

Abstract
Two separate commercial products of kratom [Mitragyna speciosa (Korth.) Havil. Rubiaceae] were used to generate reference standards of its indole and oxindole alkaloids. While kratom has been studied for over a century, the characterization data in the literature for many of the alkaloids are either incomplete or inconsistent with modern standards. As such, full 1H and 13C NMR spectra, along with HRESIMS and ECD data, are reported for alkaloids 1-19. Of these, four new alkaloids (7, 11, 17, and 18) were characterized using 2D NMR data, and the absolute configurations of 7, 17, and 18 were established by comparison of experimental and calculated ECD spectra. The absolute configuration for the N(4)-oxide (11) was established by comparison of NMR and ECD spectra of its reduced product with those for compound 7. In total, 19 alkaloids were characterized, including: the indole alkaloid mitragynine (1) and its diastereoisomers speciociliatine (2), speciogynine (3) and mitraciliatine (4); the indole alkaloid paynantheine (5) and its diastereoisomers isopaynantheine (6) and epiallo-isopaynantheine (7); the N(4)-oxides mitragynine-N(4)-oxide (8), speciociliatine-N(4)-oxide (9), isopaynantheine-N(4)-oxide (10), and epiallo-isopaynantheine-N(4)-oxide (11); the 9-hydroxylated oxindole alkaloids speciofoline (12), isorotundifoleine (13) and isospeciofoleine (14); and the 9-unsubstituted oxindoles corynoxine A (15), corynoxine B (16), 3-epirhynchophylline (17), 3-epicorynoxine B (18), and corynoxeine (19). With the ability to analyze the spectroscopic data of all of these compounds concomitantly, a decision tree was developed to differentiate these kratom alkaloids based on a few key chemical shifts in the 1H and/or 13C NMR spectra.
Graphical Abstract

The scientific literature on the chemical composition of kratom [Mitragyna speciosa (Korth.) Havil. Rubiaceae] dates back nearly 100 years, when the most studied alkaloid from this plant, mitragynine (1), was reported in 1921.1 The introduction to that paper concludes with the following prescient statements: “According to Redley………Mitragyne [sic] speciosa is used in Perak against the opium habit, whilst, according to Dr. P.P. Laidlaw, mitragynine is a local anaesthetic, which finding is of interest, since the alkaloid contains an ester grouping.” At that time, there was uncertainty on the use of kratom. Interestingly, a century later, this uncertainty continues, including in the scientific literature, the popular press, and even government regulatory bodies. Many questions persist such as: does kratom ameliorate pain? can it be used as an opioid substitute? does it relieve opioid withdrawal symptoms? does its use precipitate trips to the emergency room? is kratom harmful, harmless, or somewhere in between? Addressing these questions requires a thorough understanding of the chemical composition of kratom.
In the past decade there has been an explosion in the biomedical literature regarding at least some of the above questions, particularly case reports (Figure S1). There are reports that speak to its ability to address pain, broadly defined.2–5 However, there are a similar number that suggest that it has opioid-like properties, and thus, may be an addiction liability.6–15 Towards that end, the US Food and Drug Administration has threatened to classify kratom as a schedule I controlled substance more than once, although at the writing of this paper, it has not done so.16–19
As part of a project to study the potential for interactions between herbal medicines and drugs,20–24 our team was charged with generating reference standards of the kratom alkaloids. In doing so, we found many challenges and opportunities, largely due to gaps in the literature. Extensive chemical studies have been performed over the last century, resulting in the isolation of at least 54 alkaloids from the plant (see Figure S2), starting with the isolation of the main alkaloid, mitragynine (1), in 1921 by Ellen Field.1 More than four decades later, and over a series of three manuscripts, Becket et al. and Zacharias et al. worked on the isolation, structure elucidation, and absolute configuration of 1.25–27 Contemporaneously, the mitragynine diastereoisomers speciociliatine (2) and speciogynine (3) were reported.28 A dozen years later, the fourth diastereoisomer, mitraciliatine (4), was described from the leaves of M. speciosa.29 Like many alkaloids, a majority of isolation studies on new compounds from kratom were carried out between the 1960s and 1980s.30–32 However, due to the growing popularity of this plant near the end of the last century, there was a renewed interest, resulting in the isolation of additional alkaloids (particularly minor constituents, which could be isolated due to advances in HPLC).33, 34 Despite this, most of the pharmacological studies on constituents from kratom have focused on mitragynine (1) and 7-hydroxymitragynine (see Figure S1). In general, the tetracyclic indole and pentacyclic oxindole alkaloids isolated from M. speciosa embody a few variations, that, collectively, show the interrelatedness of these compounds (see Figure S2). Some key motifs of the 54 known compounds from M. speciosa include a hydroxy or methoxy group at C-9, unsaturation at C-3, C-5 or C-18, hydroxylation at C-7, and various configurations at C-3, C-7 and/or C-20 (see Figure S2).30–32
It was necessary to isolate standards for the alkaloids present in the plant, because even though there are over 50 kratom alkaloids reported, only 13 are commercially available (Figure S2). Moreover, in some cases we found that the quality of the commercial standards was not optimal, being mixtures, or were not even the correct compound.35, 36 From a pharmacological testing point of view, this is a serious problem, especially for scientists that don’t have the training or collaborations to verify structural identity, as it could perpetuate erroneous findings in the literature. For these reasons, we strove to develop a series of reference standards, being able to isolate and characterize 19 kratom alkaloids (1-19), including four new analogues (7, 11, 17, and 18). In doing so, we quickly realized that the reported spectroscopy data for some of the known compounds were limited, as is the case for many natural products.37–41 Thus, we took the opportunity to contribute by analyzing the NMR, HRESIMS, and ECD data for 1-19, comparing the key differences between analogues.
RESULTS AND DISCUSSION
While there are some challenges associated with acquiring kratom plant material,42 two different sources could be obtained on the kg scale, which were termed ‘Green Maeng Da’ and ‘White Jongkong’ by the suppliers. These samples were slightly different in appearance, with the former being ground leaf fragments (internally coded as K49) and the latter being a fine powder (internally coded as K52) (see Figure S6). The taxonomy of these was examined using a combination of molecular methods, specifically DNA barcoding and maximum likelihood phylogenetic analysis. For the purpose of molecular identification, the most widely recommended loci were analyzed, including the plastid intergenic spacer trnH-psbA, matK, and the nuclear ribosomal internal transcribed spacer (ITS), since these three regions have been designated as the official DNA barcodes for the plant kingdom.43, 44 Of these, trnH-psbA is a widely used plastid marker, which displays a high rate of insertion and deletion and, as such, is widely used for plant barcoding studies.45–47 All three regions indicated that samples K49 and K52 could be unequivocally identified as Mitragyna speciosa. Maximum likelihood analysis of the partial matK region placed both commercial samples in a strongly supported clade (100% bootstrap support) with the published sequences for M. speciosa (Figure S3). Importantly, this analysis included a partial matK sequence from a recently sequenced genome of M. speciosa (BioProject: PRJNA325670). In addition, a BLAST search of matK using the BOLDSYSTEMS database (version 4) showed that samples K49 and K52 had ≥ 99% sequence similarity with M. speciosa (Figures S4 and S5). As a confirmatory measure, maximum likelihood analysis using the ITS region also indicated that both samples formed a strongly supported clade (≥ 99% bootstrap support) with published sequence data of M. speciosa (Figure S6). Based on uncorrected p distances calculated for trnH-psbA, there was ≥ 99% sequence similarity with sequences of M. speciosa. This means that samples K49 and K52 were more similar to sequences of M. speciosa and more dissimilar to sequences of M. diversifolia, M. hirsuta, and M. rotundifolia (Tables S1 and S2). In short, three different methods all indicated that these materials were derived from M. speciosa. The sequence data were deposited in GenBank: matK: MT114409 and MT114408, trnH-psbA: MT114410 and MT114411, and ITS: MT111840, MT111841, MT111842, and MT111843.
Even though the sources of kratom (i.e. K49 and K52) were demonstrated to be M. speciosa based on DNA barcoding, chromatographic profiles generated via UPLC-HRESIMS (Figure S7) showed clear differences in the profile of secondary metabolites. For instance, mitragynine (1) was the most abundant alkaloid in the K49 materials, while the K52 materials contained higher levels of speciofoline (12) and related alkaloids. While those differences may be somewhat surprising, varied chemical composition for different sources of M. speciosa has been documented, where plant age, environmental factors, and processing conditions impact on the suite of kratom alkaloids.48, 49 For example, in one published study the content of 1 derived from a Thai specimen was approximately five times higher than that derived from a Malaysian specimen.50 Additionally, alkaloid differences between M. speciosa grown in Asia/Africa vs the United States have been noted.49, 51 Thus, for the purpose of generating structurally diverse kratom alkaloid reference standards, we selected sample K49 for the isolation of indole-type alkaloids and sample K52 for the isolation of oxindole-type alkaloids using an alkaloid partitioning scheme. The indole alkaloids mitragynine (1), speciociliatine (2), speciogynine (3), mitraciliatine (4), paynantheine (5),52 isopaynantheine (6)29, epiallo-isopaynantheine (7), mitragynine-N(4)-oxide (8),53 speciociliatine-N(4)-oxide (9),53 isopaynantheine-N(4)-oxide (10),54 and epiallo-isopaynantheine-N(4)-oxide 11 were isolated from the Green Maeng Da product (K49; see Figure S8), while the oxindole alkaloids speciofoline (12),55 isorotundifoleine (13),56 isospeciofoleine (14),57 corynoxine A (15),58, 59 corynoxine B (16),59 3-epirhynchophylline (17), 3-epicorynoxine B (18), and corynoxeine (19) were isolated from the White Jongkong product (K52; see Figure S9). Details for the structure elucidation for these compounds, including the identification of four new kratom alkaloids (i.e. 7, 11, 17, and 18) are presented below, using a suite of spectroscopic and spectrometric techniques (HRESIMS), including ECD. In doing so, a decision tree was developed that can be used to differentiate between these kratom alkaloids based on a few key signals in the 1H and/or 13C NMR spectra (Figure 1). This flow diagram was designed to be helpful for those with only a limited understanding of structure elucidation, focusing on key signals that serve to differentiate between the various kratom alkaloid chemotypes.
Figure 1.
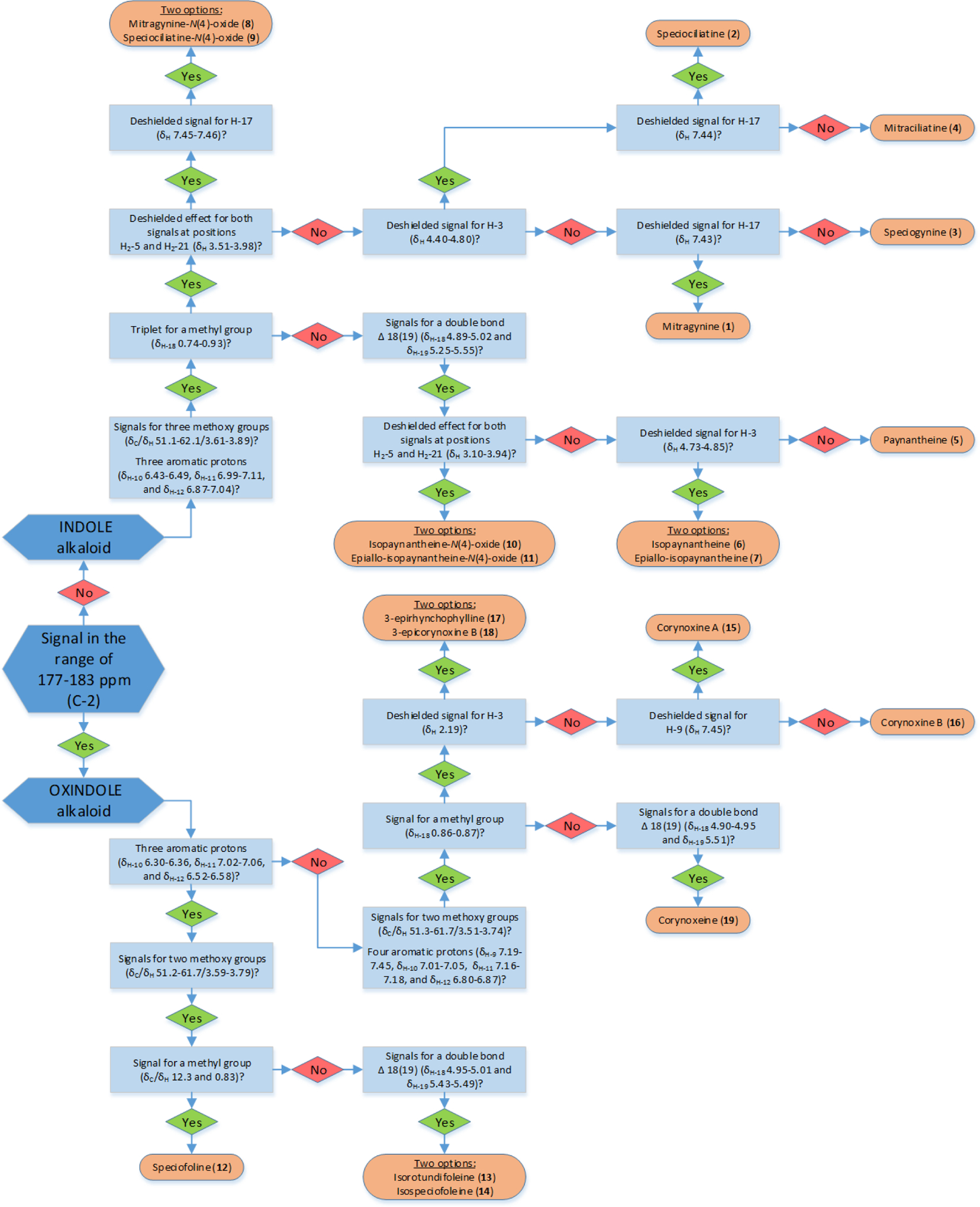
Decision tree for differentiating between 1-19 based on key NMR signals. As a single starting point, the 13C NMR signal for the lactam moiety is used to distinguish between oxindole (present) or indole (absent) kratom alkaloids. Once the broad structural class is determined, 1H NMR data can be used via a flowchart as a guide to elucidate the structure of these alkaloids.
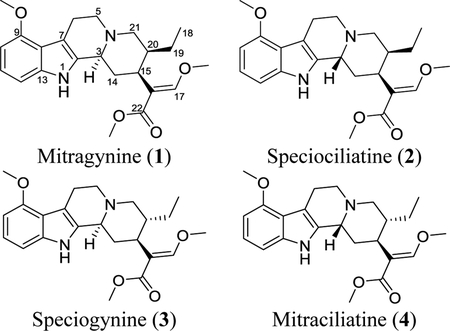
Indole Alkaloids: Mitragynine (1) and Its Diastereoisomers (2–4).
Four diastereoisomers with a molecular formula of C23H30N2O4 were isolated, specifically mitragynine (1), speciociliatine (2), speciogynine (3), and mitraciliatine (4), all of which were verified via HRESIMS data, where the protonated molecule displayed m/z 399.2274, 399.2273, 399.2274, and 399.2275 ions, respectively (Figures S10–S13). While the absolute amounts likely vary in different kratom plant materials, the quantity of compound 1 isolated was at least 20 times greater than that for each of the other diastereoisomers (i.e. >450 mg of 1 vs ~20 mg of 2, 3, or 4). The 1H and 13C NMR data matched with those reported previously,60 and key characteristics included: 1) eight sp2 signals in the 13C NMR spectrum assignable to the indole moiety (C-2 and C-7 to C-13); 2) two doublets (δH-10 6.44–6.49 and δH-12 6.87–7.01) and one triplet (δH-11 6.99–7.07), all corresponding to the indole moiety; 3) signals for the vinylic proton at C-17 (δC/δH 160.2–160.7/7.32–7.44); 4) three methoxy groups attached to C-9, C-17, and C-22 (δC/δH 51.1–61.9/3.66–3.89); and 5) the methyl group at C-19 (δC/δH 11.1–13.0/0.74–0.89). Importantly, a thorough analysis of the differences between the characteristic signals permitted the unequivocal identification of compounds 1-4. For example, as Lee and colleagues described,61 when a deshielded proton singlet assignable to H-17 appeared at δH 7.43 or 7.44 (i.e. compounds 1 and 2, respectively), it was indicative of the α orientation of H-20. Alternatively, a more shielded chemical shift for H-17 (i.e. δH 7.32 and 7.35 for compounds 4 and 3, respectively), indicated the β orientation for H-20.61 Additionally, the orientation of H-20 could be confirmed orthogonally via the chemical shift of C-18, at δC of 12.5 or 13.0 (i.e. compounds 2 and 1, respectively) for the α orientation and δC of 11.1 (i.e. for both 3 and 4) for the β orientation. On the other hand, the presence of a broad singlet at δH 4.40 or 4.80 (i.e. compounds 2 and 4) agreed with the β orientation for H-3, but a signal at δH 3.20 or 3.21 (i.e. compounds 1 and 3) indicated the opposite orientation for the same proton (i.e., H-3α). Indeed, the Δδ H-3 values of the α and β orientations depend on the dihedral angle and the distance between H-3 and the nitrogen lone pair. An anti-coplanar arrangement, as seen in the axial H-3 of the trans fused system in compounds 1 or 3 (i.e., α orientation), leads to shielding relative to the syn-coplanar arrangement of the equatorial proton (i.e., β orientation) in compounds 2 or 4, which have a dihedral angle of ~60° with the nitrogen lone pair. The donation of electron density into the antibonding orbital (i.e., n ➔ σ*) of the axial C–H bond (i.e., α orientation) effectively results in the weakening of that bond and strong shielding of the proton (Figure S18).62, 63 Key NMR signals for 1-4 are outlined (Table 1), and in addition, full 1D NMR spectra and Tables are reported in the Supporting Information (Figures S14–S17 and Table S4). Moreover, ECD spectra presented an additional approach for confirming the absolute configuration for these corynanthe-type alkaloids. It has been established that the Cotton effect between 250–300 nm could be used to establish the orientation of H-3, where a negative Cotton effect correlates with the β orientation (i.e., compounds 2 and 4) and a positive Cotton effect for the α orientation for H-3 (i.e., compounds 1 and 3; see Figure S19).61
Table 1.
Characteristic 1H and 13C NMR Data for Compounds 1–4 (CDCl3, 400 and 100 MHz, Respectively)a
| mitragynine (1) | speciociliatine (2) | speciogynine (3) | mitraciliatine (4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| position | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) |
| 3 | 61.3 | CH | 3.20, d (8.4) | 54.7 | CH | 4.40, bs | 61.9 | CH | 3.21, m | 53.9 | CH | 4.80, s |
| 10 | 99.9 | CH | 6.45, d (7.7) | 99.8 | CH | 6.47, d (7.7) | 99.8 | CH | 6.44, d (7.8) | 99.7 | CH | 6.49, d (7.6) |
| 11 | 122.0 | CH | 7.00, t (7.9) | 122.4 | CH | 7.02, t (8.0) | 122.4 | CH | 6.99, t (7.9) | 122.6 | CH | 7.07, t (7.9) |
| 12 | 104.3 | CH | 6.90, d (8.1) | 104.5 | CH | 6.91, d (8.1) | 104.5 | CH | 6.87, d (8.0) | 104.9 | CH | 7.01, d (8.0) |
| 17 | 160.7 | CH | 7.43, s | 160.6 | CH | 7.44, s | 160.4 | CH | 7.35, bs | 160.2 | CH | 7.32, s |
| 18 | 13.0 | CH3 | 0.87, t (7.3) | 12.5 | CH3 | 0.89, t (7.9) | 11.1 | CH3 | 0.85, t (7.2) | 11.1 | CH3 | 0.74, t (7.0) |
| 9-OCH3 | 55.5 | CH3 | 3.87, s | 55.3 | CH3 | 3.88, s | 55.4 | CH3 | 3.85, s | 55.3 | CH3 | 3.89, s |
| 17-OCH3 | 61.7 | CH3 | 3.73, s | 61.7 | CH3 | 3.78, s | 61.9 | CH3 | 3.72, s | 61.8 | CH3 | 3.77, s |
| 22-OCH3 | 51.5 | CH3 | 3.71, s | 51.6 | CH3 | 3.66, s | 51.1 | CH3 | 3.72, s | 51.5 | CH3 | 3.68, s |
| NH | 7.74, bs | 8.00, bs | 7.94, bs | 8.98, bs | ||||||||
See Table S4 for a full comparison for the 1H and 13C NMR data for 1–4 (Supporting Information).
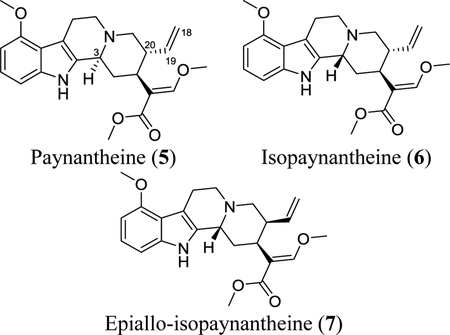
Indole Alkaloids: Paynantheine and Its Diastereoisomers (Compounds 5–7).
The structures of paynantheine (5) and isopaynantheine (6) were established by comparisons with literature data.57 Their molecular formulae were confirmed by HRESIMS as C23H28N2O4, where the protonated molecule showed an m/z of 397.2119 and 397.2117, respectively (Figures S20 and S21). The NMR data of 5 and 6 resembled those for compounds 1-4, sharing the previously enumerated characteristic signals for the indole ring. The main difference was the absence of signals for the C-20 ethyl moiety, and its replacement for a vinylic moiety, as evidenced by two doublets of doublets (δH 4.89–5.01) and a doublet of doublet of doublets or a doublet of triplets (δH 5.25–5.57), consistent with a Δ18(19) double bond (δC-18 115.7–116.7 and δC-19 137.9–139.4). As with 1-4, the orientation of H-3 was deduced from chemical shift data, where the H-3α orientation was noted for 5 (δH 3.20), while the more deshielded shift in 6 (δH 4.73) indicated the H-3β orientation. These assignments were supported by the ECD spectra, where 5 and 6 had opposite Cotton effects at 250–300 nm. Specifically, a negative Cotton effect confirmed the β orientation (i.e. 6) and a positive Cotton effect confirmed the α orientation (i.e. 5) for H-3 (see Figure S32). Comparisons of the key NMR signals for compounds 5 and 6 are shown in Table 2, and the full 1D NMR spectroscopic data are reported in the Supporting Information (Figures S23–S24 and Table S5).
Table 2.
Characteristic 1H and 13C NMR Data for Compounds 5–7 (CDCl3, 400 and 100 MHz, Respectively)a
| paynantheine (5) | isopaynantheine (6) | epiallo-isopaynantheine (7) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Position | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) |
| 3 | 60.1 | CH | 3.20, m | 53.8 | CH | 4.73, bs | 53.9 | CH | 4.85, bs |
| 10 | 99.9 | CH | 6.45, d (7.8) | 99.7 | CH | 6.49, d (7.6) | 99.7 | CH | 6.49, d (7.8) |
| 11 | 122.1 | CH | 7.00, t (7.9) | 122.6 | CH | 7.07, t (7.9) | 122.9 | CH | 7.08, t (7.9) |
| 12 | 104.3 | CH | 6.88, d (8.0) | 105.0 | CH | 7.02, d (8.1) | 105.0 | CH | 7.01, d (8.1) |
| 17 | 160.0 | CH | 7.33, s | 160.2 | CH | 7.28, s | 160.3 | CH | 7.28, s |
| 18 | 115.7 | CH2 | 5.01, dd (17.3, 2.0) 4.96, dd (10.4, 2.1) |
116.7 | CH2 | 4.96, dd (17.2, 1.8) 4.89, dd (10.2, 1.8) |
117.2 | CH2 | 4.98, dd (17.3, 1.7) 4.91, dd (10.3, 1.8) |
| 19 | 139.4 | CH | 5.55, dt (17.9, 9.3) | 137.9 | CH | 5.29, ddd (18.0, 10.3, 8.3) | 137.1 | CH | 5.27, ddd (18.0, 10.3, 8.3) |
| 9-OCH3 | 55.4 | CH3 | 3.88, s | 55.3 | CH3 | 3.89, s | 55.3 | CH3 | 3.88, s |
| 17-OCH3 | 61.7 | CH3 | 3.78, s | 61.7 | CH3 | 3.76, s | 61.7 | CH3 | 3.76, s |
| 22-OCH3 | 51.5 | CH3 | 3.69, s | 51.4 | CH3 | 3.67, s | 51.5 | CH3 | 3.67, s |
| NH | 7.85, s | 8.91, s | 9.12, s | ||||||
See Table S5 for a full comparison for the 1H and 13C NMR data for 5–7 (Supporting Information).
Compound 7 was isolated as an optically active yellow powder ( = +41.4, c 0.1 CHCl3), with a molecular formula of C23H28N2O4, deduced by HRESIMS, where the protonated molecule showed an m/z of 397.2115 (Figure S22). The 1H and 13C NMR data of 7 resembled those of compounds 5 and 6, sharing all the characteristic signals for an indole alkaloid with Δ18(19) unsaturation (see Tables 2 and S5, and Figures S23–S25). These observations, along with differences in chromatographic retention times (5: 7.5 min, 6: 7.0 min, and 7: 9.0 min; see Figure S34), suggested that 7 was a new diastereoisomer of 5 and 6. The HSQC data, along with COSY and HMBC correlations, were used to elucidate the structure of 7 (Figures S26–S28). For example, three isolated spin systems (specifically, H-10/H-11/H-12, H2-5/H2-6, and H-3/H2-14/H-15/H-20/H2-21/H-19/H2-18) and key HMBC correlations (i.e. H-3 to C-2/C-7, H-17 to C-15/C-22, H2-18 to C-20, CH3O-9 to C-9, CH3O-17 to C-17, and CH3O-22 to C-22) were used to confirm the skeleton for the proposed structure (Figure 2).
Figure 2.
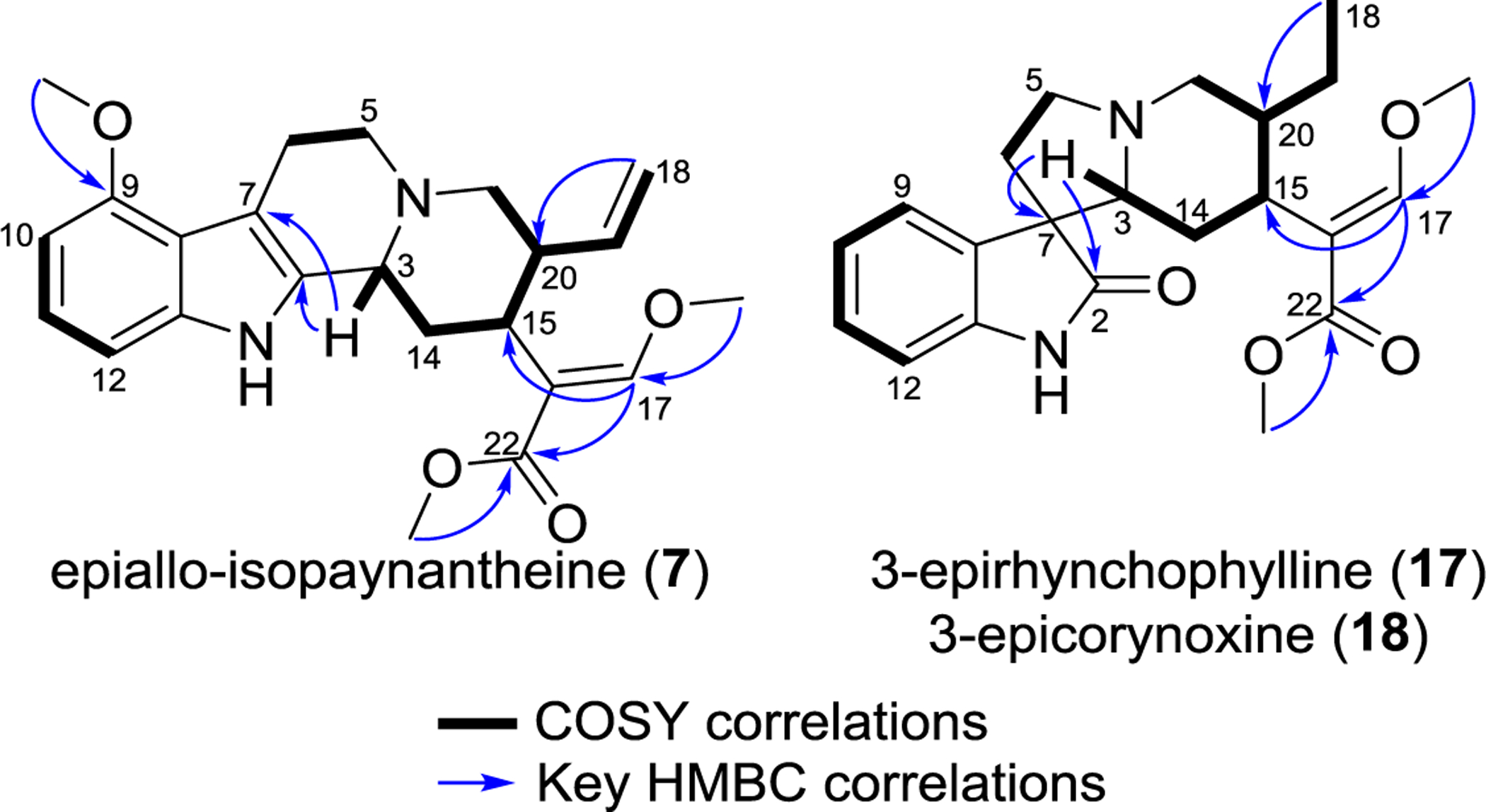
Key correlations observed in the COSY and HMBC spectra for compounds 7, 17, and 18. The latter two have the same 2D structure and thus the same correlations.
To establish the absolute configuration of 7, several complementary lines of reasoning were used. For instance, the deshielded signal at 4.85 ppm suggested H-3β, since that was similar to the chemical shift for the same position in compounds 2 (δH-3 4.40), 4 (δH-3 4.80), and 6 (δH-3 4.73), all of which have been established to be H-3β. Moreover, biogenetic assumptions were used for the C-15 configuration, since all the indole alkaloids from M. speciosa (42 compounds, to date) have the H-15α orientation.32 Those considerations narrowed the possibilities for compound 7 to either the pseudo (i.e., 3β, 15α, and 20β) or epiallo (i.e., 3β, 15α, and 20α) orientation.61 The known compound, isopaynantheine (6), has the pseudo configuration, and thus, compound 7 is the 20-epimer of 6 (i.e. H-20α), yielding an overall configuration of epiallo; those considerations were used to suggest the trivial name epiallo-isopaynantheine (7). In addition, ECD spectroscopy was used as an orthogonal probe of the proposed configuration. The spectrum for the epiallo configuration (i.e. 3β, 15α, and 20α) was calculated using a time-dependent DFT (TDDFT) method at the B3LYP/6–31G+(d) level of theory, demonstrating concordance, including the aforementioned negative Cotton effect at ca. 275 nm that confirmed the H-3β orientation (Figure 3). The NOESY spectra of compounds 6 and 7 were compared as well (Figures S29–S31). However, these data were inconclusive, since clear differences could not be discerned. This could be explained based on atomic distances, which were measured for the most stable conformers; in all cases these distances were < 5 Å (Figure S31). Collectively, the amalgamation of NMR and ECD data, and biogenetic considerations were used to establish the (3R,15S,20S) absolute configuration of the new indole alkaloid, epiallo-isopaynantheine (7).
Figure 3.
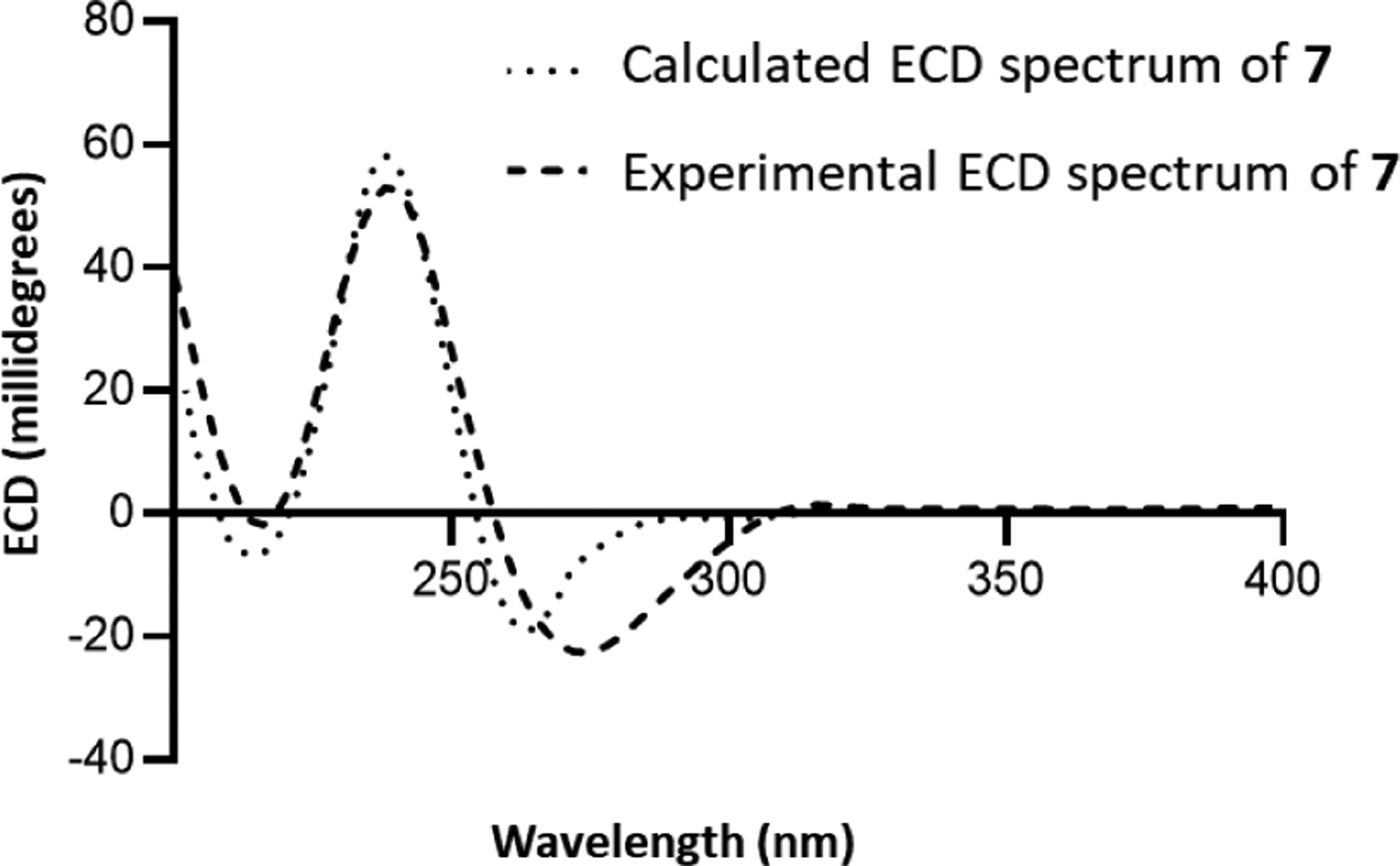
Experimental ECD spectrum of 7 in CH3OH at 0.2 mg/mL (dashed line) and calculated ECD spectrum of (3R,15S,20S)-7 (dotted line).
N(4)-oxides of Indole Alkaloids (8–11).
Compounds 8 and 9 had identical molecular formulae (C23H30N2O5), as determined by HRESIMS where the protonated molecules displayed an m/z of 415.2234 (8) and 415.2237 (9) (see Figure S35 and S36). This indicated that 8 and 9 had one extra oxygen atom compared to the diastereoisomers 1-4. 1H and 13C NMR spectra of both had strong similarities with the spectroscopic data reported for mitragynine-N(4)-oxide, sharing the characteristic NMR signals for the N(4)-oxide indole alkaloids (δH-10 6.43, δH-11 6.99–7.03, δH-12 6.92–6.95, δC-17/δH-17 161.3–161.5/7.45–7.46, δC-18/δH-18 11.8–12.7/0.85–0.93, δC-21/δH-21 68.5–68.6/3.53–3.72, δC-5/δH-5 65.8–65.9/3.51–3.98, and three methoxy groups δC/δH 51.6–62.1/3.61–3.86).30 The main difference between the spectra for compounds 8 and 9, relative to those discussed above for 1-4, was deshielding of the signals attributed to H-3, H2-5, and H2-21, which was caused by a coordinate covalent bond in the N(4)-oxide group. There is an important distinction with the N-oxides, relative to the other kratom alkaloids, in that the ΔδH-3 value is small (~0.12 ppm) and does not correlate with the orientation of H-3. As noted in the discussion of 1-4, the ΔδH-3 values of diastereoisomers mainly depend on the extent of interaction between the nitrogen free electron pair and the C–H antibonding orbitals. However, for the N-oxides, such an effect is not possible, since the electron pair is shared with the oxygen (i.e., N+−O– group). For the N-oxides, the positive charge on the nitrogen has a strong electron-withdrawing inductive effect, resulting in decreased electron density regardless whether the α-protons are axial or equatorial. In both compounds, the deshielded signal for the vinylic proton at δH-17 7.45–7.46 indicated the α orientation for H-20, analogous to compounds 1 and 2. In addition, H-15 was presumed to have the α orientation, based on the biogenetic considerations discussed for compound 7. Therefore, 8 and 9 could have the allo (i.e., 3α, 15α, and 20α) or the epiallo (i.e., 3β, 15α, and 20α) configurations. This illustrates another advantage of analyzing the ECD spectra of kratom alkaloids, particularly the Cotton effect at ca. 275 nm. Compounds 8 and 9 had opposite effects at ca. 275 nm, where a positive Cotton effect correlated with the H-3α orientation (i.e. 8) and a negative Cotton effect indicated the H-3β orientation (i.e. 9). In turn, these data allowed us to assign these compounds as mitragynine-N(4)-oxide (8)30 and speciociliatine-N(4)-oxide (9). This is the first report of NMR characterization data for 9, and key NMR signals for 8 and 9 are collated in Table 3, with full 1D NMR data reported in the Supporting Information (see Figures S39 and S40 and Table S6).
Table 3.
Characteristic 1H and 13C NMR Data for Compounds 8–11 (CDCl3, 400 and 100 MHz, Respectively)a
| mitragynine-N(4)-oxide (8) | speciociliatine-N(4)-oxide (9) | isopaynantheine-N(4)-oxide (10) | epiallo-isopaynantheine-N(4)-oxide (11) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| position | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) |
| 3 | 66.9 | CH | 5.16, s | 66.0 | CH | 5.04, d (12.5) | 69.2 | CH | 5.04, bs | 69.6 | CH | 4.91, bs |
| 10 | 99.7 | CH | 6.43, d (7.7) | 99.8 | CH | 6.43, d (7.7) | 99.9 | CH | 6.49, d (7.8) | 99.8 | CH | 6.48 d (7.7 |
| 11 | 123.3 | CH | 7.03, t (7.9) | 122.7 | CH | 6.99, t (7.9) | 123.8 | CH | 7.11, t (7.9) | 123.6 | CH | 7.10, t (7.9) |
| 12 | 105.2 | CH | 6.95, d (8.1) | 106.3 | CH | 6.92, bs | 105.1 | CH | 7.03, d (8.2) | 105.2 | CH | 7.04, d (8.1) |
| 17 | 161.5 | CH | 7.45, s | 161.3 | CH | 7.46, s | 160.5 | CH | 7.31, s | 160.5 | CH | 7.30, s |
| 18 | 12.7 | CH3 | 0.93, t (7.2) | 11.8 | CH3 | 0.85, t (7.4) | 118.0 | CH2 | 5.02, dd (17.5, 1.0) 4.95, dd (10.2, 1.7) |
117.8 | CH2 | 5.02, d (17.2) 4.94, dd (10.3, 1.6) |
| 9-OCH3 | 55.2 | CH3 | 3.85, s | 55.3 | CH3 | 3.86, s | 55.3 | CH3 | 3.88, s | 55.3 | CH3 | 3.87, s |
| 17-OCH3 | 62.1 | CH3 | 3.80, s | 61.9 | CH3 | 3.85, s | 62.0 | CH3 | 3.80, s | 62.0 | CH3 | 3.79, s |
| 22-OCH3 | 51.6 | CH3 | 3.61, s | 51.8 | CH3 | 3.68, s | 51.6 | CH3 | 3.68, s | 51.6 | CH3 | 3.68, s |
| NH | 9.46, s | - | 8.84, s | - | ||||||||
See Table S6 for a full comparison for the 1H and 13C NMR data for 8-11 (Supporting Information).
A similar set of observations were used to assign the structures of compounds 10 and 11, which were isolated as orange powders with a molecular formula of C23H28N2O5 as calculated by HRESIMS via a protonated molecule with an m/z of 413.2079 (10) and 413.2077 (11) (Figures S37 and S38). For instance, the NMR data for 10 and 11 were similar to those reported for isopaynantheine-N(4)-oxide (Table 3). These NMR data also resembled those of 8 and 9, where the main difference was the lack of the C-20 ethyl group (10 and 11) that was replaced with a vinylic group (see Table 3). In both cases, the signals for the vinylic proton appeared at δH-17 7.30–7.31, which was similar to the shifts in compounds 6 and 7. Again, ECD data were helpful, where the negative Cotton effects between at ca. 275 nm correlated with the H-3β orientation for both 10 and 11 (Figure S47). Additionally, a comparison of the ECD spectra of compounds 10 and 6 showed striking similarities; the same was true when comparing compounds 11 and 7 (see Figure S46). Therefore, compound 10 had the pseudo configuration (3β, 15α, and 20β; i.e., as seen in 6) and 11 had the epiallo configuration (3β, 15α, and 20α; i.e., as seen in 7). To confirm the absolute configuration of the new compound 11, the method described by Shellard and colleagues53 was used. Briefly, 11 was reduced by addition of H2SO4, and the reaction product should correspond to the indole alkaloid without the oxide. Indeed, the 1H NMR spectrum for the reduced product of 11 matched the spectrum of 7 (Figure S46). Collectively, these data permitted the characterization of these compounds as isopaynantheine-N(4)-oxide (10) and the new kratom alkaloid, epiallo-isopaynantheine-N(4)-oxide (11). The spectroscopic data for both are reported in the Supporting Information (Figures S41–S45 and Table S6).
Oxindole Alkaloids: 9-Hydroxylated Oxindoles Like Speciofoline (12–14).
Compound 12 had a molecular formula of C22H28N2O5, based on the protonated HRESIMS molecule with an m/z of 401.2067 (Figure S48). Comparison of the NMR data with key NMR signals in the 1H and 13C spectra reported previously permitted the identification of this compound as speciofoline (12) (Table 4).55, 56 This compound, which is one of the main alkaloids present in kratom (e.g. >55 mg isolated in this study), was first reported in 1963.55 We have included the full spectroscopic data for 12 to serve as a reference point when analyzing the spectroscopic data for some of the more minor oxindole alkaloids. For instance, the C-ring (i.e., the pyrollidine ring) of 12 is connected to the γ-lactam (i.e., B-ring) through a C-7 spiro carbon, and this results in characteristic signals in the 13C NMR spectrum at δC 180.2 and 57.3 for the carbonyl in the oxindole ring and the spiro C-7 carbon, respectively (Table 4); the lactam signal, in particular, serves to quickly differentiate between indole and oxindole alkaloids (see Figure 1). Other important NMR signals, which are similar to those discussed above for the indole alkaloids, include: 1) signals for the indole system (δH-10 6.35, δH-11 7.05 and δH-12 6.53); 2) a vinylic H-17 (δC/δH 159.8/7.38); 3) two methoxy groups at C-17 and C-22 (δC/δH 61.3/3.79 and δC/δH 51.7/3.65, respectively), and 4) a methyl group (δC-18/δH-18 12.3/0.83). Key NMR signals for 12 are tabulated (Table 4), and the full 1D NMR spectroscopic data are reported in the Supporting Information (Figure S51 and Table S7).
Table 4.
Characteristic 1H and 13C NMR Data for Compounds 12–14 (CDCl3, 500 and 125 MHz, Respectively)a
| speciofoline (12) | isorotundifoleine (13) | isospeciofoleine (14) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Position | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) |
| 2 | 180.2 | C | 177.7 | C | 179.1 | C | |||
| 3 | 63.8 | CH | 3.06, dd (11.7, 3.6) | 66.6 | CH | 3.39, dd (12.5, 3.6) | 68.7 | CH | 2.68, dd (11.6, 3.2) |
| 7 | 57.3 | C | 55.9 | C | 57.5 | C | |||
| 9 | 154.6 | C | 155.1 | C | 154.6 | C | |||
| 10 | 111.9 | CH | 6.35, d (7.6) | 112.1 | CH | 6.30, dd (7.6, 0.8) | 111.4 | CH | 6.36, dd (7.6, 0.7) |
| 11 | 129.6 | CH | 7.05, t (8.0) | 129.2 | CH | 7.02, dd (8.3, 7.7) | 129.6 | CH | 7.06, dd (8.3, 7.7) |
| 12 | 101.1 | CH | 6.53, d (8.3) | 100.3 | CH | 6.52, dd (8.5, 0.8) | 101.0 | CH | 6.58, d (8.3) |
| 17 | 159.8 | CH | 7.38, s | 159.8 | CH | 7.27, s | 159.9 | CH | 7.19, s |
| 18 | 12.3 | CH3 | 0.83, d (7.4) | 116.4 | CH2 | 5.01, m 4.96, m |
116.4 | CH2 | 4.97, m 4.95, m |
| 19 | 24.2 | CH2 | 1.21, dqd (14.0, 6.8, 2.8) | 138.8 | CH | 5.43, dt (18.7, 9.1) | 138.6 | CH | 5.49, dt (17.3, 9.5) |
| 17-OCH3 | 61.3 | CH3 | 3.79, s | 61.7 | CH3 | 3.79, s | 61.5 | CH3 | 3.71, s |
| 22-OCH3 | 51.7 | CH3 | 3.65, s | 51.3 | CH3 | 3.68, s | 51.2 | CH3 | 3.59, s |
| NH | 8.44, s | 7.45,s | 7.60, s | ||||||
See Table S7 for a full comparison for the 1H and 13C NMR data for 12-14 (Supporting Information).
Additionally, two other 9-hydroxylated oxindole alkaloids were isolated and characterized as isorotundifoleine (13) and isospeciofoleine (14). The molecular formula for both isomers was C22H26N2O5 as measured by HRESIMS based on protonated molecules with m/z values of 399.1909 and 399.1906, respectively (Figures S49 and S50). The structures were elucidated by comparing the NMR data with those reported previously (Table 4),56, 57 and the full assignment of the 1H and 13C NMR spectroscopic data for both are reported in the Supporting Information (Figures S52 and S53, and Table S7); this is the first report of comprehensive NMR data for 13. The NMR data for both 13 and 14 resembled those described above for speciofoline (12), with a major difference being the absence of the methyl group (i.e., C-18 in 12 at δC/δH 12.3/0.83), which was replaced by the Δ18(19) double bond (δC/δH 116.4/4.95–5.01 and δC/δH 138.6–138.8/5.43–5.49, respectively, for 13 and 14). As with the indole alkaloids, ECD spectra were instrumental in assigning the configuration of these compounds, especially for analyzing the C-7 spiro center. According to the literature, the negative Cotton effect at 290 nm confirmed the (7R) configuration in compounds 12 and 13, and the positive Cotton effect agreed with the (7S) configuration in compound 14. Additionally, the negative Cotton effect around 250 nm confirmed the H-3β orientation in compounds 12 and 14, whereas the opposite Cotton effect demonstrated the H-3α orientation in 13. These results agreed with the rules for ECD spectra for the 9-hydroxylated oxindole alkaloids, and a calculated ECD spectrum was generated for 12 as a confirmatory measure (see Figure S54).56, 64
Oxindole Alkaloids: 9-Unsubstituted Oxindoles Such as Corynoxine A (15–19).
Two closely related isomers, 15 and 16, were isolated with a molecular formula of C22H28N2O4, based on HRESIMS protonated molecules with m/z values of 385.2119 (i.e. 15) and 385.2127 (i.e. 16) (Figures S55 and S56). The compounds were characterized using 1H and 13C NMR data as corynoxine A (15) and corynoxine B (16), based on concordance with published data for synthetic standards of these compounds (Tables 5 and S8 and Figures S60 and S61).65 In addition, the NMR data for 15 and 16 resembled those described for 12, with the main difference being replacement of the C-9 phenolic group with an aromatic proton, which resulted in a pattern of two doublets and two triplets of doublets for the protons in the oxindole ring (H-9 to H-12, see Table 5). Both 15 and 16 had the allo configuration (i.e., 3α, 15α, and 20α), with opposite configurations at the spiro C-7, being (7S) for 15 and (7R) for 16. As Trager and colleagues reported,64 the configuration at C-7 can be deduced based on the chemical shift for the H-9 signal. For instance, when presuming the allo configuration, the presence of a deshielded signal for H-9 (δH 7.45) confirmed the (7S) configuration for 15 and the (7R) configuration in 16 due to the more shielded signal for H-9 (δH 7.19). While 15 was stable, an interesting challenge with compound 16 was its interconversion to 15 over a period of about 8 days in CDCl3 (see Figure S62). As reported previously,66 the epimerization process increases under acidic conditions via an intermolecular Mannich reaction, resulting in a ratio of ~70:30 of 15 and 16, respectively (see Figure S62). As such, only the ECD spectrum of 15 is reported, supporting the (7S) and the allo configurations based on negative Cotton effects around 290 nm and 250 nm (Figure S76).
Table 5.
Characteristic 1H and 13C NMR Data for Compounds 15–19 (CDCl3, 500 and 125 MHz, Respectively)a
| corynoxine A (15) | corynoxine B (16) | 3-epirhynchophylline (17) | 3-epicorynoxine B (18) | corynoxeine (19) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| position | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) | δC | type | δH (J in Hz) |
| 2 | 182.4 | C | 182.3 | C | 181.2 | C | 182.2 | C | 181.0 | C | |||||
| 3 | 73.2 | CH | 2.41, dd (11.3, 2.7) |
76.5 | CH | 2.24, bd (11.2) |
77.4 | CH | 2.19, dd (11.4, 2.4) |
77.0 | CH | 2.19, d (10.9) |
75.2 | CH | 2.30, dd (11.3, 2.5) |
| 9 | 125.0 | CH | 7.45, d (7.4) |
123.2 | CH | 7.19, d (7.4) |
123.3 | CH | 7.20, d (7.5) |
123.4 | CH | 7.20, d (7.8) |
123.5 | CH | 7.22, d (7.8) |
| 10 | 122.5 | CH | 7.05, td (7.6, 1.0) |
122.5 | CH | 7.01, td (7.5, 1.0) |
122.5 | CH | 7.02, td (7.6, 1.0) |
122.9 | CH | 7.02, td (7.5) |
122.7 | CH | 7.05, td (7.6, 1.0) |
| 11 | 127.4 | CH | 7.17, td (7.7, 1.3) |
127.9 | CH | 7.16, td (7.7, 1.0) |
127.8 | CH | 7.16, td (7.7, 1.2) |
128.1 | CH | 7.17, t (7.7) |
128.0 | CH | 7.18, td (7.7, 1.2) |
| 12 | 109.5 | CH | 6.86, d (7.7) |
109.5 | CH | 6.87, d (7.7) |
109.1 | CH | 6.81, d (7.7) |
109.6 | CH | 6.80, d (7.7) |
109.2 | CH | 6.82, d (7.7) |
| 18 | 13.0 | CH3 | 0.87, t (7.4) |
13.4 | CH3 | 0.86, t (7.4) |
13.5 | CH3 | 0.86, t (7.4) |
13.3 | CH3 | 0.86, t (7.3) |
115.6 | CH2 | 4.95, ddd (17.2, 2.01, 0.8) 4.90, dd (10.2, 2.1) |
| 19 | 19.4 | CH2 | 1.10, dqd (15.7, 7.8, 2.9) 1.65, dq (14.3, 7.2) |
19.3 | CH2 | 1.77, ddq (14.2, 11.1, 7.1) 1.18, m |
19.3 | CH2 | 1.78, ddq (13.6, 11.2, 7.2) 1.18, dq (14.7, 7.7) |
19.8 | CH2 | 1.80, m 1.17, dq (14.5, 7.3) |
139.6 | CH | 5.51, dt (18.0, 9.1) |
| 17-OCH3 | 61.2 | CH3 | 3.51, s | 61.6 | CH3 | 3.57, s | 61.7 | CH3 | 3.67, s | 61.7 | CH3 | 3.69, s | 61.7 | CH3 | 3.74, s |
| 22-OCH3 | 51.3 | CH3 | 3.59, s | 51.4 | CH3 | 3.61, s | 51.4 | CH3 | 3.63, s | 51.4 | CH3 | 3.63, s | 51.4 | CH3 | 3.62, s |
| NH | 8.40, s | 8.91, s | 7.62, s | 7.33, s | 7.53, s | ||||||||||
See Table S8 for a full comparison for the 1H and 13C NMR data for 15-19 (Supporting Information).
Compounds 17 and 18 were isolated as optically active powders ( −30 (c 0.1, CHCl3) and −46 (c 0.1, CHCl3), respectively). Their molecular formulae were established as C22H28N2O4 based on the HRESIMS protonated molecule with m/z values of 385.2119 (i.e. 17) and 385.2120 (i.e. 18) (Figures S57 and S58). The 1H and 13C NMR spectra of these compounds were similar to those of 15 and 16 (Table 5). These data, along with their different retention times (17: 2.76 min and 18: 2.87 min) (see Figures S57 and S58), suggested that 17 and 18 were new isomers of 15 and 16. The HSQC data, along with COSY and HMBC correlations, were used to confirm the proposed structures. In both cases, three isolated spin systems (specifically H-9/H-10/H-11/H-12, H2-5/H2-6, and H-3/H2-14/H-15/H-20/H2-21/H2-19/H3-18) and key HMBC correlations (i.e. H-3 to C-2/C-7, H-17 to C-15/C-22, H3-18 to C-20, CH3O-17 to C-17, and CH3O-22 to C-22) were used to confirm the 2D structures of 17 and 18 (see Figures 2 and S63–S65 and S69–S71). Similar to the discussion for 6 and 7, the correlations observed in the NOESY spectra for 17 and 18 were not useful to determine the relative configuration for these compounds (Figures S67 and S73). The absolute configuration of these molecules was proposed based on ECD experiments, following the rules described for the 9-unsubstituted oxindole alkaloids. In particular, the related isomer rhyncophylline served as a reference, since the crystal structure for this compound has been published and the ECD data were well described.64, 67 For instance, both 17 and 18 had the (7R) configuration, based on positive Cotton effects around 290 nm, and the H-3β orientation, as noted by positive Cotton effects around 250 nm (Figure S76). Using biogenetic reasoning for the H-15α orientation, along with the evidence observed in the ECD data, we deduced that compounds 17 and 18 should have the pseudo (i.e., 3β, 15α, and 20β) or epiallo (i.e., 3β, 15α, and 20α) configurations. To evaluate that presumption, the ECD spectra for the diastereoisomers at C-20 were calculated using a time-dependent DFT (TDDFT) method at the B3LYP/6–31G+(d) level of theory. The results showed that the calculated ECD spectrum for H-20β matched with the ECD spectrum for 17, and similarly, the calculated ECD spectrum for an analogue with the H-20α orientation matched with the ECD spectrum for compound 18 (Figure 4). NMR data, biogenetic considerations, and the examination of ECD spectra were used to establish the absolute configuration of the new oxindole alkaloids, (3R,7R,15S,20R)-3-epirhynchophylline (17) and (3R,7R,15S,20S)-3-epicorynoxine B (18).
Figure 4.
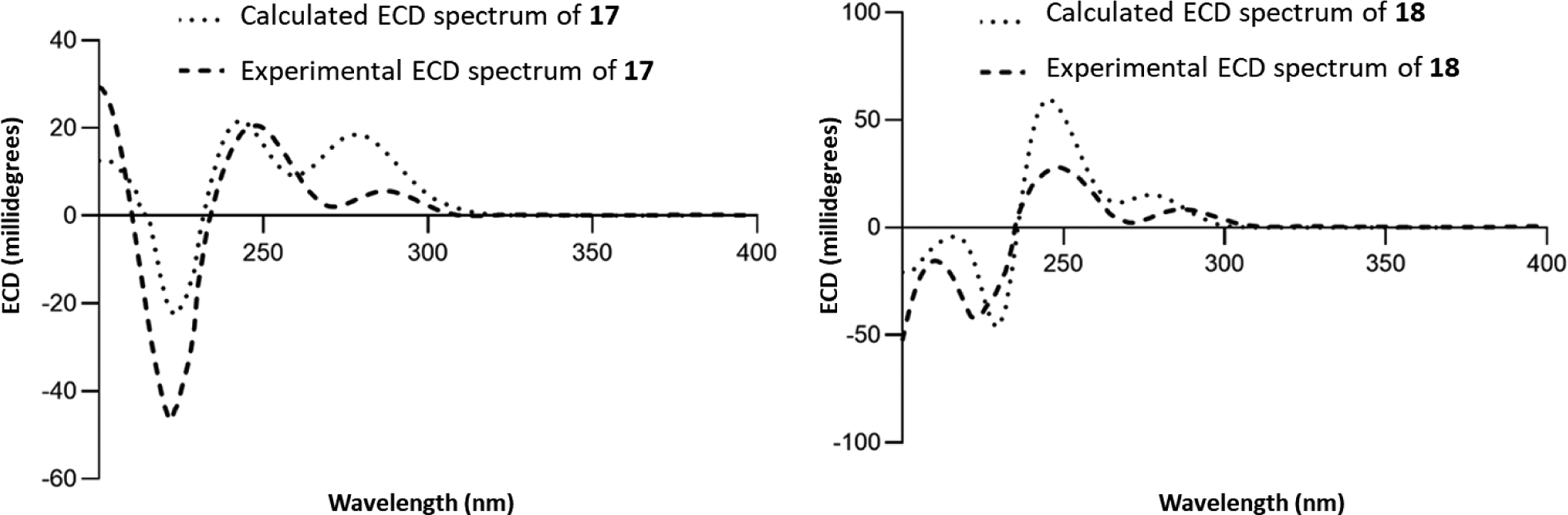
A) Experimental ECD spectra of 17 in CH3OH at 0.2 mg/mL (dashed lines) and calculated ECD spectrum of (3R,7R,15S,20R)-17 (doted lines). B) Experimental ECD spectra of 18 in CH3OH at 0.2 mg/mL (dashed lines) and calculated ECD spectrum of (3R,7R,15S,20S)-18 (doted lines).
Compound (19) was isolated as a white powder, and its molecular formula was established as C22H26N2O4, based on the HRESIMS protonated molecule with an m/z of 383.1964 (Figure S59). The NMR data for this compound showed strong similarities with the data of 15-18, where the main differences were the absence of the signals for the C-20 ethyl group, which was replaced by a vinylic group (Tables 5 and S8). The NMR data (Figure S75 and Table S8) matched with those reported for corynoxeine.68 ECD data corroborated the H-3α orientation based on the negative Cotton effect around 250 nm, and the (7R) configuration based on the positive Cotton effect around 290 (Figure S76).
To add complementary data to the structural characterization of the kratom alkaloids, an attempt was made to analyze compounds 1, 6, 7, 15, 17, and 18 by VCD, so as to examine a range of both indole and oxindole alkaloids. However, in all cases the similarities between calculated and experimental data were low, ranging between 57.7 to 70.0% (Table S9). In contrast, prominent recent VCD results published in this Journal report similarities ranging between 74.1 to 98.5%.69–73 In particular, we were surprised at the lack of concordance with the data for mitragynine (1), since it has been so well characterized, including by X-ray crystallography.27 Thus, the purity of 1, 6, 7, 15, 17, and 18, were examined after the completion of the VCD studies, and indeed, it was obvious that the compounds decomposed during the experiments (see Figure S77). Based on the 1H NMR spectra (data not shown), we hypothesize that epimers were formed, likely as a result of the VCD data being recorded at elevated temperatures in CHCl3 for up to 18 hours; the epimerization potential of these compounds has been reported.66 While disappointing, we determined that analysis of VCD spectra is not helpful for this class of compounds, at least with current technologies, due to their lack of stability under the analytical conditions.
In summary, 19 reference standards were isolated from kratom plant materials, including the two new indole alkaloids 7 and 11 and the two new oxindole alkaloids 17 and 18. To characterize their structures, including absolute configuration, a combination of mass spectrometry and NMR and ECD spectroscopy were utilized. An added benefit of analyzing the NMR spectroscopy data for a suite of closely related compounds was that it facilitated the development of a decision tree (Figure 1). Using only a few key NMR signals, even a relative novice should be able to narrow the possible structures for at least these kratom alkaloids, and an expert is encouraged to examine a full suite of NMR data in relation to those reported in the Supporting Information. It is our hope that improvements in the structural characterization of the kratom alkaloids will result in more clarity in the biomedical literature on this interesting class of compounds.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations were obtained in CHCl3 using a Rudolph Research Autopol III polarimeter. UV and ECD spectra were acquired on a Varian Cary 100 Bio UV-Vis spectrophotometer and an Olis DSM 17 CD spectrophotometer (Olis), respectively. A ChiralIR-2X dual-source, dual-polarization-modulated FT-VCD spectrometer was used to carry out IR and VCD measurements. NMR experiments were conducted using either a JEOL ECA-500 NMR spectrometer operating at 500 MHz for 1H and 125 MHz for 13C or a JEOL ECS-400 NMR spectrometer equipped with a high sensitivity JEOL Royal probe operating at 400 MHz for 1H and 100 MHz for 13C; the residual solvent signals were utilized for referencing. HRESIMS was performed on a Thermo LTQ Orbitrap XL mass spectrometer equipped with an electrospray ionization source. UPLC was carried out on a Waters Acquity system with data collected and analyzed using Thermo Xcalibur software. HPLC was carried out using a Varian ProStar HPLC system equipped with ProStar 210 pumps and a ProStar 335 photodiode array detector (PDA), with data collected and analyzed using Galaxie Chromatography Workstation software (version 1.9.3.2). For preparative HPLC, a series of Phenomenex columns were used, including a Kinetex C18 (5 μm; 250 × 21.2 mm), a Luna CN (5 μm; 250 × 21.2 mm) and a Luna PFP (5 μm; 250 × 21.2 mm) all at a 20 mL/min flow rate. For UPLC, a Waters BEH C18 column (1.7 μm; 50 × 2.1 mm) was used with a 0.6 mL/min flow rate. Flash chromatography was performed on a Teledyne ISCO CombiFlash Rf 200 using various sizes of Silica Gold columns and monitored by UV and evaporative light-scattering detectors.
Plant Material, DNA Extraction, PCR and Sequencing for Molecular Identification.
The commercial kratom products used to prepare extracts [i.e. Green Maeng Da (K49, batch: M-1005, 4 kg) and White Jongkong (K52, batch: BN-1006, 1 kg)] were identified by DNA barcoding. The manufacturers reported these as both from Southeast Asia, with K49 being chipped leaf materials and K52 being a fine powder. Samples were handled using methods described previously for the DNA barcoding of commercial mushroom products.74 Briefly, one scintillation vial was made from each product (see Figure S6), where the vial was filled with plant material from the top and middle layers of the commercial product, and these were labeled with product name, lot number, and batch number and stored at ambient temperature in a bench drawer. For DNA extractions, approximately 5 mg of plant powder was taken from the scintillation vial and transferred to a bashing bead tube with DNA lysis buffer provided by Zymo Research Quick-DNA plant/seed miniprep DNA extraction kit. DNA was extracted using procedures outlined in the Zymo Research plant DNA extraction kit. The plastid gene matK was PCR amplified with primer combination matK-xf and matK-MALP;75, 76 for psbA-trnH primer combination psbA – trnH,77 and for nuclear ITS, primer combination ITS-u1and ITS-u478 was used for PCR and sequencing. All PCRs were amplified on an Applied Biosystems Veriti thermal cycler using PuReTaq Ready-To-Go PCR Beads (GE Healthcare) with the above primers. The PCR reaction was carried out in 25 μL containing 5 μL template DNA, 2.5 μL BSA, 2.5 μL 50% DMSO, and 1 μL of each forward and reverse primer at a concentration of 10 μM. The rest of the volume was made up to 25 μL by adding molecular biology grade water from Fisher Scientific. PCR thermocycler parameters for all fragments are outlined (Table S3). The PCR products were then run on an ethidium bromide-stained 1% agarose gel (Fisher Scientific) along with a 1 kb DNA ladder (Promega) to estimate the size of the amplified band. PCR products were purified using a Wizard SV Gel and PCR Clean-up System. Sanger sequencing of the purified PCR products was performed at Eurofins Genomics (http://www.operon.com/default.aspx) using BigDye Terminator v3.1 cycle sequencing. The sequencing was accomplished bidirectionally using both strands with the same primers used for the PCR amplification. Sequences were generated on an Applied Biosystems 3730XL high-throughput capillary sequencer. For both sequencing reactions, approximately 15 μL of PCR template was used along with 2 μM sequencing primers. Sequences were assembled with Sequencher 5.3 (Gene Codes), optimized, and then corrected manually when necessary; the latter step was to ensure that the computer algorithm was assigning proper base calls. Each sequence fragment was subjected to an individual NCBI GenBank, Basic Local Alignment Search Tool (BLAST) search as well as the BOLDSYSTEMS database to verify identity.
BLAST Search and DNA Barcoding.
For identification of kratom plant material via DNA Barcoding, one plastid region (matK) was utilized for plant identification by BLAST searching against the BOLDSYSTEMS database version 4. In addition, the sequences of matK were compared against an authentic partial sequence of matK from the Mitragyna speciosa genome assembly (Center for Food Safety and Applied Nutrition (CFSAN), part of the USDA). Uncorrected p-distances were calculated in Geneious, a bioinformatics desktop software package (http://www.biomatters.com), for sequences obtained from the psbA-trnH plastid region, which is considered as one of the most variable regions of angiosperm plants, as well as for nuclear ITS region for the published sequence data from GenBank for Mitragyna spp.79–81 including M. speciosa and the newly obtained sequences in this study. The p-distances were obtained by dividing the number of nucleotide differences by the total number of nucleotides being compared. An arbitrary cut off proxy of ≥ 98–100% was applied as a criterion to designate similar species; therefore, to be considered the same species based on psbA-trnH and ITS sequence comparison, the taxa being compared would have ≥ 98% sequence similarity.
Molecular Phylogenetic Analysis.
Maximum likelihood analysis was performed separately for both the nuclear ITS and the plastid matK region to place the kratom samples into a phylogenetic framework with published sequences of M. speciosa. Methods for maximum likelihood analysis have been outlined previously.82 Nauclea officinalis (Rubiaceae) was used as an outgroup taxon in both nuclear ITS and matK analyses.
Extraction and Isolation of Kratom Alkaloids from Green Maeng Da.
The kratom plant material (Green Maeng Da, 4 kg; Figure S8) was extracted with 10 L of CHCl3-CH3OH (1:1) and 500 mL of 10% aqueous KOH by maceration over 24 h at room temperature. The mixture was filtered, and the solvent was evaporated under reduced pressure. The dried extract was reconstituted in a solution of 1M HCl and hexanes (1:1), transferred into a separatory funnel, and shaken vigorously. The hexanes phase was drawn off, the pH of the aqueous phase was adjusted to 9.0 with dropwise addition of concentrated NH4OH, and the alkaloids were extracted as the free base with CHCl3; after washing with neutral water, the organic phase was dried to yield 12 g of the alkaloid extract. This material was fractionated by normal phase flash chromatography using a silica column (120 g) and a gradient solvent system of hexanes-CHCl3-CH3OH at a flow rate of 85 mL/min over 67 min to yield 11 pooled fractions.
Fraction 4 (500 mg) was subjected to reverse phase HPLC using a CN column and a gradient system of 40:60 to 100:0 of CH3OH-H2O (10 mM of NH4OAc in both phases) over 20 min with a flow rate of 20 mL/min. This process yielded eight subfractions, and fraction 8 was identified as compound 1 (450.5 mg).
Fraction 9 (1.1 g) was fractionated by normal phase flash chromatography using a silica column (12 g) and a gradient system of hexanes-EtOAc-CH3OH using a flow rate of 30 mL/min to generate four subfractions. Subfraction 2 (40 mg) was purified by preparative HPLC over a CN column using a gradient of 70:30 to 100:0 of CH3OH-H2O (10 mM of NH4OAc in both phases) over 20 min with a flow rate of 20 mL/min. This process yielded compounds 4 (13.1 mg) and 6 (15.20 mg). Subfraction 4 (800 mg) was subjected to flash chromatography using a silica column (12 g) via a gradient of CHCl3-CH3OH (10 mM of NH4OAc in both phases) over 60 min with a flow rate of 30 mL/min to generate five fractions (F9–4_1 through F9–4_5). Fraction F9–4_3 was subjected to preparative HPLC over a Kinetex column using a gradient of 60:40 to 70:30 of CH3OH-H2O (10 mM of NH4OAc in both phases) over 30 min with a flow rate of 20 mL/min; three fractions were collected and the first fraction was characterized as compound 2 (20.5 mg). Fraction F9–4_4 (300 mg) was subjected to flash chromatography using a silica column (4 g) and a gradient of hexanes-acetone-CH3OH over 20 min at a flow rate of 18 mL/min to generate eight fractions. The second fraction was resolved by preparative HPLC over a CN column using a gradient of 50:50 to 100:0 of CH3OH-H2O (10 mM of NH4OAc in both phases) over 20 min with a flow rate of 20 mL/min; this process yielded 16.5 mg of compound 7. The seventh fraction (16 mg) from F9–4_4 was subjected to preparative HPLC using a CN column and a gradient of 40:60 to 90:10 of CH3OH-H2O (10 mM of NH4OAc in both phases) over 20 min with a flow rate of 20 mL/min; two fractions were collected and the first fraction was characterized as compound 11 (2.3 mg). Fraction F9–4_5 (40 mg) was subjected to preparative HPLC over a Kinetex column with a gradient of 50:50 to 100:0 of CH3OH-H2O (10 mM of NH4OAc in both phases) over 35 min with a flow rate of 20 mL/min; the third fraction was identified as 8 (10.5 mg).
Fraction 6 (680 mg) was subjected to a second fractionation using flash chromatography over a silica column (12 g) and a gradient of CHCl3-CH3OH (10 mM of NH4OAc in both phases) over 60 min with a flow rate of 30 mL/min to generate five subfractions. Subfraction 1 (47 mg) was subjected to preparative HPLC over a CN column and a gradient system of 55:45 to 100:0 CH3OH-H2O (10 mM of NH4OAc in both phases) over 25 min; the first and second fractions were identified as 5 (9.83 mg) and 3 (18.33 mg), respectively.
Fraction 8 (1.3 g) was fractionated by flash chromatography using a silica column (12 g) with a gradient of CHCl3-CH3OH (10 mM of NH4OAc in both phases) over 25 min with a flow rate of 30 mL/min to generate four subfractions. Subfractions 3 (12.8 mg) and 4 (7.0 mg) were purified by preparative HPLC over a CN column and a gradient system of 50:50 to 100:0 CH3OH-H2O (10 mM of NH4OAc in both phases) over 15 min at a flow rate of 20 mL/min to yield compounds 9 (5.7 mg) and 10 (4.5 mg), respectively.
Extraction and Isolation of Kratom Alkaloids from White Jongkong.
The plant material (White Jongkong, 1 kg; Figure S9) was extracted exhaustively using the same procedure described above, to obtain 1.0 g of the dried extract. Normal phase flash chromatography was used to separate the extract using a silica column (24 g) and a gradient system of hexanes-CHCl3-CH3OH over 52 min at a flow rate of 35 mL/min to generate 13 fractions. Fractions 3 (100.0 mg) and 4 (80.0 mg) were purified, separately, by preparative HPLC using a PFP column and a gradient of 70:30 to 100:0 CH3OH-H2O (10 mM of NH4OAc in both phases) over 20 min at a flow rate of 20 mL/min to yield 38.2 mg of 15 (from fraction 3), and separately, 52.5 mg of 16 and 3.5 mg of 13 (from fraction 4).
Fraction 5 (80.5 mg) was subjected to preparative HPLC over a PFP column using a gradient of 70:30 to 100:0 CH3OH-H2O (10 mM of NH4OAc in both phases) over 20 min at a flow rate of 20 mL/min to generate nine subfractions, and subfraction 4 was identified as compound 12 (55.0 mg).
Fractions 6 (156.2 mg) and 7 (84.1 mg) were combined based upon their similar HPLC chromatographic profiles. The resulting fraction (240.3 mg) was resolved by flash chromatography over a silica column (12 g) using a gradient of hexanes-CHCl3-CH3OH over 25 min at a flow rate of 18 mL/min to generate eleven subfractions. Subfraction 2 (10.0 mg) was subjected to semipreparative HPLC over a PFP column using a gradient of 50:50 to 100:0 CH3OH-H2O (10 mM of NH4OAc in both phases) over 20 min at a flow rate of 4.0 mL/min, yielding 17 (2.0 mg). Subfraction 4 (15.0 mg) was subjected to preparative HPLC over a PFP column using a gradient of 60:40 to 100:0 CH3OH-H2O (10 mM of NH4OAc in both phases) over 20 min at a flow rate of 20.0 mL/min, yielding 14 (3.6 mg). Subfraction 5 (13.0 mg) was subjected to preparative HPLC over a PFP column using a gradient of 50:50 to 100:0 CH3OH-H2O (10 mM of NH4OAc in both phases) over 20 min at a flow rate of 20.0 mL/min to generate four subfractions, where subfractions 2 and 3 were identified as 19 (1.7 mg) and 18 (1.1 mg), respectively.
Epiallo-isopaynantheine (7):
Yellowish solid powder. +41 (c 0.1, CHCl3); UV (CH3OH) λmax (log ε) 233 (3.7), 339 (2.9) nm; 1H and 13C NMR see Table 2 and Table S5; HRESIMS m/z 397.2115 [M + H]+ (calcd for C23H29N2O4, 397.2127).
Epiallo-isopaynantheine-N(4)-oxide (11):
Orange powder. +33 (c 0.1, CHCl3); UV (CH3OH) λmax (log ε) 219 (3.2), 244 (3.9) nm; 1H and 13C NMR see Table 3 and Table S6; HRESIMS m/z 413.2077 [M + H]+ (calcd for C23H29N2O5, 413.2076).
3-Epirhyncophylline (17):
White powder. −30 (c 0.1, CHCl3); UV (CH3OH) λmax (log ε) 219 (3.3), 244 (3.6) nm; 1H and 13C NMR see Table 5 and Table S8; HRESIMS m/z 385.2119 [M + H]+ (calcd for C22H29N2O4, 385.2127).
3-Epicorynoxine B (18):
White powder. −46 (c 0.1, CHCl3); UV (CH3OH) λmax (log ε) 217 (3.4), 246 (3.7) nm; 1H and 13C NMR see Table 5 and Table S8; HRESIMS m/z 385.2120 [M + H]+ (calcd for C22H29N2O4, 385.2127).
Computational Methods.
The minimum energy structures were built with Spartan’10 software (Wavefunction Inc., Irvine, CA, USA). The conformational analysis was performed using the Monte Carlo search protocol under the MMFF94 molecular mechanics force field.
For the ECD prediction, the resulting conformers were minimized using the DFT method at the B3LYP/6–311G+(2d,p) level of theory. Then, the time-dependent DFT (TDDFT) method at the B3LYP/6–31G+(d) level of theory was employed for ECD calculations. The calculated excitation energy (nm) and rotatory strength (R) in dipole velocity (Rvel) and dipole length (Rlen) forms were simulated into an ECD curve. All calculations were performed employing the Gaussian’09 program package (Gaussian Inc., Wallingford, CT, USA).
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by the National Institutes of Health/National Center for Complementary and Integrative Health via the Center of Excellence for Natural Product Drug Interaction Research (NaPDI Center, U54 AT008909), including an Administrative Supplement for Validation Studies of Analytical Methods for Dietary Supplements and Natural Products. We thank our colleagues from UNC Greensboro (S. Knowles, Z. Al Subeh, and K. Cank) for evaluating the NMR decision tree.
Footnotes
Supporting Information
The Supporting Information is available free of charge.
1H NMR, 13C NMR and HRESIMS data for compounds 1-19. 2D NMR data (COSY, HSQC and HMBC) for new compounds 7, 11, 17 and 18. ECD data for compounds 1-14 and 16-19.
REFERENCES
- (1).Field E J. Chem. Soc. Perkin Trans. I 1921, 119, 887–891. [Google Scholar]
- (2).Vermaire DJ; Skaer D; Tippets W A. A. Pract 2019, 12, 103–105. [DOI] [PubMed] [Google Scholar]
- (3).Schmuhl KK; Gardner SM; Cottrill CB; Bonny AE Subst. Abus 2019, 1–4. [DOI] [PubMed] [Google Scholar]
- (4).Coe MA; Pillitteri JL; Sembower MA; Gerlach KK; Henningfield JE Drug Alcohol. Depend 2019, 202, 24–32. [DOI] [PubMed] [Google Scholar]
- (5).Buresh M J. Addict. Med 2018, 12, 481–483. [DOI] [PubMed] [Google Scholar]
- (6).Corkery JM; Streete P; Claridge H; Goodair C; Papanti D; Orsolini L; Schifano F; Sikka K; Korber S; Hendricks A J. Psychopharmacol 2019, 1102–1123. [DOI] [PubMed] [Google Scholar]
- (7).Murthy P; Clark D Paediatr. Child. Health 2019, 24, 12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mackay L; Abrahams R Can. Fam. Physician 2018, 64, 121–122. [PMC free article] [PubMed] [Google Scholar]
- (9).Cumpston KL; Carter M; Wills BK Am. J. Emerg. Med 2018, 36, 166–168. [DOI] [PubMed] [Google Scholar]
- (10).Castillo A; Payne JD; Nugent K Proc. (Bayl. Univ. Med. Cent.) 2017, 30, 355–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Galbis-Reig D Wmj 2016, 115, 49–52; quiz 53. [PubMed] [Google Scholar]
- (12).Karinen R; Fosen JT; Rogde S; Vindenes V Forensic. Sci. Int 2014, 245, e29–e32. [DOI] [PubMed] [Google Scholar]
- (13).Forrester MB J. Addict. Dis 2013, 32, 396–400. [DOI] [PubMed] [Google Scholar]
- (14).Holler JM; Vorce SP; McDonough-Bender PC; Magluilo J Jr.; Solomon CJ; Levine B J. Anal. Toxicol 2011, 35, 54–9. [DOI] [PubMed] [Google Scholar]
- (15).McWhirter L; Morris S Eur. Addict. Res 2010, 16, 229–31. [DOI] [PubMed] [Google Scholar]
- (16).The New York Times April 17, 2019. https://www.nytimes.com/2019/04/17/us/kratom-overdose-deaths.html (accessed October 25, 2019)
- (17).U.S. Food & Drug Administration February 21, 2018. https://www.fda.gov/news-events/press-announcements/fda-oversees-destruction-and-recall-kratom-products-and-reiterates-its-concerns-risks-associated (accessed October 25, 2019)
- (18).United States Drug Enforcement Administration August 30, 2016. https://www.dea.gov/press-releases/2016/08/30/dea-announces-intent-schedule-kratom (accessed October 25, 2019)
- (19).Forbes October 13, 2016. https://www.forbes.com/sites/davidkroll/2016/08/30/dea-to-place-kratom-mitragynine-on-schedule-i-premature-move-may-compromise-research-benefits/#679fe47d2b8f (accessed October 25, 2019)
- (20).Johnson EJ; González-Peréz V; Tian D-D; Lin YS; Unadkat JD; Rettie AE; Shen DD; McCune JS; Paine MF Drug Metab. Dispos 2018, 46, 1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tian D-D; Kellogg JJ; Okut N; Oberlies NH; Cech NB; Shen DD; McCune JS; Paine MF Drug Metab. Dispos 2018, 46, 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gufford BT; Chen G; Lazarus P; Graf TN; Oberlies NH; Paine MF Drug Metab. Dispos 2014, 42, 1675–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kellogg JJ; Paine MF; McCune JS; Oberlies NH; Cech NB Nat. Prod. Rep 2019, 36, 1196–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Brantley SJ; Gufford BT; Dua R; Fediuk DJ; Graf TN; Scarlett YV; Frederick KS; Fisher MB; Oberlies NH; Paine MF CPT: Pharmacometrics Syst. Pharmacol 2014, 3, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Beckett AH; Shellard EJ; Tackie AN Planta Med 1965, 13, 241–246. [Google Scholar]
- (26).Beckett A; Shellard E; Phillipson J; Lee CM Planta Med 1966, 14, 277–288. [DOI] [PubMed] [Google Scholar]
- (27).Zacharias D; Rosenstein R; Jeffrey G Acta Crystallogr. A 1965, 18, 1039–1043. [Google Scholar]
- (28).Beckett AH; Shellard EJ; Phillipson JD; Lee CM Planta Med 1966, 14, 277–288. [DOI] [PubMed] [Google Scholar]
- (29).Shellard E; Houghton P; Resha M Planta Med 1978, 34, 253–263. [Google Scholar]
- (30).Takayama H; Ishikawa H; Kurihara M; Kitajima M; Aimi N; Ponglux D; Koyama F; Matsumoto K; Moriyama T; Yamamoto LT; Watanabe K; Murayama T; Horie SJ Med. Chem 2002, 45, 1949–1956. [DOI] [PubMed] [Google Scholar]
- (31).Takayama H Chem. Pharm. Bull 2004, 52, 916–928. [DOI] [PubMed] [Google Scholar]
- (32).Takayama H; Kitajima M; Kogure N Curr. Org. Chem 2005, 9, 1445–1464. [Google Scholar]
- (33).Marston A; Hostettmann K Nat. Prod. Rep 1991, 8, 391–413. [Google Scholar]
- (34).Stead P, Isolation by Preparative HPLC In Natural Products Isolation, Cannell RJP, Ed. Humana Press: Totowa, NJ, 1998; pp 165–208. [Google Scholar]
- (35).Betz JM; Brown PN; Roman MC Fitoterapia 2011, 82, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Li S; Yuan W; Deng G; Wang P; Yang P; Aggarwal B Pharm. Crop 2011, 2, 28–54. [Google Scholar]
- (37).Napolitano JG; Lankin DC; Chen SN; Pauli GF Magn. Reson. Chem 2012, 50, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Niemitz M; Laatikainen R; Chen SN; Kleps R; Kozikowski AP; Pauli GF Magn. Reson. Chem 2007, 45, 878–882. [DOI] [PubMed] [Google Scholar]
- (39).Seebacher W; Simic N; Weis R; Saf R; Kunert O Magn. Reson. Chem 2003, 41, 636–638. [Google Scholar]
- (40).Sy-Cordero AA; Pearce CJ; Oberlies NH J. Antibiot 2012, 65, 541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kao D; Flores-Bocanegra L; Raja HA; Darveaux BA; Pearce CJ; Oberlies NH Phytochemistry 2020, 172, 112238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).https://www.fda.gov/news-events/public-health-focus/fda-and-kratom (accessed January 20, 2020)
- (43).Hollingsworth ML; Andra Clark A; Forrest LL; Richardson J; Pennington RT; Long DG; Cowan R; Chase MW; Gaudeul M; Hollingsworth PM Mol. Ecol. Resour 2009, 9, 439–457. [DOI] [PubMed] [Google Scholar]
- (44).Kress WJ; Wurdack KJ; Zimmer EA; Weigt LA; Janzen DH Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kress WJ; Erickson DL PLoS one 2007, 2, e508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Li X; Yang Y; Henry RJ; Rossetto M; Wang Y; Chen S Biol. Rev 2015, 90, 157–166. [DOI] [PubMed] [Google Scholar]
- (47).Ebihara A; Nitta JH; Ito M PLoS one 2010, 5, e15136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Brown PN; Lund JA; Murch SJ J. Ethnopharmacol 2017, 202, 302–325. [DOI] [PubMed] [Google Scholar]
- (49).Raffa RB, Kratom and Other Mitragynines: the Chemistry and Pharmacology of Opioids From a Non-opium Source CRC Press: 2014. [Google Scholar]
- (50).Takayama H; Kurihara M; Kitajima M; Said IM; Aimi N Tetrahedron 1998, 54, 8433–8440. [Google Scholar]
- (51).León F; Habib E; Adkins JE; Furr EB; McCurdy CR; Cutler SJ Nat. Prod. Commun 2009, 4, 907–910. [PMC free article] [PubMed] [Google Scholar]
- (52).Beckett A; Shellard E; Phillipson J; Lee CM J. Pharm. Pharmacol 1965, 17, 753–755. [DOI] [PubMed] [Google Scholar]
- (53).Shellard EJ; Hougton PJ; Resha M Planta Med 1978, 33, 223–227. [Google Scholar]
- (54).Cao X-F; Wang J-S; Wang X-B; Luo J; Wang H-Y; Kong L-Y Phytochemistry 2013, 96, 389–396. [DOI] [PubMed] [Google Scholar]
- (55).Beckett AH; Lee CM; Tackie AN Tetrahedron Lett 1963, 4, 1709–1714. [Google Scholar]
- (56).Hemingway SR; Houghton PJ; Phillipson JD; Shellard EJ Phytochemistry 1975, 14, 557–563. [Google Scholar]
- (57).Ali Z; Demiray H; Khan IA Tetrahedron Lett 2014, 55, 369–372. [Google Scholar]
- (58).Cu N Bull. Soc. Chim. Fr 1957, 1292–1294. [Google Scholar]
- (59).Shellard E Planta Med 1978, 34, 26–36. [Google Scholar]
- (60).Kitajima M; Misawa K; Kogure N; Said IM; Horie S; Hatori Y; Murayama T; Takayama H J. Nat. Med 2006, 60, 28–35. [Google Scholar]
- (61).Lee CM; Trager WF; Beckett AH Tetrahedron 1967, 23, 375–385. [DOI] [PubMed] [Google Scholar]
- (62).1H NMR Properties of Piperidine Derivatives In Studies in Organic Chemistry, Rubiralta M; Giralt E; Diez A, Eds. Elsevier: 1991; Vol. 43, pp 34–87. [Google Scholar]
- (63).Alkorta I; Elguero J Magn. Reson. Chem 2004, 42, 955–961. [DOI] [PubMed] [Google Scholar]
- (64).Trager W; Lee CM; Phillipson J; Haddock R; Dwuma-Badu D; Beckett A Tetrahedron 1968, 24, 523–543. [DOI] [PubMed] [Google Scholar]
- (65).Xu J; Shao L-D; Li D; Deng X; Liu Y-C; Zhao Q-S; Xia C J. Am. Chem. Soc 2014, 136, 17962–17965. [DOI] [PubMed] [Google Scholar]
- (66).Berner M; Tolvanen A; Jokela R, Acid-catalysed Epimerization of Bioactive Indole Alkaloids and Their Derivatives In Studies in Natural Products Chemistry, Atta ur R, Ed. Elsevier: 2001; Vol. 25, pp 3–42. [Google Scholar]
- (67).Laus G; Wurst K Helv. Chim. Acta 2003, 86, 181–187. [Google Scholar]
- (68).Wanner MJ; Ingemann S; van Maarseveen JH; Hiemstra H Eur. J. Org. Chem 2013, 2013, 1100–1106. [Google Scholar]
- (69).Reinhardt JK; Klemd AM; Danton O; De Mieri M; Smieško M; Huber R; Bürgi T; Gründemann C; Hamburger M J. Nat. Prod 2019, 82, 1424–1433. [DOI] [PubMed] [Google Scholar]
- (70).Bustos-Brito C; Joseph-Nathan P; Burgueño-Tapia E; Martínez-Otero D; Nieto-Camacho A; Calzada F; Yépez-Mulia L; Esquivel B; Quijano L J. Nat. Prod 2019, 82, 1207–1216. [DOI] [PubMed] [Google Scholar]
- (71).El-Kashef DH; Daletos G; Plenker M; Hartmann R; Mándi A; Kurtán T; Weber H; Lin W; Ancheeva E; Proksch P J. Nat. Prod 2019, 82, 2460–2469. [DOI] [PubMed] [Google Scholar]
- (72).Cao F; Meng Z-H; Mu X; Yue Y-F; Zhu H-J J. Nat. Prod 2019, 82, 386–392. [DOI] [PubMed] [Google Scholar]
- (73).Arreaga-González HM; Rodríguez-García G; del Río RE; Ferreira-Sereno JA; García-Gutiérrez HA; Cerda-García-Rojas CM; Joseph-Nathan P; Gómez-Hurtado MA J. Nat. Prod 2019, 82, 3394–3400. [DOI] [PubMed] [Google Scholar]
- (74).Raja HA; Baker TR; Little JG; Oberlies NH Food Chem 2017, 214, 383–392. [DOI] [PubMed] [Google Scholar]
- (75).Dunning LT; Savolainen V J. Linn. Soc., Bot 2010, 164, 1–9. [Google Scholar]
- (76).Ford CS; Ayres KL; Toomey N; Haider N; Van Alphen Stahl J; Kelly LJ; Wikström N; Hollingsworth PM; Duff RJ; Hoot SB J. Linn. Soc., Bot 2009, 159, 1–11. [Google Scholar]
- (77).Pawar RS; Handy SM; Cheng R; Shyong N; Grundel E Planta Med 2017, 83, 921–936. [DOI] [PubMed] [Google Scholar]
- (78).Cheng T; Xu C; Lei L; Li C; Zhang Y; Zhou S Mol. Ecol. Resour 2016, 16, 138–149. [DOI] [PubMed] [Google Scholar]
- (79).Löfstrand SD; Krüger Å; Razafimandimbison SG; Bremer B Syst. Bot 2014, 39, 304–315. [Google Scholar]
- (80).Razafimandimbison SG; Bremer B Am. J. Bot 2002, 89, 1027–1041. [DOI] [PubMed] [Google Scholar]
- (81).Tnah L; Lee S; Tan A; Lee C; Ng K; Ng C; Farhanah ZN Food Control 2019, 95, 318–326. [Google Scholar]
- (82).Raja HA; Miller AN; Pearce CJ; Oberlies NH J. Nat. Prod 2017, 80, 756–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.