertes J, Galanty Y, Forment JV, Clague MJ, Urbé S, and Jackson SP (2014). Systematic characterization of deubiquitylating enzymes for roles in maintaining genome integrity. Nat. Cell Biol. 16, 1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Akopian D, and Rape M (2018). Principles of Ubiquitin-Dependent Signaling. Annu. Rev. Cell Dev. Biol 34, 18–19. [DOI] [PubMed] [Google Scholar]
- Ohtake F, Tsuchiya H, Saeki Y, and Tanaka K (2018). K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc. Natl. Acad. Sci. U. S. A. E1401–E1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei-Amponsa V, Sridharan V, Tandon M, Evans CN, Klarmann K, Cheng KT, Lack J, Chari R, Walters KJ (2020). Impact of losing hRpn13 Pru or UCHL5 on proteasome clearance of ubiquitinated proteins and RA190 cytotoxity. Mol. Cell. Biol. 40, e00122–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peth A, Nathan JA, and Goldberg AL (2013). The ATP costs and time required to degrade ubiquitinated proteins by the 26 S proteasome. J. Biol. Chem. 288, 29215–29222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham GH, Rana ASJB, Korkmaz EN, Trang VH, Cui Q, and Strieter ER (2016). Comparison of native and non-native ubiquitin oligomers reveals analogous structures and reactivities. Protein Sci. 25, 456–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Poot SAH, Tian G, and Finley D (2017). Meddling with Fate: The Proteasomal Deubiquitinating Enzymes. J. Mol. Biol. 429, 3525–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic D, Vucic D, and Dikic I (2014). Ubiquitination in disease pathogenesis and treatment. Nat Med 20, 1242–1253. [DOI] [PubMed] [Google Scholar]
- Qiu XB, Ouyang SY, Li CJ, Miao S, Wang L, and Goldberg AL (2006). hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO J. 25, 5742–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randles L, Anchoori RK, Roden RBS, and Walters KJ (2016). The proteasome ubiquitin receptor hrpn13 and its interacting deubiquitinating enzyme Uch37 are required for proper cell cycle progression. J. Biol. Chem. 291, 8773–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries LK, Sander B, Deol KK, Letzelter MA, Strieter ER, and Lorenz S (2019). Analysis of ubiquitin recognition by the HECT ligase E6AP provides insight into its linkage specificity. J. Biol. Chem. 294, 6113–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahtoe DD, vanDijk WJ, ElOualid F, Ekkebus R, Ovaa H, and Sixma TK (2015). Mechanism of UCH-L5 Activation and Inhibition by DEUBAD Domains in RPN13 and INO80G . Mol. Cell 57, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samant RS, Livingston CM, Sontag EM, and Frydman J (2018). Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature 563, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-pulido L, Kong L, and Ponting CP (2012). A common ancestry for BAP1 and Uch37 regulators. Bioinformatics 28, 1953–6. [DOI] [PubMed] [Google Scholar]
- Schägger H (2006). Tricine-SDS-PAGE . Nat. Protoc. 1, 16–23. [DOI] [PubMed] [Google Scholar]
- Schreiner P, Chen X, Husnjak K, Randles L, Zhang N, Elsasser S, Finley D, Dikic I, Walters KJ, and Groll M (2008). Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature 453, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Chen X, Elsasser S, Stocks BB, Tian G, Lee BH, Shi Y, Zhang N, De Poot SAH, Tuebing F, et al. (2016). Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science. 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, and Komander D (2016). Ubiquitin modifications. Cell Res. 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, Usher JL, Kueck AF, Gladkova C, Mevissen TET, Pruneda JN, Skern T, and Komander D (2020). Insights into ubiquitin chain architecture using Ub-clipping. Nature 572, 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang VH, Valkevich EM, Minami S, Chen Y, Ge Y, and Strieter ER (2012). Nonenzymatic Polymerization of Ubiquitin: Single-Step Synthesis and Isolation of Discrete Ubiquitin Oligomers. Angew. Chemie - Int. Ed. 51, 13085–13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkevich EM, Guenette RG, Sanchez NA, Chen YC, Ge Y, and Strieter ER (2012). Forging isopeptide bonds using thiol-ene chemistry: Site-specific coupling of ubiquitin molecules for studying the activity of isopeptidases. J. Am. Chem. Soc. 134, 6916–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkevich EM, Sanchez NA, Ge Y, and Strieter ER (2014). Middle-Down mass spectrometry enables characterization of branched ubiquitin chains. Biochemistry 53, 4979–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderLinden RT, Hemmis CW, Schmitt B, Ndoja A, Whitby FG, Robinson H, Cohen RE, Yao T, and Hill CP (2015). Structural Basis for the Activation and Inhibition of the UCH37 Deubiquitylase . Mol. Cell 57, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen CF, Baker PR, Chen PL, Kaiser P, and Huang L (2007). Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46, 3553–3565. [DOI] [PubMed] [Google Scholar]
- Wertz IE, Newton K, Seshasayee D, Kusam S, Lam C, Zhang J, Popovych N, Helgason E, Schoeffler A, Jeet S, et al. (2015). Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 528, 370–375. [DOI] [PubMed] [Google Scholar]
- Wicks SJ, Haros K, Maillard M, Song L, Cohen RE, Ten Dijke P, and Chantry A (2005). The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-β signalling. Oncogene 24, 8080–84. [DOI] [PubMed] [Google Scholar]
- Wicks SJ, Grocott T, Haros K, Maillard M, Ten Dijke P, and Chantry A (2006). Reversible ubiquitination regulates the Smad/TGF-β signalling pathway. Biochem. Soc. Trans. 34, 761–763. [DOI] [PubMed] [Google Scholar]
- Worden EJ, Dong KC, and Martin A (2017). An AAA Motor-Driven Mechanical Switch in Rpn11 Controls Deubiquitination at the 26S Proteasome . Mol. Cell 67, 799–811. [DOI] [PubMed] [Google Scholar]
- Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, Washburn MP, Conaway RC, Conaway JW, and Cohen RE (2006). Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat. Cell Biol. 8, 994–1002. [DOI] [PubMed] [Google Scholar]
- Yao T, Song L, Jin J, Cai Y, Takahashi H, Swanson SK, Washburn MP, Florens L, Conaway RC, Cohen RE, et al. (2008). Distinct Modes of Regulation of the Uch37 Deubiquitinating Enzyme in the Proteasome and in the Ino80 Chromatin-Remodeling Complex . Mol. Cell 31, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau R, and Rape M (2016). The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586. [DOI] [PubMed] [Google Scholar]
- Yau RG, Doerner K, Castellanos ER, Haakonsen DL, Werner A, Wang N, Yang XW, Martinez-Martin N, Matsumoto ML, Dixit VM, et al. (2017). Assembly and Function of Heterotypic Ubiquitin Chains in Cell-Cycle and Protein Quality Control. Cell 171, 918–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhai B, Gygi SP, Goldberg AL (2015) mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as autophagy. Proc. Natl. Acad. Sci. U. S. A. 112, 15790–15797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, Kuehn HS, Wang H, Yang D, Park YH, et al. (2016). Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. U. S. A. 113, 10127–10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1. Data for Ub AQUA, ITC, Middle-down MS, PRM and AHA Analysis.
(1A) Ub peptides analyzed in this study (related to Figure 2E–G & S3E). Peptide name, peptide sequence, accurate m/z of precursor ion are shown. SDS-PAGE analysis of samples analyzed in this study.
(1B) Standard curves of Ub AQUA peptides. Raw and analyzed data of ten accurate mass AQUA peptides are shown (related to Figure 2E–G & S3E).
(1C) Ub AQUA HR/AM analysis of HMW chains analyzed in this study. Raw and analyzed data are shown for duplicate runs (related to Figure 2E–G & S3E).
(1D) ITC analysis of UCH37•RPN13 and UCH37 C88A•RPN13 complexes with K48-linked and K6/K48-linked Ub chains (related to Figure 4 & S4).
(1E) Middle-down MS analysis of HMW K6/K48, K11/K48, and K48/K63 chains with UCH37•RPN13, WT Ptsm, and UCH37•RPN13-replenish Ptsm (related to Figure 5E & S5F–H).
(1F) MS2 analysis of K-ε-GG pulldown for K11/K48-Ubn-UBE2S-UBD and list of transition ions used for PRM analysis (related to Figure 6A).
(1G) PRM analysis for the degradation of K11/K48-Ubn-UBE2S-UBD with each indicated Ptsm (related to Figure 6D–E & S6C–F).
(1H) AHA labeling densitometry. Raw and analyzed data of 3 individual bioreps are shown (related to Figure 7E–F).
Data Availability Statement
Original/source data are available on Mendeley data (https://data.mendeley.com/datasets/pv8t3hbg4t/1).