

Abstract
Heparin is a naturally occurring glycosaminoglycan from livestock, principally porcine intestine, and is clinically used as an anticoagulant drug. A limitation to heparin production is that it depends on a single animal species and potential problems have been associated with animal-derived heparin. The contamination crisis in 2008 led to a search for new animal sources and the investigation of non-animal sources of heparin. Over the past 5 years, new animal sources, chemical, and chemoenzymatic methods have been introduced to prepare heparin-based drugs. In this review, we describe advances in the preparation and synthesis of heparin and related products.
Keywords: heparin, LMWH, chemoenzymatic synthesis, synthetic heparin, bioengineered heparin
Graphical Abstract
Introduction
Heparin is a natural polysaccharide derived from animal tissues and has been widely used in clinics as an anticoagulant for over 80 years [1,2]. Similar to many other natural products, such as hormones or neurotransmitters, heparin was discovered accidentally [3]. The heparin story started around a century ago. By the end of the 19th century, an enzyme inhibitor, called antithrombin (AT), was suggested to exhibit anticoagulant activity [4]. Heparin, isolated from dog liver, was discovered by Jay McLean in 1916 [5]. By the late 1930s, heparin was shown to be an effective anticoagulant in the presence of a plasma component called ‘heparin-cofactor’ [6]. The presence of a relationship between heparin-cofactor and AT was understood during the 1950s and it was suggested that AT activity was catalyzed by heparin [7,8]. Pure AT was isolated for the first time in 1968 by Abildgaard [9] and, finally, during the early 1980s following extensive research, a unique pentasaccharide, corresponding to the AT-binding site in heparin, was characterized [10,11].
Heparin, the most negatively charged biological molecule, is a highly sulfated member of the glycosaminoglycan (GAG) family. The GAG family comprises heparin, heparan sulfate (HS), chondroitin sulfate (CS), dermatan sulfate (DS), and keratan sulfate (KS) [12]. Heparin and HS comprise 1,4-glycosidically linked D-glucosamine (GlcN) and uronic acid (UA) residues. The UA residue is either α-L-iduronic acid (IdoA) or β-D-glucuronic acid (GlcA) and these residues can be sulfated at their 2-position (i.e., Ido2S and GlcA2S). The β-D-glucosamine moiety can be substituted with an N-sulfo (GlcNS) or an N-acetyl (GlcNAc) group. These glucosamine residues can also be O-sulfated at the 6-position (i.e., GlcNAc6S and GlcNS6S). In addition, these glucosamine residues can be 3-O-sulfated (GlcNS3S, GlcNS3S6S, GlcNAc3S, and GlcNAc3S6S). HS is considerably less sulfated than heparin and has lower anticoagulant activity [13].
The heparin polysaccharide (Figure 1) comprises disaccharide repeating units, including a major repeating unit that is trisulfated (TriS), →4) α-L-IdoA2S (1→4) α-D-GlcNS6S (1→). The polydispersity and structural variations of its chains make heparin a heterogeneous and negatively charged polysaccharide [14]. This heterogeneity gives heparin the possibility of interacting with many different proteins, resulting in a variety of biological activities. Unfractionated heparin (UFH), obtained from tissue, is a large polysaccharide with a molecular weight (MW) ~20 kDa and can contain an uncommon pentasaccharide sequence motif, GlcNAc/NS6S → GlcA → GlcNS3S6S → IdoA2S → GlcNS6S, which is responsible for the specific binding of these chains to the serine protease inhibitor, AT, resulting in the inhibition of factor IIa (thrombin) and factor Xa (FXa). The sulfo group at the C3 position of the central glucosamine residue is crucial for the interaction with AT [15]. Such pentasaccharide sequences are randomly distributed throughout heparin chains and only a fraction of these UFH chains contain these pentasaccharide sequence motifs.
Figure 1.
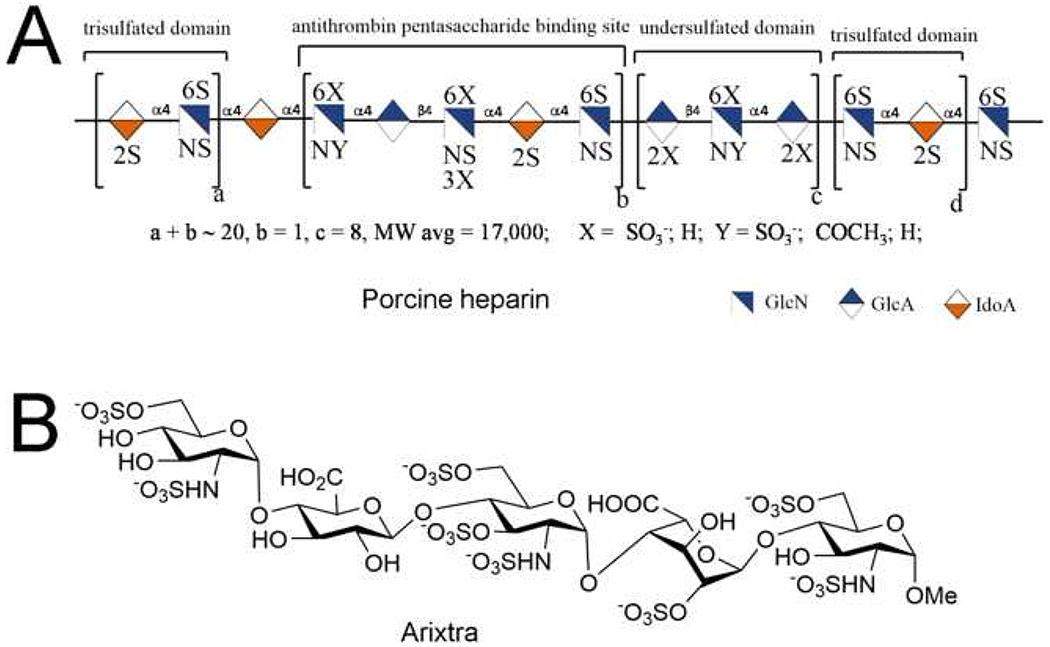
Structure of heparin derived from porcine intestine and chemically synthesized ultralow-molecular-weight heparin (ULMWH), Arixtra®. (a) The generalized symbolic structure of a typical chain present in porcine intestinal UFH. (b) The chemical structure of Arixtra®, a synthetic ULMWH containing an AT-binding site. Abbreviation: MW, molecular weight.
Heparin is a well-known, essential anticoagulant drug used in extracorporeal therapy and in surgery as well as in the prevention and treatment of deep venous thrombosis (DVT) and other coagulation abnormalities, such as pulmonary embolism (PE). Heparin interacts with a large number of heparin-binding proteins and receptors, selectively binding to these proteins and receptors and regulating their functions. As a result of these heparin-biomacromolecule interactions, heparin presents numerous additional biological and pharmacological activities, such as antilipidemic, tumor growth inhibition, regulating angiogenesis, and antimicrobial, antiparasitic, and antiviral activities [2,16–20].
Heparin biosynthesis
Heparin and HS proteoglycans (PGs) are biosynthesized in the endoplasmic reticulum (ER) and Golgi by a common pathway in a dynamic and complex process that requires the concerted action of as many as 22 enzymes [21–25]. Biosynthesis of the heparin PG core protein, serglycin, occurs in the ER [26]. The initial steps in the synthesis of a HS/heparin GAG chain is the formation of the linkage tetrasaccharide (xylose-galactose-galactose-glucuronic acid, with glucuronic acid at the nonreducing end and xylose at the reducing) (Figure 2). The construction of this linker is initiated by the coupling of a xylose to the serine through the action of xylosyltransferase (XylT)-1 or -2, followed by the sequential addition of two galactose by galactosyltransferase (GalT)-1 and -2 and GlcA by a glucuronosyltransferase [27]. Once the construction of the linker on the core protein is completed, the addition of monosaccharides to its nonreducing end is accomplished by three isoforms of the EXT glycosyltransferase family.
Figure 2.
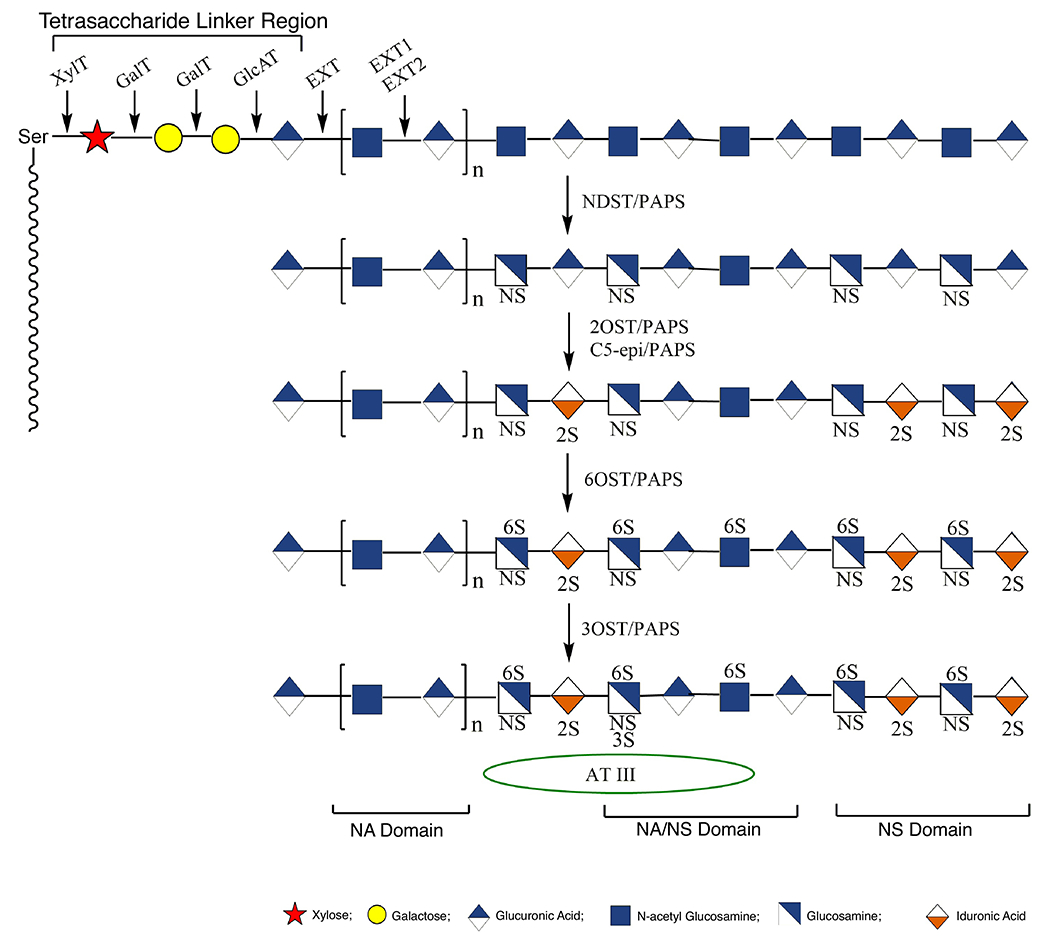
Biosynthesis of heparin/heparin sulfate (HS) occurring in the endoplasmic reticulum (ER) and the Golgi.
The polymerization of polysaccharide chains occurs by adding a GlcA followed by a GlcNAc residue to the chain by EXT1 and EXT2, respectively [28]. The GAG backbone is further modified, because chain elongation takes place through the action of additional Golgi enzymes. First, the N-acetyl groups are removed and replaced with N-sulfo groups by one of the four N-deacetylase/N-sulfotransferase (NDST) isoforms. NDST-2 has been shown to have specificity for highly modifying the GAG chains on the serglycin core protein, making it an essential enzyme for the synthesis of mast cell heparin [29]. Further modification of the HS/heparin chains, after this sulfation step, occurs in close proximity to these regions, leading to highly sulfated domains [27]. Once the N-sulfate modification has occurred, uronosyl C5-epimerase (C5-epi) epimerizes most of the GlcA residues to IdoA residues, followed by the conversion of most IdoA and a small number of the GlcA residues to their 2-O-sulfo forms. The C5-epi and 2-O-sulfotransferase enzymes both have only a single isoform and co-localize in the Golgi [30,31]. Next, the C6 hydroxyl group of either GlcNAc or GlcNS is 6-O-sulfated by one of three isoforms of the 6-O-sulfotransferase enzymes (6-OST-1,-2,-3) giving rise to the highly sulfated TriS domains found frequently in heparin but rarely in HS. Finally, one of the seven isoforms of 3-O-sulfotransferase (3-OST-1) acts to introduce a sulfate on C3-hydroxyl group of GlcNAc and GlcNS residues [31].
UFH, LMWH and ULMWH
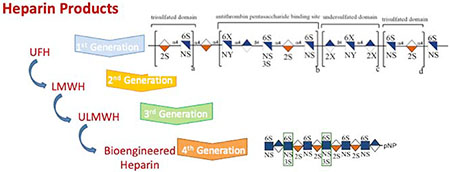
There have been four generations of heparin products. The first-generation heparin, UFH, is extracted from porcine intestinal mucosal tissue or bovine lung or intestine [average MW (MWavg) ~19 000 Da]. The second-generation heparin product, low-molecular-weight heparin (LMWH), is prepared through the controlled chemical or enzymatic depolymerization of UFH and has a MWavg ~3500–6000 Da. The third generation of heparin products, ultra (U)LMWH, principally includes chemically synthesized products [29]. The fourth generation of heparin products are chemoenzymatically and bioengineered heparins [32]. Currently, select members of the first three generations of heparin products are marketed and have been approved by the US Food and Drug Administration (FDA). These include UFH prepared from porcine intestine, several LMWHs (all derived from porcine UFH) and the ULMWH Arixtra® (fondaparinux) (Figure 1b) prepared through chemical synthesis (MW 1508.3) [33].
UFH
Commercially available UFH is isolated from animal tissues. Purification of heparin from animal tissues, which is the only source for the industrial commercial production of UFH and LMWHs, is an old process. Commercial production methods of pharmaceutical-grade heparins are strictly protected as nonpublically available industrial secrets. The heparin manufacturing processes aim to maximize the yield of the highly charged heparin chains present in a starting material containing other less highly sulfated GAGs, such as HS and DS, without resulting in chain degradation caused by the applied process conditions. Typical industrial processes can be divided into five steps [34]: (i) collection and stabilization starting material; (ii) digestion and release of heparin from proteoglycans; (iii) capture and recovery of the heparin; (iv) purification and bleaching; and (v) isolation and drying.
The procedure begins with the immersion of the cleaned intestine in a salt solution (brine), and then the mucosa is scraped from the intestines. Mucosa or whole porcine intestines, called ‘hashed porcine guts’, can be used for heparin production. Hashed guts and mucosa are preserved with 0.5–3.5% sodium metabisulfite or another suitable oxygen scavenger to prevent spoilage and oxidation. Digestion steps aim to liberate the heparin from the mast cells and requires enzymatic treatment (i.e., proteases) or chemical treatment (i.e., acidic or basic conditions at high temperatures). The capture step aims to distinguish heparin from other biopolymers (i.e., DNA, proteins/peptides, CS, DS, and HS) using soluble quaternary ammonium cations or anion exchange resins. At this stage, crude heparin is obtained but still contains impurities, such as nucleic acids, other GAGs, and a variety of pathogens. Precipitation using organic solvents, such as methanol, ethanol, propanol, or acetone, recovers and concentrates the crude heparin and bleaching using oxidation reagents removes endotoxins, and is followed by isolation and drying [34].
Animal-sourced heparins are impacted by many factors associated with animal husbandry. Heparin biosynthesis results in a variety of chain lengths and modification patterns, such as sequences with different patterns of sulfation and C5-UA epimers [35]. After animal tissue extraction, porcine intestinal mucosa contains other GAGs, including HS, that have a different charge state from that of heparin. The mixture of GAGs is passed through an anion-exchange resin to isolate heparin by separating GAGs based on their charge density. Even the most advanced purification methods currently used are not capable of making pure heparin, without the presence of other GAGs. In 2007–2008, the adulteration of crude porcine intestinal heparin with a toxic semisynthetic oversulfated chondroitin sulfate (OSCS) resulted in a heparin contamination crisis causing more than 100 deaths in the USA and disrupting the heparin supply chain [36]. Bioactive substances, such as viruses, prions, and heparin-binding growth factors, can be found in animal extracts, also representing potential concerns associated with animal-derived heparin. Given that commercial heparin is a highly complex, very heterogeneous mixture of polysaccharide chains, it is often not possible to completely purify UFH. Moreover, because LMWH and ULMWH are often prepared from animal-sourced UFH, there has been a movement to develop heparin products coming from non-animal sources.
UFH was once commonly used in the prevention and treatment of DVT and PE. It is used safely for the treatment of patients with renal failure and, if necessary, its effects can be neutralized using protamine [37]. Although it was effective for this purpose, it has pharmacokinetic, biophysical, and biological limitations. Given that UFH is highly charged and has a relatively high MW, it cannot pass through membranes, and is administered parenterally, primarily intravenously (i.v.). UFH is still used in kidney dialysis and in heart-lung machines. Heparin is a preferred anticoagulant because it is inexpensive and often more reliable than oral anticoagulants. Heparin has other advantages in that it does not pass through the placenta and has very short onset of action [38]. In addition to its anticoagulant activity, heparin also exhibits other pharmacological properties, such as antilipidemic actions, tumor growth inhibition, regulating angiogenesis, and antimicrobial, antiparasitic, anti-inflammatory and antiviral activities [2,16–20,39].
LMWH
LMWH is obtained as a result of the controlled depolymerization of larger UFH chains by chemical or enzymatic techniques. LMWH comprises 12–22 monosaccharide units (MWavg ~5000 Da). Similar to UFH, LMWH inactivates FXa in a dose-dependent fashion. Given their longer chain length, UFH chains do not discriminate between the inhibition of thrombin and factor Xa. Moreover, UFH binds to a greater variety of heparin-binding proteins than does LMWH. Studies based on the modeling of the ternary heparin/antithrombin/thrombin complex and crystallography studies suggested that the thrombin-binding site is required at the nonreducing end of AT-binding site for heparin to inhibit thrombin [40,41]. Thrombin inhibition is heparin chain size dependent, and a dodecasaccharide sequence linked to the pentasaccharide sequence represents the shortest chain length required for the ternary heparin/antithrombin/thrombin complex [42]. LMWHs have little effect on thrombin, because most LMWH do not contain sufficient saccharide units for the formation of a ternary complex. Whereas the anti-FXa/anti-FIIa activity ratio for UFH is 1, this ratio varies between 2 and 5 for LMWHs.
LMWH differs from UFH in MW, plasma clearance, tissue factor pathway inhibitor (TFPI) release, and bioavailability. The elimination half-life of LMWH is 3–6 h after subcutaneous (s.c.) administration and, unlike UFH, is dose independent. Therefore, LMWHs can be administered in an appropriate dose by weight without requiring laboratory monitoring. There are several advantages of LMWHs to UFH: (i) they exhibit high s.c. bioavailability; (ii) they have a longer half-life and less binding to plasma proteins, endothelial cells, and macrophages; (iii) they can be used as one or two doses daily; (iv) they generally show a continuous antithrombotic effect without requiring laboratory monitoring; (v) they bind less to platelet factor 4 (PF4) reducing heparin-induced immune thrombocytopenia (HIT); and (vi) they show less osteoclast activation and a lower frequency of osteopenia.
LMWH is as effective as UFH in the prevention and treatment of venous thromboembolism and is also used in stroke and unstable angina. In anticoagulant therapy after mechanical heart valve replacement and in mechanical valve cases where oral anticoagulant is contraindicated, LMWH is as effective as UFH. LMWH can only be used at low doses in patients with renal failure. The indications of LMWHs have been expanded over time to include thromboprophylaxis in high-risk abdominal surgery and in medical patients, treatment of DVT, PE, acute coronary syndromes (ACS), and superficial vein thrombophlebitis [43]. The anticoagulant effect of LMWH cannot be completely neutralized with protamine. It has been shown that homogeneous LMWHs must contain at least 12-mer oligosaccharide chains to be fully neutralized of their anticoagulant effects by protamine [44].
ULMWH
ULMWHs are synthetic, s.c. bioavailable products, corresponding to five to ten saccharide units. Given their expense and limited clinical applications, ULMWH still represents a small percentage of clinically used LMWH and UFH [45]. ULMWHs show greatly reduced risks of HIT because of their reduced chain length. The only clinically approved ULMWH, fondaparinux (Arixtra®) (Figure 1b), introduced in 2003, is a pentasaccharide containing the AT-binding domain of heparin and a selective inhibitor of FXa [46]. After a daily dose, peak levels are reached in 2 h, showing strong binding to AT and weak binding to other plasma proteins. Fondaparinux has a prolonged half-life and duration of action and shown a better biosafety profile compared with LMWH. The non-animal origin of fondaparinux reduces the risk of impurities increasing its clinical use. After successful clinical development, fondaparinux became the first synthetic heparin product for thromboprophylaxis in patients undergoing orthopedic surgery [47]. However, the high cost of treatment using fondaparinux results from the high-cost and time-consuming manufacturing process, limiting its availability. Moreover, there are several clinical applications (i.e., in kidney dialysis and heart-lung machines) where fondaparinux cannot be substituted for UFH.
Chemical synthesis and depolymerization of heparin/HS and analogs
Chemical synthesis of ULMWH
The chemical synthesis of oligosaccharides is challenging because it requires repetitive steps of protection, activation, coupling, and deprotection reactions. Moreover, intensive purification is required between these steps to remove undesirable isomers, side products, and excess reagents [48]. For example, fondaparinux synthesis originally required ~60 chemical steps in a reported overall yield of 0.1% [46]. Over the past three decades, several synthetic groups have synthesized fondaparinux dealing with the challenges of this multistep chemical synthesis, including glycosylation reactions with low yield and low stereoselectivity [49–55]. Zhao et al. reported a preactivation-based, one-pot glycosylation method with high stereoselectivity, simplified purification, and improved the synthetic efficiency [56]. Recently, Wong and coworkers developed a programmable one-pot synthesis of fondaparinux using designed thioglycoside building blocks with well-defined relative reactivity values (RRVs) for α-selective glycosylation (Figure 3). The careful selection of orthogonal protecting groups reduced the number of synthetic steps and eliminated multiple purification steps. This total synthesis of fondaparinux resulted in a 4.2% overall yield [57].
Figure 3.
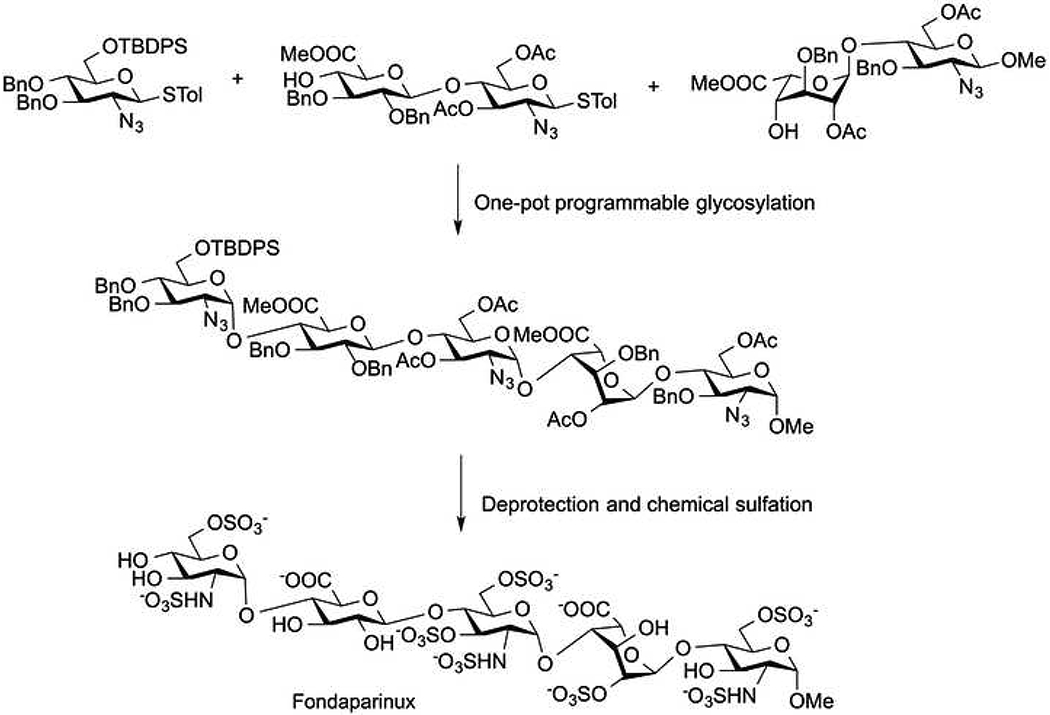
Programmable one-pot synthesis of fondaparinux.
Chemical and enzymatic depolymerization of UFH to prepare LMWHs
The preparation of LMWHs is based on three different reaction mechanisms for the depolymerization of UFH. Heparin is not stable under oxidative conditions and, thus, its depolymerization can rely on oxygen-containing oxidative reagents, such as hydrogen peroxide with or without metal ions, or by treatment radiation (i.e., ionizing γ-radiation or light). Ardeparin and parnaparin are approved and/or marketed drugs prepared using oxidative depolymerization. A two-step deaminative degradation of UFH is another means of oxidative depolymerization. In deaminative depolymerization, UFH is treated with a nitrosating reagent, such as nitrous acid or isoamilnitrite, followed by the use of a reducing reagent, such as sodium borohydride. This method affords an anhydromannitol residue at the reducing end of each newly formed chain. Dalteparin and nadroparin are examples of products synthesized using this deaminative LMWH process.
β-Elimination can rely on both chemical and enzymatic methods. Chemical β-elimination first involves the formation of the quaternary ammonium or benzethonium salt of UFH. The resulting organic solvent soluble salt is then esterified with benzyl chloride to afford the heparin benzyl ester. Treatment with base results in β-elimination at the benzyl ester of iduronic acid residues (with or without 2-O-sulfo groups). Proper control of these process conditions affords a clinically approved LMWH, enoxaparin. The relatively harsh conditions of the chemical β-elimination reaction results in process artifacts in many of the product chains, including a 1,6-anhydro residue at the reducing end of the chain. Enzymatic β-eliminative depolymerization relying on heparin lyase isolated from Flavobacterium heparinum has also been used to make LMWHs, including tinzaparin, under milder conditions.
Heparin lyase I (heparinase I, HepI) is specific for heparin and acts at →4)-α-D-GlcNS6S(1→4)-α-L-IdoA2S(1→, but is also highly selective for →4)-α-D-GlcNS3S6S(1→4)-α-L-IdoA2S(1→, a linkage present in the AT-binding site. Heparin II (heparinase II, HepII) is less selective, cutting at many linkages in heparin and HS. The third member of this family, heparin lyase III (heparinase III, HepIII) cleaves primarily next to GlcA in HS. Bohlmann and coworkers reported new bacterially sourced β-endoglucuronidase (a hydrolase), heparanase Bp in 2015, from the pathogenic bacteria Burkholderia pseudomallei [58]. Heparanase Bp has been prepared as a recombinant Escherichia coli-expressed enzyme with high purity. Heparanase Bp acts endolytically on both HS and low sulfated domains in heparin and is unable to cleave either at IdoA or at GlcA residues in high sulfated domains. Heparanase Bp, or a related hydrolase, might represent a new reagent for the preparation of LMWH [59].
Recently published methods to prepare LMWHs
LMWH can be prepared either using chemical or enzymatic methods, but these can be costly in terms of the use of enzymatic reactions or lead to side reactions in the case of chemical methods, resulting in low efficiency [60]. Thus, there has been a need to develop new methods that are easy to apply, safe, inexpensive, and efficient for the preparation of LMWH.
Photodepolymerization of UFH relies on the use of a catalyst, such as titanium oxide, to produce hydroxyl radicals (•OH). This approach affords excellent yields of LMWH and titanium oxide, an inexpensive and relatively nontoxic catalyst that can be easily removed by filtration [61]. Liquid chromatography-mass spectrometry (LC-MS) and nuclear magnetic resonance (NMR) studies confirmed that heparin can be depolymerized by photolysis through mechanisms of random scission of glycosidic linkages. This photochemical reaction produces LMWHs without damaging the main core structure and leads to a decrease in the amount of sulfo groups (Figure 4).
Figure 4.
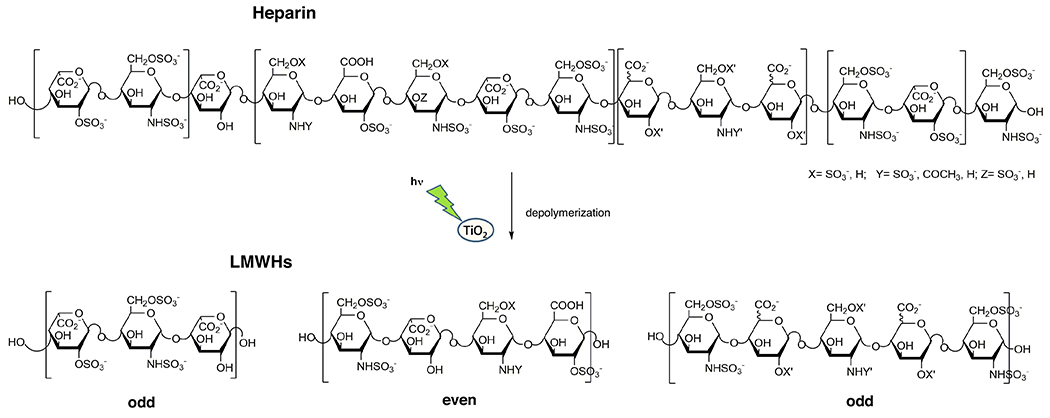
Photochemical depolymerization of heparin. Photochemical reaction with >370 nm light in presence of TiO2 in water produces low-molecular-weight heparin (LMWH) containing a complex mixture of both even and odd numbered chains. This figure contains a few possible example of these chains.
Ultrasound is an effective and environmentally friendly technique that has been used to depolymerize a variety of polysaccharides, including dextran, starch, chitosan, hydroxyl propyl methyl cellulose, carboxyl methyl cellulose, high methoxyl pectin, Guar gum, and carrageenan gum [62,63]. Hydroxyl radicals are an important reactive oxygen species that can rapidly react with many organic compounds, attacking groups in these molecules, disrupting their structures, and degrading the resulting compounds. Ultrasound produces an acoustic cavitation that can form hydroxyl radicals, which are able to break glycosidic bond in polysaccharides [64]. Ultrasound power can be combined with chemicals or enzymes to more efficiently produce •OH radicals, enhancing their reactive potential. A hydrogen peroxide (H2O2)-catalyzed radical hydrolysis reaction assisted by an ultrasonic wave has been widely used to depolymerize polysaccharides [65,66].
Combining physical ultrasonic treatment with a chemical Fenton reaction, called sono-Fenton, has been used for the preparation of LMWHs [67]. The Fenton system is a combination of ferrous ion-hydrogen peroxide that increases the production of •OH radicals. Uronic acid residues in heparin are the most susceptible region attacked by •OH radicals formed in sono-Fenton processes. Additionally, results of both activated partial thromboplastin time (APTT) and thrombin time (TT) assays of newly prepared LMWHs by sono-Fenton process showed that the APTT of LMWHs is higher than that of UFH, and anticoagulant activity evaluations suggested that LMWHs exhibited their activity mainly through the intrinsic coagulation pathway. Moreover, the anti-FXa and anti-FIIa activity results of one of the prepared LMWH were 118 IU/mg and 45 IU/mg, respectively, with the anti-FXa/anti-FIIa ratio of 2.6 comparable to that of commercial nadroparin. Thus, the sono-Fenton system might be useful for the rapid preparation of LMWHs that have relatively high anticoagulant activity, reduced adverse effects, and a preserved AT pentasaccharide binding sequence. H2O2-catalyzed free radical reactions can also be applied in the presence of copper (II) to increase depolymerization of the polysaccharide [68]. However, the difficulty of controlling H2O2 titration rate and pH changes decreases the reaction efficiency. Removal of the metal catalyst at the end of the reaction is also an important challenge. The use of ascorbic acid in place of metal catalyst can be used to overcome these disadvantages.
LMWHs were successfully prepared by physicochemical depolymerization of heparin using a H2O2/ascorbic acid free radical reaction combined with ultrasonic waves [69]. By optimizing reaction conditions, including the concentration of ascorbic acid, reaction temperature, and intensity of ultrasonic power, LMWHs were obtained with good yields. More importantly, structural analysis of these LMWHs showed that the products maintained the major structure of (1→4)-linked glucosamine and iduronic acid, suggesting that the primary structure and the sulfate esters were preserved after the depolymerization. Moreover, this environmentally friendly and mild process could be applied to the large-scale production of LMWHs, which will be required for industrial application. Cytotoxicity of such LMWHs can be quickly evaluated in vitro using A549 cell lines with the MTT method.
Nonporcine-sourced LMWHs
First-generation heparins are of animal origin. Primarily pigs, along with cows and sheep, have been using for the manufacture of UFH. Until the mid-1990s in the USA, UFH was often prepared from bovine tissues for clinic use and was approved by the FDA. Bovine heparin products were voluntarily withdrawn from the market as a result of the prevalence of mad cow disease (bovine spongiform encephalopathy, BSE) in the UK during the late 1980s. Since the mid-1990s, porcine intestinal heparin has been the only approved UFH in the USA and Europe [2]. Currently, >50% of heparin used worldwide is produced in China. Although there are promising new pharmaceutical heparins in development, currently, with the exception of synthetic fondaparinux, only the porcine-derived UFH (made in China) and LMWH (made from UFH) are produced on a large scale in an economically viable manner. Given its importance and indispensable features, the production of most heparin productions from a single country and a single source endangers the global heparin supply. The heparin supply chain was severely disrupted in 2007–2008 because of the contamination crisis caused by the adulteration of crude porcine intestinal heparin with a toxic semisynthetic oversulfated chondroitin sulfate (OSCS) [70]. The African swine fever (ASF) epidemic is a current problem in Europe and Asia and threatens swine herds in these regions and, thus, the world could face a global heparin shortage. Given the absence of a cure or an effective vaccine against it, the rapid spread of ASF decimated China’s swine herd in 2019. It is estimated that pig production in China will decrease by 33% in 2020, compared with 2018 [71]. All these developments have led regulatory agencies and researchers to consider alternative sources for heparins, such as the development of bioengineered heparins, synthetic heparins, or the re-introduction of bovine heparin onto the pharmaceutical market.
Studies of the structure and activity of bovine, ovine, and porcine-sourced heparins showed that ovine and porcine heparins exhibit a similar structural and activity profile. Bovine heparins do exhibit some different structures and a lower specific activity (units/mg) compared with porcine intestinal heparins [72,73]. The similarities and differences between porcine heparin and bovine intestinal and bovine lung heparins are well studied [74]. The MW of bovine intestinal heparin is slightly lower than that of porcine heparin; it is also more polydispersed, less highly sulfated, and more heterogeneous. Bovine intestinal heparin also has a lower content of GlcNS3S6S residues and higher content of GlcA residues compared with porcine intestinal heparin. Bovine lung heparin also has a lower MW than bovine intestinal heparin. However, unlike bovine intestinal heparin, bovine lung heparin is more highly sulfated compared with porcine intestinal heparin [75]. There is also a difference in the biological activity of bovine-sourced and porcine-sourced heparins. Porcine intestinal heparin have significantly higher activity than bovine-sourced heparins [74,76]. The latter also need to be used in significantly higher doses to achieve the same antithrombotic effect as porcine heparin. Higher doses of protamine as antidote are also required to neutralize the anticoagulant effect of bovine intestinal heparin [77].
The preparation of the dalteparin LMWH using an animal source other than porcine intestine was recently successfully carried out [78]. Dalteparin prepared from bovine UFH was compared with dalteparin prepared from porcine UFH using an integrated analytical approach. Bovine lung heparin and ovine intestinal heparin were deaminatively depolymerized with nitrous acid followed by reduction of sodium borohydride to obtain LMWHs. The resulting dalteparins were recovered by methanol precipitation for analysis.
The safety and efficacy of LMWHs are closely related to the composition and the sequence of their oligosaccharide chains. Depolymerization reaction conditions, especially nitrous acid concentration and reaction time, need to be optimized to control the chain length and distribution of the newly formed oligosaccharide products. The alcohol concentration used to precipitate the LMWH can also impact the size of the oligosaccharides recovered (or discarded). More diluted concentrations of both nitrous acid and methanol, used in the precipitation of bovine-derived LMWH, are required than for porcine-derived LMWH. Careful control of these conditions can lead to bovine and ovine LMWHs that meet European Pharmacopeia (EP) and USP specifications (i.e., acceptable MW values >5600 Da; appropriate percentages of chains with MW <3000 Da and also MW >8000 Da). Extensive 1D and 2D-NMR spectroscopy showed that the percentage of GlcA in starting bovine lung UFH is lower than that of porcine and ovine counterparts. When dalteparin analogs prepared from these porcine, bovine, and ovine UFHs were examined, it was determined that the percentage of GlcNAc and GlcA residues was lowest in the bovine lung LMWH compared with porcine and ovine LMWHs. Hydrophilic interaction LC-electrospray ionization-MS (HILIC-ESI–MS) facilitates intact chain analysis of LMWHs and clearly demonstrated that bovine lung LMWH oligosaccharides were more highly sulfated and contained a significantly lower percentage of GlcNAc residues than the corresponding oligosaccharide components of porcine and ovine LMWHs, which confirmed the NMR analysis results. The average anti-FIIa and anti-FXa activities of bovine lung LMWHs were at the lower levels of EP dalteparin monograph specifications. By contrast, ovine intestinal LMWH was more similar to porcine LMWH with regard to both structure and bioactivity [78].
Bovine lung-derived LMWHs have also been prepared by benzylation and alkaline depolymerization as an enoxaparin analog [79]. In the preparation of a USP enoxaparin, an alkaline depolymerization reaction often uses 4 M NaOH to breakdown heparin benzyl esters at 55°C in 2 h. USP enoxaparin has an average MW of 4500 Da with a range of 3800–5000 Da. Approximately 20% (15–25%) of the enoxaparin chains contain a 1,6-anhydro derivative at their reducing ends. Given that the MW of bovine lung heparin is significantly lower than that of porcine intestine heparin, milder reaction conditions are required for alkaline depolymerization. A concentration of 3.5 M NaOH and a reaction temperature of 50°C are required to prepare LMWH from bovine intestinal UFH. However, the reaction time needs to be adjusted to 6 h to obtain 1,6-anhydro derivatives within the ranges described in the USP enoxaparin monograph.
Other structural and biological comparisons between porcine intestines and bovine lung LMWHs have shown that, in bovine LMWHs, oligosaccharide components have slightly larger MWs, contain significantly less 1,6-anhydro mannopyranose, contain fewer AT-binding sites, and exhibit lower anti-FIIa and anti-FXa activities. Similar results have been reported for LMWHs prepared from bovine lung and bovine intestinal heparin compared with commercial enoxaparin [80]. Bovine lung enoxaparin met USP specifications for anti-FXa but not for anti-FIIa activities. By contrast, bovine intestinal LMWHs exhibited comparable anti-FXa and anti-FIIa activities. A wide variation in anti-FIIa activity was identified among LMWH prepared from bovine intestine, but anti-FXa/anti-FIIa ratios were within USP specifications. Although the potency of both bovine-sourced heparins are lower than porcine mucosal heparin, studies suggest that LMWHs derived from these sources by chemical β-elimination is biosimilar to its porcine counterparts [81].
Bioengineering UFH, LMWH, and ULMWH
Chemoenzymatic synthesis, which mimics the biosynthetic pathway of heparin, is a promising strategy, combining chemical and enzymatic methods, although chemoenzymatic synthesis overcomes many of the challenges encountered in chemical synthesis by benefiting from the advantages of enzymatic synthesis. The utility of enzymes as catalysts in carbohydrate synthesis provides high stereoselectivity and regioselectivity without the need for protecting group manipulations in glycosylation reactions [82,83]. Using a variety of heparin/HS biosynthetic enzymes under mild, environmentally friendly conditions, chemoenzymatic synthesis is an efficient method for preparing structurally heterogeneous heparin polysaccharides and structurally homogeneous LMWH and ULMWH oligosaccharides [44,84,85].
Bioengineered heparosan
Heparosan, an unsulfated GAG comprising a linear copolymer of repeating units α-1,4 linked GlcNAc-GlcUA, is the precursor of heparin and HS [86] and is biosynthesized by bacteria such as E. coli K5 and Pasteurella multocida capsular polysaccharides. Research has focused on the metabolic engineering of E. coli K5 to produce LMW heparosan for the chemoenzymatic synthesis of LMWH. Deletion of the α-1,2-glucosyltransferase encoding waaR in E. coli increased the productivity of LMW heparosan. These results suggest that it is possible to produce a LMW heparosan from engineered E. coli [87].
Environmentally sustainable microbial platforms can serve as an alternative metabolic engineering cyanobacteria platform for the photoautotrophic production of heparosan from CO2 through the introduction of P. multocida heparosan synthase 2 (PmHS2) [81]. Bacillus megaterium has been metabolically engineered to produce heparosan using a T7 RNA polymerase (T7 RNAP) expression system, which has been co-opted for the control of PmHS2. These heparosan products displayed a different range of MW products compared with traditional E. coli K5 products, diversifying its potential applications and facilitating increased product utility [88]. In another study, the heterologous expression of E. coli K5 kfiC and N-acetylglucosaminyltransferase (kfiA) glycosyltransferase genes enhanced heparosan production. NMR analysis confirmed that the chemical structure of B. megaterium-derived heparosan was identical to E. coli K5 heparosan and its MW was in range of ~31–60 kDa, confirming its potential as a precursor for heparin synthesis. The engineered B. megaterium yielded a maximum LMW heparosan concentration of 394 mg/l in a batch bioreactor and of 1.32 g/l in fed-batch fermentation. These studies provide efficient processes to produce heparosan from nonpathogenic B. megaterium [88,89].
Chemoenzymatic synthesis of heparins by modification of heparosan
Chemoenzymatic semisynthesis of heparin or HS usually begins with heparosan prepared from the K5 strain of E. coli. The biosynthesis pathway of heparin can be reproduced through the in vitro modification of heparosan with recombinant enzymes [90]. These recombinant biosynthetic enzymes can usually be actively expressed and produced on a large scale and have been successfully used as biocatalysts in heparin and HS synthesis [91,92].
The most problematic enzyme in the preparation of bioengineered heparin is N-deacetylase/N-sulfotransferases (NDST). Although four human NDST isoforms that have been distributed in various tissues and cells have been identified to date, only the use of NDST-1 and −2 isoforms has been demonstrated for the enzymatic synthesis of heparin [93]. The N-sulfotransferase (NST) domain of NDST can be effectively expressed in E. coli with a lack of the N-deacetylase activity. NDST-2 in a baculovirus expression and rat NDST-1 in Saccharomyces cerevisiae have been successfully expressed and their enzymatic catalysis of the N-deacetylation/N-sulfation reaction of heparosan and HS were reported on a microgram scale [94,95]. The use of recombinant NDSTs as biocatalysts is limited because they can only be expressed at low levels in yeast or insect cells [95,96].
Instead of using NDSTs, chemical N-deacetylation and N-sulfation reactions are often incorporated into chemoenzymatic heparin synthesis. Heparosan is first N-deacetylated using NaOH, followed by treatment with trimethylamine-sulfur trioxide for the N-sulfation step to prepare the intermediate N-sulfoheparosan [97]. The ratio of N-sulfation to N-acetylation and MW of the heparin derivatives obtained can be controlled by the optimization of the chemical reaction conditions [97,98].
Another key step in heparin/HS biosynthesis is the epimerization of GlcA into IdoA. D-glucuronyl C-5 epimerase (C5-epi) catalyzes the conversion of both a GlcA to an IdoA residue and IdoA to a GlcA residue. That two-way conversion produces a polysaccharide that contains both GlcA and IdoA residues. A recent study showed that C5-epi catalyzes the reversible and irreversible conversion of GlcA to IdoA, depending on the context of the site at which C5-epi acts (Figure 5) [99]. This reversibility depends on the positioning of the N-acetylation and O-sulfation in a heparin oligosaccharide. The presence of an adjacent GlcNS at epimerization site (−1 position) allows C5-epi to bind to the substrate for both reversible and irreversible C5-epimerization, whereas a GlcNAc at −1 position prevents the binding of the enzyme and C5 epimerization cannot occur. A GlcNS, a GlcN, or no sugar residue at −3 of the epimerization site results in reversible epimerization. However, GlcNAc at the −3 position prevents conversion of newly formed IdoA back to a GlcA residue. After the formation of an IdoA residue within the polysaccharide backbone, it can be locked in place through the action of 2-O-sulfotransferase (2-OST) to form IdoA2S. Given that IdoA2S is not a substrate for C5-epi, no reverse epimerization takes place even if there is a GlcNS at position −1 [100].
Figure 5.
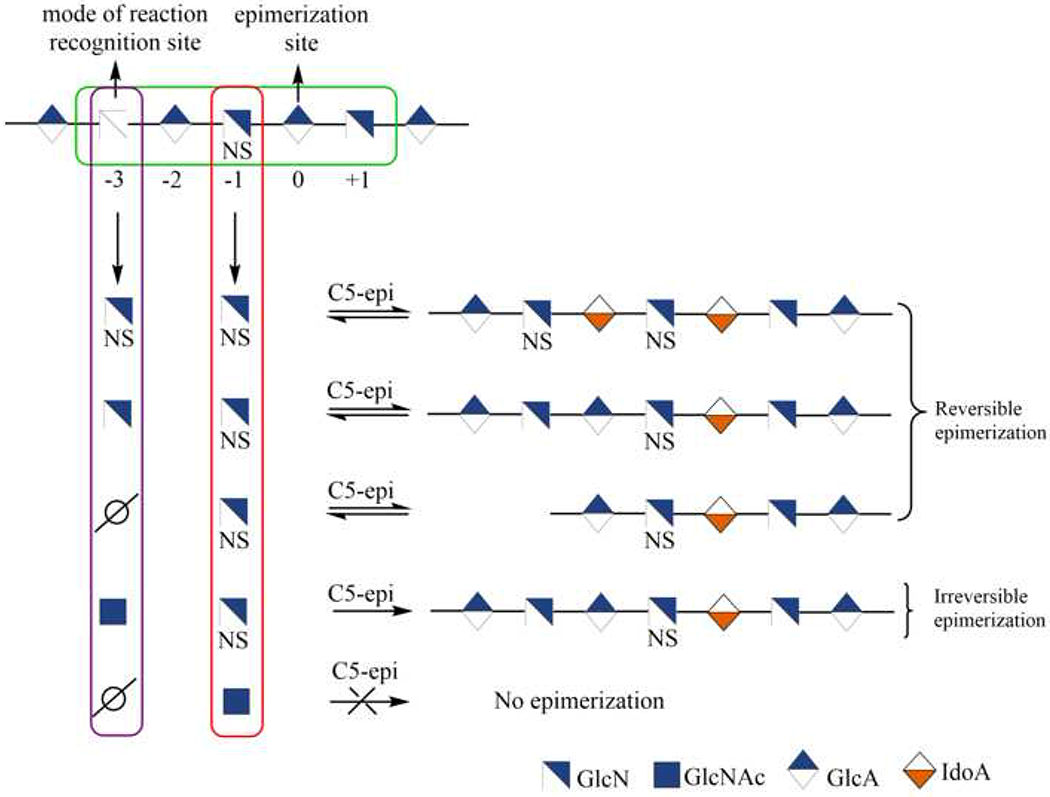
Mode of action of C5-epimerase.
O-sulfation is a crucial modification for the anticoagulant activity of heparin. A biocatalytic approach using 6-O-sulfotransferease (6-OST) and 3-O-sulfotransferase (3-OST) is required for the chemoenzymatic synthesis of heparin and HS because of the high conversion efficiency of these enzymes and the mild reaction conditions [101]. In humans, there are three 6-OST isoforms (6-OST1, 6-OST2, and 6-OST3), and seven isoforms of 3-OST (3-OST-1–3-OST-7) transfer sulfo groups to the 6- and 3-positions of GlcN, respectively. Given that 6-OST1, 6-OST2, and 6-OST-3 show similar substrate specificity patterns, a combination of 6-OST isoforms (such as 6-OST1 and 6-OST3) has been widely used to catalyze 6-O-sulfation. Fed-batch fermentation has been used to produce active sulfotransferase enzymes, 6-OST1 and 6-OST3 in E. coli [92,102]. The GlcA residues adjacent to GlcNS6S and GlcNAc6S units are not substrates for the C5-epi. Thus, if heparosan is treated with 6-OST-1 or 6-OST-3, after N-acetylation/N-sulfation and before epimerization reaction, the resulting modified heparosan cannot serve as a substrate for C5-epi. Once C5-epimerization and then 2-O-sulfation is completed, IdoA2S-containing modified heparosan is ready to be treated with the 6OSTs to afford the major TriS disaccharide-repeating unit of heparin, IdoA2S-GlcNS6S [103].
The final step in the preparation of heparin with anticoagulant activity requires treatment with 3-OST. The different 3-OST isoforms exhibit different substrate specificities. The 3-OST-1 isoform catalyzes 3-O-sulfation to a GlcNS6X or GlcNAc6X residue linked to a GlcA/IdoA residue at the nonreducing end to afford an AT pentasaccharide-binding site [104]. The level of 3-O-sulfation correlates to the anticoagulant activity of heparin; thus, monitoring 3-OST-1 sulfation is crucial in the synthesis of bioengineered heparin. A high-throughput sensing platform based on ELISA and enzymatic signal amplification has been developed that allows the in-process monitoring of 3-OST sulfation in the last step of bioengineering heparin synthesis [105].
Synthetic LMWH and ULMWH
GAG oligosaccharides have also been synthesized using backbone elongation on monosaccharide or disaccharide acceptors followed by chemoenzymatic modifications [31,33]. Monosaccharide and disaccharide derivatives have been used as acceptors, greatly facilitating the development of chemoenzymatic synthesis. The GlcA (1→4) anhydromannose disaccharide and two monosaccharides, β-glucuronide with p-nitrophenyl (p-NP) and p-aminophenyl N-(6-azidohexanamidyl) (pNA-N3), have been used as acceptors in chemoenzymatic synthesis. Acceptors can be further modified to introduce a ‘click’ reactive azide group or amino group at the reducing end, allowing for conjugation, p-NP or pNA-N3 groups enable easy detection, purification, and modification with a fluorescent tag or biotin of the resulting oligosaccharides [106].
The donors in the chemoenzymatic synthesis of GAGs, uridine diphosphate (UDP) monosaccharides, are transferred to glycosyl receptors by glycosyltransferase or synthase. Two bacterial glycosyltransferases, an N-acetylglucosaminyltransferase (KfiA) from E. coli K5 strain and PmHS2 from P. multocida, are used as substitutes for EXT1 and EXT2 to build the HS/heparin backbone. Both KfiA and PmHS2 can be readily expressed in E. coli [107,108]. UDP-GlcA and UDP-GlcNAc are the natural donor substrates required in the chemoenzymatic synthesis of HS. Although most of the HS biosynthetic enzymes have been efficiently prepared from E. coli, only an active N-sulfotransferase (NST) domain of NDST can be easily expressed in E. coli.
The synthesis of the HS backbone containing GlcNS residues represented a major problematic step because of the lack of efficient recombinant NDST. An unnatural sugar nucleotide, UDP-GlcNTFA (uridine diphosphate N-trifluoroacetyl glucosamine), has been used as an excellent substrate for KfiA in the synthesis of carbohydrates of an O-linked glycoprotein with modest yield [109,110]. In chemoenzymatic HS/heparin synthesis, the trifluoroacetyl group of the GlcNTFA residue, introduced using UDP-GlcNTFA, was treated under mild alkaline conditions and N-sulfated using N-sulfotransferase to obtain a GlcNS residue (Figure 6a). The use of UDP-GlcNTFA provides a means of introducing a GlcNS residue precisely at the desired position within an oligosaccharide [111]. Applying this strategy, structurally homogeneous ULMWHs, LMWHs, and heterogeneous heparin-like polysaccharides have been synthesized [44,111–113].
Figure 6.
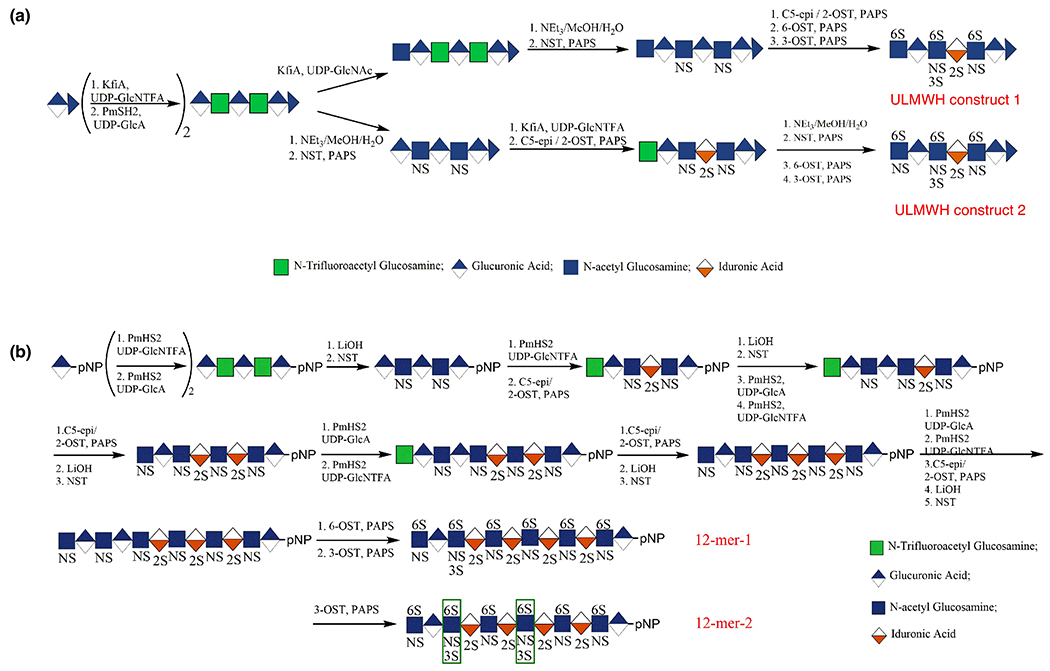
Chemoenzymatic synthesis of low-molecular-weight heparins.
Chen et al. reported a one-pot three-enzyme system to produce UDP-6N3GlcNAc [114]. Wang et al. developed a chemoenzymatic approach involving E. coli GlcNAc1-P uridyltransferase (GlmU) catalysis to obtain UDP-6N3GalNAc and UDP-GalNAc [115]. Given the presence of 1 → 4 linkages in the HS/heparin family, the C4 position of the UDP-donor offers an interesting target for modification. By chemically modifying the monosaccharide before the addition of a UDP moiety, the structure of the final polysaccharide chain can be manipulated. Unnatural chemically modified nucleotide sugars UDP-4N3-GlcNAc and UDP-4N3-GalNAc, UDP-4-deoxy-4-fluoro-N-acetylglucosamine (UDP-4FGlcNAc), and UDP-4-deoxy-4-fluoro-N-acetylgalactosamine (UDP-4FGalNAc) have been prepared using both chemical and chemoenzymatic syntheses. UDP-4N3-GlcNAc and UDP-4N3-GalNAc were then tested for incorporation into hyaluronan, heparosan, or chondroitin using polysaccharide synthases, and UDP-4N3-GlcNAc at the nonreducing end of the sugar chain served as a chain termination substrate in HA and heparin. Conjugation with Alexa Fluor 488 DIBO alkyne using ‘click chemistry’, showed that this approach can be used for labeling and detecting glycosaminoglycans [116]. Similar results were obtained when PmHS1 was used with UDP-4FGlcNAc, resulting in the incorporation of a single 4FGlcNAc at the nonreducing end of the acceptor serving as a chain terminator [117].
Starting from commercially available p-nitrophenyl glucuronide (GlcApNP), two dodecasaccharides (Figure 6b) were synthesized chemoenzymatically using UDP-sugar donors, a glycosyltransferase, sulfotransferases, and an epimerase, with an overall yield of ~10% [118]. These dodecasaccharides are structurally homogeneous, containing an N-sulfo-6-O-sulfo glucosamine (GlcNS6S) residue at their nonreducing end, a glucuronic acid (GlcA) residue at the reducing end, and one or two GlcNS3S6S residues in the middle of the structure. When the chain elongation was completed, six 6-O-sulfo groups were installed into each dodecasaccharide in a single reaction step.
There have been some concerns over the scalability of chemoenzymatic synthesis of LMWH. Completing the compound 6-O-sulfation in a single step, eliminating the need to purify from a partial 6-O-sulfated 12-mer mixture, facilitated the purification of the product, allowing it to be obtained with high yield at the gram scale. Recombinant C5-epi and 2-OST expressed in insect cells using the baculovirus expression system instead of E. coli were used to obtain these LMWHs. These recombinant C5-epi and 2-OST from insect cells are more active but more expensive than the same enzymes expressed in E. coli. Hence, the excellent expression of C5-epi and 2-OST offsets the higher costs of culturing insect cells. High-performance liquid chromatography (HPLC), high-resolution MS and 1D and 2D-NMR analyses confirmed the structure and purity of synthesized LMWHs. The in vitro anticoagulant activity and its protamine neutralization were compared with that of UFH and to two other FDA-approved heparin drugs, fondaparinux and enoxaparin. In vitro experiments demonstrated that the anticoagulant activity of the dodecasaccharides could be reversed using protamine. Additional activity and toxicity studies suggest that a synthetic homogeneous oligosaccharide can replace animal-sourced LMWHs [118].
The main disadvantage of this approach is that, because of the GlcApNP used as the starting material, it contains a structural feature at the reducing end of the carbohydrate chain of the target molecule that is unnatural. The elimination of pNP from chemoenzymatically synthesized HS oligosaccharides using ceric ammonium nitrate has been investigated [119]. Using a modified Smith degradation, unnatural moieties are formed at the reducing end of the resulting degradation products. Treatment with periodate oxidation followed by Smith degradation or alkaline elimination results in the selective cleavage of vicinal diol-containing glucuronic acid residues, affording heparin pentasaccharides rich in TriS disaccharide units with a completely natural structure. Recently, this strategy was used to prepare a heparin nonasaccharide, containing an internal 2-sulfoiduronic acid residue uniformly 13C-labeled, with a natural structure [120]. This was the first publication in which these two degradation approaches have been applied to the preparative synthesis of a natural heparin oligosaccharide (Figure 7). Importantly, no desulfation was detected during the synthesis. This chemoenzymatically synthesized 13C-labeled compound was intravenously administered to septic and nonseptic mice. This study confirmed that oligosaccharides containing these highly sulfated domains selectively targeted and penetrated the hippocampal blood-brain barrier following sepsis, where they might influence spatial memory formation [120].
Figure 7.
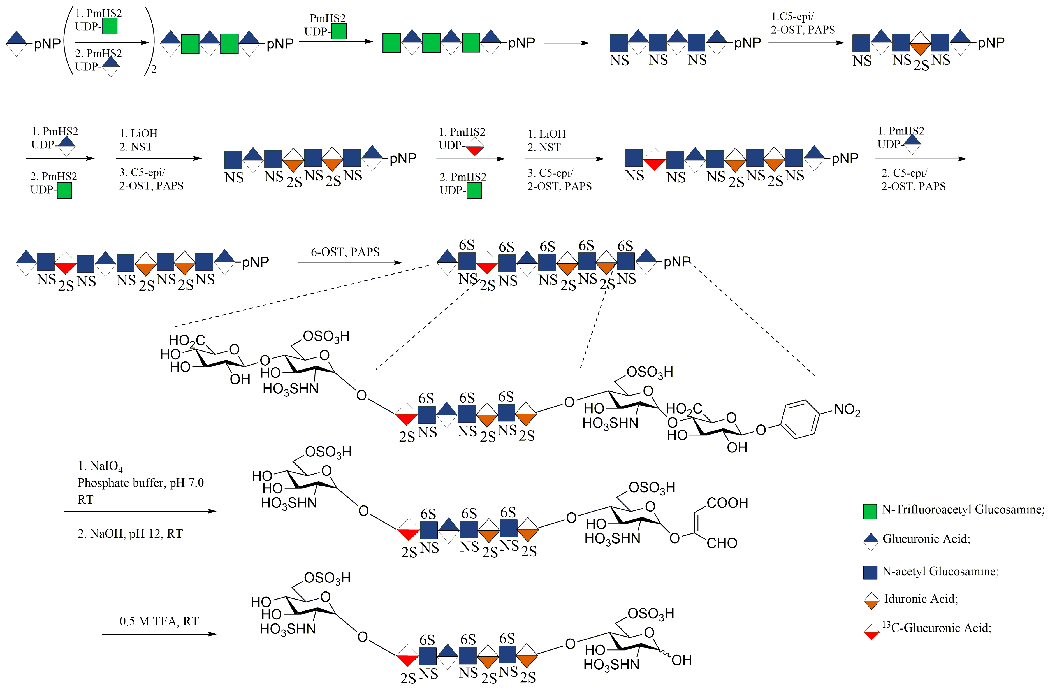
Chemoenzymatic synthesis of the low-molecular-weight heparin (LMWH) containing an internal (13C)IdoA2S residue; GlcA-pNP cleavage via alkaline elimination and Smith degradation.
As a part of an ongoing effort to introduce more natural group on acceptor, a HS tetrasaccharide intermediate was prepared which contained an O-methylglycoside at its reducing end, similar to that found in fondaparinux. Importantly, this was the first use of GlcNAc-OMe as an acceptor in PmHS2-catalyzed glycosylation and efficiently prepared a HS key tetrasaccharide in seven steps with an overall yield of 7.1% [121].
The development of new heparin products and new biochemical and pharmacological analysis are major challenges in the GAG field. The stable isotope labeling (SIL) strategy is a unique way to produce and investigate the roles of heparin in metabolic pathways. Selective partial isotopic labeling of UFH was previously reported [122,123]. A SIL heparin with 13C and 15N labeling was synthesized chemoenzymatically a decade ago [85]. A recently synthesized heparin nonasaccharide, containing a uniformly 13C-labeled internal 2-sulfoiduronic acid residue, allowed the tracking of its distribution in septic mice using MS [120]. Recently, SIL was applied to the first preparation of stable isotope enriched perdeuteroheparin (‘heavy’ heparin), from microbially produced perdeuteroheparosan [124]. Perdeuteroheparosan chemically deacetylated and N-sulfonated, using NaOH and a trimethylamine-sulfurtrioxide complex, respectively. Simultaneous epimerization and 2-O-sulfation of this chemically derived intermediate, yielded perdeutero-2-O-sulfo-N-sulfoheparosan. This intermediate was sulfated through the action of recombinant 6-OST and 3-OST enzymes, to generate perdeuteroheparin with similar disaccharide content to pharmaceutical heparin. Using heavy heparin, SIL disaccharide standards were prepared and a nonradioactive NMR assay for glucuronosyl-C5-epimerase was developed [125].
One-pot chemical synthesis of heparin oligosaccharides from sugar building blocks is used for simplified heparin synthesis, and successful synthesis was reported with reasonable overall yields [126]. A combinatorial study of multienzyme, one-pot, in intro biocatalytic synthesis resulted in heparin products similar to pharmaceutical heparin. Following this chemoenzymatic process, N-sulfoheparosan was first prepared by partial chemical N-deacetylation/N-sulfonation of bioengineered heparosan, followed by simultaneously treatment of N-sulfoheparosan with C5-epimerase and 2-OST, and low levels of 6-OST, followed by 3-OST. Optimization of the enzyme/substrate ratio is required to control of the structure of the final bioengineered heparin. This approach allows the production of anticoagulant heparin without time consuming work-up processes of all the intermediates and requires only the purification and characterization of the final product with a moderate yield [127].
Concluding remarks
Anticoagulant therapy is a vital treatment method in the prophylaxis and treatment of thromboembolic diseases. Heparin and heparin products are globally used indispensable anticoagulants. With the latest developments in the fields of synthesis, biotechnology, metabolic engineering and analysis, methods for the preparation of new structurally defined heparin derivatives with high yields are rapidly emerging. Over the past 5 years, new chemical and chemoenzymatic methods have been introduced to prepare LMWH, ULMWH, and bioengineered heparins. All these advances are important to extend our understanding of the structural differences between porcine and bovine heparins, and to develop new, safer, and improved animal and/or non-animal sourced heparins and related products using cutting-edge chemoenzymatic and metabolic engineering technologies. Preclinical and clinical studies are required to determine pharmacokinetic and pharmacodynamic properties and toxicity profiles of the heparin derivatives obtained using chemoenzymatic methods. However, the successful synthesis of heparin oligosaccharides provides structurally defined carbohydrates for advancing heparin research and presents the potential to introduce new therapeutic agents to the clinic to treat antitumoral, anti-inflammatory, and infectious diseases.
Highlights.
Metabolic engineering of heparosan, heparin and heparan sulfate
Chemical and chemoenzymatic synthesis of low molecular weight heparins
Development of new heparin products
Acknowledgments
This research is funded by the US National Institutes of Health in the form of grants DK111958 and CA231074 to R.J.L. and by TUBITAK-TURKEY (Bideb-2219 programme) funding to S.B.
Biographies
Author Biographies

Sultan N. Baytas
Sultan N. Baytas received her PhD in medicinal chemistry from Gazi University in 2002, did her postdoctoral studies at Rensselaer Polytechnic Institute, and is currently a professor of pharmaceutical chemistry at Gazi University, Faculty of Pharmacy. Her research activities include the design, discovery, and development of new molecules for pathologies associated with infections, thrombosis, cancer development, and inflammation. She is currently on sabbatical leave, working with Robert J. Linhardt’s group at Rensselaer Polytechnic Institute focusing on the chemoenzymatic synthesis of glycosaminoglycans.

Robert J. Linhardt
Robert J. Linhardt received his PhD in chemistry from Johns Hopkins University in 1979 and did his postdoctoral studies at Massachusetts Institute of Technology. He is currently the Anne and John Broadbent, Jr.’59 Senior Constellation Chair in Biocatalysis and Metabolic Engineering at Rensselaer Polytechnic Institute. His research focuses on glycoscience, and he is an expert on glycosaminoglycans and their synthesis, biology, and analysis. He has received multiple honors, including the National Academy of Inventors (NAI) Fellow, American Chemical Society Horace S. Isbell, Claude S. Hudson, and Melville L. Wolfrom Awards, the AACP Volwiler Research Achievement Award, the Society of Glycobiology Karl Meyer Award, and the Scientific American 10.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Teaser: We review recent advances in the field of preparation and synthesis of indispensable anticoagulant drugs, heparin, and related products.
References
- 1.Onishi A et al. (2016) Heparin and anticoagulation. Front. Biosci 21, 1372–1392 [DOI] [PubMed] [Google Scholar]
- 2.Mulloy B et al. (2016) Pharmacology of heparin and related drugs. Pharmacol. Rev 68, 76–141 [DOI] [PubMed] [Google Scholar]
- 3.Spadarella G et al. (2020) From unfractionated heparin to pentasaccharide: paradigm of rigorous science growing in the understanding of the in vivo thrombin generation. Blood Rev. 39, 100613. [DOI] [PubMed] [Google Scholar]
- 4.Contejean C (1895) Recherches sur les injections intraveineuses de peptone et leur influence sur la coagulabilite du sang chez le chien. Arch. Physiol. Norm. Pathol 7 45–53 [Google Scholar]
- 5.McLean J (1916) The thromboplastic action of cephalin. Am. J. Physiol 41 250–257 [Google Scholar]
- 6.Brinkhous KM et al. (1939) The inhibition of blood clotting: an unidentified substance which acts in conjunction with heparin to prevent the conversion of prothrombin into thrombin. Am. J. Physiol 125, 683–687 [Google Scholar]
- 7.Waugh DF and Fitzgerald MA (1956) Quantitative aspects of antithrombin and heparin in plasma. Am. J. Physiol 184, 627–639 [DOI] [PubMed] [Google Scholar]
- 8.Monkhouse FC et al. (1955) Studies on the antithrombin and heparin cofactor activities of a fraction absorbed from plasma by aluminium hydroxide. Circ. Res 3, 397–402 [DOI] [PubMed] [Google Scholar]
- 9.Abildgaard U (1968) Highly purified antithrombin III with heparin cofactor activity prepared by disc electrophoresis. Scand. J. Clin. Lab. Invest 21, 89–90 [DOI] [PubMed] [Google Scholar]
- 10.Choay J et al. (1981) Structural studies on a biologically active hexasaccharide obtained from heparin. Ann. N.Y. Acad. Sci 370, 644–649 [DOI] [PubMed] [Google Scholar]
- 11.Thunberg L et al. (1982) Further characterisation of the antithrombin-binding sequence in heparin. Carbohydr. Res 100, 393–410 [DOI] [PubMed] [Google Scholar]
- 12.Liu H et al. (2009) Lessons learned from the contamination of heparin. Nat. Prod. Rep 26, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rabenstein DL (2002) Heparin and heparan sulfate: structure and function. Nat. Prod. Rep 19, 312–331 [DOI] [PubMed] [Google Scholar]
- 14.Comper WD (1981) Heparin (and Related Polysaccharides): Structural and Functional Properties, Gordon and Breach Science Publishers [Google Scholar]
- 15.Linhardt RJ (2003) Cloude S. Hudson Award address in carbohydrate chemistry. Heparin: structure and activity. J. Med. Chem 46, 2551–2554 [DOI] [PubMed] [Google Scholar]
- 16.McCrea K et al. (2014) Removal of carbapenem-resistant enterobacteriaceae (CRE) from blood by heparin-functional hemoperfusion media. PLoS ONE 9, e114242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vieira TC et al. (2014) Heparin binding confers prion stability and impairs its aggregation. FASEB J. 28, 2667–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Boer SM et al. (2012) Heparan sulfate facilitates Rift Valley fever virus entry into the cell. J. Virol 86, 13767–13771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okona-Mensah KB et al. (1998) Inhibition of serum and transforming growth factor beta (TGF-beta1)-induced DNA synthesis in confluent airway smooth muscle by heparin. Br. J. Pharmacol 125, 599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mousa SA et al. (2006) Anti-metastatic effect of a non-anticoagulant low-molecular-weight heparin versus the standard low molecular-weight heparin, enoxaparin. Thromb. Haemost 96, 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Höök M et al. (1975) Biosynthesis of heparin. Studies on the microsomal sulfation process. J. Biol. Chem 250, 6065–6071 [PubMed] [Google Scholar]
- 22.Lidholt K et al. (1989) Biosynthesis of heparin. Relationship between the polymerization and sulphation processes. Biochem. J 261, 999–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitelock JM and Iozzo RV (2005) Heparan sulfate: a complex polymer charged with biological activity. Chem. Rev 105, 2745–2764 [DOI] [PubMed] [Google Scholar]
- 24.Fu L et al. (2016) Bioengineered heparins and heparan sulfates. Adv. Drug Delivery Rev 97, 237–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang T et al. (2020) Chemoenzymatic synthesis of ultralow and low-molecular weight heparins. Biochim. Biophys. Acta 1868, 140301. [DOI] [PubMed] [Google Scholar]
- 26.Stevens RL and Adachi R (2007) Protease-proteoglycan complexes of mouse and human mast cells and importance of their betatryptase-heparin complexes in inflammation and innate immunity. Immunol. Rev 217, 155–167 [DOI] [PubMed] [Google Scholar]
- 27.Sugahara K and Kitagawa H (2002) Heparin and heparan sulfate biosynthesis. IUBMB Life 54, 163–175 [DOI] [PubMed] [Google Scholar]
- 28.Farrugia BL et al. (2015) Can we produce heparin/heparan sulfate biomimetics using ‘Mother-Nature’ as the gold standard? Molecules 20, 4254–4276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kreuger J and Kjellén L (2012) Heparan sulfate biosynthesis: regulation and variability. J. Histochem. Cytochem 60, 898–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinhal MAS et al. (2001) Enzyme interactions in heparan sulfate biosynthesis: uronosyl 5-epimerase and 2-O-sulfotransferase interact in vivo. Proc. Natl. Acad. Sci. U. S. A 98, 12984–12989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J and Linhardt RJ (2014) Chemoenzymatic synthesis of heparan sulfate and heparin. Nat. Prod. Rep 31, 1676–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoppensteadt D et al. (2005) Basic and clinical differences of heparin and low molecular weight heparin treatment In Chemistry and Biology of Heparin and Heparan Sulfate (Garg HG et al. , eds), pp. 583–606, Elsevier [Google Scholar]
- 33.Linhardt RJ and Liu J (2012) Synthetic heparin. Curr. Opin. Pharmacol 12, 217–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Meer J-Y. et al. (2017) From farm to pharma: an overview of industrial heparin manufacturing methods. Molecules 22, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeAngelis PL et al. (2013) Chemoenzymatic synthesis of glycosaminoglycans: re-creating, re-modeling and re-designing nature’s longest or most complex carbohydrate chains. Glycobiology 23, 764–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guerrini M et al. (2008) Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat. Biotechnol 26, 669–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirsh J et al. (2007) Beyond unfractionated heparin and warfarin. Circulation 116, 552–560 [DOI] [PubMed] [Google Scholar]
- 38.Heres EK et al. (2001) The clinical onset of heparin is rapid. Anesth. Analg 92, 1391–1395 [DOI] [PubMed] [Google Scholar]
- 39.Young E (2008) The anti-inflammatory effects of heparin and related compounds. Thrombosis Research 122, 743–752 [DOI] [PubMed] [Google Scholar]
- 40.Grootenhuis PDJ et al. (1995) Rational design of synthetic heparin analogues with tailor-made coagulation factor inhibitory activity. Nat. Struct. Biol 2, 736–739 [DOI] [PubMed] [Google Scholar]
- 41.Jin L et al. (1997) The anticoagulant activation of antithrombin by heparin. Proc. Natl Acad. Sci. U. S. A 94, 14683–14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petitou M et al. (1999) Synthesis of thrombin-inhibiting heparin mimetics without side effects. Nature 398, 417–422 [DOI] [PubMed] [Google Scholar]
- 43.Choay J et al. (1981) Oligosaccharides de faible poids moléculaire présentant une activité inhibitrice du facteur Xa en milieu plasmatique. Ann. Pharm. Fr 39, 37–44 [PubMed] [Google Scholar]
- 44.Xu Y et al. (2014) Homogeneous low-molecular-weight heparins with reversible anticoagulant activity. Nat. Chem. Biol 10, 248–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dickinson DM et al. (2015) Chemoenzymatic synthesis of heparin In Glycoscience: Biology and Medicine (Taniguchi N et al. , eds), pp. 419–426, Springer [Google Scholar]
- 46.Petitou M and van Boeckel CAA (2004) Synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew. Chem. Int. Ed 43, 3118–3133 [DOI] [PubMed] [Google Scholar]
- 47.Turpie AG et al. (2002) Fondaparinux vs enoxaparin for the prevention of venous thromboembolism in major orthopedic surgery: a meta-analysis of 4 randomized double-blind studies. Arch. Intern. Med 162 1833–1840 [DOI] [PubMed] [Google Scholar]
- 48.Mende M et al. (2016) Chemical synthesis of glycosaminoglycans. Chem. Rev 116, 8193–8255 [DOI] [PubMed] [Google Scholar]
- 49.Duchaussoy P et al. (1991) First total synthesis of the antithrombin III binding site of porcine mucosa heparin. Bioorg. Med. Chem. Lett 1, 99–102 [Google Scholar]
- 50.Petitou M et al. (1991) A new, highly potent, heparin-like pentasaccharide fragment containing a glucose residue instead of a glucosamine. Bioorg. Med. Chem. Lett 1, 95 [Google Scholar]
- 51.Lin F et al. (2013) Synthesis of fondaparinux: modular synthesis investigation for heparin synthesis. Carbohydr. Res 371, 32–39 [DOI] [PubMed] [Google Scholar]
- 52.Chang C-H. et al. (2014) Synthesis of the heparin-based anticoagulant drug fondaparinux. Angew. Chem., Int. Ed 53, 9876–9879 [DOI] [PubMed] [Google Scholar]
- 53.Li T et al. (2014) Total synthesis of anticoagulant pentasaccharide fondaparinux. ChemMedChem 9 1071–1080 [DOI] [PubMed] [Google Scholar]
- 54.Dai X et al. (2016) Formal synthesis of anticoagulant drug fondaparinux sodium. J. Org. Chem 81, 162–184 [DOI] [PubMed] [Google Scholar]
- 55.Ding Y et al. (2017) Efficient and practical synthesis of Fondaparinux. Bioorg. Med. Chem. Lett 27, 2424–2427 [DOI] [PubMed] [Google Scholar]
- 56.Jin H et al. (2019) Preactivation-based, iterative one-pot synthesis of anticoagulant pentasaccharide fondaparinux sodium. Org. Chem. Front 6, 3116–3120 [Google Scholar]
- 57.Dey S et al. Programmable one-pot synthesis of heparin pentasaccharide fondaparinux. Org. Lett 22, 4638–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bohlmann L et al. (2015) Functional and structural characterization of a heparanase. Nat. Chem. Biol 11, 955–957 [DOI] [PubMed] [Google Scholar]
- 59.Yu Y et al. (2019) Specificity and action pattern of heparanase Bp, a β-glucuronidase from Burkholderia pseudomallei. Glycobiology 29, 572–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Linhardt RJ and Gunay NS (1999) Production and chemical processing of low molecular weight heparins. Semin. Thrombos. Hemostas 25, 5–16 [PubMed] [Google Scholar]
- 61.Higashi K et al. (2012) Photochemical preparation of a novel low molecular weight heparin. Carbohydr. Polym 87, 1737–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma X et al. (2016) Synergistic effect and mechanisms of combining ultrasound and pectinase on pectin hydrolysis. Bioprocess Technol. 7, 1249–1257 [Google Scholar]
- 63.Ogutu FO et al. (2015) Ultrasonic modification of selected polysaccharides-review. J. Food. Process. Technol 6, 446 [Google Scholar]
- 64.Ribeiro AR et al. (2015) An overview on the advanced oxidation processes applied for the treatment of water pollutants defined in the recently launched Directive 2013/39/EU. Environ. Int 75, 33–51 [DOI] [PubMed] [Google Scholar]
- 65.Achour O et al. (2013) Ultrasonic-assisted preparation of a low molecular weight heparin (LMWH) with anticoagulant activity. Carbohydr. Polym 97, 684–689 [DOI] [PubMed] [Google Scholar]
- 66.Petit A-C. et al. (2007) Ultrasonic depolymerization of an exopolysaccharide produced by a bacterium isolated from a deep-sea hydrothermal vent polychaete annelid. Ultrason. Sonochem 14, 107–112 [DOI] [PubMed] [Google Scholar]
- 67.Zhi Z et al. (2019) Preparation of low molecular weight heparin using an ultrasound-assisted Fenton-system. Ultrasonics Sonochem. 52 184–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li J et al. (2016) Depolymerization of fucosylated chondroitin sulfate with a modified Fenton-system and anticoagulant activity of the resulting fragments. Mar. Drugs 14, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shen X et al. (2019) Development of low molecular weight heparin by H2O2/ascorbic acid with ultrasonic power and its antimetastasis property. Int. J. Biol. Macromol 133, 101–109 [DOI] [PubMed] [Google Scholar]
- 70.Fareed J et al. (2019) Porcine mucosal heparin shortage crisis! What are the options? Clin. Appl. Thrombos. Hemostas 25, 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haley M and Gale F (2020) African Swine Fever shrinks pork production in China, swells demand for imported pork. Amber Waves Mag. July 30 [Google Scholar]
- 72.Hoppensteadt D et al. (2015) Resourcing of heparin and low molecular weight heparins from bovine, ovine, and porcine origin. Studies to demonstrate the biosimilarities. Blood 126, 4733 [Google Scholar]
- 73.Monakhova YB et al. (2018) Authentication of animal origin of heparin and low molecular weight heparin including ovine, porcine and bovine species using 1D NMR spectroscopy and chemometric tools. J Pharm Biomed Anal. 149, 114–119 [DOI] [PubMed] [Google Scholar]
- 74.Fu L et al. (2013) Structural characterization of pharmaceutical heparins prepared from different animal tissues. J. Pharm. Sci 102, 1447–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Santos GR et al. (2014) Structural and functional analyses of bovine and porcine intestinal heparins confirm they are different drugs. Drug Discov. Today 19, 1801–1807 [DOI] [PubMed] [Google Scholar]
- 76.Tovar AM et al. (2013) Bovine and porcine heparins: different drugs with similar effects on human haemodialysis. BMC Res. Notes 6, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jeske W et al. (2019) Bovine mucosal heparins are comparable to porcine mucosal heparin at USP potency adjusted levels. Front. Med 5, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xie S et al. (2018) Preparation of low molecular weight heparins through nitrous acid degradation from bovine and ovine heparins. Carbohydr. Polym 197, 83–91 [DOI] [PubMed] [Google Scholar]
- 79.Guan Y et al. (2016) Comparison of low molecular weight heparins prepared using bovine lung heparin and porcine intestine heparin as starting materials. J. Pharm. Sci 105, 1843–1850 [DOI] [PubMed] [Google Scholar]
- 80.Liu X et al. (2017) Comparison of low molecular weight heparins prepared from bovine heparins with enoxaparin. Clin. Appl. Thrombos. Hemostas 23, 542–553 [DOI] [PubMed] [Google Scholar]
- 81.St. Ange K et al. (2016) Analysis of heparins derived from bovine tissues and comparison to porcine intestinal heparins. Clin. Appl. Thrombos. Hemostas 22, 520–527 [DOI] [PubMed] [Google Scholar]
- 82.Gijsen HJ et al. (1996) Recent advances in the chemoenzymatic synthesis of carbohydrates and carbohydrate mimetics. Chem. Rev 96, 443–473 [DOI] [PubMed] [Google Scholar]
- 83.Karst NA and Linhardt RJ (2003) Recent chemical and enzymatic approaches to the synthesis of glycosaminoglycan oligosaccharides. Curr. Med. Chem 10, 1993–2031 [DOI] [PubMed] [Google Scholar]
- 84.Chen J et al. (2005) Enzymatically redesigning of biologically active heparan sulfate. J. Biol. Chem 280, 42817–42825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Z et al. (2008) Solution structure of chemoenzymatically synthesized heparin and its precursors. J. Am. Chem. Soc 130, 12998–13007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jin P et al. (2016) Efficient biosynthesis of polysaccharides chondroitin and heparosan by metabolically engineered Bacillus subtilis. Carbohydr. Polym 140 424–432 [DOI] [PubMed] [Google Scholar]
- 87.Huang H et al. (2016) Recombinant Escherichia coli K5 strain with the deletion of waaR gene decreases the molecular weight of the heparosan capsular polysaccharide. Appl. Microbiol. Biotechnol 100 7877–7885 [DOI] [PubMed] [Google Scholar]
- 88.Williams A et al. (2019) Metabolic engineering of Bacillus megaterium for heparosan biosynthesis using Pasteurella multocida heparosan synthase, PmHS2. Microb. Cell Factor 18, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nehru G et al. (2020) Production and characterization of low molecular weight heparosan in Bacillus megaterium using Escherichia coli K5 glycosyltransferases. Int. J. Biol. Macromol 160, 69–76 [DOI] [PubMed] [Google Scholar]
- 90.Chappell EP and Liu J (2013) Use of biosynthetic enzymes in heparin and heparan sulfate synthesis. Bioorg. Med. Chem 21, 4786–4792 [DOI] [PubMed] [Google Scholar]
- 91.Raedts J et al. (2013) A novel bacterial enzyme with D-glucuronyl C5-epimerase activity. J. Biol. Chem 288, 24332–24339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Restaino OF et al. (2013) High cell density cultivation of a recombinant E. coli strain expressing a key enzyme in bioengineered heparin production. Appl. Microbiol. Biotechnol 97, 3893–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li YJ et al. (2018) Characterization of heparan sulfate N-deacetylase/N-sulfotransferase isoform 4 using synthetic oligosaccharide substrates. Biochim. Biophys. Acta 1862, 547–556 [DOI] [PubMed] [Google Scholar]
- 94.Kuberan B et al. (2003) Enzymatic synthesis of antithrombin III-binding heparan sulfate pentasaccharide. Nat. Biotechnol 21, 1343–1346 [DOI] [PubMed] [Google Scholar]
- 95.Saribas AS et al. (2004) Production of N-sulfated polysaccharides using yeast-expressed N-deacetylase/N-sulfotransferase-1 (NDST-1). Glycobiology 14, 1217–1228 [DOI] [PubMed] [Google Scholar]
- 96.Dou W et al. (2015) Role of deacetylase activity of N-deacetylase/N-sulfotransferase 1 in forming N-sulfated domain in heparan sulfate. J. Biol. Chem 290, 20427–20437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Z et al. (2011) Control of the heparosan N-deacetylation leads to an improved bioengineered heparin. Appl. Microbiol. Biotechnol 91, 91–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang Z et al. (2011) Response surface optimization of the heparosan N-deacetylation in producing bioengineered heparin J. Biotechnol 156, 188–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sheng J et al. (2012) Uncovering biphasic catalytic mode of C5-epimerase in heparan sulfate biosynthesis. J. Biol. Chem 287, 20996–21002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Paul P et al. (2012) Recent advances in sulfotransferase enzyme activity assays. Anal. Bioanal. Chem 403, 1491–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lange B et al. (2016) Towards keratan sulfate – chemoenzymatic cascade synthesis of sulfated N-acetyllactosamine (LacNAc) glycan oligomers. Adv. Synth. Catal 358, 584–596 [Google Scholar]
- 102.Zhang J et al. (2014) High cell density cultivation of a recombinant Escherichia coli strain expressing a 6-O-sulfotransferase for the production of bioengineered heparin. J. Appl. Microbiol 118, 92–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sterner E et al. (2014) Assays for determining heparan sulfate and heparin O-sulfotransferase activity and specificity. Anal Bioanal. Chem. Commun 406, 525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Z et al. (2017) Synthesis of 3-O-sulfated oligosaccharides to understand the relationship between structures and functions of heparan sulfate. J. Am. Chem. Soc 139, 5249–5256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin L et al. (2019) High-throughput method for in process monitoring of 3-O-sulfotransferase catalysed sulfonation in bioengineered heparin synthesis. Anal. Biochem 586, 113419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang X et al. (2020) Chemoenzymatic synthesis of glycosaminoglycans. Acc. Chem. Res 53, 335–346 [DOI] [PubMed] [Google Scholar]
- 107.Chen M et al. (2006) Determination of the substrate specificities of N-acetyl-d-glucosaminyltransferase. Biochemistry 45, 12358–12365 [DOI] [PubMed] [Google Scholar]
- 108.Sismey-Ragatz AE et al. (2007) Chemoenzymatic synthesis with distinct Pasteurella heparosan synthases. J. Biol. Chem 282, 28321–28327 [DOI] [PubMed] [Google Scholar]
- 109.Sala RF et al. (1998) UDP-N-trifluoroacetylglucosamine as an alternative substrate in N-acetylglucosaminyltransferase reactions. Carbohydr. Res 306, 127–136 [DOI] [PubMed] [Google Scholar]
- 110.Liu R et al. (2010) Chemoenzymatic design of heparan sulfate oligosaccharides. J. Biol. Chem. Rev 285, 34240–34249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu Y et al. (2011) Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science Transl. Med 334, 498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sterner E et al. (2014) Fibroblast growth factor-based signaling through synthetic heparan sulfate block copolymers studied using high-cell density 3D cell printing. J. Biol. Chem, 289, 9754–9765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chandarajoti K et al. (2014) De novo synthesis of a narrow size distribution low-molecular-weight heparin. Glycobiology 24, 476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen Y et al. (2011) One-pot three-enzyme synthesis of UDP-GlcNAc derivatives. Chem. Commun 47, 10815–10817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wen L et al. (2018) Chemoenzymatic synthesis of unnatural nucleotide sugars for enzymatic bioorthogonal labeling. ACS Catal. 8, 7659–7766 [Google Scholar]
- 116.Zhang X et al. (2017) Synthesis of 4-azido-N-acetylhexosamine uridine diphosphate donors: clickable glycosaminoglycans. J. Org. Chem 82, 9910–9915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schultz VL et al. (2017) Chemoenzymatic synthesis of 4-fluoro-N-acetylhexosamine uridine diphosphate donors: chain terminators in glycosaminoglycan synthesis. J. Org. Chem 82, 2243–2248 [DOI] [PubMed] [Google Scholar]
- 118.Xu X et al. (2017) Synthetic oligosaccharides can replace animal-sourced low-molecular weight heparins. Science Transl. Med 9, eaan5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cai C et al. (2013) Toward the chemoenzymatic synthesis of heparan sulfate oligosaccharides: oxidative cleavage of p-nitrophenyl group with ceric ammonium salts. Tet. Lett 54, 4471–4474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang X et al. (2019) Circulating heparin oligosaccharides rapidly target the hippocampus in sepsis, potentially impacting cognitive functions Proc. Natl. Acad. Sci. U. S. A 116, 9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang X et al. (2019) Chemoenzymatic synthesis of heparan sulfate tetrasaccharide from a N-acetyl-α-D-glucosamine-Omethylglycoside acceptor. Tet. Lett 60, 911–915 [Google Scholar]
- 122.Prÿchoux et al. (2015) C5-Epimerase and 2-O-Sulfotransferase Associate in vitro to Generate Contiguous Epimerized and 2-O-Sulfated Heparan Sulfate Domains. ACS Chem. Biol 10, 1064–1071 [DOI] [PubMed] [Google Scholar]
- 123.Pomin VH et al. (2010) Characterization of glycosaminoglycans by 15N NMR spectroscopy and in vivo isotopic labeling. Anal. Chem 82, 4078–4088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cress BF et al. (2019) Heavy heparin: a stable isotope-enriched, chemoenzymatically-synthesized, poly-component drug. Angew. Chem. Int. Ed 58, 5962–5966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vaidyanathan D et al. (2020) Elucidating the unusual reaction kinetics of D-glucuronyl C5-epimerase. Glycobiology. Published online April 18, 2020 10.1093/glycob/cwaa1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Polat T and Wong C-H (2007) Anomeric reactivity-based one-pot synthesis of heparin-like oligosaccharides. J. Am. Chem. Soc 129, 12795–12800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bhaskar U et al. (2015) Combinatorial one-pot chemoenzymatic synthesis of heparin. Carbohydr. Polym 122, 399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]