

Summary
Aberrant activation of Wnt signaling triggered by mutations in either Adenomatous Polyposis Coli (APC) or CTNNB1 (β-catenin) is a hallmark of colorectal cancers (CRC). As part of a program to develop epigenetic regulators for cancer therapy, we developed carboxamide-substituted benzhydryl amines (CBAs) bearing either aryl or heteroaryl groups that selectively targeted histone lysine demethylases (KDMs) and functioned as inhibitors of the Wnt pathway. A biotinylated variant of N-((5-chloro-8-hydroxyquinolin-7-yl) (4-(diethylamino)phenyl)-methyl)butyramide (CBA-1) identified KDM3A as a binding partner. KDM3A is a Jumonji (JmjC) domain-containing demethylase that is significantly upregulated in CRC. KDM3A regulates the demethylation of histone H3's lysine 9 (H3K9Me2), a repressive marker for transcription. Inhibiting KDM3 increased H3K9Me2 levels, repressed Wnt target genes, and curtailed in vitro CRC cell proliferation. CBA-1 also exhibited in vivo inhibition of Wnt signaling in a zebrafish model without displaying in vivo toxicity.
Subject Areas: Biological Sciences, Biochemistry, Biochemical Mechanism
Graphical Abstract
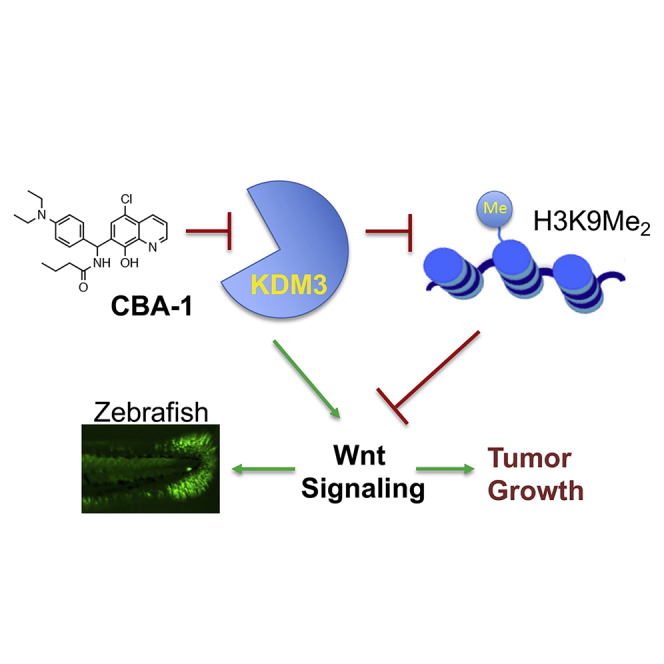
Highlights
-
•
A class of carboxamide-substituted benzhydryl amine (CBA) Wnt inhibitors
-
•
A biological active, biotinylated CBA to identify KDM3A as a direct target
-
•
CBA-1 interacted with the Mn2+ ion in the JmjC domains of KDM3A/3B
-
•
CBA-1 inhibited Wnt signaling in colon cancer cells and in zebrafish models
Biological Sciences; Biochemistry; Biochemical Mechanism
Introduction
Colorectal cancer (CRC) is an unrelenting cause of worldwide, cancer-related mortality with unfortunately high levels in the United States. Many colorectal cancers involve dysfunctional protein participants in the Wnt signaling pathway driven by mutations in either the Adenomatous Polyposis Coli (APC) gene or the CTNNB1 (β-catenin) gene (Anastas and Moon, 2013; Kinzler and Vogelstein, 1996; Nusse and Clevers, 2017; Zhong and Virshup, 2020). In normal cells, β-catenin undergoes phosphorylation by casein kinase-1α (CK1α) and glycogen synthase kinase-3 (GSK3) and the phosphorylated β-catenin subsequently recruits multiprotein Skp1-Cullin-F-box (SCF) Ring-type E3 ligase (β-TrCP) that promotes the ubiquitination of β-catenin and initiates its proteasomal degradation (Anastas and Moon, 2013; Nusse and Clevers, 2017). In CRC cells, the APC and β-catenin mutations not only prevent this normal β-catenin phosphorylation and ubiquitination but also promote abnormal β-catenin stabilization, translocation, and nuclear accumulation (Liu et al., 1999, 2002; Yang et al., 2006). In the nucleus, β-catenin binds T cell factor/lymphoid enhancer-binding factor (TCF/LEF) and its co-activators, such as CBP/p300 and Bcl9, and activates the transcription of Wnt target genes, including many oncogenes (Anastas and Moon, 2013; Nusse and Clevers, 2017). The crucial role played by Wnt signaling in CRC progression makes it a challenging but viable target for the development of new antineoplastic agents (Anastas and Moon, 2013; Barker and Clevers, 2006; Garber, 2009; Zhong and Virshup, 2020).
Many reported inhibitors target upstream events in the Wnt signaling pathway and induce β-catenin degradation (Chen et al., 2009; Huang et al., 2009; Liu et al., 2013). For example, a tankyrase inhibitor, XAV939, stabilizes Axin and induces β-catenin degradation (Huang et al., 2009). Porcupine (PORCN) inhibitors, IWP2 and LSK-974, inhibit Wnt processing and secretion. Although these inhibitors affect Wnt signaling in normal cells or cancer cells with wild-type APC, Axin, and β-catenin, they are less effective for many CRC cells containing Wnt pathway mutations than for those cancer cells lacking these mutations. To address this problem, we seek to develop Wnt inhibitors targeting key steps that lie downstream of β-catenin, such as β-catenin nuclear translocation and β-catenin-mediated gene expression (Lyou et al., 2017), or to develop inhibitors of mitochondrial oxidative phosphorylation that also repress Wnt signaling (Zhang et al., 2019). Others, who also recognized this need, seek to develop Wnt inhibitors that alter the β-catenin/TCF interaction (Lee et al., 2013; Lepourcelet et al., 2004; Schneider et al., 2018), the β-catenin-Bcl9 interactions (Feng et al., 2019; Wisniewski et al., 2016), or the β-catenin/CBP interaction (Emami et al., 2004; Lenz and Kahn, 2014).
Histone methylation events on various lysine residues either activate or repress transcription (Greer and Shi, 2012; Hyun et al., 2017). The generation of H3K4Me3 by histone lysine methyltransferase complexes (KMTs) that contains MLL1/2, ASH2L, BRBP5, WDR5, and other proteins leads to Wnt activation (Sierra et al., 2006). ASH2L interacts with β-catenin and recruits the MLL/1/2 complex to Wnt target genes (Gu et al., 2010). The methylation of H3K79 and H4K20 also correlates with Wnt activation. Dot1L, the mammalian homolog of Dot1 that is a SAM-dependent KMT, regulates the methylation of H3K79Me2 and H3K79Me3, and both of these methylated histones participates in Wnt activation (Mahmoudi et al., 2010). In the intestine, Dot1L undergoes recruitment to the TCF/β-catenin complex through its co-factor, AF10, and these events regulate Wnt signaling in intestinal stem cells. In addition to the MLL1/2 and Dot1L KMTs, Set8 regulates Wnt signaling through H4K20 mono-methylation (Li et al., 2011). Inhibitors for MLL1/2 (e.g., an MLL1/WDR5 inhibitor called MM-102 [Karatas et al., 2013]), Dot1L (e.g., EPZ-5676 [Daigle et al., 2013]), and Set8 (e.g., Ryuvidine [Blum et al., 2014]) are commercially available, but the initial expectations for these inhibitors as promising drugs for the treatment of leukemia are unfortunately offset by their limited effects on Wnt signaling and CRC proliferation, probably because of cell-type dependency or the redundancy of KMTs.
On the other hand, demethylation events by histone demethylases (KDMs) also regulate the levels and patterns of methylation and thereby affect chromatin remodeling and gene expression. Inhibition of KDMs may lead to a net increase in histone methylation patterns at specific lysine residues (Cloos et al., 2008; Jambhekar et al., 2017; Klose et al., 2006), leading, for example, to increased methylation of H3K9 or H3K27 that in turn represses transcription. The first reported KDM is LSD1 or KDM1A (Shi et al., 2004) that belongs, along with a related demethylase called KDM1B, to the so-called type 1 family of KDMs that contains a flavin adenine dinucleotide (FAD)-dependent amine oxidase (Jambhekar et al., 2017; Kooistra and Helin, 2012). The second type of KDMs contains a Jumonji C (JmjC) domain (Jambhekar et al., 2017; Klose et al., 2006) and embrace seven families of human JmjC domain-containing KDMs with specific demethylase activities (Klose et al., 2006; Kooistra and Helin, 2012). In the course of a program designed to develop new epigenetic regulators as antineoplastic agents (Sviripa et al., 2014; Zhang et al., 2013), we now report a family of carboxamide-substituted benzhydryl amines (CBAs) as KDM3A/3B inhibitors that selectively induce elevated levels of H3K9 methylation that in turn inhibit the Wnt signaling pathway in cell and zebrafish models.
Results
High-Throughput Screening
To identify novel Wnt regulators by high-throughput screening, we assembled a stable HEK293T cell line containing the TOPFlash reporter (Zhang et al., 2019). Since mutations in the Wnt pathway often activated β-catenin, it was important to inhibit Wnt signaling downstream of β-catenin. We activated Wnt signaling by treating cells with lithium chloride that inhibited GSK3 and stabilized β-catenin (Hedgepeth et al., 1997), dual processes that mimicked the constitutively active Wnt signaling found in cancer cells. Starting from a compound library previously available from the Drug Discovery Center at University of Cincinnati (Cincinnati, OH, USA) as well as CBAs synthesized in-house using standard, chemical methodology, we identified CBA-1 that inhibited Wnt signaling at 500 nM concentrations.
To validate the screening results, we synthesized and purified CBA-1 (Figure 1A). Heating a 1:1 mixture of 4-(N,N-diethylamino)benzaldehyde and 5-chloroquinolin-8-ol with a 4-fold excess of butyramide at 130°C for 2 h and quenching with isopropanol furnished a precipitate of N-((5-chloro-8-hydroxyquinolin-7-yl) (4-(diethylamino)phenyl)-methyl)butyramide (CBA-1) (Figure 1A) in good yield and pure form, unlike commercial samples of CBA-1 that were contaminated with ca. 5% of an impurity, 7,7'-((4-(diethylamino)phenyl)methylene)bis (5-chloroquinolin-8-ol). This contaminant derived from the condensation of one equivalent of 4-(N,N-diethylamino)benzaldehyde with two equivalents of 5-chloroquinolin-8-ol.
Figure 1.

Validation of a Leading Wnt Inhibitor
(A) Synthesis of CBA-1 from 5-chloroquinolin-8-ol (1 eq), butyramide (4 eq), and 4-diethylaminobenzaldehyde (1 eq), 130°C, 2 h followed by the addition of isopropanol to induce precipitation (71% yield).
(B) Effects of CBA-1 (3 μM) and XAV939 (5 μM) on TOPFlash reporter activity (∗p < 0.0001; #p < 0.01; n = 3).
(C) CBA-1 repressed Wnt target genes expression in LS174T CRC cells. Densitometry was analyzed using three western blot images (∗p < 0.05; n = 3).
(D) CBA-1 inhibited CRC cell proliferation (∗p < 0.001; #p < 0.05; n = 3).
We tested the activity of synthesized CBA-1 using TOPFlash reporter assay. Similar to the screening results, CBA-1 inhibited Wnt signaling induced by either LiCl or Wnt protein (Figure 1B). As a control, a well-known Wnt inhibitor, XAV939 (i.e., 3,5,7,8-tetrahydro-2-[4-(trifluoromethyl)phenyl]-4H-thiopyrano [4,3-d]pyrimidin-4-one, Sigma-Aldrich, MO), strongly inhibited Wnt-activated, but not LiCl-activated, TOPFlash reporter (Figure 1B), a result that indicated that CBA-1 and XAV939 have different mechanisms in inhibiting Wnt signaling. Treatment of LS174T CRC cells with CBA-1 repressed the expression of Wnt target genes (Figure 1C) and inhibited cell proliferation (Figure 1D), as expected given the importance of Wnt signaling in CRC.
XAV939 inhibited tankyrase, stabilized Axin, and promoted β-catenin degradation that thereby inhibited Wnt signaling. On the other hand, LiCl inhibited GSK3 that effected β-catenin phosphorylation and degradation and thereby activated Wnt signaling. When β-catenin N-terminal phosphorylation was blocked by either a serine/threonine mutation or GSK3 inhibitors, β-catenin became resistant to either XAV939- or Axin-induced degradation. Because CBA-1 inhibited LiCl-induced Wnt signaling, it was reasonable to assume that it inhibited Wnt signaling downstream of β-catenin, most likely by blocking β-catenin activity in the nucleus. To further analyze the activity of CBA-1, we treated mouse colon cancer organoids isolated from Apcf/+/KrasLSL−G12D/Villin-Cre mouse model (Figure S1A) (Wen et al., 2017). CBA-1 significantly inhibited colon cancer organoids formation (Figure S1B), suggesting that CBA-1 could inhibit colon cancer cells with APC and K-ras mutations.
CBA-1 Regulates Histone Methylation
CBA-1 showed no clear effects on the assembly of β-catenin/TCF (Figure 2A). Consequently, we hypothesized that CBA-1 inhibited Wnt signaling epigenetically by altering chromatin remodeling, and we probed the effects of CBA-1 on histone methylation. In both HEK293T and LS174T colorectal cancer cells, CBA-1 significantly increased the levels of H3K9Me2 (Figures 2B and 2C) but had no effect on H3K4Me2, outcomes that suggested that CBA-1 inhibited specific histone demethylase(s) and thereby promoted increased repressive methylation levels of histones and ultimately inhibited transcription. Indeed, chromatin immunoprecipitation (ChIP) assay with an anti-H3K9Me2 Ab found that CBA-1 increased the levels of H3K9Me2 on the promoters of Wnt target genes, c-Myc and Cyclin B1 (Figure 2D).
Figure 2.

CBA-1 Regulates Histone Methylation
(A) CBA-1 did not inhibit the binding between β-catenin and TCF4. FLAG-tagged β-catenin and HA-tagged TCF4 were transfected separately into HEK293T cells. Equal amounts of β-catenin and TCF4 were mixed and immunoprecipitated by an anti-FLAG antibody. TCF4 was analyzed by an anti-HA antibody.
(B and C) CBA-1 significantly increased H3K9Me2 and H3K27Me3 in HEK293T and LS174T cells. Densitometry was analyzed using three western blot images (∗p < 0.05; n = 3).to new sentences: (B) CBA-1 significantly increased H3K9Me2 and H3K27Me3 in HEK293T cells. Densitometry was analyzed using three western blot images (∗p < 0.05; n = 3). (C) CBA-1 significantly increased H3K9Me2 and H3K27Me3 in LS174T cells. Densitometry was analyzed using three western blot images (∗p < 0.05; n = 3).
(D) CBA-1 increased H3K9Me2 on the promoters of Wnt target genes. The promoter sequences were analyzed by both standard PCR (left) and qPCR (right) (∗p < 0.01; n = 3).
(E) KDM3A, a potential target of CBA-1, overexpressed in CRC. Tumor: red. Normal tissue: gray.
(F) High levels of KDM3A correlated with poor CRC patient survival.
Target Identification
Because it was reported that KDM3A regulated H3K9Me2 in cancer cells (Li et al., 2017; Wang et al., 2019; Yoo et al., 2020), we analyzed the expression of KDM3A in CRC. The mRNA levels of KDM3A were upregulated in CRC (Figure 2E), and high levels of KDM3A expression also correlated with poor survival (Figure 2F). These findings suggested that KDM3A represented a previously unappreciated target for drug intervention in CRC. Because relatively few prior studies focused on KDM3A in CRC and because it has no specific FDA-approved inhibitor, we sought additional evidence to support the direct interaction of CBA-1 with KDM3A before launching additional synthetic, structure-activity relationship (SAR) studies.
Among the approaches for evaluating this direct interaction, we designed and synthesized biotinylated analogs of CBA-1 in which we attached D-biotin through a polyethylene glycol (PEG) spacer to the C-8 hydroxyl group in the 5-chloroquinolin-8-ol in CBA-B1 (Figure S2); the N,N-diethylamino group in CBA-B2 (Figure 3); and the carboxamide moiety in 4-(((5-chloro-8-hydroxyquinolin-7-yl) (4-morpholinophenyl)methyl)amino)-4-oxobutanoic acid in CBA-B3 (Figure S2). As a prerequisite for pull-down studies, we determined that CBA-B2 retained sufficient biological activity relative to CBA-1 as determined independently by evaluating Wnt inhibition (Figures 3A and 3B). A streptavidin-bead pull-down assay using HEK293T cell lysates and CBA-B2 identified KDM3A as a binding partner (Figure 3C). After incubating the lysate with the streptavidin-CBA-B2-loaded beads, washing thoroughly, and evaluating the eluate by western blotting, we detected KDM3A, a finding that suggested that CBA-1 bound endogenous KDM3A. Consistent with this binding result, a dose-response study suggested that CBA-1 inhibited KDM3A activity in vitro at low micromolar levels (Figure 3D). Knocking down KDM3A by shRNA also inhibited Wnt target gene expression in LS174T CRC cells (Figure 3E) and inhibited Wnt-mediated TOPFlash reporter activity in HEK293T cells (Figure 3F), outcomes that validated KDM3A as a cancer-relevant target. These studies corroborated the CBA-1 inhibition of KDM3A as the key event in the observed Wnt inhibition. KDM3A shRNA also inhibited CRC cell proliferation (Figure 3G), a finding that linked in vitro inhibition of cell proliferation to the inhibition of Wnt signaling requiring KDM3A. These findings are consistent with a previous report that JMJD1A (KDM3A) promotes CRC growth and metastasis by enhancing Wnt signaling (Peng et al., 2018).
Figure 3.

CBA-1 Interacts with KDM3A
(A) Synthesis of CBA-B2, a biological active biotinylated version of CBA-1; Legend: a, 4-fluorobenzaldehyde, ethyl piperidine-4-carboxylate, K2CO3, dimethyl sulfoxide, 120°C, 3 h (63% yield); b, 2 M NaOH, 48 h, 25°C (100% yield); c, N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-5-(2-oxohexahydro-1H-thieno [3,4-d]imidazol-4-yl)pentanamide (1 eq), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (1.5 eq), 1-hydroxybenzotriazole (1.5 eq), triethylamine (1.5 eq), 12 h, 25°C (57% yield); d, 5-chloroquinolin-8-ol (1 eq), butyramide (6 eq), 150°C, 5 h (57% yield).
(B) CBA-B2 inhibited Wnt signaling (∗p < 0.0001; n = 3).
(C) KDM3A was pulled down from HEK293T cell lysates by CBA-B2 and streptavidin beads.
(D) Dose-responses of CBA-1 on the enzymatic activities of KDM3A.
(E) Knocking down of KDM3A reduced Wnt target genes expression in LS174T CRC cells. Densitometry was analyzed using three western blot images (∗p < 0.05; n = 3).
(F) Knocking down of KDM3A decreased Wnt-induced TOPFlash reporter activity (∗p < 0.01; #p < 0.05; n = 3).
(G) Knocking down of KDM3A inhibited LS174T CRC cell proliferation (∗p < 0.0001; n = 3).
Molecular Modeling Studies
In the absence of an X-ray crystal structure of KDM3A, we turned to molecular modeling studies of a related histone demethylase. We utilized a BLAST search to determine that KDM3A exhibited 89% sequence similarity and 60% sequence identity to another family member, KDM3B (Figure S3A), that also possessed a JmjC domain and that had a high-resolution crystal structure (PDB: 4C8D). We utilized molecular dynamics to construct a reasonable representation of KDM3A using the structure of KDM3B as a departure point. According to the RCSB database, the coordination of the manganese ion in the KDM3B active site involved water and N-oxalylglycine, and we assumed similar coordination in the modeled structure for KDM3A. In the homology model of KDM3A that emerged from these studies, the binding pose of the modeled structure of KDM3A (Figure 4A) predicted that CBA-1 coordinated directly to the Mn2+ ion site. An expanded view (Figure 4B) of the coordination of Mn2+ by CBA-1 showed that nitrogen of the quinoline ring in CBA-1 coordinated with the Mn2+ at 2.8 Å. The backbone atoms of KDM3A also coordinated with Mn2+at 2.2 and 2.1 Å. We noted interesting hydrophobic interactions (Figure 4C) between portions of CBA-1 and residues L1120, V1123, and F1280 that resided within 4 Å of CBA-1. Finally, various polar and charged residues (Figure 4D) surrounded CBA-1 within 3 Å and defined the arrangement of functional groups in CBA-1 that provided the specificity seen in its binding to KMD3A. The binding pose shown in Figure 4 has a binding free energy of −12.3 kcal/mol.
Figure 4.

Molecular Modeling of the Binding Site of KDM3A with CBA-1
(A) Global view of the binding of CBA-1 to KDM3A. Cartoon model representations are shown of the KDM3A in cyan. CBA-1 is shown in stick model and colored yellow.
(B) Local view of the Mn2+-binding site including CBA-1. Residues involved in the binding site are shown with stick model and colored the same color as its protein backbone. Dashed lines represent the coordination with distances shown close to the respective lines.
(C) Shown here is KDM3A with a focus on the hydrophobic interactions between CBA-1 and residues of KDM3A.
(D) Shown here is CBA-1 surrounded by residues of KDM3A that are within 4 Å of the inhibitor.
The JmjC domains of KDM3A and KDM3B are very similar (Figure S3A). CBA-1 also inhibited KDM3B (Figure S3B), but the inhibition activity for KDM3B was poorer than its inhibition activity for KDM3A (Figure 3D). We also analyzed the binding pose of the inhibitor CBA-1 with the crystal structure of KDM3B (Figure S3C). The global view of the structure suggested that the inhibitor bound and coordinated with Mn2+ in the JmjC domain. The CBA-1 inhibitor and the three residues from KDM3B coordinated with the manganese cation (Figure S3D). A delta nitrogen from H1689 and H1560 of KDM3B coordinated with Mn2+ at 2.3 Å. A third residue, namely, D1562 of KDM3B, coordinated with Mn2+ through two oxygens from the carboxylic group at 2.1 Å. Lastly, the inhibitor CBA-1 coordinates with Mn2+ through a cyclic nitrogen at 3.9 Å. Neighboring residues of KDM3B promoted the binding of the inhibitor to the domain with hydrophobic and basic side-chain interactions (Figure S3E). These residues are F1720, I1683, and H1648, which can be found to be within 3 Å of CBA-1 (Figure S3F); no other important interactions between CBA-1 and KDM3B were noted.
Although a specific KDM3A inhibitor was not available commercially, other KDM inhibitors target the JmjC domain. For example, GSK-J1 and GSK-J4 were originally described as specific inhibitors for H3K27 demethylases JMJD3/KDM6B (UTX) (Kruidenier et al., 2012). Further studies found that these KDM inhibitors in fact had broad activities in inhibiting JmjC domain in KDMs, probably with differential preferences for different KDMs (Heinemann et al., 2014). We found, for example, that GSK-J4, but not GSK-J1, inhibited Wnt signaling (Figure S4), a finding that suggested that GSK-J4 like CBA-1 also inhibited KDM3A. GSK-J1 was not active in this cell-based assay, possibly because of its restricted cell permeability (Kruidenier et al., 2012). However, the facile absorption and metabolism of GSK-J4 to GSK-J1 in the cells (Kruidenier et al., 2012) suggested GSK-J1 is the actual, active KDM3A inhibitor.
We also analyzed the binding pose of the inhibitor GSK-J1 with the modeled structure of KDM3A (Figures 4 and S5) that we developed. A global view of the protein with the Mn2+ ion and GSK-J1 (Figure S5A) showed that GSK-J1 bound to KDM3A at the Mn2+-binding site with coordination between GSK-J1 and the Mn2+ ion (Figure S5B). A nitrogen from GSK-J1 coordinated with the Mn2+ ion at a distance of 2.8 Å. KDM3A residues coordinated with Mn2+ at 2.2 and 2.1 Å. There were no other important interactions between GSK-J1 and KDM3A other than the coordination with Mn2+ as shown in alternate views of surrounding residues within 3 Å of GSK-J1 (Figures S5C and S5D). The binding pose for this structure in Figure S5 has a binding free energy of −6.6 kcal/mol. The binding affinity of CBA-1 to KDM3A is greater than that of GSK-J1, a finding consistent with the TOPFlash reporter assay that indicated that CBA-1 was a better Wnt inhibitor than either GSK-J1 or GSK-J4 (Figure S4B).
In Vivo Studies with CBA-1
To validate the in vivo activity of CBA-1, we employed a Wnt/β-catenin zebrafish reporter line, Tcf/Lef-miniP:dGFP(Scaffidi et al., 2002), in which GFP expression occurred mostly in the tail of the larvae where Wnt signaling is active during development (Figure 5A). The effects of CBA-1 on GFP expression were analyzed using the following agents: DMSO (vehicle) and GSK3 inhibitor called Bio (6-bromoindirubin-3′-oxime, Sigma-Aldrich, MO, USA). CBA-1 inhibited Wnt signaling in the tail (Figure 5B). As a control, Bio inhibited GSK3, stabilized β-catenin, and thus activated Wnt signaling (Figure 5C). Wnt signaling is also critical for zebrafish tail fin regeneration after amputation. CBA-1 treatment reduced Wnt-dependent tail regeneration in amputated tails of adult Tcf/Lef-miniP:dGFP zebrafish and led to a decrease in GFP expression at the regenerating edge of the tail fin compared with DMSO control (Figures 5F–5L). Since some compounds may affect the reporter or tail fin regeneration through nonspecific or even toxic effects, we performed an eye rescue experiment to further test if CBA-1 specifically inhibits the Wnt signaling in zebrafish (Figures 5D and 5E). Wnt signaling also mediates eye development in zebrafish, in which excessive Wnt signaling has deleterious effects in early embryos. Bio treatment resulted in a “no-eye” phenotype in zebrafish embryos, CBA-1 rescued Bio-induced eye defects (Figure 5D), and CBA-1 rescued Bio-induced eye defects in a dose-responsive manner (Figure 5E). At both 5 and 10 μM concentrations, CBA-1 was able to rescue completely the loss-of-eye phenotype. Additionally, we observed no obvious toxicity or embryo deaths with up to 20 μM concentrations of CBA-1.
Figure 5.

CBA-1 Inhibited Wnt Signaling in Zebrafish Models
(A) TCF/LEF-GFP zebrafish model. GFP was expressed in the tails where there is active Wnt signaling.
(B) CBA-1 (10 μM) inhibited GFP expression in the tails.
(C) Bio (1 μM), a GSK3 inhibitor and Wnt activator, significantly induced GFP expression in the TCF/LEF-GFP zebrafish model. Scale bar, 0.25 mm.
(D) Representative images of no eye phenotype seen with Bio (1 μM) treatment and eye rescue phenotype seen with Bio (1 μM) + CBA-1 (10 μM) treatment. Scale bar, 0.5 mm.
(E) Eye development assay. Wnt activation by Bio inhibited eye development (no eye phenotype). CBA-1 inhibited Bio-induced Wnt signaling, leading to partial (one eye) or full (two eyes) rescue of the eye development in a dose-dependent manner. Numbers above bars represent total number of fish per group.
(F) Uncut tail of TCF/LEF GFP sheer zebrafish. Scale bar, 2 mm.
(G) Tail immediately post amputation.
(H and I) Partial tail regeneration and GFP fluorescence were observed in DMSO-treated zebrafish. Arrowhead depicts tail regeneration.
(J and K) A complete loss of GFP fluorescence and lack of tail regeneration were observed in CBA-1 (5 μM)-treated fish.
(L) CBA-1 treatment leads to a significant decrease in length of tail regeneration. ∗∗∗p = 0.0003.
Discussion
The inhibition of Wnt signaling is an important goal for the development of new therapeutics for colon cancer treatment (Anastas and Moon, 2013; Nusse and Clevers, 2017). Histone acetylases and histone methyltransferases play central roles in chromatin remodeling and hence transcriptional activation of Wnt target genes. Small-molecule inhibitors that target these epigenetic mechanisms are attractive agents for regulating transcription and developing new antineoplastic drugs, particularly in the case of CRC where Wnt signaling is important (Patnaik and Anupriya, 2019; Polakis, 2012). In this study, we identified N-((5-chloro-8-hydroxyquinolin-7-yl) (4-(diethylamino)phenyl)-methyl)butyramide (CBA-1), a representative member of a family of small-molecule, epigenetic Wnt inhibitors for a specific histone demethylase, KDM3A, that foreclosed on Wnt signaling by increasing histone methylation at H3K9, a repression marker for gene transcription.
A high-throughput screening program using a stable cell line containing a TOPFlash reporter first identified this carboxyamide-substituted benzhydryl amine CBA-1 as a Wnt signaling inhibitor. We compared CBA-1 with XAV939, a well-known tankyrase inhibitor that regulated β-catenin stability. CBA-1 significantly inhibited LiCl-induced Wnt signaling, suggesting that CBA-1 did not affect β-catenin stability but inhibited β-catenin activity (Figure 1B). CBA-1 also had no effects on β-catenin/TCF/complex (Figure 2A), a finding that indicated that CBA-1 inhibited Wnt signaling by affecting chromatin remodeling. We analyzed different histone methylation markers and found that CBA-1 induced histone methylation on H3K9Me2 (Figures 2B–2D) that controlled transcriptional silencing. A biologically active, biotinylated variant, namely, CBA-B2 (Figures 3A–3C), led to a successful pull-down experiment that confirmed KDM3A as the target of CBA-1. Additional validation of this outcome emerged from shRNA suppression of KDM3A that inhibited, as expected, Wnt signaling (Figures 3E–3G) and pointed to KDM3A as a crucial regulatory element in the Wnt signaling pathway.
Although we identified KDM3A as a direct target of CBA-1 and an important regular of Wnt signaling, we do not expect that KDM3A only regulates Wnt signaling. As we know, the well-established Wnt inhibitor XAV939 targets tankyrase (Huang et al., 2009) and ICG-001 targets CBP (Emami et al., 2004). These enzymes regulate many proteins and are also not specific for Wnt signaling. However, our findings suggested that Wnt signaling can be efficiently inhibited by small-molecule KDM3A inhibitors, such as CBA-1. CBA-1 inhibited colon cancer cells with APC or β-catenin mutations (Figure 1D) and also inhibited colon cancer organoids with APC and K-ras mutation (Figure S1B). KDM3A was overexpressed in CRC, and the expression levels of KDM3A inversely correlated with patient survival (Figures 2E and 2F). KDM3A is required for both growth and metastasis of CRC (Peng et al., 2018), further suggesting that KDM3A is an important target for CRC treatment.
We recognize that KDM3A may not be the only target and structural variations in CBA-1 may affect other JmjC domain-containing histone demethylases, particularly other members of KDM3 (Li et al., 2017). As demonstrated in Figure 3F, CBA-1 further decreased the Wnt reporter activity inhibited by KDM3A shRNA, suggesting that CBA-1 may inhibit other KMD (s) that also regulate the Wnt signaling. Indeed, CBA-1 inhibited KDM3B activity as well (Figure S3). We also recognize that inhibitors for other histone demethylases, structurally unrelated to CBA-1, may also inhibit KDM3A through binding events at locales other than the site identified in our molecular modeling study. For example, we compared the KDM6 inhibitors GSK-J1/J4 with CBA-1. Although less potent that CBA-1, GSK-J1/J4 also inhibited Wnt signaling (Figure S4). Docking studies suggest that CBA-1 and GSK-J1 coordinated the manganese Mn2+ ion in the active site of KDM3A, but CBA-1 has additional hydrophobic interactions with residues L1120, V1123, and F1280 and thus exhibited a much higher binding affinity (Figures 4 and S5) than the binding affinity of GSK-J1. The docking study explained the finding that CBA-1 was superior to GSK-J1/J4 in KDM3A/Wnt inhibition (Figures S4 and S5). CBA-1 coordinated with Mn2+ at a long distance in KDM3B (3.9 Å) than the distance in KDM3A (2.8 Å) (Figures 4 and S3). This could be one of the reasons that CBA-1 inhibited KDM3A more significantly than KDM3B.
To test the activity of CBA-1 in vivo, we utilized a zebrafish Wnt/β-catenin reporter line that indicated that CBA-1 inhibited Wnt-mediated GFP expression in the tails where the Wnt pathway was most active (Figure 5). CBA-1 inhibited tail fin regeneration, a process regulated by Wnt signaling. During embryogenesis, excess Wnt signaling also blocked eye development in zebrafish. CBA-1 rescued the “no-eye phenotype” induced by a Wnt activator Bio with no toxicity to developing animals, an important observation that suggested that CBA-1 specifically inhibited Wnt signaling in vivo and that also suggested that CBA-1 is a promising leading compound for the development of clinically useful Wnt inhibitors. These zebrafish models provide high-throughput methods to screen Wnt inhibitors and to test the toxicity of drug candidates in vivo.
Given the high similarity of the KDM structures, particularly the JmjC domains, development of small-molecule inhibitors that target only a specific JmjC domain-containing KDM remains as a scientific challenge. For example, GSK-J1/J4 were originally developed as specific KDM6 inhibitors (Kruidenier et al., 2012) but were later reported as broad KDM inhibitors (Heinemann et al., 2014). Other reported “specific” KDM inhibitors may also have multiple targets. As we discussed above, CBA-1 may also inhibit other JmjC domain-containing KDMs that regulate H3K9 methylation and Wnt signaling. In addition, CBA-1 increased H3K27 methylation (Figures 2B and 2C), suggesting that it may inhibit cancer cell growth by targeting multiple JmjC domain-containing KDMs. Although a highly specific inhibitor that only targets a single enzyme is often prized as a molecular probe, an inhibitor that targets a family of functionally redundant enzymes has value in therapeutic drug development. Epigenetic drugs targeting a subfamily of KDMs may be a more efficient approach for cancer treatment. CBA-1 not only inhibited Wnt signaling in cancer cells but may also inhibit Wnt signaling in normal cells (Figure 3F), suggesting that it may have potential toxic effects in animals and should be further optimized through structure-activity relationship (SAR) studies. Nonetheless, the identification of CBA-1 as KDM3A and KDM3B inhibitors and the utility of molecular modeling of CBAs in the KMD3A/3B construct provide the means for structure-guided, future SAR studies that will develop CBA inhibitors targeting specific KDMs for Wnt inhibition and cancer treatment.
Limitations of the Study
In this study, we developed a family of carboxamide-substituted benzhydryl amines (CBAs) as histone lysine demethylase (KDM) inhibitors. Specifically, we found that CBA-1 inhibited KDM3A/3B and increased the levels of H3K9Me2. We also found that CBA-1 inhibited Wnt signaling in vitro in colon cancer cells and in vivo in zebrafish models. However, the specificity of CBA-1 on different KDMs should be further examined in the future. It is also important to perform additional SAR studies to further optimize CBA-1 for future clinical development.
Resource Availability
Lead Contact
Further information and requests should be directed to the Lead Contact, Professor Chunming Liu (chunming.liu@uky.edu)
Materials Availability
Materials are available from the corresponding authors on request.
Data and Code Availability
This study did not generate computer code. All data and analytical methods are available in the main text or in Supplemental Information.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We are grateful to Professor Randall Moon for the Super 8× TOPFlash and 8× FOPFlash plasmids and Professor Tianyan Gao for mouse colon cancer organoids. C.L. and D.S.W. were supported by NIH R01 CA172379 from the National Institutes of Health and by NIH UL1 TR000117 from the National Institutes of Health to the University of Kentucky's Center for Clinical and Translational Science. D.S.W. was also supported in part by the Office of the Dean of the College of Medicine, the Center for Pharmaceutical Research and Innovation in the College of Pharmacy, the Department of Defense (DoD) Prostate Cancer Research Program Award W81XWH-16-1-0635 [Grant Log# PC150326P2], and NIH P30 RR020171 from the National Institute of General Medical Sciences to L. Hersh. V.M.S. was supported by grant IRG 16-182-28 from the American Cancer Society. J.S.B. and M.G.H. are supported by NIH R37 CA227656 and T32 CA165990, respectively. This study is also supported by Markey Cancer Center (P30 CA177558).
Author Contributions
Conceptualization, C.L. and D.S.W.; Methodology, W.Z., V.M.S., Y.X., T.Y., and M.G.H.; Data Analysis, W.Z., V.M.S., J.S.B., and C.-G.Z.; Supervision, C.L., D.S.W., J.S.B., and C.-G.Z.; Writing, C.L. and D.S.W.
Declaration of Interests
C.L. and D.S.W. have partial ownership in a for-profit venture, Epionc, Inc., that seeks to develop small-molecule inhibitors for cancer treatment. In accord with University of Kentucky policies, C.L. and D.S.W. have disclosed this work to the University of Kentucky's Intellectual Property Committee and to a Conflict of Interest Oversight Committee in accord with University of Kentucky policies.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101795.
Contributor Information
David S. Watt, Email: dwatt@uky.edu.
Chunming Liu, Email: chunming.liu@uky.edu.
Supplemental Information
References
- Anastas J.N., Moon R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- Barker N., Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- Blum G., Ibanez G., Rao X., Shum D., Radu C., Djaballah H., Rice J.C., Luo M. Small-molecule inhibitors of SETD8 with cellular activity. ACS Chem. Biol. 2014;9:2471–2478. doi: 10.1021/cb500515r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Dodge M.E., Tang W., Lu J., Ma Z., Fan C.W., Wei S., Hao W., Kilgore J., Williams N.S. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009;5:100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloos P.A., Christensen J., Agger K., Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–1140. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S.R., Olhava E.J., Therkelsen C.A., Basavapathruni A., Jin L., Boriack-Sjodin P.A., Allain C.J., Klaus C.R., Raimondi A., Scott M.P. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–1025. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami K.H., Nguyen C., Ma H., Kim D.H., Jeong K.W., Eguchi M., Moon R.T., Teo J.L., Kim H.Y., Moon S.H. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected] Proc. Natl. Acad. Sci. U S A. 2004;101:12682–12687. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng M., Jin J.Q., Xia L., Xiao T., Mei S., Wang X., Huang X., Chen J., Liu M., Chen C. Pharmacological inhibition of beta-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating Treg cells. Sci. Adv. 2019;5:eaau5240. doi: 10.1126/sciadv.aau5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber K. Drugging the Wnt pathway: problems and progress. J. Natl. Cancer Inst. 2009;101:548–550. doi: 10.1093/jnci/djp084. [DOI] [PubMed] [Google Scholar]
- Greer E.L., Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B., Watanabe K., Dai X. Epithelial stem cells: an epigenetic and Wnt-centric perspective. J. Cell Biochem. 2010;110:1279–1287. doi: 10.1002/jcb.22650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgepeth C.M., Conrad L.J., Zhang J., Huang H.C., Lee V.M., Klein P.S. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev. Biol. 1997;185:82–91. doi: 10.1006/dbio.1997.8552. [DOI] [PubMed] [Google Scholar]
- Heinemann B., Nielsen J.M., Hudlebusch H.R., Lees M.J., Larsen D.V., Boesen T., Labelle M., Gerlach L.O., Birk P., Helin K. Inhibition of demethylases by GSK-J1/J4. Nature. 2014;514:E1–E2. doi: 10.1038/nature13688. [DOI] [PubMed] [Google Scholar]
- Huang S.M., Mishina Y.M., Liu S., Cheung A., Stegmeier F., Michaud G.A., Charlat O., Wiellette E., Zhang Y., Wiessner S. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- Hyun K., Jeon J., Park K., Kim J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017;49:e324. doi: 10.1038/emm.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambhekar A., Anastas J.N., Shi Y. Histone lysine demethylase inhibitors. Cold Spring Harb. Perspect. Med. 2017;7:a026484. doi: 10.1101/cshperspect.a026484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatas H., Townsend E.C., Cao F., Chen Y., Bernard D., Liu L., Lei M., Dou Y., Wang S. High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J. Am. Chem. Soc. 2013;135:669–682. doi: 10.1021/ja306028q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler K.W., Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Klose R.J., Kallin E.M., Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- Kooistra S.M., Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell. Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- Kruidenier L., Chung C.W., Cheng Z., Liddle J., Che K., Joberty G., Bantscheff M., Bountra C., Bridges A., Diallo H. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E., Madar A., David G., Garabedian M.J., Dasgupta R., Logan S.K. Inhibition of androgen receptor and beta-catenin activity in prostate cancer. Proc. Natl. Acad. Sci. U S A. 2013;110:15710–15715. doi: 10.1073/pnas.1218168110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz H.J., Kahn M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci. 2014;105:1087–1092. doi: 10.1111/cas.12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepourcelet M., Chen Y.N., France D.S., Wang H., Crews P., Petersen F., Bruseo C., Wood A.W., Shivdasani R.A. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004;5:91–102. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- Li J., Yu B., Deng P., Cheng Y., Yu Y., Kevork K., Ramadoss S., Ding X., Li X., Wang C.Y. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/beta-catenin signalling. Nat. Commun. 2017;8:15146. doi: 10.1038/ncomms15146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Nie F., Wang S., Li L. Histone H4 Lys 20 monomethylation by histone methylase SET8 mediates Wnt target gene activation. Proc. Natl. Acad. Sci. U S A. 2011;108:3116–3123. doi: 10.1073/pnas.1009353108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Kato Y., Zhang Z., Do V.M., Yankner B.A., He X. beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc. Natl. Acad. Sci. U S A. 1999;96:6273–6278. doi: 10.1073/pnas.96.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Li Y., Semenov M., Han C., Baeg G.H., Tan Y., Zhang Z., Lin X., He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Liu J., Pan S., Hsieh M.H., Ng N., Sun F., Wang T., Kasibhatla S., Schuller A.G., Li A.G., Cheng D. Targeting Wnt-driven cancer through the inhibition of porcupine by LGK974. Proc. Natl. Acad. Sci. U S A. 2013;110:20224–20229. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyou Y., Habowski A.N., Chen G.T., Waterman M.L. Inhibition of nuclear Wnt signalling: challenges of an elusive target for cancer therapy. Br. J. Pharmacol. 2017;174:4589–4599. doi: 10.1111/bph.13963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi T., Boj S.F., Hatzis P., Li V.S., Taouatas N., Vries R.G., Teunissen H., Begthel H., Korving J., Mohammed S. The leukemia-associated Mllt10/Af10-Dot1l are Tcf4/beta-catenin coactivators essential for intestinal homeostasis. PLoS Biol. 2010;8:e1000539. doi: 10.1371/journal.pbio.1000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R., Clevers H. Wnt/beta-Catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Patnaik S., Anupriya Drugs targeting epigenetic modifications and plausible therapeutic strategies against colorectal cancer. Front. Pharmacol. 2019;10:588. doi: 10.3389/fphar.2019.00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K., Su G., Ji J., Yang X., Miao M., Mo P., Li M., Xu J., Li W., Yu C. Histone demethylase JMJD1A promotes colorectal cancer growth and metastasis by enhancing Wnt/beta-catenin signaling. J. Biol. Chem. 2018;293:10606–10619. doi: 10.1074/jbc.RA118.001730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012;4:a008052. doi: 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P., Misteli T., Bianchi M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Schneider J.A., Craven T.W., Kasper A.C., Yun C., Haugbro M., Briggs E.M., Svetlov V., Nudler E., Knaut H., Bonneau R. Design of peptoid-peptide macrocycles to inhibit the beta-catenin TCF interaction in prostate cancer. Nat. Commun. 2018;9:4396. doi: 10.1038/s41467-018-06845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Lan F., Matson C., Mulligan P., Whetstine J.R., Cole P.A., Casero R.A., Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Sierra J., Yoshida T., Joazeiro C.A., Jones K.A. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 2006;20:586–600. doi: 10.1101/gad.1385806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sviripa V.M., Zhang W., Balia A.G., Tsodikov O.V., Nickell J.R., Gizard F., Yu T., Lee E.Y., Dwoskin L.P., Liu C., Watt D.S. 2',6'-Dihalostyrylanilines, pyridines, and pyrimidines for the inhibition of the catalytic subunit of methionine S-adenosyltransferase-2. J. Med. Chem. 2014;57:6083–6091. doi: 10.1021/jm5004864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.Y., Long Q.Y., Tang S.B., Xiao Q., Gao C., Zhao Q.Y., Li Q.L., Ye M., Zhang L., Li L.Y., Wu M. Histone demethylase KDM3A is required for enhancer activation of hippo target genes in colorectal cancer. Nucleic Acids Res. 2019;47:2349–2364. doi: 10.1093/nar/gky1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y.A., Xing X., Harris J.W., Zaytseva Y.Y., Mitov M.I., Napier D.L., Weiss H.L., Mark Evers B., Gao T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017;8:e2593. doi: 10.1038/cddis.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski J.A., Yin J., Teuscher K.B., Zhang M., Ji H. Structure-based design of 1,4-dibenzoylpiperazines as beta-catenin/B-cell lymphoma 9 protein-protein interaction inhibitors. ACS Med. Chem. Lett. 2016;7:508–513. doi: 10.1021/acsmedchemlett.5b00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Zhang W., Evans P.M., Chen X., He X., Liu C. Adenomatous polyposis coli (APC) differentially regulates beta-catenin phosphorylation and ubiquitination in colon cancer cells. J. Biol. Chem. 2006;281:17751–17757. doi: 10.1074/jbc.M600831200. [DOI] [PubMed] [Google Scholar]
- Yoo J., Jeon Y.H., Cho H.Y., Lee S.W., Kim G.W., Lee D.H., Kwon S.H. Advances in histone demethylase KDM3A as a cancer therapeutic target. Cancers (Basel) 2020;12:1098. doi: 10.3390/cancers12051098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Sviripa V., Chen X., Shi J., Yu T., Hamza A., Ward N.D., Kril L.M., Vander Kooi C.W., Zhan C.G. Fluorinated N,N-dialkylaminostilbenes repress colon cancer by targeting methionine S-adenosyltransferase 2A. ACS Chem. Biol. 2013;8:796–803. doi: 10.1021/cb3005353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Sviripa V.M., Kril L.M., Yu T., Xie Y., Hubbard W.B., Sullivan P.G., Chen X., Zhan C.G., Yang-Hartwich Y. An underlying mechanism of dual Wnt inhibition and AMPK activation: mitochondrial uncouplers masquerading as Wnt inhibitors. J. Med. Chem. 2019;62:11348–11358. doi: 10.1021/acs.jmedchem.9b01685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z., Virshup D.M. Wnt signaling and drug resistance in cancer. Mol. Pharmacol. 2020;97:72–89. doi: 10.1124/mol.119.117978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate computer code. All data and analytical methods are available in the main text or in Supplemental Information.