

Summary
Understanding the dynamic transcriptional landscape throughout organ development will provide a template for regenerative therapies. Here, we generated a single-cell RNA sequencing atlas of murine submandibular glands identifying transcriptional profiles that revealed cellular heterogeneity during landmark developmental events: end bud formation, branching morphogenesis, cytodifferentiation, maturation, and homeostasis. Trajectory inference analysis suggests plasticity among acinar and duct populations. We identify transcription factors correlated with acinar differentiation including Spdef, Etv1, and Xbp1, and loss of Ybx1, Eno1, Sox11, and Atf4. Furthermore, we characterize two intercalated duct populations defined by either Gfra3 and Kit, or Gstt1. This atlas can be used to investigate specific cell functions and comparative studies predicting common mechanisms involved in development of branching organs.
Subject Areas: Biological Sciences, Developmental Biology, Systems Biology, Transcriptomics
Graphical Abstract
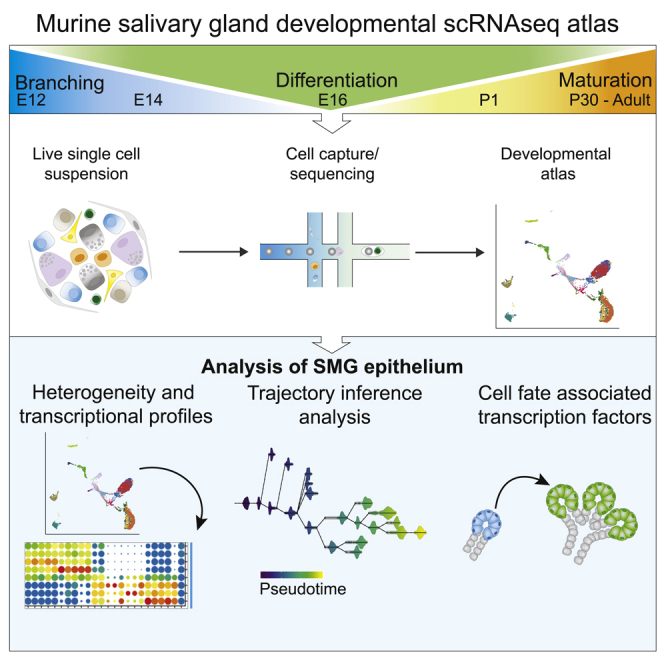
Highlights
-
•
Generated scRNAseq atlas of E12, E14, E16, P1, P30, and adult SMG
-
•
Smgc and Bpifa2 define two proacinar populations in the developing SMG
-
•
Loss of Ybx1, Eno1, Sox11, and Atf4 associated with acinar phenotype
-
•
Gstt1 defines Kit-negative and sexually dimorphic intercalated duct cells
Biological Sciences; Developmental Biology; Systems Biology; Transcriptomics
Introduction
In salivary glands and other developmentally similar branching organs like mammary gland, pancreas, kidney, lung, and lacrimal glands epithelial heterogeneity and cell lineages are not well understood. Developmentally, branching organs undergo a sequence of key events that are conserved across tissues, namely, initiation, tubulogenesis, branching morphogenesis, cytodifferentiation, and homeostasis (Goodwin and Nelson, 2020). These processes require coordinated regulation of proliferation, differentiation, and cell interactions with their microenvironment (Patel and Hoffman, 2014), which are also critical for regeneration. Proper characterization of the heterogeneous branching epithelium and identification of the molecular factors involved in the transition between development stages will inform therapeutic strategies aiming to regenerate injured organs.
Mouse submandibular gland (SMG) development begins at embryonic day (E) 11 with an epithelial placode forming within a condensed mesenchyme. Epithelial progenitors form an end bud at E12 that gives rise to all epithelial cells in the adult gland (Athwal, et al., 2019). Branching morphogenesis begins ~E13.5 and continues throughout fetal development and cell differentiation of proacinar and myoepithelial cells (MECs) begins ~E16. At postnatal day 1 (P1), functional acinar differentiation is required for saliva secretion and further acinar and ductal differentiation continues postnatally (Hauser and Hoffman, 2015; Patel and Hoffman, 2014; Tucker, 2007). The specific cell types and molecular factors required for the progression through these stages and those involved in lineage commitment and specification of terminally differentiated cells such as secretory acinar cells are not well known. This is of clinical relevance because acinar cells are lost or permanently damaged during autoimmune disease or as a consequence of cancer therapies (Jensen, et al., 2019) and little is known about the regulation of acinar development and regeneration.
Single-cell RNA sequencing (scRNAseq) has made it possible to discover new and rare cell types, explore cellular heterogeneity, and identify cell-fate-determining factors during developmental trajectories (Hedlund and Deng, 2018; Zappia, et al., 2018). Recently, scRNAseq analyses in branching organs including the kidney (Combes, et al., 2019), lung (Angelidis, et al., 2019; Reyfman, et al., 2019), pancreas (Qadir, et al., 2020; Byrnes, et al., 2018; Sznurkowska, et al., 2018), mammary gland (Bach, et al., 2017), and many others have helped characterize tissue-specific heterogeneity and cell lineages during specific developmental stages or physiological conditions (i.e., embryonic development, aging, or injury). scRNAseq offers a unique opportunity to investigate acinar and duct development in salivary glands to identify cells that may be primed to produce acinar cells and define key transcription factors (TFs) involved in cell-fate decisions.
Here, we generate a scRNAseq resource of key stages of SMG development (E12, E14, E16, P1, P30, and adult). This atlas provides a transcriptional signature of cells at each developmental stage and highlights the epithelial heterogeneity of the gland. Furthermore, we show the potential of this resource through computational analysis to evaluate developmental trajectories, identify TFs potentially involved in acinar and duct differentiation, and characterize subpopulations of discrete cell types in anatomically defined compartments of the gland. This atlas can be coupled with other scRNAseq databases to predict cell functions and identify biologically conserved processes across multiple tissues. The database is a useful resource for the field to identify target genes and understand organogenesis in developmental and regenerative studies.
Results
Generation of Transcriptional Atlas Capturing Landmark Events during SMG Development
We generated six individual scRNAseq libraries of murine SMG development using the 10X Genomics platform (Figures 1A, 1B, and S1). We chose embryonic stages corresponding to primary end bud formation (E12), branching morphogenesis (E14), and onset of cell differentiation (E16), as well postnatal days 1 and 30 (P1, P30) and adult glands (10 months). Unsupervised clustering and differential expression analyses were performed with the SEURAT package (Stuart, et al., 2019; Butler, et al., 2018) to identify cell populations. Embryonic epithelial populations were identified by expression of Epcam (all epithelium), Krt14 and Krt5 (basal duct), Krt19 (duct), Sox9 and Aqp5 (end bud and proacinar), and Acta2 (myoepithelial). Postnatal epithelium was annotated according to expression of known cell type-specific markers summarized in Figure S1. Non-epithelial clusters were identified by expression of Pecam1 (endothelial), Tubb3 (nerves), Ncam1 (glial), Alas2+ (erythroid), Adgre1 (macrophages), Kit (mast cells), Nkg7 and Gzma (natural killer cells), and Acta2+Epcam- (smooth muscle). A cluster of undefined mitotic epithelial cells was also observed at P1. Subsequent data integration was performed for embryonic and postnatal stages individually for visualization and to adjust for potential batch effects between samples (Figure 1B). The proportion of epithelial (Epcam+) and non-epithelial cells (Vim+) recovered was consistent with immunostaining for these proteins (Figures 1C and 1D), and as expected, the number of epithelial clusters increased throughout development reflecting epithelial specialization (Figures 1E and 1F). The transcriptional profiles for all identified cell populations are provided in Data S1. Using this resource, we aimed to (1) dissect the heterogeneity of the SMG epithelium and identify cell type-specific markers, (2) to identify putative cell-fate-determinant TFs involved in acinar and ductal cell specification and differentiation, and (3) to characterize epithelial cell subpopulations.
Figure 1.

scRNAseq Analysis of Murine SMG Development
(A and B) Single-cell suspensions from embryonic and postnatal SMG were used to build scRNAseq libraries. Data integration of embryonic and postnatal stages is shown in separate UMAPs colored by developmental stage.
(C) UMAPs showing expression of Epcam.
(D) Immunostaining of Epcam (green) and Vimentin (red). Scale bars, 20 μm.
(E) Clusters were annotated based on expression of known markers.
(F) Table showing cell numbers within each population from each stage.
Transcriptional Heterogeneity Identified in Embryonic SMG Epithelium
To identify putative epithelial subpopulations, epithelial cells from each developmental stage were separated bioinformatically and re-clustered for downstream analysis maintaining their assigned cell annotations from Figure 1E. We anticipated unique heterogeneity across developmental stages; thus, individual analyses were performed for each stage. To choose an optimal resolution for unsupervised clustering, we tested 12 different resolution values and compared their performance using clustree package in R (not shown). We then chose a resolution that accurately discriminated cell types according to their previously assigned annotations.
Unsupervised clustering of E12, E14, and E16 epithelium resulted in seven to nine clusters (Figure 2A). Immunofluorescence images and UMAPs of Krt5 and Krt14 expression are shown to confirm that unsupervised clustering accurately represents the spatiotemporal expression of these progenitor markers throughout development (Figures 2B–2D). As expected, spatial separation between clusters was increased at E16 likely due to ongoing cell specification, which was also evident by expression of the proacinar marker Aqp5 in clusters 1, 4, and 5 (Figures 2A and 2D).
Figure 2.

SEURAT Analysis of Embryonic SMG Epithelium
(A) From the embryonic integrated library, epithelial clusters from individual stages were separated and re-clustered with SEURAT. Individual UMAPs are shown, and labeled outlines indicate main cell types.
(B–D) Representative images of Krt5 (green) and Krt14 (red) whole-mount staining of embryonic SMG and complimentary UMAPs. Additional UMAPs are shown for Sox10, Krt19, Aqp5, and Acta2. Scale bars, 50 μm.
(E–G) Dot plots with scaled expression (color of the dot) and percentage of expression (size of the dot) of the top five genes for each cluster from Figure 2A. A representative gene from each group is shown in a UMAP.
As the number of resulting clusters is dependent on the chosen resolution for analysis, it does not necessarily indicate the number of discrete subpopulations. Therefore, we also performed differential expression analysis of all clusters to cluster-specific gene expression. The top five expressed markers per cluster are shown in Figures 2E–2G, and their complete transcriptional profile is provided in Data S2. Observed epithelial heterogeneity at E12 was primarily due to the proliferative state of cells as shown by expression of cell cycle S phase (Ung) and G2/M phase (Aurka, Mki67) markers (Figures S2A and S2B). Proliferative end bud cells were found in cluster 2, whereas proliferative Krt19+ duct cells were in cluster 4. At E14, proliferative end bud cells were represented by cluster 2 but a discrete cluster of proliferative duct cells was not identified (Figures S2C and S2D). In addition, cluster 3 at E14 identified a subpopulation of end bud cells characterized by expression of Cldn10, which was distinct from end bud cells in clusters 0 and 1 characterized by Actg1 expression (Figure 2F). These are likely to represent the inner and outer layers of the end bud, respectively (Figure S2G). At E16, proliferative end bud, basal duct, and MECs were found in clusters 5, 8, and 7, respectively (Figures 2G, S2E, and S2F). In addition, end bud cells were also divided in two major groups characterized by expression of Muc19 and its splice isoform Smgc in cluster 1, and Bpifa2 (parotid secretory protein or Psp) and Dcpp1 in clusters 4 and 5 (Figure 2G). Although DEGs were enriched in a cluster or cell population, these cell-defining genes were often expressed elsewhere to lesser degrees.
At all stages, there were undefined cell clusters (clusters 5 at E12, 4 at E14, and 6 at E16) expressing mesenchymal markers Col1a1, Col1a3a1, as well as relatively low levels of Krt14, Krt19, Sox10, and Epcam (Figures 2E–2G and S2). In addition, cluster 6 at E12 is defined by high expression of genes such as Prdx1, Ppia, and Eif4a1 that are broadly expressed within many cell clusters in both epithelial and non-epithelial cells (Figure 2E). As cell doublets were removed during analyses, these undefined clusters may represent either uncommitted cells, or cells that are undergoing epithelial to mesenchymal transition, or vice versa. They may be homologous to a KRT17+ matrix producing epithelial cell associated with pulmonary fibrosis (Habermann, et al., 2020). Further analysis of these cells is required to understand their identity.
scRNAseq of Postnatal SMG Reveals Two Related but Distinct Proacinar Populations
Clustering of epithelial cells from postnatal glands resulted in 10 epithelial clusters at P1 and P30 and 12 clusters in adult gland. Most of the previous annotations remained in a single cluster, but proacinar and acinar cells were represented by multiple discrete clusters at all postnatal stages, highlighting their heterogeneity (Figures 3A and 3B). The top five DEGs per cluster and representative UMAP of a gene from each cluster are shown (Figures 3C–3E). Duct populations at P1 were defined by high keratin expression and fell into two main groups: basal duct (BD) in cluster 4 (Krt14+ Krt17+) and differentiating ducts that express Krt19 and Krt7 in cluster 2 (Figure 3C). In the P30 and adult gland, well-separated duct clusters represented Kit+ intercalated duct (ID) cells, striated ducts (SD), Ascl3+ cells, and BD cells, all of which had unique transcriptional signatures (Figures 3C and 3D). Granular convoluted tubules (GCTs) were also in a discrete cluster at P30 (cluster 5) but were mixed with SD cells in the adult dataset (cluster 2), which was exclusively from female glands. MECs were in cluster 9 at P1 and cluster 8 in the adult gland (Figures 3C and 3E), and were defined by expression of Acta2, Myh11, and Cnn1. At P30, MECs clustered together with BD cells (Figures 3A and 3D), likely due to the low number of MECs recovered (Figure 1E). For further analyses, MECs at P30 were manually annotated based on expression of Acta2 and Cnn1. Proliferation in P30 and adult gland was limited to a few cells in multiple clusters (Figures S3C–S3F). Last, few undefined cells that expressed the mesenchymal genes Col1a1 and Col3a1 were scattered across multiple clusters at all postnatal stages (Figure S3).
Figure 3.

SEURAT Analysis of Postnatal SMG Epithelium
(A) From the postnatal integrated library, epithelial clusters from individual stages were separated and re-clustered with SEURAT. Individual UMAPs are shown, and labeled outlines indicate main cell types.
(B) UMAPs of expression of Bhlha15, Aqp5, and Smgc in P1 and P30 SMG.
(C–E) Dot plots with scaled expression (color of the dot) and percentage of expression (size of the dot) of the top five genes for each cluster from Figure 3A. A representative gene from each group is shown in a UMAP.
The mouse SMG is known to produce both serous and mucous secretions, and the greatest heterogeneity for identified cell types was observed within proacinar and acinar cells (Figures 3A and 3B). Proacinar cells at P1 were found across seven clusters that fell into three major groups based on top DEGs: Smgc+ proacinar (clusters 0, 3, and 6), Bpifa2+ proacinar (clusters 1, 7, and 8), and mitotic cells (cluster 5) (Figures 3A, 3C, S3A, and S3B). Individual clusters within the first two groups shared similar transcriptional profiles with varying expression levels of their defining genes. Mature acinar cells defined by expression of Aqp5 and Bhlha15 (Mist1) were found in clusters 0, 2, and 7 in P30 (Figure 3D) and clusters 0 and 4 in adults (Figure 3E). They expressed varying levels of both serous and mucous secretory genes suggesting a seromucous phenotype (Nelson, et al., 2013; Kivela, et al., 1999; Mirels, et al., 1998). These included prolactin-induced protein (Pip), lactoperoxidase (Lpo), mucin-like 1 and 2 (Mucl1, Mucl2), proline-rich lacrimal 1 (Prol1, also known as Muc10), and carbonic anhydrase 6 (Car6) (Figures 3C, 3D, 4A, and 4C). Notably, Bpifa2 was expressed in a discrete population (clusters 3 and 10, Figures 3A and 3E), which co-expressed Dcpp genes in adult SMG (Figure 4B). Immunostaining showed that Bpifa2 delineates serous cells in parotid gland and was detected in some Lpo+ acinar cells in adult SMG (Figure 4D). This population was not found in our P30 data and was not detected by immunostaining in P30 SMG from males and females (not shown); however, Bpifa2 expression was detected by qPCR in postnatal glands (Figure 4D). Taken together, this suggests that Bpifa2+ cells in adult SMG may represent an additional population of serous-like acinar cells. Thus, we refer to Bpifa2+ cells in adult gland as serous acinar cells and Car6+ Prol1+ Lpo+ cells as seromucous acinar cells.
Figure 4.

Expression of Serous and Mucous Genes in Acinar Cells
(A and B) Epithelium from P30 and adult SMG colored and annotated by cell type with UMAPs showing expression of serous and mucous markers alongside.
(C) Immunostaining of Lpo (green), Prol1 (red), and Mist1 (white) in P30 SMG (top panel) and Bpifa2 (Red) in adult PG and SMG (bottom panel).
(D) Left qPCR graph shows decreased expression of Bhlha15 and Bpifa2 in P20 SMG normalized to P1 SMG. Graph on the right shows qPCR of selected serous and mucous genes in P30 and P180 female SMG normalized to P20. Data are represented as mean ± SEM and asterisks denote statistical significance (p < 0.05) compared with baseline (n = 3, two-tailed t test).
Trajectory Inference Analysis Predicts Plasticity across Epithelial Populations
To further explore possible lineage relationships and developmental trajectories, we integrated epithelial cells from all stages (Figure 5A) maintaining cell type annotations. Four regions could be identified in the resulting integrated UMAP, which corresponded to Smgc+, acinar, basal, and specialized duct cells, respectively. Resolution between individual populations was greatly reduced, likely because of the influence of embryonic cell types, which were spread throughout the UMAP projection. Next, we performed trajectory inference (TI) analysis with Dynverse package in R (Saelens, et al., 2019), which aligns cells along an unbiased pseudotemporal trajectory called pseudotime based on their transcriptional similarities. Dynverse compares >50 TI methods and chooses the most appropriate one to evaluate individual cell transcriptomes in a given dataset to infer their order in pseudotime. Here, TI was performed using PAGA-tree algorithm (Wolf, et al., 2019). Although pseudotime is determined in an unbiased manner, we manually selected E12 epithelial cells as the origin of the trajectory because they are the most primitive population in our dataset.
Figure 5.

Trajectory Inference Analysis of SMG Epithelium
(A) Integrated and re-clustered epithelial cells from all stages shown in UMAP. Left panel is colored by stage and right panel by cell type.
(B and C) TI analysis using the PAGA-tree algorithm in Dynverse package. The determined pseudotemporal trajectory is colored by stage (B) and pseudotime score (C).
(D) Distribution of specific cell types along the trajectory is indicated by color and labels.
The inferred pseudotime trajectory accurately represented the biological progression of developmental stages (Figures 5B–5D) with embryonic populations receiving the lowest pseudotime scores. However, the resulting trajectory did not reflect the natural developmental progression of specific cell types according to reported lineage-tracing studies. For instance, lineage tracing of Kit, Krt5, and Krt14 in the adult SMG shows that these populations are lineage-restricted during homeostasis (Kwak, et al., 2018; May, et al., 2018; Kwak, et al., 2016), whereas our predicted trajectory situated Kit+ cells at a branching point preceding Smgc+ and seromucous acinar cells (Figure S4). Nonetheless, plasticity of multiple salivary gland cell types including myoepithelial, Kit+, and Krt5+ cells has been reported under severe damage (Ninche, et al., 2020). Thus, our TI analysis may be more informative of potential plasticity rather than lineage trajectories. Accordingly, primitive cell populations like end buds and Krt19+ ducts were often found together in multiple branch points and closely related cell types remained in similar branches (Figure 5D). MECs and BD cells from E16 onward were in terminal branches with low pseudotime scores, likely because of their early specification. Similarly, a fraction of end bud cells from E14 and E16 were in terminal branches. This is not surprising as not every cell from a given developmental stage is expected to function as a progenitor or to maintain plasticity.
Notably, Krt19+ duct and mitotic cells from E16 and P1 were localized at the branching point from which all other postnatal populations were derived, except for BD and MECs (Figure 5D). The most striking result was the separation between P1 proacinar cells and acinar populations, which were expected to appear in a continuous branch. Although it is possible that batch effect between developmental stages may influence these observations, this could also reflect the differences within the acinar compartment between neonatal and mature glands described in the previous section (Figure 4). The significance of our TI analysis to predict plasticity between populations will require further studies. Nonetheless, because of our consistent observations regarding complexity and heterogeneity in the acinar lineage, we next aimed to determine the TFs potentially involved in acinar cell development.
Identification of Transcription Factors Involved in Acinar Specification and Differentiation
To identify genes associated with acinar cell development, we performed Pearson correlation analysis across our integrated dataset (Figure 6A) to find genes correlated with expression of Bhlha15. Bhlha15 is a well-known acinar gene that encodes the TF Mist1, which is regulated by Xbp1 in acinar cells (Lo, et al., 2017; Hess et al., 2016; Huh, et al., 2010). Not surprisingly, the genes with the strongest correlations to Bhlha15 were the acinar markers Lpo (0.38), Aqp5 (0.37), and Pip (0.36) (Data S3). We specifically focused on TFs and found that Creb3l1 (0.30), Spdef (0.27), Etv1 (0.23), Creb3l4 (0.20), and Xbp1 (0.19) were the most positively correlated TFs, whereas Eno1 (−0.34), Ybx1 (−0.32), and Sox11 (−0.32) were the most negatively correlated TFs (Data S3). The 10 strongest positive and negative correlated TFs with Bhlha15 are shown in Figure 6B.
Figure 6.

Transcriptional Regulators of Acinar Differentiation
(A) UMAP of integrated SMG epithelium highlighting end bud, proacinar, and acinar populations colored by cell type.
(B) Analysis of TFs correlated with Bhlha15. Color scale represents correlation scores (p < 0.05).
(C) Heatmap showing scaled expression of Bhlha15-correlated genes in end bud, proacinar, and acinar clusters. The colored bars represent developmental stages.
(D) Schematic summarizing gene expression changes of selected genes throughout development. Blue area shows genes that decrease in expression, whereas red indicates genes that increase during acinar differentiation.
(E) Violin plots of top differentially expressed TFs between P1 Smgc+ proacinar and P30 Smgc+ cells. Color scale consistent with Figure 6C.
(F) Venn diagram of the comparison between defining genes for P1 proacinar populations and seromucous acinar cells at P30.
(G) Violin plots of top differentially expressed TFs between P1 Bpifa2+ proacinar and P30 seromucous acinar cells. Color scale consistent with Figure 6C.
We hypothesized that TFs positively correlated with Bhlha15 would become expressed after cytodifferentiation begins at E16 (Nelson, et al., 2013), and that in turn, negatively correlated genes would be downregulated. Indeed, when viewed as a heatmap along with Bhlha15, positively correlated genes were enriched after E16 (Figure 6C). To investigate if these TFs were involved in specific developmental transitions, we compared related populations from contiguous developmental stages. The top five enriched TFs (if present) for each transition are shown as violin plots, and the complete list is provided in Data S4. Not surprisingly, higher expression of positive Bhlha15-correlated genes (Ehf, Spdef, and Xbp1) occurred at E16 (Figure S5A), whereas negative Bhlha15-correlated genes (Eno1, Sox11 Ybx1, Etv4, and Hmga2) progressively decreased in expression during development (Figures 6D, S5A, and S5B).
Our earlier analysis showed two distinct proacinar populations defined by Smgc and Bpifa2 expression. Interestingly, similar TFs were decreased in expression at P1 for both populations including Sfpq, Hnrnpk, Cebpb, and Ybx1 (Figure S5B). In contrast Etv1 and Zbtb20 were enriched in P1 Bpifa2+ proacinar cells, whereas Atf3, Arid5b, and Hopx were enriched in Smgc+ proacinar cells (Figure S5B). Smgc+ cells from P30 continued to show enriched expression of Junb, Cebpd, Fos, Atf3, and Egr1 postnatally, but the acinar-correlated gene Spdef was decreased (Figure 6E). Because serous acinar cells were not identified at P30, we could not determine TFs potentially involved in this transition from P1 SMG.
The postnatal maturation of seromucous acinar cells is not well understood, and TI analysis did not predict a direct lineage relationship with proacinar cells. Thus, it is unclear which of the two populations of proacinar cells at P1 (Smgc+ and Bpifa2+), if either, give rise to seromucous acinar cells. To predict a potential lineage relationship, we compared the transcriptional profile of proacinar cells and seromucous acinar cells to identify transcriptional similarities. Only 4 of the 267 defining genes for Smgc+ proacinar cells were also enriched in seromucous acinar cells at P30, whereas 50% of the defining genes for Bpifa2+ proacinar cells were enriched in seromucous cells (Figure 6F). Accordingly, 20 of the 26 seromucous acinar-defining genes were enriched in Bpifa2+ proacinar cells suggesting that Bpifa2+ proacinar cells are more closely related to mature seromucous acinar cells in mouse SMG. Differential expression analysis between Bpifa2+ proacinar and seromucous acinar cells showed reduced expression of the TFs Jun, Ybx1, Nfkbia, Atf4, and Jund in mature acinar cells (Figure 6G).
Taken together with our previous analyses, these data suggest that Bpifa2+ and Smgc+ proacinar cells may differentiate from a common progenitor around E16 but subsequently undergo distinct developmental programs: one that shares the transcriptional signature of mature acinar cells (Bpifa2+) and one that undergoes further differentiation into a distinct phenotype (Smgc+). Future lineage tracing experiments are required to confirm these predictions.
Putative TFs Involved in Duct-Acinar Differentiation Include Atf3 and Jun
Krt5+ cells have also been reported to generate acinar cells upon severe injury (Weng, et al., 2018), and lineage tracing studies have shown that Krt5+ BD cells act as progenitors for GCTs and SD cells (Aure, et al., 2019). Little is understood about the factors driving BD cell differentiation and maturation, as well as those involved in their plasticity to regenerate acinar cells. Thus, using a similar strategy to our previous analysis of the acinar lineage, we aimed to identify TF potentially involved in development of duct populations (Figure 7A).
Figure 7.

Transcriptional Regulators of Duct Differentiation
(A) UMAP of integrated SMG epithelium highlighting duct populations colored by cell type.
(B and D) Differential expression analysis between pairs of clusters from contiguous developmental stages as indicated by the color scheme and cell type labels. Top differentially expressed TFs (p < 0.05) are shown. TFs are sorted by fold change.
(C) Venn diagram of the comparison between defining genes for basal duct in embryonic versus postnatal stages.
(E) Differential expression analysis from BD to SD, GCTs, and acinar cells. Top differentially expressed TFs (p < 0.05) are shown.
Early development of Krt19+ ducts involved increased expression of Cebpd, Klf6, and Tfcp2l1 at both E14 and E16 (Figure 7B). Interestingly, decreased expression of Ybx1, Hmga2, Sox11, and Eno1 also occurred, similar to our observations of end bud development (Figure S5A). Analysis of BD populations across developmental stages showed that 70% of BD-defining genes at embryonic stages (156 of 225) are maintained postnatally but the number of defining genes increases 5-fold to 1,135 genes in postnatal BD cells (Figure 7C), suggesting further specialization. The top TFs involved in BD development are shown in Figure 7D. To explore putative transitions from BD to GCT, SD, and acinar cells in adult SMG, we performed differential expression analysis between these populations (Figure 7E). Bola3 and Id2 were enriched in SD compared with BD cells, and Carhsp1, Mafb, Bola3, Eno1, and Phb were enriched in GCT compared with BD. Last, Bhlha15, Etv1, and Creb3l1 were the only enriched TFs in acinar cells compared with BD populations, suggesting that they are potentially involved in the plasticity of Krt5+ basal cells to generate acinar cells. The genes Junb, Nfkbia, and Klf6 were enriched in BD compared with GCT, SD, and acinar cells, suggesting that these genes may be important for the BD phenotype. Other TFs enriched in BD cells included Atf3 and Fos.
Many of the TFs involved in duct development were present in the developmental transitions of the acinar lineage (Figure 5), including Ybx1, Eno1, Atf3, and Jun. Notably, Ybx1 and Eno1 were decreased during early differentiation of both duct and acinar lineages and may therefore represent factors involved in global epithelial differentiation not exclusive to a specific lineage. In contrast, Atf3 and Jun were decreased in the transition from either proacinar cells at P1 or BD cells to acinar cells. In pancreas, Atf3 activation is associated with loss of acinar cell phenotype (Fazio, et al., 2017). Thus, these results warrant further investigation to determine whether downregulation of Atf3 and Jun is required for acinar specification and maturation. The complete list of DEGs for each transition is provided in Data S4.
Subpopulations of ID in Mouse SMG Are Defined by Expression of Gstt1 and Gfra3
Smgc, a splice variant of Muc19, has been described as an embryonic mucin in mucous proacinar cells (Das, et al., 2009). It is hormonally regulated and declines during postnatal development persisting only in female SMG (Kusakabe, et al., 2016). Our analyses consistently showed that Smgc delineated a discrete population in the P30 and adult glands, separate from acinar cells (Figure 3B). Accordingly, we found Smgc localized to IDs adjacent to Mist1+Aqp5+ acinar cells in P30 and adults but did not express Mist1, unlike Smgc+ cells at P1 (Figure 8A). Surprisingly, although Smgc+ protein was not detected in male SMG by immunostaining, our P30 scRNAseq, which contained male and female glands showed a cluster of Smgc+ cells in the male SMG (Figure S6A).
Figure 8.

Subpopulations of SMG ID Are Defined by Expression of Smgc, Gstt1, and Gfra3
(A) Immunofluorescence staining showing localization of Smgc (green) in P1 and P30 SMG. Proacinar and acinar cells are labeled with Mist1 and Aqp5 (red).
(B) qPCR for genes enriched in Smgc+ (top panel) and Kit+ ID cells (bottom panel). Data are normalized to Rsp29 and age-matched female SMG (dotted line). Data are represented as mean ± SEM, and asterisks show statistical significance (p < 0.05) compared with age-matched female controls (n = 3; two-tailed t test).
(C) Immunofluorescence staining showing expression of Gstt1 (red) in Smgc+ cells (green, top panel) and Nkcc1+ cells (green, lower panel) in P30 SMG from male and female mice.
(D) UMAPs showing selected DEGs in Kit+ ID cells in P30 glands.
(E) Immunofluorescence shows distinct ID populations with no overlap between Gfra3, Gstt1, and Krt14 protein.
(F) In situ hybridization showing expression and localization of Gfra3, Prol1, Nkd2, Aqp5, and Esp18 mRNA in P30 SMG. Scale bars, 20 μm.
Sexual dimorphism in mouse SMG by P25 is well documented but little is known about transcriptional differences at single cell level (Mukaibo, et al., 2019; Gresik, 1994). Thus, we performed differential expression analysis between male and female cell populations using our P30 dataset. The complete list of identified sexually dimorphic genes is provided in Data S5. Surprisingly, GCTs, which hold the most distinct morphologic difference, did not show a significant number of DEGs with the exception of Smgc, which was detected in female GCT cells (Figures S6B and S6C). On the other hand, the Smgc+ population was the most sexually dimorphic at a transcriptional level (Figure S6C). To date, Smgc is the only known marker for this population. Therefore, to better characterize these sexually dimorphic ID cells, we evaluated DEGs identified in our analysis. Dcdc2a was enriched in female Smgc+ ID, whereas Serpinb11 was enriched in males, and Gstt1 was a common gene in both sexes (Figure S6D). Sex-dependent expression of these genes was detected by PCR between P20 and P30 and became more pronounced over time (Figure 8B). Cdkn1c was enriched in females in our scRNAseq data, but the gene was also detected in males by qPCR (Figures 8B and S6D). Gstt1 protein was also detected in IDs of both male and female glands (Figure 8C). We propose that Gstt1 is a defining gene for this ID population in both males and females.
The ID segment has long been proposed to contain a reservoir of progenitors with regenerative potential and several reports have identified Kit as a marker for an ID subpopulation (Kwak, et al., 2016; Katsumata, et al., 2009; Man, et al., 2001). Indeed, we identified a specific cluster of Kit+ cells in the P30 and adult SMG. The Kit+ ID population was defined by expression of Gfra3, Nkd2, and Esp18 (Figures 8D–8F). These markers were not sexually dimorphic, and Gfra3 protein was detected within a subset of ID cells adjacent to and distinct from Gstt1+ IDs and in adult glands, consistent with scRNAseq data (Figure 8E). Coexpression and localization with Nkd2 and Esp18 mRNA was confirmed by in situ hybridization (Figure 8F). Gfra3+ ID cells did not overlap with Krt14+ cells (Figure 8E), indicating that they are also a distinct population. Interestingly, Kit, Nkd2, and Esp18 are coexpressed in the same clusters at P1, but Gfra3 is not expressed in the epithelium (Figure S6E). qPCR analysis of intact SMGs shows that expression of Gfra3 and Nkd2 is markedly increased at P8 (Figure S6F), suggesting that differentiation of these cells occurs around this stage. Gfra3/Ret signaling occurs in nerves following binding of the neurotrophic factor artemin (Baloh, et al., 1998). As expected, in situ hybridization showed that Gfra3 was co expressed with its co-receptor Ret in the parasympathetic ganglia of P1 mice (Figure S7). However, in the adult gland where Gfra3 is expressed in the ID epithelium, we found that Ret was broadly expressed in acinar cells, but not in the ID (Figure S7), suggesting a different signaling mechanism in epithelial cells that remains to be determined.
Our observations indicate that Gstt1+ and Gfra3+ cells define two ID subpopulations with potentially distinct functions. We compared the defining genes for both populations, and found that they share a third of their transcriptional profile (Figure 9A). Interestingly, the top defining genes for Gfra3+ ID are also expressed by Krt19+ ducts from P1, whereas the top defining genes for Gstt1+ ID cells are shared by Smgc+ proacinar cells (Figure 9A, heatmap). KEGG pathway analysis showed that both populations have functions related to fluid sheer stress and tight junctions, which may be related to their duct phenotype (Figure 9B). Gstt1+ cells were also enriched for genes associated with estrogen and MAPK signaling, whereas the top pathways in Gfra3+ cells were antigen processing, leukocyte migration, and cancer pathways.
Figure 9.

Subpopulations of SMG ID Are Defined by Expression of Smgc, Gstt1, and Gfra3
(A) Venn diagram of the comparison between defining genes for Gstt1+ and Gfra3+ ID cells. Top 15 defining genes for each population are shown in the heatmap.
(B) Pathway analysis of defining genes from ID populations.
(C–E) UMAP of SMG ID cells integrated with selected populations from the Tabula Muris colored by tissue of origin and cell type as indicated in the legend.
(F) UMAP showing expression of Krt18, Krt8, and Cldn4.
To gain further insight into the potential biological functions of these cell types, we combined Gstt1+ and Gfra3+ ID cells with the Tabula Muris scRNAseq database, which contains cells from multiple non-salivary tissues including mammary gland, lung, trachea, pancreas, bladder, and kidney (Tabula Muris, et al., 2018). We integrated and re-clustered cells with SEURAT to identify transcriptional similarities between salivary gland ID and other cell types. Salivary ID cells were in two of the resulting 22 unsupervised clusters, namely, clusters 2 and 6 (Figures S8A and S8B). Cluster 2 also contained luminal cells from bladder and mammary gland, connecting tubule cells from the kidney, and duct pancreatic cells, according to the original annotations from the Tabula Muris (Figures 9C–9E, S8C, and S8D), whereas cluster 6 contained alpha, beta, delta, and polypeptide pancreatic (PP) cells; mammary T cells; and mesenchymal bladder cells. Evaluation of the defining genes for clusters 2 and 6 revealed variation in gene expression in subsets of cells within these clusters (Figure S8E), likely reflecting tissue-specific differences. Nonetheless, we identified a small group of genes that were conserved across most cells from clusters 2 and 6, including Krt8, Krt18, Epcam, and Cldn4 (Figures 9F and S8E). In addition, Wfdc2, Krt7, Foxq1, Sfn, Krt19, and a riken gene (1600029D21Rik) were widely expressed across cells in cluster 2 (Figure S8E). Given that most cell types in cluster 2 are luminal or ductal in nature, it is possible that these markers are involved in duct or luminal specification across tissues. Future investigation using genetic approaches will be needed to determine whether these markers are involved in specification of Gstt1+ and Gfra3+ ID cells in salivary glands and to determine whether ID cells share functional similarities with luminal and duct cells in other tissues.
Cell type annotations in the P30 and adult SMG were updated based on our findings and are provided along with their defining genes as supplementary material (Data S6). Ready-to-use SEURAT files are available from https://figshare.com/s/01778d0ed37fabf61b8e (embryonic SMG integrated) and https://figshare.com/s/134e9898ef8a20ff5c68 (postnatal SMG integrated).
Discussion
A Resource for Development and Salivary Gland Research
Although previous scRNAseq studies in salivary glands have started to uncover heterogeneity at defined timepoints (Sekiguchi, et al., 2020; Oyelakin, et al., 2019; Song, et al., 2018), a major advance of our scRNAseq resource is the inclusion of discrete developmental landmarks that will inform future studies on the mechanisms that regulate the development of branching organs, particularly salivary glands. These data add to our previous microarray analysis of SMG development and are available in the Salivary Gland Molecular Anatomy Project (SGMAP, https://sgmap.nidcr.nih.gov). The SGMAP includes microarray and bulk RNAseq of salivary gland from mice and humans. Incorporation of this resource and other available scRNAseq datasets from parotid gland and salivary tumors (Praktiknjo, et al., 2020) to the SGMAP will allow for more comprehensive analyses available to the scientific community.
One caveat of our atlas is that stromal and neuronal cell populations were not well-represented in the adult gland, despite attempts to isolate these cells using multiple dissociation techniques and digestion enzymes. This limitation has been reported in other scRNAseq studies and is potentially due to the morphological complexity and abundance of extracellular matrix surrounding these cell types. One way to circumvent this issue is through single nuclei isolation to recover populations hard to dissociate (Nguyen, et al., 2019), which may provide a complementary technique for future studies seeking to build upon the SGMAP. Nonetheless, the strengths of this resource are highlighted through the identification of cell-defining genes and heterogeneity in the SMG epithelium throughout development, and we further demonstrate its potential applications by identifying TFs (Ybx1, Eno1, Sox11 and Atf4) and cell populations potentially involved in acinar cell differentiation. We predict a lineage relationship between Bpifa2+ proacinar cells and seromucous acinar cells, and we characterized two distinct subpopulations of ID cells: a Kit+ population defined by Gfra3 and a proacinar-derived subset characterized by Gstt1 with sexually dimorphic Smgc co-expression in females and high Serpinb11 co-expression in males.
SMG scRNAseq Atlas Will Inform Regenerative Approaches
Irreversible loss of acinar cells occurs during treatment of head and neck cancer with radiation and as a consequence of autoimmune diseases, such as Sjögren syndrome (Jensen, et al., 2019). This leads to hyposalivation, dry mouth, and decreased quality of life with only palliative therapies available (Chibly, et al., 2014). As a result, a major goal in the field is to identify cell populations and cell-fate-determinant factors that drive specification and development of acinar cells to design effective regenerative therapies.
Mist1 is a well-known marker of acinar differentiation necessary and sufficient to induce and maintain secretory cell architecture of the stomach, pancreas, and salivary glands (Lo, et al., 2017). Consistent with previous reports, our dataset revealed the presence of a seromucous population of acinar cells with high expression of the Mist1 gene Bhlha15, and other secretory markers including Car6, Lpo, and Prol1. This population was transcriptionally similar to Bpifa2+ proacinar cells that appeared at E16, suggesting a potential lineage relationship. Bpifa2+ proacinar cells persisted at P1, and they have also been identified at P5 (Nelson, et al., 2013). Bpifa2 was absent at P30, similar to a report showing lack of Bpifa2 staining in P20 SMG (Nelson, et al., 2013). These finding may suggest that seromucous differentiation from Bpifa2+ proacinar cells occurs between P5 and P20. Interestingly, a serous-like population characterized by expression of Bpifa2 and Dcpp genes reappeared in adult SMG. A similar cluster was also described in a single-cell RNA study of salivary gland cancer (Praktiknjo, et al., 2020).
Lineage tracing of Bhlha15 (Mist1) demonstrated the ability of salivary acinar cells to undergo self-renewal with little or no input from progenitors (Aure, et al., 2015); however, Bhlha15 KO mice are still able to produce acinar cells, indicating that additional factors are required for acinar specification (Lo, et al., 2017; Huh, et al., 2010; Pin, et al., 2001). Our analysis revealed TFs that become upregulated at the onset of acinar differentiation and correlate with Bhlha15 expression, including Spdef, Xbp1, and Etv1. Interestingly, Spdef is required for terminal differentiation of mucous secreting cells in lungs and the intestinal tract (Horst, et al., 2010; Chen, et al., 2009), and Xbp1 is upstream of Mist1 and is associated with the unfolded protein response, which is prominent in secretory cells (Hess et al., 2016). In addition to these genes, we observed that acinar differentiation was characterized by a gradual downregulation of numerous TFs, including Ybx1, Eno1, Atf3, Jun, and Atf4. This is consistent with a study showing that Atf3 inhibits acinar specification (Fazio, et al., 2017).
After severe damage, Krt5+-expressing cells have shown limited ability to repopulate acinar cells (Weng, et al., 2018), but the regulatory mechanisms remain unknown. Our SMG atlas allows us to speculate about the potential factors involved in acinar cell generation from Krt5+ basal cells, which included Bhlha15, Etv1, Atf3, and Jun. Further research into the contribution of these TFs to acinar development and regeneration post-injury is warranted.
Characterization of Salivary ID Subpopulations
It has been widely speculated that ID cells may have varying degrees of plasticity to give rise to acinar cells following injury due to their proliferative capacity, label-retaining properties, and Kit expression (Kwak, et al., 2016; Katsumata, et al., 2009; Man, et al., 2001). Kit lineage tracing failed to demonstrate contribution to acinar cells during homeostasis (Kwak, et al., 2018), whereas recent studies suggest plasticity of multiple populations after severe injury (Ninche, et al., 2020). In this regard, it is interesting that Kit+ ID cells are defined by expression of Gfra3, a neurotrophic factor receptor that could potentially modulate their function upon binding to its cognate ligand artemin, which is expressed in the neurons within the gland. A precedent for the potential therapeutic effects of neurotrophic factors in injured salivary glands has already been demonstrated in vivo with administration of GDNF or Neurturin, which bind Gfra1 and Gfra2 receptors and improve saliva secretion post-irradiation (Ferreira, et al., 2018; Xiao, et al., 2014; Knox, et al., 2013). This is also reminiscent of acinar regeneration of SMGs in vivo after experimental duct ligation, which requires input from the nerves (Carpenter, et al., 2009). Whether innervation of ID plays a role in regeneration through Gfra3 remains to be explored.
Our data also allowed us to characterize an ID population defined by Gstt1 with sexually dimorphic expression of Smgc in females and Seprinb11 in males indicating that these cells have sex-dependent functions. This is supported by previous studies that report a pheromone-like function of Smgc when secreted into saliva of female mice (Isogai, et al., 2018). Whether Gstt1+ or Gfra3+ ID cells have the ability to repopulate acinar cells in the adult gland remains to be determined. Our atlas provides with cell-specific markers in these populations that will inform future studies.
In summary, this transcriptional atlas will benefit future studies to understand the mechanisms of SMG development, functions of specific cell populations, and lineage relationships and will enable comparisons with other tissues to identify evolutionarily conserved or unique cell types and functions. A combination of scRNAseq with lineage tracing models informed by the results in this study will be essential to answer questions about lineage and regeneration (Wagner and Klein, 2020). This atlas can be built upon by the addition of scRNAseq analysis of salivary gland disease and damage models, such as duct ligation, radiation, and partial gland extirpation, which will allow comparison of the cells that regenerate the gland after damage in each of these models.
Limitations of the Study
Due to the dissociation technique, few mesenchymal cells are represented in P30 and adult SMG, and neuronal cells were not recovered from all postnatal samples including P1, P30, and adult SMG. In addition, current methods for trajectory analysis like the one used in this study, are not yet fully optimized for complex datasets where divergent developmental processes are expected (i.e., multiple lineages) across multiple time points. Thus, results from trajectory analysis should be carefully interpreted and further validated.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alejandro Chibly (chiblyaa@nih.gov).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession number for the single-cell RNAseq libraries reported in this paper is GEO: GSE150327. The code used for analysis is provided in Data S7 and also available through github: https://github.com/chiblyaa/Salivary-Gland-Development. Ready-to-use SEURAT objects are also available via figshare:
Embryonic SMG integrated dataset: https://doi.org/10.6084/m9.figshare.13157687.
Postnatal SMG integrated dataset: https://doi.org/10.6084/m9.figshare.13157726.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank Dr. Daniel Martin, Dr. Robert Morell, Dr. Erich Boger from the Genomics and computational biology core (GCBC) and Dr. R. Sekiguchi for helpful discussions. This work used the NIDCR Veterinary Resources Core (ZIC DE000740-05) and computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). The GCBC funds were from the NIDCD Division of Intramural Research/NIH (DC000086 to the GCBC). The study was supported by the Intramural Research Program of the National Institute of Dental and Craniofacial Research, NIH.
Author Contributions
Conceptualization, methodology, writing and editing, B.R.H., M.H.A., M.P.H., and A.M.C.; Software, A.M.C., M.C.K., and GCBC; Resources, M.P.H. and A.M.C.; Visualization, B.R.H., M.H.A., and A.M.C.; Data Curation, Project Administration, and Supervision, A.M.C.
Declaration of Interests
The authors declare no competing interests.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101838.
Contributor Information
Matthew P. Hoffman, Email: mhoffman@dir.nidcr.nih.gov.
Alejandro M. Chibly, Email: chiblyaa@nih.gov.
Supplemental Information
References
- Angelidis I., Simon L.M., Fernandez I.E., Strunz M., Mayr C.H., Greiffo F.R., Tsitsiridis G., Ansari M., Graf E., Strom T.M. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019;10:963. doi: 10.1038/s41467-019-08831-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athwal H.K., Murphy G., 3rd, Tibbs E., Cornett A., Hill E., Yeoh K., Berenstein E., Hoffman M.P., Lombaert I.M.A. Sox10 regulates plasticity of epithelial progenitors toward secretory units of exocrine glands. Stem Cell Reports. 2019;12:366–380. doi: 10.1016/j.stemcr.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aure M.H., Konieczny S.F., Ovitt C.E. Salivary gland homeostasis is maintained through acinar cell self-duplication. Dev. Cell. 2015;33:231–237. doi: 10.1016/j.devcel.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aure M.H., Symonds J.M., Mays J.W., Hoffman M.P. Epithelial cell lineage and signaling in murine salivary glands. J. Dent. Res. 2019;98:1186–1194. doi: 10.1177/0022034519864592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach K., Pensa S., Grzelak M., Hadfield J., Adams D.J., Marioni J.C., Khaled W.T. Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing. Nat. Commun. 2017;8:2128. doi: 10.1038/s41467-017-02001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloh R.H., Tansey M.G., Lampe P.A., Fahrner T.J., Enomoto H., Simburger K.S., Leitner M.L., Araki T., Johnson E.M., Milbrandt J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRα3–RET receptor complex. Neuron. 1998;21:1291–1302. doi: 10.1016/s0896-6273(00)80649-2. [DOI] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes L.E., Wong D.M., Subramaniam M., Meyer N.P., Gilchrist C.L., Knox S.M., Tward A.D., Ye C.J., Sneddon J.B. Lineage dynamics of murine pancreatic development at single-cell resolution. Nat. Commun. 2018;9:3922. doi: 10.1038/s41467-018-06176-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter G.H., Khosravani N., Ekstrom J., Osailan S.M., Paterson K.P., Proctor G.B. Altered plasticity of the parasympathetic innervation in the recovering rat submandibular gland following extensive atrophy. Exp. Physiol. 2009;94:213–219. doi: 10.1113/expphysiol.2008.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Korfhagen T.R., Xu Y., Kitzmiller J., Wert S.E., Maeda Y., Gregorieff A., Clevers H., Whitsett J.A. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J. Clin. Invest. 2009;119:2914–2924. doi: 10.1172/JCI39731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibly A.M., Nguyen T., Limesand K.H. Palliative care for salivary gland dysfunction highlights the need for regenerative therapies: a review on radiation and salivary gland stem cells. J. Palliat. Care Med. 2014;4:1000180. doi: 10.4172/2165-7386.1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes A.N., Phipson B., Lawlor K.T., Dorison A., Patrick R., Zappia L., Harvey R.P., Oshlack A., Little M.H. Development; 2019. Correction: Single Cell Analysis of the Developing Mouse Kidney Provides Deeper Insight into Marker Gene Expression and Ligand-Receptor Crosstalk; p. 146. [DOI] [PubMed] [Google Scholar]
- Das B., Cash M.N., Hand A.R., Shivazad A., Culp D.J. Expression of Muc19/Smgc gene products during murine sublingual gland development: cytodifferentiation and maturation of salivary mucous cells. J. Histochem. Cytochem. 2009;57:383–396. doi: 10.1369/jhc.2008.952853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio E.N., Young C.C., Toma J., Levy M., Berger K.R., Johnson C.L., Mehmood R., Swan P., Chu A., Cregan S.P. Activating transcription factor 3 promotes loss of the acinar cell phenotype in response to cerulein-induced pancreatitis in mice. Mol. Biol. Cell. 2017;28:2347–2359. doi: 10.1091/mbc.E17-04-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira J.N.A., Zheng C., Lombaert I.M.A., Goldsmith C.M., Cotrim A.P., Symonds J.M., Patel V.N., Hoffman M.P. Neurturin gene therapy protects parasympathetic function to prevent irradiation-induced murine salivary gland hypofunction. Mol. Ther. Methods Clin. Dev. 2018;9:172–180. doi: 10.1016/j.omtm.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin K., Nelson C.M. Development; 2020. Branching Morphogenesis; p. 147. [DOI] [PubMed] [Google Scholar]
- Gresik E.W. The granular convoluted tubule (GCT) cell of rodent submandibular glands. Microsc. Res. Tech. 1994;27:1–24. doi: 10.1002/jemt.1070270102. [DOI] [PubMed] [Google Scholar]
- Habermann A.C., Gutierrez A.J., Bui L.T., Yahn S.L., Winters N.I., Calvi C.L., Peter L., Chung M.I., Taylor C.J., Jetter C. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020;6:eaba1972. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser B.R., Hoffman M.P. Regulatory mechanisms driving salivary gland organogenesis. Curr. Top Dev. Biol. 2015;115:111–130. doi: 10.1016/bs.ctdb.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedlund E., Deng Q. Single-cell RNA sequencing: technical advancements and biological applications. Mol. Aspects Med. 2018;59:36–46. doi: 10.1016/j.mam.2017.07.003. [DOI] [PubMed] [Google Scholar]
- Hess D.A., Strelau K.M., Karki A., Jiang M., Azevedo-Pouly A.C., Lee A.H., Deering T.G., Hoang C.Q., MacDonald R.J., Konieczny S.F. MIST1 links secretion and stress as both target and regulator of the UPR. Mol. Cell Biol. 2016;23:2931–2944. doi: 10.1128/MCB.00366-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst D., Gu X., Bhasin M., Yang Q., Verzi M., Lin D., Joseph M., Zhang X., Chen W., Li Y.P. Requirement of the epithelium-specific Ets transcription factor Spdef for mucous gland cell function in the gastric antrum. J. Biol. Chem. 2010;285:35047–35055. doi: 10.1074/jbc.M110.164541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh W.J., Esen E., Geahlen J.H., Bredemeyer A.J., Lee A.H., Shi G., Konieczny S.F., Glimcher L.H., Mills J.C. XBP1 controls maturation of gastric zymogenic cells by induction of MIST1 and expansion of the rough endoplasmic reticulum. Gastroenterology. 2010;139:2038–2049. doi: 10.1053/j.gastro.2010.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai Y., Wu Z., Love M.I., Ahn M.H., Bambah-Mukku D., Hua V., Farrell K., Dulac C. Multisensory logic of infant-directed aggression by males. Cell. 2018;175:1827–1841.e17. doi: 10.1016/j.cell.2018.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen S.B., Vissink A., Limesand K.H., Reyland M.E. Salivary gland hypofunction and xerostomia in head and neck radiation patients. J. Natl. Cancer Inst. Monogr. 2019;2019:lgz016. doi: 10.1093/jncimonographs/lgz016. [DOI] [PubMed] [Google Scholar]
- Katsumata O., Sato Y., Sakai Y., Yamashina S. Intercalated duct cells in the rat parotid gland may behave as tissue stem cells. Anat. Sci. Int. 2009;84:148–154. doi: 10.1007/s12565-009-0019-0. [DOI] [PubMed] [Google Scholar]
- Kivela J., Parkkila S., Parkkila A.K., Leinonen J., Rajaniemi H. Salivary carbonic anhydrase isoenzyme VI. J. Physiol. 1999;520 Pt 2:315–320. doi: 10.1111/j.1469-7793.1999.t01-1-00315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox S.M., Lombaert I.M., Haddox C.L., Abrams S.R., Cotrim A., Wilson A.J., Hoffman M.P. Parasympathetic stimulation improves epithelial organ regeneration. Nat. Commun. 2013;4:1494. doi: 10.1038/ncomms2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe Y., Shindo Y., Kawai T., Takahashi Y., Kobori M., Inoue H., Saito I. Sex-based differences in smgc expression in the submandibular gland of C57BL/6 mice. Pathobiology. 2016;83:287–294. doi: 10.1159/000446000. [DOI] [PubMed] [Google Scholar]
- Kwak M., Alston N., Ghazizadeh S. Identification of stem cells in the secretory complex of salivary glands. J. Dent. Res. 2016;95:776–783. doi: 10.1177/0022034516634664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak M., Ninche N., Klein S., Saur D., Ghazizadeh S. c-Kit(+) cells in adult salivary glands do not function as tissue stem cells. Sci. Rep. 2018;8:14193. doi: 10.1038/s41598-018-32557-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo H.G., Jin R.U., Sibbel G., Liu D., Karki A., Joens M.S., Madison B.B., Zhang B., Blanc V., Fitzpatrick J.A. A single transcription factor is sufficient to induce and maintain secretory cell architecture. Genes Dev. 2017;31:154–171. doi: 10.1101/gad.285684.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man Y.G., Ball W.D., Marchetti L., Hand A.R. Contributions of intercalated duct cells to the normal parenchyma of submandibular glands of adult rats. Anat. Rec. 2001;263:202–214. doi: 10.1002/ar.1098. [DOI] [PubMed] [Google Scholar]
- May A.J., Cruz-Pacheco N., Emmerson E., Gaylord E.A., Seidel K., Nathan S., Muench M.O., Klein O., Knox S.M. Development; 2018. Diverse Progenitor Cells Preserve Salivary Gland Ductal Architecture after Radiation Induced Damage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirels L., Hand A.R., Branin H.J. Expression of gross cystic disease fluid protein-15/Prolactin-inducible protein in rat salivary glands. J. Histochem. Cytochem. 1998;46:1061–1071. doi: 10.1177/002215549804600910. [DOI] [PubMed] [Google Scholar]
- Mukaibo T., Gao X., Yang N.Y., Oei M.S., Nakamoto T., Melvin J.E. Sexual dimorphisms in the transcriptomes of murine salivary glands. FEBS Open Bio. 2019;9:947–958. doi: 10.1002/2211-5463.12625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson D.A., Manhardt C., Kamath V., Sui Y., Santamaria-Pang A., Can A., Bello M., Corwin A., Dinn S.R., Lazare M. Quantitative single cell analysis of cell population dynamics during submandibular salivary gland development and differentiation. Biol. Open. 2013;2:439–447. doi: 10.1242/bio.20134309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M.Q., Le Pichon C.E., Ryba N. Stereotyped transcriptomic transformation of somatosensory neurons in response to injury. Elife. 2019;8:e49679. doi: 10.7554/eLife.49679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninche N., Kwak M., Ghazizadeh S. Development; 2020. Diverse Epithelial Cell Populations Contribute to the Regeneration of Secretory Units in Injured Salivary Glands; p. 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyelakin A., Song E.A.C., Min S., Bard J.E., Kann J.V., Horeth E., Smalley K., Kramer J.M., Sinha S., Romano R.A. Transcriptomic and single-cell analysis of the murine parotid gland. J. Dent. Res. 2019;98:1539–1547. doi: 10.1177/0022034519882355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V.N., Hoffman M.P. Salivary gland development: a template for regeneration. Semin. Cell Dev. Biol. 2014;25-26:52–60. doi: 10.1016/j.semcdb.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin C.L., Rukstalis J.M., Johnson C., Konieczny S.F. The bHLH transcription factor Mist1 is required to maintain exocrine pancreas cell organization and acinar cell identity. J. Cell Biol. 2001;155:519–530. doi: 10.1083/jcb.200105060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praktiknjo S.D., Obermayer B., Zhu Q., Fang L., Liu H., Quinn H., Stoeckius M., Kocks C., Birchmeier W., Rajewsky N. Tracing tumorigenesis in a solid tumor model at single-cell resolution. Nat. Commun. 2020;11:991. doi: 10.1038/s41467-020-14777-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadir M.M.F., Alvarez-Cubela S., Klein D., van Dijk J., Muniz-Anquela R., Moreno-Hernandez Y.B., Lanzoni G., Sadiq S., Navarro-Rubio B., Garcia M.T. Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proc. Natl. Acad. Sci. U. S. A. 2020;117:10876–10887. doi: 10.1073/pnas.1918314117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyfman P.A., Walter J.M., Joshi N., Anekalla K.R., McQuattie-Pimentel A.C., Chiu S., Fernandez R., Akbarpour M., Chen C.I., Ren Z. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saelens W., Cannoodt R., Todorov H., Saeys Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 2019;37:547–554. doi: 10.1038/s41587-019-0071-9. [DOI] [PubMed] [Google Scholar]
- Sekiguchi R., Martin D., Genomics and Computational Biology Core, Yamada K.M. Single-Cell RNA-seq identifies cell diversity in embryonic salivary glands. J. Dent. Res. 2020;99:69–78. doi: 10.1177/0022034519883888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E.C., Min S., Oyelakin A., Smalley K., Bard J.E., Liao L., Xu J., Romano R.A. Genetic and scRNA-seq analysis reveals distinct cell populations that contribute to salivary gland development and maintenance. Sci. Rep. 2018;8:14043. doi: 10.1038/s41598-018-32343-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902 e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznurkowska M.K., Hannezo E., Azzarelli R., Rulands S., Nestorowa S., Hindley C.J., Nichols J., Gottgens B., Huch M., Philpott A. Defining lineage potential and fate behavior of precursors during pancreas development. Dev. Cell. 2018;46:360–375.e5. doi: 10.1016/j.devcel.2018.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabula Muris Consortium. Overall coordination. Logistical coordination. Organ collection and processing. Library preparation and sequencing. Computational data analysis. Cell type annotation. Writing group. Supplemental text writing group. Principal investigators Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562:367–372. doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker A.S. Salivary gland development. Semin. Cell Dev. Biol. 2007;18:237–244. doi: 10.1016/j.semcdb.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Wagner D.E., Klein A.M. Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 2020;21:410–427. doi: 10.1038/s41576-020-0223-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng P.L., Aure M.H., Maruyama T., Ovitt C.E. Limited regeneration of adult salivary glands after severe injury involves cellular plasticity. Cell Rep. 2018;24:1464–1470.e3. doi: 10.1016/j.celrep.2018.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Hamey F.K., Plass M., Solana J., Dahlin J.S., Gottgens B., Rajewsky N., Simon L., Theis F.J. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019;20:59. doi: 10.1186/s13059-019-1663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao N., Lin Y., Cao H., Sirjani D., Giaccia A.J., Koong A.C., Kong C.S., Diehn M., Le Q.T. Neurotrophic factor GDNF promotes survival of salivary stem cells. J. Clin. Invest. 2014;124:3364–3377. doi: 10.1172/JCI74096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappia L., Phipson B., Oshlack A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. PLoS Comput. Biol. 2018;14:e1006245. doi: 10.1371/journal.pcbi.1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the single-cell RNAseq libraries reported in this paper is GEO: GSE150327. The code used for analysis is provided in Data S7 and also available through github: https://github.com/chiblyaa/Salivary-Gland-Development. Ready-to-use SEURAT objects are also available via figshare:
Embryonic SMG integrated dataset: https://doi.org/10.6084/m9.figshare.13157687.
Postnatal SMG integrated dataset: https://doi.org/10.6084/m9.figshare.13157726.