

Abstract
Up to 30% of all breast cancer cases may be inherited and up to 85% of those may be due to segregation of susceptibility genes with low and moderate risk [odds ratios (OR) ≤ 3] for (mostly peri- and post-menopausal) breast cancer. The majority of low/moderate-risk genes, particularly those with minor allele frequencies (MAF) of < 30%, have not been identified and/or validated due to limitations of conventional association testing approaches, which include the agnostic nature of Genome Wide Association Studies (GWAS). To overcome these limitations, we used a hypothesis-driven integrative genomics approach to test the association of breast cancer with candidate genes by analyzing multi-omics data. Our candidate-gene association analyses of GWAS datasets suggested an increased risk of breast cancer with ERCC6 (main effect: 1.29 ≤ OR ≤ 2.91, 0.005 ≤ p ≤ 0.04, 11.8 ≤ MAF ≤ 40.9%), and implicated its interaction with ERCC8 (joint effect: 3.03 ≤ OR ≤ 5.31, 0.01 ≤ pinteraction ≤ 0.03). We found significant upregulation of ERCC6 (p = 7.95 × 10–6) and ERCC8 (p = 4.67 × 10–6) in breast cancer and similar frequencies of ERCC6 (1.8%) and ERCC8 (0.3%) mutations in breast tumors to known breast cancer susceptibility genes such as BLM (1.9%) and LSP1 (0.3%). Our integrative genomics approach suggests that ERCC6 may be a previously unreported low- to moderate-risk breast cancer susceptibility gene, which may also interact with ERCC8.
Subject terms: Cancer, Genetics, Systems biology
Introduction
Breast cancer is the most commonly-reported cancer and the second leading cause of cancer death among women in the United States (US). Approximately, 1 in 8 women in the US will develop breast cancer in their lifetime and 1 in 35 will die from the disease1. There will be an estimated 276,480 new cases of invasive breast cancer and 42,170 deaths due to breast cancer among US women in 20201. Breast cancer is a complex disorder with genetic and environmental factors playing synergistic roles in its etiology.
Up to 30% of all breast cancer cases in the general population may be due to inherited susceptibility factors2. About 15% of inherited breast cancer cases may be due to segregation of rare germline mutations [minor allele frequency (MAF) ≤ 1%] in high-penetrance [i.e., relative risk (RR) > 10] susceptibility genes such as BRCA1, BRCA2, TP53, and PTEN2,3. Medium-penetrance (3 ≤ RR ≤ 10) genes such as CHEK2, ATM, PALB2 (suggested by some as being high-penetrance4,5), BRIP1 (disputed as a breast cancer susceptibility gene by some6) have also been identified and account for about 8% of inherited breast cancers cases2. Nearly all high- and medium-penetrance genes were identified through family-based genetic epidemiologic investigations and gene resequencing7. Early (pre-menopausal) onset of the disease is a well-established hallmark of inherited breast cancers that are due to segregation of mutations in high- and medium-penetrance genes. Low-penetrance [RR or odds ratio (OR) < 3] genes, such as FGFR2, CASP8, MAP3K1 and LSP1, with MAF of about 30% in the general population and accounting for about 15% of inherited breast cancer cases have also been identified through genome-wide association studies (GWAS)7. Susceptibility genes accounting for the remaining of about 62% of inherited breast cancers (mostly believed to be low- to medium-penetrance genes leading to peri- or post-menopausal breast cancer) are yet to be identified and/or validated (Fig. 1).
Figure 1.

Breast cancer susceptibility.
Inherent limitations of GWAS, namely their agnostic nature necessitating stringent p-value thresholds, multiple-testing corrections, and exceedingly large sample size requirements for detecting small effects and gene–gene interactions, may be responsible for the apparent inability to identify and replicate the remainder of low- to medium-penetrance breast cancer susceptibility genes, particularly those with MAF < 30%. Integrative approaches enable extraction of deeper biological insights than what can be achieved through single-dimensional or conventional analyses. Inspired by our hypothesis-driven integrative genetic epidemiologic investigations of DNA repair disorders, which enabled us to decipher the biologic mechanisms that underlie the association between mutations in a subset of DNA repair genes and certain clinical outcomes associated with these disorders8,9, we designed a similar hypothesis-driven integrative approach to identify susceptibility genes and gene variants that influence the risk of common (i.e., peri- and post-menopausal) breast cancer. We report on the analysis of raw multiomics data from several relevant GWAS, transcriptome, and somatic mutation datasets to test the association of breast cancer with two candidate genes, ERCC6 and ERCC8.
Results
Candidate gene analysis of genome-wide association studies (GWAS) datasets
Cancer Genetic Markers of Susceptibility (CGEMS)
The demographic characteristics of subjects in the CGEMS10,11 dataset analyzed in our study (1089 cases, 1093 controls) are described in the Methods section and were published in our previous report12. All ERCC6 SNPs analyzed in CGEMS were in Hardy–Weinberg Equilibrium (HWE) and in strong Linkage Disequilibrium (LD) among the controls (Supplementary Fig. 1A). Both ERCC8 SNPs were also in HWE and their LD pattern among the controls is shown in Supplementary Fig. 1B.
Three ERCC6 SNPs, rs3750751 (NC_000010.11:g.49457882C > T), rs3750749 (NC_000010.11:g.49476182A > G), and rs4253082 (NC_000010.11:g.49509540C > T), were associated with an increased risk for breast cancer in CGEMS (Table 1). Statistically significant increased risks were found for homozygotes for the variant allele at rs3750751 (OR 2.91, 95% CI 1.05–8.06, p = 0.04), heterozygotes at rs3750749 (OR 1.40, 95% CI 1.07–1.82, p = 0.01), and heterozygotes at rs4253082 (OR 1.32, 95% CI 1.09–1.61, p = 0.005). The trend (i.e., increasing number of variant alleles) was also significant (OR 1.36, 95% CI 1.05–1.75, p = 0.02) for rs3750749 (Table 1).
Table 1.
ERCC6 and ERCC8 single nucleotide polymorphisms (SNPs) significantly associated with breast cancer in at least one breast cancer genome-wide association study (GWAS) dataset used as secondary data in our study.
| SNP | Alleles | Odds ratio (OR), 95% confidence interval (CI), p-value | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CGEMSa | NSABPb | WHIc | BPC3d | |||||||
| Case (N = 1089) | Control (N = 1093) | Case (N = 430) | Control (N = 822) | Case (N = 465) | Control (N = 1394) | Case (N = 977) | Control (N = 1026) | |||
| rs3750751 | 0 | GG | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| 1 | GA | 0.99 (0.79–1.25), 0.96 | 0.63 (0.45–0.88), 0.01 | NA | 1.35 (1.01–1.80), 0.04 | |||||
| 2 | AA | 2.91 (1.05–8.06), 0.04 | 2.53 (0.59–10.97), 0.21 | NA | 2.90 (0.52–16.07), 0.22 | |||||
| Trend | 1.10 (0.89–1.35), 0.37 | 0.74 (0.54–1.00), 0.05 | NA | 1.38 (1.05–1.81), 0.02 | ||||||
| rs2229760 | 0 | GG | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| 1 | GA | 1.08 (0.89–1.31), 0.43 | 1.40 (1.08–1.83), 0.01 | NA | 0.88 (0.70–1.09), 0.24 | |||||
| 2 | AA | 1.03 (0.80–1.33), 0.80 | 1.28 (0.90–1.81), 0.16 | NA | 1.15 (0.84–1.56), 0.39 | |||||
| Trend | 1.03 (0.91–1.16), 0.68 | 1.17 (0.99–1.38), 0.07 | NA | 1.02 (0.88–1.19), 0.76 | ||||||
| rs3750749 | 0 | TT | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| 1 | TC | 1.40 (1.07–1.82), 0.01 | 1.00 (0.69–1.46), 1.00 | NA | 0.86 (0.64–1.15), 0.31 | |||||
| 2 | CC | 1.06 (0.21–5.28), 0.94 | 0.40 (0.05–3.43), 0.40 | NA | 0.20 (0.02–1.73), 0.14 | |||||
| Trend | 1.36 (1.05–1.75), 0.02 | 0.94 (0.66–1.34), 0.74 | NA | 0.81 (0.61–1.07), 0.14 | ||||||
| rs4253082 | 0 | GG | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| 1 | GA | 1.32 (1.09–1.61), 0.005 | 1.04 (0.80–1.34), 0.79 | 1.27 (0.99–1.64), 0.06 | 1.24 (0.64–2.38), 0.52 | |||||
| 2 | AA | 0.79 (0.44–1.41), 0.43 | 0.81 (0.34–1.93), 0.64 | 0.98 (0.45–2.11), 0.96 | 1.27 (0.67–2.40), 0.46 | |||||
| Trend | 1.17 (0.99–1.39), 0.06 | 0.91 (0.71–1.16), 0.44 | NA | 1.05 (0.87–1.28), 0.59 | ||||||
| rs2228528 | 0 | GG | 1.00 | 1.00 | 1.00 | 1.00 | ||||
| 1 | GA | NA | 1.04 (0.80–1.35), 0.77 | 1.29 (1.01–1.66), 0.04 | 0.99 (0.79–1.24), 0.91 | |||||
| 2 | AA | NA | 0.82 (0.35–1.93), 0.64 | 1.02 (0.47–2.21), 0.95 | 0.82 (0.43–1.56), 0.55 | |||||
| Trend | NA | 1.00 (0.80–1.26), 0.98 | NA | 0.96 (0.79–1.16), 0.69 | ||||||
| rs1012553 | 0 | TT | 1.00 | 1.00 | 1 | 1.00 | ||||
| 1 | TA | NA | NA | 1.35 (1.07–1.71), 0.01 | NA | |||||
| 2 | AA | NA | NA | 1.13 (0.69–1.84), 0.63 | NA | |||||
| Trend | NA | NA | 1.20 (1.00–1.44), 0.05 | NA | ||||||
NA Not analyzed due to missing SNPs or subjects (i.e., cells containing 0 subjects).
aCancer Genetic Markers of Susceptibility10,11: Our analysis of raw data involved Caucasian women ≥ 55 years of age using unconditional logistic regression adjusting for family history of breast cancer.
bNational Surgical Adjuvant Breast and Bowel Project (NSABP) Prevention Trials (P-114 and P-213): Our analysis of raw data involved Caucasian women ≥ 50 years of age using conditional logistic regression maintaining matching criteria set by the original study investigators (i.e., age at trial entry, time in the study, history of lobular carcinoma in situ, and 5-year predicted breast cancer risk based on the Gail model).
cWomen's Health Initiative (WHI)15,16 Hormone Therapy Trials data was used to create a nested case–control dataset of women diagnosed with invasive breast cancer ≥ 50 years of age (N = 465) and healthy controls (N = 1394) frequency-matched to the cases based on age in 3:1 control to case ratio: Our analysis of raw data involved Caucasian women ≥ 50 years of age using unconditional logistic regression adjusting for family history of breast cancer, parity, oral contraceptive use, breast feeding and body mass index.
dBreast and Prostate Cancer Cohort Consortium (BPC3)17,18: Our analysis of raw data involved Caucasian women ≥ 50 years of age using unconditional logistic regression adjusting for family history of breast cancer.
Haplotype analysis revealed borderline significant association of breast cancer with ERCC6 Hap 5 (OR 1.30, 95% CI 1.00–1.69, p = 0.048) (Supplementary Table 2). Diplotype analysis revealed statistically-significant association of breast cancer with three diplotypes in ERCC6; these included 2/5 (OR 2.65, 95% CI 1.14–6.17, p = 0.024), 1/6 (OR 3.89, 95% CI 1.42–10.66, p = 0.008), and 4/4 (OR 2.92, 95% CI 1.04–8.22, p = 0.042) (Supplementary Table 2).
None of the ERCC8 SNPs, haplotypes or diplotypes were associated with a statistically significant increased risk of breast cancer in CGEMS. Joint effect analysis revealed increased risk of breast cancer with the ERCC8 1/1 and ERCC6 2/5 diplotype combination (OR 3.03, 95% CI 1.16–7.91, p = 0.024) and the ERCC8 1/1 and ERCC6 1/6 diplotype combination (OR 3.27, 95% CI 1.17, 9.18, p = 0.024) compared with the reference category (Table 2A). Gene–gene interaction analysis revealed statistically-significant interaction between ERCC6 and ERCC8 at the diplotype level (pinteraction = 0.010) (Table 2A).
Table 2.
Joint effect analysis of ERCC6 and ERCC8 diplotypes.
| ERCC8 | A. Cancer Genetic Markers of Susceptibility (CGEMS) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ERCC6 | ||||||||||||||||||
| Hap0/Hap0 | Hap2/Hap5 | Hap1/Hap6 | ||||||||||||||||
| Control | Case | Adjusted OR (95% CI) | p-Value | Control | Case | Adjusted OR (95% CI) | p-Value | Control | Case | Adjusted OR (95% CI) | p-Value | |||||||
| n | % | n | % | n | % | n | % | n | % | n | % | |||||||
| Hap1/Hap1 | 162 | 78.3 | 163 | 69.10 | 1 | 6 | 2.9 | 17 | 7.2 | 3.03 (1.16–7.91) | 0.024 | 5 | 2.4 | 16 | 6.8 | 3.27 (1.17–9.18) | 0.024 | |
| Hap1/Hap2 | 18 | 8.7 | 9 | 3.80 | 0.50 (0.22, 1.14) | 0.100 | 1 | 0.5 | 1 | 0.4 | 1.11 (0.07,17.89) | 0.943 | NA | NA | ||||
| Hap1/Hap3 | 9 | 4.3 | 15 | 6.40 | 1.72 (0.73, 4.05) | 0.217 | 1 | 0.5 | 2 | 0.8 | 1.92 (0.17,21.59) | 0.599 | NA | NA | ||||
| ERCC8 | B. Women’s Health Initiative (WHI) Hormone Therapy Trials | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ERCC6 | ||||||||||||||||||
| Hap0/Hap0 | Hap2/Hap4 | |||||||||||||||||
| Control | Case | Adjusted OR (95% CI) | p-Value | Control | Case | Adjusted OR (95% CI) | p-value | |||||||||||
| n | % | n | % | n | % | n | % | |||||||||||
| Hap0/Hap0 | 81 | 41.10 | 20 | 29.00 | 1 | 5 | 2.50 | 5 | 7.20 | 5.31 (1.22–23.09) | 0.026 | |||||||
| Hap0/Hap1 | 34 | 17.30 | 18 | 26.10 | 2.13 (0.95–4.77) | 0.066 | 2 | 1.00 | 1 | 1.40 | 2.23 (0.18–27.51) | 0.533 | ||||||
| Hap0/Hap2 | 25 | 12.70 | 10 | 14.50 | 2.06 (0.80–5.30) | 0.136 | 3 | 1.50 | 1 | 1.40 | 1.47 (0.14–15.84) | 0.749 | ||||||
| Hap0/Hap3 | 23 | 11.70 | 4 | 5.80 | 0.97 (0.28–3.33) | 0.965 | 3 | 1.50 | 3 | 4.30 | 5.43 (0.88–33.63) | 0.069 | ||||||
| Hap0/Hap4 | 13 | 6.60 | 1 | 1.40 | 0.25 (0.03–2.11) | 0.201 | NA | NA | NA | NA | NA | NA | ||||||
| Hap0/Hap5 | 7 | 3.60 | 5 | 7.20 | 5.09 (1.23–21.03) | 0.025 | 1 | 0.50 | 1 | 1.40 | 2.66 (0.15–46.84) | 0.505 | ||||||
| ERCC8 | C. Breast and prostate cancer cohort consortium (BPC3) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ERCC6 | ||||||||||||||||||
| Hap1/Hap1 | Hap5/Hap1 | |||||||||||||||||
| Control | Case | Adjusted OR (95% CI) | p-Value | Control | Case | Adjusted OR (95% CI) | p-value | |||||||||||
| n | % | n | % | n | % | n | % | |||||||||||
| Hap1/Hap1 | 55 | 25.82 | 50 | 27.78 | 1 | 29 | 13.62 | 9 | 5.00 | 0.33 (0.12–0.88) | 0.027 | |||||||
| Hap2/Hap1 | 53 | 24.88 | 63 | 35.00 | 1.45 (0.77–2.71) | 0.248 | 24 | 11.27 | 14 | 7.78 | 0.51 (0.21–1.23) | 0.136 | ||||||
| Hap2/Hap2 | 22 | 10.33 | 12 | 6.67 | 0.52 (0.20–1.40) | 0.197 | 7 | 3.29 | 13 | 7.22 | 1.64 (0.53–5.08) | 0.391 | ||||||
| Hap1/Hap3 | 5 | 2.35 | 5 | 2.78 | 1.28 (0.31–5.31) | 0.732 | 7 | 3.29 | 4 | 2.22 | 0.16 (0.02–1.42) | 0.100 | ||||||
| Hap1/Hap4 | 8 | 3.76 | 7 | 3.89 | 1.25 (0.30–5.16) | 0.754 | 3 | 1.41 | 3 | 1.67 | 0.65 (0.10–4.34) | 0.660 | ||||||
CGEMS: p-value for interaction = 0.010 (Wald Test, Chi-Square 19.98).
WHI: p-value for interaction = 0.034 (Wald Test, Chi-Square 31.66).
BPC3: p-value interaction = 0.047 (Wald Test, Chi-Square 21.21).
NA Not analyzed due to missing diplotypes or subjects (i.e., cell containing 0 subjects).
AUnconditional logistic regression analysis adjusted for family history of breast cancer.
BUnconditional logistic regression adjusted for family history of breast cancer, parity, oral contraceptive use, number of months of breast feeding, and body mass index.
CUnconditional logistic regression models adjusted for family history of breast cancer and consent group [i.e., three cohorts that make up our BPC3 dataset, namely Prostate, Lung, and Colorectal Cancer (PLCO), European Prospective Investigation into Cancer and Nutrition (EPIC), and Polish Breast Cancer Study (PBCS) and restricted to Caucasian ≥ 50 years of age.
National Surgical Adjuvant Breast and Bowel Project (NSABP)
The study design and subject characteristics of the NSABP13,14 dataset used in our study (430 cases, 822 controls) are described in the Methods section. All ERCC6 and ERCC8 SNPs analyzed in our NSABP dataset were in HWE and in strong LD among the controls (Supplementary Fig. 1C,D).
One ERCC6 SNP, rs2229760 (NC_000010.11:g.49472987G > A) was associated with an increased risk for breast cancer among heterozygotes (OR 1.40, 95% CI 1.08–1.83, p = 0.010) (Table 1). No statistically-significant increased risks were found with any ERCC8 SNPs or ERCC6 and ERCC8 Haplotypes and diplotypes (Supplementary Table 3). There were no significant interactions detected between ERCC6 and ERCC8 diplotypes in NSABP.
Women’s Health Initiative (WHI): Hormone herapy Trials
The study design and subject characteristics of the original WHI dataset15,16 and the subset dataset used in our study (465 cases and 1394 controls)12 have been published and are described in the Methods section. All ERCC6 and ERCC8 SNPs typed on these subjects were in HWE and in strong LD among the controls (Supplementary Fig. 1E,F). The most common haplotypes of ERCC6 and ERCC8 among the controls are also depicted in Supplementary Fig. 1E,F.
Statistically significant increased risk of breast cancer in WHI was found with heterozygotes at ERCC6 rs1012553 (NC_000010.11:g.49532097A > T) (OR 1.35, 95% CI 1.07–1.71, p = 0.01) and rs2228528 (NC_000010.11:g.49524234C > T) (OR 1.29, 95% CI 1.01–1.66, p = 0.04) (Table 1). Haplotype analysis revealed statistically-significant increased risk of breast cancer with ERCC6 Hap 2 (OR 1.36, 95% CI 1.03–1.78, p = 0.03) (Supplementary Table 4). Diplotype analysis revealed statistically-significant increased risk of breast cancer with diplotype 2/4 (OR 2.48, 95% CI 1.11–5.55, p = 0.03) (Supplementary Table 4).
None of the ERCC8 SNPs, haplotypes or diplotypes were associated with an increased risk of breast cancer at a statistically-significant level. Joint effect analysis revealed increased risk of breast cancer with the ERCC8 0/0 and ERCC6 2/4 diplotype combination (OR 5.31, 95% CI 1.22–23.09, p = 0.026) and the ERCC8 0/5 and ERCC6 0/0 diplotype combination (OR 5.09, 95% CI 1.23, 21.03, p = 0.025) compared with the reference category (Table 2B). Gene–gene interaction analysis revealed statistically-significant interaction between ERCC6 and ERCC8 at the diplotype level (pinteraction = 0.034) (Table 2B).
Breast and Prostate Cancer Cohort Consortium (BPC3)
The demographic characteristics of subjects in the BPC317,18 dataset used in our study (977 cases, 1026 controls) are described in the Methods section. All ERCC6 and ERCC8 SNPs typed on these subjects were in HWE and in strong LD among the controls (Supplementary Fig. 1G,H). The most common haplotypes for ERCC6 (Supplementary Fig. 1G) and ERCC8 (Supplementary Fig. 1H) among the controls are also depicted.
One ERCC6 SNP, rs3750751, was associated with a significantly increased risk (OR 1.35, 95% CI 1.01–1.80, p = 0.04) of breast cancer among heterozygotes in BPC3 (Table 1), and the trend (i.e., increasing number of variant alleles) was also significant (OR 1.38, 95% CI 1.05–1.81, p = 0.02). This SNP was associated with an even higher risk of breast cancer (OR 1.88, 95% CI 1.17–3.02, p = 0.009) in stratified analysis in one of the three cohorts that made up our BPC3 dataset, namely the Polish Breast Cancer Study (PBCS) (Table 3). Haplotype and diplotype analysis did not reveal any statistically-significant associations in the entire BPC3 dataset; however, when stratified by cohort, significant association of ERCC6 Hap 6 (OR 1.65, 95% CI 1.02–2.68, p = 0.04) with breast cancer was detected in PBCS (results not shown).
Table 3.
Stratified analysis of ERCC6 rs3750751 in the three cohorts of our Breast and Prostate Cancer Consortium (BPC3) dataset, namely Prostate, Lung, and olorectal Cancer (PLCO), European Prospective Investigation into Cancer and Nutrition (EPIC), and Polish Breast Cancer Study (PBCS).
| Study | SNP rs3750751 |
Alleles | Case (N = 255) | Case % | Control (N = 340) | Control % | OR (95% CI) | p-value |
|---|---|---|---|---|---|---|---|---|
| PLCO | GG | 211 | 82.75 | 283 | 83.24 | 1 | ||
| GA | 40 | 15.70 | 55 | 16.18 | 1.04 (0.66–1.63) | 0.87 | ||
| AA | 4 | 1.57 | 2 | 0.59 | 2.78 (0.50–15.39) | 0.25 | ||
| EPIC | Case (N = 368) | Case % | Control (N = 354) | Control % | ||||
| GG | 303 | 82.34 | 294 | 83.05 | 1 | |||
| GA | 65 | 17.66 | 58 | 16.38 | 1.25 (0.64–2.45) | 0.51 | ||
| AA | 0 | 0.00 | 2 | 0.56 | NA | NA | ||
| PBCS | Case (N = 354) | Case % | Control (N = 332) | Control % | ||||
| GG | 298 | 84.18 | 302 | 90.96 | 1 | |||
| GA | 56 | 15.82 | 30 | 9.04 | 1.88 (1.17–3.02) | 0.009 | ||
| AA | 0 | 0.00 | 0 | 0.00 | NA | NA |
Unconditional logistic regression models adjusted for family history of breast cancer and restricted to Caucasian subjects ≥ 50 years.
NA Not analyzed due to small number of subjects (cells with 0 value).
None of ERCC8 SNPs, haplotypes or diplotypes were associated with a statistically-significant increased risk of breast cancer (Supplementary Table 5). Gene–gene interaction analysis revealed borderline-significant interaction between ERCC6 and ERCC8 at the diplotype level (pinteraction = 0.047) (Table 2C).
Analysis of gene-expression microarray datasets
Individual analysis of raw data from two gene expression microarray datasets, one containing invasive ductal breast cancers (IDBC) and normal adjacent tissue, GSE1078019, and one from a case–control study of IDBC, E-TAMB-27620, revealed significant upregulation of ERCC6 and ERCC8. In GSE10780, upregulation of ERCC6 (fold change = 1.2, p = 1.5 × 10–5) and ERCC8 (fold change = 1.3, p = 1.8 × 10–8) was comparable to that of BRCA1 (fold change = 1.5, p = 1 × 10–15) (Fig. 2). Meta-analysis of GSE1078015,16 and E-TABM 27615,16 also revealed significant upregulation of ERCC6 (p = 7.95 × 10–6) and ERCC8 (p = 4.67 × 10–6).
Figure 2.

Individual analysis of gene expression microarray dataset GSE10780 containing invasive ductal breast cancer (IDBC) cases (n = 42) and control samples (n = 143) among peri- and post-menopausal women.
Analysis of TCGA dataset
Results of our analysis of TCGA21 (https://portal.gdc.cancer.gov/) breast tumor data revealed that 95% of genes mutated in breast tumors had mutation frequencies that were below 1% (Fig. 3a). All known breast cancer susceptibility genes, however, had mutation frequencies which were greater than 1%. We found that the mutation frequency of ERCC6 (1.8%) in breast tumors was similar to those of known breast cancer susceptibility genes, such as BRCA1 (2.9%), BRCA2 (2.9%), BLM (1.9%), FGFR2 (1.5%), and CHEK2 (1.0%) (Fig. 3a). The mutation frequency of ERCC8 (0.3%) in breast tumors was also similar to some of the other known breast cancer susceptibility genes such as STK11 (0.3%) and LSP1 (0.3%) (Fig. 3a).
Figure 3.
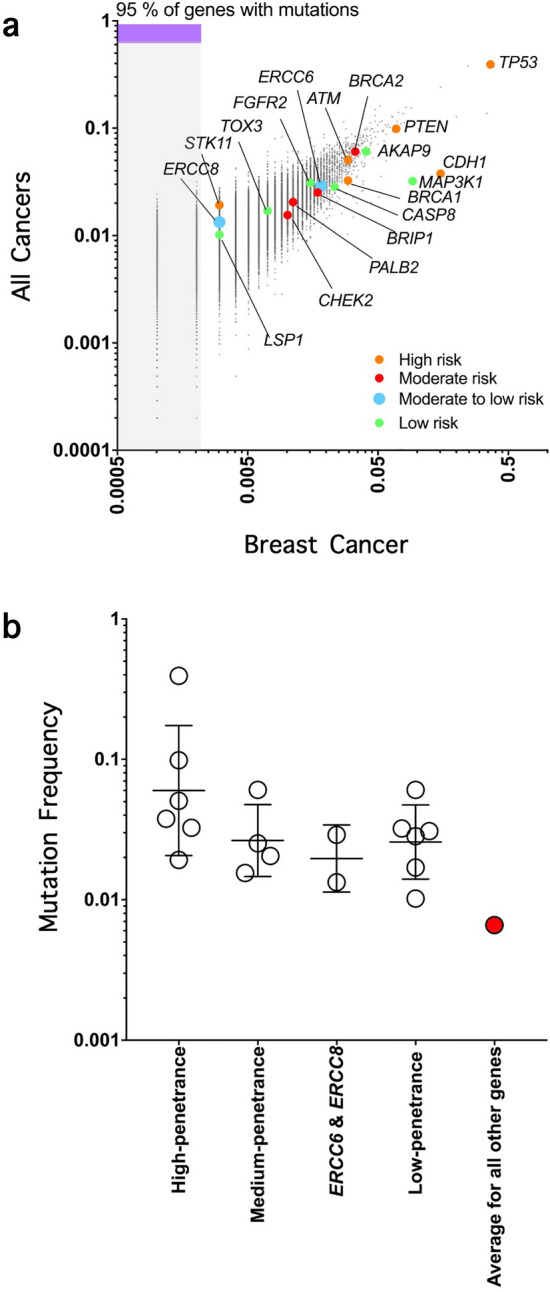
(a,b) Frequency of somatic mutations in ERCC6 and ERCC8 in comparison with known breast cancer susceptibility genes in the Cancer Genome Atlas (TCGA) Dataset. (a) Mutation frequency in all cancers versus breast cancer. (b) Mutation frequencies of breast cancer susceptibility genes in TCGA breast cancers.
Using TCGA breast tumor data, we compared mutation frequencies of ERCC6 and ERCC8 to those of breast cancer susceptibility genes with high-penetrance (BRCA1, BRCA2, TP53, PTEN, CDH1 and STK11), medium-penetrance (BLM, ATM, CHEK2, BRIP1, and PALB2), and low-penetrance (FGFR2, MAP3K1, CASP8, TOX3, and LSP1), and to the rest of the genome. This analysis revealed that high-penetrance breast cancer susceptibility genes had higher mutation frequencies than medium- and low-penetrance genes (Fig. 3b), which had similar mutation frequencies to each other and to ERCC6 and ERCC8 (Fig. 3b). Analysis of TCGA breast tumors also revealed a higher mutation frequency for all known breast cancer susceptibility genes and for ERCC6 and ERCC8 compared to the average for all other genes (Fig. 3b).
Using data on all cancers in TCGA, we compared the ratio of high-impact mutations (stop-gained, frame-shift variant, splice-acceptor variant, splice-donor variant, start-lost, stop-lost) to moderate-impact mutations (missense variant, in-frame deletion, in-frame insertion, protein-altering variant, splice-region variant, incomplete terminal codon variant) in ERCC6 and ERCC8 versus the known high-, medium- and low-penetrance breast cancer susceptibility genes versus a group of size-matched (i.e., matched based on gene length) control genes (i.e., genes not involved in breast cancer susceptibility, which included those coding for immunoglobulin and T-cell receptors as well as olfactory receptors). We identified 200 moderate-impact and 41 high-impact mutations in ERCC6 and similar numbers in ERCC8. The ratios of high- to moderate-impact mutations for ERCC6 (0.20) and ERCC8 (0.22) were similar to those for BRCA1 (0.30), BRCA2 (0.30), CHEK2 (0.28), and BLM (0.20) (Fig. 4a). Somatic mutation landscape of ERCC6, ERCC8, and the BRCA genes are depicted in Fig. 4b (obtained from https://www.cbioportal.org/), and show the location of two of our significant ERCC6 SNPs located in exons, rs2228528 and rs2229760, with respect to the reported mutations in TCGA.
Figure 4.

(a,b) Mutation analysis of selected genes in all cancers in the Cancer Genome Atlas (TCGA) dataset. (a) Ratio of high- to moderate-impact mutations in ERCC6 and ERCC8 in comparison to known breast cancer susceptibility genes and control genes. (b) Somatic mutation landscape of ERCC6, ERCC8, BRCA1 and BRCA2.
Discussion
Using a hypothesis-driven (candidate-gene) integrative genetic epidemiologic approach to analysis of raw multi-omics data, we tested the association of ERCC6 and ERCC8 with peri- and post-menopausal breast cancer. Our candidate-gene association study involving individual analysis of four GWAS datasets (containing total of 2882 cases and 4397 controls) found 30% to threefold increased risk of breast cancer (OR 1.30 with rs2228528 in WHI to OR 2.91 with rs3750751 in CGEMS) conferred by six ERCC6 variations. The MAF of these six ERCC6 variations ranged from 11.8 for rs3750751 to 40.9% for rs4838519 in the general Caucasian population [based on MAF reported in the NCBI (https://www.ncbi.nlm.nih.gov/snp/)].
Our findings were consistent at the SNP, haplotype and diplotype level within each dataset. In CGEMS, the associated ERCC6 haplotype, Hap 5, contained the variant alleles at two of the associated SNPs, rs3750749 and rs4253082. Furthermore, Hap 5 was one of the haplotype pairs in diplotype 2/5, which was associated with nearly threefold increase in risk. One of the other ERCC6 diplotypes associated with nearly threefold increase in risk in CGEMS, 4/4, contained the variant allele at the third associated SNP, rs375075. Similarly, the associated haplotype in the WHI, Hap 2, contained the variant alleles at the two associated SNPs, namely rs2228528 and rs1012553; Hap 2 was one of the haplotype pairs in diplotype 2/4, which was associated with 2.5-fold increased risk in this dataset. In BPC3, Hap 6 was the only haplotype associated with a borderline significant increased risk and was also the only common haplotype that contained the variant allele at the associated SNP, rs3750751.
The results of joint effect analyses were also compatible with main effect analyses within each dataset in that ERCC6 diplotypes with main effects were those highlighted in joint effects analyses. For example, in CGEMS, joint effect analysis highlighted diplotype 2/5, which had a significant main effect in diplotype analysis. Similarly, in WHI, joint effect analysis highlighted diplotype 2/4. Even though it is difficult to compare associated haplotypes and diplotypes across different datasets (due to different genotyping platforms and different SNPs typed in each dataset as well as LD between typed and untyped SNPs), consistency can also be noted across the analyzed datasets. One ERCC6 SNP (rs3750751) was significant in 2 of 3 GWAS where it was typed (this SNP is discussed in detail below).
In order to gain additional support for our findings, we conducted analyses of transcriptome and TCGA data. In general, somatic transcript levels and mutation frequencies do not necessarily parallel germline mutation profiles. Therefore, our findings from these analyses, although consistent with the results of our analyses of GWAS datasets, should be interpreted only as additional evidence. Analysis of transcriptome datasets showed statistically- significant upregulation of ERCC6 and ERCC8 in breast cancer, which is notable given the large sample size (total of 65 breast cancer samples and 153 normal tissue samples in the meta-analysis and 42 breast cancer samples and 143 normal tissue control samples in GSE10780 in individual analysis). The similarity of ERCC6 (and ERCC8) to BRCA1 in terms of both expression level and fold-change estimate is also noteworthy. BRCA1 levels are normally elevated in tumor biopsies from breast cancer patients who do not carry a germline mutation in the BRCA122. This is beleived to be due to the involvement of BRCA1 in cell cycle control and DNA repair processes23.
We also assessed protein expression levels of ERCC6 and ERCC8 in breast cancer using the Broad Institute proteome database (https://prot-shiny-vm.broadinstitute.org:3838/CPTAC-BRCA2016/) and found elevated expression of both, similar to some of our other comparison proteins such as CHEK2 (Data not shown).
Analysis of 522 TCGA breast cancers (with mutation information on 17,243 genes) also provided supporting evidence for involvement of ERCC6 as it revealed similar frequency of mutations and similar ratio of high- to low-impact mutations in ERCC6 and ERCC8 compared to known breast cancer susceptibility genes such as BRCA1, BRCA2, CHEK2, and BLM.
Although, the main effects of ERCC8 variations on breast cancer risk did not reach statistical significance in some of our GWAS datasets, we cannot rule out a main or modifying effect for ERCC8 given our findings of possible interaction between ERCC6 and ERCC8 in three of four GWAS analyzed. The findings from our analyses of transcriptome and TCGA datasets further suggest a potential main or modifying effect for ERCC8 in breast cancer susceptibility.
One ERCC6 SNP (rs3750751) was significant in 2 of 3 GWAS where it was typed. While heterozygotes at this SNP had 35% increased risk in BPC3 (and 88% in PBCS, 1 of 3 cohorts pulled together to form our BPC3 dataset), homozygotes for the variant allele at rs3750751 had close to threefold increased risk of breast cancer in CGEMS. The frequency of heterozygotes at ERCC6 rs3750751 ranged from 14 to 16% among the controls in our GWAS datasets. The homozygote variant frequency for this SNP was less than 1% among controls in our datasets. The minor allele frequency (MAF) of rs3750751 was 7.2–8.6% among the controls in our datasets (Supplementary Table 1) and has been reported as 11.8% in the general Caucasian US population (https://www.ncbi.nlm.nih.gov/snp/).
ERCC6 rs3750751 has not been previously implicated in breast cancer risk, however it has been reported to be associated with an increased risk of bladder cancer in one study24. rs3750751 lies in the 3′ untranslated region (3′-UTR), which has been proposed to be involved in post-transcription regulation of protein expression25. Recent studies implicate the role of micro RNA (miRNA)s in such regulation, suggesting that SNP variants located in 3′ UTRs may destroy or create miRNA binding sites26, thus influencing tumor susceptibility27. Each miRNA may be able to repress hundreds of gene targets post-transcriptionally, therefore, they are powerful regulators of gene expression28. miRNAs are involved in regulating a diverse set of biological processes including growth, differentiation and apoptosis29. The other ERCC6 variations associated with breast cancer in our study were rs3750749, rs4253082, and rs1012553, all located in the introns, and rs2228528 and rs2229760, which are missense variants in the exons. An interaction with smoking (pack-years) was noted for rs4253082 in one bladder cancer study30 and a possible association of rs2228528 with muscle-invasive bladder tumors was noted in another study31. To establish the role of these SNPs in cancer susceptibility and elucidate the underlying mechanism of carcinogenesis, functional assays are needed.
Analysis of different breast cancer GWAS to date have led to associations with 182 variants2,10,11,32–43, a few of which have been validated. One validated low-penetrance breast cancer susceptibility gene, FGFR210, has been found to be associated with 26% increased risk of breast cancer (OR 1.26, 95% CI 1.23–1.30) and has a MAF of > 30% in the general population. In comparison, our findings suggest 30% to threefold increased risk of breast cancer for ERCC6 variations with MAF of 11.8–40.9% in the general population. Lack of identification of ERCC6 in previous studies may be due to inherent limitations of GWAS and candidate gene studies. A major limitation of GWAS is that they are agnostic and exploratory, hence the high probability of false positive associations and the need for very large sample sizes for detection of weak main effects and gene–gene interactions. To alleviate the multiple-testing large false positive burden, stringent threshold levels for significance (i.e., p < 0.0001 for main effects and p < 10–8 for interactions) have been recommended for GWAS. This stringent threshold levels may lead to many true positive associations being missed. Candidate-gene approaches, which study association between variations in a small number of genes with the risk of disease, are more powerful since they are hypothesis-driven and have a lower probability of false-positive associations (hence, may be guided by conventional significance threshold levels). However, selection of suitable candidate genes is challenging and may be imprecise.
Selection of ERCC6 and ERCC8 as candidates for our integrative genomic study reported here was based on our previous genetic epidemiologic studies of DNA repair disorders44–46, which led to the hypothesis that genes involved in the nucleotide excision repair (NER) pathway may be involved in breast cancer susceptibility. ERCC6 and ERCC8 code for the main components of the transcription-coupled (TC) repair sub-pathway of the NER, which repairs damage to actively-transcribed regions of DNA47,48 caused by ultraviolet radiation, chemicals and free radicals49. Free radicals and reactive oxygen species (ROS) are associated with oxidative stress, a mechanism relevant to cancer. It has been observed that TC-NER is induced as the result of damage caused by oxidative stress, and that ERCC6 accumulates at sites of locally induced oxidative damage in vivo in a transcription-dependent manner50. It has also been observed that cells with mutations in ERCC6 and ERCC8 are sensitive to ROS51. The effect of ERCC6 and ERCC8 variants analyzed in our study on TC-NER is not known. However, given that TC-NER is a critical protective pathway against genotoxic agents52, it is plausible that certain variations in ERCC6 and ERCC8 may affect protein function, and repair efficiency of TC-NER, hence increase susceptibility to cancer.
In our study, we found some evidence for interaction between ERCC6 and ERCC8 diplotypes. Our findings of interaction are biologically plausible as these genes work together in the same biological pathway and sub-pathways. Our results suggest that TC-NER and global genome (GG)-NER may be good candidates for future pathway-based association studies of breast cancer.
The public health significance of this study stems from its potential to provide leads for inclusion of additional low- to moderate-risk breast cancer susceptibility genes (upon replication and proper validation in clinical studies) in the panel of susceptibility genes for risk scores41,53. Currently, genetic screening and counseling regarding personalized preventive and management strategies (such as increased surveillance, chemoprevention and prophylactic surgery) are available to individuals from families and ethnic groups with identified high- and medium-penetrance mutations3. Multigene panel tests for breast cancer exist and are offered to women who meet the criteria for clinical genetic testing such as early age of onset, bilateral and/or triple-negative disease, and a family history54. In theory, incorporation of additional low- and moderate-risk genes into multigene panel tests, following substantial validation and efficacy studies, may improve prevention strategies.
Strengths of our study include its unique integrative nature and uniform application of inclusion/exclusion factors to all GWAS and transcriptome datasets. One limitation of our study, which was related to the secondary nature of our datasets, was lack of information on individual estrogen receptor (ER) and progesterone receptor (PR) status in all datasets and inclusion of only ER-negative cases in BPC3. Stratifying by receptor status may modify the associations observed in our study or identify new associations. Nevertheless, the fact that our findings with respect to ERCC6 associations were consistent in all four datasets despite the differences in ER and PR status of the subjects further strengthens our results.
Another limitation of our study (also due to the secondary nature of our datasets) was lack of information on all risk-modifying environmental and reproductive/behavioral variables. We were limited by the variables which were provided to us as part of each dataset. While we had information on nearly all confounders in WHI, we were only provided with 5-year age categories and family history information in CGEMS. Despite this limitation, we utilized all information provided to us on all reproductive and behavioral variables in order to properly adjust our odds ratios for the effect of confounders. Our future studies to replicate and confirm these findings would need to involve primary data collection of pre-, peri- and post-menopausal breast cancer cases. Future studies would also need to validate and extend our findings to other populations and racial/ethnic groups.
Conclusions
Using a hypothesis-driven integrative genetic epidemiologic approach to analysis of multi-omics data, we propose that ERCC6 may be a previously-unreported low- to moderate-risk breast cancer susceptibility gene, and that it may interact with ERCC8. Our results suggest that NER may be a good candidate for future pathway-based association studies of breast cancer. Our findings have the potential to provide a deeper insight into the genetic basis of common breast cancer and, following proper replication and validation, generate leads for improved prevention methods.
Materials and methods
Candidate gene and single nucleotide polymorphism (SNP) selection
Based on our previous genetic epidemiologic studies of DNA repair disorders44–46, ERCC6 and ERCC852, were selected as part of a panel of candidate genes for association studies with breast cancer. All SNPs within ERCC6 and ERCC8 (including those in the upstream and downstream regulatory regions) which were typed in selected GWAS datasets (described below) in at least 80% of the subjects were used for analysis. After quality control exclusions, our roster of SNPs within the two candidate genes included 12 ERCC6 and 2 ERCC8 SNPs in CGEMS, 17 ERCC6 and 3 ERCC8 SNPs in NSABP, 14 ERCC6 and 8 ERCC8 SNPs in WHI, and 16 ERCC6 and 3 ERCC8 SNPs in BPC3. Information about these SNPs along with their chromosomal positions [obtained from the reference sequence in the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/snp/) Build 38.p12] and physical locations within the gene are listed in Supplementary Table 1.
Selection and statistical analysis of genome-wide association studies (GWAS) datasets
We selected case–control GWAS datasets which contained cases with invasive ductal breast cancer (IDBC) diagnosed peri- or post-menopause (≥ 50 years of age) and healthy controls (with no personal history of breast cancer) appropriately-matched to the cases based on a number of criteria by the original investigators. Race was restricted to Caucasian to avoid population stratification55.
Raw data from four GWAS datasets fitting our inclusion criteria were downloaded from the National Institutes of Health (NIH) after obtaining approvals from the corresponding data repository committees and stored on a computer and/or the server in RM’s laboratory at the University at Albany. The datasets analyzed in this study included the Cancer Genetic Markers of Susceptibility (CGEMS)10,11 (1089 cases, 1093 controls), a nested case–control (430 cases, 822 controls) within the National Surgical Adjuvant Breast and Bowel Project (NSABP)13,14, a nested case–control (465 cases and 1394 controls) within the Women's Health Initiative (WHI)15,16 Hormone Therapy Trials, and a nested case–control (977 cases, 1026 controls) within the Breast and Prostate Cancer Cohort Consortium (BPC3)17,18. Institutional Review Board (IRB) approval was obtained from University at Albany [by RM] for analyses of secondary datasets (Protocol #11-298 and #11-E-177) reported in this paper. Detailed information about each secondary dataset is provided below.
Raw data from CGEMS10,11, a case–control study nested within the Nurse’s Health Study (NHS) cohort containing genotype information at 528,178 single nucleotide polymorphism (SNP) loci on 1145 post-menopausal women of European ancestry with invasive breast cancer and 1,142 controls was downloaded from the NIH [accession #6175-13 for version phs000147/GRU]. Detailed information about the design and original analyses of CGEMS data is available from published reports33. For this dataset, we restricted the analysis to cases diagnosed at ≥ 55 years of age (N = 1089) and frequency-matched controls (N = 1093). Besides 5-year age categories, the only other phenotype variable available to us on these subjects was presence or absence of a first-degree family history of breast cancer.
Raw data from a nested case–control study within the PGRN-RIKEN Mayo National Surgical Adjuvant Breast and Bowel Project (NSABP) Prevention Trials (P-114 and P-213) were downloaded from the NIH (accession #10821-11 for version phs000305/APTC). This dataset contained genotype information at 601,273 SNPs on 592 breast cancer cases and 1171 controls of European (Caucasian) ancestry. We restricted our analysis to women diagnosed with breast cancer ≥ 50 years of age (N = 430) and appropriately-matched controls (N = 822). In NSABP13,14, controls were matched to cases based on several factors by the original study investigators; the matching factors included age at trial entry, time in the study, history of lobular carcinoma in situ, and 5-year predicted breast cancer risk based on the Gail model.
Raw data from the Women's Health Initiative (WHI)15,16 Hormone Therapy Trials containing genotype information at 1,051,295 SNPs [in Genomics and Randomized Trials Network (GARNET) subgroup of WHI] and 733,202 SNPs [in Women’s Health Initiative Memory Study (WHIMS) group] on a total of 10,634 subjects (4894 in GARNET and 5740 in WHIMS) was downloaded [NIH accession #11295-10 for version phs000200/HMB-IRB and #11296-10 for phs000200/HMB-IRB-NPU]. We created a nested case–control dataset of women diagnosed with invasive breast cancer ≥ 50 years of age (N = 465) and healthy controls (N = 1394) frequency-matched to the cases based on age in 3:1 control to case ratio. Variables provided to us on WHI subjects included a number of confounders of breast cancer risk such as family history of breast cancer, parity, oral contraceptive use, breast feeding, and body mass index, which we adjusted for in our analyses. The demographic characteristics of the 465 cases in our study and the entire control population that gave rise to the 1394 controls included in our analyses were published in our previous report12.
Raw data from the Breast and Prostate Cancer Cohort Consortium (BPC3)17,18 was downloaded from the NIH [accession #40019-3 for version phs000812/HMB-PUB-MDS, #40020-3 for phs000812/CADM, and #40021-3 for phs000812/DS-BOED-MDS]. The parent BPC3 GWAS included cases and controls from eight studies, but the subset BPC3 dataset which was made available to us contained genotype information at 550,000 loci on 1309 ER-negative breast cancer cases and 1351 appropriately-matched controls from three of the original BPC3 cohorts, namely the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, the European Prospective Investigation into Cancer and Nutrition (EPIC), and the Polish Breast Cancer Study (PBCS). We restricted our analysis to breast cancer cases diagnosed ≥ 50 years of age (N = 977) and appropriately-matched healthy controls (N = 1026) in this subset BPC3-PLCO/EPIC/PBCS dataset, referred to as BPC3 in this paper. Phenotype variables provided to us in this dataset included family history of breast cancer, which was adjusted for in our analyses.
For each gene, linkage disequilibrium (LD) patterns and deviations from Hardy–Weinberg Equilibrium (HWE) were assessed among the controls in each dataset using Haploview56 and a χ2 test of independence. Individual haplotypes and diplotypes (haplotype pairs) were determined for all subjects using PHASE57,57. Logistic regression models were used to calculate odd ratios (OR) and 95% confidence intervals (CI) for association between breast cancer and each SNP, haplotype and diplotype, while adjusting for covariates. Unconditional logistic regression was used for analyses of CGEMS, WHI and BPC3 data, while conditional logistic regression was used to analyze NSABP. The CGEMS, NSABP, and WHI datasets contained combination of both estrogen receptor (ER)-positive and -negative cases where as the BPC3 dataset made accessible to us contained ER-negative cases only. All datasets contained combination of progesterone (PR)-positive and –negative cases. The individual ER and PR status of cases were not made available to us for any of the GWAS datasets. Joint effects of ERCC6 and ERCC8 diplotypes on risk of breast cancer were examined and multiplicative interaction at the diplotype level was tested by assessing statistical significance of the interaction terms in the logistic regression models using the Wald test. All association testing was done using SAS version 9.3 (SAS Institute, Cary, NC) and SPSS version 24 (IBM SPSS Statistics).
Selection and statistical analysis of gene expression microarray datasets
Raw data from two gene expression microarray datasets, GSE1078019 and E-TABM 27620, containing normal and cancerous breast tissue, were downloaded from Gene Expression Omnibus (GEO) and Array Express, respectively, and used for meta-analysis. As per the inclusion criteria, both datasets ascertained cases of IDBC among peri- and post-menopausal Caucasian women. Control samples in GSE1078019 contained normal adjacent tissue from breast cancer cases. Controls in E-TABM 27620 had no personal history of breast cancer and contributed histologically normal breast tissue. Other inclusion criteria were presence of more than five samples per group separated by more than two standard deviations (SD) difference between the groups in Principle Component Analysis (PCA) using first two axes of the PCA. GSE10780 contained 42 IDBC cases and 143 control samples and E-TAMB-276 contained 23 IDBC cases and 10 healthy controls.
The data was geometric-mean normalized and analyzed using non-parametric T-test. Meta-analysis was conducted on GSE1078019 and E-TABM-27620 by calculating the p-values for each gene in each experiment using student’s t-test. P-values for genes with discordant fold differences between datasets were changed to “one” and Fisher’s method was used to calculate meta-analysis p-values. Fold changes were averaged. All statistical analyses of gene expression microarray data were done using Partek Genomics version 6.6 (Partek Inc., St. Louis, MO, USA).
Statistical analysis of the cancer genome atlas (TCGA) dataset
We sought to validate our findings by examining frequency of somatic mutations in The Cancer Genome Atlas (TCGA)21. We analyzed TCGA breast tumor data by comparing frequency of mutations in ERCC6 and ERCC8 with the frequencies in known breast cancer susceptibility genes and the rest of the genome. We also compared the ratio of high-impact mutations (stop-gained, frame-shift variant, splice-acceptor variant, splice-donor variant, start-lost, stop-lost) to moderate-impact mutations (missense variant, in-frame deletion, in-frame insertion, protein-altering variant, splice-region variant, incomplete terminal codon variant) in ERCC6 and ERCC8 versus several comparison groups in the TCGA. One comparison group included all known high-, medium- and low-penetrance breast cancer susceptibility genes. Another comparison set included a group of size-matched (i.e., matched based on gene length) control genes (i.e., genes not involved in breast cancer susceptibility, which included those coding for immunoglobulin and T-cell receptors as well as olfactory receptors). All analyses of the TCGA data were done through the TCGA portal21 (https://portal.gdc.cancer.gov/).
Additionally, we created the somatic mutation landscape of ERCC6, ERCC8, and the BRCA genes using https://www.cbioportal.org/) and assessed protein expression levels of ERCC6 and ERCC8 in breast cancer using the Broad Institute proteome database (https://prot-shiny-vm.broadinstitute.org:3838/CPTAC-BRCA2016/).
Supplementary information
Acknowledgements
We thank all Primary Investigators, study participants, research personnel and administrative staff at dbGaP, GEO and TCGA for access to the GWAS, transcriptome and proteome datasets used in our study. Technical support provided by UAlbany’s Information Technology Services was instrumental to the success of our study. We also appreciate technical assistance by Hector Martinez at UAlbany. This study was supported in part by a grant to the Center for Social and Demographic Analysis (CSDA) at University at Albany from National Institutes of Child Health and Human Development (NICHD), NIH (R24 HD044943) and a Seed Grant Faculty Research Award to RM from CSDA. Opinions, findings and conclusions expressed here are those of the authors and do not necessarily reflect the views of the funding agencies.
Author contributions
R.M. conceived the hypothesis and methodology of this integrative genetic epidemiologic investigation, supervised and/or helped with all data analyses, interpreted the findings, and drafted the manuscript. H.S.T. conducted statistical analysis of all GWAS datasets. N.Z. helped with initial analysis of the CGEMS and NSABP datasets, and with creation of the WHI subset dataset analyzed in our study. C.S. helped with preliminary individual analysis of gene expression microarray datasets. S.F. helped with the analysis of BPC3 dataset. A.D. contributed to the study design, conducted all individual and meta-analysis of gene expression microarray datasets, analyzed the TCGA data, and helped with interpretation of findings. All authors contributed to and/or approved the manuscript.
Data availability
All datasets analyzed in this study are available from the relevant NIH data repositories mentioned in the text as follows: Database for Genotypes and Phenotypes (dbGaP) (https://dbgap.ncbi.nlm.nih.gov/), GEO (https://www.ncbi.nlm.nih.gov/geo/), and TCGA (https://www.cancer.gov/tcga). All data analysis programs/software used in this study are outlined in the respective statistical analysis sub-sections of the “Materials and methods” and “Results” sections.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-77037-7.
References
- 1.American Cancer Society. Cancer Facts & Figures 2020. (American Cancer Society, Atlanta, 2020). [DOI] [PMC free article] [PubMed]
- 2.Wendt C, Margolin S. Identifying breast cancer susceptibility genes—A review of the genetic background in familial breast cancer. Acta Oncol. 2019;58(2):135–146. doi: 10.1080/0284186X.2018.1529428. [DOI] [PubMed] [Google Scholar]
- 3.Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science. 2014;343(6178):1466–1470. doi: 10.1126/science.1251827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkas K, Roberts J, et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014;371(6):497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3(9):1190–1196. doi: 10.1001/jamaoncol.2017.0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Easton DF, Lesueur F, Decker B, Michailidou K, Li J, Allen J, et al. No evidence that protein truncating variants in BRIP1 are associated with breast cancer risk: Implications for gene panel testing. J. Med. Genet. 2016;53(5):298–309. doi: 10.1136/jmedgenet-2015-103529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foulkes WD. Inherited susceptibility to common cancers. N. Engl. J. Med. 2008;359(20):2143–2153. doi: 10.1056/NEJMra0802968. [DOI] [PubMed] [Google Scholar]
- 8.Moslehi R, Mills JL, Signore C, Kumar A, Ambroggio X, Dzutsev A. Integrative transcriptome analysis reveals dysregulation of canonical cancer molecular pathways in placenta leading to preeclampsia. Sci. Rep. 2013;3:2407. doi: 10.1038/srep02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moslehi R, Ambroggio X, Nagarajan V, Kumar A, Dzutsev A. Nucleotide excision repair/transcription gene defects in the fetus and impaired TFIIH-mediated function in transcription in placenta leading to preeclampsia. BMC Genom. 2014;15:373. doi: 10.1186/1471-2164-15-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007;39(7):870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haiman CA, Chen GK, Vachon CM, Canzian F, Dunning A, Millikan RC, et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nat. Genet. 2011;43(12):1210–1214. doi: 10.1038/ng.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moslehi R, Freedman E, Zeinomar N, Veneroso C, Levine PH. Importance of hereditary and selected environmental risk factors in the etiology of inflammatory breast cancer: A case-comparison study. BMC Cancer. 2016;16:334. doi: 10.1186/s12885-016-2369-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: The NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. 2006;295(23):2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- 14.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, et al. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J. Natl. Cancer Inst. 1998;90(18):1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 15.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women's Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 16.Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women's Health Initiative randomized controlled trial. JAMA. 2004;291(14):1701–1712. doi: 10.1001/jama.291.14.1775. [DOI] [PubMed] [Google Scholar]
- 17.Hunter DJ, Riboli E, Haiman CA, Albanes D, Altshuler D, Chanock SJ, et al. A candidate gene approach to searching for low-penetrance breast and prostate cancer genes. Nat. Rev. Cancer. 2005;5(12):977–985. doi: 10.1038/nrc1754. [DOI] [PubMed] [Google Scholar]
- 18.Maas P, Barrdahl M, Joshi AD, Auer PL, Gaudet MM, Milne RL, et al. Breast cancer risk from modifiable and nonmodifiable risk factors among white women in the United States. JAMA Oncol. 2016;2(10):1295–1302. doi: 10.1001/jamaoncol.2016.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen DT, Nasir A, Culhane A, Venkataramu C, Fulp W, Rubio R, et al. Proliferative genes dominate malignancy-risk gene signature in histologically-normal breast tissue. Breast Cancer Res. Treat. 2010;119(2):335–346. doi: 10.1007/s10549-009-0344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng AS, Culhane AC, Chan MW, Venkataramu CR, Ehrich M, Nasir A, et al. Epithelial progeny of estrogen-exposed breast progenitor cells display a cancer-like methylome. Cancer Res. 2008;68(6):1786–1796. doi: 10.1158/0008-5472.CAN-07-5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao GF, Parker JS, Reynolds SM, Silva TC, Wang LB, Zhou W, et al. Before and after: Comparison of legacy and harmonized TCGA genomic data commons' data. Cell Syst. 2019;9(1):24 e10–34 e10. doi: 10.1016/j.cels.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Zhang J, Zhang Y, Deng Q, Liang H. Expression and mutations of BRCA in breast cancer and ovarian cancer: Evidence from bioinformatics analyses. Int. J. Mol. Med. 2018;42(6):3542–3550. doi: 10.3892/ijmm.2018.3870. [DOI] [PubMed] [Google Scholar]
- 23.Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95(11):866–871. doi: 10.1111/j.1349-7006.2004.tb02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michiels S, Laplanche A, Boulet T, Dessen P, Guillonneau B, Mejean A, et al. Genetic polymorphisms in 85 DNA repair genes and bladder cancer risk. Carcinogenesis. 2009;30(5):763–768. doi: 10.1093/carcin/bgp046. [DOI] [PubMed] [Google Scholar]
- 25.Matoulkova E, Michalova E, Vojtesek B, Hrstka R. The role of the 3' untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012;9(5):563–576. doi: 10.4161/rna.20231. [DOI] [PubMed] [Google Scholar]
- 26.Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3' untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68(20):8535–8540. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iuliano R, Vismara MF, Dattilo V, Trapasso F, Baudi F, Perrotti N. The role of microRNAs in cancer susceptibility. Biomed. Res. Int. 2013;2013:591931. doi: 10.1155/2013/591931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005;3(3):e85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan BM. microRNAs in cancer susceptibility. Adv. Cancer Res. 2017;135:151–171. doi: 10.1016/bs.acr.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Xing J, Dinney CP, Shete S, Huang M, Hildebrandt MA, Chen Z, et al. Comprehensive pathway-based interrogation of genetic variations in the nucleotide excision DNA repair pathway and risk of bladder cancer. Cancer. 2012;118(1):205–215. doi: 10.1002/cncr.26224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savina NV, Nikitchenko NV, Kuzhir TD, Rolevich AI, Krasny SA, Goncharova RI. The cellular response to oxidatively induced DNA damage and polymorphism of some DNA repair genes associated with clinicopathological features of bladder cancer. Oxid. Med. Cell Longev. 2016;2016:5710403. doi: 10.1155/2016/5710403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahmed S, Thomas G, Ghoussaini M, Healey CS, Humphreys MK, Platte R, et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat. Genet. 2009;41(5):585–590. doi: 10.1038/ng.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antoniou AC, Wang X, Fredericksen ZS, McGuffog L, Tarrell R, Sinilnikova OM, et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat. Genet. 2010;42(10):885–892. doi: 10.1038/ng.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447(7148):1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fletcher O, Johnson N, Orr N, Hosking FJ, Gibson LJ, Walker K, et al. Novel breast cancer susceptibility locus at 9q31.2: Results of a genome-wide association study. J. Natl. Cancer Inst. 2011;103(5):425–435. doi: 10.1093/jnci/djq563. [DOI] [PubMed] [Google Scholar]
- 36.Stacey SN, Manolescu A, Sulem P, Thorlacius S, Gudjonsson SA, Jonsson GF, et al. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat. Genet. 2008;40(6):703–706. doi: 10.1038/ng.131. [DOI] [PubMed] [Google Scholar]
- 37.Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, Cox DG, et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1) Nat. Genet. 2009;41(5):579–584. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 2010;42(6):504–507. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng W, Long J, Gao YT, Li C, Zheng Y, Xiang YB, et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat. Genet. 2009;41(3):324–328. doi: 10.1038/ng.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013;45(4):353–361. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yanes T, Young MA, Meiser B, James PA. Clinical applications of polygenic breast cancer risk: A critical review and perspectives of an emerging field. Breast Cancer Res. 2020;22(1):21. doi: 10.1186/s13058-020-01260-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michailidou K, Lindstrom S, Dennis J, Beesley J, Hui S, Kar S, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551(7678):92–94. doi: 10.1038/nature24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferreira MA, Gamazon ER, Al-Ejeh F, Aittomaki K, Andrulis IL, Anton-Culver H, et al. Genome-wide association and transcriptome studies identify target genes and risk loci for breast cancer. Nat. Commun. 2019;10(1):1741. doi: 10.1038/s41467-018-08053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moslehi R. Breast and ovarian cancer risk among heterozygotes for xeroderma pigmentosum nucleotide excision repair gene mutations. Curr. Oncol. 2009;16(5):93. [Google Scholar]
- 45.Moslehi R, Kumar A, Mills JL, Ambroggio X, Signore C, Dzutsev A. Phenotype-specific adverse effects of XPD mutations on human prenatal development implicate impairment of TFIIH-mediated functions in placenta. Eur. J. Hum. Genet. 2012;20(6):626–631. doi: 10.1038/ejhg.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moslehi R, Signore C, Tamura D, Mills JL, Digiovanna JJ, Tucker MA, et al. Adverse effects of trichothiodystrophy DNA repair and transcription gene disorder on human fetal development. Clin. Genet. 2010;77(4):365–373. doi: 10.1111/j.1399-0004.2009.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boetefuer EL, Lake RJ, Fan HY. Mechanistic insights into the regulation of transcription and transcription-coupled DNA repair by Cockayne syndrome protein B. Nucleic Acids Res. 2018;46(15):7471–7479. doi: 10.1093/nar/gky660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bradsher J, Auriol J, Proietti de Santis L, Iben S, Vonesch JL, Grummt I, et al. CSB is a component of RNA pol I transcription. Mol. Cell. 2002;10(4):819–829. doi: 10.1016/S1097-2765(02)00678-0. [DOI] [PubMed] [Google Scholar]
- 49.Spivak G. Nucleotide excision repair in humans. DNA Repair (Amst.) 2015;36:13–18. doi: 10.1016/j.dnarep.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014;15(7):465–481. doi: 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- 51.Carbone M, Arron ST, Beutler B, Bononi A, Cavenee W, Cleaver JE, et al. Tumour predisposition and cancer syndromes as models to study gene-environment interactions. Nat. Rev. Cancer. 2020;20:533–549. doi: 10.1038/s41568-020-0265-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karikkineth AC, Scheibye-Knudsen M, Fivenson E, Croteau DL, Bohr VA. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017;33:3–17. doi: 10.1016/j.arr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mavaddat N, Michailidou K, Dennis J, Lush M, Fachal L, Lee A, et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 2018;104:21–34. doi: 10.1016/j.ajhg.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Obeid EI, Hall MJ, Daly MB. Multigene panel testing and breast cancer risk: Is it time to scale down? JAMA Oncol. 2017;3(9):1176–1177. doi: 10.1001/jamaoncol.2017.0342. [DOI] [PubMed] [Google Scholar]
- 55.Barnholtz-Sloan JS, McEvoy B, Shriver MD, Rebbeck TR. Ancestry estimation and correction for population stratification in molecular epidemiologic association studies. Cancer Epidemiol. Biomark. Prev. 2008;17(3):471–477. doi: 10.1158/1055-9965.EPI-07-0491. [DOI] [PubMed] [Google Scholar]
- 56.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 57.Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 2005;76(3):449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68(4):978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets analyzed in this study are available from the relevant NIH data repositories mentioned in the text as follows: Database for Genotypes and Phenotypes (dbGaP) (https://dbgap.ncbi.nlm.nih.gov/), GEO (https://www.ncbi.nlm.nih.gov/geo/), and TCGA (https://www.cancer.gov/tcga). All data analysis programs/software used in this study are outlined in the respective statistical analysis sub-sections of the “Materials and methods” and “Results” sections.