

Abstract
Natural products are an enduring source of chemical information useful for probing biologically relevant chemical space. Toward gathering further structure activity relationship (SAR) information for a particular natural product, synthetic chemists traditionally proceeded first by a total synthesis effort followed by the synthesis of simplified derivatives. While this approach has proven fruitful, it often does not incorporate hypotheses regarding structural features necessary for bioactivity at the synthetic planning stage, but rather focuses on the rapid assembly of the targeted natural product; a goal that often supersedes the opportunity to gather SAR information en route to the natural product. Furthermore, access to simplified variants of a natural product possessing only the proposed essential structural features necessary for bioactivity, typically at lower oxidation states overall, is sometimes non-trivial from the original established synthetic route. In recent years, several synthetic design strategies were described to streamline the process of finding bioactive molecules in concert with fathering further SAR studies for targeted natural products. This Review article will briefly discuss traditional retrosynthetic strategies and contrast them to selected examples of recent synthetic strategies for the investigation of biologically relevant chemical space revealed by natural products. These strategies include: diversity-oriented synthesis (DOS), biology-oriented synthesis (BIOS), diverted-total synthesis (DTS), analogue-oriented synthesis (AOS), two-phase synthesis, function-oriented synthesis (FOS), and computed affinity/dynamically ordered retrosynthesis (CANDOR). Finally, a description of pharmacophore-directed retrosynthesis (PDR) developed in our laboratory and initial applications will be presented that was initially inspired by a retrospective analysis of our synthetic route to pateamine A completed in 1998.
Graphical Abstract
1. Introduction
The total synthesis of natural products dates back to Wohler’s synthesis of urea in 18281 and has reached amazing heights providing access to complex natural products such as quinine, strychnine, palytoxin, taxol, and many others as the arsenal of synthetic strategies and methods available to chemists continues to expand. A powerful and unified strategy utilized to realize elegant and concise solutions to the total synthesis of complex natural products is retrosynthetic analysis. First articulated by Corey in The Logic of Chemical Synthesis,2 retrosynthetic analysis has become a cornerstone to modern synthetic endeavors allowing for the deconvolution of complex natural products into increasingly simplified intermediates and eventually commercially available starting materials. The increased capabilities realized through retrosynthesis fostered the generation of ideas which encompass an “ideal synthesis”, a term used by Hendrickson3 which has evolved to include: concise and convergent strategies (step economy),4 decreasing reactant waste throughout a synthesis (atom economy),5 and eliminating unnecessary redox manipulations (redox economy).6
The concept of an ideal synthesis is one that all chemists aspire to achieve; however, the completion of a complex natural product is not always considered simultaneously with collection of information on functionality required for a natural product’s biological effects. This two-fold goal can become quite challenging, requiring a balance between synthetic efficiency and gained structure activity relationship (SAR) data. This is best articulated in Wender’s function-oriented synthesis strategy, elaborated further below, and indeed a likely inspiration for many of the strategies discussed in this review.4b-d Given the high percentage of drugs that were inspired by natural product scaffolds, or are natural products themselves,7 the continued harvesting of the vast information content available from natural products and their cellular receptors is essential. While historically most large pharmaceutical companies had a large presence in natural product research into the early 1990’s, this has slowly diminished to a point that few companies (e.g. Eisai Inc., Novartis) maintain a strong presence for the study of natural products as initial hits toward new drug leads. To gather SAR information while maintaining synthetic efficiency, natural product chemists have developed various strategies that are the subject of this Review. Following a brief introduction to synthetic library strategies that may have contributed to the demise of natural products but later returned to natural products for inspiration, we will conclude with a more in-depth analysis of our recently described pharmacophore-directed retrosynthesis (PDR) strategy illustrated through applications to gracilin A and rameswaralide.
2. Strategies for Probing Chemical Space through Total Synthesis
Before describing various recent synthetic strategies developed to marry total synthesis with SAR profile development of a given natural product, the role that small molecule libraries have played in probing chemical space should be mentioned. Of particular relevance to the topic of this review is the development of natural-product inspired libraries.
2.1. Small Molecule Libraries
Over the past ~30 years, the synthesis of small molecule libraries has become commonplace to aid in drug discovery and development when searching for novel hit compounds to a known cellular target via high throughput screening. Recent challenges with this approach stem from limited structural diversity among molecules within these libraries, which contain a high percentage of simple leads only differing in peripheral functionality often with similar 2-dimensional topologies. To address these issues, streamlined approaches toward the generation of more diverse, sp3-rich chemical libraries including those inspired by natural products were developed that could increase the probability of identifying initial hit compounds that can be further developed into lead compounds.
2.1.1. Schreiber’s Diversity-Oriented Synthesis (DOS)
The process of generating a diverse small molecule library can be an onerous task especially if not carried out in an efficient manner. Diversity-oriented synthesis (DOS) attempts to address this problem by allowing for programmed synthesis of libraries including split-pool synthesis (one bead-one compound libraries)8 containing extensive diversity to cover a large volume of 3D chemical space, useful for the identification of initial hit compounds for specific protein targets.8b, 8e, 9 The sp3 rich nature results in “natural product-like”10 libraries with increased structural diversity leading to a higher probability of identifying hits for a large set of biological targets. The reader is directed to multiple reviews on DOS,8b, 8e, 9-11 however, a brief overview is included to demonstrate the progression of small molecule libraries to provide a comparison to NP-inspired libraries created using biology-oriented synthesis (BIOS).
Reagent-based DOS involves the use of simple, commercially available building blocks and reagents to install varied functionality thereby diverging, ideally in under 5 steps, to achieve synthetic efficiency and diverse carbo-skeletal cores. These cores undergo further diversification, allowing for simultaneous access to a complex library containing substantial diversity.8b, 8e, 11a, 11b, 11d, 12 Alternatively, a substrate-based approach to DOS can be undertaken wherein substrates with “pre-encoded skeletal information” can be utilized through similar chemical transformations to arrive again at a diverse library of complex molecules.8f It is important to note that in DOS there is no specified target molecule. Given no predetermined target molecule, there is no limit on the reactivity utilized or types of scaffolds synthesized through the process, allowing for synthesis of a wide array of molecules and vast coverage of 3D chemical space (Scheme 1a).8b As such, the overarching goal of DOS is to provide a library with extensive diversity which can be utilized to find molecules to target various proteins in a HTS screening campaign.11c
Scheme 1.

a) Schreiber’s diversity-oriented synthesis (DOS) providing complex functionally and skeletally diverse molecules. b) Waldmann’s biology-oriented synthesis (BIOS) providing access to libraries based on “privileged” natural product skeletons.
A more recent example displaying the power of DOS can be seen through a collaborative effort to find drug leads for Chagas disease, specifically the chronic phase of this affliction. At the time of publication (2014), and still today, there are two main treatments for the acute phase of Chagas disease, benznidazole (1) and nifurtimox (not shown), both having numerous side effects (Figure 2). To address this, a library consisting of 100,000 diverse small molecules synthesized through DOS were subjected to the standard workflow commonly found in traditional medicinal chemistry. Phenotypic HTS followed by SAR studies of hits led to improved activity and selectivity eventually resulting in a lead compound, ML341 (2), which displays much greater trypanocidal activity compared to benznidazole (1) (40 nM vs 6.6 μM, respectively, Figure 2).12 This example shows how DOS can serve as a useful tool to provide a library of novel small molecules as initial hits in the drug discovery process for any cellular target when high-throughput screening is a viable option.
Figure 2.
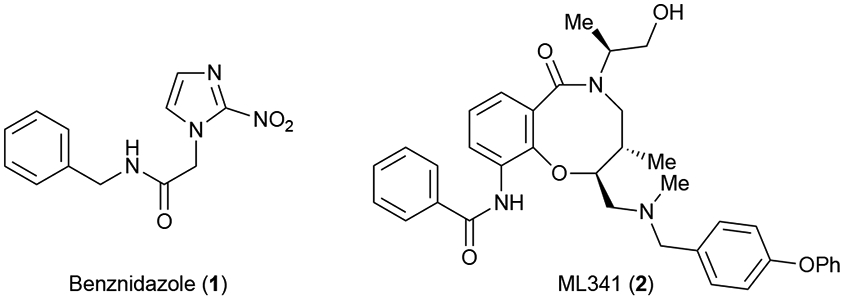
Current front-line treatment for Chagas disease and drug lead realized from DOS library noting the stark contrast in molecular complexity especially in number of stereocenters and sp3-carbons.
2.1.2. Waldmann’s Biology-Oriented Synthesis (BIOS)
An alternative approach to library synthesis was disclosed by Waldmann termed biology-oriented synthesis (BIOS, Scheme 1b). In contrast to DOS’s forward synthetic approach to library synthesis, BIOS utilizes target-oriented synthesis (TOS) to design libraries based on the notion that natural products are “privileged” structures; therefore, their core structures have pre-determined functionality leading to a higher probability of displaying bioactivity.9, 13
As such, natural products are utilized as target molecules to inspire BIOS library synthesis. A retrosynthesis is thus devised to allow access to a focused library of analogues which contain a similar skeleton to the natural product (Scheme 1b). Because the blueprint for all molecules within the library is focused on the natural product skeleton, the library lacks the extensive diversity found in a DOS library. However, the target-oriented nature of BIOS requires the synthesis of fewer molecules within a library to realize an initial hit molecule.13a, 13b
An example of BIOS in action is shown through Waldmann’s work on an oxepane based library.14 The core oxepane was chosen due to its prevalence in many bioactive natural products including the allelopathic and phytotoxic heliannuol B (3) and C (not shown), the antitumor sodwanone S (5), and a contraceptive, zoapatanol (4) (Scheme 2a). Thus, an efficient synthesis (one pot, 4-8 steps) of oxepane containing derivatives via a key common core oxepane 8 amenable to extensive derivatization was devised. After synthesizing ~91 derivatives the group explored the bioactivity of these privileged structures. Given the widespread activity of oxepane containing natural products, the group studied their activity in various cell-based assays and found that the most promising activity was toward Wnt signaling. Chemical proteomics studies revealed that the bioactivity likely resulted from interaction with Vangl1, a target not previously modulated by small molecules.14
Scheme 2.

a) Example natural products which show interesting bioactivity and contain an oxepane core. b) Short synthesis of oxepane core to allow rapid synthesis of a library of privileged oxepane containing scaffolds. c) Selected compounds resulting from oxepane based library which show intriguing activation of Wnt signaling (values shown are ED50 in the reporter gene assay).14
Waldmann and co-workers further realized the power of privileged core frameworks present in natural products to complete fragment-based design libraries termed pseudonatural products.15 These libraries result from the coupling of natural product-based fragments allowing the synthesis of new natural product-like entities to further fill biologically relevant chemical space. For a more in depth look into BIOS readers are directed to the following reviews.13a, 13b, 16
This brief overview of small molecule library synthesis highlights some key factors differentiating DOS and BIOS. DOS strives to indiscriminately synthesize many molecules that are structurally diverse, allowing the resulting library to be used to find initial compound hits for multiple known protein targets in a phenotypic screen or a biochemical screen for a particular cellular protein target. In BIOS, given that libraries are inspired by a natural product, they tend to have limited structural diversity based on a privliaged core structure and can be utilized when attempting to identify hit compounds for known or unknown protein targets.
2.2. Biology-Inspired, Target-Oriented Synthesis
Target-oriented synthesis is a primary arena for the discovery of novel bioactive small molecules and while practitioners currently set out to complete a total synthesis of a natural product along with development of an SAR profile, typically the gathering of such SAR information is considered following completion of a total synthesis. Historically, retrosynthetic analysis is applied to a natural product target to identify the most efficient and thereby most elegant synthesis, involving novel disconnections or synthetic strategies of a given natural product. While this approach has indeed led to numerous elegant and economic syntheses of complex natural products, the optimized routes are not always amenable to the collection of SAR information. To address this fundamental pitfall, many groups have devised strategies to more efficiently collect bioactivity data of natural products and their analogues as part of their total synthesis efforts. These strategies are discussed in detail below.
2.2.1. Danishefsky’s Diverted Total Synthesis (DTS)
Danishefsky’s diverted total synthesis (DTS) seeks to develop an efficient synthetic route to a natural product of interest utilizing traditional retrosynthetic analysis, working backwards to simple building blocks proceeding through one or more advanced intermediates in the usual manner. Following completion of a total synthesis, these advanced intermediates can serve as starting points for the synthesis of natural product analogues, alleviating the need to design a new synthetic strategy for SAR studies (Scheme 3).17 The recognition of DTS adds further value to total synthesis by providing a platform for enabling SAR efforts to build on a successful total synthesis campaign. Applications of DTS are agnostic to any knowledge of the cellular target.
Scheme 3.

A generic view of diverted total synthesis (DTS).
2.2.2. Vanderwal’s analogue-oriented synthesis (AOS)
Myers developed a convergent building block strategy to allow rapid access to a diverse array of antibiotics related to erythromycin18 possessing features reminiscent of both DOS and DTS. This was later termed analogue-oriented synthesis (AOS) by the Vanderwal group in their synthetic efforts toward lissoclimide analogues. AOS is again a target-oriented synthetic approach using retrosynthesis to devise routes to advanced intermediates, amenable to the synthesis of natural product analogues. However, in contrast to DTS, in the single example of AOS to date much more initial information regarding the cellular target of lissoclimide was known. The group had previously developed a biomimetic semi-synthesis of chlorolissoclimide (12, X = H, Y = Cl, R1 = H, R2 = OH, Scheme 4), 19 which allowed the formation of an X-ray co-crystal with the eukaryotic 60S ribosome, resulting in the identification of the binding pocket and basis for its bioactivity. 20 This enabled specific SAR questions to be posed by the Vanderwal group during initial development of the synthetic design toward lissoclimide derivatives. Thus, a synthetic strategy was devised to gain access to one or more advanced intermediates possessing orthogonal functionality, namely 13, 16, and 17 (Scheme 4). These intermediates enabled site-specific modification including substituent and functional group variations in addition to stereochemical modifications to answer specific SAR questions. As a result, the group noted key interactions with the 60S ribosome and gained increased knowledge of the SAR surrounding the lissoclimides, as translation-elongation inhibitors.20 Thus, while AOS has relationships with DTS, it is best suited for natural products in which specific SAR questions toward an established cellular target are being probed at the outset of a synthetic effort, as in the case of lissoclimide.
Scheme 4.

Vanderwal’s analogue-oriented synthesis applied to the lissoclimides.
2.2.3. Baran’s Two-Phase Synthesis
More recently, Baran and co-workers have disclosed their two-phase approach to the synthesis of bioactive natural products which attempts to mimic biological systems’ “cyclase” and “oxidase” phases. As such, a total synthesis takes on two phases, the first to access the carbon skeleton of the target natural product (cyclase phase) which through various site selective oxidations can be converted into not only the target natural product, but also related natural products and analogues of increasing oxidation (oxidase phase). Ideally, this will provide straightforward access to analogues and natural products of varying oxidation en route to the target natural product.21 Like DTS and AOS, this approach will increase knowledge of the target natural product families’ SAR profile while the required site-selective nature during the oxidase phase provides a playground for the development of new reactions to gain access to the desired targets selectively.
To date, the group has reported two synthetic endeavours employing this strategy; namely, the total synthesis of eudesmane terpenes21-22 and Taxol® (21).21, 23 The Baran laboratory validated their two-phase synthesis approach to the widely studied Taxol.® Despite many total synthesis efforts and years of clinical use, previous strategies toward Taxol® had not provided intermediates readily amenable to analogue synthesis. However, the Baran group found that by using the carbocyclic core, namely taxidienone (22) and precursor diketone 20, from the cyclase phase, an oxidase phase employing various strategies ultimately provided not only access to Taxol® (21), but also variants with differing oxidation states including taxadiene (23), decinnamoyltaxinine E (24a), taxabaccatin III (24b), and taxuyunnanine D (25) as well as non-natural derivatives all contributing to novel SAR information for the taxane family of natural products (Scheme 5b).
Scheme 5.

(a) A generic look at Baran's two-phase synthesis involving initial construction of the carbon skeleton followed by site-selective oxidation and functionalization to access intermediates and natural products of various oxidation state. (b) Application of two-phase synthesis to Taxol® providing access to various members of the taxane family.
2.2.3. Wender’s function-oriented synthesis (FOS)
The Wender group was one of the first to bring the function of a natural product to the forefront in synthetic planning. While the aforementioned approaches largely focus ultimately on natural product total synthesis, they aim to incorporate changes in a given synthetic route to access bioactive analogues. However, the complexity of natural products often results in only small quantities of bioactive natural products and can be obtained only after significant optimization. Wender and his group sought to address this issue by utilizing a strategy they termed function-oriented synthesis (FOS), wherein the function (interactions of a given bioactive small molecule) is considered early in a synthetic endeavor. Under this framework, the pharmacophoric elements of a natural product are identified using various techniques including the analysis of X-ray crystal structures of ligand-protein complexes and computational modelling of the natural product and simplified derivatives to ensure similar binding to the protein target. These studies ideally result in the realization of a simplified structure that retains only those elements important for bioactivity which is subsequently targeted for synthesis. An example of this approach entails the overlapping of the putative pharmacophore of two or more disparate natural products, as can be found in studies of bryostatin I, comparing competitive binders, phorbol ester and 1,2-diacyl-syn-glycerol resulting in a simplified equipotent or more potent analogue which can be synthesized more efficiently and in sufficient quantities to be useful.4b-d
One of the Wender group’s early and successful endeavours in application of FOS was toward studies of dynemicin (26), a member of a larger enediyne family of antitumor antibiotics known to induce DNA cleavage. The active species is unmasked post reductive epoxide cleavage, allowing a conformational change that is favorable for Bergman cyclization,24 resulting in the formation of arene diyl 28 (Figure 3). The arene diyl subsequently abstracts hydrogen atoms from proximal DNA strands resulting in strand cleavage.25 Thus the “functional” array of dynemicin is made up of the enediyne moiety as well as the epoxide/amine trigger which provides the conformational change necessary for the desired reactivity. The combination of these moieties was proposed to represent the key pharmacophoric elements of dynemicin (26). Thus, the simplified analogue enediyne 27 was designed and synthesized devoid of the A, B and E rings present in the natural product. The group utilized a carbamate protecting group (R) to remove electron density from the aniline functionality, acting as the trigger for enediyne 27 as opposed to the reduction required for dynemicin (26). Upon cleavage by acid, photochemical, or other conditions (dependent on N-protecting group) the increased electron density of the nitrogen triggers the epoxide opening, trapping of the resulting carbocation with a nucleophile, and Bergman cyclization to form arene diyl 28. Biological studies of the dynemicin mimic 27 (R= o-NO2PhCH2) revealed its ability to break both single and double stranded DNA in the presence of photoactivation. Requiring only 8 steps to synthesize and displaying comparable reactivity to the natural product dynemicin (26), this is a great example of the power of FOS.26
Figure 3.
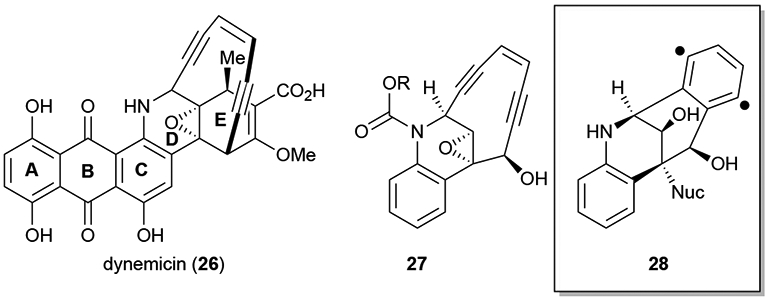
Dynemicin (26) and a FOS designed dynemicin mimic 27 and the targeted reactive arene diyl species involved in DNA scission by these enediyne antitumor antibiotics.
2.3.2. Shenvi’s Computed Affinity / Dynamically Ordered Retrosynthesis (CANDOR)
The Shenvi group has recently disclosed a strategy towards the synthesis of analogues of bioactive natural products which show inherent chemical and metabolic instability within their core structure termed computed affinity/dynamically ordered retrosynthesis (CANDOR). CANDOR changes the notion of a singular target molecule and instead looks to minimally perturb the initial target structure to simplify the molecule’s synthesis from both a feasibility and stability perspective, utilizing the inspiration of natural products as initial hits which can be developed into a drug lead. Once an idealized target has been identified, in silico techniques are utilized to determine if the target retains sufficient binding to the target protein. Synthesis is then commenced to access the new target molecule following traditional retrosynthetic analysis, which ideally displays improved properties both in terms of inherent reactivity and bioactivity. As a result of improved chemical properties of the designed analogue, the synthetic route can be utilized to complete analogue synthesis and develop an SAR profile.27 This strategy is based on similar ideology to FOS, however, less emphasis is placed on synthesizing analogues which maintain only necessary functionality, and instead focus is placed on minimal changes to a natural product’s complete structure which can aid in synthesis through stabilization while also gaining SAR information.
The Shenvi group initially disclosed this strategy in their efforts toward the synthesis of 20-nor-Salvinorin A 30 (Scheme 6). Salvinorin A (29), a natural product being pursued as an alternative to addictive opioids, has inherent instability which hinders lead optimization. The Shenvi group attributed this instability to 1,3-diaxial strain between the two axial methyl groups (29, Scheme 6). By way of dynamic ordered retrosynthesis, they imagined that simple deletion of one of the methyl groups would reduce the strain and improve the molecule’s stability. As such, in silico studies seemed to show similar binding to the target protein and thus a 10-step synthesis of 20-nor-Salvinorin A 30 commenced. The brevity of the sequence and increased stability resulting from the deleted methyl group allowed for reasonable scale synthesis and permitted the first studies of the SAR surrounding the aromatic furan moiety.27b It is important to note that in utilizing CANDOR, a prerequisite is knowledge of the target protein to allow verification that modifications to the target structure do not significantly impede binding. By simply modifying the bioactive natural product (removing a methyl group), the Shenvi group successfully addressed the instability problems associated with this natural product, while simplifying the synthesis and increasing SAR data for this neuroactive small molecule. Overall, CANDOR provides the potential of finding analogues which have enhanced drug-like properties which is often overlooked as a key parameter for a useful drug lead.
Scheme 6.
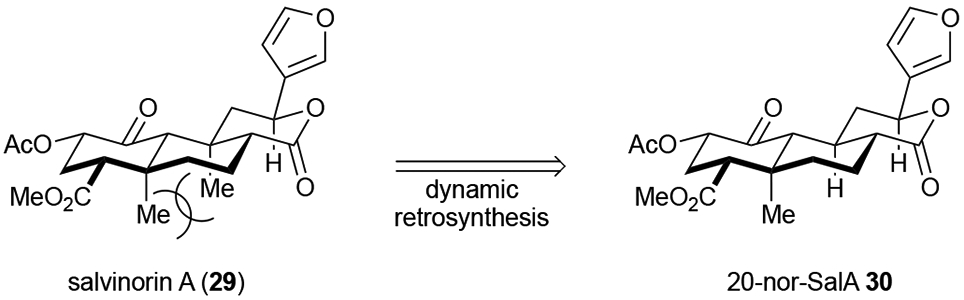
Salvinorin A as an example case for improvement on chemical properties aiding in synthesis of bioactive natural product analogues using CANDOR.
3. Pharmacophore-Directed Retrosynthesis (PDR)
Recently, we have started to approach our total synthesis efforts quite differently than in the past ~27 years of our independent research program. While we have always been driven by the synthesis of biologically active natural products toward gaining a molecular level understanding of their biological effects, our approach rarely strayed from classic total synthesis ideology, particularly with regard to traditional retrosynthetic analysis. We recently gained perspective for the shortcomings of this approach in hindsight due to our extensive and continued studies of the immunosuppressive and antitumor marine natural product, pateamine A (PatA, 31, Figure 4),28 a natural product first isolated by Munro and Blunt from a sponge off the New Zealand shores.29 We were initially drawn to PatA due to its novel structure, reported immunosuppressive activity, yet unknown mechanism of action. PatA was one of our group’s earliest success stories in delivering a reasonably efficient total synthesis, performing preliminary minimal SAR, and ultimately determining its mechanism of action through synthesis of a biotin conjugate. Through a fruitful collaboration with the Liu Group (John Hopkins), we determined that PatA exerts its potent antiproliferative activity by binding to elongation initiation factor-4A (eIF4A) resulting in inhibition of cap-dependent eukaryotic protein translation. Formation of the PatA-eIF4A complex stalls the translation initiation complex on mRNA in vitro leading to protein synthesis inhibition, stress granule formation, and ultimately apoptosis.28f-h, 28j However, we did not anticipate that our journey with PatA almost 25 years later would lead to the concept outlined herein, namely pharmacophore-directed retrosynthesis (PDR). We completed a total synthesis of PatA in 199828a and later that year we reported an improved synthetic route which in collaboration with the Liu group provided our first biological studies of PatA and related analogues.28b It took an additional six years before further studies were published detailing the design, synthesis, and bioactivity of PatA analogues, most notably DMDAPatA (33), which we proposed possessed the required pharmacophoric elements for PatA’s bioactivity reminiscent and certainly inspired by Wender’s FOS ideas. During our initial bioactivity studies of PatA and derivatives, we found that Boc-PatA (32) had only a ~6-fold decrease (0.3 vs 2.1 nM) in the IL-2 assay, which we were employing at the time to pursue the reported immunosuppressive activity, relative to the natural product. This led us to hypothesize that PatA consisted of a less conformationally constrained region (red, ‘scaffolding domain’, Figure 4) which could tolerate alteration, while modification of the more conformationally rigid region (‘binding domain’, blue) resulted in significantly diminished bioactivity. Thus, a convergent route to des-methyl, des-amino PatA (DMDAPatA) devoid of two of four total stereocenters was developed building on our established synthesis of PatA through application of DTS. Bioactivity studies subsequently revealed that DMDAPatA actually displayed similar bioactivity to the natural product.28c As our studies of PatA and its analogues continue to this day including collaborative animal studies for various cancers,28i the initial finding of DMDAPatA’s comparable activity to PatA with a greatly simplified structure led to an intriguing retrospective question. Could the design and synthesis of DMDAPatA have been conceived earlier and thus synthesized en route to pateamine A rather than many years later? This strategy, which we have termed ‘pharmacophore-directed retrosynthesis (PDR),’ builds on concepts first introduced by Wender to bring function to the forefront of any target-oriented synthesis project. Our continued interest in total synthesis of bioactive natural products, particularly those with unknown biological receptors, led us to conceive of PDR. To restate a question posited in our first disclosure of the concept of PDR: “Can the total synthesis of natural products, in particular with limited SAR or unknown or unconfirmed cellular targets, be more closely aligned to proposed biological activity during the retrosynthetic planning stages?”30 A total synthesis following PDR principles would allow the gathering of valuable SAR information during the course of a total synthesis since multiple intermediates possessing the putative pharmacophore would be accessed en route to the natural product. This ultimately increases the potential of identifying simplified equipotent versions much earlier in a total synthesis effort.
Figure 4.

PatA served as inspiration for development of pharmacophore-directed retrosynthesis. (IC50 values for the interleukin-2 reporter gene assay using the same population of Jurkat cells.)
We termed the strategy pharmacophore-directed retrosynthesis (PDR) to emphasize the importance of considering the pharmacophore or “pharmacophoric” elements of a natural product at the retrosynthetic planning stage, thus allowing the design of a total synthesis wherein the putative pharmacophore is synthesized first, followed by more elaborate derivatives with increasing structural complexity en route to the natural product. PDR aims to utilize Wender’s notion of bringing function to the forefront of synthesis (FOS) while employing the logic of retrosynthesis to target simplified derivatives bearing the proposed pharmacophore. This approach has the potential to reveal simplified analogues much earlier in a total synthesis effort with the important caveat that for many natural products, but certainly not all,26b, 31 the entire structure may indeed be required to obtain comparable bioactivity to the natural product (Scheme 7). However, application of PDR enables one to determine this requirement prior to completion of the natural product which in some cases may be available in quantities useful for subsequent control experiments in biological studies.
Scheme 7.

Overview of various synthetic strategies bring biological function to the forefront including those employed to utilize bioactive natural products as starting points for biological studies building on ideas of FOS with comparison to PDR. The latter enables gathering of SAR information en route to a given natural product via target-oriented synthesis.
PDR was developed to be applied to natural products in which only minimal information is known regarding the structural features required for bioactivity and limited or no information on its putative cellular target(s). If this information is known, one can apply the aforementioned strategies such as AOS, FOS, or CANDOR to identify simplified, more equipotent derivatives. To apply PDR to a given natural product, one must first develop a hypothesis for the ‘pharmacophore’ or ‘minimal structural requirements for bioactivity’ for a particular natural product or class of natural products. It is important to note that within PDR, the term ‘pharmacophore’ is more loosely used compared to what a medicinal chemist might use as a definition taking into account all hydrogen-bonding, hydrophobic interaction, π–π interactions, etc. Thus, application of PDR does not require prior identification of the cellular target nor docking experiments to reveal the actual pharmacophore; however, computational methods could be used to ascertain the major conformation and exposed hydrogen-bonding donor/acceptors of the natural product. Furthermore, the absence of cellular target identity precludes the use of both structure and ligand-based modelling to aid in actual pharmacophore determination as in FOS. Thus, to apply PDR, the development of a hypothesis for a ‘pharmacophore’ is required and can be informed by several considerations: i) overall structural and conformational analysis of the natural product with chemical intuition, ii) existing SAR data of natural product congeners or biosynthetic precursors often provided by isolation chemists, iii) the bioactivity of structurally related natural products, and in the easiest application iv) the presence of reactive functionality which may covalently modify cellular targets (e.g. β-lactones, epoxides, aldehydes).
With a proposed pharmacophore in hand, a retrosynthesis is designed to access the hypothesized pharmacophore early in the total synthesis effort, and subsequent elaboration to more complex pharmacophore-containing intermediates enables gathering of SAR information at a much earlier stage of total synthesis efforts (Scheme 7). The forward synthesis can proceed in stages. Stage I seeks to synthesize the simplest form of the proposed pharmacophore. Ideally, the target from stage I will be utilized as a synthon to access more complex derivatives in Stage II, however this may not always be practical, especially in cases where the proposed pharmacophore is reactive or unstable. Finally, natural product derivatives synthesized in Stage II can be utilized as intermediates for the synthesis of the natural product and closely related derivatives (Stage III). The derivatives resulting from both Stages I and II and the natural product will then serve as the platform providing valuable SAR information regarding the natural product’s bioactivity. Inevitably, this will lead to further questions regarding the motifs and moieties present on the synthetic derivatives and natural product. Fortunately, as previously mentioned, the PDR approach towards synthesis provides multiple advanced intermediates which can be utilized for a final stage (Stage IV) to SAR ‘gap fill’ utilizing Danishefsky’s DTS without the need to develop a new synthetic strategy to allow for derivatization. The stages not only help to provide a near term target, but they also help facilitate concurrent synthetic and biological studies.
Importantly, as with many of the approaches outlined in Section 2, PDR prioritizes the collection of SAR information for natural products; therefore, while the economies of synthesis are still of concern, it holds lower priority, as displayed in the primarily linear routes required to utilize PDR for full benefit. To date, we have applied PDR to two natural products that indeed led to the identification of simplified natural product derivatives possessing in some cases more potent activity compared to the natural product, providing initial proof of concept for the utility of PDR.
3.1. PDR Applied to the Gracilins
The gracilins, isolated from Spongionella gracilis,32 are a family of natural products, many of which contain a unique bis-acetoxy furanose (Figure 5, red), wherein gracilin A was reported to possess both neuroprotective and immunosuppressive activity.30, 32-33 Given that only minimal information regarding SAR and no prior synthetic work was available for these natural products, we targeted gracilin A (34) for application of PDR to interrogate these two bioactivities in a collaborative effort with the Botana group (Universidad de Santiago de Compastella) who had reported potential interactions of these sponge isolates, particularly gracillin A, with the cyclophilins.
Figure 5.

a. Members of the spongiane family of diterpene natural products including gracilin A (34). b. Macfarlandin E (39) and t-butyl derivative 40 bearing a related masked 1,4-ketoaldehyde and shown to undergo Paal-Knorr pyrrole synthesis with lysine in simulated physiological conditions.
As described above, the first step of applying PDR to a target molecule is the development of a hypothesized pharmacophore. In the case of gracilin A and related congeners what readily stood was the common bis-acetoxy furanose moiety (Figure 5a) which can be seen as a masked 1,4-dialdehyde that upon hydrolysis could engage protein targets through Schiff base formation and further condensation, Paal-Knorr pyrrole synthesis.34 In the case of macfarlandin E (39, Figure 5b), the Overman group had demonstrated that a simplified t-butyl derivative 40, possessing a similar masked 1,4-dialdehyde engaged lysine in a Paal-Knorr condensation under simulated physiological conditions.35 Furthermore, computational studies provided evidence that the bis-acetoxy motif of gracilin A (34) and a structurally related natural product aplysulphurin-1 (not shown) binds divalent cations including Ca2+, which might be related to their bioactivity. Thus, these lines of evidence led to our hypothesis that the pharmacophore of gracilin A is the bis-acetoxy furanose which became our initial synthetic target for stage I. We therefore developed a retrosynthetic analysis that would intercept multiple intermediates bearing the hypothesized bis-acetoxy furanose pharmacophore (red). Specifically, simple tetrahydrofuran 43 (Stage I), the simplified bicycle 42 (Stage Iand tricycle 41 (Stage III, Scheme 8) were initially targeted for synthesis through application of PDR.
Scheme 8.

Various stages of PDR applied to gracilin A.
In Stage I, oxidation and reduction of furan gave the minimal pharmacophore as a mixture of diastereomers 43/45 (Scheme 9) which were assayed together. In practice, the reactivity of the bis-acetoxy furanose did not allow for maintenance of this moiety throughout the sequence, rather it was introduced in just a few steps from various intermediates. The core of the gracilins was available in optically active form employing our recently disclosed Diels-Alder lactonization organocascade to afford the bicyclic TBS enol ether 46.36 This key intermediate provided access to the simple bicyclic core 42 through diol 47, and also the tricyclic derivative 41 via lactone 48. The tricyclic derivative 41, obtained as a separable mixture of diastereomers and regioisomers, possessed all functionality found in gracilin A (34) with the exception of the exocyclic alkylidene. Given the availability of gracilin A (34), its synthesis was not required as a comparator for biological studies, precluding the need to complete Stage III in this case.
Scheme 9.

An overview of route used to complete stage I and II of the gracilin PDR route, showing intermediates both used for testing and as starting points for stage IV DTS.
A compelling case for application of PDR to natural products came from the fact that a simplified derivative of gracilin A displayed nanomolar activity toward cyclophilin A as determined by surface plasmon resonance (SPR) (KD 5.34 ± 1.68 nM) measurements which was ~500X more potent than gracillin A (Figure 6). Our studies further revealed the necessity of the cyclohexyl moiety since highly simplified bis-acetoxy furanoses 42 and 4330 were unsurprisingly inactive while an interesting interplay between the C10-quaternary carbon stereochemistry and the alkene regiochemistry was revealed by careful HPLC separation of all four diastereomers of tricyclic derivatives 41 that were not initially optimized for diastereoselectivity nor regioselectivity.
Figure 6.

Selected bioactivity data resulting from PDR applied to the gracilins highlighting the importance of C10 stereochemisty and the superfluous exocyclic alkene.
As expected, a number of new questions were generated upon initial synthesis of gracilin A derivatives through application of PDR. To expand the SAR profile through ‘gap-filling’ and to specifically answer questions regarding selectivity for immunosuppressive vs neuroprotective activity of the gracilin derivatives through interaction with either CypD versus CypA, we made use of DTS with intermediates previously synthesized. This enabled a more comprehensive view of structural features that led to the greatest selectivity between CypD and CypA and therefore selective neuroprotection or immunosuppression.25
3.2. PDR Applied to Rameswaralide
Rameswaralide (56) is a cembranoid natural product first isolated from the soft coral Sinularia dissecta in 199837 and later from the related Sinularia inelegans (Figure 7). The latter study reported the absolute stereochemistry of rameswaralide as determined by X-ray crystallography.38 The cembranoid natural products isolated are notoriously complex, highly oxygenated and caged, containing a unique 5,5,7,6 fused ring system in the case of rameswaralide (56) and the related norcembranoid ineleganolide (55),39 while other related members contain a 5,5,6,7 fused ring system.40 Little information has been collected on the bioactivity of the members within this family. While the bioactivity of ineleganolide has been disputed, the 5,5,7-fused (blue) ABC tricyclic core-containing natural products have generally displayed greater cytotoxicity compared to those members possessing a 5,5,6-fused (red) tricyclic ABC core.37-39, 41 Limited availability of these natural products has precluded more extensive bioactivity studies.
Figure 7.

Cembranoid and norcembranoid natural products related to rameswaralide.
The bioactivity and complexity of these molecules has stimulated interest for several synthetic studies including those toward rameswaralide (56),42 ineleganolide (55),43 scabrolides A & B (52),44 and yonarolide (51).45 In addition, biomimetic strategies toward ineleganolide (55) and sinulochmodin C (54)46 were reported. However, despite these extensive synthetic efforts, only the biomimetic synthesis of ineleganolide (55) and sinulochmodin C (54) by Pattenden46 and the recent de novo synthesis of scabrolide A (52) by the Stoltz group44 were successful. A desire to gain further SAR information for this bioactive Sinularia family of natural products while also gaining access to further quantities of these rare natural products made them an ideal opportunity to further explore and apply our PDR strategy as described below.42d
As described above, with only minimal SAR studies of the Sinularia diterpenes mainly coming from continued isolation and bioassaying of family members, the challenge of applying PDR in this context is clear. However, we decided to focus our efforts on rameswaralide since it contains what we hypothesize to be the more bioactive 5,5,7-fused ABC tricyclic core and fewer synthetic efforts have been reported for this diterpene. Other possible functional arrays for inclusion in a proposed pharmacophore are the α,β-unsaturated γ-lactone present in rameswaralide (56), a moiety known to be reactive as a Michael acceptor; however, this is not conserved across the family of natural products nor is the C-ring tertiary alcohol nestled in the concave face of this caged molecule masked as a tetrahydrofuran in ineleganolide (55). We hypothesized that the ABC core may play a prominent role in bioactivity while the D rings displaying greater variations in structure may be less important. We therefore developed a PDR strategy that involved synthesis of a 5,5,6- tricyclic ABC core which could then be elaborated through ring expansion to the 5,5,7 via ring expansion followed by annulations of various D rings including that found in rameswaralide (Scheme 10). This strategy would provide SAR information for the two simplified ABC cores 57/58 in Stages I and II of PDR and annulation of D rings in Stage III would deliver rameswaralide and closely related congeners. Further SAR gap filling would also be possible through application of DTS employing the advanced tricyclic cores 57/58.
Scheme 10.

PDR applied to rameswaralide (56) guided by the proposed pharmacophore consisting of the common 5,5,7-fused ABC ring system to be accessed in Stages I and II of PDR via the 5,5,6-fused ring system common to other family members.
We therefore planned our PDR route toward rameswaralide to enable access to the 5,5,6 core skeleton 58 followed by ring expansion to the 5,5,7 core skeleton 57 (Scheme 10). D-Ring annulation would then allow access to a minimally functionalized tetracycle which could be converted to rameswaralide (56) along with related congeners. Our synthetic strategy proceeding through both 5,5,6- and 5,5,7-fused ABC tricyclic cores, setting up Stages I and II of PDR to potentially allow access to several members of this natural product family and further SAR gap filling, enabled through DTS employing advanced tricycles 57 and 58.
Our application of PDR to rameswaralide led to a linear strategy enabling gradual and systematic increases in complexity throughout the course of our synthetic studies maximizing SAR information gathering. Our Diels-Alder lactonization (DAL) organocascade36 was well suited to provide rapid access to the protected 5,5,6-tricyclic core 63, proceeding in 77% yield with high diastereoselectivity (>20:1) (Scheme 11). There is an additional benefit of a PDR approach to total synthesis which is enhanced further if attempts are made to minimize protecting groups.47 Namely, undesired products typically only used to understand side-reactions for optimization purposes, may in fact bear the proposed pharmacophore and can add to the overall SAR profile. This was first realized in the key DAL step which under our initial conditions, elimination of the tertiary alcohol and hydrolysis of the enol ether, resulted in the formation of enone 61. This by-product corresponds to the core of yonarolide (51) which through subsequent dehydrogenation with IBX delivered an interesting dienone 62 for biological testing while also serving as a starting point for an ongoing synthesis of yonoralide by an undergraduate student in our group. We were ultimately able to optimize the DAL to provide the tricyclic enol ether 63 in 77% yield while avoiding elimination of the tertiary alcohol. Transposition of the enol ether provided enol ether 64 which allowed access to the first targeted pharmacophore, tricyclic ketone 58. To access the targeted 5,5,7-tricyclic core, we developed a cyclopropanation/ring expansion strategy. Once again, failed experiments, in this case a silver-mediated ring expansion, led to additional tricyclic derivatives 67 and 68 that were assayed for bioactivity. Pyranone 67 which bears some resemblance to the core of ineleganolide (55) was formed through spontaneous oxa-Michael addition of the tertiary alcohol onto the generated cycloheptenone pointing to the caged nature of these molecules. We ultimately achieved the desired ring expansion to provide epoxide 69 which bears resemblance to the core of rameswaralide and is currently serving as a useful intermediate toward D-ring annulation. Through application of DTS, we also synthesized an additional enone-bearing derivative 65 (core of scabrolide A) from ketone 58 for testing.42d
Scheme 11.

Optimized synthetic sequence toward a potential rameswaralide precursor 69 involving a key Diels-Alder lactonization to deliver tricycle 63. Unanticipated reactions leading to by-products in a PDR strategy coupled with minimizing protecting groups and redox manipulation can result in direct access to interesting derivatives for bioactivity testing e.g. truncated ABC tricyclic derivatives 61, 67, 68.
Stage I and preliminary Stage II of our recently disclosed PDR approach to rameswaralide delivered several tricyclic derivatives that were assayed by the Liu Group (Johns Hopkins) to assess cytotoxicity toward three cancer cell lines (HCT116, colon; MDA-MB-231, breast; and A549, lung) along with a non-cancerous cell line (human umbilical vein cells, HUVEC) to assess selectivity. Several of our intermediates possessed moieties known to result in non-selective toxicity including Michael acceptors and an epoxide. Despite this, we acquired some useful initial SAR information and some unexpected selective cytotoxicity (Figure 8). Some broader lessons can be gleaned from derivatives tested thus far. To date, our hypothesis of greater bioactivity associated with 5,5,6 vs 5,5,7 holds true when comparing dienones 62 and 68. Furthermore, we found that not unexpectedly, epoxy bromo enone 69 displayed the most potent cytotoxicity potentially due to the two electrophilic moieties present. However, what was unexpected was the cell line selectivity toward the HCT116 colon cancer cell line for both dienone 62 and bromoenone 69 (3-10X selectivity over A549 cells). It is also interesting to note that two simplified derivatives were already more cytotoxic against A549 cells compared to rameswaralide (56, IC50 of 67 ± 3.7 μM) however this may due to the presence of Michael acceptors.38 We recognize that these simplified intermediates may not share the same protein target(s) as the natural products themselves, but as we push to complete the PDR approach to rameswaralide (56) we expect to shed light on this question through competitive binding studies with the natural products themselves.
Figure 8.

Biological data obtained from preliminary stage I & II analogues showing increased potency compared to rameswaralide and intriguing selectivity toward HCT116 cells. aRameswaralide was previously assayed only against the A549 cell line (IC50 of 67 ± 3.7 μM).35
There is still much to learn from the application of PDR to rameswaralide (56); however, through Stage I and preliminary Stage II studies, we have managed to identify three bioactive simplified variants and we will expand upon our understanding of their overall SAR upon completing Stages III and, if needed, application of DTS in Stage IV. Application of PDR to rameswaralide (56) nicely demonstrates the goal of synthesizing increasingly complex structures en route to the natural product and a goal of PDR of ‘testing as you go’ rather than after a total synthesis is completed. In addition, this synthesis demonstrated that side-reactions can provide useful probes to broader SAR information when principles of PDR are applied in conjunction with minimizing protecting groups and redox manipulations.
3.3. PDR Applied to Ophiobolin A
Ophiobolin A (71, OpA),48 belonging to a large family of highly bioactive, fungal-derived sesterterpenoids49 has drawn extensive interest from the synthetic community owing to its synthetically challenging and intriguing 5,8,5,5 ring system and both its unique selectivity toward cancer stem cells (CSCs)50 and ability to induce paraptosis.51 Thus, OpA is a great candidate to apply PDR in an attempt to gain a better understanding of structure-activity relationships inducing paraptosis in particular, why OpA and ophiobolin B (72, OpB)52 exhibit CSC selectivity, and ultimately to determine putative cellular targets of the ophiobolins.
Widespread interest in the ophiobolins has led to some preliminary SAR data,49, 53 which combined with our group’s recent work on OpA and derivatives54 suggested the importance of retaining the integrity of the A and B ring to maintain the desired biological effects since both 3-deoxy and 6-epi-OpA49 both exhibited decreased potency. Furthermore, OpA contains a 1,4-ketoaldehyde moiety appended to the A ring which has the potential to react with primary amines (lysine) through a Paal-Knorr pyrrole reaction.55 Given the necessity of the 1,4-keto aldehyde embedded in a 5,8-bicyclic (AB) ring system, the C3-hydroxyl, and C6-relative stereochemistry, we proposed that their combination constitutes the hypothetical pharmacophore of OpA (Figure 9, red). This led us to propose the following PDR route wherein the simplest form of the pharmacophore, monocyclic 1,4-keto aldehyde 75 is synthesized first in Stage I, followed by simplified bicyclic and tricyclic derivatives (74 and 73 respectively) which all contain the proposed pharmacophore and can provide valuable SAR information on both activity and selectivity of OpA toward CSCs. We recently described Stage I and II toward OpA54 and Stage III will be the completion of OpA and closely related congeners synthesized in route to OpA followed by SAR gap filling as needed utilizing DTS in Stage IV.
Figure 9.

Structure of ophiobolins A & B. showing proposed pharmacophore (red).
Applying PDR to OpA takes on a slightly different form than that described for both gracilin A (34) and rameswaralide (56). Optimal PDR again takes a more linear approach such that each stage can commence from the target of the previous stage (i.e. the product of Stage I is the starting material for Stage II). In the case of gracilin A, the reactivity of the proposed pharmacophore prevented application of PDR in this manner while upon application to rameswaralide, we have for the most part been able to follow this framework. In the case of OpA, it is not reactivity but rather synthetic feasibility and efficiency that results in an inability to, in a strict sense, complete the application of PDR in a “linear” fashion. However, we imagined that the chemistry needed to complete the total synthesis, namely forming the central 8-membered B-ring, synthesizing the A-ring involving a diastereoselective conjugate addition/protonation could also be utilized in the synthesis of simplified tricyclic derivatives (e.g. 73 respectively) in Stage II. Therefore, Stage II would not only provide probes for bioactivity studies, but would also serve to model the chemistry that would be used to complete tricyclic derivatives and importantly the total synthesis. This has resulted in the adaptation of the slogan “more than a model” in our group (sung to the tune of “More than a Feeling” by Boston), where model systems, which have been utilized extensively by synthetic chemist to explore late stage, novel, or challenging transformations,56 have the added benefit of enabling access to simplified natural product derivatives and therefore SAR studies through application of PDR.
We have recently disclosed the completion of Stage I and initial Stage II studies57 in which application of PDR enabled optimization of the A-ring synthesis involving a substrate-controlled conjugate addition and facially-selective protonation, key for installation of the critical C6-stereocenter. This one pot process enabled would ultimately enable annulation of the B and C rings, and closure of the 8-membered ring using Nakada’s RCM strategy.58 Again, these transformations serve as model studies for eventual application to synthesis of tricyclic derivatives and ultimately OpA while also providing interesting simplified derivatives for biological studies toward developing an SAR profile. The synthetic studies conducted in Stage II of PDR enabled us to address multiple challenges that were solved and these will ultimately be applied to tricyclic derivatives. Particularly, it was found that a typical cuprate addition of neopentyl iodide 76 into enone 78 was not possible and instead a higher order cuprate 77 was required. This unfortunately also revealed that mono and di substituted alkene variations of cuprate 77 quickly underwent 5-exo-trig cyclization. Furthermore, it was found that the terminal alkene a des-phenyl variant of the trisubstituted alkene 80 quickly isomerized to give the corresponding conjugated enone. Thus, the knowledge gained through these studies was compiled to devise the synthesis of OpA bicyclic derivatives (Scheme 13) and is also proving extremely useful in informing the chemistry to synthesize both tricyclic derivatives and the natural product itself.57
Scheme 13.
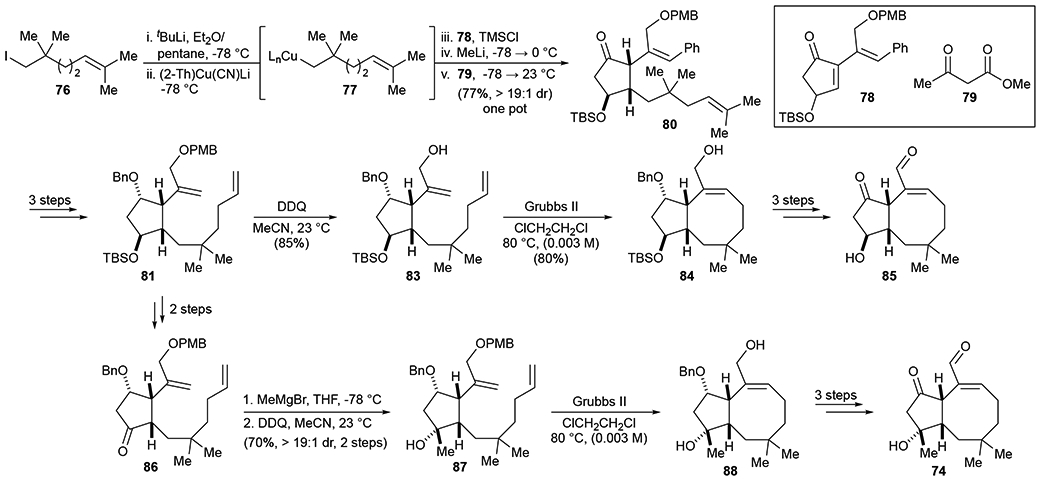
Optimized synthesis of Stage II bicyclic target 74 and related bicycle 85.
Beyond the extensive synthetic knowledge gleaned from the synthesis of the bicyclic derivative 74, we also obtained some interesting biological activity from the bicyclic derivatives of OpA. Given the ease of accessing the epimeric C3-des-methyl bicyclic derivative bearing a secondary alcohol, bicyclic derivative 85, this was also synthesized to provide information regarding the importance of the C3 alcohol stereochemistry. OpA, bicyclic derivatives 74 and 85, and the proposed highly simplified ketoaldehyde 75 were all tested in two cell lines, a non-cancerous cell line (MCF10A) and a breast cancer cell line (MDA-MB-231) to assess the cytotoxicity of the synthesized derivatives as well as their selectivity (Scheme 14). While both bicyclic derivatives (74 and 85) displayed low micromolar cytotoxicity, they did not show the 3-fold selectivity displayed by OpA. The bioactivity for bicyclic derivatives supports the hypothesis that the 1,4-ketoaldehyde embedded in a 5,8-bicyclic ring system constitutes a major portion of the pharmacophore as proposed, however the lack of the C ring and attendant substituents likely decreases the selectivity of these bicyclic derivatives. It was also interesting to find that the stereochemistry and degree of substitution at C3 is not vital for these bicyclic derivatives, as both secondary alcohol 85 and tertiary alcohol 74 displayed similar activities. Therefore, it will be illuminating to explore C3-substitution on future OpA derivatives throughout the remainder of Stage II and into Stage IV of PDR.
Scheme 14.

Biological activity resulting from Stage I and initial Stage II application of PDR to Ophiobolin A (71).
By applying a PDR route toward OpA, we successfully developed a reliable synthetic approach to construct a functionalized A ring, strategies to annulate the B ring, including conditions to perform the diastereoselective conjugate addition/protonation to set the key C6-stereocenter. Application of PDR enabled crucial model studies to optimize the ring-closing metathesis to install the 8-membered B-ring. Importantly, this work also serves as model studies to develop chemistry which will likely be suitable to construct tricyclic derivatives of OpA and closely related congeners of OpA including truncated variants, not easily accessible from the natural product itself. This application of PDR to OpA leading to simplified bicyclic variants, which again are in fact model studies for eventual total synthesis, provides support for the proposed OpA pharmacophore and demonstrates the concept of “more than a model.”
4. Summary and Outlook
The enduring utility of natural products will continue to be realized for many years to come, particularly in human medicine. Due to the sheer structural and stereochemical complexity of varied natural products, chemists have gravitated toward synthetic strategies with a goal of coupling their synthetic efforts with the harvesting of their rich information content in an economic and time efficient manner. Wender’s function-oriented synthesis (FOS) encouraged chemists to begin marrying their synthetic efforts with biological function. Danishefsky’s diverted-total synthesis (DTS) approach recognized the value of key intermediates in a total synthesis effort as starting points for medicinal chemistry programs. Baran’s two-phase synthesis, building on elements of biosynthesis has great potential to explore in a truly medicinal chemistry approach, the bioactivity of increasingly complex structures toward natural products as recently exemplified by their two-phase synthesis of Taxol® (21). However, the continued development of broadly applicable but selective CH oxidation methods will be required to realize the full potential of this approach. Analogue-oriented synthesis from the labs of Myers and Vanderwal, which builds on known cellular targets of a given natural product, guides analog synthesis providing a means to design derivatives likely to have bioactivity related to the original natural product. CANDOR takes into account the often-overlooked parameter of pharmacokinetic properties of natural products into the design of natural product derivatives. Our lab’s pharmacophore-directed retrosynthesis (PDR) strategy, ideally suited to natural products with little to no SAR information or information regarding cellular targets, employs the logic of retrosynthesis to guide the discovery of equipotent simplified natural product derivatives at a much earlier stage in a total synthesis effort. This is achieved by developing a hypothesis of a natural product’s pharmacophore, loosely applied, and using this hypothesis to guide the retrosynthesis by targeting the proposed pharmacophore first, followed by elaboration of that structure with increasingly complexity to access the natural product or possibly an equipotent derivative of the natural product along the way which may preclude the need to synthesize the natural product. This may especially be prudent in cases where it is available in reasonable quantities from natural sources. We described in this review three initial case studies of PDR involving gracilin A (34) and preliminary application to rameswaralide (56) and ophiobolin A (71), demonstrating that this strategy can indeed lead to bioactive simplified derivatives of a given natural product en route to a total synthesis. It is important to note that simplified bioactive natural products have the potential to serve as starting points for fragment-based design59 to further develop initial hit compounds into potential lead compounds.
While to date we have realized success with the PDR strategy, the development of hypotheses regarding a natural product’s actual pharmacophore, absent an obvious electrophilic moiety, remains a primary challenge but one that can be informed by various strategies as described above. However, the great potential to identify simplified derivatives of a natural product through application of PDR would seem to balance any concern about having chosen the ‘incorrect pharmacophore.’ Our lab will continue to push the frontiers of total synthesis through the lens of PDR to gather biological information en route to complex natural products. Furthermore, we will continue to evaluate the efficacy of this approach, expand its applicability, and test its limits in our ongoing and future total synthesis efforts toward harvesting the rich and enduring potential of natural products to serve as lead molecules for drug discovery.
Figure 1.

Approximate timeline for the evolution of various synthetic strategies marrying natural product total synthesis and biological studies, including the gathering of structure-activity relationship information.
Scheme 12.
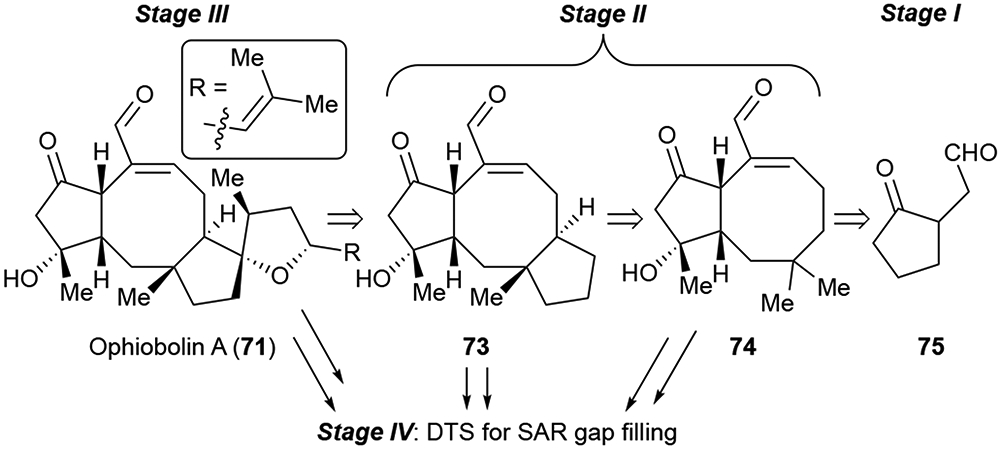
PDR approach to ophiobolin A (71) guided by the 1,4-ketoaldehyde embedded in the 5,8-bicyclic (AB) ring system as the proposed pharmacophore.
Acknowledgements
We thank the National Institute of General Medical Sciences at NIH (GM134910, GM052964) for generous support of our group’s research program including work leading to the concept of PDR described herein. A Ruth L. Kirschstein Predoctoral Fellowship (F31 GRANT12692796, to N.J.T) is also gratefully acknowledged.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Dedicated to Profs. Murray H. G. Munro and John W. Blunt for their incredible marine natural product isolation feats including pateamine A.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Wöhler F, Ann. Phys. (Berlin), 1828, 87, 253–256. [Google Scholar]
- 2.Corey EJ and Cheng XM, The Logic of Chemical Synthesis, Wiley, 1989. [Google Scholar]
- 3.Hendrickson JB, J. Am. Chem. Soc, 1975, 97, 5784–5800. [Google Scholar]
- 4.(a) Wender PA, Croatt MP and Witulski B, Tetrahedron, 2006, 62, 7505–7511; [Google Scholar]; (b) Wender PA, Verma VA, Paxton TJ and Pillow TH, Acc. Chem. Res, 2008, 41, 40–49; [DOI] [PubMed] [Google Scholar]; (c) Wender PA, Tetrahedron, 2013, 69, 7529–7550; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wender PA, Nat. Prod. Rep, 2014, 31, 433–440. [DOI] [PubMed] [Google Scholar]
- 5.(a) Trost BM, Science, 1991, 254, 1471. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Angew. Chem. Int. Ed, 1995, 34, 259–281 [Google Scholar]; (c) Trost BM, Acc. Chem. Res, 2002, 35, 695–705. [DOI] [PubMed] [Google Scholar]
- 6.Newhouse T, Baran PS and Hoffmann RW, Chem. Soc. Rev, 2009, 38, 3010–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newman DJ and Cragg GM, J. Nat. Prod, 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- 8.(a) Tan DS, Foley MA, Shair MD and Schreiber SL, J. Am. Chem. Soc, 1998, 120, 8565–8566; [Google Scholar]; (b) Schreiber SL, Science, 2000, 287, 1964–1969; [DOI] [PubMed] [Google Scholar]; (c) Furka À, Sebestyén F, Asgedom M and Dibó G, Int. J. Pept. Protein Res, 1991, 37, 487–493; [DOI] [PubMed] [Google Scholar]; (d) Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM and Knapp RJ, Nature, 1991, 354, 82–84; [DOI] [PubMed] [Google Scholar]; (e) Burke MD and Schreiber SL, Angew. Chem. Int. Ed, 2004, 43, 46–58; [DOI] [PubMed] [Google Scholar]; (f) Burke MD, Berger EM and Schreiber SL, J. Am. Chem. Soc, 2004, 126, 14095–14104 [DOI] [PubMed] [Google Scholar]; (g) Sternson SM, Louca JB, Wong JC and Schreiber SL, J. Am. Chem. Soc, 2001, 123, 1740–1747 [DOI] [PubMed] [Google Scholar]; (h) Lee D, Sello JK and Schreiber SL, Org. Lett, 2000, 2, 709–712. [DOI] [PubMed] [Google Scholar]
- 9.Galloway WRJD, Isidro-Llobet A and Spring DR, Nat. Commun, 2010, 1, 80. [DOI] [PubMed] [Google Scholar]
- 10.Pavlinov I, Gerlach EM and Aldrich LN, Org. Biomol. Chem, 2019, 17, 1608–1623. [DOI] [PubMed] [Google Scholar]
- 11.(a) Nielsen TE and Schreiber SL, Angew. Chem. Int. Ed, 2008, 47, 48–56; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) O' Connor CJ, Beckmann HSG and Spring DR, Chem. Soc. Rev, 2012, 41, 4444–4456; [DOI] [PubMed] [Google Scholar]; (c) Schreiber SL, Chem. Eng. News, 2003, 81, 51–61 [Google Scholar]; (d) Spring DR, Org. Biomol. Chem, 2003, 1, 3867–3870. [DOI] [PubMed] [Google Scholar]
- 12.Dandapani S, Germain AR, Jewett I, le Quement S, Marie J-C, Muncipinto G, Duvall JR, Carmody LC, Perez JR, Engel JC, Gut J, Kellar D, Siqueira-Neto JL, McKerrow JH, Kaiser M, Rodriguez A, Palmer MA, Foley M, Schreiber SL and Munoz B, ACS Med. Chem. Lett, 2014, 5, 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Wetzel S, Bon RS, Kumar K and Waldmann H, Angew. Chem. Int. Ed, 2011, 50, 10800–10826; [DOI] [PubMed] [Google Scholar]; (b) van Hattum H and Waldmann H, J. Am. Chem. Soc, 2014, 136, 11853–11859; [DOI] [PubMed] [Google Scholar]; (c) Kumar K and Waldmann H, Angew. Chem. Int. Ed, 2009, 48, 3224–3242. [DOI] [PubMed] [Google Scholar]
- 14.Basu S, Ellinger B, Rizzo S, Deraeve C, Schürmann M, Preut H, Arndt H-D and Waldmann H, Proc. Natl. Acad. Sci. USA, 2011, 108, 6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karageorgis G, Foley DJ, Laraia L and Waldmann H, Nat. Chem, 2020, 12, 227–235. [DOI] [PubMed] [Google Scholar]
- 16.(a) Kaiser M, Wetzel S, Kumar K and Waldmann H, Cell. Mol. Life Sci, 2008, 65, 1186–1201; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bon RS and Waldmann H, Acc. Chem. Res, 2010, 43, 1103–1114; [DOI] [PubMed] [Google Scholar]; (c) Karageorgis G and Waldmann H, Synthesis, 2018, 51, 55–66; [Google Scholar]; (d) Wolfram W, Tobias JZ, Markus K and Herbert W, Biological Chemistry, 2010, 391, 491–497.20030592 [Google Scholar]
- 17.Wilson RM and Danishefsky SJ, J. Org. Chem, 2006, 71, 8329–8351. [DOI] [PubMed] [Google Scholar]
- 18.Seiple IB, Zhang Z, Jakubec P, Langlois-Mercier A, Wright PM, Hog DT, Yabu K, Allu SR, Fukuzaki T, Carlsen PN, Kitamura Y, Zhou X, Condakes ML, Szczypiński FT, Green WD and Myers AG, Nature, 2016, 533, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quinn RK, Könst ZA, Michalak SE, Schmidt Y, Szklarski AR, Flores AR, Nam S, Horne DA, Vanderwal CD and Alexanian EJ, J. Am. Chem. Soc, 2016, 138, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Könst ZA, Szklarski AR, Pellegrino S, Michalak SE, Meyer M, Zanette C, Cencic R, Nam S, Voora VK, Horne DA, Pelletier J, Mobley DL, Yusupova G, Yusupov M and Vanderwal CD, Nat. Chem, 2017, 9, 1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishihara Y and Baran PS, Synlett, 2010, 2010, 1733–1745. [Google Scholar]
- 22.(a) Chen K and Baran PS, Nature, 2009, 459, 824–828; [DOI] [PubMed] [Google Scholar]; (b) Chen K, Ishihara Y, Galan MM and Baran PS, Tetrahedron, 2010, 66, 4738–4744. [Google Scholar]
- 23.(a) Mendoza A, Ishihara Y and Baran PS, Nat. Chem, 2012, 4, 21–25; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wilde NC, Isomura M, Mendoza A and Baran PS, J. Am. Chem. Soc, 2014, 136, 4909–4912; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yuan C, Jin Y, Wilde NC and Baran PS, Angew. Chem. Int. Ed, 2016, 55, 8280–8284; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kanda Y, Nakamura H, Umemiya S, Puthukanoori RK, Murthy Appala VR, Gaddamanugu GK, Paraselli BR and Baran PS, J. Am. Chem. Soc, 2020, 142, 10526–10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones RR and Bergman RG, J. Am. Chem. Soc, 1972, 94, 660–661. [Google Scholar]
- 25.Shiraki T and Sugiura Y, Biochemistry, 1990, 29, 9795–9798. [DOI] [PubMed] [Google Scholar]
- 26.(a) Wender PA and Zercher CK, J. Am. Chem. Soc, 1991, 113, 2311–2313; [Google Scholar]; (b) Wender PA, Zercher CK, Beckham S and Haubold EM, J. Org. Chem, 1993, 58, 5867–5869. [Google Scholar]
- 27.(a) Huffman BJ and Shenvi RA, J. Am. Chem. Soc, 2019, 141, 3332–3346; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Roach JJ, Sasano Y, Schmid CL, Zaidi S, Katritch V, Stevens RC, Bohn LM and Shenvi RA, ACS Cent. Sci, 2017, 3, 1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Rzasa RM, Shea HA and Romo D, J. Am. Chem. Soc, 1998, 120, 591–592; [Google Scholar]; (b) Romo D, Rzasa RM, Shea HA, Park K, Langenhan JM, Sun L, Akhiezer A and Liu JO, J. Am. Chem. Soc, 1998, 120, 12237–12254; [Google Scholar]; (c) Romo D, Choi NS, Li S, Buchler I, Shi Z and Liu JO, J. Am. Chem. Soc, 2004, 126, 10582–10588; [DOI] [PubMed] [Google Scholar]; (d) Low W-K, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick WC, Romo D and Liu JO, Mol. Cell, 2005, 20, 709–722; [DOI] [PubMed] [Google Scholar]; (e) Dang Y, Kedersha N, Low W-K, Romo D, Gorospe M, Kaufman R, Anderson P and Liu JO, J. Biol. Chem, 2006, 281, 32870–32878; [DOI] [PubMed] [Google Scholar]; (f) Low W-K, Dang Y, Bhat S, Romo D and Liu JO, Chem. Biol, 2007, 14, 715–727; [DOI] [PubMed] [Google Scholar]; (g) Low WK, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Rzasa RM, Shea HA, Li S, Park K, Ma G, Romo D and Liu JO, in Methods in Enzymology, Academic Press, 2007, vol. 431, pp. 303–324; [DOI] [PubMed] [Google Scholar]; (h) Dang Y, Low W-K, Xu J, Gehring NH, Dietz HC, Romo D and Liu JO, J. Biol. Chem, 2009, 284, 23613–23621; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Kuznetsov G, Xu Q, Rudolph-Owen L, TenDyke K, Liu J, Towle M, Zhao N, Marsh J, Agoulnik S, Twine N, Parent L, Chen Z, Shie J-L, Jiang Y, Zhang H, Du H, Boivin R, Wang Y, Romo D and Littlefield BA, Mol. Cancer Ther, 2009, DOI: 10.1158/1535-7163.MCT-08-1026, 1535–7163.MCT-1508–1026; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Low W-K, Li J, Zhu M, Kommaraju SS, Shah-Mittal J, Hull K, Liu JO and Romo D, Bioorganic & Medicinal Chemistry, 2014, 22, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Northcote PT, Blunt JW and Munro MHG, Tetrahedron Lett., 1991, 32, 6411–6414. [Google Scholar]
- 30.Abbasov ME, Alvariño R, Chaheine CM, Alonso E, Sánchez JA, Conner ML, Alfonso A, Jaspars M, Botana LM and Romo D, Nat. Chem, 2019, 11, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Wender PA, Baryza JL, Brenner SE, Clarke MO, Craske ML, Horan JC and Meyer T, Curr. Drug Discov. Technol, 2004, 1, 1–11; [DOI] [PubMed] [Google Scholar]; (b) Towle MJ, Salvato KA, Budrow J, Wels BF, Kuznetsov G, Aalfs KK, Welsh S, Zheng W, Seletsky BM, Palme MH, Habgood GJ, Singer LA, DiPietro LV, Wang Y, Chen JJ, Quincy DA, Davis A, Yoshimatsu K, Kishi Y, Yu MJ and Littlefield BA, Cancer Research, 2001, 61, 1013; [PubMed] [Google Scholar]; (c) Posner GH and O'Neill PM, Acc. Chem. Res, 2004, 37, 397–404. [DOI] [PubMed] [Google Scholar]
- 32.Rateb ME, Houssen WE, Schumacher M, Harrison WTA, Diederich M, Ebel R and Jaspars M, J. Nat. Prod, 2009, 72, 1471–1476. [DOI] [PubMed] [Google Scholar]
- 33.(a) Leirós M, Sánchez AJ, Alonso E, Rateb EM, Houssen EW, Ebel R, Jaspars M, Alfonso A, and Botana ML, Mar. Drugs, 2014, 12; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Leirós M, Alonso E, Rateb ME, Houssen WE, Ebel R, Jaspars M, Alfonso A, and Botana LM, Neuropharmacology, 2015, 93, 285–293 [DOI] [PubMed] [Google Scholar]; (c) Sánchez JA, Alfonso A, Leirós M, Alonso E, Rateb ME, Houssen WE, Ebel R, Tabudravu J and Botana LM, Pharmacol. Res, 2016, 107, 407–414. [DOI] [PubMed] [Google Scholar]
- 34.Kornienko A and La Clair JJ, Nat. Prod. Rep, 2017, 34, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schnermann MJ, Beaudry CM, Egorova AV, Polishchuk RS, Sütterlin C and Overman LE, Proc. Natl. Acad. Sci. USA, 2010, 107, 6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.(a) Abbasov ME, Hudson BM, Tantillo DJ and Romo D, J. Am. Chem. Soc, 2014, 136, 4492–4495; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Abbasov ME, Hudson BM, Tantillo DJ and Romo D, Chem. Sci, 2017, 8, 1511–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramesh P, Reddy NS, Venkateswarlu Y, Reddy MVR and Faulkner DJ, Tetrahedron Lett., 1998, 39, 8217–8220. [Google Scholar]
- 38.Chitturi BR, Tatipamula VB, Dokuburra CB, Mangamuri UK, Tuniki VR, Kalivendi SV, Bunce RA and Yenamandra V, Tetrahedron, 2016, 72, 1933–1940. [Google Scholar]
- 39.(a) Duh C-Y, Wang S-K, Chia M-C and Chiang MY, Tetrahedron Lett., 1999, 40, 6033–6035; [Google Scholar]; (b) Cui W-X, Yang M, Li H, Li S-W, Yao L-G, Li G, Tang W, Wang C-H, Liang L-F and Guo Y-W, Bioorganic Chemistry, 2020, 94, 103350. [DOI] [PubMed] [Google Scholar]
- 40.(a) Tseng Y-J, Ahmed AF, Dai C-F, Chiang MY and Sheu J-H, Org. Lett, 2005, 7, 3813–3816; [DOI] [PubMed] [Google Scholar]; (b) Iguchi K, Kajiyama K and Yamada Y, Tetrahedron Lett, 1995, 36, 8807–8808; [Google Scholar]; (c) Sheu J-H, Ahmed AF, Shiue R-T, Dai C-F and Kuo Y-H, J. Nat. Prod, 2002, 65, 1904–1908; [DOI] [PubMed] [Google Scholar]; (d) Appa Rao KMC, Krishna MM and Anjaneyulu V, J. Chem. Res., (S), 1995, 188–189. [Google Scholar]
- 41.(a) Thao NP, Nam NH, Cuong NX, Quang TH, Tung PT, Dat LD, Chae D, Kim S, Koh Y-S, Kiem PV, Minh CV and Kim YH, Bioorg. Med. Chem. Lett, 2013, 23, 228–231 [DOI] [PubMed] [Google Scholar]; (b) Lillsunde K-E, Festa C, Adel H, De Marino S, Lombardi V, Tilvi S, Nawrot AD, Zampella A, Souza L, Auria VM and Tammela P, Mar. Drugs, 2014, 12 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lakshmi V and Kumar R, Nat. Prod. Res, 2009, 23, 801–850; [DOI] [PubMed] [Google Scholar]; (d) Kamel HN and Slattery M, Pharm. Biol, 2005, 43, 253–269; [Google Scholar]; (e) Cheng S-Y, Chuang C-T, Wen Z-H, Wang S-K, Chiou S-F, Hsu C-H, Dai C-F and Duh C-Y, Bioorganic & Medicinal Chemistry, 2010, 18, 3379–3386 [DOI] [PubMed] [Google Scholar]; (f) Ahmed AF, Su J-H, Kuo Y-H and Sheu J-H, J. Nat. Prod, 2004, 67, 2079–2082. [DOI] [PubMed] [Google Scholar]
- 42.(a) Pattenden G and Winne JM, Tetrahedron Lett, 2009, 50, 7310–7313 [Google Scholar]; (b) Srikrishna A and Dethe DH, Org. Lett, 2004, 6, 165–168 [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Nguyen HM and Koradin C, Tetrahedron Lett, 2010, 51, 6232–6235 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Truax NJ, Ayinde S, Van K, Liu JO and Romo D, Org. Lett, 2019, 21, 7394–7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.(a) Craig IIRA, Roizen JL, Smith RC, Jones AC, Virgil SC and Stoltz BM, Chem. Sci, 2017, 8, 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Horn EJ, Silverston JS and Vanderwal CD, J. Org. Chem, 2016, 81, 1819–1838; [DOI] [PubMed] [Google Scholar]; (c) Moeller KD, Synlett, 2009, DOI: 10.1055/s-0028-1088126, 1208–1218; [DOI] [Google Scholar]; (d) Roizen JL, Jones AC, Smith RC, Virgil SC and Stoltz BM, J. Org. Chem, 2017, 82, 13051–13067; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tang F and Moeller KD, J. Am. Chem. Soc, 2007, 129, 12414–12415; [DOI] [PubMed] [Google Scholar]; (f) Tang F and Moeller KD, Tetrahedron, 2009, 65, 10863–10875. [Google Scholar]
- 44.Hafeman NJ, Loskot SA, Reimann CE, Pritchett BP, Virgil SC and Stoltz BM, J. Am. Chem. Soc, 2020, DOI: 10.1021/jacs.0c02513. [DOI] [PubMed] [Google Scholar]
- 45.(a) Deng M, Zhang X, Li Z, Chen H, Zang S and Liang G, Org. Lett, 2019, 21, 1493–1496; [DOI] [PubMed] [Google Scholar]; (b) Ueda Y, Abe H, Iguchi K and Ito H, Tetrahedron Lett, 2011, 52, 3379–3381. [Google Scholar]
- 46.Li Y and Pattenden G, Tetrahedron, 2011, 67, 10045–10052. [Google Scholar]
- 47.Young IS and Baran PS, Nat. Chem, 2009, 1, 193–205. [DOI] [PubMed] [Google Scholar]
- 48.Nozoe S, Morisaki M, Tsuda K, Iitaka Y, Takahashi N, Tamura S, Ishibashi K and Shirasaka M, J. Am. Chem. Soc, 1965, 87, 4968–4970. [DOI] [PubMed] [Google Scholar]
- 49.(a) Masi M, Dasari R, Evidente A, Mathieu V and Kornienko A, Bioorg. Med. Chem. Lett, 2019, 29, 859–869; [DOI] [PubMed] [Google Scholar]; (b) Tian W, Deng Z and Hong K, Mar. Drugs, 2017, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Najumudeen AK, Jaiswal A, Lectez B, Oetken-Lindholm C, Guzmán C, Siljamäki E, Posada IMD, Lacey E, Aittokallio T and Abankwa D, Oncogene, 2016, 35, 5248–5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.(a) Morrison R, Lodge T, Evidente A, Kiss R and Townley H, Int J Oncol, 2017, 50, 773–786; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim IY, Kwon M, Choi M-K, Lee D, Lee DM, Seo MJ and Choi KS, Oncotarget, 2017, 8, 106740–106752 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bury M, Girault A, Mégalizzi V, Spiegl-Kreinecker S, Mathieu V, Berger W, Evidente A, Kornienko A, Gailly P, Vandier C and Kiss R, Cell Death Dis., 2013, 4, e561–e561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canonica L, Friecchi A, Kienle UG and Scala A, Tetrahedron Lett., 1966, 7, 1329–1333. [Google Scholar]
- 53.Au TK, Chick WSH and Leung PC, Life Sci., 2000, 67, 733–742. [DOI] [PubMed] [Google Scholar]
- 54.Reisenauer KN, Tao Y, Song S, Patel SD, Ingros A, Sheesley P, Masi M, Boari A, Evidente A, Kornienko AV, Romo D and Taube J, bioRxiv, 2020, DOI: 10.1101/2020.05.06.079343, 2020.2005.2006.079343. [DOI] [Google Scholar]
- 55.(a) Dasari R, Masi M, Lisy R, Ferderin M, English LR, Cimmino A, Mathieu V, Brenner AJ, Kuhn JG, Whitten ST, Evidente A, Kiss R and Kornienko A, Bioorg. Med. Chem. Lett, 2015, 25, 4544–4548; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kong Au T and Chow Leung P, Plant Physiology, 1998, 118, 965; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chidley C, Trauger SA, Birsoy K and O'Shea EK, eLife, 2016, 5, e14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sierra MA and de la Torre MC, Dead Ends and Detours: Direct Ways to Successful Total Synthesis, Wiley, 2004. [Google Scholar]
- 57.Tao Y, Reisenauer K, Taube J and Romo D, Org. Lett, 2020, DOI: 10.1021/acs.orglett.0c02938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsuna K, Noguchi N and Nakada M, Angew. Chem. Int. Ed, 2011, 50, 9452–9455; [DOI] [PubMed] [Google Scholar]; Tsuna K, Noguchi N and Nakada M, Chem. Eur. J, 2013, 19, 5476–5486. [DOI] [PubMed] [Google Scholar]
- 59.Erlanson DA, McDowell RS and O'Brien T, J. Med. Chem, 2004, 47, 3463–3482. [DOI] [PubMed] [Google Scholar]