

Abstract
Purpose
This study determines if δ-opioid receptor agonist (i.e. SNC-121)-induced epigenetic changes via regulation of histone deacetylases (HDACs) for retinal ganglion cell (RGC) neuroprotection in glaucoma model.
Methods
Intraocular pressure was raised in rat eyes by injecting 2M hypertonic saline into the limbal veins. SNC-121 (1 mg/kg; i.p.) was administered to the animals for 7 days. Retinas were collected at days 7 and 42, post-injury followed by measurement of HDAC activities, mRNA, and protein expression by enzyme assay, quantitative real-time PCR (qRT-PCR), Western blotting, and immunohistochemistry.
Results
The visual acuity, contrast sensitivity, and pattern electroretinograms (ERGs) were declined in ocular hypertensive animals, which were significantly improved by SNC-121 treatment. Class I and IIb HDACs activities were significantly increased at days 7 and 42 in ocular hypertensive animals. The mRNA and protein expression of HDAC 1 was increased by 1.33 ± 0.07-fold and 20.2 ± 2.7%, HDAC 2 by 1.4 ± 0.05-fold and 17.0 ± 2.4%, HDAC 3 by 1.4 ± 0.06-fold and 17.4 ± 3.4%, and HDAC 6 by 1.5 ± 0.09-fold and 15.1 ± 3.3% at day 7, post-injury. Both the mRNA and protein expression of HDACs were potentiated further at day 42 in ocular hypertensive animals. HDAC activities, mRNA, and protein expression were blocked by SNC-121 treatment at days 7 and 42 in ocular hypertensive animals.
Conclusions
Data suggests that class I and IIb HDACs are activated and upregulated during early stages of glaucoma. Early intervention with δ-opioid receptor activation resulted in the prolonged suppression of class I and IIb HDACs activities and expression, which may, in part, play a crucial role in RGC neuroprotection.
Keywords: glaucoma, neuroprotection, HDACs, epigenetics, opioids
Glaucoma is a slowly progressive disease in which retinal ganglion cells (RGCs) die slowly and this leads to irreversible blindness. The global prevalence of glaucoma is estimated to reach 122 million by the year 2040.1 The primary open angle glaucoma (POAG) is the most common form of the glaucoma worldwide. The current existing therapeutic modalities focus on lowering the elevated intraocular pressure (IOP), which is considered to be a primary risk factor for glaucoma.2,3 However, patients undergoing glaucoma therapy continue to lose vision as a result of RGC death suggesting IOP is not the only causative factor for RGC death in glaucoma. Based on existing literature and work done in last decade in our laboratory, glaucoma is now considered to be a multifactorial disease in which biomechanical stress,3 epigenetic changes,4−6 mitochondrial dysfunction,7 pro-inflammatory cytokines,8−11 deprivation of neurotrophic factors, and oxidative stress12 play crucial roles. Hence, there is a need to understand the cellular and molecular mechanisms potentially playing roles in glaucoma pathology. This information will lead to the development of a better neuroprotective therapy for glaucoma.
Epigenetic modification appears to be a promising novel approach to modulate cellular function in neurodegenerative diseases,13 however, it remains poorly defined in glaucoma. Protein acetylation is a dynamic process, which is controlled by two enzymes called histone deacetylases (HDACs) and histone acetyltransferases (HATs). Studies have shown impairment in the acetylation homeostasis in numerous neurodegenerative diseases.14−16 In the eyes, studies have also shown disturbance in acetylation during optic neuropathies.17 More specifically, hypoacetylation of histones is observed in the apoptotic loss of RGCs during retinal degeneration.6 Further, increased activity of HDACs in ocular injury models, such as the optic nerve crush, ischemia/reperfusion, and glaucoma model, have been shown.4,5,18,19
Earlier, we have shown that activation of δ-opioid receptor by a selective ligand (i.e. SNC-121 for 7 days’ treatment) provides RGC neuroprotection for a prolonged period of time.8,20−22 We have shown that multiple pathways and effector proteins, including p38 MAP kinase, NF-κB, caspases, nitric oxide, iNOS, TNF-α, and matrix metalloproteinases play crucial roles in the RGC neurodegeneration, whereas PI3K/Akt signaling pathway was crucial for RGC neuroprotection.8,21−23 However, we were intrigued by the observation that early intervention with the δ-opioid receptor agonist, SNC-121, for 7 days resulted in RGC survival up to 42 days in our chronic rat glaucoma model. This led us to believe that δ-opioid receptor-mediated RGC neuroprotection occurs via epigenetic changes. In the present study, we determined the effects of SNC-121 on HDAC enzyme activity and expression of mRNA and protein in normal and ocular hypertensive animals. Our data demonstrated that class I and IIb HDACs activity and expression were altered during glaucoma pathology, which were reversed by activation of the δ-opioid-receptor by SNC-121. To the best of our knowledge this is the first report in which we have shown that δ-opioid receptor activation regulates the activities and expression of class I and IIb HDACs comprehensively in a chronic rat glaucoma model.
Materials and Methods
Animals
Equal numbers of adult male and female Brown Norway rats (2–5 months of age; 150–350 grams) were used in this study. These animals were obtained from Charles River laboratory (San Diego, CA, USA). Rats were maintained under a cycle of 12-hours of light and 12-hours of dark throughout the studies. Animals were anesthetized using a cocktail of ketamine (75 mg/kg body weight) and xylazine (10 mg/kg body weight) and body temperature was maintained at 37°C using a heating pad. All animal handling was performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The study protocol was approved by the Animal Care and Use Committee at the Medical University of South Carolina.
Development of Glaucoma Model by Hypertonic Saline Injection
Baseline IOP was recorded using a Tonolab tonometer prior to hypertonic saline injection. IOP was elevated by injecting 50 µl of 2M hypertonic saline into the limbal veins, as described earlier.8,21,22 An antibacterial ointment (Neomycin) was applied at the injection site of each animal to prevent any possible bacterial infections. IOP readings were taken every week after IOP elevation. Animals that had elevation in IOP more than 25% were included in the experimental design. IOP measurements were performed between 9 AM and 11 AM at each time point.
Drug Preparation
Delta (δ)-opioid-receptor agonist, SNC-121 dihydrochloride (Cat # SC-204291; Santa Cruz Biotechnology, Dallas, TX, USA), was dissolved in PBS and administered in Brown Norway rats. SNC-121 (1 mg/kg body weight) was administered intraperitoneally (i.p.) 30 minutes post-hypertonic saline injection. The drug administration (150–200 µl) was continued for 7 days, once daily at the same time (i.e. 9 AM to 11 AM).
Pattern Electroretinogram Recordings
Three baselines for pattern electroretinograms (ERGs) were recorded prior to the hypertonic saline injection and then every 2 weeks after the injury. A pattern ERG electrode was placed on the corneal surface using a micromanipulator on undilated pupils. A visual stimulus generated by black and white alternating contrast reversing bars (mean luminance, 50 cd m‒2; spatial frequency, 0.033 cycle/deg; contrast, 99%; and temporal frequency, 1 Hz) was aligned with the projection of the pupil using the UTAS-2000 (LKC Technologies, Gaithersburg, MD, USA). Each pattern-ERG was an average of 150 sweeps at an interval of 1 second, with measurements made between a peak and adjacent trough of the waveform as described earlier.8,9,21,22
Optokinetic Response Measurements
Visual acuity and contrast sensitivity of Brown Norway rats were measured by observing their optomotor responses to moving sine-wave gratings using OptoMotry (Cerebral Mechanics Inc., Lethbridge, AB, Canada) in a mask fashion as described by others.24 Conscious rats were placed individually on a central elevated pedestal without any restraint surrounded by a square array of computer monitors that display stimulus grating. Animals were allowed to adjust for 5 to 10 minutes in the chamber prior to any measurements. Rats reflexively respond to rotating vertical gratings by moving their head in the direction of the grating rotation. The movement of head was monitored via an overhead closed-circuit camera. All animals were tested under conditions with a mean luminescence of 52 cd m‒2. Visual acuity was measured by finding the spatial frequency threshold of each animal at a constant speed (12 degrees/second) and contrast (100%) with a staircase procedure that systematically increased the spatial frequency of the grating until the animal no longer exhibited detectable responses. Contrast sensitivity was determined by taking the reciprocal of the contrast threshold at a fixed spatial frequency (0.131 cycle/degree) and speed (12 degrees/second). Contrast of the pattern was decreased systematically in a staircase manner until the animal stopped responding. The changes in the optokinetic response (OKR) were measured at days 0 and 42, prior and post-injury, respectively.
Histone Deacetylases Activity Assay
The enzyme activity of HDACs was measured using the 4-methylcoumarin-7-amide (AMC), a fluorophore conjugated to acetylated lysine substrates, Boc-Lys(Ac)-AMC (Cat # K-6405, Aurum Pharmatech) or (Boc-Lys(Tfa)-AMC (Cat # I-1985, Bachem), as described previously.4,25 Retina extract was prepared using lysis buffer (50 mM Tris-base, 10 mM EDTA, 0.5 mM sodium orthovanadate, 0.5% sodium deoxycholic acid, 1% Triton X-100, and cocktail protease inhibitor). Equal amounts of retina protein extract were incubated with 100 µM substrate in HDAC buffer (50 mM Tris-Cl pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, and 0.1 mg/mL bovine serum albumin) in 96-well black bottom plates at room temperature for 1 hour. For inhibition of total HDAC activity, retina extracts were pre-incubated with Trichostatin A (1µM) before adding the substrates. Baseline fluorescence was determined before the addition of peptidase enzyme, trypsin. Following incubation, trypsin was added in excess (5 µg/reaction) to cleave the conjugated AMC to fluoresce. The amount of fluorogenic AMC generated was measured with a standard fluorospectrometer (Ex/Em at 355/460). The Boc-Lys(Ac)-AMC is a specific substrate to class I and IIb HDAC 1, 2, 3, and 6; whereas Boc-Lys(Tfa)-AMC is a specific substrate for class IIa and IV HDAC 4, 5, 7, 8, 9, 10. and 11.
Western Blotting
Equivalent amounts of protein (10–20 µg) from retina lysates were separated in a Nu PAGE 10% Bis-Tris gel (Thermo Fisher Scientific, Carlsbad, CA, USA) and proteins were transferred to nitrocellulose membranes, as described earlier.8,22 The membranes were blocked with 5% non-fat dry milk for 1 hour followed by incubation with primary antibodies (anti-HDAC 1 [Cell Signaling Technology; Cat # D5C6U], anti-HDAC 2 [Cell Signaling Technology; Cat # D6S5P], anti-HDAC 3 [Cell Signaling Technology; Cat # 7G6C5], or anti-HDAC 6 [Cell Signaling Technology; Cat # D21B10] at 1:1000 dilutions, or β-actin [Sigma Aldrich; Cat # A5316] at 1:4000 dilutions) for 16 hours at 4°C. After washing, the membranes were incubated for 1 hour at room temperature with the appropriate secondary antibodies (Anti-rabbit IgG, HRP-linked; Cell Signaling Technology; Cat # 7074; anti-mouse IgG, HRP-linked; Cell Signaling Technology; Cat # 7076 dilution 1:5000). Prestained molecular weight magic markers (Thermo Fisher Scientific) were run along with the samples to identify the molecular weight of proteins. For chemiluminescent detection, the membranes were treated with enhanced chemiluminescent (ECL) reagent for 1 minute (Super Signal; Thermo Scientific, Rockford, IL, USA), and the signal was captured using a Biorad Versadoc imaging system (Biorad, Hercules, CA, USA).
RNA Extraction and Quantitative Real Time PCR
Total RNA was isolated from rat retina using TRIzol reagent (Cat # 15596-026; Life Technologies, Grand Island, NY, USA). Template cDNA was prepared from 1 µg of total RNA from different samples using iScript cDNA synthesis Kit (Cat # 1708891; Bio-Rad Laboratories, Inc., Hercules, CA, USA). The mRNA levels for HDAC 1, 2, 3, and 6 were analyzed by quantitative real time PCR (qRT-PCR) using SYBR Green (Cat # 172527; Bio-Rad Laboratories, Inc., Hercules, CA, USA) and a Bio-Rad iCycler system, according to the manufacturer's manual. Conditions for qRT-PCR were: 95°C for 30 seconds followed by two-step 40 cycles of 95°C for 10 seconds and 60°C for 30 seconds. The relative gene expression is calculated based on the comparative threshold cycle (ΔΔCt) method. The level of the housekeeping gene β-actin was used as a reference control for the normalization of RNA differences among the samples. The sequence of specific primers used for each HDAC were: HDAC 1: Forward 5′-GCGAGCAAGATGGCGCAGACT-3′, Reverse 5′-GTGAGGCTTCATTGGGTGCCCT-3′; HDAC-2: Forward 5′-CTCCGGGCTGTCCTTGCTGC-3′, Reverse 5′-GCCGCCTCCTTGACTGTACGC-3′; HDAC 3: Forward 5′-ACCAGGCCTCCCAGCATGACA-3′, Reverse 5′-CCGGGAAACACAGGGCAGTCG-3′; HDAC 6: Forward 5′-TGTGGCTGCCCGCTATGCAC-3′, Reverse 5′-GGGGCCAGAACCGACCATGC-3′, and β-Actin: Forward 5′-ATGGTCAACCCCACCGTGT-3′, and Reverse 5′-CGTGTGAAGTCACCACCCT-3′. The primers were synthesized by Integrated DNA Technology (IDT, Coralville, IA, USA).
Immunohistochemistry
Rat eyes were enucleated on days 7 and 42, post-injury, and placed in 4% paraformaldehyde (PFA) for 12 to 16 hours at 4°C. The eyes were then transferred to 30% sucrose for 24 hours followed by embedding the eyes in OCT on dry ice. Cryosections (7–8 µm) were cut at −30°C and stored at −20°C until used for immunohistochemistry. Tissue sections were rinsed with Tris-buffered saline (TBS) three times at room temperature followed by permeabilization using 0.5% Triton X-100 in TBS. The tissues were blocked with 5% BSA in TBS for 1 hour and then rinsed again with TBS followed by incubation with primary antibodies (anti-HDAC 1, anti-HDAC 2, anti-HDAC 3, or anti-HDAC 6 at 1:100 dilution; Cell Signaling Technology, Danvers, MA, USA) for 24 hours at 4°C. After washing, the tissues were incubated with secondary antibody (Alexa Fluor 488 at 1:500 dilution; Thermo Fisher Scientific, Eugene, OR, USA) for 1 hour at room temperature. The sections were observed under a bright-field microscope equipped with epifluorescence, and digitized images were captured by a digital camera (Zeiss), as described earlier.8,9,21,22
Statistical Analysis
Data are presented in this manuscript as ± standard error of mean (SEM) from three or more independent experiments as indicated by n. Where “n” refers to biological replicates. Power analysis of variance from previous in vivo studies, including pattern-ERG studies demonstrates that a group size of 6 is sufficient to detect a 20% change in functional assays and measurements of changes in the enzyme assays, RT-PCR, and Western blot analysis. Statistical analyses included mean, standard error, confidence interval, limits, linear regression, 1-way ANOVA for multiple group comparison, and a t-test for grouped or paired data analysis of variance. We performed statistical analysis using 1-way ANOVA followed by a post hoc Tukey test for multiple comparisons (GraphPad Software, Inc., San Diego, CA, USA). A P value ≤ 0.05 was considered significant.
Results
Functional Deficits in Response to Glaucomatous Injury
To assure that hypertonic saline injection resulted in the expected elevation in IOP and functional deficits, we measured changes in the IOP every week, pattern ERGs, and OKR at day 0 (prior to injury) and day 42 (post ocular injury). As shown in Figure 1a, a significant increase in IOP was observed in ocular hypertensive eyes by day 7, which remained elevated until day 42 post-injury. Similar to our previously published reports,8,21,22 pattern ERGs were significantly declined in ocular hypertensive animals. The declines in pattern ERGs were reversed in SNC-121 treated animals (Fig. 1b), whereas SNC-121 itself had no effects on IOP and pattern ERGs in normal eyes, as shown in our earlier studies.8,22 Earlier, we have also shown that RGC numbers were significantly declined in ocular hypertensive animals and such losses of RGCs were blocked by SNC-121 treatment.8 To further confirm that glaucomatous injury leads to vison loss or impairments, we measured changes in the visual acuity and contrast sensitivity using OKRs. As shown in Figure 1c, visual acuity in ocular hypertensive animals was significantly (P < 0.0001) decreased at day 42 post-injury when it was compared with day 0 (prior to injury). Interestingly, we found that SNC-121 treatment increased visual acuity significantly (P = 0.0137) in ocular hypertensive animals when it was compared with ocular hypertensive animals (without drug treatment) at day 42 post-injury. Furthermore, we measured contrast threshold at day 42 (post-injury) and compared them with day 0 (prior to injury) as shown in Figure 1d. Contrast sensitivity is reciprocally proportional to the contrast threshold. Contrast threshold was significantly (P < 0.0001) increased in ocular hypertensive animals at day 42, suggesting that animals had less contrast sensitivity in this group. As we expected, contrast threshold was decreased significantly (P = 0.0021) in SNC-121 treated ocular hypertensive animals when it was compared with ocular hypertensive animals (without drug treatment) at day 42 post-injury. These data suggest that SNC-121 treatment enhances both visual acuity and contrast sensitivity in ocular hypertensive animals.
Figure 1.

Intraocular pressure (a), pattern ERG (b), and OKR (c, d) in rats. IOP of conscious animals was measured at every week using Tonolab. Pattern-ERG, spatial acuity, and contrast threshold were measured at day 0 (prior) and 42 (post), prior and post injury. Brown Norway rats were treated with 1 mg/kg SNC-121 (i.p.) 1 hour after hypertonic saline injections, and subsequently once daily for 7 days. Data are expressed as mean ± SE. *, # P < 0.05; ***P < 0.001; ****P < 0.0001; n = 7–8.
Histone Deacetylases Activity in the Retina
HDACs’ activity was measured to identify the major HDACs that are contributing in retina HDAC activity. We found that 95.48 ± 0.8% of histone deacetylase activity is due to class I and IIb HDACs (e.g. HDAC 1, 2, 3, and 6), whereas, only 4.6 ± 0.8% HDAC activity was contributed by class IIa and IV (e.g. HDAC 4, 5, 7, 8, 9, 10, and 11; Fig. 2a). Additionally, basal activity of class I and IIb HDAC was significantly inhibited by SNC-121 (25.8 ± 1.4%, P = 0.009) treatment (Fig. 2b). As shown in Figure 2a, HDAC activity was completely abrogated (96%, n = 4; P < 0.0001) in the presence of a pan-HDACs inhibitor, Trichostatin A (TSA). Based on these findings, we focused our subsequent studies on HDAC 1, 2, 3, and 6. We did not measure class III (e.g. sirtuins 1–7) activity in this study.
Figure 2.

Measurement of histone deacetylases (HDACs) activity (a), and effects of SNC-121 on HDAC activity (b) in retina extracts obtained from normal Brown Norway rats. Enzyme activity for selective HDAC was measured using specific conjugated fluorophore acetylated lysine substrates, Boc-Lys(Ac)-AMC or Boc-Lys(Tfa)-AMC for HDAC 1, 2, 3, and 6 or HDAC 4, 5, 7 to 11, respectively, as shown in a. To determine the specificity of assay, equal amount of retina extract (1 µg) was pre-incubated with vehicle (DMSO) or 1µM Trichostatin A (TSA) for 15 minutes before adding specific substrates as shown in a. Additionally, we measured the effects of SNC-121 treatment on HDAC 1, 2, 3, and 6 activity using Boc-Lys(Ac)-AMC as substrate in normal animals as shown in b. Normal Brown Norway rats were treated with 1 mg/kg SNC-121 (i.p.) once a day for 7 days. On day 7, animals were euthanized and retinas were collected and analyzed for HDAC class I and IIb enzyme activity. Equal amount of retina extract (1 µg) was incubated with specific substrate. After 1 hour, trypsin was added and the HDAC activity was quantified by detection of released fluorescent amino-methoxy cumarin (Ex/Em at 355/460). Data are expressed as mean ± SE. ****P < 0.0001; n = 6.
Regulation of HDAC Activity by δ-Opioid Receptor Activation in Rat Glaucoma Model
In our earlier studies, we have shown that 7 days of δ-opioid receptor agonist (SNC-121) treatment provides prolonged RGC neuroprotection.8,22 To understand the mechanistic aspects of such prolonged neuroprotection, we measured class I and IIb HDAC activity at days 7 and 42 post-injury. The HDAC activity was significantly increased in ocular hypertensive eyes by 17.0 ± 2.6% (n = 11–16; P < 0.0001) on day 7 (Fig. 3a) and 31.9 ± 5.5% (n = 8–12; P < 0.0001) and on day 42 (Fig. 3b) when compared with the normal eyes of untouched animals. Interestingly, we found a continued increase in HDAC activity up to 42 days in the ocular hypertensive eyes, suggesting that HDAC activity progresses consistently along with glaucoma progression. To determine if δ-opioid receptor activation inhibits class I and IIb HDAC activity, we treated animals with SNC-121 (1 mg/kg; i.p. injection; once a day) for 7 days and measured class I and IIb activity on days 7 and 42 post-injury. At both time points, the increase in class I and IIb HDAC activity was fully blocked in 7 days SNC-121 treated animals (Figs. 3a, 3b). Intriguingly, the 7 days δ-opioid agonist treatment was able to maintain the suppression of the class I and IIb HDAC activity until day 42, as shown in Figure 3b.
Figure 3.

Effect of SNC-121 on HDACs activity in retina at days 7 and 42 post-injury. Activities of specific HDAC 1, 2, 3, and 6 were measured in the retina extracts at days 7 (a) and 42 (b) post-injury using Boc-Lys(Ac)-AMC as substrate. The HDAC activity was quantified by the detection of released fluorescent amino-methoxy cumarin (Ex/Em at 355/460). Data are expressed as mean ± SE. ****P < 0.0001; n = 8–16.
Sex is one of the biological variables, hence we also determined if changes in HDAC activity in response to glaucomatous injury were sex-dependent. HDAC activity was increased in both male and female rats at day 7 and 42 and fully blocked in 7 days in SNC-121 treated animals. However, changes in HDAC activities were not sex-dependent because we did not find any statistically significant differences in HDAC class I and IIb activity between the male and female animals (data not shown).
Delta Opioid Receptors Mediated Changes in the mRNA and Protein Expression of Class I and IIb HDACs in the Retina
We measured changes in the mRNA expression of class I and IIb HDACs (e.g. HDAC 1, 2, 3, and 6) using selective primers for each HDAC, as described in the Methods section. The mRNA expression of HDAC 1 was increased by 1.33 ± 0.07-fold (n = 7–9; P < 0.001; Fig. 4a), HDAC 2 by 1.4 ± 0.05-fold (n = 7–8; P < 0.0001; Fig. 5a), HDAC 3 by 1.4 ± 0.06-fold (n = 7–10; P < 0.0001; Fig. 6a), and HDAC 6 by 1.5 ± 0.09-fold (n = 7–9; P < 0.0001; Fig. 7a) at day 7 post-injury. Furthermore, the mRNA expression of HDAC 1 was increased by 1.22 ± 0.04-fold (n = 3–4; P < 0.05; see Fig. 4b), HDAC 2 by 1.33 ± 0.06-fold (n = 3–4; P < 0.05; see Fig. 5b), HDAC 3 by 1.5 ± 0.08-fold (n = 3–4; P < 0.01; see Fig. 6b), and HDAC 6 by 1.45 ± 0.16-fold (n = 3–4; P < 0.05; see Fig. 7b) at day 42 in ocular hypertensive animals. Administration of SNC-121 (i.p. injection) for 7 days (once a day) completely abrogated the increased in the mRNA expression of HDAC 1, 2, 3, and 6 and this suppressing effect remained active until day 42 post-injury.
Figure 4.

Measurement of changes in mRNA and protein expression of HDAC 1. Changes in mRNA and protein expression of HDAC 1 by SNC-121 treatment in normal and ocular hypertensive animals at day 7 (a, c) and day 42 (b, d) post-injury. Total RNA was extracted from the retina using TRIzol reagent and 1 µg RNA was used to synthesize cDNA. The mRNA levels were examined by quantitative RT-PCR using primers specific to HDAC 1 and relative gene expression was calculated based on the comparative threshold cycle (Ct) method, normalized to housekeeping genes, β-actin a and b. Retina lysates (10 µg) were analyzed by Western blotting using anti-HDAC 1 and anti-β-actin antibodies. The band intensities were measured using chemiluminescent reagent and Versadoc imaging system c and d. Data expressed as mean ± SE. *P < 0.05; ***P < 0.001; ****P < 0.0001; n = 3–23.
Figure 5.

Measurement of changes in mRNA and protein expression of HDAC 2. Measurement of changes in mRNA and protein expression of HDAC 2 by SNC-121 treatment in normal and ocular hypertensive animals at day 7 (a, c) and day 42 (b, d) post-injury. Total RNA was extracted from retina using TRIzol reagent and 1 µg RNA was used to synthesize cDNA. The mRNA levels were examined by quantitative RT-PCR using primers specific to HDAC 2 and relative gene expression was calculated based on the comparative threshold cycle (Ct) method, normalized to housekeeping genes, β-actin a and b). Retina lysates (10 µg) were analyzed by Western blotting using anti-HDAC 2, and anti-β-actin antibodies. The band intensities were measured using chemiluminescent reagent and Versadoc imaging system c and d. Data expressed as mean ± SE. *P < 0.05; ****P < 0.0001; n = 3–26.
Figure 6.

Measurement of changes in mRNA and protein expression of HDAC 3. Measurement of changes in mRNA and protein expression of HDAC 3 by SNC-121 treatment in normal and ocular hypertensive animals at day 7 (a, c) and day 42 (b, d) post-injury. Total RNA was extracted from retina using TRIzol reagent and 1 µg RNA was used to synthesize cDNA. The mRNA levels were examined by quantitative RT-PCR using primers specific to HDAC 3 and relative gene expression was calculated based on the comparative threshold cycle (Ct) method, normalized to housekeeping genes, β-actin a and b. Retina lysates (20 µg) were analyzed by Western blotting using anti-HDAC 3, and anti-β-actin antibodies. The band intensities were measured using chemiluminescent reagent and Versadoc imaging system c and d. Data expressed as mean ± SE. **P < 0.01; ***P < 0.001; ****P < 0.0001; n = 3–23.
Figure 7.

Measurement of changes in mRNA and protein expression of HDAC 6. Measurement of changes in mRNA and protein expression of HDAC 6 by SNC-121 treatment in normal and ocular hypertensive animals at day 7 (a, c) and day 42 (b, d) post-injury. Total RNA was extracted from retina using TRIzol reagent and 1 µg RNA was used to synthesize cDNA. The mRNA levels were examined by quantitative RT-PCR using primers specific to HDAC 6 and relative gene expression was calculated based on the comparative threshold cycle (Ct) method, normalized to housekeeping genes, β-actin a and b. Retina lysates (10 µg) were analyzed by Western blotting using anti-HDAC 6, and anti-β-actin antibodies. The band intensities were measured using chemiluminescent reagent and Versadoc imaging system c and d. Data expressed as mean ± SE. *P < 0.05; **P < 0.01; ****P < 0.0001; n = 3–21.
We also measured changes in the protein expression of class I and IIb HDACs using whole retina extract and selective antibodies for each HDAC. We found that elevation of IOP increases the protein expression of HDAC 1 by 20.2 ± 2.7% (n = 19–23; P < 0.0001; see Fig. 4c), HDAC 2 by 17.0 ± 2.4% (n = 23–31; P < 0.0001; see Fig. 5c), HDAC 3 by 17.4 ± 3.4% (n = 21–23; P < 0.0001; see Fig. 6c), and HDAC 6 by 15.1 ± 3.3% (n = 22–24; P < 0.0001; see Fig. 7c) at day 7 post-injury, which remained elevated above basal level up to 42 days post-injury (Figs. 4d–7d). The upregulation in class I and IIb HDAC protein expression at days 7 and 42 was completely blocked in SNC-121 treated ocular hypertensive animals (see Figs. 4c–7c, 4d–7d). Furthermore, SNC-121 treatment for 7 days reduced the protein expression of HDAC 1 by 39.9 ± 6.03% (n = 4; P < 0.001), HDAC 2 by 17.9 ± 4.3% (n = 3–4; P < 0.01), HDAC 3 by 41.5 ± 6.0% (n = 3–4; P < 0.001), and HDAC 6 by 32.4 ± 4.7% (n = 3–4; P < 0.001) in normal animals (Fig. 8).
Figure 8.

Effects of SNC-121 treatment on class I and IIb HDACs in normal animals. Brown Norway rats were treated with 1 mg/kg SNC-121 (i.p.) once a day for 7 days. No ocular injury was performed in this group of animals. On day 7, the animals were euthanized and the retinas were collected and analyzed for HDAC 1 (a), HDAC 2 (b), HDAC 3 (c), and HDAC 6 (d) protein expression by Western blotting using selective antibodies for each HDAC. The band intensities were measured using chemiluminescent reagent and Versadoc imaging system. Data expressed as mean ± SE. **P < 0.01; ***P < 0.001; n = 3–4.
Immunohistochemistry of Class I and IIb HDACs in the Retina
Retina sections of normal, ocular hypertensive, and SNC-121 treated ocular hypertensive animals were analyzed for HDAC immunostaining using selective antibodies for each HDAC. As shown in Figures 9 and 10, there was a mild basal immunostaining of HDAC 1 in the RGC layer, nerve fiber (NF) layer, and inner nuclear layer (INL), which was remarkably increased in ocular hypertensive animals. We have seen a robust increase in HDAC 1 immunostaining in the NF, INL, and RGC layers at day 7 in ocular hypertensive animals. Interestingly, HDAC 1 immunostaining continued to increase in these inner retinal layers at day 42 in ocular hypertensive animal. At both time points (7 and 42 days), HDAC 1 immunostaining was reduced by SNC-121 treatment (see Fig. 9, 10). Similarly, a mild and diffused immunostaining of HDAC 2 was seen in NF and RGC layers, which was clearly increased in NF and RGC layers at days 7 and 42 post-injury. We also noticed a moderately increase in immunostaining of HDAC 2 in INL at day 7, which was robustly increased at day 42 post-injury in ocular hypertensive animals (see Figs. 9, 10). The majority of the HDAC 2 immunostaining was localized to the nuclei and it was colocalized with dapi (data not shown). At both time points (7 and 42 days), HDAC 2 immunostaining was significantly reduced in SNC-121 treated animals. A diffused cytosolic immunostaining of HDAC 3 and HDAC 6 was also seen in normal retina, which was clearly increased in nuclei in ocular hypertensive animals. As shown in Figures 9 and 10, both nuclear and cytosolic immunostaining for HDAC 3 and HDAC 6 were increased in ocular hypertensive animals, which was reduced remarkably in SNC-121 treated animals.
Figure 9.
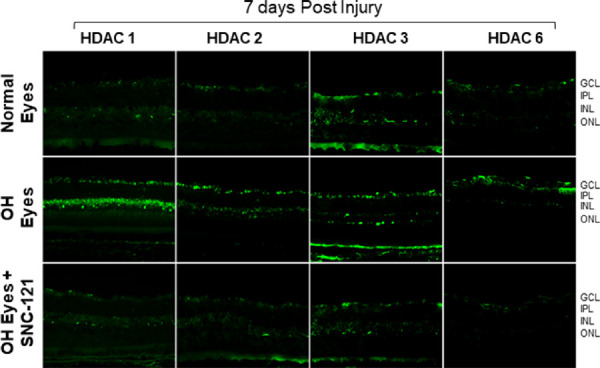
Immunohistochemistry of HDAC 1, 2, 3, and 6 in the retina at day 7 post-injury. Animals were euthanized and eyes were enucleated at day 7 post hypertonic saline injections. Cryosections were immunostained by a selective anti-HDAC 1, anti-HDAC 2, anti-HDAC 3, or anti-HDAC 6 antibodies. There was no positive staining when primary antibodies were omitted (not shown). Data are a representation of at least six independent experiments. Comparable immunostaining for each HDAC was seen in the retina of other animals (magnification times 20). GCL, ganglion cells layer; IPL, inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer.
Figure 10.
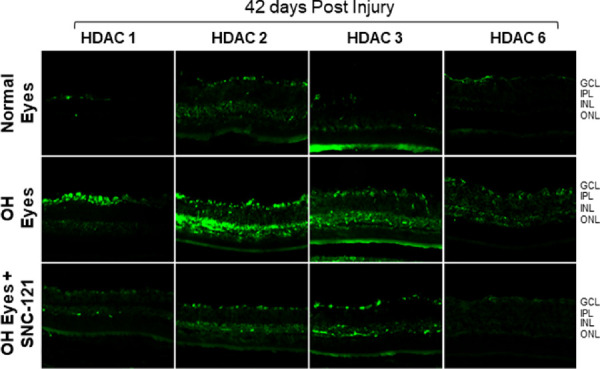
Immunohistochemistry of HDAC 1, 2, 3, and 6 in the retina at day 42 post-injury. Animals were euthanized and eyes were enucleated at day 42 post hypertonic saline injections. Cryosections were immunostained by a selective anti-HDAC 1, anti-HDAC 2, anti-HDAC 3, or anti-HDAC 6 antibodies. There was no positive staining when primary antibodies were omitted (not shown). Data are a representation of at least six independent experiments. Comparable immunostaining for each HDAC was seen in the retina of other animals (magnification times 20). GCL, ganglion cells layer; IPL, inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer.
Discussion
Epigenetics is a biological and cellular process that involves heritable and nonheritable alterations in a gene without changes in genetic sequence. It deals with changes in gene expression via DNA methylation, histone acetylation, and noncoding RNAs. Chromatin remodeling has gained tremendous attention in the field of neurodegenerative diseases in regulating transcription of numerous genes and it has a robust potential in the development of neuroprotective strategies. The process of acetylation and deacetylation targets both histones and nonhistones substrates that regulate the transcription of target genes. Histone deacetylation through activation of HDACs compacts chromatin structure and represses the transcription process. HDACs have been known to be the potential therapeutic targets in numerous diseases from neurodegeneration to cancer.26 There are 18 mammalian HDACs, which are divided into 4 classes based on their sequences and structural similarities to yeast deacetylases. Class I (HDAC 1, 2, 3, and 8), class II a (HDAC 4, 5, 7, and 9), class IIb (HDAC 6 and 10), class III (sirtuins 1–7), and class IV (HDAC 11). Class I and II HDACs are considered transcription repressors because they bind to DNA as a corepressor recruited by transcription factors. In general, HDAC 1, 2, and 3 function as part of the transcription silencing complex to deacetylate histone residues at the nucleosomes,27 whereas HDAC 6 is exclusively cytoplasmic and deacetylates nonhistone substrates, such as α-tubulin, Hsp-90, and β-catenin.28 Class III HDACs or sirtuins family are known to regulate mitochondrial activity.29 The role of class IV (HDAC 11) is not well established, however, limited studies have shown the involvement of HDAC 11 in carcinogenesis and the expression modulation of interleukin 10.30
HATs and HDACs function antagonistically to control protein acetylation homeostasis. The equilibrium between HATs and HDACs activities is tightly regulated in neurons of the brain under physiological conditions and we assume the existence of a similar scenario in the eye. However, the impairment of acetylation homeostasis results in a shift toward deacetylation, which is a prominent molecular feature of numerous neurodegenerative diseases.16 If such impairments in acetylation exist during glaucoma, pathology remains largely unknown. In other systems, studies have shown that HDAC inhibitors provide significant beneficial effects in cardiovascular diseases,31 kidney disease,32 stroke and hemorrhage,33,34 and in chronic neurodegenerative diseases.14 Additionally, in vitro studies have also shown that HDAC inhibitors protect neurons from glutamate-induced excitotoxicity35 and oxidative stress.36 Mechanistically, HDAC inhibitors protect neurons from oxidative stress via the upregulation of the cell cycle inhibitor p21(waf1/cip1)36 and p53/PUMA signaling pathways.37 More specifically, HDAC 2 has been associated with cancer, diverse neurodegenerative diseases, and neuronal toxicity.38 Durham et al.39 reported pharmacological inhibition or selective knockdown of HDAC 1 or HDAC 2 can suppress the expression of IL-6 and TNF-α in LPS-activated murine microglial cells. Studies have also shown that HDAC 2 forms a physical complex with Forkhead box O3a (FOXO3a), which plays a crucial role in FOXO3a-dependent gene transcription and oxidative stress-induced neuronal apoptosis.40 In the eyes, enhanced RGCs survival by the treatment with broad spectrum HDAC inhibitors, TSA, and Valproic acid (VPA) suggest the involvement of multiple isoforms of HDACs in the neuroprotection in acute ischemia5 and chronic glaucoma models.4,6 Studies have also shown that combined ablation of HDAC 1 and 2 promotes survival of axotomized RGCs in an acute optic nerve injury model via alteration in the acetylation of P53-PUMA.18 However, the involvement of individual HDAC isoform in δ-opioids mediated RGC neuroprotection remains fully unknown.
In this paper, we have shown that class I and IIb HDACs are major (over 95%) contributors for HDAC activity in the retina under physiological conditions, which is in agreement with a previous report.41 Studies have shown that women are at a higher risk for angle closure glaucoma, but there is no clear sex predilection for open angle glaucoma.42 There are some reports in human glioma showing that the risk of occurrence is more in men than women in older ages (72–74 years of age).43 To determine if the δ-opioids mediated effects are sex-dependent, we have analyzed data in male and female rats separately. However, we did not see any sex-specific changes in the HDAC activities in the ocular hypertensive and SNC-121 treated ocular hypertensive animals, suggesting that sex does not play a significant role in HDAC activity as a result of ocular injury or by δ-opioids treatment.
Our initial thoughts that δ-opioid receptor activation could lead to the epigenetic changes for RGC neuroprotection were supported by the data presented in this paper. We have shown that class I and IIb HDACs activities at both early (i.e. day 7) and delayed (day 42) time points were significantly increased in ocular hypertensive animals. This increase in HDAC activity was completely inhibited when animals were treated with SNC-121 for 7 days. We were surprised to see a continued suppression in HDAC activity until day 42, whereas SNC-121 treatment was stopped at day 7. These findings provide experimental support that 7 days of δ-opioid treatment causes epigenetic changes via regulation of HDACs that may subsequently be responsible for RGC neuroprotection. This claim is supported by the functional data in which SNC-121 treatment provides RGC neuroprotection as measured by pattern ERGs and visual acuity. We have seen almost full recovery in pattern ERGs and partial but statistically significant recovery in OKR by SNC-121 treatment. The differences in the functional outcomes using two different procedures could be due to the experimental condition (e.g. conscious animals versus anesthetized animals), effects of cornea thickness, and/or efficiency/sensitivity of the procedure (i.e. pattern ERG versus OKR). To shed more lights, we also measured mRNA and protein expression of HDAC 1, HDAC 2, HDAC 3, and HDAC 6 in the retina of normal, ocular hypertensive, and SNC-121 treated ocular hypertensive eyes. We found a significant increase in the mRNA transcript and protein expression of HDAC 1, 2, 3, and 6 at days 7 and 42 post-injury. The treatment with SNC-121 decreased mRNA and protein expression of class I and IIb HDACs in ocular hypertensive eyes. Interestingly, both mRNA and protein expression remained suppressed up to 42 days post-injury, whereas SNC-121 treatment was stopped at day 7 post-injury. Our findings clearly suggest that HDAC upregulation occurs during the early stages of glaucoma pathology and they remain elevated during the progression of this disease, which is suppressed by initial activation of δ-opioid receptors. We have used immunohistochemistry to identify the tissues/cells that express each HDAC in normal, ocular hypertensive, and SNC-121 treated ocular hypertensive animals. Our immunohistochemistry data show that HDAC 1 and 2 were localized to the nuclei whereas HDAC 3 and 6 were expressed more in the cytoplasm. We have seen mild immunostaining in normal retina for HDAC 1, 2, 3, and 6, which was remarkably increased in ocular hypertensive animals. More specifically, we have seen a robust increase in HDAC 1 immunostaining in the NF, INL, and RGC layers of ocular hypertensive animals. The immunostaining for HDAC 2 and 3 was also remarkably increased in the NF and RGC layers but they were moderately increased in the INL of ocular hypertensive animals. In contrast, immunostaining for HDAC 6 was increased only in the NF and RGC layers. These observations clearly indicated that each HDAC responds to glaucomatous injury differently. Interestingly, all HDACs of class I and IIb were remarkably increased in the NF and RGC layers, suggesting their potential involvement in glial cell activation and RGC death. Our data also suggest that bipolar, horizontal cell, and amacrine cells could also be affected in glaucomatous injury because HDAC 1, 2, and 3 expression is clearly increased in the INL. Further studies are warranted to investigate this novel finding. Similar to Western blotting data and mRNA expression, both class I and IIb HDAC immunostaining was remarkably reduced by SNC-121 treatment in ocular hypertensive animals.
In the eyes, limited evidence suggests the involvement of HDACs in retina degeneration. For example; mRNA expression of HDAC 1, 2, and 3 was increased in acute optic nerve injury.6,19 Genetic ablation of HDAC 3 does not seem to play a large role in early gene silencing in RGCs, but HDAC 3 knockdown has been shown to stop histones deacetylation and attenuate subsequent RGC death after axonal injury.19 The class I and II HDAC inhibitor VPA reduces RGC death by day 8 after acute optic nerve injury.44 Additionally, TSA attenuated RGC soma degeneration in DBA/2J mouse model without any axonal degeneration.45 VPA and sodium butyrate (SB) reduces RGC death in vitro via reduction of HDAC activity.46 Pharmacological inhibition of HDAC 3 specific activity by RGFP966 was able to preserve histone H4 acetylation and RGC survival after acute optic nerve injury.47 HDAC 3 has been reported to translocate to the nucleus from the cytoplasm of RGCs following injury in both the optic nerve crush and NMDC-induced excitotoxicity rodent models.19 Selective inhibitors of HDAC 1, 2, and 3 (MS-275) protect RGCs in optic nerve injury model.48 Overexpression of HDAC 2 resulted in increased in glial cell activity (e.g. expression of GFAP, Iba-1, and iNOS) in the ischemic retina.49 Genetic and pharmacological methods have shown that inhibition of HDAC 6 promotes the survival and regeneration of neurons in neurodegenerative disease.50 HDAC 6 deacetylates its substrates, including α-tubulin, Hsp90, and cortactin, and then forms a complex with other partner proteins and involved in numerous biological processes, such as cell migration and cell to cell interaction.51 These published reports provide useful information regarding neurodegenerative roles of HDACs, however, it remains in question that how and which subtype of HDAC plays a crucial role in RGC degeneration in glaucoma. Overall, studies using HDAC inhibitors or genetic ablation do not provide similar outcomes, which indicate that genetic deletion of certain HDACs may be compensated by other HDACs. Hence, it remains unclear how each HDAC plays a crucial role in the silencing of genes and/or function of proteins that are detrimental for RGC during glaucoma. Overall, we have seen relatively small but statistically significant changes in class I and IIb HDACs activities and expression in the retina of ocular hypertensive animals. These findings indicated the involvement of other epigenetic events, such as DNA methylation, which could also be parallelly active for RGC death in glaucoma. Another factor could be the study duration, we focused our study for 6 weeks and there is a possibility that HDAC expression and RGC function may deteriorate further beyond 6 weeks. We might see more appreciable changes in the HDAC activities and expression beyond 6 weeks in ocular hypertensive animals. It is also important to emphasize that we have used young animals (2–3 months old), however, glaucoma is an age-dependent disease. This is very likely that young animals are more resistant to ocular insult and strong endogenous compensatory neuroprotective pathways counteract to mitigate occur hypertension-induced injury. These are some of the possibilities for mild changes and open a wide array of unanswered questions. These critical questions will require additional studies using current glaucoma model for the young and old animals for a longer duration study (i.e. 12 weeks or longer), and a different age-dependent glaucoma model (i.e. DBA/2J). In addition, other epigenetic events, such as DNA methylation, is equally important and its role in RGC death remains unknown. Future studies should also be focusing to understand the role of DNA methylation in RGC death and if they are also regulated by δ-opioids.
The molecular mechanisms that are involved in the upregulation of HDACs remains largely unknown. Earlier, we have shown that pro-inflammatory cytokines are upregulated during glaucomatous injury in chronic glaucoma model8,9 and acute ischemia model.5,52 We also have shown that a pan inhibitor of HDAC (TSA) blocks ischemia/reperfusion induced TNF-α production5 and δ-opioid receptor activation provides RGC neuroprotection in chronic glaucoma model via inhibiting the production of nitric oxide, iNOS, TNF-α, and the secretion of matrix metalloproteinases.8,21,22 Moreover, numerous signaling mediators, including p38, MAP Kinase, NF-κB, and caspases, play crucial role in the RGC neurodegeneration in our glaucoma models.8,21−23 Studies from other laboratories have also suggested that inhibition of pro-inflammatory cytokines can be attributed to suppression of transcriptional activation by HDAC inhibition that will ultimately suppress cytokines production.39 In in vitro studies, we have shown that TNF-α activates and phosphorylates NF-ĸB/IĸB signaling pathway in ONH astrocytes, which could lead to the production of cytokines that are detrimental for RGCs.23 Based on our previous studies, we speculate that NF-ĸB/IĸB signaling could be involved in the HDAC upregulation during glaucomatous injury. However, the involvement of other transcription factors cannot be ruled out, their involvement in this process will be investigated in our future studies.
In conclusion, regulation of HDACs activities in the retina may be beneficial during glaucoma, however, a global prevention of deacetylation of nonhistone targets may be detrimental. Therefore, the development of selective HDAC inhibitors will be vital to develop an effective therapy to target detrimental HDAC for glaucoma therapy. To develop a HDAC selective therapy, it is important to understand the role of each HDAC in glaucoma progression. This manuscript provides insights for the changes occurring in the activity, mRNA, and protein expression of class I and IIb HDACs at an early and delayed time point during glaucoma progression. In this study, we provide evidence that histone deacetylase activity, particularly HDAC 1, 2, 3, and 6 increased at day 7 and remained elevated for up to 42 days post-injury. This study also provides significant information for the first time that δ-opioids regulate class I and IIb HDACs activities, mRNA, and protein expression in the retina of chronic rat glaucoma model. Taken together, these results provide clues that the elevation of HDAC activity and/or their expression in the retina could be detrimental for the survival of RGCs. However, the mechanism by which acetylation homeostasis is disturbed and how it is coupled to RGC degeneration remains unclear. Future studies will address these important aspects of epigenetics in glaucoma.
Acknowledgments
Supported by NIH/NEI Grant EY-027355 (S.H.). This funding body helped in designing of study, collection, analysis, and interpretation of data and in writing of this manuscript.
Disclosure: S.A.H. Zaidi, None; W. Guzman, None; S. Singh, None; S. Mehrotra, None; S. Husain, None
References
- 1. Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014; 121: 2081–2090. [DOI] [PubMed] [Google Scholar]
- 2. Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. 2012; 31: 152–181. [DOI] [PubMed] [Google Scholar]
- 3. Boland MV, Quigley HA. Risk factors and open-angle glaucoma: classification and application. J Glaucoma. 2007; 16: 406–418. [DOI] [PubMed] [Google Scholar]
- 4. Alsarraf O, Fan J, Dahrouj M, Chou CJ, Yates PW, Crosson CE. Acetylation preserves retinal ganglion cell structure and function in a chronic model of ocular hypertension. Invest Ophthalmol Vis Sci. 2014; 55: 7486–7493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crosson CE, Mani SK, Husain S, Alsarraf O, Menick DR. Inhibition of histone deacetylase protects the retina from ischemic injury. Invest Ophthalmol Vis Sci. 2010; 51: 3639–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pelzel HR, Schlamp CL, Nickells RW. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010; 11: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Osborne NN, Nunez-Alvarez C, Joglar B, Del Olmo-Aguado S. Glaucoma: focus on mitochondria in relation to pathogenesis and neuroprotection. Eur J Pharmacol. 2016; 787: 127–133. [DOI] [PubMed] [Google Scholar]
- 8. Abdul Y, Akhter N, Husain S. Delta-opioid agonist SNC-121 protects retinal ganglion cell function in a chronic ocular hypertensive rat model. Invest Ophthalmol Vis Sci. 2013; 54: 1816–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Husain S, Abdul Y, Crosson CE. Preservation of retina ganglion cell function by morphine in a chronic ocular-hypertensive rat model. Invest Ophthalmol Vis Sci. 2012; 53: 4289–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang X, Luo C, Cai J, et al.. Neurodegenerative and inflammatory pathway components linked to TNF-alpha/TNFR1 signaling in the glaucomatous human retina. Invest Ophthalmol Vis Sci. 2011; 52: 8442–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Husain S, Abdul Y, Potter DE. Non-analgesic effects of opioids: neuroprotection in the retina. Curr Pharm Des. 2012; 18: 6101–6108. [DOI] [PubMed] [Google Scholar]
- 12. Peters D, Bengtsson B, Heijl A. Lifetime risk of blindness in open-angle glaucoma. Am J Ophthalmol. 2013; 156: 724–730. [DOI] [PubMed] [Google Scholar]
- 13. Didonna A, Opal P.. The promise and perils of HDAC inhibitors in neurodegeneration. Ann Clin Transl Neurol. 2015; 2: 79–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferrante RJ, Kubilus JK, Lee J, et al.. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003; 23: 9418–9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003; 22: 6537–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saha RN, Pahan K.. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006; 13: 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alkozi HA, Franco R, Pintor JJ. Epigenetics in the eye: an overview of the most relevant ocular diseases. Front Genet. 2017; 8: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lebrun-Julien F, Suter U.. Combined HDAC1 and HDAC2 depletion promotes retinal ganglion cell survival after injury through reduction of p53 target gene expression. ASN Neuro. 2015; 7(3): 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmitt HM, Pelzel HR, Schlamp CL, Nickells RW. Histone deacetylase 3 (HDAC3) plays an important role in retinal ganglion cell death after acute optic nerve injury. Mol Neurodegener. 2014; 9: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Husain S. Delta opioids: neuroprotective roles in preclinical studies. J Ocul Pharmacol Ther. 2018; 34: 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Husain S, Abdul Y, Singh S, Ahmad A, Husain M. Regulation of nitric oxide production by δ-opioid receptors during glaucomatous injury. PLoS One. 2014; 9: e110397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Husain S, Ahmad A, Singh S, Peterseim C, Abdul Y, Nutaitis MJ. PI3K/Akt pathway: a role in delta-opioid receptor-mediated RGC neuroprotection. Invest Ophthalmol Vis Sci. 2017; 58: 6489–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akhter N, Nix M, Abdul Y, Singh S, Husain S. Delta-opioid receptors attenuate TNF-α-induced MMP-2 secretion from human ONH astrocytes. Invest Ophthalmol Vis Sci. 2013; 54: 6605–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Annamalai B, Nicholson C, Parsons N, et al.. Immunization against oxidized elastin exacerbates structural and functional damage in mouse model of smoke-induced ocular injury. Invest Ophthalmol Vis Sci. 2020; 61: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zaidi S, Thakore N, Singh S, et al. . Histone deacetylases regulation by δ-opioids in human optic nerve head astrocytes. Invest Ophthalmol Vis Sci. 2020;61(11):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009; 10: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen HP, Zhao YT, Zhao TC. Histone deacetylases and mechanisms of regulation of gene expression. Crit Rev Oncog. 2015; 20: 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li G, Jiang H, Chang M, Xie H, Hu L. HDAC6 alpha-tubulin deacetylase: a potential therapeutic target in neurodegenerative diseases. J Neurol Sci. 2011; 304: 1–8. [DOI] [PubMed] [Google Scholar]
- 29. Rehan L, Laszki-Szczachor K, Sobieszczanska M, Polak-Jonkisz D. SIRT1 and NAD as regulators of ageing. Life Sci. 2014; 105: 1–6. [DOI] [PubMed] [Google Scholar]
- 30. Gupta R, Ambasta RK, Kumar P. Pharmacological intervention of histone deacetylase enzymes in the neurodegenerative disorders. Life Sci. 2020; 243: 117278. [DOI] [PubMed] [Google Scholar]
- 31. Rawal S, Munasinghe PE, Nagesh PT, et al.. Down-regulation of miR-15a/b accelerates fibrotic remodelling in the type 2 diabetic human and mouse heart. Clin Sci (Lond). 2017; 131: 847–863. [DOI] [PubMed] [Google Scholar]
- 32. Pili R, Quinn DI, Hammers HJ, et al.. Immunomodulation by entinostat in renal cell carcinoma patients receiving high-dose interleukin 2: a multicenter, single-arm, phase I/II trial (NCI-CTEP #7870). Clin Cancer Res. 2017; 23: 7199–7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007; 321: 892–901. [DOI] [PubMed] [Google Scholar]
- 34. Sinn DI, Kim SJ, Chu K, et al.. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis. 2007; 26: 464–472. [DOI] [PubMed] [Google Scholar]
- 35. Kanai H, Sawa A, Chen RW, Leeds P, Chuang DM. Valproic acid inhibits histone deacetylase activity and suppresses excitotoxicity-induced GAPDH nuclear accumulation and apoptotic death in neurons. Pharmacogenomics J. 2004; 4: 336–344. [DOI] [PubMed] [Google Scholar]
- 36. Langley B, D'Annibale MA, Suh K, et al.. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 2008; 28: 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brochier C, Dennis G, Rivieccio MA, et al.. Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J Neurosci. 2013; 33: 8621–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guan JS, Haggarty SJ, Giacometti E, et al.. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009; 459: 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Durham BS, Grigg R, Wood IC. Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J Neurochem. 2017; 143: 214–224. [DOI] [PubMed] [Google Scholar]
- 40. Peng S, Zhao S, Yan F, et al.. HDAC2 selectively regulates FOXO3a-mediated gene transcription during oxidative stress-induced neuronal cell death. J Neurosci. 2015; 35: 1250–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Z, Zang C, Cui K, et al.. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009; 138: 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vajaranant TS, Nayak S, Wilensky JT, Joslin CE. Gender and glaucoma: what we know and what we need to know. Curr Opin Ophthalmol. 2010; 21: 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Crabb DP, Saunders LJ, Edwards LA. Cases of advanced visual field loss at referral to glaucoma clinics - more men than women? Ophthalmic Physiol Opt. 2017; 37: 82–87. [DOI] [PubMed] [Google Scholar]
- 44. Biermann J, Grieshaber P, Goebel U, et al.. Valproic acid-mediated neuroprotection and regeneration in injured retinal ganglion cells. Invest Ophthalmol Vis Sci. 2010; 51: 526–534. [DOI] [PubMed] [Google Scholar]
- 45. Pelzel HR, Schlamp CL, Waclawski M, Shaw MK, Nickells RW. Silencing of Fem1cR3 gene expression in the DBA/2J mouse precedes retinal ganglion cell death and is associated with histone deacetylase activity. Invest Ophthalmol Vis Sci. 2012; 53: 1428–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Biermann J, Boyle J, Pielen A, Lagreze WA. Histone deacetylase inhibitors sodium butyrate and valproic acid delay spontaneous cell death in purified rat retinal ganglion cells. Mol Vis. 2011; 17: 395–403. [PMC free article] [PubMed] [Google Scholar]
- 47. Schmitt HM, Schlamp CL, Nickells RW. Targeting HDAC3 activity with RGFP966 protects against retinal ganglion cell nuclear atrophy and apoptosis after optic nerve injury. J Ocul Pharmacol Ther. 2018; 34: 260–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chindasub P, Lindsey JD, Duong-Polk K, Leung CK, Weinreb RN. Inhibition of histone deacetylases 1 and 3 protects injured retinal ganglion cells. Invest Ophthalmol Vis Sci. 2013; 54: 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sung MS, Heo H, Eom GH, et al.. HDAC2 regulates glial cell activation in ischemic mouse retina. Int J Mol Sci. 2019; 20: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rivieccio MA, Brochier C, Willis DE, et al.. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc Natl Acad Sci USA. 2009; 106: 19599–19604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seidel C, Schnekenburger M, Dicato M, Diederich M. Histone deacetylase 6 in health and disease. Epigenomics. 2015; 7: 103–118. [DOI] [PubMed] [Google Scholar]
- 52. Husain S, Liou GI, Crosson CE. Opioid receptor activation: suppression of ischemia/reperfusion-induced production of TNF-α in the retina. Invest Ophthalmol Vis Sci. 2011; 52: 2577–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]