

Summary
Tepotinib is an oral MET inhibitor approved for metastatic non-small cell lung cancer (NSCLC) harboring MET exon 14 (METex14) skipping mutations. Examining treatment-naive or tepotinib-resistant cells with MET amplification or METex14 skipping mutations identifies other receptor tyrosine kinases (RTKs) that co-exist in cells prior to tepotinib exposure and become more prominent upon tepotinib resistance. In a small cohort of patients with lung cancer with MET genetic alterations treated with tepotinib, gene copy number gains of other RTKs were found at baseline and affected treatment outcome. An Src homology 2 domain-containing phosphatase 2 (SHP2) inhibitor delayed the emergence of tepotinib resistance and synergized with tepotinib in treatment-naive and tepotinib-resistant cells as well as in xenograft models. Alternative signaling pathways potentially diminish the effect of tepotinib monotherapy, and the combination of tepotinib with an SHP2 inhibitor enables the control of tumor growth in cells with MET genetic alterations.
Subject Areas: Treatment, Cancer
Graphical Abstract
Highlights
-
•
Tepotinib is a selective MET inhibitor with efficacy for METex14-positive NSCLC
-
•
Receptor tyrosine kinase signaling is activated in response to targeted therapies
-
•
SHP2 is a downstream effector of receptor tyrosine kinase signaling
-
•
SHP2 inhibition may enhance the antitumor activity of tepotinib
Treatment; Cancer
Introduction
The treatment paradigm of non-small cell lung cancer (NSCLC) has dramatically changed owing to an improved understanding of cancer biology followed by the introduction of agents targeting specific molecular pathways (Rosell et al., 2013). After the discovery of epidermal growth factor receptor (EGFR) mutations in lung adenocarcinomas (Lynch et al., 2004; Sordella et al., 2004), which correlate with response to EGFR tyrosine kinase inhibitors (TKIs) (Karachaliou et al., 2018b), several other genes related to the pathogenesis and treatment of NSCLC, including anaplastic lymphoma kinase (ALK), rearrangement of the ROS proto-oncogene 1 (ROS1), v-raf murine sarcoma viral oncogene homolog B1 (BRAF), rearranged during transfection (RET), and neurotrophic tyrosine receptor kinase (NTRK), have been discovered (Herbst et al., 2018; Rosell et al., 2020).
Mesenchymal-epithelial transition (MET or hepatocyte growth factor receptor, HGFR) is a tyrosine kinase receptor normally activated following binding to its ligand, HGF (Rong et al., 1994; Shen et al., 2000). Grb2, Shc, Src, and the p85 regulatory subunit of phosphatidylinositol 3-kinase (PI3K) are transducers that directly or indirectly, through the scaffolding protein Gab1, interact with the docking sites of MET (Pelicci et al., 1995; Weidner et al., 1996). This interaction leads to activation of the PI3K/AKT, MAPK/ERK, and STAT3 oncogenic signaling pathways (Lai et al., 2014). MET signaling is deregulated in diverse tumors types, including lung cancer, via MET overexpression, genomic amplification, autocrine/paracrine ligand stimulation, translocations, point mutations, and alternative splicing (Gherardi et al., 2012). MET amplification and exon 14 (METex14) skipping mutations are primary oncogenic drivers (Reungwetwattana and Ou, 2015) reported in 2%–4% and 3%–4% of NSCLC, respectively (Awad et al., 2016; Tong et al., 2016). MET amplification may also drive acquired resistance to EGFR TKIs in 5%–20% of EGFR-mutant lung adenocarcinomas (Pilotto et al., 2017; Tong et al., 2016; Turke et al., 2010).
Several compounds targeting MET signaling have been described, including TKIs and monoclonal antibodies (mAbs) against MET or HGF mAbs. Tepotinib and capmatinib are highly selective ATP-competitive MET TKIs, which have demonstrated preliminary efficacy in ongoing phase II trials in patients with NSCLC harboring METex14 skipping mutations (Paik et al., 2020; Wolf et al., 2020). Tepotinib (TEPMETKO) is approved in Japan for the treatment of unresectable, advanced or recurrent NSCLC with METex14 skipping alterations, whereas capmatinib (TABRECTA, Novartis) is approved by the Food and Drug Administration for patients with metastatic NSCLC with confirmed METex14 skipping mutations. However, as is the case with other targeted therapies, it is expected that MET-altered tumors will inevitably develop resistance to MET TKIs. Acquired resistance to receptor tyrosine kinase (RTK) inhibitors commonly occurs either via the development of mutations in the target that hinder drug binding or via the activation of alternate RTKs, which maintain downstream signal transduction (Gusenbauer et al., 2013; Karachaliou et al., 2018a; Rikova et al., 2007; Stommel et al., 2007). Primary or acquired resistance to MET inhibitors are still mechanistically unexplained, warranting further investigation (Fujino et al., 2019).
Src homology 2 domain-containing phosphatase 2 (SHP2), encoded by the PTPN11 gene, is a widely expressed cytoplasmic tyrosine phosphatase. SHP2 controls MAPK, PI3K/AKT, and JAK/STAT signaling downstream of diverse RTKs, including MET, to promote cell proliferation, differentiation, and survival (Karachaliou et al., 2019). The attractiveness of SHP2 as a target for therapeutic intervention in cancer has led to the development of several allosteric SHP2 inhibitors, which are currently undergoing preclinical or early clinical evaluation (Shen et al., 2020). Furthermore, combination with inhibitors of SHP2 was shown to overcome resistance of preclinical cancer models to targeted therapies such as MEK (Fedele et al., 2018).
We used comprehensive genomic, transcriptional, and proteomics profiling to demonstrate that the activation of alternate RTKs represents a mechanism of acquired tepotinib resistance in multiple independent MET-altered human cancer cell lines. Of importance, we report that the baseline presence of copy number gains in alternate RTKs affected the outcome of tepotinib treatment in patients with NSCLC positive for MET amplification or METex14 skipping mutations. Pharmacological inhibition of SHP2 synergized with tepotinib in MET-altered cells in vitro and in vivo and delayed the emergence of tepotinib resistance. These findings provide the first comprehensive insights into the mechanisms of resistance to MET TKIs and suggest that combination of these agents with inhibitors of SHP2 may represent an attractive strategy to improve the outcome of patients.
Results
Generation of EBC-1 and Hs746T Cells with Acquired Resistance to Tepotinib
To understand the molecular mechanisms of tepotinib resistance, we exposed the EBC-1 (NSCLC, MET amplified) and Hs746T (gastric cancer, METex14 mutated/MET amplified) human cell lines to tepotinib to generate resistant cell lines. After 12 months of continuous exposure of EBC-1 cells to increasing concentrations of tepotinib (up to 5 μmol/L), two resistant cell lines, TR1 and TR2, were generated. A single Hs746T tepotinib-resistant cell line TR3 was generated following 12 months of exposure of Hs746T cells to increasing tepotinib concentrations up to 6 μmol/L. Next-generation sequencing (NGS) of all three resistant cell lines did not reveal any acquired resistant mutation in the MET gene compared with the parental ones (data not shown).
Prior to further molecular profiling, TR cell lines were cultured in the absence of tepotinib in order to prevent treatment-induced bias. The maintenance of drug resistance during cell line passaging was monitored using viability assays. Calculation of the individual 50% inhibitory concentration (IC50) values revealed that TR1 and TR2 cells (low passage) were less sensitive to tepotinib than the parental EBC-1 cell line upon an exposure time of 3 and 6 days (Figures 1A and S1A, and Table S1). Similar results were obtained for TR3 cells (low passage), which were less sensitive to tepotinib than the parental Hs746T cell line upon an exposure time of 3 and 6 days (Figures 1B and S1B, and Table S1). Higher-passage TR1 and TR2 cells regained sensitivity to tepotinib, which was in an IC50 range comparable with that of parental EBC-1 cells (Figures 1C and S1C, and Table S1), whereas TR3 cells maintained tepotinib resistance during passaging (Figures 1D and S1D, and Table S1). Based on these findings, subsequent experiments were performed using low passages of TR1, TR2, and TR3 cells.
Figure 1.

Generation of EBC-1 and Hs746T Cells with Acquired Resistance to Tepotinib
(A–D) Dose-response curves for tepotinib in parental tepotinib-sensitive (EBC-1, Hs746T) and tepotinib-resistant (TR1, TR2, TR3) cell lines with (A and B) low passage number (#5) and (C and D) high passage number (#12) upon exposure for 6 days. Data are expressed as the mean ± SEM percentage fluorescent values relative to the corresponding DMSO control. Each experiment was performed in technical replicates (n = 6). TR1, tepotinib-resistant EBC-1 cell line #1; TR2, tepotinib-resistant EBC-1 cell line #2; TR3, tepotinib-resistant Hs746T #1; SEM, standard error of the mean.
Differential Gene Expression Profiling of Tepotinib-Resistant Cell Lines
We next conducted targeted gene expression profiling (GEP) of parental and TR EBC-1 and Hs746T cell lines using the 770 gene Nanostring Pan-Cancer panel (Nanostring Technologies, Seattle, WA), which covers genes pertaining to 13 cancer-associated signaling pathways. Independent RNA samples from each TR (n = 3) cell line, parental EBC-1 cells (n = 3), and parental Hs746T cells (n = 6) were used for the analyses. The differentially expressed genes (DEGs) in TR1, TR2, and TR3 that exhibited a log2-fold change >1.0 or < −1.0 and P < 0.05 compared with the parental cell lines are shown in Figures 2A–2E and S2. In total, 32 genes were upregulated, and 34 genes downregulated in TR1 and TR2 compared with EBC-1 cells. Relative to Hs746T cells, 48 genes were upregulated and 44 downregulated in TR3 cells. Three independent analysis methods (limma, nSolver, DESeq2) were used to confirm the differential expression results (Figures S2C–S2D).
Figure 2.

Differential Gene Expression Profiling of Tepotinib-Resistant Cell Lines
Digital gene expression quantification was performed using NanoString nCounter applying NanoString nCounter PanCancer Pathways Panel Assay (NanoString Technologies). The indicated relative expression data represent normalized and unsupervised clustering of parental and tepotinib-resistant cell lines. Horizontal rows represent individual genes and vertical columns represent individual cell lines. The color scale at the bottom of each heatmap depicts the corresponding gene expression levels, red indicating elevated expression, green indicating reduced expression. The DEGs in TR1, TR2, and TR3 that exhibited a log2-fold change >1.0 or <-1.0 and p< 0.05 compared to the parental cell lines are shown. Heatmap clustering of selected genes representative for (A) oncogenic signaling pathways or (B) RTKs comparing expression in EBC-1 cells (blue bars) with TR1 (purple bars) and TR2 (red bars). Heatmap clustering of selected genes representative for (C and E) oncogenic signaling pathways or (D) RTKs comparing expression in Hs746T cells (red bars) with TR3 (purple bars). RTKs, receptor-tyrosine kinases; TR1, tepotinib-resistant EBC-1 cell line #1; TR2, tepotinib-resistant EBC-1 cell line #2; TR3, tepotinib-resistant Hs746T #1; DEGs, differentially expressed genes.
Relative to EBC-1 cells, TR1 and TR2 exhibited increased expression of MAP3K5, NFKB1, NFKBIA, AKT3, NF1, TP53, PIK3CA, PTEN, NOTCH1, TGFB3, EGFR, TNFAIP3, IGF1R, FGFR2, LIFR, TGFB2, TGFR2, IL6R, PDGFA, and TNFRSF10C (Figures 2A and 2B). FGFR4 and NUMBL were only upregulated in TR1, whereas STAT1, MAP3K1, JUN, MYC, CREB3L4, MAPK1, MAPK8, RB1, MAP3K8, DUSP10, and CD40 were only upregulated in TR2 (Figures 2A and 2B). TR1 and TR2 exhibited decreased expression of MAPK9, EIF4EBP1, CTNNB1, JAK1, PTPN11, DUSP4, MAP2K4, TNFRSF10B TNFRSF10A, VEGFC, and MET (Figures 2A and 2B). FGF11 and TGFB1 were downregulated only in TR1, whereas HES1, CREB5, MAP3K13, JAK2, MAP2K2, KRAS, FAS, LIF, TNFRSF10D, TNFSF10, and VEGFA were only downregulated in TR2 (Figures 2A and 2B).
Relative to Hs746T, TR3 cells exhibited increased expression of HES1, KRAS, TP53, STAT4, CREB3L1, NFKBIA, STAT1, JAK2, DUSP10, AKT3, MAPK3, MAP2K4, MAP3K14, MAPK8IP2, RAF1, MAP2K1, BRAF, EGF, FGFR1, VEGFA, TGFB2, FGF11, TNFSF10, LIFR, FGF9, PDGFD, AR, and VEGFC (Figures 2C–2E). TR3 exhibited decreased expression of PIK3R3, NF1, PIK3CA, CREB5, STAT3, CTNNB1, PIK3R1, JUN, MAP3K5, MAPK8, DUSP6, DUSP5, MAPK1, MAP3K12, GRB2, LIF, EGFR, MET, TNFAIP3, TGFBR2, NOTCH1, FGF13, and IL6R (Figures 2C–2E).
Together, these data indicate that TR cells derived from the MET-amplified EBC-1 cell line exhibit increased expression of RTKs including EGFR and FGFR2. In contrast, TR3 cells demonstrated differential expression of downstream signaling nodes including AKT3, KRAS, RAF1, and BRAF. These data suggest that tepotinib resistance may be acquired by divergent mechanisms.
Acquired Resistance to Tepotinib Is Associated with the Activation of Multiple Oncogenic Signaling Nodes
We next investigated the phosphorylation status of RTKs and downstream signaling nodes in tepotinib-resistant cells using phospho-protein arrays.
Tyrosine phosphorylated MET (HGFR) levels were comparable in the parental EBC-1, TR1, and TR2 cell lines (Figures 3A and 3C and Table S2A). Meanwhile, MET phosphorylation was decreased in TR3 cells compared with the parental Hs746T control (Figures 3B and 3D and Table S2B). Levels of phospho-EGFR were elevated in TR1 and TR2 compared with EBC-1, which contrasted to decreased phospho-EGFR levels in TR3 relative to Hs746T. Among other ErbB family members, phosphorylated ErbB3 was elevated in TR1 and TR2 compared with EBC-1, whereas phospho-ErbB4 was increased in TR3 relative to Hs746T. Expression of the TAM-family RTK Axl has been described to limit the antitumor efficacy of EGFR TKIs in EGFR-mutated tumors (Graham et al., 2014; Karachaliou et al., 2018a). We observed robust phosphorylation of Axl in EBC-1 and Hs746T cells, which was further elevated in TR2 cells. In contrast, TR3 cells demonstrated decreased levels of phospho-Axl as well as phospho-Mer, a second TAM-family member. Of interest, phosphorylated Ret was detected in EBC-1 cells and its TR derivatives, whereas phospho-PDGFRα was elevated in TR3 compared with Hs746T.
Figure 3.

Acquired Resistance to Tepotinib Is Associated with the Activation of Multiple Oncogenic Signaling Nodes
(A) Results of phospho-RTK array analysis in parental tepotinib-sensitive (EBC-1, Hs746T) and tepotinib-resistant (TR1, TR2, TR3) cell lines. The phosphorylation status of 49 RTKs was assessed in each cell line using the Proteome Profiler Human Phospho-RTK Array Kit. Reference spots in the corners (A1, A2, A23, A24, F1, F2) represent positive controls, and PBS spots (F23, F24) represent negative controls for determination of background values.
(B) Phospho-kinase array analyses in parental tepotinib-sensitive (EBC-1, Hs746T) and tepotinib-resistant (TR1, TR2, TR3) cell lines. The phosphorylation status of 43 kinases and 2 related proteins was assessed in each cell line using the Proteome Profiler Human Phospho-Kinase Array Kit. Reference spots in the corners (A1, A2, A17, A18, G1, G2) represent positive controls, and PBS spots (G9, G10, G17, G18) represent negative controls.
(C) Heatmap of phospho-RTK array data indicating the absolute integrated pixel density values.
(D) Heatmap of phospho-kinase array data indicating the absolute integrated pixel density values. TR1, tepotinib-resistant EBC-1 cell line #1; TR2, tepotinib-resistant EBC-1 cell line #2; TR3, tepotinib-resistant Hs746T #1.
All three TR cells exhibited increased phosphorylation of AKT1/2/3 (S473 and T308) and PRAS40 (T246) compared with the parental cell lines (Figures 3C and 3D and Table S2A and S2B). Interestingly, phosphorylation of Src (Y419), Yes (Y426), STAT3 (Y705 and S727), FAK (Y397), and the MAPK-pathway proteins c-Jun (S63) and CREB (S133) were increased in TR1 and TR2 cells compared with EBC-1 but not in the TR3 cells compared with Hs746T.
Together with the expression data described above, our findings suggest that hyperactivation of the RTKs, particularly those belonging to the ErbB family, as well as downstream signaling components, is altered in tepotinib-resistant cells.
Molecular and Clinical Profile of Patients with NSCLC with METex14 Skipping Mutations or MET Amplification Treated with Tepotinib
Between March 2017 and April 2020, six patients with METex14 skipping mutations and four with MET amplification were treated with tepotinib in the Dr. Rosell Oncology Institute, Dexeus University Hospital, QuironSalud Group, Barcelona, Spain. The baseline clinical and molecular characteristics of the patients are summarized in Figure 4A. All patients had lung adenocarcinoma, except case #8 who had a large cell carcinoma, and all patients had stage IV disease, except case #6 who had stage IIIB before tepotinib initiation. Seven patients were treated with tepotinib within the phase II VISION study (ClinicalTrials.gov number, NCT02864992) (Paik et al., 2020), whereas cases #3, #9, and #10 received tepotinib under a compassionate use program. Case #3 had brain metastases and case #9 had leptomeningeal carcinomatosis prior to therapy initiation. Most patients (6 of 10) received tepotinib as first-line therapy. Two patients received tepotinib as second-line therapy after platinum-based chemotherapy, and two patients received tepotinib as third-line therapy after platinum-based chemotherapy and nivolumab (Figure 4A). In case #1, the METex14 skipping mutation was detected in tissue RNA using the Oncomine Focus Assay (OFA; 52-gene) in the central laboratory of the VISION study, whereas in case #9 and case #10, METex14 skipping mutations were detected using the Nanostring nCounter platform (Teixido et al., 2018) in the Pangaea Oncology, Laboratory of Oncology, Quirón Dexeus University Hospital, Barcelona, Spain. For all other cases, molecular analysis was performed using the NGS panel Guardant360 (73-gene) on circulating tumor DNA. Tumor burden changes in reference to baseline (%) at the time point of best overall response in nine patients ranged from −70.97 to +3.73 (median −38.30) (Figure 4B). For the four patients who achieved stable disease, the median change in tumor burden in reference to baseline (%) at the time point of best overall response was −10.89 (range −20.00 to +3.73) (Figure 4B).
Figure 4.

Molecular and Clinical Profile of Patients with NSCLC with METex14 Skipping Mutations or MET Amplification Treated with Tepotinib
(A) Oncoprint describing the baseline clinical characteristics of each patient.
(B) Waterfall plot of tumor burden change at best overall response in nine patients. Dashed lines represent the thresholds for partial response (−30%) and progressive disease (+20%).
(C) Oncoprint representing the alterations detected in each patient. Individual samples are represented as columns and individual genes are represented as rows. Identified mechanisms of resistance are annotated with x.
(D) The radiological tumor evolution in a patient with METex14 skipping mutation without co-occurring genetic alterations. Left: computed tomography (CT) scans at baseline indicate a lung lesion and pleural infiltration. Right: CT scans indicate an almost complete response after two cycles of tepotinib.
M, male; F, female; d, days; L, line; PR, partial response; SD, stable disease; NE, non-evaluable; amp, amplification; VAF, variant allelic frequency; GCN, gene copy number.
Amplification or mutation of the ErbB family members EGFR and HER2, amplification of PDGFRα, and FGFR1 and activating KRAS mutations were detected in the four MET amplified patients (case #3, case #4, case #5, and case #6, respectively). BRAF and CDK6 amplification was detected in three of the four MET amplified patients. This may potentially be the result of a common genetic event, given the proximity of CDK6, MET, and BRAF on chromosome 7q (7q21.2, 7q31.2, and 7q34, respectively) (Le et al., 2018). At baseline, case #5 harbored MET amplification and an activating KRAS mutation, as well as amplification of EGFR, PDGFRα, and FGFR1 (Figure 4C). This patient received tepotinib for only one cycle and succumbed to his disease in less than 1 month after treatment initiation. Among the other MET-amplified patients, case #6 exhibited co-occurring PDGFRα amplification, TP53, ARID1A, NOTCH1, and MET mutations, and received tepotinib therapy for more than 7 months. Case #4 had co-occurring EGFR and PDGFRα amplification, as well as TP53 and ARID1A mutations. The patient had stable disease with tepotinib and remained on therapy for 5 months (Figure 4C). Finally, case #3 was a MET-amplified patient without any co-occurring gene copy number gains of other RTKs but harbored several co-occurring mutations, including NRAS mutation and loss of NF1 and TP53. Case #3 achieved a partial response to tepotinib but remained on therapy for only 4 months (Figure 4C). Interestingly, case #8 harbored a METex14 skipping mutation in the absence of concomitant genetic alterations and this patient almost achieved a complete response after 2 months of tepotinib therapy (Figure 4D).
Among the six patients with METex14 skipping mutations, case #9 exhibited a concomitant EGFR amplification at baseline and has achieved stable disease after five cycles of tepotinib therapy. In keeping with the observations in TR cell lines described above, patients with METex14 skipping mutations had less copy number gains of other genes, compared with the patients with MET amplification (Figure 4C). In addition, the duration of tepotinib therapy was shorter in the MET-amplified patients compared with patients with METex14 skipping mutations (Figures 4A–4C).
The above clinical observations indicate that patients with lung cancer with MET alterations, particularly MET amplification, may harbor other co-occurring genetic alterations, especially gene copy number gains of other RTKs, that may maintain downstream signaling independent of MET inhibition.
MET and SHP2 Blockade Inhibit the Proliferation and Growth of Cells in Models Exhibiting MET Genetic Alterations
Resistance to targeted RTK inhibitors has previously been described to occur via the compensatory re-establishment of downstream signaling cascades by alternate RTKs (Karachaliou et al., 2019; Shen et al., 2020). SHP2 is a core component of a signaling multi-protein complex that plays an essential role in diverse RTK and cytokine receptor signaling pathways and is associated with resistance to different targeted therapies (Fedele et al., 2018; Graham et al., 2014; Salgia, 2017). Based on our findings and previous knowledge we examined the effects of the combination of SHP2 and MET inhibitors in MET-altered cell lines.
First, we assessed the effect of tepotinib in treatment-naive cell lines exhibiting either high-level MET amplification (EBC-1, MNK-45, NCI-H1993), low-level MET amplification (NCI-H441), or both METex14 skipping mutation and MET amplification (Hs746T) (Wu et al., 2013) (Table S3). Tepotinib efficiently decreased the viability of cells with high-level MET amplification but had no effect on the NCI-H441 cell line (Figures 5A–5D and S3A). In addition, we used the same cell lines to investigate the effect of two previously published allosteric SHP2 inhibitors, henceforth referred as SHP2i_01 (Nichols et al., 2018) and SHP2i_02 (International Patent Application Publication No.: WO2020/033828 A1, Example #10b) (Sun et al., 2020). The MEK1/2 inhibitor pimasertib (von Richter et al., 2016) was used as a control for MAPK pathway inhibition. As shown in Figures 5A–5D and S3A, each of the cell lines was less sensitive to the SHP2 inhibitors and pimasertib compared with tepotinib monotherapy.
Figure 5.

MET and SHP2 Blockade Inhibit the Proliferation and Growth of Cells in Models Exhibiting MET Genetic Alterations
(A–D) Dose-response curves for cells treated with tepotinib, SHP2i_01, SHP2i_02, or pimasertib for 6 days in 2D. Data are expressed as the mean ± SEM percentage fluorescent values relative to the corresponding DMSO control.
(E–H) Cells grown in 2D were treated with a 6 x 6 combination matrix of tepotinib and SHP2i_01, SHP2i_02, or pimasertib for 6 days. The combination index (CI < 1) was determined using the Loewe combination method.
(I–L) Dose-response curves for tepotinib, SHP2i_01, SHP2i_02, and pimasertib in MET-altered cell lines.
(M) Cells grown in 3D were treated with a 6 x 6 combination matrix of tepotinib and SHP2i_02 for 6 days. The combination index (CI < 1) was analyzed using the Loewe combination method.
(N) Antitumor activity of tepotinib and SHP2i_02 as monotherapy and combination therapy in EBC-1 xenografts. 10 mice were included in each treatment group. Each compound was applied once daily via oral gavage. Data are expressed as the mean ± SEM. d, days; SEM, standard error of the mean.
We next explored the effects of combination of tepotinib with SHP2i_01, SHP2i_02, or pimasertib in viability assays using the EBC-1, Hs746T, MKN-45, and NCI-H1993 cell lines cultured in 2D. Data analysis using Loewe combination method (Loewe, 1953) revealed synergism based on three distinct parameters (combination index <1, synergy score >2, negative excess volume) between tepotinib and the SHP2 inhibitors in each cell line (Figures 5E–5H, S3B, and S3C, and Table S4). Synergy was also observed between tepotinib and pimasertib.
We next assessed the effect of tepotinib combined with SHP2i_02 in cells grown as 3D spheroids. Tepotinib and SHP2i_02 demonstrated comparable IC50 values in EBC-1, Hs746T, and NCI-H1993 cells grown in 2D and 3D. In contrast, MKN-45 cells were less sensitive to tepotinib and SHP2i_02 in 3D (Figures 5I–5L and Table S3). In keeping with our observations using 2D cultures, tepotinib and SHP2i_02 synergistically reduced the proliferation of 3D spheroids of the Hs746T, MKN-45, and NCI-H1993 cell lines (Figures 5M, S3B, and S3C, and Table S4). In contrast, synergy was not observed with EBC-1 spheroids, which may potentially be explained by exceptional potency of tepotinib monotherapy in this setting.
Finally, we explored the in vivo effects of the combination of tepotinib with SHP2 inhibitors. Mice bearing EBC-1 xenografts were treated orally with a vehicle control, SHP2i_02, or tepotinib in monotherapy or the combination of SHP2i_02 with tepotinib (Figure 5N). In line with the findings in the 3D in vitro assay for EBC-1, tepotinib monotherapy (125 mg/kg, once daily) demonstrated high efficacy, exemplified by complete tumor regression. At a lower dose of 5 mg/kg once daily, tepotinib delayed the growth of EBC-1 tumors. A comparable degree of tumor growth delay was observed with SHP2i_02 monotherapy at 50 mg/kg, once daily. The combination of SHP2i_02 (50 mg/kg, once daily) with tepotinib (5 mg/kg, once-daily) induced regression of EBC-1 tumors.
Taken together, these in vitro and in vivo findings suggest that SHP2 inhibition potentiates the efficacy of tepotinib in treatment-naive cancer cells harboring MET alterations.
SHP2 Inhibition Delays the Emergence of Resistance to Tepotinib in MET-Amplified Cells
We next examined whether SHP2 inhibition could overcome the emergence of tepotinib resistance in the EBC-1 cell line. Considering the exquisite sensitivity of EBC-1 cells to tepotinib shown above, as well as the persistent reduction of MET phosphorylation even following tepotinib withdrawal (Figure 6A), in vitro cycles of tepotinib treatment and withdrawal were established that resulted in the eradication of most EBC-1 cells, while still enabling the emergence of drug-resistant cells over the course of several weeks of culture (treatment cycles depicted in Figure 6B). Tepotinib-resistant cells appeared after 3 weeks of culture on treatment cycle “a.” The addition of SHP2i_2 (200 nmol/L) to this treatment cycle delayed the emergence of resistant cells by 2 weeks, whereas higher SHP2i_2 doses (400 and 800 nmol/L) completely prevented the emergence of resistant cells (Figure 6C). When using treatment cycle “b,” tepotinib-resistant cells were first observed after 4 weeks of culture. The addition of SHP2i_2 (200, 400, 800 nmol/L) to this cycle prevented the emergence of resistant cells (Figure 6D). EBC-1 cells grew under treatment with SHP2i_02 monotherapy (Figure 6E).
Figure 6.

SHP2 Inhibition Delays the Emergence of Resistance to Tepotinib in MET-Amplified Cells
(A) Western blot analysis of the effect of tepotinib treatment at the indicated concentrations for 24 h followed by culture in the absence of tepotinib for 1, 3 or 6 days. Beta actin served as a loading control.
(B) Schematic depiction of the experimental treatment cycle ‘a’ and ‘b’.
(C) Real-time proliferation of EBC-1 cells during exposure cycles to tepotinib and SPH2i_02 for 24 h followed by SPH2i_02 for 6 days, compared to tepotinib alone.
(D) Real-time proliferation of EBC-1 cells during exposure cycles to tepotinib alone for 24 h followed by SPH2i_02 for 6 days, compared to tepotinib alone.
(E) Real-time proliferation upon exposure of SPH2i_02 alone. For (C–E) real-time proliferation was determined based on weekly measurements of cell surface confluency using IncuCyte S3 system. d, days; h, hours.
SHP2 Inhibition Reverts Established Resistance to Tepotinib in MET-Altered Cells
The NanoString and phospho-protein array analyses described above implicated the overexpression and activation of RTKs and downstream signaling pathways in acquired tepotinib resistance. As SHP2 is a critical regulator of RTK signaling via the MAPK, PI3K/AKT, and STAT3 pathways (Grossmann et al., 2010), we next examined the effects of SHP2 inhibitors in combination with tepotinib in established TR cell lines.
First, we assessed the effect of tepotinib, SHP2i_01, SHP2i_02, as well as pimasertib on cell viability of the TR1, TR2, and TR3 cell lines compared with their parental counterparts. Consistent with data shown above (Figure 1A), TR1 and TR2 cells were resistant to tepotinib (Figure S4A and Table S5). EBC-1, TR1, and TR2 cells showed equivalent sensitivity to SHP2i_01 and SHP2i_02 (Figures S4B and S4C). In agreement with results shown above (Figure 1B), tepotinib decreased the viability of parental Hs746T cells but not of TR3 cells (Figure S4E). In contrast to the findings in the EBC-1 cells and its TR derivatives, TR3 cells demonstrated reduced sensitivity to SHP2i_01 and SHP2i_02 compared with parental Hs746T cells (Figures S4F and S4G). Pimasertib sensitivity was found to be reduced in each of the TR cell lines compared with the parental cells (Figures S4D and S4H).
We next evaluated the effect of tepotinib and SHP2 inhibitors in monotherapy and combination on the expression and phosphorylation of MET and ERK in the EBC-1-derived TR2 and Hs746T-derived TR3 cell lines. Exposure to 10 nmol/L tepotinib monotherapy fully inhibited MET phosphorylation at all tested time points in both TR2 and TR3 cells (Figure 7A). This suggests that tepotinib resistance in these cells was not due to suboptimal MET inhibition. Comparable effects of MET phosphorylation were observed when tepotinib was combined with 1 μmol/L of SHP2i_01 or SHP2i_02. Tepotinib and the SHP2 inhibitors reduced ERK phosphorylation when used as monotherapies in TR2 cells. In the same cell line, the combination of tepotinib with either SHP2i_01 or SHP2i_02 fully abrogated ERK phosphorylation at all tested time points. Similarly, the combination of tepotinib with SHP2 inhibitors in the TR3 cell line led to a more pronounced suppression of phospho-ERK compared with each monotherapy. In this cell line, recovery of ERK phosphorylation was observed at later time points, even under combined tepotinib and SHP2 inhibitor treatment.
Figure 7.

SHP2 Inhibition Reverts Established Resistance to Tepotinib in MET-Altered Cells
(A) Western blot analysis of the effect of tepotinib with or without SHP2i_01 or SHP2i_02 at the indicated concentrations and treatment times on phospho-MET and phospho-ERK1/2 levels in the tepotinib-resistant cell lines TR2 and TR3. GAPDH expression was monitored to control for equivalent loading of protein in each gel lane.
(B and C) Clonogenic assays in tepotinib-resistant cell line TR1 to determine the effect of the combination of tepotinib with SHP2i_01. A total of 47 images were analyzed per well, and data are shown as the mean ± SEM.
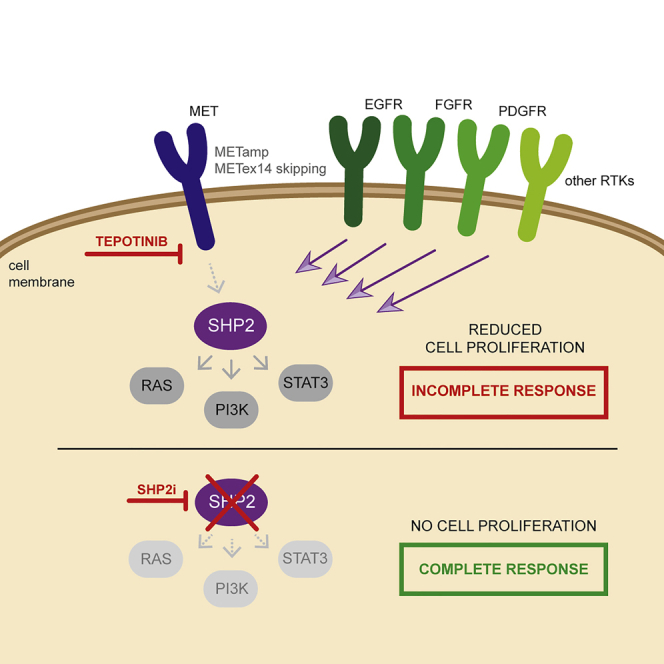
(D) Schematic of the effects of tepotinib as monotherapy or in combination with an SHP2 inhibitor. Inhibition of MET via tepotinib leads to suppression of the oncogenic MAPK, PI3K/AKT, and STAT3 signaling pathways. Resistance to tepotinib is associated with the upregulation of alternate RTKs such as EGFR and AXL, which activate SHP2 to reactivate MAPK, PI3K/AKT, and STAT3 signaling. Combined inhibition of MET and SHP2 prevents the emergence of resistance to tepotinib by inhibiting reactivation of MAPK, PI3K/AKT, and STAT3, thereby sustaining antitumor efficacy. TR1, tepotinib-resistant EBC-1 cell line #1; TR2, tepotinib-resistant EBC-1 cell line #2; TR3, tepotinib-resistant Hs746T #1; h, hours; RTK, receptor tyrosine kinase; SEM, standard error of the mean.
Lastly, we performed a modified clonogenic outgrowth assay using confluency as a readout to investigate whether SHP2 inhibition could revert tepotinib resistance. TR1 was exposed to tepotinib (10 nmol/L) with or without SHP2i_01 (1 μmol/L) for 3 weeks. Consistent with earlier findings, SHP2i_01 monotherapy reduced growth of TR1 colonies, whereas tepotinib had no effect on them (Figures 7B and 7C). The combination of these compounds significantly reduced colony growth compared with SHP2i_01 monotherapy.
Overall, these findings suggest that the combination of tepotinib with an SHP2 inhibitor represents a promising strategy to overcome resistance in tumors harboring MET amplification and, potentially, METex14 skipping mutations as depicted in Figure 7D.
Discussion
After the initial discovery of the MET signaling pathway through the generation of an oncogenic fusion translocated promoter region (TPR)-MET in a human osteosarcoma cell line, in the 1980s (Soman et al., 1991), MET has been established as a potent oncogene that contributes to different stages of carcinogenesis (Koch et al., 2020). In NSCLC, MET deregulation has been documented to occur via protein overexpression, gene amplification, or gene mutations, with the METex14 skipping mutation being the most common mutation (Kong-Beltran et al., 2006; Salgia, 2017). Thus, MET is an attractive target in NSCLC and MET targeted therapies have demonstrated efficacy in lung tumors with MET alterations (Koch et al., 2020). Tepotinib is an oral, potent, highly selective MET inhibitor (Johne et al., 2020) that has been approved in Japan for the treatment of patients with unresectable, advanced, or recurrent NSCLC with METex14 skipping alterations (Paik et al., 2020), whereas it is under clinical investigation in combination with EGFR TKIs for patients with EGFR-mutant lung adenocarcinomas and MET amplification as an acquired resistance mechanism (NCT03940703). The present work provides molecular evidence that resistance to tepotinib is mediated by bypass signaling via other RTKs and the activation of downstream signaling nodes. The addition of an SHP2 inhibitor to tepotinib delayed the emergence of tepotinib resistance in a treatment-naive lung cancer cell line with MET amplification, whereas the combination of tepotinib with an SHP2 inhibitor abrogated the growth of cells with acquired resistance to tepotinib.
Mechanisms of resistance to MET TKIs are heterogeneous and may also differ based on the class of MET TKI (type I or II) (Backes et al., 2008; Gherardi et al., 2012; Recondo et al., 2020). For instance, secondary MET kinase domain mutations such as D1228V, or high-level MET amplification, have been described as mechanisms of resistance to MET TKIs (Collie et al., 2019; Recondo et al., 2020), and sequential treatment with a structurally different MET TKI was found to be effective in overcoming resistance in these settings (Recondo et al., 2020). Guo et al. explored both primary and acquired resistance to MET inhibitors such as crizotinib in patients with NSCLC with METex14 skipping mutations (Guo et al., 2019). Crizotinib response was improved in patients displaying baseline tumor MET expression compared with patients in which MET expression was not detected. In the same study, acquired resistance via secondary MET kinase domain mutations was uncommon, whereas bypass pathways of acquired resistance such as EGFR amplification or inactivation of the RAS negative regulator RASA1 were more frequently detected (Guo et al., 2019). PI3K pathway alterations, like PIK3CA mutations or loss of PTEN, have also been reported to confer primary resistance to MET TKIs in NSCLC with METex14 skipping mutations (Jamme et al., 2020). Recondo et al. reported that both MET kinase domain secondary mutations or amplification of the METex14 mutant allele and KRAS mutations, KRAS, EGFR, HER3, or BRAF amplifications were associated with resistance to MET inhibitors (Recondo et al., 2020).
In our study, TR cells derived from the MET-amplified EBC-1 cell line (TR1 and TR2) demonstrated significantly upregulated expression of EGFR but downregulation of MET. The phosphorylation of EGFR, as well as Axl, was also increased in the same TR cell lines. In contrast, the TR3 cell line, derived from the METex14 skipping mutation positive and MET-amplified Hs746T cell line, exhibited downregulation of EGFR and MET and upregulation of KRAS. Meanwhile, tyrosine phosphorylation of EGFR, MET, and Axl was decreased in TR3 cells. Compensatory pathways related to alteration of critical proteins including mTOR, FGFR1, EGFR, STAT3, and COX-2 have been previously described in MET inhibitor-resistant glioblastoma tumors (Cruickshanks et al., 2019). In our study, all three TR cell lines exhibited activation of nodes of the PI3K/AKT cascade, whereas MAPK activation was only observed in the EBC-1-derived TR cell lines. We report co-occurring genomic alterations in 10 patients with advanced lung cancer with MET genetic alterations, highlighting the most common events and identifying that co-expression of RTKs other than MET is associated with shorter duration of tepotinib therapy.
In several cell lines with MET alterations, we found the combination of tepotinib with an SHP2 inhibitor or a MEK inhibitor (pimasertib) to more efficiently abrogate cell growth compared with each monotherapy. It was previously shown that SHP2 activity drives adaptive resistance to MEK inhibitors in different tumor types (Fedele et al., 2018), which could be prevented by the combination of inhibitors of SHP2 and the MAPK-pathway nodes RAF or MEK (Ahmed et al., 2019). Most importantly, we found that the addition of an SHP2 inhibitor to tepotinib delayed the emergence of resistance in the EBC-1 MET-amplified cell line. These findings suggest that the upfront addition of an SHP2 inhibitor may prolong the duration of response of patients with MET-amplified NSCLC to tepotinib. In keeping with the critical role of SHP2 in signaling downstream of diverse RTKs (Ahmed et al., 2019), TR cell lines demonstrating upregulation and/or hyperactivation of RTKs were found to be sensitive to the combination of an SHP2 inhibitor with tepotinib. SHP2 inhibitors are currently in clinical testing as monotherapy or in combination with other targeted therapies including MEK inhibitors. Preliminary clinical data of an SHP2 inhibitor monotherapy study show acceptable tolerability and initial signs of clinical activity (Ou et al., 2020).
Our study provides the first indication that the activation of RTKs other than MET, as well as downstream signaling components, is a key event in the development of acquired resistance to tepotinib, a MET TKI that is expected to become a standard treatment option for patients with NSCLC with METex14 skipping mutations. The addition of an SHP2 inhibitor is hypothesized to suppress the reactivation of oncogenic signaling by alternate RTKs or downstream signaling nodes during the development of tepotinib resistance. Overall, our study provides a roadmap for the clinical implementation of this combination strategy in order to improve the depth of tepotinib response and delay the emergence of tepotinib resistance in patients with MET-altered tumors.
Limitations of the Study
The study has some limitations. First, the small sample size of patients with NSCLC with MET alterations treated with tepotinib limits us from drawing strong conclusions. Second, in vitro and in vivo experiments with alternate dosing schedules for SHP2 inhibition are needed to help elucidate tolerable treatment schedules in patients. Besides toxicity, alternate dosing may avoid potential feedback reactivation of ERK upon continuous suppression of the MAPK pathway. Still, the data presented herein provide important biological insights that might help to orient future clinical trials with the combination of tepotinib plus SHP2 inhibitors for patients with NSCLC with METex14 skipping mutations or MET amplification.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Niki Karachaliou (niki.karachaliou@merckgroup.com).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate datasets or analyze codes.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Jacqueline Buttler and Catherine Eichhorn for technical guidance and support with long-term in vitro combination study as well as Stephanie Karl and Daniel Kuntze for technical assistance with the in vivo combination studies.
Author Contributions
Conceptualization: L.P. and N.K.; Methodology: L.P., F.J., C.R., N.L., D.M., and N.K.; Investigation: L.P., F.J., C.R., S.V., R.R., K.M.S., and N.K.; Formal analysis: L.P., F.J., C.R., S.V., N.L., R.R., K.M.S., and N.K.; Writing – Original Draft: L.P., R.R., and N.K.; Writing – Review & Editing: L.P., F.J., C.R., S.V., C.S., N.L., M.S., D.M., M.K., C.E., A.B., R.R., K.M.S., and N.K.; Visualization: L.P. and N.K.; Supervision: F.J., N.L., R.R., K.M.S., and N.K.
Declaration of Interests
L.P., F.J., C.R., C.S., N.L., M.S., D.M., M.K., C.E., A.B., K.M.S., and N.K. are employees of Merck KGaA. A.B. and M.S. hold stocks in Merck KGaA. No other potential conflict of interest relevant to this article was reported.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101832.
Supplemental Information
References
- Ahmed T.A., Adamopoulos C., Karoulia Z., Wu X., Sachidanandam R., Aaronson S.A., Poulikakos P.I. SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep. 2019;26:65–78.e5. doi: 10.1016/j.celrep.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad M.M., Oxnard G.R., Jackman D.M., Savukoski D.O., Hall D., Shivdasani P., Heng J.C., Dahlberg S.E., Janne P.A., Verma S. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-met overexpression. J. Clin. Oncol. 2016;34:721–730. doi: 10.1200/JCO.2015.63.4600. [DOI] [PubMed] [Google Scholar]
- Backes A., Zech B., Felber B., Klebl B., Muller G. Small-molecule inhibitors binding to protein kinase. Part II: the novel pharmacophore approach of type II and type III inhibition. Expert Opin. Drug Discov. 2008;3:1427–1449. doi: 10.1517/17460440802580106. [DOI] [PubMed] [Google Scholar]
- Collie G.W., Koh C.M., O'Neill D.J., Stubbs C.J., Khurana P., Eddershaw A., Snijder A., Mauritzson F., Barlind L., Dale I.L. Structural and molecular insight into resistance mechanisms of first generation cMET inhibitors. ACS Med. Chem. Lett. 2019;10:1322–1327. doi: 10.1021/acsmedchemlett.9b00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshanks N., Zhang Y., Hine S., Gibert M., Yuan F., Oxford M., Grello C., Pahuski M., Dube C., Guessous F. Discovery and therapeutic exploitation of mechanisms of resistance to MET inhibitors in glioblastoma. Clin. Cancer Res. 2019;25:663–673. doi: 10.1158/1078-0432.CCR-18-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele C., Ran H., Diskin B., Wei W., Jen J., Geer M.J., Araki K., Ozerdem U., Simeone D.M., Miller G. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 2018;8:1237–1249. doi: 10.1158/2159-8290.CD-18-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino T., Kobayashi Y., Suda K., Koga T., Nishino M., Ohara S., Chiba M., Shimoji M., Tomizawa K., Takemoto T. Sensitivity and resistance of MET exon 14 mutations in lung cancer to eight MET tyrosine kinase inhibitors in vitro. J. Thorac. Oncol. 2019;14:1753–1765. doi: 10.1016/j.jtho.2019.06.023. [DOI] [PubMed] [Google Scholar]
- Gherardi E., Birchmeier W., Birchmeier C., Vande Woude G. Targeting MET in cancer: rationale and progress. Nat. Rev. Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- Graham D.K., DeRyckere D., Davies K.D., Earp H.S. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer. 2014;14:769–785. doi: 10.1038/nrc3847. [DOI] [PubMed] [Google Scholar]
- Grossmann K.S., Rosario M., Birchmeier C., Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Adv. Cancer Res. 2010;106:53–89. doi: 10.1016/S0065-230X(10)06002-1. [DOI] [PubMed] [Google Scholar]
- Guo R., Offin M., Brannon A.R., Chow A., Delasos L., Somwar R., Wilkins O., Scott K., Tian Y., Cecchi F. MET inhibitor resistance in patients with MET exon 14-altered lung cancers. J. Clin. Oncol. 2019;37(15–suppl):9006. [Google Scholar]
- Gusenbauer S., Vlaicu P., Ullrich A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene. 2013;32:3846–3856. doi: 10.1038/onc.2012.396. [DOI] [PubMed] [Google Scholar]
- Herbst R.S., Morgensztern D., Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- Jamme P., Fernandes M., Copin M.C., Descarpentries C., Escande F., Morabito A., Gregoire V., Jamme M., Baldacci S., Tulasne D. Alterations in the PI3K pathway drive resistance to MET inhibitors in NSCLC harboring MET exon 14 skipping mutations. J. Thorac. Oncol. 2020;15:741–751. doi: 10.1016/j.jtho.2020.01.027. [DOI] [PubMed] [Google Scholar]
- Johne A., Scheible H., Becker A., van Lier J.J., Wolna P., Meyring M. Open-label, single-center, phase I trial to investigate the mass balance and absolute bioavailability of the highly selective oral MET inhibitor tepotinib in healthy volunteers. Invest. New Drugs. 2020;38:1507–1519. doi: 10.1007/s10637-020-00926-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachaliou N., Cardona A.F., Bracht J.W.P., Aldeguer E., Drozdowskyj A., Fernandez-Bruno M., Chaib I., Berenguer J., Santarpia M., Ito M. Integrin-linked kinase (ILK) and src homology 2 domain-containing phosphatase 2 (SHP2): novel targets in EGFR-mutation positive non-small cell lung cancer (NSCLC) EBioMedicine. 2019;39:207–214. doi: 10.1016/j.ebiom.2018.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachaliou N., Chaib I., Cardona A.F., Berenguer J., Bracht J.W.P., Yang J., Cai X., Wang Z., Hu C., Drozdowskyj A. Common Co-activation of AXL and CDCP1 in EGFR-mutation-positive non-smallcell lung cancer associated with poor prognosis. EBioMedicine. 2018;29:112–127. doi: 10.1016/j.ebiom.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachaliou N., Fernandez-Bruno M., Paulina Bracht J.W., Rosell R.J.T.C.R. EGFR first- and second-generation TKIs—there is still place for them in EGFR -mutant NSCLC patients. 2018;2018:S23–S47. doi: 10.21037/tcr.2018.10.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J.P., Aebersold D.M., Zimmer Y., Medova M. MET targeting: time for a rematch. Oncogene. 2020;39:2845–2862. doi: 10.1038/s41388-020-1193-8. [DOI] [PubMed] [Google Scholar]
- Kong-Beltran M., Seshagiri S., Zha J., Zhu W., Bhawe K., Mendoza N., Holcomb T., Pujara K., Stinson J., Fu L. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66:283–289. doi: 10.1158/0008-5472.CAN-05-2749. [DOI] [PubMed] [Google Scholar]
- Lai A.Z., Cory S., Zhao H., Gigoux M., Monast A., Guiot M.C., Huang S., Tofigh A., Thompson C., Naujokas M. Dynamic reprogramming of signaling upon met inhibition reveals a mechanism of drug resistance in gastric cancer. Sci. Signal. 2014;7:ra38. doi: 10.1126/scisignal.2004839. [DOI] [PubMed] [Google Scholar]
- Le X., Puri S., Negrao M.V., Nilsson M.B., Robichaux J., Boyle T., Hicks J.K., Lovinger K.L., Roarty E., Rinsurongkawong W. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC. Clin. Cancer Res. 2018;24:6195–6203. doi: 10.1158/1078-0432.CCR-18-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung. 1953;3:285–290. [PubMed] [Google Scholar]
- Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W., Harris P.L., Haserlat S.M., Supko J.G., Haluska F.G. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Nichols R.J., Haderk F., Stahlhut C., Schulze C.J., Hemmati G., Wildes D., Tzitzilonis C., Mordec K., Marquez A., Romero J. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol. 2018;20:1064–1073. doi: 10.1038/s41556-018-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou S.I., Koczywas M., Ulahannan S., Janne P., Pacheco J., Burris H., McCoach C., Wang J.S., Gordon M., Haura E. A12 the SHP2 inhibitor RMC-4630 in patients with KRAS-mutant non-small cell lung cancer: preliminary evaluation of a first-in-man phase 1 clinical trial. J. Thorac. Oncol. 2020;15:S15–S16. [Google Scholar]
- Paik P.K., Felip E., Veillon R., Sakai H., Cortot A.B., Garassino M.C., Mazieres J., Viteri S., Senellart H., Van Meerbeeck J. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N. Engl. J. Med. 2020;383:931–943. doi: 10.1056/NEJMoa2004407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicci G., Giordano S., Zhen Z., Salcini A.E., Lanfrancone L., Bardelli A., Panayotou G., Waterfield M.D., Ponzetto C., Pelicci P.G. The motogenic and mitogenic responses to HGF are amplified by the Shc adaptor protein. Oncogene. 1995;10:1631–1638. [PubMed] [Google Scholar]
- Pilotto S., Carbognin L., Karachaliou N., Ma P.C., Rosell R., Tortora G., Bria E. Tracking MET de-addiction in lung cancer: a road towards the oncogenic target. Cancer Treat. Rev. 2017;60:1–11. doi: 10.1016/j.ctrv.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Recondo G., Bahcall M., Spurr L.F., Che J., Ricciuti B., Leonardi G.C., Lo Y.C., Li Y.Y., Lamberti G., Nguyen T. Molecular mechanisms of acquired resistance to MET tyrosine kinase inhibitors in patients with MET exon 14-mutant NSCLC. Clin. Cancer Res. 2020;26:2615–2625. doi: 10.1158/1078-0432.CCR-19-3608. [DOI] [PubMed] [Google Scholar]
- Reungwetwattana T., Ou S.H. MET exon 14 deletion (METex14): finally, a frequent-enough actionable oncogenic driver mutation in non-small cell lung cancer to lead MET inhibitors out of "40 years of wilderness" and into a clear path of regulatory approval. Transl. Lung Cancer Res. 2015;4:820–824. doi: 10.3978/j.issn.2218-6751.2015.12.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikova K., Guo A., Zeng Q., Possemato A., Yu J., Haack H., Nardone J., Lee K., Reeves C., Li Y. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Rong S., Segal S., Anver M., Resau J.H., Vande Woude G.F. Invasiveness and metastasis of NIH 3T3 cells induced by Met-hepatocyte growth factor/scatter factor autocrine stimulation. Proc. Natl. Acad. Sci. U S A. 1994;91:4731–4735. doi: 10.1073/pnas.91.11.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell R., Bivona T.G., Karachaliou N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet. 2013;382:720–731. doi: 10.1016/S0140-6736(13)61715-8. [DOI] [PubMed] [Google Scholar]
- Rosell R., Karachaliou N., Arrieta O. Novel molecular targets for the treatment of lung cancer. Curr. Opin. Oncol. 2020;32:37–43. doi: 10.1097/CCO.0000000000000590. [DOI] [PubMed] [Google Scholar]
- Salgia R. MET in lung cancer: biomarker selection based on scientific rationale. Mol. Cancer Ther. 2017;16:555–565. doi: 10.1158/1535-7163.MCT-16-0472. [DOI] [PubMed] [Google Scholar]
- Shen D., Chen W., Zhu J., Wu G., Shen R., Xi M., Sun H. Therapeutic potential of targeting SHP2 in human developmental disorders and cancers. Eur. J. Med. Chem. 2020;190:112117. doi: 10.1016/j.ejmech.2020.112117. [DOI] [PubMed] [Google Scholar]
- Shen Y., Naujokas M., Park M., Ireton K. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell. 2000;103:501–510. doi: 10.1016/s0092-8674(00)00141-0. [DOI] [PubMed] [Google Scholar]
- Soman N.R., Correa P., Ruiz B.A., Wogan G.N. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc. Natl. Acad. Sci. U S A. 1991;88:4892–4896. doi: 10.1073/pnas.88.11.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordella R., Bell D.W., Haber D.A., Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Stommel J.M., Kimmelman A.C., Ying H., Nabioullin R., Ponugoti A.H., Wiedemeyer R., Stegh A.H., Bradner J.E., Ligon K.L., Brennan C. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- Sun Y., Meyers B.A., Czako B., Leonard P., Mseeh F., Harris A.L., Wu Q., Johnson S., Parker C.A., Cross J.B. Allosteric SHP2 inhibitor IACS-13909 overcomes EGFR-dependent and EGFR-independent resistance mechanisms towards osimertinib. Cancer Res. 2020;80:4840–4853. doi: 10.1158/0008-5472.CAN-20-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixido C., Gimenez-Capitan A., Molina-Vila M.A., Peg V., Karachaliou N., Rodriguez-Capote A., Castellvi J., Rosell R. RNA analysis as a tool to determine clinically relevant gene fusions and splice variants. Arch. Pathol. Lab Med. 2018;142:474–479. doi: 10.5858/arpa.2017-0134-RA. [DOI] [PubMed] [Google Scholar]
- Tong J.H., Yeung S.F., Chan A.W., Chung L.Y., Chau S.L., Lung R.W., Tong C.Y., Chow C., Tin E.K., Yu Y.H. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin. Cancer Res. 2016;22:3048–3056. doi: 10.1158/1078-0432.CCR-15-2061. [DOI] [PubMed] [Google Scholar]
- Turke A.B., Zejnullahu K., Wu Y.L., Song Y., Dias-Santagata D., Lifshits E., Toschi L., Rogers A., Mok T., Sequist L. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Richter O., Massimini G., Scheible H., Udvaros I., Johne A. Pimasertib, a selective oral MEK1/2 inhibitor: absolute bioavailability, mass balance, elimination route, and metabolite profile in cancer patients. Br. J. Clin. Pharmacol. 2016;82:1498–1508. doi: 10.1111/bcp.13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner K.M., Di Cesare S., Sachs M., Brinkmann V., Behrens J., Birchmeier W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature. 1996;384:173–176. doi: 10.1038/384173a0. [DOI] [PubMed] [Google Scholar]
- Wolf J., Seto T., Han J.Y., Reguart N., Garon E.B., Groen H.J.M., Tan D.S.W., Hida T., de Jonge M., Orlov S.V. Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung cancer. N. Engl. J. Med. 2020;383:944–957. doi: 10.1056/NEJMoa2002787. [DOI] [PubMed] [Google Scholar]
- Wu W., Bi C., Credille K.M., Manro J.R., Peek V.L., Donoho G.P., Yan L., Wijsman J.A., Yan S.B., Walgren R.A. Inhibition of tumor growth and metastasis in non-small cell lung cancer by LY2801653, an inhibitor of several oncokinases, including MET. Clin. Cancer Res. 2013;19:5699–5710. doi: 10.1158/1078-0432.CCR-13-1758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or analyze codes.